

Abstract
The hippocampus is the most common seizure focus in people. Within the hippocampus, aberrant neurogenesis plays a critical role in the initiation and progression of epilepsy in rodent models, but it is unknown whether this also holds true in humans. To address this question, we used immunofluorescence on control healthy hippocampus and surgical resections from mesial temporal lobe epilepsy (MTLE), plus neural stem cell cultures, and multi-electrode recordings of ex vivo hippocampal slices. We found that a longer duration of epilepsy is associated with a sharp decline in neuronal production, and persistent numbers in astrogenesis. Further, immature neurons in MTLE are mostly inactive, and are not observed in cases with local epileptiform-like activity. However, immature astroglia are present in every MTLE case and their location and activity are dependent upon epileptiform-like activity. Immature astroglia, rather than newborn neurons, therefore represent a potential target to continually modulate adult human neuronal hyperactivity.
Introduction
Adult neurogenesis is a dramatic form of brain plasticity in which newborn neurons and astrocytes modify existing neural circuitry1. This process occurs in the dentate gyrus (DG) within the hippocampus, where newborn cells contribute positively to memory and the regulation of emotion2. However, in rodent models of epilepsy, alterations to adult hippocampal neurogenesis have negative consequences. In rodents, increased cell proliferation, migration and formation of aberrant synaptic hippocampal connections can initiate epileptic seizures3–5. In addition, chronic temporal lobe seizures result in a decline of the stem cell pool, neuronal differentiation and memory impairment6–8. Newborn neurons show enhanced activity compared to mature granule neurons9, making them a potential cellular source of neuronal hyperactivity. Silencing newborn neurons or suppressing neurogenesis have been shown to reduce seizure activity; however, these effects are transient, implying other cellular sources as modulators of neuronal hyperactivity during epilepsy progression10–12. One potential source could be reactive astrocytes generated from differentiating neural stem cells, whose contribution to epilepsy is not well defined13.
The role of newborn adult cells is even less clear in people with epilepsy. Evidence exists for and against the presence of adult hippocampal neurogenesis in healthy individuals14–21 and in patients with epilepsy15,22,23. Further, no studies have documented the prevalence of human newborn neurons and immature astroglia during epilepsy progression and their effects on seizure phenotypes. Mesial Temporal Lobe Epilepsy (MTLE), the most common drug resistant form of adult epilepsy, is attributed to abnormal local firing in the hippocampus24. Pharmacoresistant MTLE patients undergo resective hippocampal surgery to prevent further seizures25. In this study, we performed histology on adult MTLE and age- and sex-matched control tissue, and cell culture experiments in MTLE to investigate altered neurogenesis and astrogenesis during disease duration. Further, we combined multi-electrode array (MEA) recording with histology to identify how adult human immature neurons and immature astroglia are altered with epileptiform activity.
Results
Adult neurogenesis in MTLE declines with disease duration
Adult neurogenesis was first benchmarked in the human hippocampus by performing immunofluorescence on postmortem tissue from control subjects using established methods14. Control subjects included in the present study belonged to an age group of 21–56 years, and had no neurologic or psychiatric diagnoses, no pathological or traumatic conditions affecting the brain, clear toxicology reports, and were free of psychiatric treatments and cognitive impairment (See methods, and Supplementary data Table 1). We analyzed the presence of immature cells (Dcx+) that acquire granule neuron identity (Prox1+) to quantify adult neurogenesis19. In 8 of 9 control cases, Dcx+ Prox1+ cells were detected on the order of 100–1200 cells per mm3 (Fig 1a, b, Supplementary data Table 1). We observed slightly fewer Dcx+ Prox1+ cells in older control subjects but the relationship with age was not statistically significant. This trend is consistent with other recent studies that have reported a modest decline in neurogenesis at advanced age in subjects without dementia14,19.
Figure 1. Adult neurogenesis in MTLE patients declines with disease duration.

(a) Dcx+ (green) Prox1+ (purple) immature neurons identified in the DG-GCL of adult human control subjects. Scale bar 10μm. (b) Number of Dcx+ Prox1+ cells/mm3 in the GCL with age in N = 9 control subjects (c) Dcx+ (green) Prox1+ (purple) immature neurons identified in the GCL of adult human MTLE cases. Scale bar 10μm. (d) Location of Dcx+ Prox1+ cells denoted as white dots in GCL (outlined by dotted lines). Scale bar 500μm. (e) Age at surgery, and (f) Disease duration in years (yr.) for cases in which Dcx+ Prox1+ cells were undetected (−) and detected (+). Unpaired two tailed t-test. (g, h) Number of Dcx+ Prox1+ cells/mm3 in the GCL with (g) age at surgery (yr.) and (h) disease duration (yr.). Two-tailed Pearson Correlation. Pearson r= −0.7491(e-h) Data points marked in red and blue are from MTLE cases in which Dcx+ Prox1+ cells were undetected (−) N = 10 and detected (+) N = 7 respectively. (i) Dcx+ (green) PSA-NCAM+ (red) immature neurons which are Prox1- (arrowhead) and Prox1+ (arrow) (purple) identified in the DG-GCL of adult human MTLE cases. Scale bar 10μm (j) Cartoon illustration of Dcx, PSA-NCAM, and Prox1 expression across stages of neuronal maturation and commitment to granule neuron identity. (k) Percentage of PSA-NCAM+ cells that are Dcx+, Prox1+ and Dcx+ Prox1+ in N = 4 cases. All the error bars represent s.e.m across individual cases.
Neurogenesis was then investigated in surgically resected hippocampus from adult patients with MTLE using immunofluorescence. Tissue processing methodology greatly influences neurogenesis detection in the human hippocampus19. We therefore ensured the viability of surgically resected tissue through live neuronal activity recordings and performed gentle fixation with 4% paraformaldehyde for 30 minutes on 400μm tissue slices. Such procedure prevented antigen masking and bypassed the requirement for any harsh antigen retrieval steps (See methods). Dcx+ Prox1+ immature neurons were detected in the DG of 7 out of 17 MTLE patients (Fig 1c, d, Extended data Fig 1a) ranging from 20 to 56 years (sex- and age-matched with the control group, Supplementary data Table 2). Patients in whom immature granule neurons were detected tended to be younger (Fig 1e); however, disease duration (time between age of disease onset and surgery) rather than age (at the time of surgery) showed a stronger effect on the number of Dcx+ Prox1+ cells (Fig 1f). The number of Dcx+ Prox1+ immature neurons did not significantly decline with age (Fig 1g). Instead, the number of new neurons exponentially declined with MTLE disease duration (Fig 1h). The slight decline in neurogenesis with age might be partially explained by the strong positive correlation between disease duration and age at surgery (Extended data Fig 1b). Consistently, we observed a linear correlation between greater number of immature neurons and older age at disease onset (Extended data Fig 1c). Further, subjects containing Dcx+ Prox1+ cells had older age at onset compared to those who did not (Extended data Fig 1d). These changes could be due to differences between the juvenile and adult brain or might reflect the observation that MTLE patients with longer disease durations possess earlier disease onset (Extended data Fig 1e). The number of new neurons did not significantly differ between males and females (Extended data Fig 1f), nor between left and right hippocampi (Extended data Fig 1g).
A morphological analysis of Dcx+ Prox1+ cells was performed to estimate the maturation stage of new neurons within MTLE. We classified early stages as cells containing small to no neurites, and later stages with progressively extending neurites (Extended Data Fig 1h, i). Most Dcx+ Prox1+ cells belonged to an early immature stage (Extended Data Fig 1i). Middle stages were present in 4 of 7 patients and more mature stages were found in 2 of 7 patients (Extended Data Fig 1j). To further confirm the newborn identity of Dcx+ Prox1+ cells, we used an additional immature neuron marker PSA-NCAM. All Dcx+ Prox1+ cells were PSA-NCAM triple positive in cases containing high levels of neurogenesis (n = 4 cases; Fig 1i–k, Extended Data Fig 1k, l). In these cases, nearly 30% of Dcx+ PSA-NCAM+ cells were negative for Prox1, and likely represented neural precursors that had not yet committed to granule neuron identity. These results in total confirm the presence of adult human neurogenesis14,16–19,21, and reveal robust immature neuron decline as an MTLE disease phenotype.
Immature astroglia persist through human MTLE progression
In addition to Dcx+ Prox1+ new neurons, we also observed a striking number of Dcx+ Prox1- cells with a stellar morphology in every MTLE case where histology was performed (n = 17/17). Dcx+ Prox1- cells were found as a prominent cellular band in the hilus near the granule cell layer (GCL) and were also present at lower levels within the GCL and molecular layer (ML) (Fig 2a–g). We did not observe a significant difference in the number of these cells based either on the age at surgery (Fig 2b–d), or the duration of disease (Fig 2e–g). However, a trend toward increasing Dcx+ stellar cell numbers was observed with age in the GCL (p = 0.0640) and ML (p = 0.0664) (Fig 2b, d), suggesting a potential age-associated influence of these cells on epilepsy progression. We then explored the identity of this immature cell type. We did not observe co-expression of stellar Dcx+ cells with markers of granule neurons (Prox1+), Mossy cells (GluR2/3+), interneurons (Gad65/67+) or microglia (Iba1+) (Extended Data Fig 2a). The stellar Dcx+ cells also did not co-express PSA-NCAM which also distinguishes them from immature neurons (Extended Data Fig 2b). Instead, in MTLE tissue all stellar Dcx+ cells co-expressed S100β and GFAP, indicating that these cells are immature astroglia (Fig 2h–j. Extended Data Fig 3a, b). Unlike mouse immature astrocytes and mature human astrocytes, human fetal astroglia are known to express Dcx26. Previous studies have also identified Dcx+GFAP+ cells ex vivo in adult primate cerebral cortex and in vitro from human adult cerebral cortex27. In addition to immature astroglia, we identified S100β+ and GFAP+ cells that were frequently Dcx- in the ML, providing evidence of more mature astrocytes in the human MTLE hippocampus. Yet, 81.8% of the total S100β+ cells and 64.8% of GFAP+ cells were Dcx+ in MTLE patients (n = 5; Fig 2i, j). Hence, Dcx+ immature astroglia exist in the adult human hippocampus and persist during MTLE progression. To evaluate whether these Dcx+ stellar astroglial cells are undergoing cell division, we stained for Ki67, a marker for cell proliferation (Extended Data Fig 2c). However, we did not detect any Dcx+ Ki67+ cells in the MTLE cases analyzed (n = 3). The striking prevalence of immature astroglia in all MTLE cases was surprising to us. We therefore investigated whether Dcx+ immature astroglia were present in control postmortem cases by co-staining for Dcx and GFAP (n = 6; Fig 2k, Extended Data Fig 3c). We did not observe any Dcx+ GFAP+ cells in the hippocampus of control subjects. Hence, the robust presence of Dcx+ immature astroglia is a pathological phenotype associated with MTLE.
Figure 2. Immature astroglia persist through human MTLE disease duration.
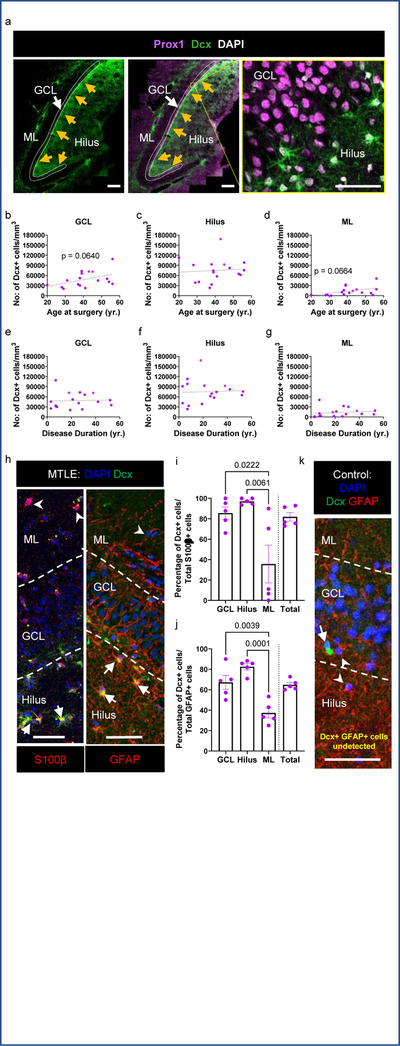
(a) Dcx+ (green) Prox1- (purple) stellar cells in the GCL (outlined by dotted lines), hilus and ML of the DG. Arrows point to the interior GCL (left 2 panels); Scale bar 500μm. Right panel shows a zoomed in view revealing that these cells are Prox1-. (b-g) Number of Dcx+ stellar cells/mm3 in GCL, hilus and ML across (b-d) age at surgery and (e-g) disease duration. N = 17. (h) Dcx+ (green) stellar cells co-express glial markers S100β (red), and GFAP (red) mostly in the hilus and GCL (marked by arrow). S100β+ Dcx- cells and GFAP+ Dcx- cells (marked by arrowhead) are present in the ML. Scale bar 50μm. (i, j) Quantification of Dcx+ cells among the total (i) S100β+ and (j) GFAP+ astroglia population. N = 5. One-way ANOVA ((i) F 3,16 = 7.309, p=0.0026, (j) (F 3,16 = 16.58, p<0.0001)) with Tukey’s multiple comparison test. (k) Dcx+ (green) GFAP+ (red) cells not detected in GCL, hilus and ML of N = 6 control cases. All the error bars represent s.e.m across individual cases.
In vitro generation of new granule neurons and astroglia
It has been postulated that Dcx and PSA-NCAM co-expressing cells re-express or keep expressing immature markers but do not necessarily represent adult-born neurons15. In addition, the large number of astroglia with an immature identity (Dcx+ S100β+, Dcx+ GFAP+) and their lack of a cell proliferation marker in MTLE patients could indicate a dedifferentiated state. To better understand whether immature neurons and astroglia can be newly generated we created neurosphere cultures and assayed neural differentiation directly from adult human MTLE patients (Fig 3a). Primary cells were isolated from both the hippocampus and adjacent cortical regions (neocortex and periallocortex) to grow neural stem cells (NSCs) in vitro (n = 5–6 patients). Cells from the hippocampal region generated neurospheres, whereas the cortex did so at very low numbers, re-affirming the hippocampus as a niche containing human NSCs (Fig 3 b,c)28,29. We then passaged NSC cultures at least once before seeding them for neural differentiation. After 3 weeks we observed that among differentiating cells (Tuj1+), a small number (10–18%) also possessed astroglial characteristics (Tuj1+ GFAP+) (n = 6; Fig 3 d,e, Extended Data Fig 4a). These results indicate that newborn cells co-expressing immature neuronal and astroglial characteristics can be generated from human MTLE hippocampus in vitro. Similar to in vivo data (Fig 2b–g), we did not observe any significant difference in the frequency of newborn astroglia generation in vitro by age at surgery nor disease duration at 3 weeks of differentiation (Fig 3 g, h). However, a longer-term differentiation of 6 weeks showed a significant increase in the frequency of Tuj1+ GFAP+ cells in MTLE cases with longer disease duration, but not by patient age (n = 5 cases; Fig 3 d, e, g, h). The increase in Tuj1+ GFAP+ cells from 3 to 6 week differentiation (Fig 3h) suggests an acquisition of glial identity during differentiation instead of a failure to downregulate immature radial glia-like cell identity. Mature astroglia (Tuj1- GFAP+) were identified at both 3 and 6 weeks differentiation, but at low frequency (0–3% of cells; Extended Data Fig 4b, c).
Figure 3: Generation of newborn granule neurons and astroglia from adult MTLE patients.

(a) Experimental approach for neurosphere generation and differentiation. Scale bar 100μm. (b) Neurospheres generated from hippocampus and adjacent cortex from the same MTLE patient. Scale bar 100μm; N = 5. (c) Quantification of the number of initial neurospheres generated per well. Paired two tailed t-test. N = 5 (d) Upper panel: Tuj1+ (green) GFAP+ (red) newborn astroglia (marked by arrows) and Tuj1+ GFAP- cells (marked by arrowheads) are present in neural differentiation cultures. Lower panel: Tuj1+ (green) Prox1+ (red) newborn granule neurons (marked by arrows) and Tuj1+ Prox1- cells (marked by arrowheads) are present in neural differentiation cultures. Scale-bar: 50μm (e) Quantification of GFAP+ and GFAP– cells among the Tuj1+ cells generated during 3-week (N = 6) and 6-week (N = 5) differentiation, One-way ANOVA F 3,18 = 5.811, p=0.0058. (f) Quantification of Prox1+ and Prox1– cells among the Tuj1+ cells generated during 3-week (N = 5) and 6-week (N = 5) differentiation. (g, h) Percentage of GFAP+ cells among the Tuj1+ cells across (g) age at surgery and (h) disease duration at 3-week (N = 6)and 6-week (N = 5) differentiation. The differences between the slopes of simple linear regression are significant. F 1,7 = 6.119, p=0.0426 (i, j) Percentage of Prox1+ cells among the Tuj1+ cells across (i) age at surgery and (j) disease duration at 3-week and 6-week differentiation (N = 5). (k) Age at surgery, and (l) Disease duration in years (yr.) for cases in which Tuj1+ Prox1+ cells were undetected (−) (N = 2) and detected (+) (N = 3). Unpaired two tailed t-test.. (m) Association between percentage of Tuj1+ cells that are Prox1+ and GFAP+ at 6-week differentiation (N = 5). Pearson Correlation. Pearson r=−0.9082 (n) Cartoon illustration of immature Tuj1+ cell differentiation into immature neurons and immature astroglia with time. All error bars represent s.e.m across individual cases.
In addition to newborn astroglial cells (Tuj1+ GFAP+), we also investigated newborn granule neuron production (Tuj1+ Prox1+) at 3 weeks and 6 weeks in vitro (Fig 3d, Extended Data Fig 4a). Prox1 expression was only identified in 1 of 5 MTLE cases analyzed at 3 weeks, yet 3 of 5 cases showed prevalent Prox1 expression in Tuj1+ cells at 6 weeks (Fig 3f). The detection of immature granule neurons both in vitro (Fig 3d: Tuj1+ Prox1+) and in vivo (Fig 1c: Dcx+ Prox1+) provides convergent evidence for the presence of adult neurogenesis in human MTLE. The prevalence of Tuj1+ Prox1+ cells showed a slightly greater decline upon longer disease duration (p = 0.0850) (Fig 3j) compared to age (p = 0.1858) (Fig 3i). Similar to our in vivo data (Fig 1 e, f), patients in whom Tuj1+ Prox1+ cells were generated in vitro were relatively younger (Fig 3k); however, disease duration contributed to a stronger reduction in newborn neuron number compared to age (Fig 3l). A comparison of astrogenesis (Tuj1+ GFAP+ cells) and neurogenesis (Tuj1+ Prox1+ cells) at 6 weeks in vitro from the same patients revealed an inverse correlation between production of these two cell types (Fig 3m). In particular, higher neurogenesis and lower astrogenesis occurs at shorter disease duration, while the converse occurs at longer disease duration (Fig 3 h, j, n). These findings imply that MTLE disease duration alters NSC fate decisions, consistent with mouse kainic acid seizure models13.
Immature neuron behavior with epileptiform activity
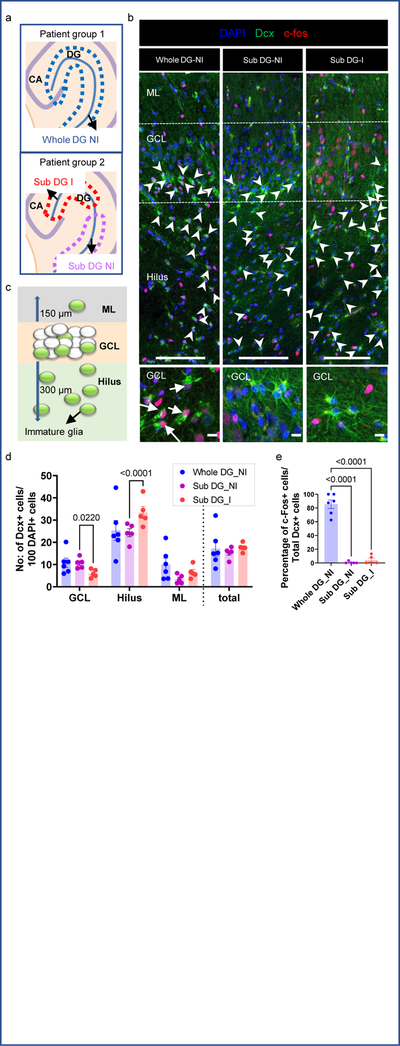
After identifying immature neurons and astroglia in MTLE hippocampus, we sought to determine their relationship with epileptiform activity. Our group has previously described MEA recordings of human hippocampal slices to distinguish regions of “inter-ictal like activity’ from those exhibiting no activity (‘non-inter-ictal like activity’)30. Inter-ictal like activity contained firing activity with a duration <200ms and a frequency <0.7 Hz, consistent with prior studies describing inter-ictal like activity in human epilepsy31,32. We recorded slices and further sub-sectioned either the MEA recorded slice or the adjacent slice for histological analysis (Fig 4a). Based on MEA recordings, MTLE patients with no MEA inter-ictal like activity inside the DG were classified as Whole DG Non- inter-ictal group (Whole DG NI, patient group 1). Patients with any MEA inter-ictal like activity within any part of the DG were considered patient group 2 (DG-I). The MEA data was sampled from several regions of the DG allowing for a detailed examination of neuronal hyperactivity. Patient group 2 is further divided into regions with inter-ictal like activity (Sub DG-I) and without inter-ictal like activity (Sub DG-NI) (Fig 4a, Extended Data Fig 5, 6). Neuronal hyperactivity and patient category were also confirmed by co-staining for immediate early genes c-fos and Arc33. The GCL of Sub DG-I patients exhibited a greater number of cells containing c-fos and Arc expression (Fig 5b Upper panel, Extended Data Fig 8b), similar to human epileptic cortex34. We then analyzed neurogenesis in both patient group 1 (Whole DG-NI, n = 7) and group 2 (DG-I, n = 5). Neurogenesis was detected in 4 out of 7 cases in the Whole DG-NI group, but not observed in 5 cases of the DG-I group (Fig 4b, c). These data indicate that neurogenesis is depleted when inter-ictal like activity is present in the DG. This absence of neurogenesis in cases with local hyperactivity also suggests that newborn neurons do not contribute to epileptiform activity at chronic stages of MTLE.
Figure 4: Newborn neuron behavior with epileptiform activity.

(a) Experimental workflow. MTLE cases cluster as whole DG non-inter-ictal-like activity (Patient Group 1: Whole DG-NI) and DG inter-ictal-like activity (Patient Group 2: DG-I). Lower panels: Multi-electrode array (MEA) recording of adult human hippocampus. Red markings indicate higher amplitude inter-ictal-like activity, yellow markings – lower amplitude and black markings – no activity. Scale bar: 500μm. (b) Fraction of patients containing neurogenesis in Whole DG-NI (N = 7) and DG-I (N = 5). Unpaired two tailed t-test. (c) Number of Dcx+ Prox1+ cells/mm3 in Whole DG-NI (N = 7) and DG-I (N = 5). (d, e) Percentage of mature Prox1+ neurons (N = 4) and immature Dcx+ PSA-NCAM+ neurons (N = 5) that co-express c-fos (d) and Arc (e). Unpaired two tailed t-test. (f) Dcx+ (green) PSA-NCAM+ (red) immature neurons stained for IEGs c-fos and Arc (purple; N = 5). Scale bar: 10μm. (g) Prox1+ (green) mature granule neurons stained for c-fos and Arc (purple; N = 4). Scale bar: 10μm. (h) Location of Dcx+ Prox1+ immature neurons as white dots in the GCL, Hilus and ML. Arrows mark cells outside the GCL. Scale bar 500μm. (i) Dcx+ Prox1+ cells in hilus and ML. Scale bar: 10μm. (j) Number of Dcx+ Prox1+ cells/mm3 in the GCL, hilus and ML (N = 7) Paired two tailed t-test. (k) Number of Dcx+ PSA-NCAM+ cells/mm3 (N = 5) Paired two tailed t-test. (l) Percentage of Dcx+ PSA-NCAM+ cells that are c-fos+ and Arc+ in the Whole DG-NI group (N = 5). (m) Dcx+ (green) PSA-NCAM+ (red) immature neurons in the hilus and ML stained for c-fos (purple). Dcx+ (green) PSA-NCAM+ (red) immature neurons stained for Arc (purple) in the hilus and ML (N = 5). Scale bar: 10μm. (n, o) Number of Dcx+ PSA-NCAM+ cells/mm3 that are c-fos −/+ (n) and Arc −/+ (o) in the Whole DG-NI group (N = 5). Two-way ANOVA ((n) F 2,24 = 10.02, p=0.0007, (o) F 2,24 = 11.65, p=0.0003) Sidak’s multiple comparison’s test. Error bars represent s.e.m across individual cases.
Figure 5: Alteration of immature astroglia behavior with epileptiform activity.
(a) Cases categorized as whole DG non-inter-ictal like activity (top panel: Whole DG-NI N = 6), and DG-inter-ictal like activity (bottom panel). In cases with DG inter-ictal like activity, sub-regions with (Sub DG-I N = 5) and without (Sub DG-NI N = 5) activity were analyzed separately. (b) Dcx (green) and c-fos (red) co-staining reveals correlations between immature cells and inter-ictal like activity. Upper panel: Increased presence of c-fos+ cells in the GCL of Sub DG-I compared to Whole DG-NI and Sub DG-NI. Dcx+ astroglia are preferentially localized to the hilus in Sub DG-I (arrowheads). Scale-bar: 100μm. Lower panel: magnified view of Dcx and c-fos co-staining. Scale-bar: 10μm. Most Dcx+ astroglial cells co-express the activity marker c-fos (arrow) in Whole DG-NI cases. (c) The GCL, adjacent hilus and ML spanning 300 and 150μm, respectively, from GCL were quantified. (d) Quantification of Dcx+ astroglia in the GCL, hilus and ML of whole DG-NI (N = 6), Sub DG-I (N = 5), and Sub DG-NI (N = 5). Two-way ANOVA (F 3,16 = 15.03, p<0.0001). Sidak’s multiple comparison’s test. (e) Quantification of immature Dcx+ astroglia activity (c-fos expression) in whole DG-NI (N = 6), Sub DG-I (N = 5), and Sub DG-NI (N = 5). One-way ANOVA (F 2,13 = 129.1, p<0.0001) Tukey’s multiple comparison’s test. All the error bars represent s.e.m across individual cases.
We then analyzed the activity of immature neurons (Dcx+ PSA-NCAM+) by co-staining for immediate early genes (IEGs) in Whole DG-NI cases defined by MEA or IEGs. Neither c-fos nor Arc were observed in any of the immature neurons (Dcx+ PSA-NCAM+) located in the GCL (n = 5 cases; Fig 4d–f). We then compared the prevalence of immature neuron activity to mature neuron activity in the GCL (n = 4) (Fig 4 d, e, g). A significantly greater number of mature Prox1+ cells expressing c-fos were detected compared to immature Dcx+ PSA-NCAM+ cells in Whole DG-NI cases (Fig 4d). A similar trend was observed for Arc expression (p = 0.0574) (n = 4; Fig 4e). The absence of detected IEG expression in immature neurons in cases without DG epileptiform activity suggests that immature neurons are largely inactive in MTLE patients.
Since new neurons undergo ectopic migration and contribute to neuronal hyperactivity in mouse models of epilepsy3–5, we further analyzed immature neurons in the hilus and ML of MTLE patients with neurogenesis detected in the GCL. Dcx+ Prox1+ cells were detected in the hilus and ML but were significantly less prevalent than in the GCL (n = 7; Fig 4 h–j, Extended data Fig 7a, b). Similarly, a significantly higher number of Dcx+ PSA-NCAM+ cells located to the GCL compared to the hilus and ML (Fig 4k) (n = 5). We next investigated whether Dcx+ PSA-NCAM+ immature neurons located in the hilus and ML were active by staining for IEGs (Fig 4 l–o, Extended data Fig 7 c, d). All analyzed Dcx+ PSA-NCAM+ cells in hilus and ML were negative for c-fos (n = 5 cases). Similarly, only a low number of Dcx+ PSA-NCAM+ cells in the hilus and ML were Arc positive, as identified in 2 of 5 cases (Fig 4 m, o). Our data suggest that the vast majority of immature neurons in the GCL, hilus and ML of MTLE patients do not exhibit heightened neuronal activity, as measured by IEG expression.
Immature astroglia behavior with epileptiform activity
The association of immature astroglia to inter-ictal like activity was then investigated. We first analyzed whether the location of immature astroglia changed with neuronal activity in MTLE patients (n =11 cases; Fig 5b, c) with and without DG inter-ictal like activity (Fig 5a). Within patient group 2, fewer immature astroglia were present in the GCL, and instead were increased in the hilus of Sub DG-I regions compared to Sub DG-NI (n = 5 cases; Fig 5b upper panel, Fig 5d). This result indicates immature astroglia migrate to the hilus upon neuronal hyperactivity, or immature astrocytes in the hilus contributes to generating seizures. We then sought to determine a relationship between immature astroglia activity and circuit-level inter-ictal like activity by staining for IEGs in the MEA-defined regions. Akin to neurons c-fos, but not Arc, expression in astrocytes marks their activity35 (Extended Data Fig 8b–d). Interestingly, we detected a strong anti-correlation between granule neuron activity and immature astroglial activity (Fig 5b lower panel, Fig 5e). Specifically, Dcx+ astroglia were c-fos+ in patient group 1 without local inter-ictal activity (n = 6 cases), whereas patient group 2 (n = 5 cases) displayed c-fos- Dcx+ astroglia in both inter-ictal like (Sub DG-I) and non-inter-ictal like (Sub DG-NI) sub-regions. We confirmed that all the Dcx+ c-fos+ cells were astroglia by co-staining for S100β (Extended data Fig 8a). The robust reciprocal activity between immature astroglia and mature granule neurons demonstrated by c-fos and MEA data suggest a functional role for immature astroglia in modulating epileptiform-like activity.
Discussion
The presence of adult neurogenesis in the human hippocampus has been contentious in both healthy14–21 and epileptic individuals15,22,23. In this study histology and cell culture provides additional complementary evidence of adult neurogenesis in the human epileptic hippocampus (Figures 1, 3), and histological confirmation in healthy individuals (Figure 1). We also find that neurogenesis dramatically declines with epilepsy duration, rather than with age in MTLE patients and compared to non-epileptic healthy controls. A higher density of immature neurons in MTLE at early disease durations (Figure 1 g–h) could also be inferred compared to healthy controls (Figure 1b). However, we do not consider it appropriate to make this quantitative comparison due to technical variations in handling control postmortem tissue and surgical MTLE tissue. The greater number of immature neurons in MTLE patients might be due to better quality of fresh surgical tissue compared to control postmortem samples. We cannot also rule out the possibility that neurogenesis is increased in early stages of MTLE as observed in mouse models4. The observed neurogenic losses in human MTLE with disease duration are, however, consistent with reports during chronic stages of rodent epilepsy models6 and more extensive than during Alzheimer’s disease progression in patients18,19. Our data in Figure 4 also shows that neurogenesis declines upon the presence of inter-ictal like activity within the dentate gyrus. Therefore, the pronounced exponential decline of neurogenesis in MTLE compared to Alzheimer’s disease19 might be attributed to active seizures in epilepsy; meanwhile, lower sub clinical epileptiform activity and other pathologies are observed in Alzheimer’s disease36,37. Instead, neurogenesis is largely preserved in cases of successful aging without cognitive decline18,19. As MTLE patients display progressive cognitive impairment38–39, and one-third of granule neurons are reported to turn over throughout the course of a lifetime16, we suggest that altered neurogenesis may contribute to convergent cognitive impairment in both epilepsy and dementia patients22.
A central remaining question is whether MTLE patients retain the potential to regain neurogenesis once lost. In mouse models heightened neuronal activity stimulates stem cell proliferation, as has been described with electroconvulsive therapy40,41, and sustained seizures may lead to exhaustion of the stem cell pool6–8. Alternatively, the stem cell pool may be maintained in people with MTLE while neurogenesis is compromised by a combination of altered NSC fate choice (Figure 3), lack of neuronal maturation (Extended Figure 1), and enhanced cell death. This latter hypothesis is supported by sustained neurosphere formation (Figure 3) and continued astrogenesis at advanced disease duration stages (Figures 2–3), despite neurogenesis depletion (Figure 1). Future studies will define latent potential for neural regeneration in the adult hippocampus.
Nonetheless, we did not detect immature neurons in MTLE cases containing epileptiform activity in the DG indicating a depletion of neurogenesis due to neuronal hyperactivity (Figure 4). In addition, immature neurons were mostly inactive and located in the GCL in cases with neurogenesis. Our data suggests that immature neurons do not serve a major function in epileptiform activity during the chronic stages of human MTLE. The majority of rodent studies have demonstrated a role for neurogenesis in the early stages of epilepsy in young mice3,5,10,12, which have much higher levels of neurogenesis compared to more mature rodents and humans42. Therefore, further studies in older mice with chronic epilepsy are necessary to understand the conserved and divergent mechanisms across species in advanced stages of disease progression. While we do not have the resolution to dissect the role of new human neurons in MTLE initiation, patients continue to experience seizures despite the loss of neurogenesis throughout disease duration. Consequently, other cell types likely make a greater contribution to epileptiform activity and disease progression11.
Since astroglia continue to be generated at later stages of epilepsy in rodent models13, we considered the possibility of astrogenesis in the human hippocampus. We observed persistent levels of immature glia throughout MTLE duration. Curiously, these cells possess the morphology and markers of astroglia (S100β, GFAP) and co-express some markers of immature neurons (Dcx, Tuj1), but do not express others (PSA-NCAM) (Figures 2, 3). Dcx+ cells co-expressing astroglial markers were not identified in the hippocampus of control individuals with no known neurological disorders. Therefore, the immature astroglia observed in this study likely represents a pathological phenotype of epilepsy caused by cell genesis or de-differentiation of mature astrocytes to an immature or fetal-like state. We harvested primary NSCs from human MTLE patients to corroborate with cell culture evidence that immature astroglia could be newborn (Figure 3)43. Differentiation of NSCs to astroglia (Tuj1+ GFAP+ cells) at 3 and 6 weeks in vitro suggests that GFAP expression in hippocampal Tuj1+ cells is acquired with maturation to an astroglial state, rather than being a remnant of radial glial identity, as observed in mice44. Further, we detected an increase in the number of Prox1+ cells from 3 to 6 weeks in vitro indicating slower human adult neuronal maturation than in mice44. We propose that adult neurogenesis in human occurs in a protracted manner akin to human neurodevelopment. Meanwhile, comparison between newborn granule neuron (Tuj1+Prox1+ cells) and hippocampal astroglia (Tuj1+ GFAP+ cells) production at 6 weeks suggests that progenitor cells from MTLE patients begin to favor an astroglial cell fate, versus a neuronal one, with increasing disease duration. Given that the majority of astroglia in the DG co-expressed Dcx (Figure 2), we also hypothesize that newborn astroglia undergo delayed maturation in MTLE patients. Yet, we cannot exclude the possibility that some mature astrocytes have de-differentiated into a reactive state.
Epileptiform activity occurs due to sudden synchronous hyperexcitation of neural circuitry. However, it remains enigmatic how an epileptiform wave is triggered. Studies using mouse and zebrafish models have identified surges in astroglial activity prior to seizure onset45,46. Astroglia slowly integrate signals from neighboring neurons causing their intracellular calcium levels to rise with time. Once calcium reaches a threshold, astroglia can broadly activate inhibitory GABAergic interneurons to coordinate switches in behavioral function47,48. In epilepsy, the loss of interneurons results in an imbalance of excitation and inhibition, elevated potassium and glutamate levels, and a greater dependence on astrocyte engagement to quickly return the neuronal microenvironment to a physiological state49. Immature astroglia have depolarized resting potentials and low Kir 4.1 channel expression50, which results in lower potassium buffering capacity and further compromises their ability to terminate synaptic transmission. Additionally, reduced activity in immature astroglia may result in low tonic inhibition from interneurons, triggering the transition from stable to pathological electrical activity in MTLE.
Our findings of a strong anti-correlation between immature astroglia activity and circuit-level inter-ictal like activity (Figure 5) therefore suggest a potential role for immature astroglia in initiating coordinated epileptiform activity. We propose that immature astroglia may serve as a “lossy capacitor” in human epilepsy51. In such a model, immature astroglia would prevent excessive excitatory neurotransmission and limit neuronal hyperactivity in granule neurons; however, once immature astroglia are unable to maintain extracellular ionic homeostasis, functional alterations in their activity leads to aberrant modulation of the network. Our observation that DG sub regions with higher inter-ictal like activity contain less immature astroglia suggests differences in local circuit regulation exist compared to lower activity sub regions (Figure 5). It is possible that inter-ictal regions are more vulnerable to neuronal hyperactivity due to the recruitment of immature astroglia from the GCL ictal zone to the hilus, indicating inter-ictal seizure generation may be a result of astroglia network reorganization. The newborn immature glial cells that we have identified in human MTLE may therefore have a role in both initiating and modulating seizure activity.
In conclusion, our study finds that adult neurogenesis occurs in healthy humans and epilepsy patients. However, neuronal production declines with MTLE disease duration and epileptiform activity in the dentate gyrus. This drop in neurogenesis might account for several cognitive and affective impairments that increase in MTLE patients during disease progression. Yet, the majority of immature neurons are inactive, and seizures continue in patients lacking neurogenesis. Therefore, immature neurons are unlikely to regulate epileptiform activity during chronic stages of adult epilepsies. Instead, immature astroglia are prevalent in epilepsy cases throughout MTLE disease duration, not detected in age-matched healthy individuals, and show altered location and activity upon local epileptiform activity. Immature astrocytes are preferentially generated at longer disease duration in vitro and slowly mature while maintaining some neuronal characteristics. These results point to a new role of adult astrogenesis in modulating seizure activity.
Materials and Methods:
Control Tissue Processing/Preservation:
Control brain tissue was obtained from the Brain Collection of the New York State Psychiatric Institute at Columbia University, which includes brain samples from the Republic of Macedonia. The New York State Psychiatric Institute (NYSPI) IRB approved the study. Brain tissue collection was conducted with IRB approval and consent obtained from all informants. At brain collection, 2 cm-thick coronal blocks of the right hemisphere were flash-frozen in liquid Freon (−20°C) and stored at −80°C. Tissue samples were fixed in formalin for neuropathological examination. On cerebellar samples pH determination were performed as pH affects protein and RNA stability52. Brain and blood toxicology exams were performed. Over 30 drugs were screened for and quantified including amphetamine, cocaine, fluoxetine, sertraline, paroxetine, fluvoxamine, amitriptyline, nortriptyline, imipramine, citalopram, clomipramine, diazepam, alprazolam, buspirone, methadone, olanzapine, clozapine and haloperidol. Routine neuropathological examination was performed on fixed slices of left hemisphere. If the subject was 45 years old or older, hippocampal formation and neocortex were stained to exclude the presence of senile plaques and neurofibrillary tangles. Neuropathology exam also excluded microvascular disease, including white matter lacunae.
The hippocampal formation was dissected from fresh frozen 2-cm coronal blocks, fixed in 4% paraformaldehyde (PFA) at 4°C for a length of time proportional to the thickness of the block, corresponding to approximately 12 hours fixation per mm of tissue thickness. It has to be noted that in a thick block the penetration of the fixative takes more time that in a thin slice like those we used for the epilepsy tissue, therefore we think this fixation is the closest possible to the one implemented for the epilepsy tissue. Blocks were then soaked in increasing sucrose concentrations starting at 10%, then increasing to 20% and finally 30%. Tissue was then sectioned at 50 μm on a sliding microtome (Microm HM440E) and stored in 40-wells boxes at −20°C in cryoprotectant (30% ethylene glycol in 0.1M PBS). During the sectioning procedure, one section every 500 microns was set aside for Nissl staining. Nissl-stained sections were later used for anatomical alignment along the dentate gyrus (DG) rostro-caudal axis of sections processed for immunocytochemistry.
Control Subject Selection and matching procedure:
All subjects are deceased. Our IRB has determined that this postmortem work is not Human Subjects Research. The only involvement of live individuals were as informants for Psychological Autopsy interviews. Subjects were diagnosed using our validated psychological autopsy53 for DSM-IV Axis I and II diagnoses. History of lifetime mood disorders and recent medication records were obtained. As part of the clinical evaluation, global functioning was measured by the Global Assessment Scale (GAS, score range 0–100, e.g.: 1–10 = danger of harming self or others, inability to maintain personal hygiene, or serious suicide attempt; 91–100 = no problems, or superior functioning in several areas, or admired by others due to positive qualities)54, and controls had all a score between 80 and 100, as appropriate for a healthy subject.
Control subjects included in the study had no neurological or psychiatric diagnoses, no pathological or traumatic condition affecting the brain, clear toxicology reports, and were free of psychiatric treatments and cognitive impairment. Exclusion criteria were: any neuropsychiatric diagnosis, positive toxicology for psychoactive drugs or alcohol, alcoholism-associated liver changes, suicide attempt history, intellectual disability, AIDS, chronic illness, positive neuropathology, undetermined death, resuscitation with pro- longed (> 10min) hypoxia, received prescription of psychotropic medications in the last three months prior to death, long agonal states, and any chronic diseases. Subjects with the presence of diseases affecting the brain were excluded using clinical data, psychological autopsy interviews and neuropathological exams. Neuropathology excluded microvascular disease, including white matter lacunae. All subjects died by sudden death and postmortem interval (PMI) was limited to 23 hours, brains with damaged tissue or low pH were excluded, due to effects of PMI and pH on brain proteins52.
Matching:
Non-psychiatric non-suicide sudden death subjects with clear toxicology for drugs and medications affecting the brain were matched for age (within 5 years) with MTLE subjects. We also matched MTLE cases and controls for rostro-caudal level of the hippocampus and used Nissl stained sections to align epilepsy cases sections with sections that were selected for the assays for the control group. We also assured groups did not differ for sex distribution. Based on this matching procedure, we selected 9 control subjects, ages 21 to 56, consisting of 3 females and 6 males. PMI ranged from 6 to 23 hours with no significant difference between aged and younger individuals (Supplementary data Table 1). The subjects’ cause of death included: 3 cardiovascular events and 6 traumatic deaths (homicide or motor vehicle accident).
Control Immunofluorescence Protocols:
Before performing immunohistochemistry, tissue was taken from floating well boxes stored at −20°C. Sections were selected to match anatomical alignment between epilepsy cases and controls. Then tissue was left equilibrating to room temperature for 2 hours. Tissue sections were further washed in 1X PBS for 1 hour, then incubated in 0.3% hydrogen peroxide (H2O2) for 10 minutes to inhibit endogenous peroxidase activity. Tissue was washed three times in 1X PBS for 10 minutes each wash. Next blocking and permeabilization was performed in 3% Bovine serum albumin (BSA), 10% Donkey serum in 0.1% Triton-X in PBS for 2 hours at room temperature. The sections were incubated with primary antibodies for 18–20 hours at 4°C in 0.3 % BSA, 1% Donkey serum and 0.1% Triton X in PBS. Following a 1-hour equilibration to room temperature, slides were washed in 0.1% Triton X in PBS for 15 minutes 3 times. Secondary antibodies from Jackson Immuno research laboratories were diluted from a stock solution of 0.6mg/ml and incubated along with 1μg/ml of DAPI (Roche, Cat No: 10236276001) for 2 hours at room temperature in 0.3 % BSA, 1% Donkey serum and 0.1% Triton X. Sections were washed 3 times in 0.1% Triton X in PBS for 15 minutes. Tissue was then incubated in 0.1% Sudan Black diluted in 70% Ethanol for 5 minutes to reduce the auto-fluorescence. Tissue was subsequently washed in 1X PBS, mounted and cover slipped with 90% glycerol in PBS.
Leica Sp8 2 Photon Imaging Acquisition for control tissue:
To quantify co-localization of cell markers, control tissue processed for immunofluorescence was scanned using a confocal laser scanning microscope (Leica TCS SP8 2-Photon, Leica Microsystems Inc.). Tiled z stacks of the entire hippocampus area were obtained by first imaging conjugated fields of view via an automated stage in conjunction with the Stage Overview function of the LAS X software (Leica Application Suite X 3.1.1.15751), and then by stitching the images of adjacent areas with the merging algorithm provided with the software. For each experimental set, image stacks were acquired applying the same parameters to each subject. All z stacks were imaged with a dry Leica 20X objective (NA 0.70, working distance 0.5 mm), with a field of view of 553.6 3 553.6 mm, a pixel size of 0.54 3 0.54 mm, optical sectioning of 2.36 mm, and a z step of 1 μm.
Human MTLE Hippocampal Tissue:
Human hippocampal tissue was obtained from 19 patients undergoing an en bloc resection of their hippocampus for the treatment of medically resistant MTLE, following strict ethical guidelines. Written consents for the surgical treatment of MTLE, tissue donation for the study as well as relevant clinical and demographic information collection (University of Southern California Institutional Review Board approval number: HS-17–00370) were obtained from all the patients prior to the surgery. No patient compensation was provided as surgical resection was part of the treatment protocol. Retrospective patient clinical record review was performed to collect relevant demographic and clinical information. De-identified patient clinical details (age upon surgery, duration of epilepsy, age of disease onset, sex, left vs right hemisphere of resection) are provided in Supplementary Data Table 2. Both male (N = 7) and female (N = 12) cases were included to examine sex as a biological variable in the study. The surgery was performed by neurosurgeons at the Keck Medical Center or Los Angeles County Hospital, University of Southern California. Surgically resected hippocampal tissue was transported to the laboratory within 30 minutes to 1 hour of surgical removal in a tube containing HypoThermosol® FRS (BioLife Solutions, Bothell, WA) solution maintained at approximately 4°C in an ice box.
Tissue Processing:
A larger slice (1–2 mm) of human tissue was first allocated for primary neurosphere culture. Following that, remaining hippocampal tissue was cut into 400 μm thick slices in chilled (4°C), oxygenated (95% O2, 5% CO2) N-methyl-D-glycamine - artificial cerebrospinal fluid (aCSF) solution (NMDG-aCSF; NMDG 93mM; KCl 2.5mM; NaH2PO4 1.2mM; NaHCO3 30mM; HEPES 20mM; Glucose 25mM; Sodium ascorbate 5mM; Thiourea 2mM; Sodium pyruvate 3mM; MgSO4 10mM; CaCl2 0.5mM) using a vibrating microtome (Leica VT1200S, Leica Biosystems, Germany). Vertical deflection of the blade was minimized with Vibrocheck technology (Leica Biosystems, Germany). Slicing parameters were set at speed 0.5–0.15 mm/s and vibration amplitude 1.5 mm. Hippocampal slices were then transported to a different laboratory for electrophysiology experiment within approximately one hour after vibratome slicing. The slices were transported in glass scintillation vials containing hypothermic biopreservation media HypoThermosol® FRS solution kept on ice at 4°C. This biopreservation approach during transportation obviated the need for continuous oxygenation as we previously reported30. Slices were then transferred to a recovery chamber and allowed to recover in warm (32°C) aCSF (NaCl 124 mM; KCl 4 mM; NaHCO3 26 mM; Glucose 10 mM; CaCl2 2mM; MgCl2 2mM) solution (30°C, 95% O2, 5% CO2) for 1 hour before multi-electrode array (MEA) recording. Following the MEA recording, slices were immediately fixed using 4% PFA for immunohistochemistry.
Inter-ictal like Activity (IIA) Detection Using Multi-Electrode Array (MEA):
Ex vivo hippocampal slice IIA was recorded using MEA1060-Inv extracellular multi-electrode array recording system and MC Rack v3.9.1 software (Multi Channel Systems, Germany) as previously reported30. Briefly, IIA was recorded in various hippocampal sub-regions using glass-based planar MEA 500/30iR-Ti with a 6×10 electrode configuration with 30–50 kΩ electrode impedance, 30 μm electrode diameter, 500 μm inter-electrode spacing and 11.25 mm2 total area (Multi Channel Systems, Germany) or an 8×8 60-electrode configuration with 30–50 kΩ electrode impedance, 30 μm electrode diameter, 1000 μm inter-electrode spacing and 45 mm2 total area (Multi Channel Systems, Germany). Pictures of the slice relative to the recording electrode location were acquired on an inverted microscope (Leica DM-IRB, Germany) with a digital image capture system (Spot RT Digital Camera, Spot Imaging, Sterling Heights, MI) and Leica Application Suite software (Leica Microsystems, Buffalo Grove, IL). Continuous oxygenated, warm (32–34°C) aCSF at a rate of 6 ml/min was circulated in the MEA ring well during tissue recording. A 5 min baseline recording was performed in aCSF followed by a 20–30 min recording in a modified aCSF solution containing high (8 mM) potassium, low (0.25 mM) magnesium and 100 μM 4-aminopyridine (4-AP) to induce IIA. To ensure recordings were collected from all hippocampal subregions, the slice was moved two to three times across the electrode array. Electrode location detecting induced IIA was then mapped to the anatomic hippocampus sub-regions using the slice photograph.
Immunohistochemistry:
Slices were pre-treated in a similar manner for methodological consistency between cases containing or not receiving MEA recordings. Briefly, slices were recovered in aCSF for 60 minutes, followed by a 15 minute treatment in modified aCSF containing high potassium (8 mM), low magnesium (0.25 mM) and 4-AP (100 μM). Slices were fixed in 4% PFA - made freshly from 16% PFA stored at −20°C - for 30 minutes at room temperature and washed thoroughly with 1x phosphate buffered saline (PBS) overnight. The slices were dehydrated using 30% sucrose in PBS at 4 °C. After 1 week, once the slice sank it was embedded in O.C.T compound (Tissue-Tek), ensuring the DG region is flat. Slices were further sub-sectioned by cryostat (Leica CM 3050 S) into 30μm and mounted to super frost plus slides. The slides were dried overnight at room temperature and transferred to −20°C for long term storage. Before performing immunohistochemistry, slides were equilibrated to room temperature for 2 hours. Slides were further washed in PBS for 1 hour, followed by blocking and permeabilization in 3% Bovine serum albumin (BSA), 10% Donkey serum in 0.1% Triton-X in PBS for 2 hours at room temperature. The sections were incubated with primary antibodies (Supplementary data Table 3) for 18–20 hours at 4°C in 0.3 % BSA, 1% Donkey serum and 0.1% Triton X in PBS. Following a 1-hour equilibration to room temperature, slides were washed in 0.1% Triton X in PBS for 15 minutes 3 times. Secondary antibodies (Supplementary data Table 3) from Jackson Immuno research laboratories were diluted from a stock solution of 0.6mg/ml and incubated along with 1μg/ml of DAPI (Roche, Cat No: 10236276001) for 2 hours at room temperature in 0.3 % BSA, 1% Donkey serum and 0.1% Triton X. Sections were washed 3 times in 0.1% Triton X in PBS for 15 minutes and mounted with cover glass. The whole DG region was imaged at 20X using a Zeiss LSM 700 confocal microscope automated tile scan.
Cell quantification:
All cell quantification was performed manually in Zeiss blue software. GCL boundaries were marked using the DAPI channel around the seemingly compact cell layer leaving a gap of two cell nuclear distance on both sides of the hilus and molecular layer. The whole section was analysed to quantify the number of rare Dcx+ Prox1+, Dcx+ PSA-NCAM+ Prox1-, Dcx+ PSA-NCAM+ Prox1+ cells. A representative sub-region in the GCL was analysed for Dcx+ Prox1- astroglial cells due to their high prevalence. An adjacent region of hilus and molecular layer spanning 300μm and 150μm respectively was also marked and analyzed. MEA recorded slices or the adjacent slices were immuno stained for immediate early genes (IEGs) Arc and c-fos to quantify the number of cell types in inter-ictal like and non-inter-ictal like regions. Regions with high MEA inter-ictal like activity in the DG also displayed high IEG expression (Figure 5b, Extended Data Fig 8b). Regions of DG-GCL with inter-ictal like activity (Sub DG-I) and spatially farthest non-inter-ictal like regions (Sub DG-NI) were compared. DG region with close proximity to an inter-ictal like hilus, CA or Subiculum, showed more IEG expression in some cases and therefore was omitted from quantification. In cases with DG inter-ictal like activity, the cell counts for Figure 2 b–g was done by averaging Sub DG-I and NI regions due to varying number of Dcx+ cells with inter-ictal like activity (Fig 5 d). Remaining statistical analysis and plots were prepared using Prism8 from GraphPad Inc.
Primary human hippocampal Cell Culture:
Human tissue sections were removed from hypothermosol and rinsed with PBS. The hippocampal region and adjacent cortical regions were separated and further minced to smaller pieces (size less than 1mm) using a blade and a scalpel in a 100mm dish. The minced tissue was transferred into a 15ml tube and centrifuged at 350g. The tissue was further digested using an enzymatic mix of 0.125% Trypsin (Gibco, Cat No: 25200056) and 1mg/ml collagenase (Gibco, Cat No: 17104–019) at 37°C for 10 minutes. The tissue was triturated and incubated for an additional 5–10 minutes as required and cells were passed through a 70μm filter. The cells were further washed in PBS, and cellular pellet was resuspended in 5 ml PBS. Equal proportions of 60% percoll (G.E. Life Sciences, Cat No: 17089102) were added to the cellular suspension. The tube was inverted to gently mix and then centrifuged at 800g for 10 minutes without brake. The top myelin layer was aspirated. The remaining solution was again centrifuged at 350g with brake for 5 minutes. Red Blood Cells (RBC) cells were removed using 1X RBC lysis buffer (Biolegend, Cat No: 420301) at room temperature for 5 minutes. After washing with PBS twice, cells were resuspended in DMEM/F12 with L-Glutamine (Gibco, Cat No: 11320033) + 1X N2 supplement (Gibco, Cat No: 17502048) + 1X B27 Supplement (Gibco, Cat No:17504044) + 1X Penstrep (Gibco, Cat No:15140–122) + 20ng/ml bFGF Human Recombinant Basic Fibroblast Growth Factor (bFGF) (Biopioneer, Cat No: HRP-0011) and 20ng/ml Human Recombinant Epidermal Growth Factor (EGF) (Biopioneer, Cat No: HRP-0012). The cells were counted using hemocytometer and seeded at a density of 80,000 cells/well of a 24 well plate. Half of the 1.5 ml media was changed every 3–4 days. Primary Neurospheres (P0) of size 50–120 μm were observed in 1–3 weeks of time. The neurospheres were further passaged using 0.125% Trypsin and 1mg/ml collagenase and seeded at a density of 40,000 cells/well of a 24 well plate. In cases of observed cell death or loose cell aggregation during the primary seeding, passaging was performed to facilitate compact neurosphere formation. Quantification of neurosphere number was performed on the initial compact sphere formation (either the primary seeding or passage 1 in the cases were P0 were only loose cell aggregates) using Leica Microsystems Inverted Laboratory Microscope DMIL LED Fluo with Leica Application Suite V4 software.
Neuronal Differentiation and Immunofluorescence staining:
Coverslips (VWR, Cat No: 89015–725) were coated with poly-L-ornithine (Sigma, Cat No: P2533) and laminin (Sigma, Cat No: L2020) for neuronal differentiation. Coverslips placed in 24 well plate were rinsed with 70% ethanol, and dried. Coverslips were first coated with 1ml of 15μg/ml polyornithine in autoclaved distilled water overnight at 37°C. They were washed twice with autoclaved distilled water and further coated with 0.5 ml of 5μg/ml laminin in PBS overnight at 4°C. The dish was equilibrated to room temperature for 1 hour before seeding cells. The cells were seeded as single cells (10,000–30,000 cells/well) in 1.5 ml differentiation media (DMEM/F12 with L Glutamine+ 1X N2 + 1X B27 + 1X Penstrep). Half of the 1.5 ml media was changed every 3–4 days. After 3 and 6 weeks of differentiation the cells were fixed with 4% PFA for 10 minutes. Following 3 washes in PBS for 10 minutes each, the cells were blocked for 1 hour using 10% Donkey serum and 0.1% triton X-100 in PBS. The cells were incubated with primary antibodies for immature neuronal (Tuj1), mature neuronal (Prox1) and glial markers (GFAP) at 4°C, overnight and secondary antibody for 2 hours at room temperature in 1% Donkey serum, and 0.1% Triton X in PBS. All the washes were performed using 0.1% Triton X in PBS. The coverslips with cells were mounted on a glass slide and imaged at 20X using Zeiss LSM 700.
Study design:
Sample size:
No statistical methods were used to pre-determine sample sizes. Instead, sample sizes of 19 MTLE patients and 9 control specimens are similar to those reported in previous publications14,18.
Data exclusion:
No data points are excluded.
Randomization:
In Fig 1, 3–5, patient classification into groups were not random but based on the presence and absence of measured phenotype (Dcx+ Prox1+ cells (Fig 1e, f), Tuj1+ Prox1+ cells (Fig 3k, l), Inter-ictal-like activity in DG (Fig 4b, c and 5d, e)). Therefore, covariates are not relevant.
Blinding:
Group allocation was determined according to experimental results described during randomization. Investigators were blinded to these groups while performing experiments, data acquisition, quantifications and data analysis. The partial exception is Figure 4 and 5 where complete blinding was not feasible during confocal image analysis due to the significantly high levels of c-fos and Arc expressed in regions with epileptiform activity. Instead, regions of analysis were marked blindly using DAPI staining to prevent any bias in counting.
Statistical Analysis:
All the statistical analysis was performed using Graphpad Prism 8. Data distribution was assumed to be normal, but this was not formally tested. Individual data points are shown in all the relevant graphs. Comparisons between two experimental groups were performed using a two tailed Students t-test. Comparisons among more than two experimental groups were performed using one-way ANOVA, followed by a Tukey’s multiple comparison post-hoc test. However, to compare simple effect in two matched variables (eg. comparisons between Sub DG-NI and DG-I from the same patient in Fig 5d), a two-way ANOVA was followed by a Sidak’s multiple comparison post-hoc test. A two-tailed Pearson’s correlation test was performed to calculate the strength of linear relationship between two variables. For Fig 1h a comparison of fits between linear regression, logarithmic, and exponential one phase decay was performed. The parameters for goodness of fit were better (R2 value closer to 1, lower RMSE) for exponential compared to logarithmic and linear regressions (Supplementary data Table 4). In Fig 4b the standard error in mean frequencies was calculated as √(p(1-p)/N) where p is the frequency of the given characteristic and N the total number of MTLE cases considered for a given characteristic. The statistical methods used for each figure is specifically noted in the figure legends.
Extended Data
Extended Data Fig. 1. Relationship among human immature neurons, age, MTLE onset and disease duration.

(a) Dcx+ (green) Prox1+ (purple) immature neuron in late stage maturation across Z stacks. Scale-bar 10μm. (b) Correlation between disease duration (yr.) and age at surgery (yr) (N = 17). Two tailed Pearson Correlation ** P ≤ 0.01. Pearson r=0.6299 (c) Number of Dcx+ Prox1 cells/mm3 in the Granule cell layer (GCL) with age of onset (yr.) (N = 17). Two tailed Pearson Correlation ** P ≤ 0.01. Pearson r=0.7021 (d) Age of onset for cases in which Dcx+ Prox1+ cells were undetected (−) (N = 10) vs detected (+) (N = 7). The graph represents s.e.m. Unpaired two tailed t-test. * P ≤ 0.05. (e) Correlation between disease duration (yr.) and age of onset (yr.) (N = 17). Pearson Correlation *** P ≤ 0.001. Pearson r=−0.7728 (b-e) Data points marked in red and blue are from cases in which Dcx+ Prox1+ cells were undetected (−) N = 10 and detected (+) N = 7 respectively. (f, g) Number of Dcx+ Prox1+ cells/mm3 in the GCL in (f) female (N = 11) vs male (N = 6) cases, and (g) left (N = 7) vs right (N = 10) hippocampus. The graph represents s.e.m. (h) Magnified view of Dcx+ Prox1+ cells ranging early, mid and late maturation states morphologically. Scale bar: 10μm. (i) Fraction of Dcx+ Prox1+ cells in various maturation stages in N = 7 cases with Dcx+ Prox1+ cells detected. One-way ANOVA (F 1.115,6.687 = 18.29, p=0.0036) Tukey’s multiple comparison’s test. * P ≤ 0.05, *** P ≤ 0.001. (j) Fraction of early, mid and late maturation stage Dcx+ Prox1+ immature neurons in each of N = 7 MTLE cases with Dcx+ Prox1+ cells detected. (k) Dcx+ (green) PSA-NCAM+ (red) cells with late maturation state morphology identified in the GCL of N = 1 MTLE case. (l) Dcx+ (green) PSA-NCAM+ (red) Prox1+ (purple) cell across Z-planes for image in Fig 1i lower panel Representative image from staining done in N = 4 cases.
Extended Data Fig. 2. Identifying Dcx+ cells as immature astroglia in MTLE tissue.

(a) Dcx+ stellar cells (green) do not co-express markers for granule neurons (Prox1) (N = 17 MTLE cases), mossy cells (GluR2/3) (N = 2 MTLE cases) inhibitory neurons (Gad65+67) (N = 2 MTLE cases) and microglia (Iba1) (N = 2 MTLE cases) stained in red. Scale bar 10μm. (b) Dcx+ (green) Prox1- (purple) stellar cells do not co-express PSA-NCAM (red), a marker for immature neuron Scale bar 10μm. (N = 4 MTLE cases) (c) Dcx+ stellar cells (green) do not co-express ki-67 (red), a marker for cell proliferation (N = 3 MTLE cases) Scale bar 10μm.
Extended Data Fig. 3. Dcx+ stellar cells co-express glial markers in MTLE patients but not in healthy controls.

(a, b) Dcx+ (Green) stellar cells co-express glial markers (a) S100β (Red), and (b) GFAP (Red) mostly in the hilus and GCL (marked by arrow). S100β + Dcx- cells and GFAP+ Dcx- cells (marked by arrowhead) are present in the ML. N = 5 MTLE cases. Scale bar 50μm. (c) Dcx+ (Green) GFAP+ (Red) cells not detected in GCL, hilus and ML of N = 5 control cases.
Extended Data Fig. 4. Neural differentiation from adult MTLE patients.

(a) Upper 3 panels: Tuj1+ (green) GFAP+ (red) newborn astroglia (marked by arrows) and Tuj1+ GFAP- cells (marked by arrowheads) present in neural differentiation cultures at 3-week (N = 6) and 6-week (N = 5) differentiation. Lower 3 panels: Tuj1+ (green) Prox1+ (red) newborn granule neurons (marked by arrows) and Tuj1+ Prox1- cells (marked by arrowheads) present in neural differentiation cultures at 3-week (N = 5) and 6-week (N = 5) differentiation. Scale-bar: 50μm (b) Tuj1- GFAP+ mature astroglia were rarely identified in N = 3 cases. Scale-bar: 50μm. (c) Percentage of Tuj1- GFAP+ mature astroglia at 3 (N = 6) - and 6-week (N = 5) differentiation. Graph represents s.e.m.
Extended Data Fig. 5. Mapping inter-ictal like activity with multi-electrode array (MEA) in DG-I cases.

(a) Illustrative 8×8 and 6×10 60-electrode MEA configuration and hippocampal slice recording of adult human MTLE tissue (4 representative cases from DG-I group). Inter-ictal like activity detected by electrodes covering the dentate gyrus (DG). Red markings overlaying the brightfield slice image and individual electrode recordings corresponds to inter-ictal like activity with comparatively higher amplitude, yellow markings – electrodes detecting comparatively lower amplitude and black markings – electrodes not detecting inter-ictal like activity.
Extended Data Fig. 6. Mapping inter-ictal like activity with multi-electrode array (MEA) in Whole DG-NI cases.

(a) Illustrative 8×8 60-electrode MEA configuration and hippocampal slice recording of adult human MTLE tissue (4 representative cases from Whole DG-NI group lacking activity in the DG). Inter-ictal like activity not detected by electrodes covering the DG. Red markings in the slice picture and MEA electrode layout indicate electrodes detecting comparatively higher amplitude inter-ictal like activity, yellow markings – electrodes detecting comparatively lower amplitude and black markings – electrodes not detecting inter-ictal like activity.
Extended Data Fig. 7. Ectopic immature neurons in MTLE patient hippocampus.

(a) Representative image of Dcx+ (green) Prox1+ (purple) cells in the Hilus (left panel) and ML (right panel). Scale bar: 50μm. (b) Distance of Dcx+ Prox1+ cell from GCL in the hilus and ML of N = 7 MTLE cases. Graph represents s.e.m. (c) Dcx+ (green) PSA-NCAM+ (red) immature neurons in hilus and ML are not positive for c-fos (Purple) in N = 5 MTLE cases. (d) Dcx+ (green) PSA-NCAM+ (red) immature neurons that are Arc- and Arc+ (Purple) identified in hilus and ML of N = 5 MTLE cases.
Extended Data Fig. 8. Immediate early genes c-fos and Arc in adult MTLE patient hippocampus.

(a) All Dcx+ (green) c-fos+ (purple) cells co-express S100β (red) (N = 2 cases) (b) Dcx (green) and Arc (red) co-staining determine the relationship between immature cells and inter-ictal like activity. Increased presence of Arc+ cells in the GCL of Sub DG-I compared to Whole DG-NI and Sub DG-NI. Dcx+ astroglia are preferentially localized to the hilus in Sub DG-I (arrowheads). Scale-bar: 100μm (c) Magnified view of Dcx and Arc co-staining. Scale-bar 10μm. Dcx+ atypical astroglial cells (green) are mostly Arc- in whole DG-NI cases and sub-regions of cases with inter-ictal like activity. (d) Quantification of Arc expression in immature Dcx+ astroglia in whole DG-NI cases (N = 6) and sub-regions of cases with inter-ictal like activity (N = 5). Data points are from individual MTLE cases, and the graph represents s.e.m.
Supplementary Material
Acknowledgements:
We thank the families that gave consent for brain tissue collection and interviews to investigators at Columbia University, the New York State Psychiatric Institute and the University of Southern California. We thank teams performing the psychological autopsy interviews and Mihran J. Bakalian for assistance with lab equipment and software. We thank the USC Neurorestoration administration and clinical research staff for their support on this study. We thank Andrew P. McMahon and members of the Bonaguidi lab for helpful discussions.
Funding:
This work was supported by the NIH (R00NS089013, R56AG064077 to M.A.B; MH83862, NS090415, MH94888 to M.B.; U01MH098937 to R.H.C; MH64168, MH098786 to A.J.D; MH40210 to V.A; MH090964 to J.J.M), Donald E. and Delia B. Baxter Foundation, L.K. Whittier Foundation, Eli and Edythe Broad Foundation (to M.A.B), USC Neurorestoration Center (to J.J.R and C.Y.L.), Rudi Schulte Research Institute (to C.Y.L.), American Foundation for Suicide Prevention SRG-0-129-12, Brain and Behavior Research Foundation Independent Investigator Grant 56388, New York Stem Cell Initiative C029157 and C023054, Dr. Brigitt Rok-Potamkin’s Foundation, the Morris Stroud III Center for Study of Quality of Life in Health and Aging (to M.B.), and American Epilepsy Society (to A.A).
Footnotes
Competing interests: J.J.M. receives royalties for commercial use of the C-SSRS from the Research Foundation for Mental Hygiene. Work by Dr. Arango related to this paper was completed when she was employed at Columbia University and the New York State Psychiatric Institute; the opinions expressed in this article are the author’s own and do not reflect the views of the National Institutes of Health, the Department of Health and Human Services, or the United States government. The remaining authors declare no competing interests.
Data Availability:
All data are available in the manuscript or supplementary materials. Individual data points for each figure are deposited in Mendeley before manuscript publication [insert link]. Any information regarding the availability of raw data, materials and methods can be directed to corresponding author.
References:
- 1.Bond AM, Ming GL & Song H Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell vol. 17 385–395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller SM & Sahay A Functions of adult-born neurons in hippocampal memory interference and indexing. Nat. Neurosci. (2019) doi: 10.1038/s41593-019-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho K-O et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 6, 6606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parent JM et al. Dentate Granule Cell Neurogenesis Is Increased by Seizures and Contributes to Aberrant Network Reorganization in the Adult Rat Hippocampus. J. Neurosci. 17, 3727–3738 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pun RYK et al. Excessive Activation of mTOR in Postnatally Generated Granule Cells Is Sufficient to Cause Epilepsy. Neuron 75, 1022–1034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hattiangady B, Rao MS & Shetty AK Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol. Dis. 17, 473–490 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Kralic JE, Ledergerber DA & Fritschy J-M Disruption of the neurogenic potential of the dentate gyrus in a mouse model of temporal lobe epilepsy with focal seizures. Eur. J. Neurosci. 22, 1916–1927 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Hattiangady B & Shetty AK Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia 49, 26–41 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marín-Burgin A, Mongiat LA, Pardi MB & Schinder AF Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science (80-. ). 335, 1238–1242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou QG et al. Chemogenetic silencing of hippocampal neurons suppresses epileptic neural circuits. J. Clin. Invest. 129, 310–323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varma P, Brulet R, Zhang L & Hsieh J Targeting seizure-induced neurogenesis in a clinically-relevant time-period leads to transient but not persistent seizure reduction. J. Neurosci. 0920–19 (2019) doi: 10.1523/jneurosci.0920-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lybrand ZR et al. A critical period of neuronal activity results in aberrant neurogenesis rewiring hippocampal circuitry in a mouse model of epilepsy. Nat. Commun. 12, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sierra A et al. Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 16, 488–503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boldrini M et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorrells SF et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spalding KL et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 153, 1219–1227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eriksson PS et al. Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Tobin MK et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 24, 974–982.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Jiménez EP et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nature Medicine vol. 25 554–560 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Cipriani S et al. Hippocampal radial glial subtypes and their neurogenic potential in human fetuses and healthy and Alzheimer’s disease adults. Cereb. Cortex 28, 2458–2478 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Knoth R et al. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One 5, e8809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coras R et al. Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain 133, 3359–3372 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Liu YWJ et al. Doublecortin expression in the normal and epileptic adult human brain. Eur. J. Neurosci. 28, 2254–2265 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Engel J & International League Against Epilepsy (ILAE). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 42, 796–803 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Wiebe S, Blume WT, Girvin JP, Eliasziw M & Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N. Engl. J. Med. 345, 311–8 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloch J et al. Doublecortin-positive cells in the adult primate cerebral cortex and possible role in brain plasticity and development. J. Comp. Neurol. 519, 775–789 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Johansson CB, Svensson M, Wallstedt L, Janson AM & Frisén J Neural Stem Cells in the Adult Human Brain. Exp. Cell Res. 253, 733–736 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Kukekov VG et al. Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp. Neurol. 156, 333–44 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Hsiao M-C et al. An in vitro seizure model from human hippocampal slices using multi-electrode arrays. J. Neurosci. Methods 244, 154–163 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Pillai J & Sperling MR Interictal EEG and the diagnosis of epilepsy. Epilepsia vol. 47 14–22 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Gabriel S et al. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J. Neurosci. 24, 10416–10430 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simonato M et al. Differential expression of immediate early genes in the hippocampus in the kindling model of epilepsy. Brain Res. Mol. Brain Res. 11, 115–24 (1991). [DOI] [PubMed] [Google Scholar]
- 34.Rakhade SN et al. A common pattern of persistent gene activation in human neocortical epileptic foci. Ann. Neurol. 58, 736–747 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Adamsky A et al. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 174, 59–71.e14 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Lam AD et al. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 23, 678–680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vossel KA et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 80, 858–870 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen A, Capelli V & Husain M Cognition and dementia in older patients with epilepsy. Brain 141, 1592–1608 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermann B, Seidenberg M, Lee EJ, Chan F & Rutecki P Cognitive phenotypes in temporal lobe epilepsy. J. Int. Neuropsychol. Soc. 13, 12–20 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Segi-Nishida E, Warner-Schmidt JL & Duman RS Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc. Natl. Acad. Sci. U. S. A. 105, 11352–11357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang MH et al. Secreted frizzled-related protein 3 regulates activity-dependent adult hippocampal neurogenesis. Cell Stem Cell 12, 215–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snyder JS Recalibrating the Relevance of Adult Neurogenesis. Trends in Neurosciences vol. 42 164–178 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Chen M et al. Neural Progenitor Cells in Cerebral Cortex of Epilepsy Patients do not Originate from Astrocytes Expressing GLAST. Cereb. Cortex 27, 5672–5682 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Bonaguidi MA et al. Noggin Expands Neural Stem Cells in the Adult Hippocampus. J. Neurosci. 28, 9194–9204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian G-F et al. An astrocytic basis of epilepsy. Nat. Med. 11, 973–981 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz Verdugo C et al. Glia-neuron interactions underlie state transitions to generalized seizures. Nat. Commun. 10, 3830 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deemyad T, Lüthi J & Spruston N Astrocytes integrate and drive action potential firing in inhibitory subnetworks. doi: 10.1038/s41467-018-06338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mu Y et al. Glia Accumulate Evidence that Actions Are Futile and Suppress Unsuccessful Behavior. Cell 178, 27–43.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 49.de Lanerolle NC, Kim JH, Robbins RJ & Spencer DD Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 495, 387–395 (1989). [DOI] [PubMed] [Google Scholar]
- 50.W S. et al. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41 Suppl 6, (2000). [DOI] [PubMed] [Google Scholar]
- 51.Nossenson N, Magal A & Messer H Detection of stimuli from multi-neuron activity: Empirical study and theoretical implications. Neurocomputing 174, 822–837 (2016). [Google Scholar]
Additional References:
- 52.Lewis DA The human brain revisited: Opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology 26, 143–154 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Kelly TM & Mann JJ Validity of DSM-III-R diagnosis by psychological autopsy: A comparison with clinician ante-mortem diagnosis. Acta Psychiatr. Scand. 94, 337–343 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Endicott J, Spitzer RL, Fleiss JL & Cohen J The Global Assessment Scale: A Procedure for Measuring Overall Severity of Psychiatric Disturbance. Arch. Gen. Psychiatry 33, 766–771 (1976). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript or supplementary materials. Individual data points for each figure are deposited in Mendeley before manuscript publication [insert link]. Any information regarding the availability of raw data, materials and methods can be directed to corresponding author.