

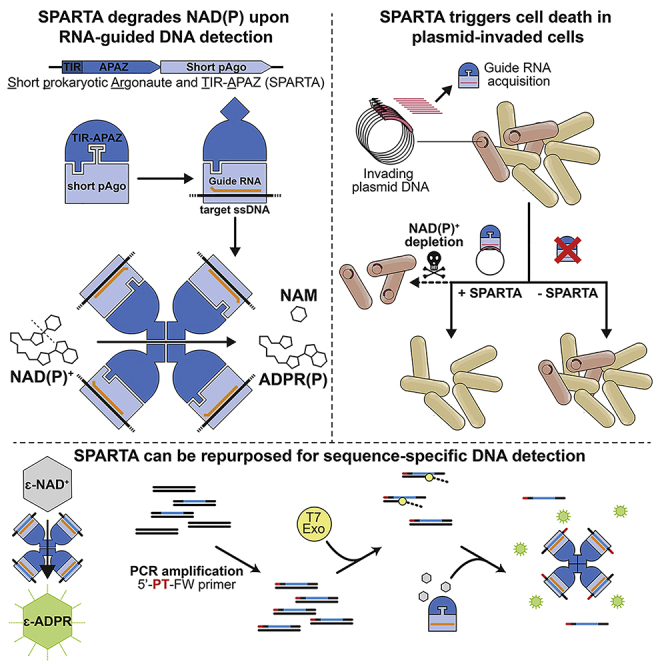
Summary
Argonaute proteins use single-stranded RNA or DNA guides to target complementary nucleic acids. This allows eukaryotic Argonaute proteins to mediate RNA interference and long prokaryotic Argonaute proteins to interfere with invading nucleic acids. The function and mechanisms of the phylogenetically distinct short prokaryotic Argonaute proteins remain poorly understood. We demonstrate that short prokaryotic Argonaute and the associated TIR-APAZ (SPARTA) proteins form heterodimeric complexes. Upon guide RNA-mediated target DNA binding, four SPARTA heterodimers form oligomers in which TIR domain-mediated NAD(P)ase activity is unleashed. When expressed in Escherichia coli, SPARTA is activated in the presence of highly transcribed multicopy plasmid DNA, which causes cell death through NAD(P)+ depletion. This results in the removal of plasmid-invaded cells from bacterial cultures. Furthermore, we show that SPARTA can be repurposed for the programmable detection of DNA sequences. In conclusion, our work identifies SPARTA as a prokaryotic immune system that reduces cell viability upon RNA-guided detection of invading DNA.
Keywords: Argonaute, APAZ, TIR, prokaryotic immunity, short pAgo, RNAi, small RNA, Ago, host defense, abortive infection
Graphical abstract
Highlights
-
•
Short prokaryotic Argonaute and TIR-APAZ proteins form a heterodimeric SPARTA complex
-
•
RNA-guided ssDNA target binding unleashes TIR domain-mediated NAD(P)ase activity
-
•
Highly transcribed multicopy invading DNAs activate SPARTA, which triggers cell death
-
•
SPARTA can be repurposed for sequence-specific NA detection
Upon RNA-guided detection of invading DNA, short prokaryotic Argonaute and the associated proteins trigger NAD(P)+ depletion, leading to the removal of invaded cells from bacterial cultures.
Introduction
Argonaute proteins recruit small (∼15–30 nucleotide [nt]) single-stranded guide oligonucleotides to bind complementary single-stranded target sequences. Eukaryotic Argonaute proteins (eAgos) use guide RNAs to bind and/or cleave target RNA, a process that underlies RNA interference (RNAi) (Peters and Meister, 2007; Vaucheret, 2008). Prokaryotes encode homologs of eAgos (pAgos) but lack other proteins required for RNAi (Shabalina and Koonin, 2008). Most characterized pAgos share a fixed four-domain composition (N-PAZ-MID-PIWI) with eAgos. These “long” pAgos interfere with the invading nucleic acids (NAs), such as plasmids (Olovnikov et al., 2013; Swarts et al., 2014a) and bacteriophages (Kuzmenko et al., 2020). Long pAgos have also been implicated in genome decatenation (Jolly et al., 2020) and stimulation of homologous recombination (Fu et al., 2019; Lee et al., 2021). In contrast to eAgos, certain long pAgos utilize single-stranded (ss)DNA guides to target invading DNA sequences (Kuzmenko et al., 2020; Swarts et al., 2014a, 2015; Zander et al., 2017). Other long pAgos use guide RNAs to act on RNA and/or DNA targets (Doxzen and Doudna, 2017; Kaya et al., 2016; Kropocheva et al., 2021; Lapinaite et al., 2018; Li et al., 2021; Olovnikov et al., 2013). While our knowledge of the function and mechanisms of long pAgos is rapidly expanding, the majority of pAgos (58%) consist of poorly studied “short” pAgos (Ryazansky et al., 2018).
Short pAgos comprise the MID and PIWI domains only (Makarova et al., 2009; Ryazansky et al., 2018; Swarts et al., 2014b). Akin to certain eAgos and long pAgos, short pAgos lack the catalytic tetrad required for target cleavage (Swarts et al., 2014b), which suggests that they are catalytically inactive. However, MID domain residues required for guide 5′-phosphate (5′-P) binding are conserved in short pAgos (Ryazansky et al., 2018), and the MID and PIWI domains alone are sufficient to facilitate guide-mediated target binding in truncated long Agos (Dayeh et al., 2018; Ma et al., 2005; Miyoshi et al., 2016; Parker et al., 2009). This suggests that small pAgos mediate 5′-P guide-mediated target binding.
Short pAgos are encoded in the same operon as (or are fused to) a domain that is named “analog of PAZ” (APAZ) (Makarova et al., 2009). However, APAZ domains are homologous to the N domain of long pAgos rather than to their Piwi-Argonaute-Zwille (PAZ) domain (Burroughs et al., 2013; Willkomm et al., 2017). APAZ domains are commonly fused to SIR2, Toll/interleukin-1 receptor (TIR)-like, or DUF4365 domains (Ryazansky et al., 2018; Swarts et al., 2014b). It was predicted that these domains are nucleases that compensate for the loss of catalytic activity of the associated short pAgos (Hegge et al., 2018), and it was recently proposed that a short pAgo complexed with a DUF4365-APAZ domain mediates RNA-guided DNA cleavage (Kim et al., 2020). However, most short pAgo-associated domains are fused to a SIR2 or TIR domain. The function and mechanisms of these short pAgos remain unexplored.
Here, we describe the role and mechanism of short prokaryotic Argonaute TIR-APAZ (SPARTA) systems. We demonstrate that short pAgo and TIR-APAZ form a heterodimeric SPARTA complex. Upon small RNA-guided ssDNA target binding, four SPARTA heterodimers form an oligomer in which the NAD(P)ase activity of the TIR domain is unleashed. In vivo, SPARTA is activated in the presence of highly transcribed multicopy plasmid DNA. This results in cellular NAD(P)+ depletion and consequentially reduces the viability of the cells harboring the invading plasmid DNA. As a result, uninvaded cells outcompete invaded cells, which drives the plasmid DNA to extinction. Finally, we show that SPARTA can be repurposed to detect DNA sequences of choice.
Results
Short pAgos cluster with APAZ-domain-containing proteins only
To determine whether short pAgos genetically cluster with proteins other than the APAZ-domain-containing proteins, we first identified 283 short pAgo homologs in the RefSeq database containing at least scaffold-level assemblies. All the identified short pAgos comprise the middle (MID) and P-element-induced wimpy testis (PIWI) domains and lack a catalytic DEDX tetrad. Short pAgos form four phylogenetic subclades (Figure 1A), which we named S1A (SIR2-APAZ-pAgo fusions), S1B (operon with SIR2-APAZ and pAgo), S2A (operons with (Mrr-)TIR-APAZ and pAgo), and S2B (operons mainly consisting of pAgo and DUF4365-APAZ or DHS-like-APAZ). We coined the term “short pAgo system” to indicate a short pAgo and its associated APAZ-domain-containing protein (clades S1B, S2A, and S2B) or a fusion thereof (clade S1A). In all subclades, the APAZ-domain-containing protein is the only protein that specifically associates with pAgo. This suggests that pAgo systems function as stand-alone systems or rely on common host factors for their activity.
Figure 1.

Phylogenetic analysis of short pAgo systems
(A) Maximum-likelihood-based phylogenetic analysis of 283 short pAgo proteins. Clade S1A: SIR2-APAZ-pAgo fusion (SPARSA-A). Clade S1B: operon with pAgo and SIR2-APAZ (SPARSA-B). Clade S2A: operon with pAgo and TIR-APAZ (SPARTA) or Mrr-TIR-APAZ. Clade S2B: operon with pAgo and APAZ fused to one of various domains, including DUF4365 or DHS-like domains. CrtAgo: Crenotalea thermophila pAgo. MapAgo: Maribacter polysiphoniae pAgo. Green circles indicate bootstrap values >75%. SIR2: InterPro IPR039444. TIR: InterPro IPR000157. Mrr: InterPro IPR007560. DUF4365: InterPro IPR025375. DHS-like: InterPro IPR029035.
(B) Tanglegram of phylogenetic trees of 56 short pAgo proteins (left) and associated TIR-APAZ proteins (right). Dashed lines indicate coevolved proteins; red lines indicate differential evolution. Acidobacter: Acidobacteria (Actinomycetota). CrtTIR-APAZ: C. thermophila TIR-APAZ. MapTIR-APAZ: M. polysiphoniae TIR-APAZ.
See also Figure S1.
We named S2A systems “SPARTA systems,” which stands for "short prokaryotic Argonaute TIR-APAZ systems." In one subclade of SPARTA systems, a putative modification requiring restriction endonuclease (Mrr) domain is fused to the TIR-APAZ protein (Mrr-TIR-APAZ; Figure 1A). Short pAgo and associated TIR-APAZ proteins coevolved in Bacteroidetes and in one lineage of Proteobacteria (Figures 1B and S1A), which suggests that they function in conjunction. In another lineage of Proteobacteria, short pAgo and TIR-APAZ show distinct evolutionary patterns. This suggests that there might be a functional difference between SPARTA systems found in distinct phylogenetic clades.
Figure S1.

Distribution and conservation of catalytic residues in SPARTA systems, related to Figure 1
(A) Circle diagram indicating bacterial phyla (abbreviated) and classes in which SPARTA systems are found. Aci, acidobacteria; Bac, bacteroidetes; Pro, proteobacteria.
(B) Multiple sequence alignment of TIR domains from SPARTA systems with various prokaryotic and eukaryotic TIR domains that demonstrate NADase activity. Green and red triangles indicate catalytic residues important for NADase activity; red triangle indicates residue substituted in CrtTIR-APAZE77A and MapTIR-APAZE77A.
(C) Multiple sequence alignment of MID domains from different prokaryotic Argonaute proteins and their preferred guide types. Green triangles indicate residues important for 5′-phosphate binding.
It has been suggested that APAZ-associated domains compensate for the lack of nuclease activity in short pAgos (Hegge et al., 2018). However, in both eukaryotic and prokaryotic immune systems, TIR domains degrade NAD(P)+ (Horsefield et al., 2019; Johnson et al., 2020; Morehouse et al., 2020; Ofir et al., 2021; Wan et al., 2019). The catalytic residues required for NAD(P)ase activity are conserved in TIR-APAZ proteins (Figure S1B), which suggests that they might have a similar mode of action. Based on the conservation of the 5′-P binding residues in the pAgo MID domain (Figure S1C), we hypothesized that in SPARTA systems, short pAgos facilitate guide-mediated target binding. Consequently, TIR-APAZ could either mediate endonuclease activity (compensating for the lack of endonuclease activity of short pAgo) or NAD(P)+ nucleosidase activity (based on homology to other TIR domains).
Short pAgo and TIR-APAZ proteins form an heterodimeric complex
To biochemically characterize SPARTA systems, we coexpressed SPARTA proteins from Maribacter polysiphoniae (MapAgo/MapTIR-APAZ) or Crenotalea thermophila (CrtAgo/CrtTIR-APAZ) in Escherichia coli (Figure 2A). These pAgo and TIR-APAZ proteins share 81% and 73% sequence identity, respectively. For both systems, N-terminally 6xHis-maltose binding protein (MBP)-tagged TIR-APAZ could be used to pull down untagged short pAgo. After the removal of the 6xHis-MBP tag, the short pAgo and TIR-APAZ of both systems remained complexed throughout multiple chromatography steps (Figure 2B). Size exclusion chromatography-multi-angle light scattering (SEC-MALS) analysis of the purified complexes reveals that short pAgo and TIR-APAZ form a heterodimeric complex with a 1:1 stoichiometry (Figure 2C).
Figure 2.

TIR-APAZ NAD(P)ase activity is controlled by short pAgo
(A) Schematic diagram of CrtSPARTA and MapSPARTA operon structure and domain organization.
(B) TIR-APAZ binds to short pAgo. SDS-PAGE of copurified TIR-APAZ and short pAgo proteins.
(C) TIR-APAZ and short pAgo form SPARTA complexes with a 1:1 stoichiometry. The molecular weight (MW) determined by size exclusion chromatography-multi-angle light scattering corresponds to the theoretical MW of heterodimeric CrtAgo-CrtTIR-APAZ (111.4 kDa) and MapAgo-MapTIR-APAZ (111.1 kDa) complexes.
(D and E) TIR-APAZ is an NADase, and short pAgo inhibits TIR-APAZ activity. LC-MS ion count (D) and spectra (E) for NAD+ after incubation with CrtSPARTA proteins. NAD+ is converted to nicotinamide (NAM) and adenosine diphosphate ribose (ADPR) upon incubation with CrtTIR-APAZ but not when incubated with CrtAgo or CrtSPARTA.
(F) Reaction structural formulas of TIR-APAZ-mediated NAD(P)+ hydrolysis. Blue dotted line: scissile bond.
See also Figure S2.
TIR-APAZ converts NAD(P)+ to NAM and ADPR(P)
To determine whether SPARTA shows nuclease activity, MapSPARTA and CrtSPARTA were incubated with 5′-phosphorylated (5′-P) 21 nt-long ssRNA or ssDNA guides and complementary ssRNA or ssDNA targets. SPARTA does not facilitate guide-mediated target cleavage for any guide/target combination (Figure S2A). Next, we investigated whether TIR-APAZ shows NAD(P)ase activity. As MapTIR-APAZ is unstable when purified in the absence of MapAgo, we focused on CrtTIR-APAZ in these experiments. LC-MS and 1H NMR spectroscopy reveal that CrtTIR-APAZ is a nucleosidase that converts NAD(P)+ into noncyclic adenosine diphosphate ribose (phosphate) (ADPR[P]) and nicotinamide (NAM) (Figures 2D–2F and S2B–S2D). Other TIR domains also convert NAD(P)+ to NAM and ADPR(P) or structural analogs thereof (cADPR or v-ADPR) (Essuman et al., 2018; Horsefield et al., 2019; Morehouse et al., 2020; Ofir et al., 2021; Wan et al., 2019). By contrast, CrtAgo and CrtSPARTA show no NADase activity (Figure 2D), suggesting that short pAgo controls the activity of TIR-APAZ.
Figure S2.

CrtTIR-APAZ converts NAD(P)+ to nicotinamide and adenosine diphosphate ribose (phosphate), related to Figure 2
(A) SPARTA does not cleave nucleic acids in a guide-dependent manner. CrtSPARTA and MapSPARTA were incubated with different guides and Cy5-labeled DNA or RNA target in a 10:10:1 molar ratio (SPARTA:guide:target). Cleavage products were resolved on denaturing (7 M) urea PAGE gels.
(B) TIR-APAZ converts NAD+ into ADPR and NAM. NMR spectra of the reaction products from NAD+ converted by CrtTIR-APAZ correspond to the spectra of nicotinamide (NAM) and adenosine diphosphate ribose (ADPR).
(C) TIR-APAZ degrades NADP+ but not NADH. NMR spectra of NADP+ (upper two panels) and NADH (lower two panels) incubated with and without CrtTIR-APAZ.
(D) TIR-APAZ shows no preference for NAD+ or NADP+. NAD+ and NADP+ were mixed in equimolar ratios and incubated with CrtTIR-APAZ. NMR spectra show that during incubation with CrtTIR-APAZ, NAD+ and NADP+ are converted with a similar reaction rate.
Guide-RNA-mediated target ssDNA binding activates SPARTA
We speculated that SPARTA could be catalytically activated upon guide-mediated target binding. MapSPARTA and CrtSPARTA were incubated with various guide/target combinations (Figure 3A) and ε-NAD+, which is converted into NAM and fluorescent ϵ-ADPR by NADase activity. Apo-SPARTA, SPARTA with guide RNA or DNA alone, and SPARTA with guide RNA and target RNA show no NADase activity (Figures 3B and S3A). By contrast, both MapSPARTA and CrtSPARTA degrade NAD+ when provided with a guide RNA and a complementary target ssDNA. When the guide RNA and ssDNA are noncomplementary, SPARTA does not degrade NAD+, implying that the activation of SPARTA is sequence specific. Alanine substitution of Glu77 in the catalytic site of the TIR domain (CrtSPARTATIR-E77A) abolishes NADase activity (Figure S3B). This confirms that the TIR domain confers the nucleosidase activity of SPARTA.
Figure 3.

SPARTA degrades NAD+ upon RNA-guided ssDNA binding
(A) Schematic representation of guide (orange) and target (gray, target sequence: blue) oligonucleotides used in (B) and (C).
(B and C) MapSPARTA or CrtSPARTA was mixed with guide and target oligonucleotides in a 1:1:2 molar ratio and incubated with ϵ-NAD+. Graphs indicate the maximum change in fluorescence over time. The average of three technical replicates is shown; error bars indicate standard deviations.
(B) SPARTA is preferentially activated upon by guide RNA-mediated ssDNA target binding. (C) SPARTA prefers 5′-P guides over 5′-OH guides. CrtSPARTA 5′-P guide samples are identical in (B) and (C).
(D) SPARTA oligomerizes upon guide RNA-mediated target ssDNA binding. MapSPARTA was incubated with guide RNA and target ssDNA in a 1:1:1 molar ratio and subjected to size exclusion chromatography-multi-angle light scattering analysis. Colored backgrounds indicate fractions that were tested for NADase activity (E). Control without nucleic acids is identical to that in Figure 2C.
(E) SPARTA is catalytically activated upon oligomerization. SEC-MALS fractions (D) were incubated with ϵ-NAD+, and change in fluorescence was determined over time.
(F) Proposed mechanism for SPARTA activation. TIR-APAZ and associated short pAgo form a catalytically inactive heterodimeric SPARTA complex. Guide RNA-mediated target ssDNA binding induces conformational changes that facilitate oligomerization. Guide/target-bound SPARTA heterodimers form a complex with a 4:4:4:4 (TIR-APAZ:short pAgo:guide:target) stoichiometry in which the NAD(P)ase activity of the TIR domain is unleashed.
See also Figure S3.
Figure S3.

SPARTA is preferentially activated upon guide RNA-mediated ssDNA target binding, related to Figure 3
(A) Kinetic data related to Figure 3B. Graph indicates total fluorescence measured over time.
(B) The catalytic mutant CrtSPARTATIR-E77A does not show NADase activity even when incubated with guide RNA and target ssDNA. Graph indicates the maximum change in fluorescence over time.
(C) Kinetic data related to Figure 3C. Graph indicates total fluorescence measured over time.
(D) Effect of incubation temperature on the activity of MapSPARTA (left) or CrtSPARTA (right) incubated with guide RNA and target ssDNA. Graph indicates end-point fluorescence intensity after 60 min of incubation.
(E and F) Effect of guide RNA length on the activity of MapSPARTA (E) or CrtSPARTA (F). Left and middle graphs indicate the maximum change in fluorescence over time. Right panels show kinetic data related to the middle panels and indicate total fluorescence measured over time.
(G and H) MapSPARTA is activated by RNA/DNA hybrids (G) and guide RNA-mediated detection of small dsDNA targets (H). Graphs indicate the maximum change in fluorescence over time. Lower panels show the kinetic data related to the upper panels and indicate total fluorescence measured over time. For all panels, CrtSPARTA, CrtSPARTATIR-E77A, or MapSPARTA was mixed with guide and target oligonucleotides in a 1:1:2 molar ratio and incubated with ϵ-NAD+. The average of three technical replicates is shown, error bars indicate standard deviations.
Albeit at lower efficiencies than guide RNA/target ssDNA, guide ssDNA/target RNA combinations also activate MapSPARTA (3.4-fold lower activity) and CrtSPARTA (1.5-fold lower activity), and guide ssDNA/target ssDNA combinations activate CrtSPARTA (3.4-fold lower activity) (Figures 3B and S3A). However, the guide 5′ group (5′-P or 5′-OH) has no effect for guide ssDNA/target RNA combinations (Figures 3C and S3C), suggesting that guide ssDNAs might not be properly bound by the MID domain. By contrast, NAD+ turnover is 8.5-fold (MapSPARTA) or 3.6-fold (CrtSPARTA) lower for 5′-OH guide RNAs compared with 5′-P guide RNAs. This implies that SPARTA is preferentially activated by 5′-P guide RNA-mediated target ssDNA binding.
Furthermore, guide RNAs that are 15–50 nt in length facilitate target ssDNA-mediated SPARTA activation (Figures S3E and S3F). This suggests that guide RNA 3′ end-binding is not required for SPARTA activation. MapSPARTA is also activated by longer RNA/DNA hybrids (Figure S3G), possibly because they resemble guide RNA/target ssDNA ligands. dsRNA and dsDNA ligands do not trigger robust SPARTA activation, but when guide RNAs are provided, short (≤60 bp) but not long (≥152 bp) dsDNA targets trigger the activation of SPARTA (Figure S3H). Possibly, their small size allows for unwinding, which facilitates recognition by the SPARTA-guide RNA complex. Combined, these results show that TIR-mediated NAD(P)ase activity of SPARTA is preferentially activated by 5′-P guide RNA-mediated binding of ssDNA targets.
SPARTA is catalytically activated upon oligomerization
As TIR-APAZ is catalytically active in the absence of pAgo, we speculated that it might be released from the SPARTA complex upon activation. However, SEC-MALS analysis of MapSPARTA pre-incubated with a guide RNA/ssDNA target NA duplex revealed two distinct elution peaks with molecular weights (MWs) larger than that of apo-MapSPARTA (Figure 3D). Conjugate analysis showed that the first peak corresponds to MapSPARTA bound to the NA duplex in a 1:1 stoichiometry (MapSPARTA:NA duplex), whereas the second peak corresponds to an oligomeric complex with a 4:4 stoichiometry. Elution fractions containing 4:4 complexes showed substantial NADase activity directly upon incubation with ε-NAD+, whereas the fraction containing 1:1 complexes only becomes active upon prolonged incubation (Figure 3E). We speculate that the 1:1 complexes oligomerize during the assay, which renders them active. In conclusion, guide RNA-mediated ssDNA target binding triggers the formation of an oligomeric complex with a 4:4:4:4 stoichiometry (TIR-APAZ:short pAgo:guide:target) in which the NAD(P)ase activity of the TIR domain is unleashed (Figure 3F).
SPARTA recruits guide RNAs to target highly expressed genes
To shed light on the biological role of SPARTA, we examined the NAs it associates with in vivo. The 5′ group-dependent 32P NA labeling revealed that MapSPARTA copurifies with 5′-P RNAs that are predominantly ∼15–25 or ∼40 nt in length (Figure 4A). MapSPARTA also copurifies with 5′-OH RNAs of various lengths and with DNAs that are >50 nt in length (Figures 4C and S4A).
Figure 4.

MapSPARTA associates with guide RNAs targeting highly transcribed genes
(A) MapSPARTA associates with 5′-phosphorylated (5′-P) small RNAs. Nucleic acids that copurified with MapSPARTA were [γ-32P]-ATP labeled, treated with DNase I or RNase A, and resolved on a denaturing polyacrylamide gel. nt, nucleotides; tRFs, tRNA fragments.
(B and C) Length distribution of small RNA (B) and DNA (C) copurified with MapSPARTA as determined by sequencing.
(D) Percentages of long RNA or small RNA reads that align to specific genomic or plasmid sequences. RNA was extracted from E. coli expressing MapSPARTA (E. coli total) or purified MapSPARTA (MapSPARTA-associated). rRNA-derived reads are excluded.
(E) Small RNA associated with MapSPARTA have a bias for 5′-adenosine bases. Nucleotide bias of small RNAs copurified with MapSPARTA (left) or extracted from E. coli (right).
(F) Correlation between MapSPARTA-associated small RNA and E. coli-extracted long RNA sequences.
(G) Correlation between MapSPARTA-associated small RNA and DNA sequences.
Figure S4.

CrtSPARTA associates with small RNAs targeting highly transcribed genes, related to Figure 4
(A) Left: CrtPARTA associates with 5′-phosphorylated (5′-P) small RNAs. Right: MapSPARTA and CrtSPARTA associate with 5′-OH nucleic acids of undefined lengths. Nucleic acids that copurified with CrtSPARTA or MapSPARTA were [γ-32P]-ATP labeled, treated with DNase I or RNase A, and resolved on a denaturing polyacrylamide gel. nt, nucleotides.
(B) Length distribution of small copurified with CrtSPARTA as determined by sequencing.
(C) Percentages of long RNA-seq or small RNA-seq reads that align to specific genomic or plasmid sequences. RNA was extracted from E. coli expressing CrtSPARTA (E. coli total) or from purified CrtSPARTA (CrtSPARTA associated). rRNA-derived reads are excluded from this graph.
(D) CrtSPARTA-associated small RNA sequences and E. coli-extracted long RNA sequences correlate (Pearson correlation coefficient r > 0.99, p < 10−99).
(E) Small RNA associated with CrtSPARTA have a bias for 5′ adenosine bases. Nucleotide bias of small RNAs copurified with MapSPARTA (left) or extracted from E. coli (right).
See also Table S1.
A preference for a specific base at the 5′ end of the guide is a general feature of Argonaute proteins (Frank et al., 2010; Jolly et al., 2020; Olovnikov et al., 2013; Swarts et al., 2014a). Indeed, small RNA-seq revealed that MapSPARTA-associated small RNAs have a bias toward an adenine base at the first (73%) and second (64%) positions at the 5′ end, which was not observed for E. coli-extracted small RNAs (Figure 4E). This implies that these small RNAs are specifically bound by MapSPARTA. No such bias was observed for long RNAs associated with MapSPARTA, which suggests that these RNAs copurify nonspecifically.
The abundance of specific sequences in the transcriptome and corresponding MapSPARTA-associated small RNA pools are strongly correlated (Figure 4F; Pearson correlation coefficient r > 0.99, p < 10−99). This implies that the abundance of RNA sequences in the transcriptome determines the chance of SPARTA being loaded with corresponding small RNAs. Of note, the MapSPARTA-associated 39–41-nt-long RNAs are tRNA fragments (tRFs). tRFs also associate with RsAgo (Olovnikov et al., 2013) and eAgos (Burroughs et al., 2013; Maute et al., 2013; Nakanishi, 2021), but the biological relevance is unclear.
The correlation between the abundance of mRNAs and MapSPARTA-associated small RNAs suggests that MapSPARTA recruits small RNAs to target highly expressed genes. Interestingly, plasmid-encoded mRNA is more abundant than genome-encoded mRNA in the transcriptome (Figure 4D), which is probably caused by a combination of strong promoters and the multicopy nature of the MapSPARTA expression plasmid (15–20 copies per cell). Of note, the high number of reads assigned to E. coli genes lacI and malE are likely transcribed from the plasmid-encoded lacI and the MBP-tag of MBP-MapTIR-APAZ, which share respectively 100% and 99.8% sequence identity to the lacI and malE on the genome of E. coli. The results of CrtSPARTA-associated RNA sequencing were almost identical to that of MapSPARTA-associated RNA (Figure S4), but the length of the associated small RNAs was less well defined and no tRF peak was observed for CrtSPARTA.
Sequencing of MapSPARTA-associated DNA confirmed that it associates with both plasmid and genomic DNA and showed a relatively weak but highly significant correlation between specific DNA and small RNA sequence abundance (Figure 4G; Pearson correlation coefficient r > 0.32, p < 10−99). In line with the observation that SPARTA is preferentially activated by 5′-P guide RNAs-mediated ssDNA in vitro, these results suggest that in vivo SPARTA recruits 5′-P guide RNAs with 5′-adenosine bases from the transcriptome to target highly transcribed DNA.
SPARTA induces cell death in the presence of plasmid DNA
Based on its association with plasmid-targeting small RNA guides in vivo, we speculated that SPARTA could detect the invading plasmid DNA and subsequently deplete NAD+ and NAD(P)+. NAD+ and NADP+ are central in various metabolic pathways, and their depletion might induce lower fitness, cell dormancy, or programmed cell death, as observed in other eukaryotic and prokaryotic immune systems (Johnson et al., 2020; Morehouse et al., 2020; Ofir et al., 2021; Wan et al., 2019).
MapSPARTA was selected for in vivo characterization in E. coli as, in contrast to CrtSPARTA, it shows robust activity at 37°C in vitro (Figure S3B). To mimic genomic expression levels, MapSPARTA was expressed from a bacterial artificial chromosome (BAC) with a copy number of 1 (Shizuya et al., 1992). NAD (NAD+ + NADH) and NADP (NADP+ + NADPH) levels, cell density, (OD600 nm) and viability (defined here as the number of colony-forming units [CFUs]) were determined at 4 h and/or 20 h after the induction of MapSPARTA expression. After 20 h, MapSPARTA expression resulted in a modest reduction of NAD (−30%, p < 0.01) and an increase in NADP (+18%, p < 0.001) compared with MapSPARTATIR-E77A expression, but no change in OD600 nm or CFU was observed (S5A–S5D). This implies that MapSPARTA expression does not lower cell viability under these conditions. By contrast, in the presence of the high copy number plasmid pUC-empty (500–700 copies per cell; Vieira and Messing, 1991), expression of BAC-encoded MapSPARTA resulted in a strong reduction of NAD (4 h: −85%, 20 h: −89%, p < 0.001), NADP (4 h: −79%, 20 h: −91%, p < 0.001) and cell viability (4 h: −27%, n.s., 20 h: −49%, p < 0.01) (Figures 5A and S5A). This shows that the presence of a high copy number plasmid triggers SPARTA-mediated NAD(P) degradation and induces cell death.
Figure 5.

Highly transcribed multicopy plasmids trigger SPARTA-mediated cell death
(A) Plasmid DNA triggers SPARTA-mediated NAD depletion and cell death. NAD levels and cell viability were determined in E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A in the presence or absence of the high copy number plasmid pUC-empty.
(B) High transcription of plasmid DNA enhances SPARTA-mediated cell death. Cell viability was determined in E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A either in the absence or in the presence of a pUC-derivate from which mRFP was expressed (pUC-mRFP), transcribed but not translated (pUC-mRFPΔRBS), or encoded but not transcribed (pUC-mRFPΔT7-ΔRBS).
(C) Mutations in T7 RNA polymerase-encoding gene facilitate the escape of SPARTA-mediated cell death. Upper panel: OD700 nm and mRFP fluorescence were measured in E. coli cultures containing pUC-mRFP and an empty BAC or a BAC expressing MapSPARTA or MapSPARTATIR-E77A. Lower panel: viable clones expressing MapSPARTA have point mutations or insertions of transposable elements (IS2) in the gene encoding T7 RNA polymerase.
(D) SPARTA activation relies on plasmid copy number and transcription. NAD levels and cell viability were determined in E. coli cultures containing a BAC expressing MapSPARTA in the absence or presence of plasmids with different origin of replications (ori) from which mRFP was transcribed (mRFPΔRBS) or encoded but not transcribed (mRFPΔT7-ΔRBS).
(E) Transcription of RNA complementary to plasmid DNA enhances SPARTA-mediated cell death. NAD levels and cell viability were determined in E. coli cultures containing a BAC expressing MapSPARTA or MapSPARTA and mRFP in the absence or presence of plasmids lacking or containing a nontranscribed mRFP gene. Graphs show the average of three (A–D) or two (E) biological replicates. Error bars indicate standard deviation. n.s. = not significant; ∗p < 0.05, ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S5.
Figure S5.

Plasmid DNA triggers SPARTA-mediated NAD(P) depletion and cell death, related to Figure 5
(A) Plasmid DNA triggers SPARTA-mediated NAD(P) depletion and cell death. NAD and NADP levels and viability were determined in E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A in the presence or absence of the high copy pUC-empty plasmid. Panels showing NAD levels at t = 4 and cell viability at t = 20 are identical to those in Figure 5A.
(B) Native RNA polymerase-mediated transcription of plasmid-encoded genes enhances SPARTA-mediated cell death. Cell viability was determined in E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A either in the absence of plasmid DNA or in the presence of a pUC-derivate from which mRFP is transcribed but not translated (pUC-mRFPXylS-Pm) or encoded but not transcribed (pUC-mRFPXylS-ΔPm). The Pm promoter induces transcription by native RNA polymerases. Graphs show the average of three biological replicates. Error bars indicate standard deviations. n.s = not significant; ∗∗p < 0.01; ∗∗∗p < 0.001.
High transcription of plasmid DNA enhances SPARTA activity
As SPARTA-associated guide RNA abundance is correlated with transcript abundance (Figure 4F), we speculated that high transcription of plasmid-encoded genes might trigger a stronger SPARTA-mediated response. To test this, BAC-encoded MapSPARTA was expressed in the presence of one of three pUC-derivates encoding monomeric red fluorescent protein (mRFP), and viability was determined 4 h after induction of MapSPARTA expression (Figure 5B). Compared with cells expressing MapSPARTA without plasmid, no significant loss of viability is observed in presence of pUC when mRFP lacks a promoter and ribosomal binding site (RBS) (pUC-mRFPΔT7-ΔRBS). By contrast, viability is drastically lowered when mRFP transcription is under the control of a T7 promoter (pUC-mRFP; −98%, p < 0.001), even when an RBS is absent (pUC-mRFPΔRBS; −92%, p < 0.001). Also, when mRFP transcription is under the control of the XylS/Pm promoter (Brautaset et al., 2009), which relies on native RNA polymerases for transcription, cell viability is reduced compared with cells in which mRFP is not transcribed (pUC-mRFPXylS/pm, −59%, p < 0.001; Figure S5B). This excludes the possibility that MapSPARTA activation relies specifically on transcription by phage-derived T7 RNA polymerase. In conclusion, high transcription of plasmid-encoded genes results in stronger SPARTA-induced cell death.
Overnight cultures of mRFP-expressing strains show bright mRFP fluorescence in the presence of MapSPARTATIR-E77A but not in the presence of MapSPARTA (−80%, p < 0.01; Figure 5C). Plating and subsequent sequencing revealed that in all viable MapSPARTA-expressing clones, the gene encoding T7 RNA polymerase was disrupted either by a point mutation or insertion of an IS2 transposable element (Siguier et al., 2006; Figure 5C). As the loss of the T7 RNA polymerase results in loss of mRFP transcription, these “escape mutants” elude SPARTA-mediated cell death.
SPARTA activation relies on plasmid copy number and transcription
SPARTA associates with genome-targeting guide RNAs (Figure 4) but does not lower cell viability in the absence of plasmids (Figure 5A). We, therefore, hypothesized that the copy number of the target DNA plays a role in the formation of activated SPARTA complexes. To test this, MapSPARTA was expressed in the presence of a plasmid with one of three different origin of replications (ori), resulting in a low (pSC101 ori; ∼5 copies), medium (pET ori, 15–20 copies) or high (pUC ori, 500–700 copies) copy number (Hasunuma and Sekiguchi, 1977; Vieira and Messing, 1991). Each of these plasmids encodes an mRFP gene without a promoter (no transcription; mRFPΔT7-ΔRBS) or with T7 promoter (transcription; mRFPΔRBS).
NAD levels and cell viability were determined respectively 4h and 20h after induction of MapSPARTA expression but were not significantly reduced in cultures with the low-copy plasmid, even when mRFP is transcribed (Figure 5D). This could explain why MapSPARTA does not lower cell viability in the absence of a plasmid DNA despite acquiring guides that target the single-copy genome of E. coli. The presence of the medium-copy plasmid results in lowered NAD (−96%, p < 0.001) and cell viability (−93%, p < 0.001) only when mRFP is transcribed. Presence of the high copy number plasmid without mRFP transcription lowers both NAD levels (84%, p < 0.001) and cell viability (49%, p < 0.001), and mRFP transcription further reduces cell viability (−96%, p < 0.01). These results imply that both plasmid copy number and transcription of plasmid-encoded genes play a role in the SPARTA activation.
However, higher plasmid copy numbers also result in more transcripts of plasmid-encoded genes. To determine whether SPARTA is activated by increased availability of target DNA independent of higher transcript abundance, T7 promoter-controlled mRFP was placed on the MapSPARTA expression BAC (copy number = 1). Transcription of mRFP from the BAC does not result in the reduction of NAD levels or viability in the absence of plasmid DNA or in the presence of a medium-copy pET plasmid, even if it encodes a nontranscribed mRFP gene (Figure 5E). While the presence of a high-copy pUC plasmid itself results in lower NAD levels (−69%, p < 0.001) and cell viability (−24%, p < 0.05), the inclusion of a nontranscribed mRFP gene on that plasmid further reduces NAD levels (−88% p < 0.001) and cell viability (−55%, p < 0.001). This implies that BAC-encoded transcripts facilitate SPARTA activation in the presence of high copy number DNA containing a sequence complementary to that transcript. We conclude that both high transcription levels and plasmid DNA abundance stimulate the formation of a higher number of activated SPARTA complexes, resulting in lower NAD levels and consequential cell death.
SPARTA confers selective pressure against plasmid DNA
Given that SPARTA induces cell death in the presence of high-copy plasmids, we investigated whether SPARTA interferes with plasmid transformation or propagation. MapSPARTA does not significantly reduce the transformation efficiency of pUC-empty or pUC-mRFPXylS/pm (Figure S6A). Possibly, SPARTA is not activated because the plasmid copy number is presumably low directly after transformation. Furthermore, SPARTA does not induce plasmid loss in E. coli cells harboring pUC-empty (Figure S6B). This suggests that SPARTA is unable to protect individual cells against invading DNA: it cannot prevent it from entering or remove it from individual cells after entry.
Figure S6.

Effect of SPARTA on plasmid DNA and bacteriophages, related to Figure 6
(A) E. coli cells expressing MapSPARTA or MapSPARTATIR-E77A or harboring an empty BAC were transformed with pUC-empty or pUC-mRFPXylS-Pm, and the transformation efficiency was calculated. The average of three biological replicates is shown; error bars indicate standard deviations.
(B) E. coli cultures harboring pUC-empty and expressing MapSPARTA or MapSPARTATIR-E77A or harboring an empty BAC were grown without selective pressure (Ampicillin) to maintain pUC-empty. The fraction of cells containing pUC-empty was determined on a daily basis for nine consecutive days. The average of four biological replicates is shown; error bars indicate standard deviations.
(C) E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A or harboring an empty BAC were grown in agar plates in the presence of various bacteriophages, and the number of plaques relative to the strain harboring an empty BAC was determined. The average of three biological replicates is shown; data points indicate results from individual replicates.
(D) E. coli cultures expressing MapSPARTA or MapSPARTATIR-E77A or harboring an empty BAC were grown in the presence of various bacteriophages (bacteriophage type indicated above each graph), and the OD600 nm was monitored over time. Bacteriophages were added to E. coli cells with the following multiplicity of infection: T1—1.4 ∗ 10−7; T4—2 ∗ 10−7; T7—1 ∗ 10−7; Lambda—3 ∗ 10−7; Nami—1 ∗ 10−7. The average of three biological replicates is shown; shadings indicate standard deviations.
(E) E. coli JM109(DE3) cultures expressing MapSPARTA or MapSPARTATIR-E77A or harboring an empty vector were grown in the presence of chronic-infecting phage M13. The number of infectious M13 phage virions excreted into the bacterial culture was determined at multiple timepoints using E. coli ER2738 in which phage M13 is lytic. The average of three biological replicates is shown; error bars indicate standard deviations.
We hypothesized that SPARTA provides immunity at the population-level instead. To test this, “mixed cultures” consisting of plasmid-free (4%) and pUC-empty-containing E. coli cells (96%) were cultivated (Figure 6A). In the presence of an empty BAC or a BAC expressing MapSPARTATIR-E77A, the percentage of pUC-empty-containing cells remains stable at around 96% (Figure 6B). By contrast, cultures expressing MapSPARTA are dominated by plasmid-free cells after 3 days (Figure 6B, p < 0.001). This implies that SPARTA provides a selective advantage for plasmid-free cells by driving the plasmid-invaded cells to extinction.
Figure 6.

SPARTA depletes plasmid-harboring cells from bacterial cultures
(A) Graphical schematic representation of the mixed-culture competition experiment (B).
(B) Cultures of MapSPARTA-expressing E. coli cells that contain pUC-empty or those that were plasmid-free were mixed in a 24:1 ratio, cultivated in LB, and the percentage of plasmid-containing cells was determined daily. Cultures of E. coli cells with an empty BAC or a BAC expressing MapSPARTATIR-E77A were used as control. Graph shows the average of six biological replicates.
See also Figure S6.
SPARTA does not provide robust defense against selected bacteriophages
Infection by lytic bacteriophages (from here: phages) leads to rapid replication (and thus high copy number) and high transcription of phage genomes (Wang et al., 1996). However, no defense is observed for E. coli strains expressing MapSPARTA or MapSPARTATIR-E77A in plaque assays with phage T1, T4, T7, Nami, and Lambda-vir (Figure S6C). Also, no protection was observed in culture collapse studies with phage T4 and T7 (Figure S6D). Culture collapse studies with phage T1 and Nami showed a small but significant (p < 0.005) reduction in collapse for strains expressing MapSPARTA or MapSPARTATIR-E77A and for phage Lambda-vir in the strain expressing MapSPARTA (p < 0.03; Figure S6D). Furthermore, both MapSPARTA and MapSPARTATIR-E77A show a small but significant (p < 0.001) reduction in the release of infectious virions from the chronic-infecting phage M13 (Figure S6E). In all cases, the observed defense is limited compared with the known anti-phage systems (Doron et al., 2018; Gao et al., 2020). The observation that the SPARTA-mediated phage defense does not rely on NAD(P)ase activity in most cases suggests that MapSPARTA is not properly activated in response to the phages used.
SPARTA can be repurposed for sequence-specific nucleic acid detection
To determine whether SPARTA could be programmed to detect NAs akin to various CRISPR-Cas effector enzymes (Chen et al., 2018; Gootenberg et al., 2017, 2018; Li et al., 2019; Steens et al., 2021), the specificity and sensitivity of CrtSPARTA and MapSPARTA were determined. CrtSPARTA activity is attenuated by single mismatches between the guide RNA and target ssDNA at guide positions 5–10 and is further lowered by double mismatches (Figure 7A). Double mismatches at middle-region positions 5–6 to 9–10 and 14–15 result in low to no detectable CrtSPARTA activity, while mismatches toward the 5′ end or 3′ end of the guide are well tolerated. MapSPARTA is less sensitive to mismatches under the tested conditions and also for MapSPARTA double mismatches in the middle-region lower activity most (Figure 7B). Both CrtSPARTA and MapSPARTA detect target ssDNA in the low nM range (Figures 7C and 7D).
Figure 7.

PARTA systems can be used for nucleic acid detection
(A and B) Effect of mismatches on SPARTA activation. CrtSPARTA (A) or MapSPARTA (B) were mixed with guide RNA and target ssDNA containing single or double mismatches and incubated with ε-NAD+. Graphs indicate the change in fluorescence over time relative to a control target ssDNA without mismatches.
(C and D) SPARTA detects target ssDNA at nM concentrations. CrtSPARTA (C) or MapSPARTA (D) were mixed with guide RNA in a 1:1 molar ratio and target ssDNA was added at different concentrations. After the addition of ε-NAD+, total fluorescence was measured over time (corrected with no target control).
(E) Schematic representation of SPARTA-mediated DNA detection. Activated SPARTA converts ε-NAD+ to fluorescent ε-ADPR. DNA is PCR amplified using a phosphorothioate (PT)-modified forward (FW) primer and a nonmodified reverse (RV) primer. PCR products are treated with T7 exonuclease (T7 exo) yielding ssDNA products detectable by SPARTA.
(F) SPARTA combined with PCR and T7 exonuclease facilitates the detection of dsDNA at aM levels. MapSPARTA was mixed with guide RNA in a 1:1 molar ratio, and PCR-amplified T7 exonuclease-digested target DNA was added. After the addition of ε-NAD+, total fluorescence was measured over time. All graphs show the average of three technical replicates. Error bars (A and B) or shadings (C, D, and F) indicate standard deviations.
See also Figure S7.
Although the sensitivity of SPARTA is comparable to the detection limit of Cas12a and Cas12b (Chen et al., 2018; Li et al., 2019), it is not sensitive enough to achieve clinically relevant attomolar detection thresholds (Jet et al., 2021; Oh et al., 2016; Pan et al., 2020; To et al., 2010). CRISPR-based detection tools are often combined with DNA amplification techniques to improve their detection limits. As a proof of concept, we enhanced SPARTA sensitivity and enabled dsDNA detection based on a previously developed method (Harrington et al., 2018; Figure 7E): to-be-detected DNA was PCR amplified using forward (FW) primers with a 5′-phosphorothioate (PT) group. Subsequent or simultaneous incubation with T7 exonuclease (T7 exo) removes the reverse (RV) strand lacking 5′-PT groups, facilitating MapSPARTA-mediated dsDNA detection at aM sensitivity (Figures 7F and S7). This shows that SPARTA can be combined with NA amplification techniques and used to detect both ssDNA and dsDNA sequences in a sequence-specific manner.
Figure S7.

SPARTA can be used to detect dsDNA, and SPARSA systems degrade NAD+in vivo, related to Figure 7
(A) SPARTA combined with colony PCR and subsequent T7 exonuclease treatment facilitates the detection of dsDNA. MapSPARTA was mixed with guide RNA in a 1:1 molar ratio, and colony PCR-amplified DNA treated with T7 exonuclease was added. After the addition of ε-NAD+, total fluorescence was measured over time. No target negative control and 100 nM ssDNA positive control are identical to those in Figure 7F.
(B) SPARTA mixed with target dsDNA, and T7 exonuclease simultaneously facilitates the detection of dsDNA at aM levels. MapSPARTA was mixed with guide RNA in a 1:1 molar ratio, and target DNA and T7 exonuclease were added. After the addition of ε-NAD+, total fluorescence was measured over time. No target negative control and 100 nM ssDNA positive control are identical to those in Figure 7F.
(C) MapSPARTA is not activated by small molecules, and small molecules do not enhance guide RNA/target ssDNA-mediated activity of MapSPARTA. MapSPARTA was mixed with guide and target oligonucleotides in a 1:1:2 molar ratio and incubated with ϵ-NAD+. Graphs indicate the maximum change in fluorescence over time.
(D) Left: schematic diagram of the operon structure and domain organization of SPARTA, SPARSA-1A, and SPARSA-1B systems. Right: SPARSA SIR2-APAZ proteins lower total NAD when heterologously expressed in E. coli. Total NAD levels were determined in E. coli cultures harboring an empty expression vector (pML1) or a pML1 expression vector encoding clade S2A CrtTIR-APAZ, clade S1A Joostella marina JomSIR2-APAZ-Ago, or clade S1B Xanthomonas vesicatoria XavSIR2-APAZ. Graphs show the average of three (A and C) or two (B) technical replicates or two biological replicates (D). Error bars and shadings indicate standard deviations.
Discussion
In this study, we describe the mechanism and function of SPARTA, a prokaryotic immune system that belongs to the short pAgo family. Short pAgo and its associated TIR-APAZ protein form a catalytically inactive heterodimeric SPARTA complex. Guide RNA-mediated binding of a target ssDNA facilitates the formation of an oligomeric SPARTA-NA complex with a 4:4:4:4 stoichiometry (TIR-APAZ:short pAgo:guide:target) with NAD(P)ase activity (Figure 3). Further structure-function studies are required to unravel the molecular mechanism for guide-mediated target binding and subsequent activation of SPARTA.
TIR domain activation by oligomerization has also been observed in the prokaryotic immune systems Pycsar and STING (Morehouse et al., 2020; Tal et al., 2021) and in-plant receptor proteins (Ma et al., 2020; Martin et al., 2020), and other prokaryotic immune systems also rely on TIR-mediated NAD+ depletion (Ka et al., 2020; Morehouse et al., 2020; Ofir et al., 2021). Some of these systems are activated by small molecules (Morehouse et al., 2020; Ofir et al., 2021; Tal et al., 2021), but SPARTA is not activated by NAD+, NADP+, NAM, ADPR(P), c-ADPR, or other cyclic monophosphate nucleotides, and these small molecules do not enhance guide RNA/target ssDNA-mediated SPARTA activation (Figure S7C). While different immune systems might rely on distinct mechanisms for invader recognition, TIR-mediated NAD(P)+ depletion is an immune strategy found in both prokaryotes and eukaryotes (Essuman et al., 2017; Hopkins et al., 2021; Horsefield et al., 2019; Killackey et al., 2019; Wan et al., 2019).
In vivo, SPARTA acquires 5′-P guide RNAs from the transcriptome (Figure 4). Guide RNAs and mRNA abundance are correlated, implying that SPARTA does not specifically recruit invading DNA-targeting guides. Similar observations were made for RsAgo, a catalytically inactive long pAgo (Olovnikov et al., 2013). This suggests that RNA-guided pAgos from different phylogenetic clades rely on similar mechanisms for guide acquisition, which remain to be determined.
Multicopy plasmid DNA trigger SPARTA-mediated NAD(P)+ depletion and consequential cell death (Figure 5), while single or low copy number plasmids do not trigger significant levels of SPARTA-mediated NAD+ degradation. This explains why SPARTA does not cause cell death when associated with genome-targeting guide RNAs. High transcription of plasmid-encoded genes or of sequences homologous to plasmid DNA enhances SPARTA-mediated NAD+ degradation and consequential cell death, presumably because SPARTA can acquire more plasmid-targeting guides from the transcriptome. In theory, this allows for the genomic transcription of RNA to program SPARTA to target complementary multicopy invader DNA. It remains to be determined whether enhanced SPARTA activation by multicopy target DNA is a consequence of higher target sequence abundance only or whether also their replication rate plays a role, as this might increase the accessibility of the target DNA.
Highly transcribed multicopy elements are not only a metabolic burden to the cell but may also indicate plasmid or viral replication and impending spread to neighboring cells. Unless high transcription is prevented through mutational escape (which might neutralize the invader), SPARTA lowers cell viability. When SPARTA triggers cell death, this could prevent the spread of the invader to protect the remainder of the bacterial population. Although this does not protect the infected cell itself, it provides population-level immunity akin to abortive infection systems (Lopatina et al., 2020). Reduced cell viability could also induce a state of dormancy in which the invader cannot replicate efficiently (Meeske et al., 2019; Page and Peti, 2016). Bacteriostasis does not only allow other immune systems to remove the invader (Dimitriu et al., 2022) but also lets uninvaded bacterial cells outcompete infected cells (Figure 6).
SPARTA provides minor protection against phages which, strikingly, does not rely on its NADase activity (Figure S6). Other catalytically inactive pAgos also interfere with invading DNA (Kuzmenko et al., 2020; Olovnikov et al., 2013). This suggests that pAgos do not critically rely on catalytic activity for DNA interference but might provide a minimal level of defense by binding invading DNA, which could hamper replication and/or transcription. In nature, many phage replication cycles are (partially) lysogenic and/or phages (temporarily) exist extrachromosomal, akin to plasmids (Correa et al., 2021). Such infection strategies might provide a larger window of opportunity for SPARTA to become activated and remove the invader DNA from the population.
We hypothesize that short pAgos from other phylogenetic clades also control the activity of their associated APAZ-fused catalytic domains. S1A and S1B short prokaryotic Argonaute SIR2-APAZ (SPARSA) systems contain a SIR2 domain, which is a predicted NADase, akin to SPARTA TIR domains. Expression of SPARSA systems in E. coli also triggers NAD depletion (Figure S7D), suggesting a similar mode of action. Certain clade S2B APAZ proteins are fused to a DHS-like domain, which is a predicted NAD/FAD-binding domain (InterPro IPR029035) (Dym and Eisenberg, 2001), and various long pAgos are encoded in operons with SIR2-like proteins (Ryazansky et al., 2018; Swarts et al., 2014b). This suggests that NADase activity is not restricted to SPARTA and SPARSA systems but might have independently evolved several times in both short and long pAgo systems.
Finally, we provide proof of concept for SPARTA-mediated detection of DNA sequences. Upon DNA detection, SPARTA NADase activity can be monitored using fluorescence or by colorimetric cycling assays. Its limit of detection is comparable with that of CRISPR-Cas-based detection tools, but SPARTA makes use of shorter RNA guides and does not require the presence of a protospacer adjacent motif in the target sequence. Combined with pre-amplification techniques, DNA can be detected at aM detection levels. We predict that DNA can also be amplified using isothermal amplification techniques and that RNA can be detected using RV transcriptase-based amplification techniques. As such, SPARTA-based tools can potentially be used for the detection of pathogens or specific mutations in DNA. In conclusion, our study elucidates the biological role and mechanism of SPARTA systems and facilitates their implementation as a NA detection tool.
Limitations of the study
While SPARTA is preferentially activated with 5′-P guide RNAs and target ssDNA in vitro and associates with 5′-P guide RNAs in vivo, it cannot be ruled out that guide ssDNA/target RNA combinations also play a role in the activation of SPARTA.
Compared with other bacterial immune systems, SPARTA systems provide relatively little (and TIR-independent) protection against some of the phages in our panel. However, many prokaryotic immune systems are effective against certain types of phages only (Doron et al., 2018; Ofir et al., 2021). Furthermore, the laboratory experiments performed are optimal for phage propagation and might be inappropriate to observe SPARTA-mediated immunity.
SPARTA-mediated immunity might further benefit from host-specific factors for guide generation and invader targeting. Furthermore, SPARTA-generated ADPR(P) and/or NAM might function as secondary messenger molecules to trigger auxiliary host-specific proteins.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli NEB 5-alpha | New England Biolabs | C2987 |
| E. coli BL21 Star (DE3) | ThermoFisher Scientific | C601003 |
| E. coli BW25113 | Coli Genetics Stock Center | CGSC#7926 |
| E. coli BW25113 (DE3) | Gift of Prof. Dr. Ruud Weusthuis (Wageningen University) (Vuoristo et al., 2015) | N/A |
| E. coli BL21 (DE3) | ThermoFisher Scientific | EC0114 |
| E. coli R10256 | Isolated at Erasmus M.C (Rotterdam, Netherlands) | Clinical isolate |
| E. coli JM109 (DE3) | Promega | P9801 |
| E. coli K12 ER2738 | New England Biolabs | E4104 |
| Bacteriophage T1 | DSMZ | 5801 |
| Bacteriophage T4 | Gift from Elizabeth Kutter’s Lab (Evergreen Phage Lab) | N/A |
| Bacteriophage T7 | Gift from Ian Molineux’s Lab (University of Texas at Austin) | N/A |
| Bacteriophage Nami | Brouns’ Lab collection (Delft University of Technology) | MZ502380.1 |
| Bacteriophage λ-vir | Brouns’ Lab collection (Delft University of Technology) | N/A |
| Bacteriophage M13 | New England Biolabs | N4040S |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth Low Salt | Duchefa Biochemie | L1703 |
| LB Agar Low Salt | Duchefa Biochemie | L1705 |
| LB broth (used for bacteriophage experiments) | Sigma Aldrich | L3022 |
| DNase I, RNase-free | Thermo Scientific | EN0521 |
| RNase A, DNase and protease-free | Thermo Scientific | EN0531 |
| T4 polynucleotide kinase | Thermo Scientific | EK0031 |
| T7 exonuclease | New England biolabs | M0263S |
| ε-NAD | BIOLOG | N 010 |
| (c)-(di/tri)NTPs | BIOLOG | Various |
| Critical commercial assays | ||
| NEBuilder HiFI DNA assembly kit | New England biolabs | E2621 |
| NAD/NADH Cell-Based Assay Kit | Cayman Chemical | 600480 |
| NAD/NADH Quantitation Kit | Sigma-Aldrich | MAK037-1KT |
| NADP/NADPH Quantitation Kit | Sigma-Aldrich | MAK038-1KT |
| Qubit™ dsDNA HS Assay Kits | Thermo Scientific | Q32851 |
| Deposited data | ||
| Raw NGS data | This study | GEO: GSE191271 |
| Raw imaging data | This study | https://dx.doi.org/10.17632/7f4gsbhrpp.1 |
| Oligonucleotides | ||
| RNA oligos | IDT | See Table S2 |
| DNA oligos | IDT | See Table S2 |
| Genomic and recombinant DNA | ||
| Crenatolea thermophila | DSM 14807 | DSM 14807 |
| Maribacter polysiphoniae | DSM 23514 | DSM 23514 |
| pET His6 MBP TEV LIC (1M) | Addgene | 29656 |
| pBeloBAC11 | New England Biolabs | E4154 (discontinued) |
| pBbS5k-RFP | Addgene | 35285 |
| pUC-21 | Addgene | 49787 |
| Plasmid DNA | This study | See Table S2 |
| Software and algorithms | ||
| HMMER | https://www.hmmer.org | v3.3.1 |
| InterProScan | (Jones et al., 2014) | v5.51-85.0 |
| MAFFT | (Katoh and Standley, 2013) | v7.475 |
| trimAI | (Capella-Gutiérrez et al., 2009) | v1.4 |
| IQtree | (Minh et al., 2020) | v2.0.4 |
| ModelFinder | (Kalyaanamoorthy et al., 2017) | N/A |
| iTOL | https://itol.embl.de (Letunic and Bork, 2021) | v6 |
| Diamond blastp | (Buchfink et al., 2021) | v2.0.7.145 |
| Cytoscape | (Shannon et al., 2003) | v3.7.1 |
| R (phylogeny) | https://www.r-project.org/ | v4.0.3 |
| Phylogram | (Wilkinson and Davy, 2018) | v2.1.0 |
| Dendextend | (Galili, 2015) | v1.15.1 |
| R (statistics) | https://www.r-project.org/ | v4.1.0 |
| BBmap (BBtools) | (Bushnell et al., 2017) | v38.90 |
| HISAT2 | (Kim et al., 2015) | v2.1.0 |
| FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | v0.11.9 |
| FeatureCounts | (Liao et al., 2014) | v2.0.1 |
| Astra | Wyatt Technology | v8.0 |
| Compass | Bruker Daltonics | v1.2 |
| TopSpin | Bruker BioSpin GmbH | V4.1.3 |
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, Daan C. Swarts (daan.swarts@wur.nl).
Materials availability
Protein expression vectors encoding MapSPARTA (pBK086) and CrtSPARTA (pBK092) have been deposited on Addgene (plasmid numbers 183145 and 183146). Other newly generated materials used in this paper will be shared upon request.
Experimental model and subject details
NEB 5-alpha E. coli
For cloning purposes, NEB 5-alpha E. coli (New England Biolabs, C2987) was used.
E. coli BL21 star (DE3) and E. coli BL21(DE3)
For protein expression and purification E. coli BL21 Star (DE3) (ThermoFisher Scientific, C601003) transformed with plasmids encoding the protein(s) of interest was used, except when expressing proteins for nucleic acid pulldown, then E. coli BL21 (DE3) (ThermoFisher Scientific, EC0114) was used.
E. coli BW25113 and E. coli BW25113(DE3)
For in vivo investigation of the role of SPARTA systems E. coli BW25113 (Coli Genetics Stock Center, CGSC#7926) transformed with bacterial artificial chromosomes (BACs) encoding the protein(s) of interest, and pUC-empty when applicable, were used. For in vivo experiments that required T7 RNA polymerase-mediated expression of RFP from pUC-mRFP or derivatives thereof, E. coli BW25113(DE3) was used (kind gift from Prof. dr. Ruud Weusthuis (Wageningen University)) (Vuoristo et al., 2015).
E. coli JM109(DE3), E. coli K12 ER2738
For phage M13 propagation and M13 plaque experiments E. coli ER2738 (NEB, E4104) and E. coli K12 JM109(DE3) (Promega, P9801) were used, respectively.
All E. coli strains were cultivated in LB Broth Low Salt (Duchefa Biochemie, L1703) or on LB agar plates (LB Broth Low Salt (Duchefa Biochemie, L1705) supplemented with 1% agar). For experiments involving bacteriophages LB (Sigma Aldrich, L3022) was used instead. All cultivations were performed at 37 °C unless stated otherwise. Antibiotics corresponding to the plasmids used in each experiment were added to the following concentrations: ampicillin (AMP): 100 μg/mL; kanamycin (KAN): 34 μg/mL, chloramphenicol (CAM): 25 μg/mL. When required, expression was induced by adding IPTG (for protein expression and purification and for expression of mRFP during in vivo assays), m-toluic acid (for expression of mRFP during in vivo assays) or arabinose (for in vivo assays) to final concentrations of 0.25 mM, 1 mM, and 0.2% w/v, respectively.
Method details
Plasmid construction
Genes encoding short pAgo systems or individual proteins thereof were amplified from genomic DNA obtained from DSMZ (Crenatolea thermophila: DSM 14807; Maribacter polysiphoniae DSM 23514; Joostella marina: DSM 19592, Xanthomonas vesicatoria: DSM 22252) or from synthetic DNA constructs ordered from Twist Bioscience. The genes were cloned in the pET-His6-MBP-TEV-LIC cloning vector (1M), which was a kind gift of Prof. Dr. Martin Jinek (University of Zurich) (Addgene #29656). In this plasmid the genes are placed under control of a T7 promoter which allowed for expression of the proteins that are linked to an N-terminal 6x-His-Maltose Binding Protein (MBP) tag by a Tobacco Etch Virus (TEV) cleavage site-containing linker.
For in vivo experiments genes were cloned in the Bacterial Artificial Chromosome (BAC) pBeloBAC11 (New England Biolabs, E4154) under control of an araBAD promoter. For M13 experiments, the low-copy number vector pBbS5k-RFP-based vector pBK133 was used (Addgene #35285). For combined expression of short Ago and TIR-APAZ proteins, an artificial operon was constructed by inserting a 45 nt spacer containing an additional ribosomal binding site (RBS) between the two genes.
For construction of plasmids (with different copy numbers) with mRFP expression pET-His6-MBP-TEV-LIC, pBK133 and pUC-21 (Addgene #49787) were used.
All cloning procedures, including minor modifications of RBSs and introducing point mutations, were performed using the NEBuilder HiFi DNA Assembly kit (New England Biolabs, E2621), subsequent DNA cleanup (Zymo) and electroporation or heat-shock transformation into NEB 5-alpha E. coli cells (New England Biolabs).
Plasmids and primers for cloning used in this study are given in Table S2.
Protein purification
Individual SPARTA proteins and SPARTA complexes with N-terminal 6x-his-MBP-tags were heterologously expressed in E. coli BL21 Star (DE3) and purified by a combination of Ni2+-affinity, Amylose-affinity, heparin cation exchange, and size exclusion chromatography. Plasmids used for protein expression were transformed into E. coli BL21 Star (DE3). A single colony was used to inoculate a 10 mL LB culture which was incubated o/n at 37 °C at 180 RPM. The o/n culture was used to inoculate 8x750 mL LB medium which was incubated at 37 °C at 150 RPM. When an OD600 nm of ∼0.25 was reached, the temperature of the incubator was switched to 18 °C and after 1 h (when OD600 nm reached ∼0.6), protein expression was induced by addition of IPTG. Protein expression took place at 18 °C for 16 hours. Cells were harvested by centrifugation at 5000 x g at 4 °C for 15 min. Cells were resuspended in NiNTA lysis buffer (500 mM NaCl, 20 mM Imidazole, protease inhibitors (200 μg/mL AEBSF, 1 μg/mL pepstatin), 20 mM HEPES pH 7.5) and stored at -80 °C until used for protein purification. After thawing, cells were lysed by sonication (QSONICA Q700A-220 sonicator with ½” tip; amp 50%, 1s ON/2s OFF, 5 minutes total ON time) and the lysate was centrifuged for 30 min at 30,000 x g at 4 °C. The clarified lysate was applied to a 5 mL HisTrap HP column (Cytiva Life Sciences). The column was washed with ∼6 column volumes (CV) NiNTA buffer A (500 mM NaCl, 5 mM Imidazole, 20 mM Tris pH 8) or until A280 nm of the eluate was below 250 a.u. The protein was eluted in NiNTA buffer B (500 mM NaCl, 350 mM Imidazole, 20 mM Tris pH 8). Fractions containing the protein of interest were pooled, and loaded on a 20 mL Amylose resin (New England Biolabs) column. The column was washed with ∼3x CV Amylose wash buffer (500 mM NaCl, 1 mM DTT, 20 mM Tris pH 8) or until the A280 nm was ∼0 a.u. The protein was eluted in Amylose elution buffer (500 mM NaCl, 1 mM DTT, 10 mM maltose, 20 mM Tris pH 8). Fractions containing the protein of interest were pooled, EDTA was added to a final concentration of 2 mM, and per TEV protease was added in a 1:50 (w/w) ratio. The sample was dialyzed in SnakeSkin dialysis tubing (10kDa MWCO, Thermo Scientific) against dialysis buffer (250 mM KCl, 1 mM DTT, 2 mM EDTA, 20 mM HEPES pH 7.5) for 16 h. Cleavage of the MBP tag by TEV was confirmed by SDS-PAGE and Coomassie Brilliant Blue (CBB) staining. The sample was diluted 1:1 with 20 mM HEPES pH 7.5 and loaded on a HiTrap Heparin column (Cytiva Life Sciences). The column was washed with 98.7% ion-exchange buffer I (100 mM KCl, 1 mM DTT, 8 mM HEPES pH 7.5) and 1.3% ion-exchange buffer II (2.5 M KCl, 1 mM DTT, 20 mM HEPES pH 7.5) until the A280 nm was ∼0. To protein was eluted with ion-exchange buffer II by applying a gradient from 1.3% to 50% over a total volume of 60 mL. Peak fractions were analyzed by SDS-PAGE and CBB staining and fractions containing the protein of interest were combined. The sample was concentrated to ∼1 mL using centrifugal filter units (Amicon) according to the protocol of the manufacturer. The sample was centrifuged for 10 min at 16,000 x g at 4°C and the protein was fractionated on a custom 200 mL Superdex 200 Prep Grade resin (Cytiva Life Sciences) column, which was equilibrated in and eluted with SEC buffer (500 mM KCl, 1 mM DTT, 20 mM HEPES pH 7.5). Peak fractions were analyzed by SDS-PAGE and CBB staining and fractions containing the protein of interest were combined. The sample was concentrated to the desired concentration using centrifugal filter units (Amicon) according to the protocol of the manufacturer. Samples were aliquoted, flash frozen in liquid nitrogen, and stored at -80 °C until further use.
Nucleic acid extraction
CrtSPARTA and MapSPARTA were purified as described above, but E. coli BL21(DE3) was used instead of E. coli BL21 Star (DE3), expression cultures (750 mL for CrtSPARTA, 8x750mL for MapSPARTA) were incubated at 37 °C throughout the procedure, and cells were harvested 3 h after induction of expression. SPARTA complexes were purified using the NiNTA and Amylose resin purification steps only. All buffer solutions used were as described above but instead contained 125 mM NaCl and they were supplemented with 2 mM MnCl2 and 2 mM MgCl2 each. After elution from the Amylose resin column and pooling, the SPARTA complex was concentrated to 10 μM using centrifugal filter units (Amicon) according to the protocol of the manufacturer. Samples were flash frozen in liquid nitrogen and stored at -80 °C until further use.
Further centrifugation steps were performed in a tabletop centrifuged cooled to 4 °C, at 16000 x g. To extract nucleic acids co-purified with the complexes, 360 μL protein solution was supplemented with EDTA (10 mM final concentration) and 40 uL proteinase K 20 mg/ml (New England Biolabs). The sample was incubated for two hours at RT. 400 μL of Roti phenol/chloroform/isoamyl alcohol was added to the mixture, followed by brief vortexing and centrifugation for 15 minutes. The upper aqueous phase was isolated and 0.1 volumes of 1M sodium acetate, 3 volumes 100 % ethanol and 10 μL linear polyacrylamide (Alfa Aesar) were added. This mixture was vortexed briefly and incubated at -20 °C for 16 hours. Samples were centrifuged for 30 min and the supernatant was removed from the pellet. The pellet was washed with 500 μL 70% ethanol (pre-cooled to -20 °C). The pellets containing the copurified nucleic acids were dried for 10 min at 50 °C, and pellets were resuspended in 50 μL free H2O.
Purified nucleic acids were [γ-32P]-ATP labelled with T4 polynucleotide kinase (PNK; Thermo Scientific) in exchange- or forward-labelling reactions. After stopping the reaction by incubation at 75 °C for 10 min, the labelled oligonucleotides were separated from free [γ-32P] ATP using a Sephadex G-25 column (GE Healthcare). Labelled nucleic acids were incubated with nucleases (RNase A, DNase and protease-free (Thermo Scientific, EN0531), or DNase I, RNase-free (Thermo Scientific, EN0521) for 30 min at 37 °C. After nuclease treatment, samples were mixed with Loading Buffer (95% (deionized) formamide, 5 mM EDTA, 0.025% SDS, 0.025% bromophenol blue and 0.025% xylene cyanol), heated for 5 min at 95 °C and resolved on 15% denaturing (7M Urea) polyacrylamide gels. Radioactivity was captured from gels using phosphor screens and imaged using a Typhoon FLA 7000 laser-scanner, GE Healthcare).
RNA and DNA sequencing and analysis
RNA was sequenced by GenomeScan (Leiden, he Netherlands). Total RNA was isolated from E. coli after 3h of MapSPARTA or CrtSPARTA expression at 37°C, or was extracted from purified MapSPARTA or CrtSPARTA complexes as described above. All small RNA and long RNA libraries were generated by GenomeScan. DNA was extracted from purified MapSPARTA complexes as described above, and pre-treated with RNase A, DNase and protease free (Thermo Scientific). The remaining DNA was purified using the MagMAX mirVana Total RNA Isolation Kit (Thermo Fisher), using the protocol of the manufacturer to isolate RNA from serum and plasma samples but excluding both DNAse treatment steps. Subsequently the samples were treated with T4 PNK (Thermo Fisher, EK0031) according to the protocol of the manufacturer, and the DNA was purified again using the MagMAX mirVana Total RNA Isolation Kit. A library of each sample was generated using the NEB Next small RNA library prep set for illumina (NEB) according to the protocol of the manufacturer, but using a DNA oligo with same sequence instead of the 5’ SR adapter. The reverse transcriptase treatment was omitted and during the PCR amplification treatment samples were first incubated for 15 minutes at 70°C without addition of SR and index primers to facilitate to facilitate dsDNA formation.
All libraries were sequenced by GenomeScan using Illumina NovaSeq6000 sequencing with paired-end reads and 150bp read length. Paired-end small RNA and DNA reads were merged and adapter sequences were trimmed using BBmap (BBtools) (Bushnell et al., 2017). Processed reads of all sequencing libraries were aligned to the genome of E. coli BL21 (GenBank: CP053602.1) and to the expression plasmid (pBK086 or pBK094) using HISAT2 (Kim et al., 2015). Length and sequence distribution of small RNAs were analysed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) using a library with determined minimum and maximum lengths of respectively 15 and 60 nucleotides. FeatureCounts (Liao et al., 2014) was used to assign reads to genomic features.
Size exclusion chromatography – multi-angle light scattering
Purified MapSPARTA complex was diluted to a 1 mg/ml concentration incubated for 15 min at 37°C either without nucleic acids, or in 1:1 molar ratio with a 5’-phosphorylated guide (oBK084) in SEC-MALS buffer (125 mM KCl, 2 mM MgCl2, 20 mM HEPES pH7.5). Subsequently, target DNA (oBK382) was added in a 1:1 molar ratio and samples were incubated for 30 min at 37°C. CrtSPARTA was diluted to a 1 mg/ml concentration in SEC-MALS buffer without nucleic acids and incubated for 30 min at 55°C. After incubation samples were resolved at RT on a Superdex 200 Increase 10/300 GL column (Cytiva Life Sciences) connected to a 1260 Infinity II HPLC system (Agilent) using SEC-MALS buffer. The eluate was subjected to Multi-Angle Light Scattering using an Optilab 1090 Differential Refractive Index detector (Wyatt Technology) and a miniDawn 1065 Multi-Angle Light Scattering system (Wyatt Technology). The data were analyzed using Astra 8.0 (Wyatt Technology). The conjugate analysis method in Astra was used to determine the contribution of protein and nucleic acid in the eluting species, using dn/dc values of 0.185 mL/g and 0.17 mL/g for protein and nucleic acid, respectively, and using theoretical extinction coefficients at 280 nm of 1.5655 mL/(mg∗cm) and 10.766 mL/(mg∗cm) for MapSPARTA and RNA/DNA duplex, respectively. Subsequently, fractions containing guide/target-complexed MapSPARTA were incubated with 25 μM ε-NAD+ to determine NADase activity, as described below.
In vitro nucleic acid cleavage assays
Duplicate or triplicate reactions were made of CrtSPARTA, CrtSPARTATIR-E77A or MapSPARTA mixed with various guide oligonucleotides and Cy5-labeled target oligonucleotides in reaction buffer on ice resulting in a total reaction volume of 15 μL with the following final concentrations: 2 μM CrtSPARTA, CrtSPARTATIR-E77A or MapSPARTA, 10 mM HEPES pH 7.5, 125 mM KCl, 2 mM MnCl2, and 2 mM MgCl2. Final guide and target concentrations were 2 μM and 0.2 μM in the same reaction mixture. First, the guide nucleic acid (ogDS01/ogDS02, Table S2) was added followed by 10 min incubation at 37 °C. Next, the target nucleic acid (oDS401/oDS403, Table S2) was added followed by 30 min incubation at 37 °C. 15 μL 2X gel loading dye (250 mM EDTA, 5% v/v glycerol, 95% v/v formamide, Bromophenol Blue) was added. Samples were incubated at 95 °C for 3 min and nucleic acids were resolved on 20% denaturing (7M Urea) polyacrylamide gels. Gels were imaged on an Ettan DIGE Imager (GE Healthcare).
In vitro NADase assays
LC-MS
Reaction mixtures with a volume of 50 μL were prepared with the following final concentrations: 0.5 μM CrtTIR-APAZ, CrtAgo or CrtSPARTA complex, 500 μM NAD+, 10 mM HEPES pH 7.5 and 125 mM KCl. The mixture was incubated for 1h at 37 °C, after which they were transferred to ice. 150 μL MilliQ H2O was added to the mixture and the mixture was subsequently filtered using centrifugal filters (Pall, 3 kDa MWCO) according to the instructions provided by the manufacturer. The flow through was collected and analyzed by LC-MS using an G1311A HPLC pump (Agilent) with G1367B WP autosampler (Agilent) and an Alltima HP C18 3μm reverse phase column (Avantor) connected to a microTOF benchtop ESI-TOF MS (Bruker Daltonics). Results were analyzed using Compass (Bruker Daltonics, V1.2).
NMR
CrtTIR-APAZ activity was measured in a total reaction volume of 600 μL with the following final concentrations: 0.5 μM 6xHis-MBP-CrtTIR-APAZ was incubated with 800 μM substrate (NAD+, NADH, or NADP) in 15 mM phosphate buffer pH 7.5, 125 mM KCl and 10% D2O at 37 °C. The reaction with NADH was performed in an anaerobic bottle flushed with nitrogen gas to minimize spontaneous degradation of NADH. NMR measurements were performed on a standard-bore 600 MHz NMR spectrometer (Bruker BioSpin GmbH). 1D Nuclear Overhauser Effect Spectroscopy (NOESY) (Williamson, 2005) experiments were conducted at RT. The 1H-1H mixing was 10 ms. A saturation of 72 Hz was applied on water during relaxation delay and mixing. NMR spectra were analysed using TopSpin 4.1.3 (Bruker BioSpin GmbH).
ϵ-NAD assays
A reaction mixture of purified SPARTA complex in SEC buffer, ε-NAD+ (Biolog, N010), RNA guide (oBK084, ogDS02) or a variant thereof, Table S2), and 5X reaction buffer (50 mM MES pH 6.5, 375 mM KCl, and 10 mM MgCl2) was prepared on ice in 96-well or 384-well plates. The mixture was incubated at room temperature for 15 min, after which DNA target (oBK090, oDS400 or a variant thereof, Table S2) was added to a final concentration of 200 nM unless otherwise indicated. For experiments with double stranded ligands, ligands were prepared by mixing forward and reverse oligonucleotides in a 1.5:1 ratio and annealed by incubation at 95°C for 2 min and gradually cooled to room temperature. Final concentrations of each component were 1 μM SPARTA complex, 25 μM ε-NAD+, 10 mM MES pH 6.5, 125 mM KCl, and 2 mM MgCl2 in a final volume of 60 μL or 20 uL. After addition of the target DNA, the plate was transferred to a preheated Synergy Neo2 or SH1M2FG plate reader (Biotek). Kinetic measurements took place at 55 °C for CrtSPARTA and 37 °C for MapSPARTA unless specified otherwise. Fluorescence intensity was measured in kinetic mode using an excitation wavelength of 310 nm and emission wavelength of 410 nm. All experiments were performed in triplicates and error bars indicate standard deviations.
For pH-range experiments, reaction mixtures were prepared as described above, but instead of 10 mM MES pH 6.5, each sample contained a final concentration of 10 mM buffer mix (BTP, MES, and citric acid in a 1:1:1 molar ratio) with a pH in the range of 4.0 to 9.5. To test whether SPARTA complex is activated by small molecules, small molecules (ADPRP, ADPR, NAD+, NADH, NADP+, NAM, cADPR, cAMP, cCMP, cGMP, cTMP, cUMP, c-diAMP, or c-triAMP) were added to a final concentration of 1 μM to reaction mixtures. For optimum temperature determination, reaction mixtures were incubated at the indicated temperatures for 60 min, and end-point fluorescence intensity was measured using excitation of wavelength of 310 nm and emission wavelength of 410 nm using a Synergy Neo2 plate reader (Biotek). All experiments were performed in triplicates and error bars indicate standard deviations.
To determine the limit of detection for dsDNA targets, initial PCR amplification was followed by detection with MapSPARTA. 35-cycle PCRs were performed either on colonies transformed with the plasmid or on a purified plasmid in MilliQ H2O. The concetration of the purified plasmid ranged from nM to aM and was determined using the Qubit™ dsDNA HS Assay Kit (ThermoFisher Scientific, Q32851). Colonies were suspended in 20 μL MilliQ H2O and incubated at 95 °C for 5 min. 50 μL PCR mixture contained 1 μL template, 25 μL Phusion High-Fidelity PCR Master Mix (ThermoFisher Scientific) and 0.5 μM of each primer (oAP030/oAP039, Table S2). The first five 5’ nucleotides of oAP039 contain phosphorothioate groups to protect that strand from degradation by T7 exonuclease (NEB, M0263). After PCR cleanup (New England Biolabs) dsDNA products were either melted at 95°C for 10 min and mixed with gRNA (oBK084, Table S2; 1:2) to form RNA:DNA hybrids or were used directly as targets in MapSPARTA detection assays containing T7 exonuclease. SPARTA reaction mixtures were prepared as described above. Reactions with T7 exonuclease were performed in the presence of 5U T7 exonuclease (New England Biolabs). All dsDNA detection reactions were incubated at 37°C. Fluorescence intensity was measured in kinetic mode every 2 minutes, using excitation wavelength of 310 nm and emission wavelength of 410 nm in a Synergy Neo2 or SH1M2FG plate reader (Biotek).
Effect of MapSPARTA on total NAD(P) levels and cell viability
Precultures of E. coli BW25113 or BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124) or an empty BAC (pBeloBAC11), as well as a pUC21-based high -copy number plasmid pUC-empty (pBK117) were grown o/n in LB supplemented with CAM and AMP in a shaker incubator at 37 °C. For experiments which includes multicopy plasmids encoding mRFP and derivates thereof (pBK144-pBK146, pBK157, pBK159-pBK161, pBK164-pBK166), homologous regions between the expression BAC and pUC-mRFP were first removed from the BAC. In that case pBK138 and pBK139 were used for expression of respectively MapSPARTA and MapSPARTATIR-E77A. For experiments to determine the effect of increased plasmid encoded target concentration (pBK179-pBK184) without increasing transcription pBK175 and pBK176 were used for expression of MapSPARTA and mRFP, or MapSPARTA only, respectively. The precultures were diluted 1:100 in 5 mL fresh LB containing CAM and AMP and were incubated in a shaker incubator at 37 °C for 3 h. When applicable, the cultures were used as input for in vivo NAD/NADH quantification using the NAD/NADH cycling assay (see below). To normalize the amount of cells, the amount of cells corresponding to 2 mL of OD600 nm = 1 was pelleted by centrifugation for 10 min at 3,500 x g. The supernatant was removed and the cell pellets were resuspended in 2 mL fresh LB supplemented with CAM, AMP, and arabinose. 10 μL of the normalized cultures was used to make a 1:2∗105 dilution in LB and 50 of this dilution μL was plated on LB agar plates supplemented with CAM and AMP. The remainder of the normalized culture was divided into two 13 mL tubes, which were incubated at 37 °C in a shaker incubator. When applicable mRFP expression was induced after one hour by adding IPTG or m-toluic acid. After 4h and 20h one of the two tubes was taken for analysis. A 1∗106 dilution was made and 60 μL was plated on LB agar plates supplemented with CAM and AMP. When applicable, the OD600 nm was again measured and samples were taken as input for in vivo total NAD quantification using an NAD/NADH cycling assay (see below) and/or in vivo total NADP quantification using an NADP/NADPH cycling assay (see below). The remaining tube after 20h was also used to measure RFP fluorescence as described below. All experiments were performed as biological triplicates, and error bars indicate standard deviations.
Effect of SPARSA on total NAD levels
Precultures of E. coli BL21 Star(DE3) containing a plasmid encoding CrtTIR-APAZ (pBK026), XavSIR2-APAZ (pBK012), JomSIR2-APAZ-pAgo (pBK014) or an empty vector (pML-1M) were grown o/n in LB supplemented with KAN in a shaker incubator at 37°C. The precultures were diluted 1:100 in 5 mL fresh LB containing KAN and were incubated in a shaker incubator at 37°C. When an OD600 nm of ∼0.4 was reached, protein expression was induced by addition of IPTG. The cultures were grown for 1.5 hours and used as input for in vivo NAD/NADH quantification using the NAD/NADH cycling assay (see below). All experiments were performed as biological triplicates, error bars indicate standard deviations.
NAD/NADH cycling assays
For NAD/NADH cycling assays after in vivo experiments (for experimental conditions see above), the OD600 nm of cultures were measured and normalized to 0.5. 0.5 mL cells were harvested by centrifuging 10 min at 3,000 x g at 4 °C. Cells were washed by adding 150 μL assay buffer of the NAD/NADH Cell-Based Assay Kit (Cayman Chemical, 600480) or PBS (when using the NAD/NADH Quantification Kit from Sigma) to the pellet, resuspending the cells, and repeating the centrifugation procedure. 150 μL assay buffer (Cayman Chemical) or NADH/NAD Extraction Buffer (Sigma) was added to the pellet and samples were stored at -80 °C until further use. To analyze the total protein amount, 17 μL of each sample was resolved by SDS-PAGE and subsequent CBB staining. The remainder of the samples were in a sonicator bath (QSONICA Q700A-220 with cup horn; amp 70%, 1s ON/4s OFF, 3 minutes total ON time) at 4 °C. After sonication, samples were filtered using centrifugal filters (Pall, 3 kDa MWCO or Merck, 10 kDa MWCO) according to the instructions provided by the manufacturer. 20 μL flowthrough was mixed with + 80 μL assay buffer (Cayman Chemical, and that mixture was used as input for the NAD/NADH Cell-Based Assay Kit (Cayman Chemical, 600480) according to the instructions provided by the manufacturer. Alternatively, 20 uL flowthrough was mixed with 30 uL NADH/NAD Extraction Buffer (Sigma, MAK037-1KT) and used as input for the NAD/NADH Quantification Kit (Sigma) according to the instructions provided by the manufacturer.
NADP/NADPH cycling assays
The same samples that were prepared for NAD cycling assays were used for NADP cycling assays. 50 μL of each sample was used as input for the NADP/NADPH Quantitation Kit (Sigma-Aldrich, MAK038-1KT) according to the instructions provided by the manufacturer.
RFP fluorescence
The effect of MapSPARTA on plasmid encoded RFP fluorescence was determined as described for the effect of MapSPARTA system on NAD/NADH and cell viability (described above). Cell densities of cells harboring plasmid pBK144 and therefore producing mRFP were determined by measuring the OD700 nm instead of OD600 nm, as mRFP interferes with OD600 nm determination (Hecht et al., 2016). To measure mRFP fluorescence, cultures from the cell viability assay (described above) were used. 20h after induction of mRFP expression a 4x dilution of the culture was made and was used to determine both the mRFP fluorescence (Excitation: 584/10, Emission: 607/10) and the OD700 nm in a Synergy Neo2 plate reader (Biotek). Additionally a 1∗106 dilution of the 20 h culture was made in LB and 30 μL was plated on LB agar plates supplemented with CAM and AMP. To assess whether the T7 RNA polymerase-encoding gene in the genome of the survivor colonies was intact, a colony PCR was performed using primers pBK405 and pBK406. PCR products were analyzed by Sanger sequencing using primers pBK406-410 (Table S2).
Effect of MapSPARTA on lasmid ransformation
E. coli BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124), or an empty BAC (pBeloBAC11), were made competent using the TSS buffer protocol. Triplicate cultures of each strain were grown o/n at 37°C in a shaker incubator. The next day the cultures were diluted 1:100 in 50 mL fresh LB and incubated for 1 h at 37°C in a shaker incubator. At that point, arabinose was added induce SPARTA expression and cultures were further incubated until an OD600 nm of 0.6 was reached. The cultures were cooled on ice for 20 min and cells were harvested by centrifugation at 3350 x g at 4 °C for 10 min. The cells were resuspended in 3 mL TSS buffer (10% w/v PEG8000, 30 mM MgCl2, 5% v/v DMSO in LB), divided in 100 μL aliquots and stored at -80 °C. For transformation duplicates of each triplicate batch of competent cells were thawed on ice, 3 ng pBK117 was added, and the cells were incubated on ice for another 30 min. Subsequently, the cells were incubated at 42 °C for 45 sec and incubated on ice for two min. Cells were diluted in 1 mL LB and 20 μL of the cell suspension was plated on LB plates supplemented with AMP, CAM and arabinose. To determine the total number of colony forming units, every sample was further diluted 1:5∗104 in LB and 30 μL of the diluted cells was plated on LB agar plates supplemented with CAM and arabinose. The plates were incubated o/n at 37 °C overnight after which colonies were counted and the fraction of CAMR colony forming units that are AMPR was determined.
Plasmid loss assay
Precultures of E. coli BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124), or an empty BAC (pBeloBAC11), as well as pUC-empty (pBK117) in LB supplemented with CAM and AMP were incubated o/n at 37 °C in a shaker incubator. The precultures were diluted 1:1000 in 1 mL fresh LB supplemented with CAM and arabinose, and further incubated at 37 °C in a shaker incubator for 24h. This procedure was repeated for nine days in a row. On days 0, 3, 6 and 9 cultures were diluted 1:106 and 30 μL of the dilution was plated in duplicate on LB agar plates supplemented with either only CAM or both CAM and AMP. The plates were incubated o/n at 37 °C after which colonies were counted and the fraction of CAMR cells that are AMPR was determined. All experiments were performed as biological quadruplicates, and error bars indicate standard deviations.
Culture competition assay
Precultures of E. coli BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124), or an empty BAC (pBeloBAC11), and either containing no additional plasmid or pUC-empty (pBK117) were inoculated in LB supplemented with CAM and AMP and were incubated in a shaker incubator at 37 °C o/n. The precultures were diluted 1:100 in 5 mL fresh LB supplemented with CAM and AMP and were incubated for another 3h at 37 °C in a shaker incubator. Subsequently, the amount of cells corresponding to 300 μL of OD600 nm = 1 was pelleted by centrifugation for 10 min at 3,500 x g. The pellet was resuspended in 300 μL LB supplemented with CAM and arabinose. Cultures containing the same BAC but with and without pBK117 plasmids were mixed in a 24:1 ratio (with plasmid:without plasmid). The cell mixture was used to inoculate 1 mL fresh LB supplemented with CAM and arabinose. These cultures were incubated for 24 h at 37 °C in a shaker incubator. Daily for three days, each culture was diluted 1:1000 into a LB supplemented with CAM and arabinose. Each day starting from the precultures on day 0, cultures were diluted 1:106 (1:4∗105 for the preculture mix) and 50 μL was plated on LB agar plates supplemented with CAM as well as CAM and AMP. The plates were incubated o/n at 37 °C overnight after which colonies were counted and the fraction of CAMR cells that are AMPR was determined. All experiments were performed as biological sextuplicates, and error bars indicate standard deviations.
Phage assays
Phage strains
E. coli phages T1 (DSMZ, 5801), T4 (Gift from Elizabeth Kutter’s Lab (Evergreen Phage Lab)), T7 (Gift from Ian Molineux’s Lab (University of Texas at Austin)), Lambda-vir (Brouns’ Lab collection (Delft University of Technology)), M13 (NEB, N4040S) and Nami (Brouns’ Lab collection (Delft University of Technology)) were used in this study. For their production, phages T1, T4, T7, and Lambda-vir were propagated in E. coli BL21-AI, as described previously (Bonilla et al., 2016), and bacteriophage Nami in its host E. coli isolate R10256 (Isolated at Erasmus M.C (Rotterdam, Netherlands)) following same procedure. Briefly, bacterial cultures at exponential growth phase (approximately 0.4 OD600 nm) were infected with a phage lysate and incubated overnight. Then, cells were collected by centrifugation and the supernatant was filtered through 0.2 μm PES membranes. When required, phages were concentrated by addition of PEG-8000 and NaCl at final concentrations of 100 mg/mL and 1 M, respectively. Subsequently, cultures were incubated overnight at 4 °C, centrifuged at 11,000 x g at 4 °C for 60 min, and the phage-containing pellet was resuspended in the desired final volume of LB media. Bacteriophage M13 was produced in E. coli JM109(DE3) as described by manufacturer (NEB #N4040S). All phage stocks were stored at 4°C before their use and titer determined as indicated below.
Phage titer determination
E. coli cultures in exponential growth phase (approximately 0.4 OD600 nm) were used to titer phages stocks. For this, 100 μL of culture were mixed with 5 mL of 0.6% LB agar at 45 °C and poured on a LB agar plate to form a bacterial lawn. Ten-fold dilutions of phages in LB were plated on the top of the bacterial lawn in 5-10 μL drops and dried for 20 min. Plates were incubated overnight up-side down at 37 °C. To determine the phage titer, the number of centers of infection (plaques) were counted. E. coli strain BW25113 was used to titer phages T1, T4, T7, Lambda-vir, and Nami. To titer phage M13, the 0.6 % LBA was supplemented with 1 mM IPTG and 200 μg/mL of X-gal, and E. coli ER2738 was used.
Bacterial survival growth curves
Overnight bacterial cultures of E. coli BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124), or an empty BAC (pBeloBAC11) were used to inoculate LB at an initial OD600 nm of 0.05. At exponential growth phase (approximately 0.4 OD600 nm), expression of SPARTA systems was induced with 0.2 % l-arabinose and then further incubated at 37 °C for 90 min. To obtain a standard initial cell density, cultures were normalized to an OD600 nm of 0.5 (corresponding to approximately 1x108 colonies forming units per mL (CFU/mL) following McFarland scale indications (Mcfarland, 1907). Then, these cultures were challenged with bacteriophages as follows. For each replicate, 190 μL of cells was dispensed into wells of flat-bottom 96-well plates and 10 μL of bacteriophages (or SM buffer for non-infection controls) was added to obtain a specific initial multiplicity of infection (MOI), as indicated in the figures. Well-plates were incubated in plate readers at 37°C with constant shaking at 280 rpm, and automatic OD600 nm reading (Epoch2 microplate reader; BioTek, Bad Friedrichshall) every 10 min during the course of the experiment. Please note that OD600 nm determined in the pre-cultures differs from that in the plate reader (Figure S7) due to different pathway lengths. All experiments were performed as biological triplicates and error bars indicate standard deviations.
Phage M13 titering over time
Bacterial cultures of E. coli JM109(DE3) containing a plasmid encoding MapSPARTA (pBK126), or MapSPARTATIR-E77A (pBK128), or an empty plasmid (pBK133), were cultivated as described above. At exponential growth phase (approximately 0.4 OD600 nm), expression of SPARTA systems was induced with 0.2 % L-arabinose and the OD600 nm was normalized to 0.05. Then, M13 phage was added at an initial MOI of 10-5 and phage titer was determined at various time points as described above. All experiments were performed as biological triplicates and error bars indicate standard deviations.
Efficiency of plating determination
To determine the efficiency of plaquing (EOP), phages were plated directly on induced cultures of E. coli BW25113 containing a BAC encoding MapSPARTA (pBK116) or MapSPARTATIR-E77A (pBK124), or an empty BAC (pBeloBAC11). For this, 200 μL of the bacterial cultures were mixed a specific, countable number of infectious particles (200-300 PFU/plate) and 4.5 mL of 0.6 % LB agar prewarmed at 45°C. The mixture was poured on top of 1 % LB agar plates to form a bacterial layer containing the infectious particles. After overnight incubation at 37 °C, the relative EOP was calculated dividing the number of plaques counted on each plate by the number of plaques formed in the strain containing the empty BAC. All experiments were performed as biological triplicates.
Phylogenetic analysis
Homologous sequences
To perform the phylogenetic analysis of Ago and APAZ-domain containing proteins, homologous sequences were retrieved from various bacterial taxa. A total of 7249 annotated genomes with chromosome, scaffold, representative or reference levels were used from the RefSeq database (O’Leary et al., 2016). HMMER (v3.3.1; hmmer.org) was used for the homology searches using an HMM model based on PIWI domain as query for Ago proteins and APAZ domain as query for the APAZ-domain containing proteins. All the resulting sequence hits i.e., 500 Ago homologs and 256 APAZ homologs (with an E-value < 0.001) were considered for further phylogenetic analysis. For all these sequences, protein domain architectures were identified using InterProScan (v5.51-85.0; Jones et al., 2014).
Phylogeny construction
Homologous sequences were aligned using MAFFT E-INS-I algorithm with a maximum of 1,000 iterations (v7.475; Katoh and Standley, 2013). Alignment positions with 30% gap threshold (-gt 0.3) were removed prior to the phylogeny construction using trimAl (v1.4; Capella-Gutiérrez et al., 2009) for both Ago and APAZ families. Phylogenetic trees were built using maximum-likelihood method implemented in IQtree (v2.0.4; Minh et al., 2020) with automatic model selection from in-built ModelFinder (Kalyaanamoorthy et al., 2017) and a maximum of 1,000 rapid bootstraps (-bb 1000 -m MFP+MERGE). LG+F+R10 and LG+F+R6 were selected as the models of evolution based on the Bayesian Information Criterion (BIC), for Ago and APAZ trees, respectively. Obtained phylogenetic trees were visualized in iTOL (v6; itol.embl.de; Letunic and Bork, 2021).
Gene neighborhood (operons) analysis
To identify APAZ-domain containing genes that are associated with short-Ago proteins in same operon, or any other gene family members even if in a different operon, five upstream as well as downstream genes of each short-Ago protein were analyzed. All-by-all similarity search was performed with all genes in the neighborhood using Diamond blastp (v2.0.7.145; Buchfink et al., 2021) with default settings (except --query-cover 80) to identify the homologous proteins. Obtained results were then visualized in Cytoscape (v3.7.1; Shannon et al., 2003) to identify the clusters of the same gene family associated with each clade of short-Ago proteins.
Tanglegram
Phylogenetic trees of SPARTA and TIR-APAZ families (saved in NEWICK format from iTOL) were loaded into R environment (v4.0.3) using read.dendrogram function of ‘phylogram’ R package (v2.1.0; Wilkinson and Davy, 2018). Later a list of dengrograms was made using dendlist and a comparison was made using tanglegram function implemented in ‘dendextend’ R package (v1.15.1; Galili, 2015).
Quantification and statistical analysis
All statistical analyses were performed in R version 4.1.0, with exception of RNA sequencing correlation analysis, which was performed in Excel.
In vitro NAD/NADH cycling assays
In vitro NAD/NADH cycling assays were performed in technical triplicates. The percentage of NAD+ consumed in each reaction was calculated as (absorption of sample / absorption of a sample containing only buffer and NAD+) ∗ 100. The averages of technical triplicates are shown in Figure 2, error bars indicate standard deviations.
ϵ-NAD assays
All assays with ssDNA targets were performed in technical triplicates, detection of dsDNA targets with different T7 exonuclease concentrations. For determining the effect of mismatches (Figures 6A and 6B), guide length preferences (Figure S10B), and buffer pH preferences (Figure S10C) the fluorescence was first corrected for background fluorescence determined using a no-target control, and change in fluorescence min-1 was determined by calculating the slopes of linear components of the kinetic curves. For the detection of mismatches (Figures 6A and 6B), the relative change in fluorescence compared to no mismatches was calculated as (change in fluorescence min-1 in sample / change in fluorescence min-1 in no mismatch control). For determining the effect of temperature (Figure S10A) fluorescence was measured after 60 min incubation. For determining the limit of detection (Figures 6C and 6D) the fluorescence was measured in kinetic mode and corrected with fluorescence of a no target control sample. For the detection of dsDNA the fluorescence was also measured in kinetic mode, but without subtracting the control (the control graph is added to the figure instead). For triplicate experiments in all panels the average are used to plot the graphs and error bars indicate standard deviations.
RNA and DNA sequences correlation analysis
Correlation between total long RNA and SPARTA-associated small RNA sequence abundance and MapSPARTA-associated small RNA and DNA sequence abundance was tested for each comparison by calculating the Pearson correlation coefficient and corresponding p-value.
In vivo assays to assess effect of pUC-empty on cells expressing MapSPARTA
All assays were performed in biological triplicates of which the averages are shown in Figures 5A and S4A, error bars indicate standard deviations. Per timepoint (0 h, 4 h and 20 h) and per dependent variable (CFU, OD600 nm, total NAD level and total NADP level) a two-way ANOVA was performed to test for significant independent and interaction effects of SPARTA system (MapSPARTA or MapSPARTATIR-E77A or empty BAC) and presence of pUC-empty. The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. Tukey’s test was used for post-hoc pairwise comparisons.
In vivo assays to assess effect of pUC-mRFP, pET-mRFP, and pC101-mRFP on cells expressing MapSPARTA
All assays were performed in biological triplicates of which the averages are shown in Figures 5A and S4A, error bars indicate standard deviations. For timepoint 4 h and as the dependent variable the total NAD level, and for timepoints 4h and 20 and as the dependent variable the CFU, two-way ANOVAs were performed to test for significant independent and interaction effects of presence of pUC-empty and T7-based transcription of mRFP. The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. Tukey’s test was used for post-hoc pairwise comparisons.
In vivo assays to assess effect of multicopy plasmids containing a nontranscribed target on cells expressing MapSPARTA and mRFP from a BAC
All assays were performed in biological duplicates of which the average and individual datapoints are shown in Figures 5A and S4A. For timepoint 4 h and as the dependent variable the total NAD level, and for timepoints 4h and 20 and as the dependent variable the CFU, two-way ANOVAs were performed to test for significant independent and interaction effects of presence of a multicopy plasmid and presence of a non-transcribed target on said plasmid. Normal distribution of the data and homogeneity of variances were assumed. Tukey’s test was used for post-hoc pairwise comparisons.
In vivo assay to assess effect of pUC-mRFP and derivatives on cells expressing MapSPARTA
Cell viability assays (measured as CFU/mL) were performed in biological triplicates of which the averages are shown in Figure 5B, error bars indicate standard deviations. Per timepoint (4 h and 20 h) a two-way ANOVA was performed to test for significant independent and interaction effects of SPARTA system (MapSPARTA, MapSPARTATIR-E77A or empty BAC) and pUC-variant (no pUC, pUC-mRFP, pUC-mRFP-ΔT7-ΔRBS or pUC-mRFP-ΔRBS). The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. Tukey’s test was used for post-hoc pairwise comparisons.
RFP fluorescence of cells expressing MapSPARTA
RFP fluorescence and OD700 nm in bacterial cultures was measured in biological triplicates of which the RFP fluorescence/ OD700 nm averages are shown in Figure 5C, error bars indicate standard deviations. A t-test was used to test for significant difference between cells expressing MapSPARTA and MapSPARTATIR-E77A. The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively.
Transformation assay
Transformation assays were performed in biological triplicates that consisted of technical replicates that were averaged. The transformation efficiency was calculated as AMPR+CAMR CFU/CAMR CFU. The average transformation efficiencies of the biological triplicates are shown in Figure S9C, error bars indicate standard deviations. One-way ANOVA was used to test for significant effect of SPARTA system on transformation efficiency (MapSPARTA, MapSPARTATIR-E77A or empty BAC). The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. As the one-way ANOVA was not significant, no post-hoc tests were performed.
Bacterial growth curves and phage M13 titering over time
Both experiments were performed in biological triplicates. The averages of the curves are shown in Figure S6, shadings (growth curves) or error bars (M13 titering over time) indicate standard deviations. For statistical analyses, the areas under the curves were calculated using the Trapezoidal rule. For every phage that was used a one-way ANOVA was performed to test for significant effect of SPARTA system on the area under the growth curve. The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. Tukey’s test was used for post-hoc pairwise comparisons.
Plasmid loss assay
Plasmid loss assays were performed in biological quadruplicates and plated in duplicates that were averaged. The fraction of AMPR cells was calculated as CFU on AMP+CAM/CFU on CAM. The average fractions of AMPR cells of the biological triplicates are shown in Figure S9B, error bars indicate standard deviations. A two-way ANOVA was performed to test for significant independent and interaction effects of SPARTA system (MapSPARTA or MapSPARTATIR-E77A) and timepoint (day 1 or day 9). The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. As the two-way ANOVA was not significant, no post-hoc tests were performed.
Culture competition assay
Culture competition assays were performed in biological sextuplets. The fraction of AMPR cells was calculated as CFU on AMP+CAM/CFU on CAM. The average fractions of AMPR cells are shown in Figure 6F, error bars indicate standard deviations. A mixed two-way ANOVA was performed to test for significant independent and interaction effects of SPARTA system (MapSPARTA or MapSPARTATIR-E77A) and timepoint (day 0, 1, 2 or 3) as within-subjects variable. The data was subjected to a Shapiro-Wilk test on linear model residuals and Levene’s test to confirm normal distribution of the data and homogeneity of variances, respectively. One-way ANOVA’s and pairwise t-tests with Bonferroni corrections were used to analyse simple main effects and for pairwise comparisons, respectively.
Acknowledgments
We are grateful to Yanzhang Luo of the MAGNEtic resonance research FacilitY (MAGNEFY) at Wageningen University for support with NMR experiments. We thank Rob Joosten, Sebas Wesseling, Jiska van de Sande, Dylan de Wit, and Imogen Oldfield for technical assistance. We thank members of the Swarts lab and the Laboratory of Biochemistry for discussion. Research in the Swarts lab was supported by a Netherlands Organization for Scientific Research (NWO) VENI grant (016.Veni.192.072) and a European Research Council (ERC) starting grant (ERC-2020-STG 948783) to D.C.S. S.J.J.B. was supported by an NWO VICI grant (VI.C.182.027) and an ERC Consolidator grant (grant agreement no. 101003229).
Author contributions
B.K., A.P., and D.C.S. designed and performed experiments. S.K.M. performed bioinformatics analyses. Bacteriophage experiments were performed by C.A.-M. under the supervision of S.J.J.B. S.L. performed SEC-MALS experiments. J.J.M.V. performed NMR and LC-MS experiments. D.C.S. supervised the project. B.K. and D.C.S. wrote the manuscript with input from all other authors.
Declaration of interests
D.C.S., B.K., and A.P. have submitted a patent application regarding the utilization of short pAgo systems for NA detection.
Published: April 4, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.03.012.
Supplemental information
Data and code availability
-
•
The unprocessed image files, raw microplate reader data, and RNA/DNA-seq analyses used to prepare the figures in this manuscript have been deposited in Mendeley Data (https://dx.doi.org/10.17632/7f4gsbhrpp.1).
-
•
The raw RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus (GEO; accession number GSE191271).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- Bonilla N., Rojas M.I., Netto Flores Cruz G.N.F., Hung S.H., Rohwer F., Barr J.J. Phage on tap—a quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ. 2016;4:e2261. doi: 10.7717/peerj.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautaset T., Lale R., Valla S. Positively regulated bacterial expression systems. Microb. Biotechnol. 2009;2:15–30. doi: 10.1111/j.1751-7915.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Reuter K., Drost H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 2021;18:366–368. doi: 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs A.M., Iyer L.M., Aravind L. Two novel PIWI families: roles in inter-genomic conflicts in bacteria and mediator-dependent modulation of transcription in eukaryotes. Biol. Direct. 2013;8:13. doi: 10.1186/1745-6150-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B., Rood J., Singer E. BBMerge—accurate paired shotgun read merging via overlap. PLoS One. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa A.M.S., Howard-Varona C., Coy S.R., Buchan A., Sullivan M.B., Weitz J.S. Revisiting the rules of life for viruses of microorganisms. Nat. Rev. Microbiol. 2021;19:501–513. doi: 10.1038/s41579-021-00530-x. [DOI] [PubMed] [Google Scholar]
- Dayeh D.M., Cantara W.A., Kitzrow J.P., Musier-Forsyth K., Nakanishi K. Argonaute-based programmable RNase as a tool for cleavage of highly-structured RNA. Nucleic Acids Res. 2018;46:e98. doi: 10.1093/nar/gky496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriu T., Kurilovich E., Łapińska U., Severinov K., Pagliara S., Szczelkun M.D., Westra E.R. Bacteriostatic antibiotics promote CRISPR-Cas adaptive immunity by enabling increased spacer acquisition. Cell Host Microbe. 2022;30:31–40.e5. doi: 10.1016/j.chom.2021.11.014. [DOI] [PubMed] [Google Scholar]
- Doron S., Melamed S., Ofir G., Leavitt A., Lopatina A., Keren M., Amitai G., Sorek R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359:eaar4120. doi: 10.1126/science.aar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxzen K.W., Doudna J.A. DNA recognition by an RNA-guided bacterial Argonaute. PLoS One. 2017;12:e0177097. doi: 10.1371/journal.pone.0177097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dym O., Eisenberg D. Sequence-structure analysis of FAD-containing proteins. Protein Sci. 2001;10:1712–1728. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K., Summers D.W., Sasaki Y., Mao X., DiAntonio A., Milbrandt J. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron. 2017;93:1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K., Summers D.W., Sasaki Y., Mao X., Yim A.K.Y., DiAntonio A., Milbrandt J. TIR domain proteins are an ancient family of NAD+-consuming enzymes. Curr. Biol. 2018;28:421–430.e4. doi: 10.1016/j.cub.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F., Sonenberg N., Nagar B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- Fu L., Xie C., Jin Z., Tu Z., Han L., Jin M., Xiang Y., Zhang A. The prokaryotic Argonaute proteins enhance homology sequence-directed recombination in bacteria. Nucleic Acids Res. 2019;47:3568–3579. doi: 10.1093/nar/gkz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., Altae-Tran H., Böhning F., Makarova K.S., Segel M., Schmid-Burgk J.L., Koob J., Wolf Y.I., Koonin E.V., Zhang F. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science. 2020;369:1077–1084. doi: 10.1126/science.aba0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A., et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P., Cofsky J.C., Kyrpides N.C., Banfield J.F., Doudna J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842. doi: 10.1126/science.aav4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasunuma K., Sekiguchi M. Replication of plasmid pSC101 in Escherichia coli K12: requirement for dnaA function. Mol. gen. genet. 1977;154:225–230. doi: 10.1007/BF00571277. [DOI] [PubMed] [Google Scholar]
- Hecht A., Endy D., Salit M., Munson M.S. When wavelengths collide: bias in cell abundance measurements due to expressed fluorescent proteins. ACS Synth. Biol. 2016;5:1024–1027. doi: 10.1021/acssynbio.6b00072. [DOI] [PubMed] [Google Scholar]
- Hegge J.W., Swarts D.C., Van Der Oost J. Prokaryotic Argonaute proteins: novel genome-editing tools? Nat. Rev. Microbiol. 2018;16:5–11. doi: 10.1038/nrmicro.2017.73. [DOI] [PubMed] [Google Scholar]
- Hopkins E.L., Gu W., Kobe B., Coleman M.P. A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front. Mol. Biosci. 2021;8:703532. doi: 10.3389/fmolb.2021.703532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsefield S., Burdett H., Zhang X., Manik M.K., Shi Y., Chen J., Qi T., Gilley J., Lai J.-S., Rank M.X., et al. NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science. 2019;365:793–799. doi: 10.1126/science.aax1911. [DOI] [PubMed] [Google Scholar]
- Jet T., Gines G., Rondelez Y., Taly V. Advances in multiplexed techniques for the detection and quantification of microRNAs. Chem. Soc. Rev. 2021;50:4141–4161. doi: 10.1039/d0cs00609b. [DOI] [PubMed] [Google Scholar]
- Johnson C.M., Harden M.M., Grossman A.D. An integrative and conjugative element encodes an abortive infection system to protect host cells from predation by a bacteriophage. Preprint at bioRxiv. 2020 doi: 10.1101/2020.12.13.422588. [DOI] [Google Scholar]
- Jolly S.M., Gainetdinov I., Jouravleva K., Zhang H., Strittmatter L., Bailey S.M., Hendricks G.M., Dhabaria A., Ueberheide B., Zamore P.D. Thermus thermophilus Argonaute functions in the completion of DNA replication. Cell. 2020;182:1545–1559.e18. doi: 10.1016/j.cell.2020.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P., Binns D., Chang H.Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka D., Oh H., Park E., Kim J.H., Bae E. Structural and functional evidence of bacterial antiphage protection by Thoeris defense system via NAD+ degradation. Nat. Commun. 2020;11:2816. doi: 10.1038/s41467-020-16703-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., Von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya E., Doxzen K.W., Knoll K.R., Wilson R.C., Strutt S.C., Kranzusch P.J., Doudna J.A. A bacterial Argonaute with noncanonical guide RNA specificity. Proc. Natl. Acad. Sci. USA. 2016;113:4057–4062. doi: 10.1073/pnas.1524385113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killackey S.A., Rahman M.A., Soares F., Zhang A.B., Abdel-Nour M., Philpott D.J., Girardin S.E. The mitochondrial nod-like receptor NLRX1 modifies apoptosis through SARM1. Mol. Cell. Biochem. 2019;453:187–196. doi: 10.1007/s11010-018-3444-3. [DOI] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.Y., Jung Y., Lim D. Argonaute system of Kordia jejudonensis is a heterodimeric nucleic acid-guided nuclease. Biochem. Biophys. Res. Commun. 2020;525:755–758. doi: 10.1016/j.bbrc.2020.02.145. [DOI] [PubMed] [Google Scholar]
- Kropocheva E., Kuzmenko A., Aravin A.A., Esyunina D., Kulbachinskiy A. A programmable pAgo nuclease with universal guide and target specificity from the mesophilic bacterium Kurthia massiliensis. Nucleic Acids Res. 2021;49:4054–4065. doi: 10.1093/nar/gkab182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmenko A., Oguienko A., Esyunina D., Yudin D., Petrova M., Kudinova A., Maslova O., Ninova M., Ryazansky S., Leach D., et al. DNA targeting and interference by a bacterial Argonaute nuclease. Nature. 2020;587:632–637. doi: 10.1038/s41586-020-2605-1. [DOI] [PubMed] [Google Scholar]
- Lapinaite A., Doudna J.A., Cate J.H.D. Programmable RNA recognition using a CRISPR-associated Argonaute. Proc. Natl. Acad. Sci. USA. 2018;115:3368–3373. doi: 10.1073/pnas.1717725115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.Z., Mechikoff M.A., Kikla A., Liu A., Pandolfi P., Fitzgerald K., Gimble F.S., Solomon K.V. NgAgo possesses guided DNA nicking activity. Nucleic Acids Res. 2021;49:9926–9937. doi: 10.1093/nar/gkab757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J. HOLMESv2: a CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019;8:2228–2237. doi: 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- Li W., Liu Y., Wang F., Ma L. A programmable pAgo nuclease with RNA target preference from the psychrotolerant bacteria Mucilaginibacter paludis. Preprint at bioRxiv. 2021 doi: 10.1101/2021.06.08.447469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lopatina A., Tal N., Sorek R. Abortive infection: bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 2020;7:371–384. doi: 10.1146/annurev-virology-011620-040628. [DOI] [PubMed] [Google Scholar]
- Ma J.-B., Yuan Y.-R., Meister G., Pei Y., Tuschl T., Patel D.J. Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature. 2005;434:666–670. doi: 10.1038/nature03514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Lapin D., Liu L., Sun Y., Song W., Zhang X., Logemann E., Yu D., Wang J., Jirschitzka J., et al. Direct pathogen-induced assembly of an NLR immune receptor complex to form a holoenzyme. Science. 2020;370:eabe3069. doi: 10.1126/science.abe3069. [DOI] [PubMed] [Google Scholar]
- Makarova K.S., Wolf Y.I., van der Oost J., Koonin E.V. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol. Direct. 2009;4:29. doi: 10.1186/1745-6150-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R., Qi T., Zhang H., Liu F., King M., Toth C., Nogales E., Staskawicz B.J. Structure of the activated ROQ1 resistosome directly recognizing the pathogen effector XopQ. Science. 2020;370:eabd9993. doi: 10.1126/science.abd9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maute R.L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R. TRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA. 2013;110:1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcfarland J. The nephelometer: an instrument for estimating the number of bacteria in suspensions used for calculating the opsonic index and for vaccines. J. Am. Med. Assoc. 1907;49:1176–1178. [Google Scholar]
- Meeske A.J., Nakandakari-Higa S., Marraffini L.A. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature. 2019;570:241–245. doi: 10.1038/s41586-019-1257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., Von Haeseler A., Lanfear R., Teeling E. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T., Ito K., Murakami R., Uchiumi T. Structural basis for the recognition of guide RNA and target DNA heteroduplex by Argonaute. Nat. Commun. 2016;7:11846. doi: 10.1038/ncomms11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morehouse B.R., Govande A.A., Millman A., Keszei A.F.A., Lowey B., Ofir G., Shao S., Sorek R., Kranzusch P.J. STING cyclic dinucleotide sensing originated in bacteria. Nature. 2020;586:429–433. doi: 10.1038/s41586-020-2719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K. Are Argonaute-associated tiny RNAs junk, inferior miRNAs, or a new type of functional RNAs? Front. Mol. Biosci. 2021;8:795356. doi: 10.3389/fmolb.2021.795356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofir G., Herbst E., Baroz M., Cohen D., Millman A., Doron S., Tal N., Malheiro D.B.A., Malitsky S., Amitai G., Sorek R. Antiviral activity of bacterial TIR domains via immune signalling molecules. Nature. 2021;600:116–120. doi: 10.1038/s41586-021-04098-7. [DOI] [PubMed] [Google Scholar]
- Oh M.D., Park W.B., Choe P.G., Choi S.-J., Kim J.-I., Chae J., Park S.S., Kim E.-C., Oh H.S., Kim E.J., et al. Viral load kinetics of MERS coronavirus infection. N. Engl. J. Med. 2016;375:1303–1305. doi: 10.1056/NEJMc1511695. [DOI] [PubMed] [Google Scholar]
- Olovnikov I., Chan K., Sachidanandam R., Newman D.K., Aravin A.A. Bacterial Argonaute samples the transcriptome to identify foreign DNA. Mol. Cell. 2013;51:594–605. doi: 10.1016/j.molcel.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S.P., Davy S.K. phylogram: an R package for phylogenetic analysis with nested lists. J. Open Source Softw. 2018;3:790. [Google Scholar]
- Page R., Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016;12:208–214. doi: 10.1038/nchembio.2044. [DOI] [PubMed] [Google Scholar]
- Pan Y., Zhang D., Yang P., Poon L.L.M., Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020;20:411–412. doi: 10.1016/S1473-3099(20)30113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Parizotto E.A., Wang M., Roe S.M., Barford D. Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol. Cell. 2009;33:204–214. doi: 10.1016/j.molcel.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L., Meister G. Argonaute proteins: mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Ryazansky S., Kulbachinskiy A., Aravin A.A. The expanded universe of prokaryotic Argonaute proteins. mBio. 2018;9:e01935. doi: 10.1128/mBio.01935-18. e01918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina S.A., Koonin E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008;23:578–587. doi: 10.1016/j.tree.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizuya H., Birren B., Kim U.J., Mancino V., Slepak T., Tachiiri Y., Simon M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P., Filée J., Chandler M. Insertion sequences in prokaryotic genomes. Curr. Opin. Microbiol. 2006;9:526–531. doi: 10.1016/j.mib.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Steens J.A., Zhu Y., Taylor D.W., Bravo J.P.K., Prinsen S.H.P., Schoen C.D., Keijser B.J.F., Ossendrijver M., Hofstra L.M., Brouns S.J.J., et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 2021;12:5033. doi: 10.1038/s41467-021-25337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D.C., Hegge J.W., Hinojo I., Shiimori M., Ellis M.A., Dumrongkulraksa J., Terns R.M., Terns M.P., Van Der Oost J. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43:5120–5129. doi: 10.1093/nar/gkv415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D.C., Jore M.M., Westra E.R., Zhu Y., Janssen J.H., Snijders A.P., Wang Y., Patel D.J., Berenguer J., Brouns S.J.J., et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014;507:258–261. doi: 10.1038/nature12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D.C., Makarova K., Wang Y., Nakanishi K., Ketting R.F., Koonin E.V., Patel D.J., Van Der Oost J. 1353 The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol. 2014;21:743–753. doi: 10.1038/nsmb.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal N., Morehouse B.B., Millman A., Stokar-avihail A., Avraham C., Fedorenko T., Yirmiya E., Herbst E., Brandis A., Mehlman T., et al. Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell. 2021;184:5728–5739.e16. doi: 10.1016/j.cell.2021.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K.K.W., Chan K.-H., Li I.W.S., Tsang T.-Y., Tse H., Chan J.F.W., Hung I.F.N., Lai S.-T., Leung C.-W., Kwan Y.-W., et al. Viral load in patients infected with pandemic H1N1 2009 influenza A virus. J. Med. Virol. 2010;82:1–7. doi: 10.1002/jmv.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucheret H. Plant ARGONAUTES. Trends Plant Sci. 2008;13:350–358. doi: 10.1016/j.tplants.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Vieira J., Messing J. New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene. 1991;100:189–194. doi: 10.1016/0378-1119(91)90365-i. [DOI] [PubMed] [Google Scholar]
- Vuoristo K.S., Mars A.E., Sangra J.V., Springer J., Eggink G., Sanders J.P.M., Weusthuis R.A. Metabolic engineering of itaconate production in Escherichia coli. Appl. Microbiol. Biotechnol. 2015;99:221–228. doi: 10.1007/s00253-014-6092-x. [DOI] [PubMed] [Google Scholar]
- Wan L., Essuman K., Anderson R.G., Sasaki Y., Monteiro F., Chung E.H., Osborne Nishimura E.O., DiAntonio A., Milbrandt J., Dangl J.L., Nishimura M.T. TIR domains of plant immune receptors are NAD+-cleaving enzymes that promote cell death. Science. 2019;365:799–803. doi: 10.1126/science.aax1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I.N., Dykhuizen D.E., Slobodkin L.B. The evolution of phage lysis timing. Evol. Ecol. 1996;10:545–558. [Google Scholar]
- Williamson M.P. In: Encyclopedia of Analytical Science. Second edition. Worsfold P., Poole C., Townshend A., editors. Elsevier; 2005. Nuclear magnetic resonance spectroscopy techniques|nuclear overhauser effect; pp. 342–349. [Google Scholar]
- Willkomm S., Oellig C.A., Zander A., Restle T., Keegan R., Grohmann D., Schneider S. Structural and mechanistic insights into an archaeal DNA-guided Argonaute protein. Nat. Microbiol. 2017;2:17035. doi: 10.1038/nmicrobiol.2017.35. [DOI] [PubMed] [Google Scholar]
- Zander A., Willkomm S., Ofer S., Van Wolferen M., Egert L., Buchmeier S., Stöckl S., Tinnefeld P., Schneider S., Klingl A., et al. Guide-independent DNA cleavage by archaeal Argonaute from Methanocaldococcus jannaschii. Nat. Microbiol. 2017;2:17034. doi: 10.1038/nmicrobiol.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The unprocessed image files, raw microplate reader data, and RNA/DNA-seq analyses used to prepare the figures in this manuscript have been deposited in Mendeley Data (https://dx.doi.org/10.17632/7f4gsbhrpp.1).
-
•
The raw RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus (GEO; accession number GSE191271).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.