

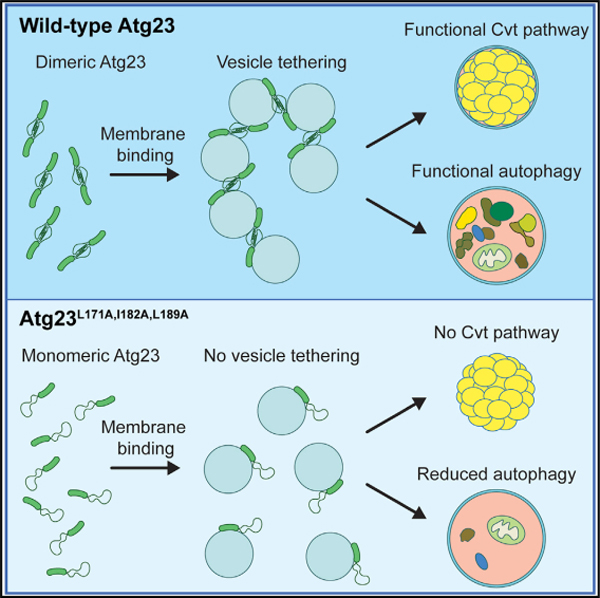
SUMMARY
Eukaryotes maintain cellular health through the engulfment and subsequent degradation of intracellular cargo using macroautophagy. The function of Atg23, despite being critical to the efficiency of this process, is unclear due to a lack of biochemical investigations and an absence of any structural information. In this study, we use a combination of in vitro and in vivo methods to show that Atg23 exists primarily as a homodimer, a conformation facilitated by a putative amphipathic helix. We utilize small-angle X-ray scattering to monitor the overall shape of Atg23, revealing that it contains an extended rod-like structure spanning approximately 320 Å. We also demonstrate that Atg23 interacts with membranes directly, primarily through electrostatic interactions, and that these interactions lead to vesicle tethering. Finally, mutation of the hydrophobic face of the putative amphipathic helix completely precludes dimer formation, leading to severely impaired subcellular localization, vesicle tethering, Atg9 binding, and autophagic efficiency.
In brief
Atg23 is an Atg9-interacting protein of unknown function required for selective autophagy and efficient nonselective autophagy. Hawkins et al. show that Atg23 is an extended dimeric membrane-binding protein capable of tethering vesicles.
Graphical Abstract
INTRODUCTION
Macroautophagy (hereafter referred to as autophagy) is an intracellular homeostasis and stress response process conserved across all eukaryotic cells. In autophagy, various cargoes, including proteins, nucleic acids, and/or entire organelles, are enveloped by an expanding membrane structure termed the phagophore, which eventually closes to form a complete double-membrane vesicle, termed the autophagosome (Bernard and Klionsky, 2013; Johansen and Lamark, 2011; Yorimitsu and Klionsky, 2005b). The outer autophagosomal membrane subsequently fuses with the vacuole in yeast (lysosome in mammals), releasing the inner membrane and its encapsulated contents into the degradative organelle to be digested into building blocks for reuse by the cell (Eskelinen et al., 2011; Mizushima et al., 2011). This process can be nonselective, as is often observed in times of starvation (nonselective autophagy; Reggiori et al., 2012), or discriminant, in which case a specific cargo is recognized by autophagy receptor proteins and enriched in the forming phagophore (selective autophagy; Gubas and Dikic, 2021; Zaffagnini and Martens, 2016).
Autophagy is a unique process in that the encapsulating vesicle is not the result of budding from an existing membrane but rather nucleates in a peri-vacuolar, cytosolic location (Mari et al., 2010). The membrane dynamics of autophagosome formation have been the focus of much research over the last decade, resulting in significant advancement of our understanding. We now know there to be several sources of lipids contributing to phagophore formation and expansion, including local lipid synthesis (Schutter et al., 2020) and lipid transfer from phagophore-adjacent membranes by Atg2 (Maeda et al., 2019; Osawa et al., 2019; Valverde et al., 2019). However, many questions remain around the longest-studied source of autophagosomal membrane: Atg9-containing vesicles. These vesicles are small (30–60 nm), single-membrane vesicles that contain the essential, transmembrane autophagy protein Atg9 and bud from peripheral membrane sources, including, but possibly not limited to, the Golgi apparatus (Mari and Reggiori, 2010; Yamamoto et al., 2012). These vesicles transit to the phagophore assembly site (PAS), where they fuse, contributing to the initial generation of the phagophore. It was originally thought that these vesicles were the primary source of membrane for autophagosomes, but it now appears that Atg9-containing vesicles contribute primarily to the early stages of phagophore nucleation and that other mechanisms are responsible for phagophore expansion, including lipid synthesis and transfer. Recent work has also demonstrated that Atg9 functions as a scramblase to move lipids from the outer to the inner leaflet of the phagophore, highlighting the essential role for Atg9 in phagophore expansion (Guardia et al., 2020; Maeda et al., 2020; Matoba et al., 2020). While much has been learned about the function of Atg9 in recent years, the transport and specific role of the vesicles that contain Atg9 are less well understood.
Atg23 is a peripheral membrane protein required for efficient nonselective autophagy and is essential for the cytoplasm-to-vacuole targeting (Cvt) pathway in yeast (Meiling-Wesse et al., 2004; Tucker et al., 2003). It is possible that Atg23 has a human homolog, but, if so, it has yet to be identified. Previous studies have shown that Atg23 binds Atg9 and that it plays a role in the accumulation of Atg9 at the peripheral sites that yield Atg9-containing vesicles (Backues et al., 2015; Legakis et al., 2007; Tucker et al., 2003). Deletion of ATG23 results in reduced trafficking of Atg9 to the PAS, but it is possible that this is an indirect consequence of impaired Atg9-containing vesicle formation (Yamamoto et al., 2012). Atg23 traffics between the PAS and numerous cytosolic peripheral sites, some, but not all, of which colocalize with Atg9 (Legakis et al., 2007; Yen et al., 2007). The cycling of Atg23 is dependent on the Atg1-Atg13 complex but, unlike the cycling of Atg9, is independent of the Atg2-Atg18 complex and the phosphatidylinositol 3-kinase complex I (Legakis et al., 2007; Reggiori et al., 2004). Despite these previous studies giving some insights into the behavior of Atg23, virtually no information has been reported on the structural or functional details of the protein. In this study, we reveal that Atg23 is an elongated homodimer and that its dimerization is mediated by a putative amphipathic helix. We also demonstrate that Atg23 binds directly to membranes and is capable of tethering vesicles in an Atg9-independent manner and with a significant contribution of electrostatic interactions. Finally, we demonstrate that membrane tethering, localization, and function of Atg23 in autophagy depend on its dimerization.
RESULTS
Bioinformatic analysis suggests that Atg23 is an α-helical protein with a putative coiled-coil domain
As there is currently no structure for Atg23, we initially used bioinformatics to gain insight into the predicted architecture of the protein. The secondary structure prediction server RaptorX Property (Wang et al., 2016) estimated Atg23 to be largely α-helical, with the C-terminal ~80 amino acids being disordered (Figure S1A). This disorder prediction aligns well with that of MFDp2 (Mizianty et al., 2013; Figure S1B). No predicted conserved domains could be found through homology searching; however, several coiled-coil predictors, including MultiCoil2 (Trigg et al., 2011), gave a high score for amino acids ~150–220 (Figure S1C). Coiled coils are composed of heptad repeats denoted as abcdefg, which feature conserved hydrophobic residues in positions a and d. The repetition of these conserved residues often results in an amphipathic helix, with both a hydrophobic (nonpolar) and hydrophilic (polar) face (Truebestein and Leonard, 2016). When the region containing amino acids 148–203 is viewed in a heptad repeat helical wheel, Atg23 displays two clear hydrophobic arms (in positions a and d; Figure S1D), suggesting that this region could potentially form a coiled-coil domain. Because several proteins involved in autophagy, such as Vps30/Atg6, Atg11, Atg16, Atg17, and the human homologs of these proteins, dimerize through coiled-coils or amphipathic helices (Chano et al., 2002; Fujioka et al., 2010; Li et al., 2012; Noda et al., 2012; Parkhouse et al., 2013; Ragusa et al., 2012; Suzuki and Noda, 2018; Yorimitsu and Klionsky, 2005a), we hypothesized that this region of Atg23 may facilitate homodimerization.
Atg23 is an extended helical homodimer
To test the hypothesis that Atg23 forms a dimer in vivo, we performed a coimmunoprecipitation (coIP) assay, where Atg23-MYC was expressed with either a protein A (PA)-encoding empty vector or Atg23-PA in the multiple knockout (MKO) background. The MKO strain lacks 25 genes encoding proteins involved in autophagy, including the endogenous ATG23 and ATG9 (Cao and Klionsky, 2008); Atg9 is the only known interacting partner of Atg23. Using this background reduces the likelihood that any interactions observed are indirectly mediated by another autophagy protein. We found that Atg23-PA was able to coimmunoprecipitate Atg23-MYC, suggesting that Atg23 does indeed dimerize in vivo (Figure 1A). To further characterize Atg23, we recombinantly expressed full-length Atg23 and characterized the protein using circular dichroism (CD). The CD spectrum of Atg23 contained two minima at 208 nm and 222 nm, characteristic of an α-helical protein and had 67.5% α-helical content (Louis-Jeune et al., 2012; Figure 1B). Proteins with primarily coiled-coil character contain a 222:208 ratio greater than 1.0 (Crooks et al., 2011). The 222:208 ratio for Atg23 was 0.97, suggesting that Atg23 likely contains some coiled-coil regions, which is in good agreement with our bioinformatic analysis. The melting curve collected at 222 nm indicated that Atg23 had two distinct unfolding events that occurred at 46°C and 61°C (Figure S2A). To determine whether recombinantly expressed and purified Atg23 is a dimer, we performed sedimentation analytical ultracentrifugation (AUC). Atg23 has a calculated monomeric molecular weight of 51.5 kDa, whereas purified Atg23 displayed a sedimentation coefficient of 3.5 by AUC corresponding to a molecular weight of 103.7 kDa (Figure 1C), indicating that Atg23 forms a dimer in solution.
Figure 1. Atg23 is an extended homodimer.
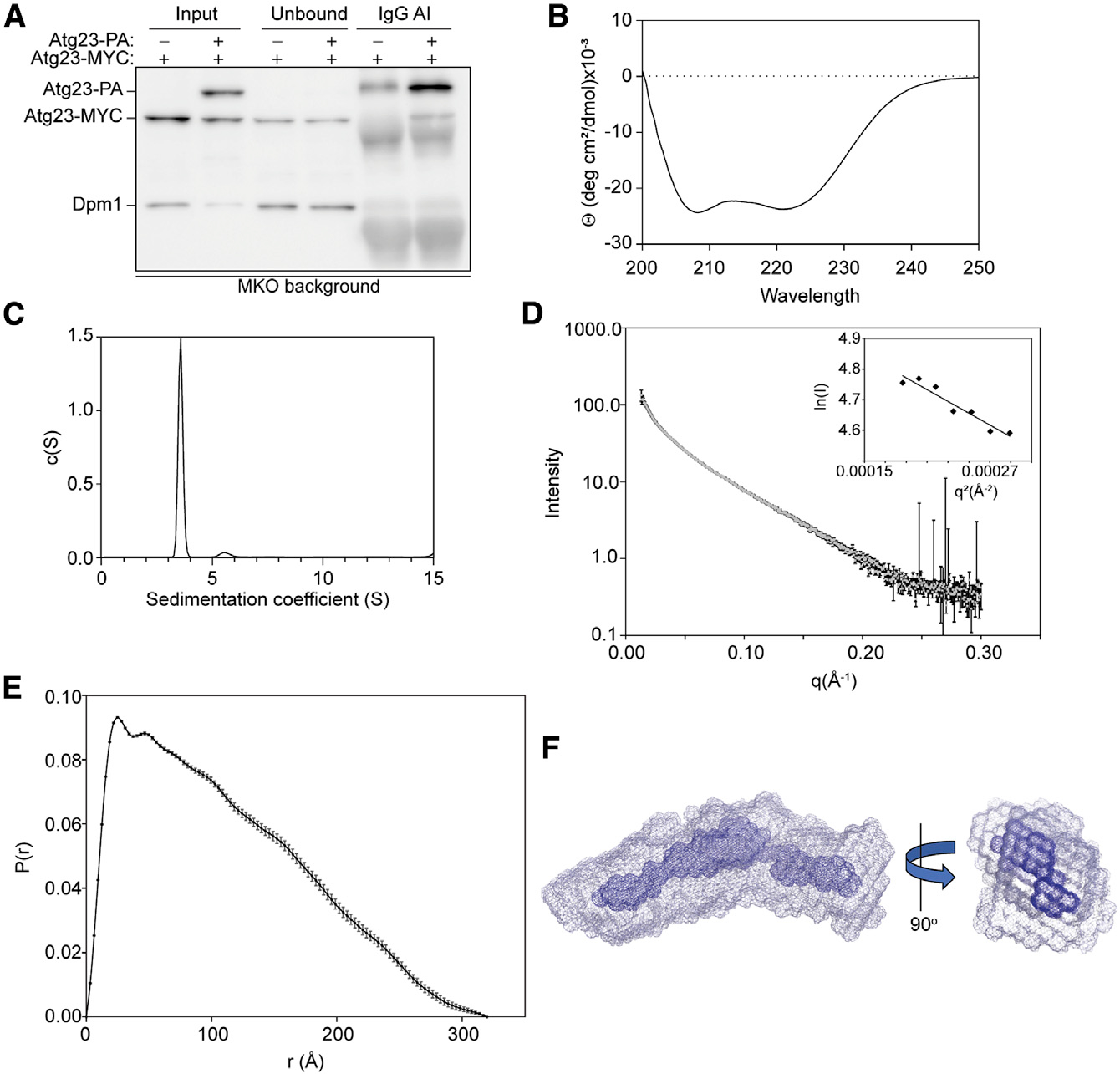
(A) Atg23-MYC with PA empty vector or Atg23-PA were overexpressed in the MKO cells under nutrient-rich conditions. Immunoglobulin G (IgG) Sepharose beads were used to affinity isolate PA. Proteins were visualized by western blot with a representative image shown.
(B) CD spectrum of Atg23 contains two minima at 208 and 222 nm.
(C) Sedimentation AUC for Atg23 (13.1 μM) yielded a single peak with a sedimentation coefficient of 3.5 and a molecular weight of 103.7 kDa.
(D) Averaged SAXS scattering curve from two independent SEC-SAXS experiments; Guinier analysis is inset. Error bars represent the standard error.
(E) A pair distance distribution function was calculated from the scattering curve in (D) using GNOM with a maximum dimension of 320 Å. Error bars represent the estimated errors from GNOM.
(F) Ten independent envelopes were calculated from the scattering curve in (D) using GASBOR. The 10 envelopes were aligned in SUPCOMB, averaged, and filtered using DAMAVER. The averaged envelope (gray) and filtered envelope (blue) are shown as mesh representations, with a 90° rotation (right).
To determine the overall shape of Atg23 and further confirm its oligomeric state, we performed two independent size-exclusion chromatography small-angle X-ray scattering (SEC-SAXS) experiments coupled with multi-angle light scattering (MALS). Because the molecular weight determined from sedimentation AUC experiments is influenced by the shape of the protein, we also used the MALS to determine a shape-independent molecular weight of Atg23. Using the MALS data, Atg23 has a molecular weight of 116.3 kDa, further confirming that it is a dimer (Figure S2B). The two independent SAXS experiments were in excellent agreement, and the final scattering curve was the average of both repeats (Figure 1D). Inspection of the low-scattering-angle Guinier plot revealed that Atg23 was monodispersed in solution with no detectable aggregation (inset of Figure 1D). The Kratky plot of Atg23 shows that the protein is primarily folded (Figure S2C). From the pair distance distribution function, Atg23 had a maximum dimension of 320 Å, while the shape most closely agreed with that of an extended rod-like structure (Figure 1E). Ten independent low-resolution envelopes were generated ab initio using GASBOR (Svergun et al., 2001). The average χ2 of the fits from the envelopes was 1.28 ± 0.16, indicating that the envelopes are in good agreement with the experimental scattering data. The 10 envelopes were aligned, averaged (gray), filtered (blue), and shown as mesh representations (Figure 1F), revealing that Atg23 is extended with a slight bend in the middle. The averaged envelope from DAMAVER (Volkov and Svergun, 2003) represents the total spread of all 10 envelopes, whereas the filtered model from DAMFILT (Volkov and Svergun, 2003) contains only the common features found within all 10 envelopes. It is important to note that the SAXS envelope generated in the absence of symmetry is consistent with a symmetric homodimer. Taken together, our in vivo and in vitro data indicate that Atg23 forms an extended homodimer.
Dimerization of Atg23 is perturbed by mutation of the predicted amphipathic helix
To test the hypothesis that the hydrophobic face of the putative amphipathic helix facilitates homodimerization, we generated the Atg23L171A,I182A,L189A mutant (Atg23[LIL]), in which two conserved leucines and one isoleucine on the hydrophobic face were mutated to alanines (Figures 2A and S1D). If these residues are involved in a coiled-coil-mediated dimerization interface, then we reasoned that these substitutions would have minimal impact on the overall tertiary structure of Atg23 while still disrupting any hydrophobic interactions formed at this putative dimerization interface. By coIP, we found that Atg23[LIL]-MYC was not affinity isolated with Atg23-PA, suggesting that mutation of the hydrophobic face of the putative amphipathic helix completely disrupted this self-interaction (Figure 2B). In support of this finding, Atg23[LIL] from MKO cell lysates ran lower by native PAGE than both Atg23 from MKO lysates and purified Atg23 (Figure 2C). Because Atg23 from the MKO strain migrated at the same position as purified Atg23, we reasoned that wild-type (WT) Atg23 from lysates was in the dimeric state and unbound to any other proteins. Thus, Atg23[LIL] likely runs faster by native PAGE due to an inability to dimerize.
Figure 2. Atg23[LIL] is a monomer.
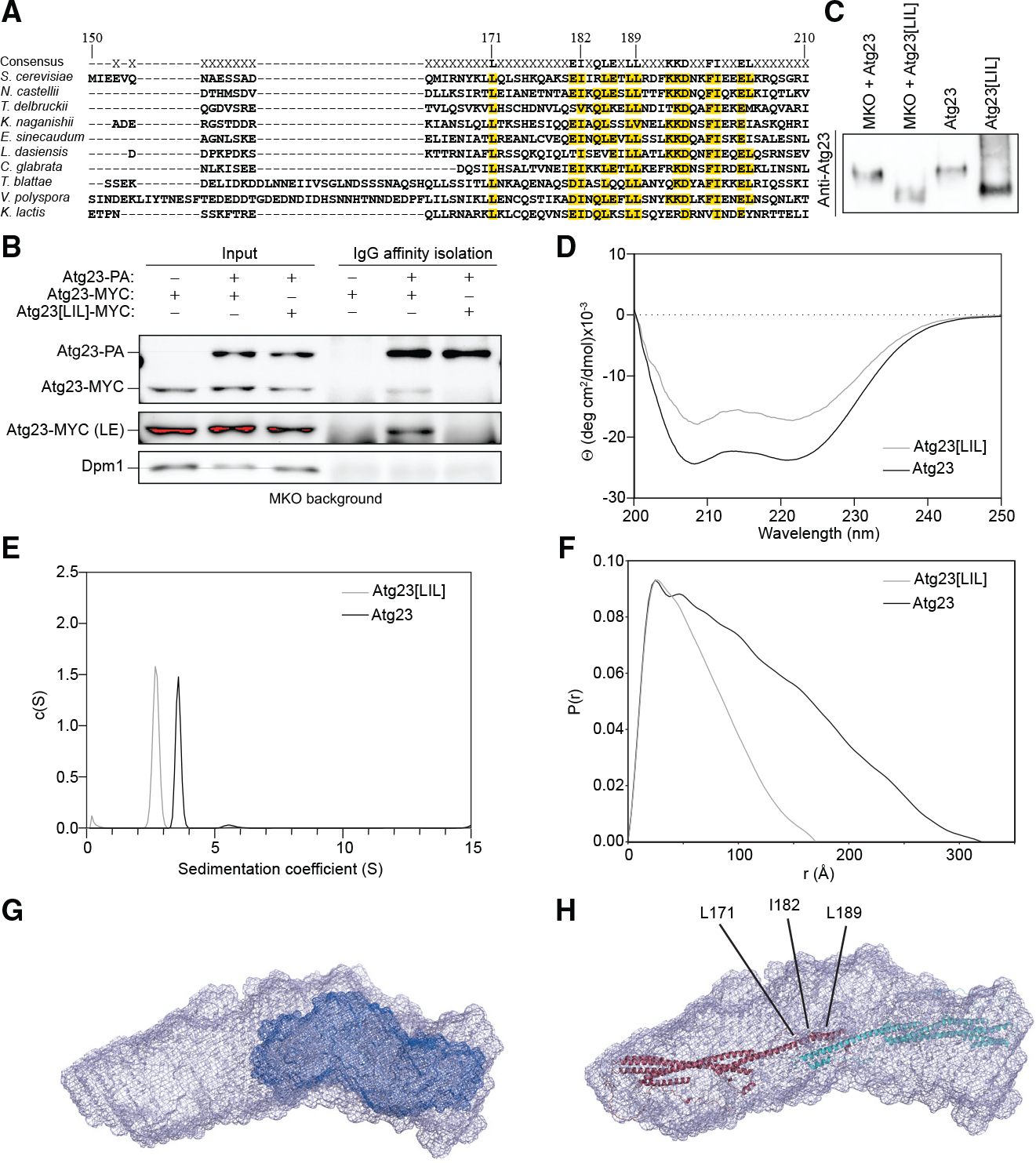
(A) Alignment of ScAtg23 amino acids 150–210 with nine Atg23 yeast species. Amino acids with greater than 50% conservation are marked in yellow. Residues mutated in Atg23[LIL] are marked.
(B) Atg23-MYC and PA empty vector, Atg23-MYC and Atg23-PA, or Atg23[LIL]-MYC and Atg23-PA were overexpressed in the MKO background under nutrient-rich conditions. IgG Sepharose beads were used to affinity isolate PA. Proteins were visualized by western blot with a representative image shown. A long exposure (LE) image was also collected.
(C) Yeast cells were grown in nutrient-rich medium, converted to spheroplasts, and lysed in native sample buffer. The resulting lysates were run on a 6% non-denaturing polyacrylamide gel. Western blotting was performed with an Atg23-specific antiserum. Purified Atg23 and Atg23[LIL] were run in lanes three and four, respectively.
(D) Overlayed CD spectra of Atg23 and Atg23[LIL]. The spectra contain two minima at 208 and 222 nm.
(E) Overlay of the sedimentation AUC for Atg23 (13.1 μM) and Atg23[LIL] (14.1 μM). Atg23[LIL] exists as a monomer with a sedimentation coefficient of 2.7 and a molecular weight of 50.9 kDa.
(F) Pair distance distribution function of Atg23 and Atg23[LIL] with maximum dimensions of 320 Å and 170 Å, respectively.
(G) Low-resolution envelopes of Atg23[LIL] (dark blue) overlayed with Atg23 WT.
(H) The AlphaFold model of Atg23 was docked using HADDOCK with L171, I182, and L189 as dimer constraints (labeled). The model was then placed in the SAXS envelope of Atg23. The two monomers are colored in red and cyan.
To verify that the Atg23[LIL] mutation specifically disrupts the dimerization interface of Atg23 and does not simply result in complete misfolding of Atg23, we purified Atg23[LIL] to high purity assessed by SDS-PAGE (Figure S3A). By size-exclusion chromatography, Atg23[LIL] eluted from a Superdex 200 column at 75 mL, whereas Atg23 eluted at 61 mL, suggesting that Atg23 [LIL] is a smaller species (Figure S3B). The CD spectrum of the Atg23[LIL] had an α-helicity of 42.3%, compared with 67.5% for the WT protein, demonstrating that the LIL mutation led to a modest reduction in secondary structure (Figure 2D). Melting curves collected at 222 nm showed that Atg23[LIL] had melting temperatures of 46°C and 60°C (Figure S3E). AUC indicated that Atg23[LIL] was monomeric with a sedimentation coefficient of 2.7 and a molecular weight of 50.9 kDa (Figure 2E). In addition, recombinant Atg23[LIL] ran at the same location by native-PAGE as Atg23[LIL] from MKO cells, demonstrating that the recombinant protein and the protein expressed in cells behaved similarly (Figure 2C).
To better determine the effects of the LIL mutation on the overall shape of Atg23, we performed SEC-SAXS with MALS. Atg23[LIL] had a molecular weight of 55.85 kDa by MALS, further confirming that Atg23[LIL] was a monomer (Figure S3F). Inspection of the low scattering Guinier region revealed that the mutant was monodispersed with no observed aggregation (Figures S3G and S3H). The pair distance distribution function of Atg23[LIL] has a maximum dimension of only 170 Å, corresponding to roughly half the length of the WT protein (Figure 2F). Low-resolution ab initio envelopes were generated for Atg23[LIL], which confirmed that Atg23[LIL] (dark blue) was roughly half the size of the WT protein (Figure 2G). The observation that Atg23[LIL] is half the length of the WT protein suggests that the coiled-coil region mediating dimerization is antiparallel.
To gain insight into the possible tertiary structure of Atg23 and how these mutations might affect its stability, we took advantage of the model of Atg23 produced using the recently released AlphaFold Protein Structure Database (Jumper et al., 2021). The Atg23 model was generated as a monomer, which is similar in size to Atg23[LIL] (Figure S3I). Intriguingly, L171, I182, and L189 all sit toward the end of a long α helix that is not packed against any other secondary structure (Figure S3J). To model the Atg23 dimer, we used two copies of the Atg23 AlphaFold model and docked them using HADDOCK (Honorato et al., 2021; van Zundert et al., 2016) with L171, I182, and L189 as constraints for the dimer interface. The top score of this docking resulted in an extended structure that was consistent with our SAXS data for Atg23 (Figure 2H). It is possible that, when these residues are mutated, this leads to not only a loss in dimerization but also a loss in the formation of this helix, which would be consistent with our CD data for Atg23 [LIL]. Based on the in vivo and in vitro data, we concluded that a mutation of the hydrophobic face of the predicted amphipathic helix directly disrupts Atg23 dimerization and displays a reduction in secondary structure but does not completely disrupt the tertiary fold of Atg23.
Atg23 binds membranes directly via electrostatic interactions
Atg23 partially associates with membranes in vivo (Figure S4), but it is not known whether this interaction is direct or facilitated by its interaction with the transmembrane protein Atg9. To test this, we performed subcellular fractionation at various salt concentrations with an MKO strain (i.e., without Atg9) expressing 6xHis-Atg23. In this assay, membranes and any proteins associated with them are pelleted, while soluble, cytosolic proteins remain in the supernatant. Atg23 was still able to associate with membranes in the absence of Atg9, and its interaction with membranes was heavily dependent on ionic strength (Figures 3A and 3B). To verify that Atg23 can directly bind membranes, we tested Atg23 membrane binding in vitro by performing liposome sedimentation assays using yeast polar lipids (YPLs) at various salt concentrations. Mirroring our in vivo subcellular fractionation results, we found that Atg23 could associate with liposomes and that this association was heavily dependent on the ionic strength of the buffer, even more so than in vivo (Figures 3C and 3D). To further explore the membrane binding preferences of Atg23, we performed liposome sedimentation with YPL liposomes of various sizes from approximately 1 μm to 72 nm in diameter. At 50 mM NaCl, Atg23 bound at similar levels to liposomes from multilamellar vesicles (MLVs) as it did to small unilamellar vesicles (SUVs), demonstrating that Atg23 was not significantly sensitive to the curvature of the membrane to which it was binding (Figures 3E and 3F).
Figure 3. Atg23 is a membrane-binding protein.
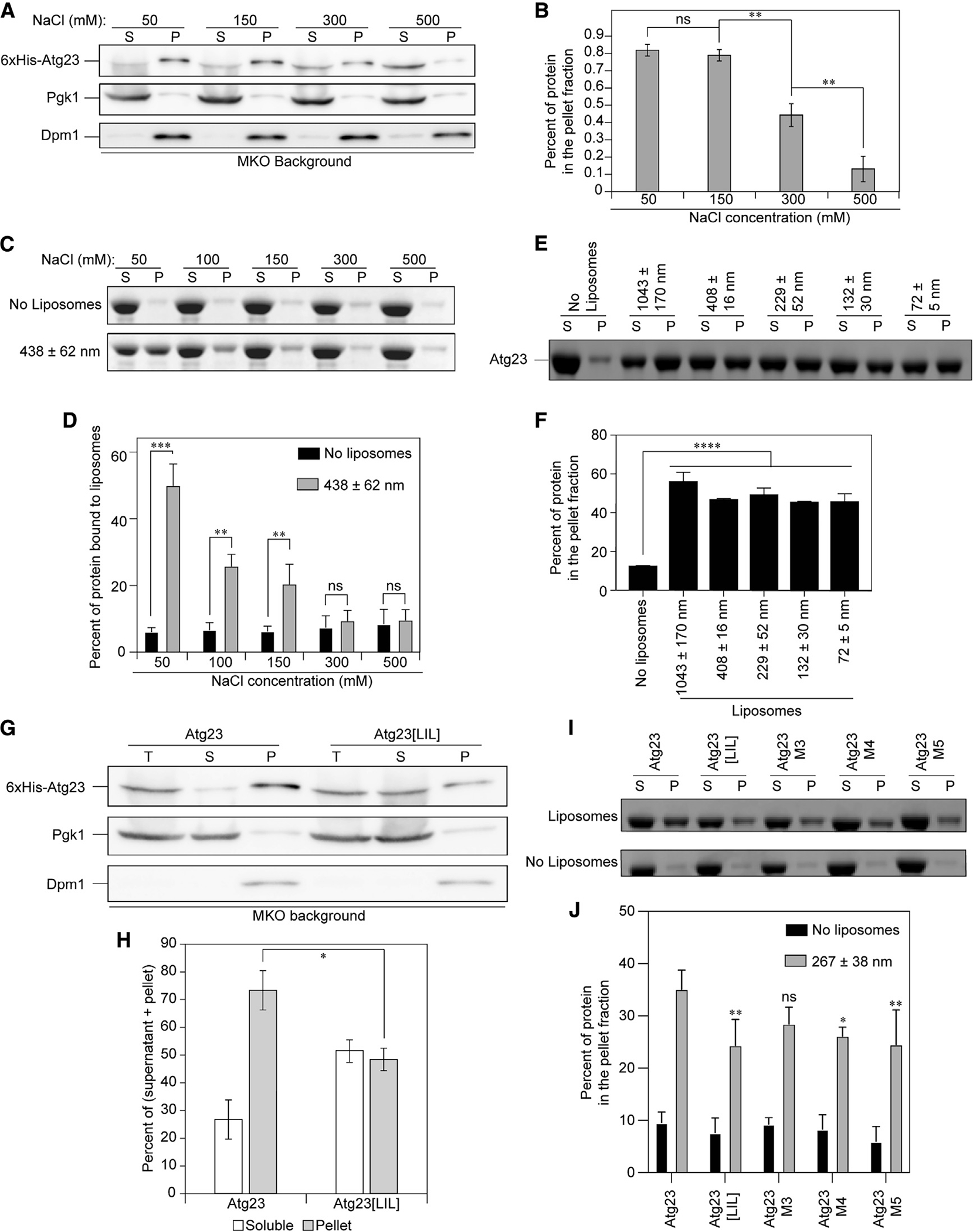
(A) 6xHis-Atg23 was overexpressed in the MKO cells and analyzed by subcellular fractionation in the presence of varying salt concentrations. “S” represents the supernatant and “P” the pellet. Pgk1 is included as a soluble protein control and the transmembrane protein Dpm1 as a membrane control.
(B) Quantification of (A). Error bars show standard error of the mean (SEM). n = 3; ns, not significant; **p < 0.01 by Tukey honestly significant difference (HSD) test.
(C) Liposome sedimentation assay with Atg23 and YPL liposomes at the indicated NaCl concentrations. Representative SDS-PAGE gels containing the S and P fractions are shown.
(D) The percent of protein in the pellet fraction in (C) was quantified by densitometry. Error bars represent the SD from three experiments. Statistical significance was determined by two-way ANOVA with Sidak’s multiple comparison test. ***p < 0.001; **p < 0.01.
(E) Liposome sedimentation assay with Atg23 and YPL liposomes of the indicated sizes in 50 mM NaCl. Representative gels containing the S and P fractions are shown.
(F) Quantification of (E) by densitometry. Error bars represent the SD from three experiments. Statistical significance was determined by ordinary one-way ANOVA with Tukey’s multiple comparison test. ****p < 0.0001 to the no-liposome control.
(G) 6xHis-Atg23 and 6xHis-Atg23[LIL] were overexpressed in the MKO cells and analyzed by subcellular fractionation. Total (T) represents the input prior to centrifugation.
(H) Quantification of (G) from five biological replicates. Error bars represent SEM; *p = 0.016 by two-sample t test.
(I) Liposome sedimentation assays with YPL liposomes and Atg23, Atg23[LIL], M3, M4, or M5 at 100 mM NaCl. Representative gels containing the S and P fractions are shown.
(J) Quantification of (I) by densitometry. Error bars represent the SD from four experiments. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparison test. **p < 0.01; *p < 0.05.
Given that Atg23 is capable of binding membranes directly, we questioned whether disrupting dimerization would have an impact on membrane binding. Extending our membrane fractionation assays to examine Atg23[LIL] revealed that membrane binding by the Atg23[LIL] mutant was significantly reduced compared with the WT but that it still retained some membrane-binding activity (Figures 3G and 3H). To verify these results in vitro, we performed liposome sedimentation assays with Atg23 and Atg23[LIL] at both 50 mM and 100 mM NaCl. Atg23[LIL] showed reduced liposome binding compared with the WT protein at both salt concentrations but still retained some ability to bind liposomes, which is in good agreement with our in vivo membrane binding data (Figures 3I, 3J, S5A, and S5B).
Mutation of positively charged residues maintains dimerization but reduces membrane binding
To disentangle Atg23 dimerization and membrane binding, we designed a series of mutants that we hypothesized would not disrupt dimerization while potentially disrupting membrane binding. Because our data suggested that the interaction between Atg23 and membranes is largely dependent on electrostatic interactions, we mutated a series of positively charged amino acids that are predicted to be on the surface of Atg23 based on the AlphaFold-predicted structure. The mutants we generated included R166E, K169E, and K176E (M1); R190E, K193E, and K194E (M2); K66E and R67E (M3); K66E, R67E, and K296E (M4); and K66E, R67E, R74E, R85E, and K296E (M5; Figure S5E). Subcellular fractionation experiments showed modest reductions in membrane association by M3, M4, and M5 in vivo (Figures S5C and S5D) and as such were pursued further in vitro. Atg23 M3, M4, and M5 were each purified and found to be dimeric based on AUC and folded based on CD (Figures S3A–S3D). Atg23 M3, M4, and M5 grew increasingly less thermostable as additional mutations were added (Figure S3E). Each of the mutants were tested for membrane binding using the liposome sedimentation assay at 50 mM and 100 mM NaCl (Figures 3I, 3J, S5A, and S5B). Atg23 M4 and M5 both exhibited reduced binding relative to WT Atg23 at both 50 mM and 100 mM NaCl. This result suggests that the mutations in Atg23 M4 and M5 lead to a partial disruption of the membrane-binding site while still retaining Atg23 dimerization.
Mutation of the amphipathic helix disrupts Atg23 cellular localization and Atg9 organization
After having identified L171A, I182A, and L189A as critical residues for dimerization, we wanted to identify the minimal mutation to fully disrupt Atg23 dimerization. Thus, single L189A (Atg23[L]) and double I182A, L189A (Atg23[IL]) point mutants were generated and coimmunoprecipitated with Atg23, which revealed that the Atg23[IL] mutation was sufficient to fully abolish Atg23 dimerization, whereas Atg23[L] was partially defective in dimerization (Figure 4A). We next decided to extend our analysis by examining the effect of the mutants on subcellular localization. Fluorophore-tagged, WT Atg23 forms several cytosolic puncta per cell, with some puncta colocalizing with Atg9 (Legakis et al., 2007). The Atg23[L] mutant resulted in a partial defect in Atg23 puncta formation, whereas the addition of the second point mutation resulted in a complete loss of Atg23 puncta (Figures 4B–4D). This observation suggests that dimerization of Atg23 is required for the formation of Atg23 puncta in cells. We note that, while Atg23 did form multiple puncta per cell in both nutrient-rich and nitrogen-starved conditions, the mutant Atg23 construct formed fewer and less intense puncta upon nitrogen starvation (Figures 4B–4D).
Figure 4. Atg23[LIL] is defective in dimerization, subcellular localization, and Atg9 puncta formation.
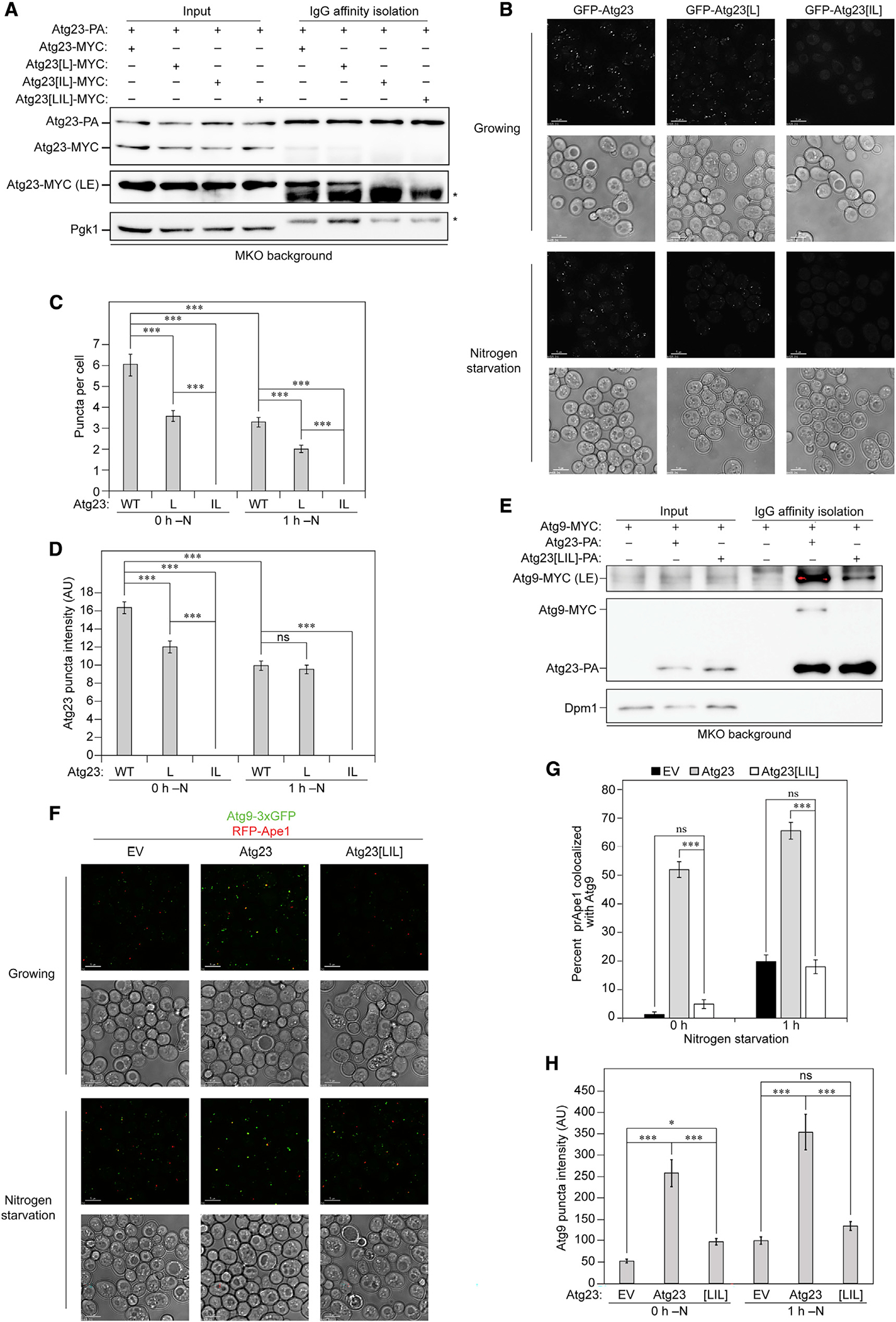
(A) Atg23-PA was overexpressed with MYC-tagged Atg23, Atg23[L], Atg23[IL], or Atg23[LIL] in the MKO background under nutrient-rich conditions. IgG Sepharose beads were used to affinity isolate PA. Proteins were visualized by western blot with representative images shown. An LE image was also collected.
(B) Representative images of endogenously expressed Atg23, Atg23[L], or Atg23[IL] chromosomally tagged with GFP were grown in either nutrient-rich or nitrogen-starvation medium for 1 h. Scale bars: 5 μm.
(C) Quantification of the average number of Atg23 puncta per cell. Error bars represent SEM. n > 50 cells; ***p < 0.001 by Tukey HSD test.
(D) Quantification of Atg23 puncta intensity represented as arbitrary intensity units (AU). Error bars represent SEM. n > 200 puncta; ***p < 0.001 by Tukey HSD test.
(E) Atg9-MYC and PA empty vector, Atg23-PA, or Atg23[LIL]-PA were overexpressed in the MKO background under nutrient-rich conditions. IgG Sepharose beads were used to affinity isolate PA. An LE image of Atg9 was also collected.
(F) Representative images of Atg9–3xGFP and RFP-Ape1 in the absence of Atg1 and presence of empty vector, Atg23, or Atg23[LIL] grown in either nutrient-rich or nitrogen-starvation medium for 1 h. Scale bars: 5 μm.
(G) Quantification of the percentage of RFP-Ape1 puncta colocalized with Atg9–3xGFP. Error bars represent SEM. n = 25 images; ***p < 0.001 by Tukey HSD test.
(H) Quantification of the Atg9 puncta intensity. Error bars represent SEM. n = 150 puncta; ***p < 0.001; *p < 0.05 by Tukey HSD test.
Because mutation of the hydrophobic face caused impaired Atg23 puncta formation and cellular localization and decreased membrane binding, we reasoned that the interaction of Atg23 [LIL] with Atg9 might also be disrupted. To test this assumption, we immunoprecipitated PA empty vector, Atg23-PA, or Atg23 [LIL]-PA in the presence of Atg9-MYC in the MKO background. We found that the interaction between Atg23 and Atg9 was almost entirely abolished upon addition of the LIL mutations, suggesting that dimerization was also required for a stable interaction with Atg9 (Figure 4E).
Given the important role of Atg23 in targeting Atg9 to the PAS, we monitored the colocalization of GFP-Atg9 and the PAS marker RFP-Ape1 in an atg1Δ strain to halt autophagy at the PAS with Atg23 or Atg23[LIL] (Figure 4F). Atg9 recruitment to the PAS was abolished in the presence of Atg23[LIL] during nutrient-rich conditions and reduced by approximately 70% in comparison to Atg23 during starvation (Figure 4G). Moreover, we saw a reduction in the intensity of Atg9 puncta with the Atg23[LIL] mutant, suggesting a defect in Atg9 organization (Figure 4H). Atg23 has been implicated in both the biogenesis and organization of Atg9-containing vesicles at the Golgi and in their anterograde trafficking to the PAS (Backues et al., 2015). To dissect Atg9 vesicle formation and organization from trafficking to the PAS, we observed Atg9–3xGFP and RFP-Ape1 in an atg11Δ strain to block anterograde trafficking downstream of Atg23, allowing us to see the effect of Atg23[LIL] on Atg9 vesicle formation and organization at the peripheral sites. Like the ATG1 deletion, Atg9 puncta intensity was significantly reduced in the presence of Atg23[LIL] in the atg11Δ strain (Figure S6), suggesting that Atg23[LIL] is defective in organizing Atg9 at the peripheral sites. Furthermore, these results support the role of Atg23 in organizing Atg9-containing vesicles at peripheral sites prior to their trafficking to the PAS.
Atg23 tethers vesicles in a dimerization-dependent manner
Because Atg23 binds membrane, forms puncta in cells, and affects Atg9 organization at peripheral sites, we hypothesized that Atg23 may act as a vesicle tether to help efficiently cluster Atg9-containing vesicles. To test this, we performed an in vitro vesicle-tethering assay in which we monitored liposome recruitment to giant unilamellar vesicles (GUVs) in the absence and presence of Atg23 (Figure 5A). We first tested whether Alexa Fluor 488-labeled Atg23 was able to bind YPL-containing GUVs. We observed that Atg23 bound to GUVs in a punctate manner (Figure S7A). Next, we monitored whether Alexa Fluor 488-labeled Atg23 was able to bind GUVs and recruit liposomes. In these experiments, we observed a strong colocalization of Alexa Fluor 488-labeled Atg23 and DiD liposomes on the surface of GUVs, suggesting that Atg23 was able to tether liposomes to GUVs (Figures 5B and 5C). In the absence of Atg23, liposomes were not readily observed to colocalize with GUVs (Figures S7B–S7D). Because the Alexa Fluor 488 dye could potentially influence the liposome-tethering properties of Atg23, we repeated the membrane-tethering assay with unlabeled Atg23 and compared it with a no-protein control (Figure 5D). To quantify liposome tethering, GUV peripheries were first marked using a segmented line in the Rhod PE channel. The fluorescence intensity traces along GUV peripheries were then plotted in the liposome DiD channel (Figures S7B–S7D). A GUV with tethered liposomes was defined as a GUV showing DiD fluorescence peaks 3-fold above the mean DiD background fluorescence. In the presence of Atg23, 79.3% of the GUVs showed strong colocalization with liposomes compared with 3.9% in the absence of Atg23, demonstrating that tethering is mediated by Atg23 (Figure 5E). Monitoring the localization of liposomes over 30 s further supported the tethering activity of Atg23 as liposomes tethered to GUVs by Atg23 remained localized to GUVs throughout the entire duration of these movies, whereas in the absence of Atg23, liposomes readily moved past GUVs (Videos S1 and S2).
Figure 5. Atg23 is a vesicle-tethering protein.
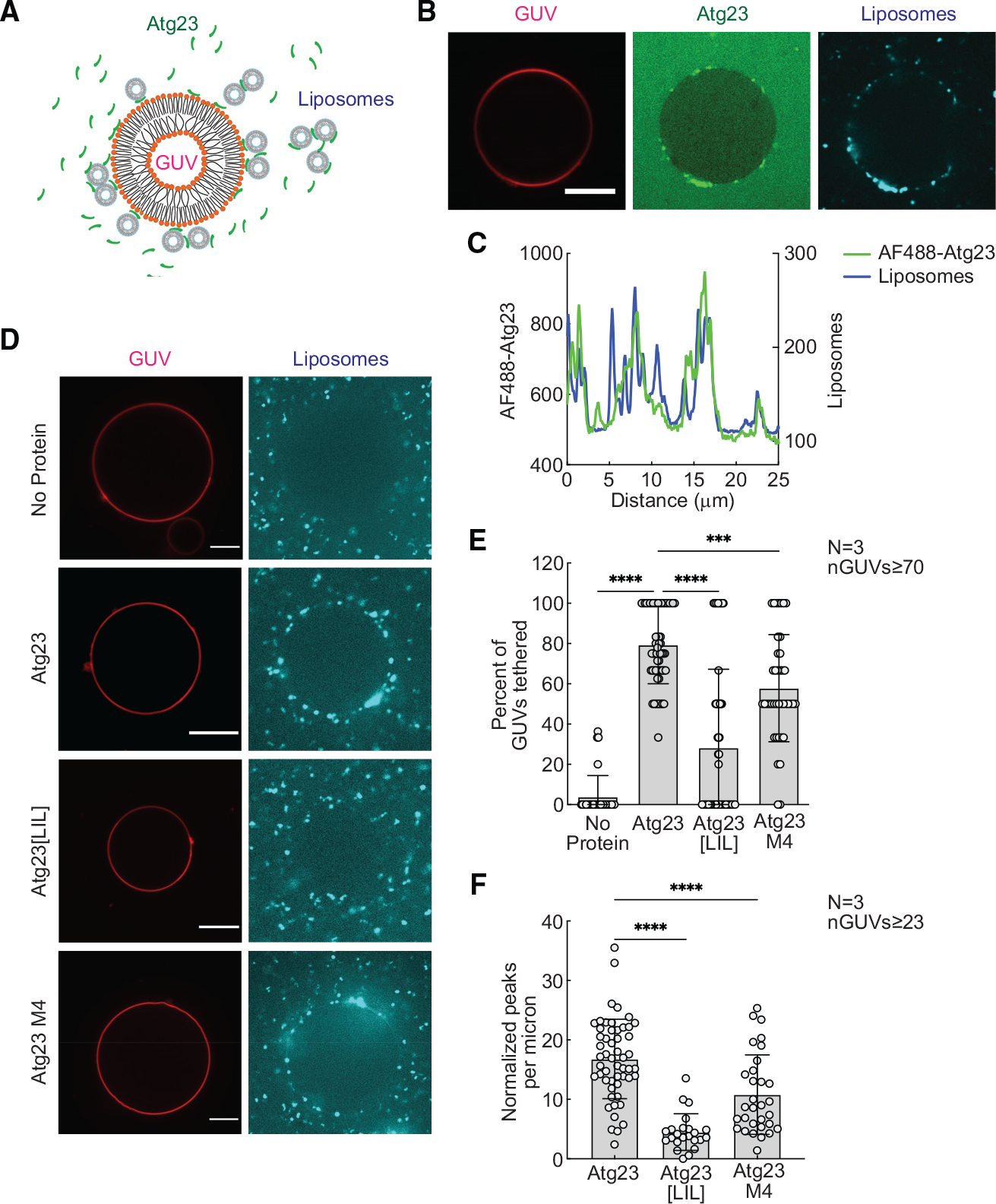
(A) Schematic representation of the tethering assay. Colocalization of Rhod-PE-GUVs and DiD-liposomes indicates vesicle tethering.
(B) GUVs were mixed with 1 μM Alexa Fluor 488 (AF488)-Atg23 and liposomes, incubated for 10 min, and imaged in a LabTek chamber. Representative images of AF488-Atg23 (green) with tethered liposomes (cyan) are shown. Scale bar: 10 μm.
(C) Intensity profile showing AF488-Atg23 and liposome fluorescence on a segmented line placed along the surface of the GUV in (B).
(D) Representative images of liposomes (cyan) tethered on GUVs (red) in the absence of protein, with unlabeled Atg23, Atg23[LIL], or Atg23 M4. Scale bars: 10 μm. Tethering was analyzed by measuring the DiD intensity profile along the entire GUV surface. GUVs with DiD intensity profiles showing peaks that were 3-fold over the mean background intensity were categorized as GUVs with tethered liposomes.
(E) Quantification showing percent GUVs with tethered liposomes.
(F) Quantification showing normalized number of DiD intensity peaks above background on GUVs with tethered liposomes.
(E and F) Data from three independent experiments were analyzed using ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ****p < 0.0001; ***p < 0.001. Error bars represent SD. n indicates number of independent repeats. nGUVs indicate total number of GUVs analyzed.
To investigate the requirement of dimerization and membrane binding for tethering, we repeated our vesicle-tethering assay with Atg23[LIL] and Atg23 M4. Atg23[LIL] was able to bind GUVs (Figures S7E and S7F) but showed a dramatic reduction in its ability to tether liposomes to GUVs with only 28.3% of GUVs showing colocalization with liposomes (Figures 5D and 5E). Atg23 M4 had a nearly identical amount of membrane binding as Atg23[LIL], but significantly higher membrane tethering than Atg23[LIL], with 57.9% of GUVs showing colocalization with liposomes. To further investigate the membrane-tethering properties of these proteins, we also quantified the number of peaks per micron on the GUVs that showed colocalization with liposomes, serving as a measure of the number of liposomes per GUV. For this quantification, Atg23, Atg23[LIL], and Atg23 M4 had 16.8, 4.5, and 10.8 peaks per micron, respectively (Figure 5F). These results indicate that, despite the similar membrane-binding properties of Atg23[LIL] and Atg23 M4 (Figures 3I and 3J), Atg23[LIL] has a larger reduction in vesicle tethering. Taken together, these data demonstrate that Atg23 dimerization is critical for membrane tethering.
Atg23[LIL] is non-functional in the Cvt pathway and nonselective autophagy
Atg23 is essential for the Cvt pathway and efficient nonselective autophagy (Meiling-Wesse et al., 2004; Tucker et al., 2003). Given the observed deficiencies upon mutating the hydrophobic face of the amphipathic helix, we tested the effect of Atg23[LIL] on autophagic activity. The Cvt pathway is a form of biosynthetic selective autophagy in which a few specific hydrolases, such as precursor aminopeptidase I (prApe1), are selectively encapsulated and delivered to the vacuole in the inactive form, where they are cleaved to form the mature, functional enzyme. Thus, the maturation of prApe1 is measured as a readout for the progression of the Cvt pathway. WT Atg23 fully complemented the atg23Δ strain, as seen by the detection primarily of the mature form of Ape1 (Figures 6A and 6B). In contrast, Atg23 [LIL] was no better than the empty vector at complementing the loss of ATG23 (Figures 6A and 6B). We used two different assays for the measurement of nonselective autophagy: the GFP-Atg8 processing assay and the Pho8Δ60 assay (Klionsky et al., 2016). The GFP-Atg8 processing assay relies on the delivery of GFP-Atg8 to the vacuole and subsequent cleavage to form free GFP, which is observed by SDS-PAGE and western blot. In the Pho8Δ60 assay, the vacuolar phosphatase Pho8 is delivered to the vacuole, where it is cleaved to the active form. The activity of a truncated form of Pho8 that is missing its transmembrane domain (rendering it an autophagic cargo rather than trafficking through the secretory pathway) is used as a readout for nonselective autophagy. By both nonselective autophagy assays, Atg23[LIL] was entirely unable to complement the deletion of ATG23, showing values no higher than the empty vector (Figures 6C–6E). This result indicates that Atg23 dimerization is critical to the function of the protein in both the Cvt pathway and nonselective autophagy.
Figure 6. Atg23 dimerization is critical for the role of Atg23 in the Cvt pathway and nonselective autophagy.
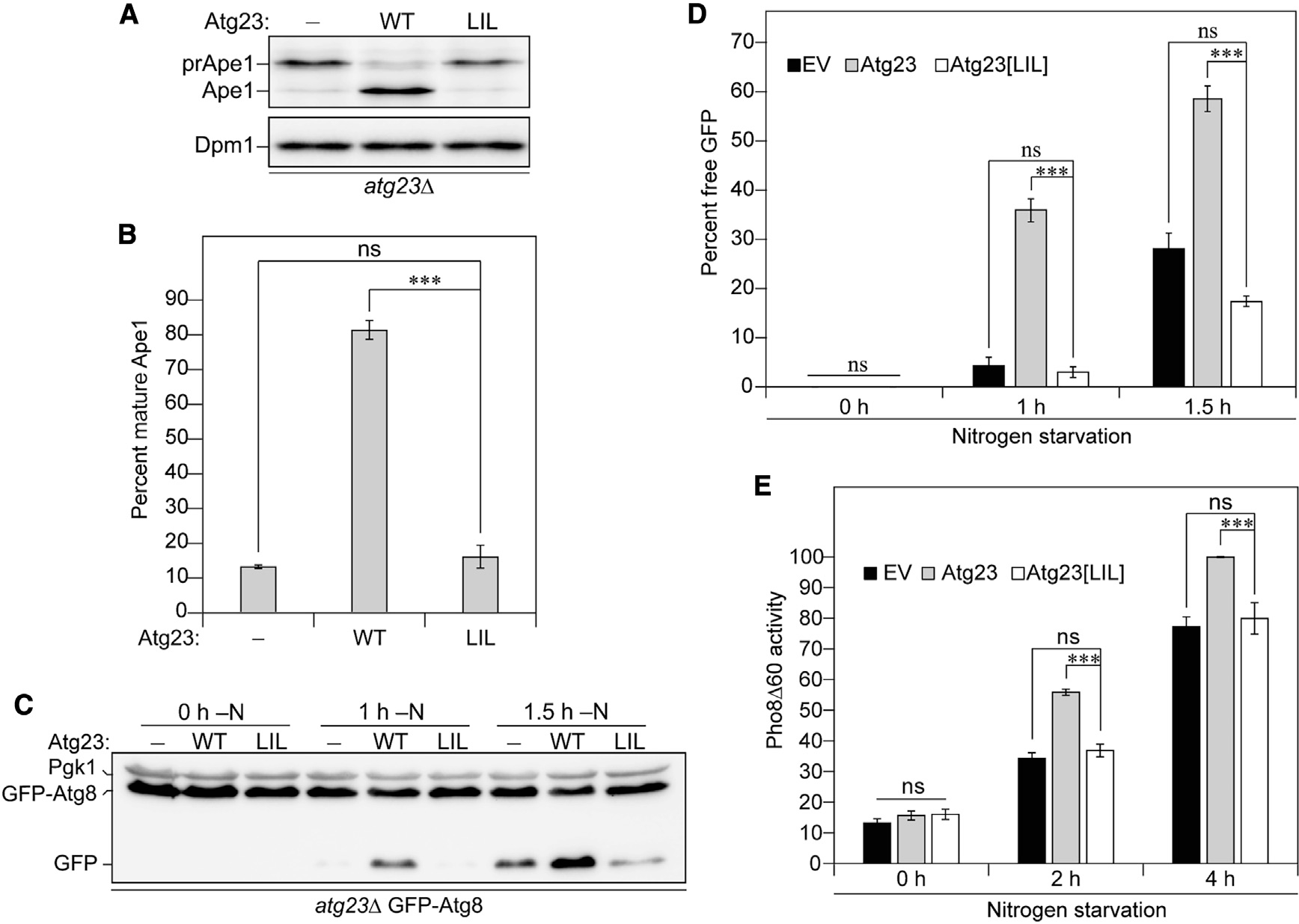
(A) atg23Δ cells exogenously expressing an empty vector, Atg23, or Atg23[LIL] under the endogenous promoter were grown overnight in nutrient-rich medium. Ape1 from cell lysates was inspected by western blot using anti-Ape1 antiserum.
(B) Quantification of (A).
(C) Empty vector, Atg23, or Atg23[LIL] driven by the endogenous promoter was expressed in atg23Δ cells also expressing GFP-Atg8 under the control of the CUP1 promoter. Cells were harvested in log phase after 0, 1, or 1.5 h of exposure to nitrogen-deficient medium. Processing of GFP-Atg8 to free GFP was assessed by western blot.
(D) Quantification of (C).
(E) Pho8Δ60 activity was assessed for atg23Δ Pho8Δ60 cells expressing empty vector, Atg23, or Atg23[LIL] under the control of the endogenous promoter. Quantification shows values as a percentage of wild-type activity after 4 h of nitrogen starvation.
(B, D, and E) Error bars represent SEM; n = 3; ***p < 0.001 by Tukey HSD test.
DISCUSSION
Since its identification in 2003, Atg23 has been described as an Atg9-binding, peripheral membrane protein that cycles between the PAS and various peripheral sites (Yen et al., 2007; Legakis et al., 2007). In addition, the deletion of ATG23 causes a complete block in the Cvt pathway and greatly hinders nonselective autophagy. However, no studies to date have investigated the structure or mechanism of Atg23. This investigation sheds light on the elusive biophysical properties and function of Atg23. We found that Atg23 is a largely α-helical homodimer. This dimerization is facilitated by a putative amphipathic helix, which is compromised upon mutation of residues L171, I182, and L189. We also demonstrated that Atg23 can directly interact with membranes via charge-dependent interactions independent of Atg9 and that it tethers vesicles. In addition to disrupting dimerization, Atg23[LIL] also severely diminishes the protein’s ability to bind Atg9 and function in both selective and nonselective autophagy. Furthermore, the mutant exhibits decreased membrane binding and dramatically reduced membrane tethering, demonstrating that dimerization is critical for its tethering ability. Lastly, we see that Atg23[LIL] greatly reduces the number of Atg9 puncta in vivo.
Taken together, we propose a model for Atg23 vesicle tethering that enhances vesicle clustering in cells (Figure 7). Atg23 is an extended helical dimer, with each monomer containing a functional Atg9- and membrane-binding site. Dimerization of Atg23 facilitates membrane tethering, enhanced membrane binding, and clustering of Atg9 vesicles in cells. Mutation of the dimerization interface of Atg23 leads to a partial unfolding of the amphipathic helix that supports Atg23 dimerization and the formation of stable monomers. Atg23[LIL] monomers retain binding for membranes and Atg9 but at significantly reduced levels, likely due to a loss of clustering supported by dimerization, which would enhance the avidity of these interactions.
Figure 7. Schematic representation of Atg23 dimerization, membrane binding, and vesicle tethering.
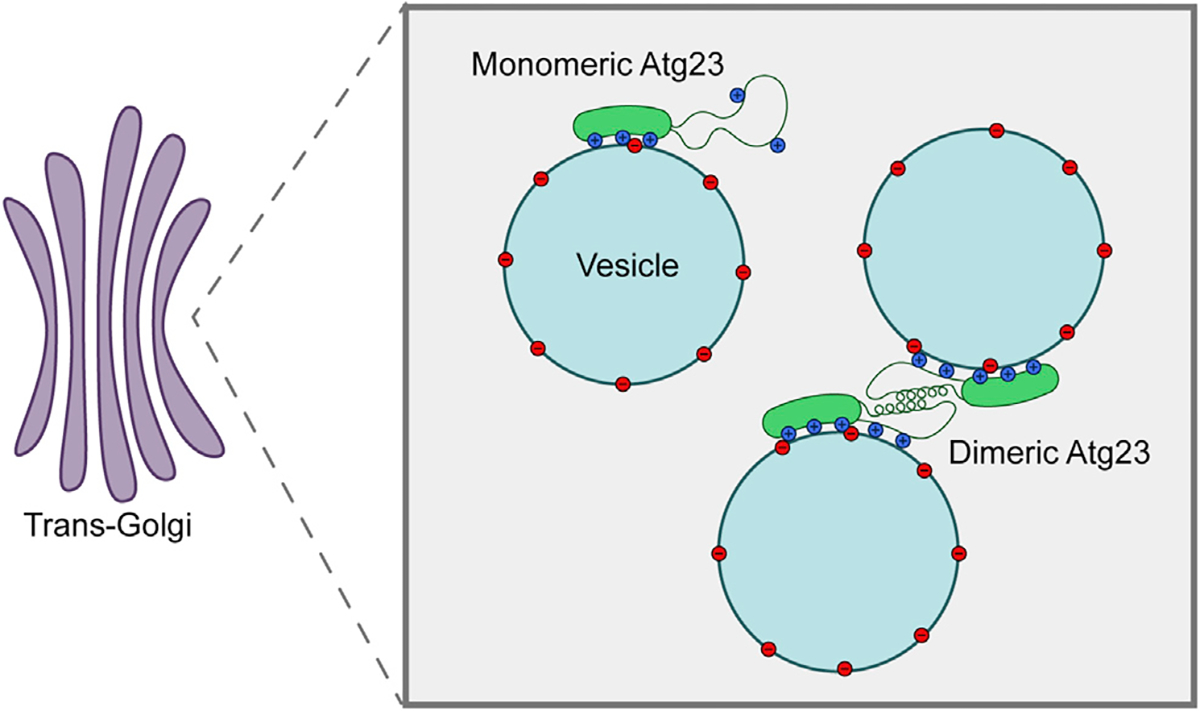
Both monomers of the protein adhere to negatively charged vesicles through several spatially dispersed residues. Dimerization of Atg23 mediated by the putative amphipathic helix results in the tethering of Atg9-containing vesicles at the trans-Golgi. Mutation of key hydrophobic residues within this putative amphipathic helix prevents dimerization as well as proper positioning of some membrane-binding residues. Thus, monomeric Atg23 is entirely deficient in subcellular localization, membrane tethering, and autophagic function while being partially deficient in membrane and Atg9 binding.
The elongated and slightly curved architecture of Atg23 bears a strong resemblance to Bin-Amphiphysin-Rvs (BAR) domains, which have demonstrated roles in autophagy (Carman and Dominguez, 2018; Popelka et al., 2017; Zhuang et al., 2013). BAR-domain proteins are coiled-coil dimeric proteins that can sense curvature of, bind to, and bend membranes. The dimerization and electrostatic membrane interaction of Atg23 also make it tempting to speculate that it is a BAR-domain protein. However, Atg23 does not appear to be sensitive to membrane curvature, and its vesicle-tethering role further distinguishes it from BAR-domain-containing proteins, suggesting that Atg23 is unlike BAR domains in its function.
There are several examples of membrane tethers in autophagy, including Atg17, which is a dimeric coiled-coil that tethers Atg9-containing vesicles at the PAS in nonselective autophagy (Noda et al., 2012; Rao et al., 2016). Like Atg17, our SAXS data suggest that Atg23 may also dimerize through an antiparallel coiled-coil region, where the monomer is roughly half the length of the dimer. This hypothesis was further supported by our docking of two monomers of Atg23 from the AlphaFold model, revealing that Atg23 may be arranged in an end-to-end fashion within the dimer similar to the Atg17 monomer in the Atg17-Atg31-Atg29 complex. Playing an analogous role to Atg17 in selective autophagy is Atg11, another dimeric coiled-coil Atg9-containing vesicle tether (Matscheko et al., 2019). Like Atg23, Atg11 and Atg17 both bind Atg9 (He et al., 2006; Sekito et al., 2009) and require dimerization for their function in autophagy. Both Atg11 and Atg17 largely function as scaffolds to mediate formation of the initiation complex at the PAS. It is intriguing that Atg23 is another Atg9-binding, vesicle-tethering protein involved in autophagy. However, further experiments will be needed to determine at what stage in autophagy Atg23-mediated vesicle tethering occurs.
Vesicle-tethering proteins are typically categorized into two classes: coiled-coil tethers and multisubunit tethering complexes. Because Atg23 dimerizes through what appears to be a coiled-coil domain, we think it belongs to the former group. One example of coiled-coil tethers is golgin proteins, which selectively tether vesicles within the secretory system by binding different interacting partners at their termini (Lowe, 2019). For example, the yeast golgin Uso1/p115 clusters COPII vesicles formed at endoplasmic reticulum exit sites (Short et al., 2005). Given that previous studies have shown that Atg9-containing vesicles are organized at peripheral sites prior to relocating to the PAS, it is reasonable to speculate that Atg23 may be performing a similar role to cluster Atg9-containing vesicles. Our work supports this hypothesis, as Atg23[LIL] shows not only a reduction in membrane tethering but also a reduction in the formation of both Atg23 and Atg9 puncta in vivo.
Limitations of the study
There are a few experimental caveats to address: as the first investigation into the membrane-binding ability of Atg23, there is no knowledge about its preferred membrane composition for binding. Therefore, we used yeast polar lipids, which likely do not represent the exact membrane composition that Atg23 binds. This paper provides structural insight into Atg23 using a variety of biophysical techniques and the AlphaFold model. AlphaFold is currently best at modeling monomers, so we generated a dimeric model based on the monomeric model. However, to understand the mechanism of membrane binding and vesicle tethering at the atomic level, a high-resolution structure of Atg23 is required. We also provide insight into the function of Atg23 as vesicle tether, but it is important to note that this technique is indirect, so quantitative conclusions cannot be drawn. One last limitation of this study is that we do not identify the precise role of vesicle tethering by Atg23 in autophagy. As such, it is currently unclear how membrane binding, tethering, and Atg9 binding by Atg23 are related and what role these different functions of Atg23 may play in autophagy. Further work will be needed to address these important questions.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Michael J Ragusa (michael.j.ragusa@dartmouth.edu).
Materials availability
Plasmids and yeast strains will be made available upon request to the Lead contact.
Data and code availability
The SAXS data for Atg23 and Atg23[LIL] were deposited in the Small Angle Scattering Biological DataBank (SASBDB) as SASDL39 and SASDL49, respectively. These data are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Strains, media and growth conditions
Strains used in this study are listed in the key resources table. Deletion (Longtine et al., 1998) and epitope-tagged (Gueldener et al., 2002) strains were constructed as described previously. Yeast cells were grown in nutrient-rich medium (YPD; 1% [wt:vol] yeast extract, 2% [wt:vol] peptone, 2% [wt:vol] glucose) or synthetic minimal medium (SMD; 0.67% yeast nitrogen base, 2% [wt:vol] glucose, and auxotrophic amino acids and vitamins). Autophagy was induced by shifting the cells to nitrogen-starvation medium (SD-N; 0.17% nitrogen base without ammonium sulfate or amino acids and 2% [wt:vol] glucose).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| α-MYC | Sigma Aldrich | C3956-100UG; RRID: AB_439680 |
| α-His | Sigma Aldrich | SAB4301134-100UL |
| α-Dpm1 | Thermo Fisher Scientific | A-6429; RRID: AB_2536204 |
| α-YFP | Takara | 632380; RRID: AB_10013427 |
| α-Pgk1 | (Baum et al., 1978) | N/A |
| α-PA | Jackson Immunoresearch | 323-005-024; RRID: AB_2315781 |
| Atg23 antiserum | This study | N/A |
| Ape1 antiserum | (Klionsky et al., 1992) | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Rosetta 2 (DE3) pLysS cells | Novagen | 71401 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Q5 mutagenesis | NEB | E0554 |
| NuPAGE 4–12% Bis-Tris gels | Invitrogen | NP0322BOX |
| LB broth | Fisher Scientific | BP1426 |
| IPTG | IBI Scientific | IB02125 |
| Triton X-100 | VWR | AAA16046-AE |
| 2-mercaptoethanol | Sigma Aldrich | M6250 |
| cOmplete Mini EDTA-free protease inhibitor tablets | Roche | 11836170001 |
| Talon resin | Clontech | 635504 |
| TCEP | VWR | 97064-848 |
| Yeast polar lipid extracts | Avanti Polar Lipids | 190001 |
| 1.0-μm Nuclepore Track-Etched membrane | VWR | 15000-610 |
| 0.4-μm Nuclepore Track-Etched membrane | Sigma Aldrich | WHA10417104 |
| 0.1-μm Nuclepore Track-Etched membrane | VWR | 15000-614 |
| Rhodamine PE | Avanti Polar Lipids | 810150 |
| Polyvinyl alcohol | Sigma Aldrich | 814894 |
| Alexa Fluor 488 | Invitrogen | A10254 |
| DiD | Thermo Fisher Scientific | D7757 |
| Agar | Formedium | AGA03 |
| Yeast extract | Formedium | YEA03 |
| Peptone | Formedium | PEP03 |
| Yeast nitrogen base | Formedium | CYN0410 |
| Acrylamide | National Diagnostics | EC-890 |
| Triton X-100 | Sigma Aldrich | X100-100ML |
| CHAPS | Anatrace | C31625GM |
|
| ||
| Critical commercial assays | ||
|
| ||
| QIAprep Spin Miniprep Kit | Qiagen | 27104 |
|
| ||
| Deposited data | ||
|
| ||
| SAXS data for Atg23 | This paper | SASBDB: SASDL39 |
| SAXS data for Atg23[LIL] | This paper | SASBDB: SASDL49 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| SEY6210 (MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 GAL) | (Robinson et al., 1988) | N/A |
| WLY176 (SEY6210 pho13Δ, PHQ8::pho8Δ60) | (Noda and Klionsky, 2008) | N/A |
| MKO (SEY6210, atg1Δ, 2Δ, 3Δ, 4Δ, 5Δ, 6Δ, 7Δ, 8Δ, 9Δ, 10Δ, 11Δ, 12Δ, 13Δ, 14Δ, 16Δ, 17Δ, 18Δ, 19Δ, 20Δ, 21Δ, 23Δ, 24Δ, 27Δ, 29Δ, 31Δ::ble) | (Cao et al., 2008) | N/A |
| KTY14 (SEY6210 atg23Δ::KAN) | (Tucker et al., 2003) | N/A |
| yWH59 (SEY6210, ATG23-mNeonGreen::TRP1) | This study | N/A |
| yWH67 (SEY6210, atg23L189A-mNeonGreen::TRP1) | This study | N/A |
| yWH68 (SEY6210, atg23I182A L189A-mNeonGreen::TRP1) | This study | N/A |
| yWH86 (WLY176, CUP1p-GFP-ATG8::LEU2, atg23Δ::URA3) | This study | N/A |
| yWH134 (SEY6210, atg1Δ, 9Δ, 23Δ, ATG9-3xGFP, RFP-APE1) | This study | N/A |
| yWH174 (SEY6210, atg1Δ, 9Δ, 23Δ, ATG9-3xGFP, RFP-APE1) | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotide information | This study | Table S1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pKL1 (pHis2_6xHis-3C_ScAtg23) | This study | N/A |
| pKL2 (pHis2_6xHis-3C_ScAtg23L171A,I182A,L189A) | This study | N/A |
| pKL137 (pHis2_6xHis-3C_ScAtg23K66E,R67E) | This study | N/A |
| pKL138 (pHis2_6xHis-3C_ ScAtg23K66E,R67E,K296E) | This study | N/A |
| pKL138 (pHis2_6xHis-3C_ ScAtg23K66E,R67E,K296E) | This study | N/A |
| pKL139 (pHis2_6xHis-3C_ ScAtg23K66E,R67E,R74E,R85E,K296E) | This study | N/A |
| pRS314 (Centromeric yeast expression plasmid, TRP1) | (Sikorski and Hieter, 1989) | N/A |
| pRS414 (Centromeric yeast expression plasmid, TRP1) | (Sikorski and Hieter, 1989) | N/A |
| pRS424 (2-micron yeast expression plasmid, TRP1) | (Sikorski and Hieter, 1989) | N/A |
| pRS426 (2-micron yeast expression plasmid, URA3) | (Sikorski and Hieter, 1989) | N/A |
| pWH1 (pRS424_pCUP1-6xHis-Atg23) | This study | N/A |
| pWH4 (pRS424_pCUP1-6xHis-Atg23L171A,I182A,L189A) | This study | N/A |
| pWH11 (pRS426_pCUP1-Atg9-MYC) | This study | N/A |
| pWH16(pRS314_pNOP1-Atg23-2xPA) | This study | N/A |
| pWH21 (pRS426_pCUP1-Atg23-MYC) | This study | N/A |
| pWH25 (pRS426_pCUP1-Atg23L171A,I182A,L189A-MYC) | This study | N/A |
| pWH67 (pRS424_pCUP1-PA) | This study | N/A |
| pWH69 (pRS414_pATG23-Atg23) | This study | N/A |
| pWH70 (pRS314_pNOP1-Atg23L171A,I182A,L189A-2xPA) | This study | N/A |
| pWH72 (pRS414_pATG23-Atg23L171A,I182A,L189A) | This study | N/A |
| pWH130 (pRS424_pCUP1-6xHis-Atg23R166E,K169E,K176E) | This study | N/A |
| pWH131 (pRS424_pCUP1-6xHis-Atg23R190E,K193E,K194E) | This study | N/A |
| pWH132 (pRS424_pCUP1-6xHis-Atg23K66E,R67E) | This study | N/A |
| pWH134 (pRS424_pCUP1-6xHis-Atg23K66E,R67E,K296E) | This study | N/A |
| pWH135 (pRS424_pCUP1-6xHis-Atg23K66E,R67E,R74E,R85E,K296E) | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji (ImageJ2 v 2.3.0) | (Schindelin et al., 2012) | https://imagej.net/software/fiji/ |
| Image Lab v 5.1 | Bio-Rad | N/A |
| Prism v 5.0 or 8.2.1 | GraphPad | https://www.graphpad.com/ |
| Dynamics v 7.1.8 | Wyatt Technology | https://www.wyatt.com/products/software/dynamics.html |
| K2D3 | (Louis-Jeune et al., 2012) | http://cbdm-01.zdv.uni-mainz.de/%7Eandrade/k2d3/ |
| SEDNTERP 20130813 Beta | (Laue et al., 1992) | http://bitcwiki.sr.unh.edu/index.php/Main_Page |
| SEDFIT v 16.1 | (Schuck et al., 2002) | N/A |
| Astra v 6.0 | Wyatt Technology | https://www.wyatt.com/products/software/astra.html |
| Primus v 3.4 | (Konarev et al., 2003) | https://www.embl-hamburg.de/biosaxs/software.html |
| GNOM v 5.0 | (Konarev et al., 2003) | https://www.embl-hamburg.de/biosaxs/software.html |
| GASBOR v 2.3 | (Svergun et al., 2001) | https://www.embl-hamburg.de/biosaxs/software.html |
| SUPCOMB v 2.3 | (Svergun et al., 2001) | https://www.embl-hamburg.de/biosaxs/software.html |
| DAMAVER v 5.0 | (Volkov and Svergun, 2003) | https://www.embl-hamburg.de/biosaxs/software.html |
| HADDOCK v 2.4 | (van Zundert et al., 2016) | https://wenmr.science.uu.nl/haddock2.4/ |
| Excel 2021 | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| R v 4.0.5 | R Project | https://www.r-project.org/ |
| Python v 3.8 | Python Software Foundation | https://www.python.org/ |
Plasmids
Plasmids used in this study are listed in the key resources table and were generated using either FAST cloning or Gibson Assembly. All plasmids were verified by DNA sequencing.
Recombinant protein expression and purification
S. cerevisiae Atg23 FL was subcloned into pHis2 (Sheffield et al., 1999) with an HRV-3C cleavage site. The mutant Atg23[LIL] was generated from Atg23 FL pHis2_3C using Q5 mutagenesis (NEB, E0554). The constructs were transformed into E. coli Rosetta 2 (DE3) pLysS cells (Novagen 71401). Cultures were grown in LB broth (Fisher Scientific, BP1426) to an OD600 of approximately 0.6 at 37°C while shaking at 220 rpm. Cultures were cooled at 4°C for 30 min. Protein expression was induced by adding 0.5 mM isopropylthio-β-D-galactopyranoside (IPTG; IBI Scientific, IB02125) and the cultures were grown for an additional 16 h at 18°C while shaking at 220 rpm. Cells were harvested and stored at −80°C. Cell pellets were thawed and resuspended in 50 mM Tris, pH 7.4, 500 mM NaCl, 1% (vol:vol) Triton X-100 (VWR, AAA16046-AE), 5 mM MgCl2, 1 mM phenylmethanesulfonyl fluoride (PMSF; VWR, 0754), 5 mM 2-mercaptoethanol (Sigma Aldrich, M6250) and cOmplete Mini EDTA-free protease inhibitor tablets (Roche, 11836170001). Cells were lysed by passing through a French Press (Thermo Electron) three times at 4°C. Lysates were cleared by centrifugation at 10,000 × g for 50 min at 4°C. The supernatant was applied to Talon resin (Clontech, 635504) pre-equilibrated with 50 mM Tris, pH 7.4, 500 mM NaCl. The resin was washed with 50 mM Tris, pH 7.4, 500 mM NaCl, 2.5 mM imidazole and eluted with 50 mM Tris, pH 7.4, 500 mM NaCl, 200 mM Imidazole. The 6xHis-tag was cleaved off the protein with HRV-3C protease at 4°C for 18 h. Fractions containing protein were pooled and applied to a HiPrep 26/10 Desalting column pre-equilibrated with 50 mM Tris, pH 7.4, 50 mM NaCl. Fractions containing protein were pooled and applied to a HiTrap Q HP anion exchange column pre-equilibrated with 50 mM Tris, pH 7.4, 50 mM NaCl and eluted with an increasing gradient of 50 mM Tris, pH 7.4, 1 M NaCl. Fractions were pooled and concentrated and applied to a HiLoad Superdex 200 PG column pre-equilibrated with 20 mM Tris, pH 7.4, 100 mM NaCl, 0.2 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP; VWR, 97064–848). Fractions from the central peak of the chromatogram containing protein were pooled and concentrated to ensure a monodispersed protein sample was used.
Subcellular fractionation
Yeast cells were grown in SMD at 30°C overnight to a final optical density of 600 nm (OD600) of 0.7 at which point 15 OD600 units of cells were harvested by centrifugation at 3000 RCF for 3 min. The cell pellet was resuspended in 400 μL lysis buffer (200 mM sorbitol, 20 mM PIPES-KOH, pH 6.8, 5 mM MgCl2, 1 mM PMSF, cOmplete EDTA-free protease inhibitor cocktail, NaCl [if applicable]). Glass beads were added, and samples were mixed by vortex at 4°C five times for one min each with two-min gaps in between. Cell debris was pelleted at 500 RCF for 10 min at 4°C and the supernatant was transferred to a new tube. The total fraction was harvested, and the remaining cell lysate solution was centrifuged at 13,000 RCF for 10 min at 4°C. The soluble fraction was harvested, and the remaining supernatant was discarded. The material remaining in the tube was kept as the pellet fraction.
Native page
Yeast cells were grown in SMD at 30°C overnight to an OD600 of 0.5; 30 OD600 units were then collected by centrifugation. Spheroplasts were prepared similarly to previously described methods (Wilson et al., 2018) and resuspended in sample buffer (10 mM Trisacetate, pH 6.9, 5 mM potassium acetate, 1 mM EDTA, 2 mM PMSF, 10% glycerol [vol:vol], 0.01% bromophenol blue [wt:vol], 0.01% xylene green [wt:vol]) with light vortexing to induce lysis. Samples were run at 80 V, 4°C using running buffer (192 mM glycine, 25 mM Tris, pH 8.2) and a 6% polyacrylamide gel lacking SDS.
Atg23 antibody production
Molecular Peptide Systems was hired for the generation of Atg23 antiserum. Two peptides, HQLMKRIGFESPLTQEK and KIGISESVVNANKNDAISKK, representing Saccharomyces cerevisiae Atg23 amino acids 229–245 and 432–451, respectively, were synthesized. These peptides were conjugated to the carrier protein, keyhole limpet hemocyanin, and injected into New Zealand White rabbits. The antibody was authenticated using purified Atg23 from E. coli.
Immunoprecipitation
Fifty OD600 units of cells were harvested after growing at 30°C overnight in SMD to a final OD600 of 0.8. Cells were resuspended in 1 mL IP buffer (Atg9-Atg23: 1× PBS, 200 mM sorbitol, 1 mM MgCl2 0.5% Triton X-100 [vol:vol], cOmplete EDTA-free protease inhibitor cocktail, 1 mM PMSF; Atg23-Atg23: 50 mM Tris, pH 7.6, 30 mM NaCl, 0.1% CHAPS [vol:vol], 10% glycerol [vol:vol], 1 mM DTT, cOmplete EDTA-free protease inhibitor cocktail, 1 mM PMSF). Glass beads were added, and samples were treated as above. Cell lysates were centrifuged at 13,000 RCF for 10 min at 4°C. Supernatants were added to 100 μL IgG Sepharose 6 Fast Flow that had been incubated with 5% milk overnight. Sepharose beads were incubated with cell lysates for two h at 4°C, at which point the beads were washed with IP buffer 6 times.
Fluorescence microscopy
Yeast cells were grown to OD600 ~ 0.5 in YPD medium and shifted to nitrogen-starvation medium for autophagy induction. Images were collected on a DeltaVision Elite deconvolution microscope (Cytiva/GE Healthcare/Applied Precision) with a 100× objective and a CCD camera (CoolSnap HQ; Photometrics). For quantification, stacks of 20 image planes were collected with a spacing of 0.2 mm to cover the entire yeast cell. Analysis was performed on an average projection of the imaging planes using ImageJ.
Precursor Ape1 and GFP-Atg8 processing assays
For prApe1 processing assays, cells were grown in nutrient-rich medium to mid-log phase (OD600 ~0.5) at which point 1 OD600 unit of cells was harvested by centrifugation. For GFP-Atg8 processing, cells expressing GFP-Atg8 under the control of the CUP1 promoter were grown to OD600 0.8 at which point cells were either harvested or shifted to SD-N medium to induce autophagy prior to harvesting. In both assays, cell lysates were subsequently harvested and analyzed by western blot.
Phosphatase activity assay
The phosphatase activity of Pho8Δ60 was assayed as described previously (Noda and Klionsky, 2008). Cells were grown in nutrient-rich medium to mid-log phase (OD600 ~0.5) at which point 1.5 OD600 unit of cells was harvested by centrifugation. Nitrogen-starvation samples were grown for a further 2 or 4 h in SD-N medium. Cells were lysed in 250 μL lysis buffer (20 mM PIPES-KOH, 0.5% Triton X-100 [vol:vol], 50 mM KCl, 100 mM KAc, 10 mM MgSO4, 10 μM ZnSO4, 1 mM PMSF) through vortexing in the presence of glass beads. 100 μL of lysis supernatant was mixed with 400 μL of reaction buffer (250 mM Tris-HCl, 0.4% Triton X-100 [vol:vol], 10 mM MgSO4, 10 μM ZnSO4, 1.25 mM p-NPP). STOP solution (1 M glycine) was added after 5 min and absorbance was measured at 420 nm. Fifty uL of lysis solution was mixed with 1 mL of BCA reaction buffer (Thermo Fisher, 23225). Absorbance was read at 560 nm after 30 min of incubation.
Liposome sedimentation assay
Yeast polar extract from S. cerevisiae (YPL; Avanti Polar Lipids, 190001) was dried under a nitrogen stream for 30 min and in a vacuum oven for 18 h. Dried liposomes were resuspended in 20 mM Tris, pH 7.4, 100 mM NaCl for size preference and Atg23 mutant experiments to a final concentration of 2.5 mg/mL. For the salt experiments, dried liposomes were resuspended in 20 mM Tris, pH 7.4. Liposomes were extruded using an Avanti Mini Extruder with the appropriate size membrane: 1.0-μm Nuclepore Track-Etched membrane (VWR, 15000–610), 0.4-μm Nuclepore Track-Etched membrane (Sigma Aldrich, WHA10417104), or 0.1-μm Nuclepore Track-Etched membrane (VWR, 15000–614). To generate small unilamellar vesicles, the resuspended liposomes were sonicated with a Fisher Scientific Sonic Dismembrator Model 500 with a microtip at 15% amplitude 1 s on and 1 s off for a total of 5 min. Vesicles were then centrifuged at 16,000 RCF for 10 min to clear any metal residue derived from the microtip. Then 25 μL of 10 μM protein was mixed with 25 μL of 2.5 mg/mL YPL liposomes. The mixture was incubated at 4°C for 60 min to allow for complex formation and centrifuged at 86,016 RCF for 40 min at 4°C using a TLA45 rotor. The supernatants were removed, and equal volumes of buffer were added to resuspend the pellets. Samples were run on Invitrogen Novex NuPAGE 4–12% Bis-Tris gels (Invitrogen, NP0322BOX) and subjected to SDS-PAGE. Bands were quantified using Image Lab v 5.1 (Bio-Rad). The supernatant and pellet band intensities were summed together to determine the total intensity of protein in each sample. The percentage of protein bound to liposomes was determined by taking the ratio of the pellet band intensity over the total intensity. Each experiment was performed in triplicate or quadruplicate as indicated in the figure legend. The results were averaged and plotted with error bars representing the standard deviations of the three or four independent experiments. Statistical analysis was performed using Prism software v 8.2.1 (GraphPad). Data were compared using an ordinary one-way ANOVA with Tukey’s multiple comparison test or using a two-way ANOVA with Sidak’s multiple comparison test. ****. p < 0.0001.
Dynamic light scattering
To determine the liposome size and homogeneity, dynamic light scattering was performed on liposomes at 2.5 mg/mL at 20°C using a DynaPro NanoStar (Wyatt Technology). Light scattering data were analyzed using Dynamics v7.1.8 (Wyatt Technology).
Circular dichroism
Secondary structure and protein thermal stability were assessed using circular dichroism (CD) using a Jasco J-185 spectrophotometer. All proteins were analyzed at 10 μM in 20 mM Tris pH 7.4, 100 mM NaCl, 0.2 mM TCEP. CD data were acquired over three accumulations from 200 to 250 nm at 20°C. Melting curves were obtained through variable temperature scans at fixed wavelength (222 nm) from 10°C to 90°C at a 1C/min ramp rate. The melting temperature Tm of each protein was estimated from the maximum of a plot of the first derivative of θ222 against temperature. Helical content was estimated from the CD spectra using K2D3 software (Louis-Jeune et al., 2012).
Analytical ultracentrifugation (AUC)
Sedimentation velocity experiments were conducted at 891,800 RCF using a Beckman Proteomelab XL-A analytical ultracentrifuge and an AN-60 rotor. The samples were monitored by absorbance at 280 nm. Both samples were in 20 mM Tris, pH 7.4, 100 mM NaCl, 0.2 mM TCEP. Atg23 WT and Atg23[LIL] were run at 13.1 μM and 14.1 μM, respectively. The solvent density (1.0029 g/mL), viscosity (0.010169 poise), and partial specific volumes that were used for the analyses, 0.735055 mL/g (Atg23 WT) and 0.735656 mL/g (Atg23[LIL]), were calculated using SEDNTERP (http://bitcwiki.sr.unh.edu/index.php/Main_Page) (Laue et al., 1992). The sedimentation coefficients and apparent molecular weights were calculated from c(s) analysis using SEDFIT (Schuck et al., 2002).
In-line size-exclusion chromatography with small-angle X-ray scattering (SEC-SAXS)
In-line SEC-SAXS was performed at SIBYLS at the Advanced Light Source (Classen et al., 2013). Atg23 WT was injected over a PROTEIN KW-802.5 SEC column equilibrated with 20 mM Tris, pH 8, 200 mM NaCl, 0.2 mM TCEP. Multi-angle light scattering (MALS) data were also collected using a Wyatt Dawn Helos MALS detector and with a Wyatt Optilab rEX refractometer. MALS data were processed in Wyatt Astra 6. Primus was used to plot scattering data and the Guinier approximation, which was used to confirm that no aggregation was present in the sample. GNOM was used to determine the pair distribution function, Pr, and maximum particle dimension, Dmax (Konarev et al., 2003). Ten independent envelopes were calculated from the scattering curve using GASBOR (Svergun et al., 2001). The ten envelopes were aligned in SUPCOMB and then averaged and filtered using DAMAVER (Kozin and Svergun, 2001; Volkov and Svergun, 2003). The averaged envelope and filtered envelopes are shown as mesh representations. The SAXS data for Atg23 and Atg23[LIL] were deposited in the SASBDB as SASDL39 and SASDL49, respectively.
Tethering assay
GUVs were made using the gel-assisted formation method (Weinberger et al., 2013). Briefly, chloroform stocks of yeast polar lipids and the fluorescent lipid Rhod PE (99:1 mol%; Avanti Polar Lipids 810150) were aliquoted in a glass tube. Chloroform was dried under a stream of nitrogen followed by drying under vacuum (1 h). The dried lipids were resuspended in fresh chloroform to make a 1 mM lipid mixture. Polyvinyl alcohol (PVA, Mw = 14500; Sigma, 814894) was dissolved in boiling water to make a 5% stock and degassed in a vacuum chamber. Twenty μL of the PVA solution was spread into a thin film on a glass coverslip kept on a heating block set to 55°C. Ten μL of the chloroform-dissolved lipid mix was then spread on the dried PVA film and kept overnight to remove the residual chloroform. The dried PVA-lipid films were peeled from the glass coverslips and transferred to a 1.5-mL microcentrifuge tube. Films were hydrated in 300 μL assay buffer (20 mM Tris, pH 7.4, 100 mM NaCl) for 1 h, and GUVs were released by gentle tapping. The PVA films were removed using a pipette.
To generate fluorescently-tagged Atg23, purified Atg23 was labeled with a 10-fold molar excess of thiol-reactive Alexa Fluor 488 (Invitrogen, A10254) for 1 h at room temperature. The labeling reaction was quenched with 1 mM DTT and the free dye was removed by extensive dialysis against 20 mM Tris, pH 7.4, 100 mM NaCl. Atg23 was resolved on a 12% SDS-PAGE gel to confirm labeling of the protein and removal of the free dye.
For tethering experiments, GUVs (200 μL) were mixed with 1 μM protein for 10 min at room temperature followed by incubation with 20 μL of 400-nm extruded liposomes containing YPL:DiD (99:1 mol%; Thermo D7757) for 10 min at room temperature. Liposomes were freshly extruded before tethering experiments. Imaging was performed using LabTek chambers. Prior to imaging, LabTek chambers were treated with 3 M NaOH for 10 min and then washed extensively with milliQ water. NaOH-treated chambers were then passivated by incubation with BSA (1 mg/mL). BSA was removed by washing with assay buffer. The GUV mix containing Atg23 and liposomes was transferred to a BSA-passivated LabTek chamber and allowed to settle for 5 min prior to imaging.
GUVs were imaged using a Nikon TiE microscope fitted with a Yokogawa CSU-W1 spinning disk system using a 100 X, 1.45 NA oil immersion objective. Images were acquired with a Photometrics Prime BSI sCMOS camera using the NIS elements software. Images were analyzed using Fiji (Schindelin et al., 2012) and statistical analysis was performed using GraphPad Prism (version 5.0a).
QUANTIFICATION AND STATISTICAL ANALYSIS
Tukey HSD tests as used in Figures 3B, 4C, 4D–4G, 4H, 6B–6D, and 6E were performed using R version 4.0.5. Student’s two-sample t-tests as used in Figures 3H, S5D, S6B, and S6C were performed using Excel 2021. The corresponding figure legends detail n values, precision measures and significance thresholds.
Statistical analysis for the liposome sedimentation assays were performed in GraphPad Prism Version 9. Statistical details of the experiments including statistical tests, standard deviations, p-values, and number of independent experiments are reported in the figure legends with their corresponding experiment. Each experiment was performed in triplicate or quadruplicate as indicated in the figure legend. The results were averaged and plotted with error bars representing the standard deviations of the three (n = 3) or four (n = 4) independent experiments. Data were compared using an ordinary one-way ANOVA with Tukey’s multiple comparison test (Figure 3F, n = 3), two-way ANOVA with Sidak’s multiple comparison test (Figure 3D, n = 3), or a two-way ANOVA with Tukey’s multiple comparison test (Figure 3J, n = 4; Figure S5B, n = 4).
Tethering experiments were performed in triplicates. The detailed statistical analyses for the tethering assay are described in Figure 5 legend. Briefly, tethering was analyzed by measuring the DiD intensity profile along the entire GUV surface. GUVs with DiD intensity profiles showing peaks that were three-fold over the mean background intensity were categorized as GUVs with tethered liposomes. Data from three independent repeats (n = 3) were pooled and plotted as a scatterplot with bar. Error bars represent standard deviation. n indicates the total number of GUVs analyzed. A minimum of 73 GUVs were analyzed for each condition to quantify percent GUVs with tethered liposomes (nGUVs≥73). A minimum of 23 GUVs were analyzed for each condition to quantify normalized peaks per micron (nGUVs≥23). Data were analyzed using ordinary one-way ANOVA with Dunnett’s multiple comparisons test. **** indicates p < 0.0001. *** indicates p < 0.0005. All analyses were performed using Graphpad Prism Version 9.1.1 (223).
Supplementary Material
Highlights.
Atg23 is an extended homodimeric helical protein and a membrane-binding protein
Atg23 is a vesicle-tethering protein
Dimerization is critical for Atg23 function in yeast
ACKNOWLEDGMENTS
This work was supported by R35GM131919 and the Protein Folding Disease FastForward Initiative, University of Michigan to D.J.K. and R35GM128663 and P20GM113132 to M.J.R. SEC-SAXS data were collected at the SIBYLS beamline (ALS LBNL in Berkeley CA), which operates through funding from the IDAT program (US DOE BES and BER contract DE-AC02-05CH11231), NIGMS P30 ALS-ENABLE (P30 GM124169), and NCI’s SBDR CA92584. We thank Ann Lavanway of the Dartmouth Life Sciences Light Microscopy Facility for microscopy training and help with data acquisition. We thank Emily Grush for her help with graphic design.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110702.
REFERENCES
- Backues SK, Orban DP, Bernard A, Singh K, Cao Y, and Klionsky DJ (2015). Atg23 and Atg27 act at the early stages of Atg9 trafficking in S. cerevisiae. Traffic 16, 172–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum P, Thorner J, and Honig L (1978). Identification of tubulin from the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U S A 75, 4962–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A, and Klionsky DJ (2013). Autophagosome formation: tracing the source. Dev. Cell 25, 116–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Cheong H, Song H, and Klionsky DJ (2008). In vivo reconstitution of autophagy in Saccharomyces cerevisiae. J. Cell Biol. 182, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, and Klionsky DJ (2008). New insights into autophagy using a multiple knockout strain. Autophagy 4, 1073–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman PJ, and Dominguez R (2018). BAR domain proteins-a linkage between cellular membranes, signaling pathways, and the actin cytoskeleton. Biophys. Rev. 10, 1587–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chano T, Ikegawa S, Kontani K, Okabe H, Baldini N, and Saeki Y (2002). Identification of RB1CC1, a novel human gene that can induce RB1 in various human cells. Oncogene 21, 1295–1298. [DOI] [PubMed] [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, et al. (2013). Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr. 46, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks RO, Rao T, and Mason JM (2011). Truncation, randomization, and selection: generation of a reduced length c-Jun antagonist that retains high interaction stability. J. Biol. Chem. 286, 29470–29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL, Reggiori F, Baba M, Kovacs AL, and Seglen PO (2011). Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy 7, 935–956. [DOI] [PubMed] [Google Scholar]
- Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, and Inagaki F (2010). Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J. Biol. Chem. 285, 1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia CM, Tan XF, Lian T, Rana MS, Zhou W, Christenson ET, Lowry AJ, Faraldo-Gomez JD, Bonifacino JS, Jiang J, et al. (2020). Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 31, 107837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubas A, and Dikic I (2021). A guide to the regulation of selective autophagy receptors. FEBS J. 289, 75–89. [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, and Hegemann JH (2002). A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, and Klionsky DJ (2006). Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J. Cell Biol. 175, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honorato RV, Koukos PI, Jimenez-Garcia B, Tsaregorodtsev A, Vaerlato M, Giachetti A, Rosato A, and Bonvin AMJJ (2021). Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front. Mol. Biosci. 8, 729513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, and Lamark T (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cueva R, and Yaver DS (1992). Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol 119, 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MH, and Svergun DI (2003). PRIMUS: a Windows PC-based system for smallangle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. [Google Scholar]
- Kozin MB, and Svergun DI (2001). Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41. [Google Scholar]
- Laue TM, Shah B, Ridgeway TM, and Pelletier SL (1992). Computer-aided interpretation of sedimentation data for proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science, Harding SE, Horton JC, and Rowe AJ, eds. (Royal Society of Chemistry; ), pp. 99–125. [Google Scholar]
- Legakis JE, Yen WL, and Klionsky DJ (2007). A cycling protein complex required for selective autophagy. Autophagy 3, 422–432. [DOI] [PubMed] [Google Scholar]
- Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, and Zhao Y (2012). Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat. Commun. 3, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Louis-Jeune C, Andrade-Navarro MA, and Perez-Iratxeta C (2012). Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80, 374–381. [DOI] [PubMed] [Google Scholar]
- Lowe M (2019). The physiological functions of the golgin vesicle tethering proteins. Front. Cell Dev. Biol. 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Otomo C, and Otomo T (2019). The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 8, e45777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Yamamoto H, Kinch LN, Garza CM, Takahashi S, Otomo C, Grishin NV, Forli S, Mizushima N, and Otomo T (2020). Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 27, 1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, and Reggiori F (2010). An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 190, 1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, and Reggiori F (2010). Atg9 reservoirs, a new organelle of the yeast endomembrane system? Autophagy 6, 1221–1223. [DOI] [PubMed] [Google Scholar]
- Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, Sugita Y, Nomura N, Iwata S, Ohsumi Y, et al. (2020). Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 27, 1185–1193. [DOI] [PubMed] [Google Scholar]
- Matscheko N, Mayrhofer P, Rao Y, Beier V, and Wollert T (2019). Atg11 tethers Atg9 vesicles to initiate selective autophagy. PLoS Biol. 17, e3000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiling-Wesse K, Bratsika F, and Thumm M (2004). ATG23, a novel gene required for maturation of proaminopeptidase I, but not for autophagy. FEMS Yeast Res. 4, 459–465. [DOI] [PubMed] [Google Scholar]
- Mizianty MJ, Peng Z, and Kurgan L (2013). MFDp2: accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord. Proteins 1, e24428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. [DOI] [PubMed] [Google Scholar]
- Noda NN, Kobayashi T, Adachi W, Fujioka Y, Ohsumi Y, and Inagaki F (2012). Structure of the novel C-terminal domain of vacuolar protein sorting 30/autophagy-related protein 6 and its specific role in autophagy. J. Biol. Chem. 287, 16256–16266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, and Klionsky DJ (2008). The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods Enzymol. 451, 33–42. [DOI] [PubMed] [Google Scholar]
- Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, Ohsumi Y, and Noda NN (2019). Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 26, 281–288. [DOI] [PubMed] [Google Scholar]
- Parkhouse R, Ebong IO, Robinson CV, and Monie TP (2013). The N-terminal region of the human autophagy protein ATG16L1 contains a domain that folds into a helical structure consistent with formation of a coiled-coil. PLoS One 8, e76237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popelka H, Damasio A, Hinshaw JE, Klionsky DJ, and Ragusa MJ (2017). Structure and function of yeast Atg20, a sorting nexin that facilitates autophagy induction. Proc. Natl. Acad. Sci. U S A 114, E10112–E10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragusa MJ, Stanley RE, and Hurley JH (2012). Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 151, 1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Perna MG, Hofmann B, Beier V, and Wollert T (2016). The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat. Commun. 7, 10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Komatsu M, Finley K, and Simonsen A (2012). Autophagy: more than a nonselective pathway. Int. J. Cell Biol. 2012, 219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Tucker KA, Stromhaug PE, and Klionsky DJ (2004). The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 6, 79–90. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, and Emr SD (1988). Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell Biol. 8, 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P, Perugini MA, Gonzales NR, Howlett GJ, and Schubert D (2002). Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutter M, Giavalisco P, Brodesser S, and Graef M (2020). Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell 180, 135–149.e14. [DOI] [PubMed] [Google Scholar]
- Sekito T, Kawamata T, Ichikawa R, Suzuki K, and Ohsumi Y (2009). Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 14, 525–538. [DOI] [PubMed] [Google Scholar]
- Sheffield P, Garrard S, and Derewenda Z (1999). Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39. [DOI] [PubMed] [Google Scholar]
- Short B, Haas A, and Barr FA (2005). Golgins and GTPases, giving identity and structure to the Golgi apparatus. Biochim. Biophys. Acta 1744, 383–395. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, and Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, and Noda NN (2018). Biophysical characterization of Atg11, a scaffold protein essential for selective autophagy in yeast. FEBS Open Bio 8, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI, Petoukhov MV, and Koch MH (2001). Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigg J, Gutwin K, Keating AE, and Berger B (2011). Multicoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PLoS One 6, e23519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truebestein L, and Leonard TA (2016). Coiled-coils: the long and short of it. Bioessays 38, 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KA, Reggiori F, Dunn WA Jr., and Klionsky DJ (2003). Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J. Biol. Chem. 278, 48445–48452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, Reinisch KM, and Melia TJ (2019). ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, and Bonvin A (2016). The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. [DOI] [PubMed] [Google Scholar]
- Volkov VV, and Svergun DI (2003). Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li W, Liu S, and Xu J (2016). RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 44, W430–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger A, Tsai FC, Koenderink GH, Schmidt TF, Itri R, Meier W, Schmatko T, Schroder A, and Marques C (2013). Gel-assisted formation of giant unilamellar vesicles. Biophys. J. 105, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ZN, Scott AL, Dowell RD, and Odorizzi G (2018). PI(3,5)P2 controls vacuole potassium transport to support cellular osmoregulation. Mol. Biol. Cell 29, 1718–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, and Ohsumi Y (2012). Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 198, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen WL, Legakis JE, Nair U, and Klionsky DJ (2007). Atg27 is required for autophagy-dependent cycling of Atg9. Mol. Biol. Cell 18, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, and Klionsky DJ (2005a). Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol. Biol. Cell 16, 1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, and Klionsky DJ (2005b). Autophagy: molecular machinery for self-eating. Cell Death Differ. 12, 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini G, and Martens S (2016). Mechanisms of selective autophagy. J. Mol. Biol. 428, 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Wang H, Lam SK, Gao C, Wang X, Cai Y, and Jiang L (2013). A BAR-domain protein SH3P2, which binds to phosphatidylinositol 3-phosphate and ATG8, regulates autophagosome formation in Arabidopsis. Plant Cell 25, 4596–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SAXS data for Atg23 and Atg23[LIL] were deposited in the Small Angle Scattering Biological DataBank (SASBDB) as SASDL39 and SASDL49, respectively. These data are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.