

Summary
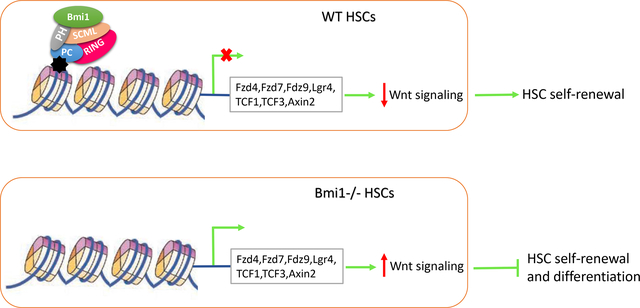
Polycomb group protein Bmi1 is essential for hematopoietic stem cell (HSC) self-renewal and terminal differentiation. However, its target genes in hematopoietic stem and progenitor cells are largely unknown. We performed gene expression profiling assays and found that genes of the Wnt signaling pathway are significantly elevated in Bmi1 null hematopoietic stem and progenitor cells (HSPCs). Bmi1 is associated with several genes of the Wnt signaling pathway in hematopoietic cells. Further, we found that Bmi1 represses Wnt gene expression in HSPCs. Importantly, loss of β-catenin, which reduces Wnt activation, partially rescues the HSC self-renewal and differentiation defects seen in the Bmi1 null mice. Thus, we have identified Bmi1 as a novel regulator of Wnt signaling pathway in HSPCs. Given that Wnt signaling pathway plays an important role in hematopoiesis, our studies suggest that modulating Wnt signaling may hold potential for enhancing HSC self-renewal, thereby improving the outcomes of HSC transplantation.
Keywords: Bmi1, HSC, self-renewal, differentiation, Wnt, β-catenin
Graphical Abstract
Introduction
Hematopoietic stem cells (HSCs) are multipotent, self-renewing progenitors that generate all mature blood cells [1–2]. In order to maintain hematopoietic homeostasis throughout the lifetime of an organism, the HSC pool must be maintained, which is achieved by the process of self-renewal [3–4]. Although practiced clinically for more than 40 years, the use of HSC transplants remains limited by the ability to expand functional HSCs ex vivo [5]. Deciphering the molecular mechanisms controlling HSC self-renewal is essential for developing clinical strategies that can enhance ex vivo HSC expansion [3–5].
Polycomb group (PcG) proteins are epigenetic gene silencers that have been implicated in stem cell maintenance and cancer development [6–11]. Genetic and biochemical studies indicate that Polycomb group proteins exist in at least two protein complexes, Polycomb repressive complex 2 (PRC2) and Polycomb repressive complex 1 (PRC1), that act in concert to initiate and maintain stable gene repression [10–11]. Bmi1, a key component of the Polycomb repressive complex 1 (PRC1), is essential for both HSC and leukemia stem cell (LSC) self-renewal [12–15]. We demonstrate that Bmi1 is a substrate of AKT and that AKT-mediated phosphorylation of Bmi1 inhibits HSC self-renewal [16]. In addition to HSC self-renewal, Bmi1 also plays key roles in multi-lineage differentiation [17]. We found that Bmi1 enhances erythroid differentiation through upregulating ribosomal genes [18]. We also found that Bmi1 maintains the self-renewal property of innate-like B lymphocytes [19]. While Bmi1 plays critical roles in hematopoiesis [12–19], its target genes in hematopoietic stem and progenitor cells (HSPCs) are largely unknown. Bmi1 is a potent negative regulator of the Ink4a/Arf locus, which encodes the cell cycle regulator and tumor suppressor p16Ink4a and p19Arf proteins [20–21]. In Bmi1−/− bone marrow (BM) cells, there is an upregulation of both p16Ink4a and p19Arf [22]. However, loss of both p16 and p19 only partially rescues the self-renewal defects of Bmi1−/− HSCs [22], suggesting that Bmi1 may regulates the expression other genes in HSPCs.
The Wnt signaling pathway has pivotal roles during the development of many organ systems, and dysregulated Wnt signaling is a key factor in the initiation of various tumors [23]. In the canonical Wnt pathway, Wnt ligand binds to its receptor Frizzled at the cell surface and inhibits glycogen synthase kinase-3β (GSK-3β)-mediated phosphorylation and degradation of β-catenin. Stabilized β-catenin then translocates to the nucleus where it binds to T cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors and induces the expression of Wnt target genes, such as Tcf and Axin2. In the absence of Wnt ligand, GSK-3β phosphorylates β-catenin and targets it for ubiquitination and degradation [23]. Canonical Wnt signaling is differentially activated during hematopoiesis [24–26], suggesting an important regulatory role for specific Wnt signaling levels [27–29]. The Adenomatous polyposis coli gene (Apc) is a negative modulator of the canonical Wnt pathway and loss of Apc in hematopoietic compartment leads to stabilization of β-catenin and activation of the Wnt signaling pathway [29–30]. By combining different targeted hypomorphic alleles and a conditional deletion allele of Apc, a gradient of five different Wnt signaling levels were obtained in vivo [26]. By analyzing the effect of different mutations of Apc on hematopoiesis, Luis and colleagues demonstrated that the canonical Wnt signaling regulates hematopoiesis in a dosage-dependent fashion: Low levels of Wnt signaling activation (2-fold increase above normal) result in the maintenance of a multipotent state, therefore resulting in increased reconstitution in stem cell transplantation assays, and that high level of Wnt signaling (more than 10-fold increase above normal) results in impaired HSC self-renewal and a block in differentiation [26]. While Wnt signaling plays an important role in hematopoiesis [24–26], how Wnt signaling is regulated in HSCs remains elusive. Identify key regulators of Wnt signaling in HSCs may lead to novel approaches to expand human HSCs ex vivo and improve transplantation efficiency.
In this study, we discovered that several genes of the Wnt signaling pathway are upregulated in Bmi1 null hematopoietic stem and progenitor cells (HSPCs). Bmi1 binds to several Wnt genes on the chromatin and represses their transcription in HSPCs. Importantly, we found that loss of β-catenin partially rescues the HSC self-renewal and differentiation defects seen in the Bmi1−/− mice. Thus, we have identified Bmi1 as a novel regulator of Wnt signaling pathway in hematopoietic stem and progenitor cells.
Materials and Methods
Mice
Global Bmi1 knockout mice were provided by Dr. Martin van Lohuizen at the Netherlands Cancer Institute, the Netherlands [20]. Conditional Bmi1 knockout mice (Bmi1F/F) in the C57BL6 background were generated at the National Institute of Biological Science, Beijing, China. The Ctnnb1F/F mice were obtained from the Jackson Labs and have been backcrossed to the C57BL6 background for at least 8 generations. Wild type C57BL/6 (CD45.2+), B6.SJL (CD45.1+) and F1 mice (CD45.2+ CD45.1+) mice were obtained from an on-site core breeding colony. All mice were maintained in the Indiana University Animal Facility according to IACUC-approved protocols.
Flow cytometry
Flow cytometry analysis of hematopoietic stem and progenitor cells was performed as described previously [31–32]. Murine hematopoietic stem and progenitor cells were identified and evaluated by flow cytometry using a single cell suspension of bone marrow mononuclear cells (BMMCs). Hematopoietic stem and progenitors are purified based upon the expression of surface markers. Bone marrow (BM) cells were obtained from femurs by flushing cells out of the bone using a syringe and phosphate-buffered saline (PBS) with 2mM EDTA. Red blood cells (RBCs) were lysed by RBC lysis buffer (eBioscience) prior to staining. Experiments were performed on FACS LSR IV cytometers (BD Biosciences) and analyzed by using the FlowJo Version 9.3.3 software (TreeStar).
Gene expression and Pathways Analyses
Transcript profiling of HSCs and MPPs from Bmi1+/+ and Bmi1−/− mice were analyzed by Agilent Whole Mouse Genome Oligo Microarrays. Raw data will be available for download from Gene Expression Omnibus (http://ncbi.nlm.nih.gov/geo/, accession number in progress). Genes whose expressions are increased or decreased more than 2-fold in Bmi1−/− cells compared to wild-type cells are shown. The Microarray data were analyzed using the Ingenuity Pathways Analysis program (Ingenuity Systems, www.ingenuity.com); to identify the pathways that met the < or > 2-fold change cutoff and were associated with a canonical pathway in the Ingenuity Pathways Knowledge base were considered for the analysis. The significance of the association between the data set and the identified canonical pathway was measured in 2 ways: (1) A ratio of the number of genes from the data set that map to the pathway divided by the total number of genes from the data set that map to the canonical pathway and (2) Fischer’s exact test, to calculate a p value determining the probability that the association between the genes in the data set and the canonical pathway is explained by chance alone.
ChIP assays
For ChIP assays, Kit+ BM cells and Baf3 cells were fixed with 1% (vol/vol) formaldehyde for 10 min at room temperature. ChIP assays were performed using the EZ-Magna ChIP A/G Kit (Millipore). Anti-Bmi1 antibody (Active Motif, AF27) and normal mouse IgG were used for immunoprecipitation. ChIP DNA was then subjected to real-time PCR analysis using primers targeting different region of genes of the canonical Wnt pathway and Ink4a/Arf locus.
Reporter assays
The Top-Flash luciferase construct encodes the firefly luciferase reporter gene under the control of a minimal CMV promoter and 8 tandem repeats of the TCF/LEF transcriptional response element (TRE). A Top-Flash mutant plasmid, containing defective TCF/LEF transcriptional response element, was used as a negative control in this assay. Each reporter is premixed with constitutively expressing Renilla luciferase, which serves as an internal control for normalizing transfection efficiencies and monitoring cell viability. 293T cells were transfected using Lipofectamine 2000 transfection reagent (ThermoFisher), according to the manufacturer’s instructions. Transfected cells were harvested 24 hours later, and processed using Dual-Luciferase Reporter Assay (Promega).
Generation of retroviruses and infection of murine HSCs and MPPs
Retroviral vectors were produced by transfection of Phoenix E cells with the MIGR1 control (MSCV-IRES-GFP) or MIGR1 full-length Bmi1 c-DNA plasmid (MSCV-Bmi1-IRES-GFP), according to standard protocols [16]. Murine HSCs and MPPs were infected with high-titer retroviral suspensions in the presence of 8 μg/mL polybrene (Sigma-Aldrich). Forty-eight hours after infection, the GFP-positive cells were sorted by FACS.
Transplantation
We transplanted 500,000 BM mononuclear cells isolated from Bmi1F/F-Ctnnb1F/F-Mx1Cre-, Bmi1+/+-Ctnnb1F/F-Mx1Cre+, Bmi1F/F-Ctnnb1+/+-Mx1Cre+, and Bmi1F/F-Ctnnb1F/F-Mx1Cre+ mice (CD45.2+) together with 250,000 competitor BM cells (CD45.1+) into lethally irradiated recipient mice (CD45.1+CD45.2+). Eight weeks following transplantation, we injected pI:pC to delete Bmi1 and/or Ctnnb1 from hematopoietic cells. Peripheral blood was obtained by tail vein bleeding every 4-week after pI:pC treatment, RBC lysed, and the PB mononuclear cells stained with anti-CD45.2 FITC and anti-CD45.1 PE, and analyzed by flow cytometry. 20 weeks following transplantation, bone marrow cells from recipient mice were analyzed to evaluate donor chimerism in bone marrows. For secondary transplantation, 3 × 106 BM cells from primary recipient mice reconstituted with Bmi1F/F-Ctnnb1F/F-Mx1Cre-, Bmi1+/+-Ctnnb1F/F-Mx1Cre+, Bmi1F/F-Ctnnb1+/+-Mx1Cre+, and Bmi1F/F-Ctnnb1F/F-Mx1Cre+ BM cells were injected into lethally irradiated F1 mice (CD45.1+CD45.2+).
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 8 software (GraphPad software, Inc). All data are presented as mean ± standard error of the mean (SEM). The sample size for each experiment are included in the figure legends. Statistical analyses were performed using unpaired, two-tailed Student’s t test where applicable for comparison between two groups, and a One-way ANOVA test or Two-way ANOVA was used for experiments involving more than two groups. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
Results
Genes of the Wnt signaling pathway are upregulated in Bmi1 null hematopoietic stem and progenitor cells
To understand how Bmi1 regulates HSC self-renewal and differentiation, we performed gene expression profiling assays (using microarray analysis and quantitative RT-PCR analysis) to identify Bmi1 target genes in HSCs (CD48−CD150+Lin−Sca1+Kit+ cells) and MPPs (CD48+CD150−Lin−Sca1+Kit+ cells). Microarray analysis revealed that several genes in the canonical Wnt signaling pathway are significantly elevated in both HSCs and MPPs from Bmi1−/− mice (Fig. 1A–B), suggesting that Wnt signaling may be activated in the absence of Bmi1. We first confirmed upregulation of several genes of the canonical Wnt pathway, including Fzd4, Fzd7, Fzd9, Tcf3, and Axin2, in Bmi1−/− HSCs compared to Bmi1+/+ HSCs using quantitative RT-PCR analysis (Fig. 1C). Fzd4, Lgr4, Lef1, Tcf3, and Axin2 are significantly upregulated in Bmi1−/− MPPs compared to Bmi1+/+ MPPs. (Fig. 1D). These findings suggest that Bmi1 may be associated with genes in the Wnt pathway, thereby repressing their expression in HSPCs.
Fig. 1.

Genes of the Wnt signaling pathway are upregulated in hematopoietic stem and progenitor cells. (A) Transcript profiling of HSCs (CD48−CD150+LSKs) from Bmi1+/+ and Bmi1−/− mice were analyzed by Agilent Whole Mouse Genome Oligo Microarrays. Genes that are differentially expressed in Bmi1−/− HSCs compared to wild-type cells are shown. We utilized Ingenuity pathways Analysis (Ingenuity Systems) to group genes into specific canonical pathways. Values are shown for three biological replicates. Color red indicates genes that are upregulated in Bmi1−/− HSCs and color blue indicates genes that are downregulated in Bmi1−/− HSCs. (B) Transcript profiling of MPPs (CD48+CD150−LSKs) from Bmi1+/+ and Bmi1−/− mice were analyzed by Agilent Whole Mouse Genome Oligo Microarrays. Genes that are differentially expressed in Bmi1−/− MPPs compared to wild-type cells are shown. We utilized Ingenuity pathways Analysis (Ingenuity Systems) to group genes into specific canonical pathways. Values are shown for three biological replicates. Color red indicates genes that are upregulated in Bmi1−/− MPPs and color blue indicates genes that are downregulated in Bmi1−/− MPPs. (C) Real-time RT-PCR analysis of gene expression in Bmi1+/+ and Bm1−/− HSCs. Data shown are relative expression as compared to Bmi1+/+ HSCs (set as 1), n = three biological replicates, *p<0.05, **p<0.01. (D) Real-time RT-PCR analysis of gene expression in Bmi1+/+ and Bm1−/− MPPs. Data shown are relative expression as compared to Bmi1+/+ MPPs (set as 1), n = three biological replicates, *p<0.05, **p<0.01, ***p<0.001.
Bmi1 is associated with Wnt genes in hematopoietic cells
To determine if Bmi1 is directly associated with genes in the Wnt pathway, we performed chromatin immunoprecipitation (ChIP) assays using an antibody against Bmi1 or normal mouse IgG. As expected, Bmi1 was associated with the Ink4a/Arf locus in both Kit+ BM and Baf3 cells (Fig. 2A–B). We detected the association of Bmi1 with the Lef1, Fzd4, and Axin2 promoters in these cells (Fig. 2A–B). Ring1b is the catalytic component of the PRC1 complex and genome-wide ChIP-seq analysis showed Ring1b binging to several Wnt target genes, including Ccnd2, Fzd7, Ckn2a and Tcf3, in murine L8057 megakaryoblastic cells (Fig. 2C) [33]. Thus, we demonstrate that Bmi1/PRC1 is associated with Wnt genes in hematopoietic cells.
Fig. 2.

Bmi1 is associated with Wnt genes in hematopoietic cells. (A) Bmi1 binds to promoters of genes of the Wnt pathway in vivo. Chromatin bound DNA from Kit+ BM cells was immunoprecipitated with a Bmi1-specific antibody or with normal mouse IgG. qRT-PCR amplification was performed on corresponding templates using primers for indicated genes, n = three biological replicates, **p<0.01. (B) Bmi1 binds to promoters of genes of the Wnt pathway in vivo. Chromatin bound DNA from Baf3 cells was immunoprecipitated with a Bmi1-specific antibody or with normal mouse IgG. qRT-PCR amplification was performed on corresponding templates using primers for indicated genes, n = three biological replicates, *p<0.05, **p<0.01. (C) Representative Ring1b and IgG ChIP-seq profiles of loci occupied by Ring1b in murine L8057 megakaryoblastic cells. It appears that Ring1b associates with several Wnt target genes, including Ccnd2, Fzd7, Cdkn2a and Tcf3, in L8057 cells.
Bmi1 represses Wnt gene expression in hematopoietic stem and progenitor cells
To determine the impact of Bmi1 expression on Wnt activation in cells, we used the Top-Flash Wnt reporter system. The Top-Flash reporter system is designed to monitor the activity of Wnt signal transduction pathway in cultured cells [34]. While Wnt3a readily activates the Wnt reporter in 293T cells, Wnt activation is efficiently repressed by ectopic Bmi1 expression, demonstrating that Bmi1 indeed can inhibit Wnt signaling activation in cultured cells (Fig. 3A).
Fig. 3.

Bmi1 regulates Wnt gene expression in hematopoietic stem and progenitor cells. (A) Bmi1 represses Wnt3a-induced Wnt reporter activation. Luciferase activity was assayed 24 hours after transfection of 293T cells. Values are means (±SEM), n = three biological replicates, *p<0.05. (B) Ectopic Bmi1 expression represses Wnt gene expression in Bmi1+/+ HSCs. Data shown are relative expression compared to control viruses (MSCV-IRES-GFP) transduced HSCs, n = three biological replicates, *p<0.05, **p<0.01. (C) Ectopic Bmi1 expression represses Wnt gene expression in Bmi1+/+ MPPs. Data shown are relative expression compared to control viruses transduced MPPs, n = three biological replicates, *p<0.05, **p<0.01. (D) Ectopic Bmi1 expression represses Wnt gene expression in Bmi1−/− HSCs. Data shown are relative expression compared to control viruses transduced Bmi1−/− HSCs, n = three biological replicates, *p<0.05, **p<0.01. (E) Ectopic Bmi1 expression represses Wnt gene expression in Bmi1−/− MPPs. Data shown are relative expression compared to control viruses transduced Bmi1−/− MPPs, n = three biological replicates, *p<0.05, ***p<0.001.
To determine whether Bmi1 represses the expression of Wnt signaling genes in HSPCs, we introduced Bmi1 (MSCV-Bmi1-IRES-GFP) or GFP (MSCV-IRES-GFP) into HSCs and MPPs purified from Bmi1+/+ and Bmi1−/− mice using retroviruses mediated transduction. 48 hours after transduction, we isolated mRNA from transduced cells (GFP+) and performed qRT-PCR assays for genes involved in the canonical Wnt signaling pathway. We found that that ectopic Bmi1 expression results in downregulation of Fzd4, Fzd7, and Lef1 expression in Bmi1+/+ HSCs compared to that of the control viruses (MSCV-IRES-GFP) transduced cells (Fig. 3B). We also found that that ectopic Bmi1 expression leads to downregulation of Fzd4, Fzd7, Fzd9, Lef1, and Axin2 expression in Bmi1+/+ MPPs compared to that of the control viruses transduced cells (Fig. 3C). In addition, ectopic Bmi1 expression represses Fzd4, Fzd7, and Fzd9 expression in Bmi1−/− HSCs compared to that of the control viruses transduced cells (Fig. 3D). Further, ectopic Bmi1 expression leads to downregulation of Fzd4, Fzd9, and Lef1 in Bmi1−/− MPPs (Fig. 3E). Thus, we demonstrate that Bmi1 represses Wnt gene expression in HSPCs.
Loss of β-catenin partially rescued HSC self-renewal and differentiation defects seen in Bmi1 null mice
The Ctnnb1 gene encodes β-catenin and loss of β-catenin reduces Wnt activation [35]. To determine the impact of β-catenin deficiency on Bmi1−/− HSPCs, we generated Bmi1F/F-Ctnnb1F/F-Mx1Cre+ mice. We transplanted 500,000 BM cells isolated from Bmi1F/F-Ctnnb1F/F-Mx1Cre-, Bmi1+/+-Ctnnb1F/F-Mx1Cre+, Bmi1F/F-Ctnnb1+/+-Mx1Cre+, and Bmi1F/F-Ctnnb1F/F-Mx1Cre+ mice (CD45.2+) together with 250,000 competitor BM cells (CD45.1+) into lethally irradiated recipient mice (CD45.1+CD45.2+). Eight weeks following transplantation, we injected pI:pC to delete Bmi1 and/or Ctnnb1 from hematopoietic cells and examined the frequency of donor-derived cells (CD45.2+) in peripheral blood every 4 weeks for twenty weeks. Conditional deletion of Ctnnb1 does not affect the repopulating ability of BM cells (Fig. 4A). While the percentage of donor-derived cells in PB of recipient mice repopulated with Bmi1−/− BM cells decreases, loss of Ctnnb1 significantly increased the engraftment of Bmi1−/− BM cells at twenty weeks following pI:pC treatment (Fig. 4A). We observed increased number of donor-derived HSCs in the BM of recipient mice repopulated with Bmi1−/−Ctnnb1−/− BM cells compared to that of the Bmi1−/− cells (Fig. 4B–C). Loss of Bmi1 resulted in decreased myeloid differentiation and increased lymphoid differentiation in the BM, whereas Ctnnb1 deficiency rescued differentiation defects seen in Bmi1−/− mice (Fig. 4D).
Fig. 4.

Loss of β-catenin partially rescued HSC self-renewal and differentiation defects seen in Bmi1 null mice. (A) Percentage of donor-derived cells (CD45.2+) in the peripheral blood of recipient mice following pI:pC treatment. n=7 mice per group, **p<0.01. (B) Percentage of donor-derived cells (CD45.2+) in the BM of recipient mice at 20 weeks following pI:pC treatment. n=4 mice per group, **p<0.01. (C) The frequency of donor-derived HSCs in the BM of recipient mice at 20 weeks following pI:pC treatment. n=4 mice per group, *p<0.05. (D) Lineage distribution of donor-derived cells in the bone marrow of primary recipient mice at 20 weeks following pI:pC treatment. n=4 mice per group, **p<0.01, ***p<0.001. (E) Percentage of donor-derived cells (CD45.2+) in the peripheral blood of recipient mice at 8 weeks following secondary transplantation. n=7–8 mice per group, ***p<0.001.
To determine the impact of genetic deletion of Ctnnb1 on Bmi1−/− HSC self-renewal, we performed secondary BM transplantation assays. We found that Bmi1−/−Ctnnb1−/− BM cells show increased engraftment compared to Bmi1−/− cells following secondary transplantation (Fig. 4E). Thus, we demonstrate that loss of β-catenin, which reduces Wnt activation, partially rescues HSC self-renewal and differentiation defects seen in Bmi1−/− mice.
Discussion
Hematopoietic stem cell (HSC) self-renewal requires a complex crosstalk between extrinsic signals from the microenvironment and the HSC-intrinsic regulators to maintain an undifferentiated state [1–4]. However, the crosstalk between signaling pathways and HSC-intrinsic regulators has not been well defined at the molecular level [1–4]. Thus, there remains a critical need to improve our understanding of the interactions between signaling pathways and HSC-intrinsic regulators and develop novel strategies that can enhance ex vivo HSC expansion and improve the efficiency and outcome of HSC transplantation [5].
The role of Wnt signaling in adult hematopoiesis has been controversial [24–25]. While loss of Wnt3a impairs HSCs self-renewal and differentiation [37–38], blocking the secretions of Wnt proteins in the hematopoietic system does not affect hematopoiesis [39]. Constitutive beta-catenin activation impairs HSC self-renewal and blocks terminal differentiation [36]; however, mice lacking β- and γ-catenin have normal stem cell self-renewal and differentiation [35]. These findings indicate canonical Wnt signaling is far more complicated than expected. Indeed, the canonical Wnt signaling appears to regulate adult hematopoiesis in a dosage-dependent manner: low level of canonical Wnt activation enhances HSC self-renewal, whereas high level of canonical Wnt activation impairs HSC self-renewal and blocks terminal differentiation [26].
Polycomb group protein Bmi1 plays an important role in cellular homeostasis by maintaining a balance between proliferation and senescence [12–15, 20]. It is often overexpressed in cancer cells and is required for stem cell self-renewal [14–15]. However, the downstream targets that mediate Bmi1 function remain elusive. To explore the mechanism by which Bmi1 enhances hematopoiesis, we performed transcript profiling assays to compare gene expression in HSCs and MPPs isolated from wild type and Bmi1−/− mice. We found that the expression of several genes of the Wnt signaling pathway was upregulated in hematopoietic stem and progenitor cells (HSPCs). Further, Bmi1 directly associates with the promoter of these genes in hematopoietic cells. Importantly, we found that loss of β-catenin partially rescued self-renewal defects see in Bmi1 null mice. Given that activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block [26, 29–30, 36], our studies uncover a novel mechanism by which Bmi1 enhances HSC self-renewal and promotes terminal differentiation,
Previous studies have implicated Bmi1 in regulating WNT signaling in some cancer cells [40–41]. BMI1 has been shown to autoactivate its own promoter via an E-box present in its promoter. Further, BMI1 activates the WNT pathway by repressing DKK family of WNT inhibitors. BMI1 mediated repression of DKK1 results in up-regulation of WNT target c-Myc, leading to transcriptional autoactivation of BMI1 [36]. This positive feedback loop regulating BMI1 expression may be relevant to the role of BMI1 in promoting cancer and maintaining stem cell phenotype [40]. For example, BMI1 is upregulated in colon cancer tissues and cell lines. Overexpression of BMI1 in primary epithelial colon cells promotes cellular growth and activates WNT pathway, whereas knocking down of BMI1 expression in colon cancer cells represses these effects [41]. Mechanistically, BMI1 activates WNT signaling in colon cancer by negatively regulating the WNT antagonist IDAX [41]. These findings suggest that Bmi1 may play a context-dependent role in regulating Wnt signaling during development and tumorigenesis.
Although BMI1 is upregulated in many cell types, very little is known about the signaling pathways that regulate its expression. Wnt signaling plays a key role in intestinal stem cells and Bmi1 has been shown to be a potential marker for intestinal stem cells [42]. The expression of Bmi1 in human colon cancers is associated with nuclear β-catenin, a hallmark for the activated Wnt signaling. Thus, these studies suggest that Wnt signaling may regulate the expression of Bmi1 in colon cancer cells [42]. Whether Wnt signaling regulates Bmi1 expression in hematopoietic stem and progenitor cells is not known, thereby awaiting future investigation.
In summary, we have identified Bmi1 as a negative regulator of canonical Wnt signaling pathway in hematopoietic stem and progenitor cells. Our studies suggest that modulating canonical Wnt signaling may hold potential for enhancing HSC self-renewal, thereby improving the outcomes of HSC transplantation,.
Acknowledgements
This work was supported by R01 HL150624, R56 DK119524, R56 AG052501, DoD W81XWH-18–1-0265 and DoD W81XWH-19–1-0575 awards to YL. This work was supported in part by the Leukemia &Lymphoma Society Translational Research Program award 6581–20 and the St. Baldrick’s Foundation Scholar Award to YL. The authors would like to acknowledge the Flow Cytometry Core and In vivo Therapeutic Core Laboratories, which were sponsored, in part, by the NIDDK Cooperative Center of Excellence in Hematology (CCEH) grant U54 DK106846. This work was supported, in part, by a Project Development Team within the ICTSI NIH/NCRR Grant Number UL1TR001108.
Footnotes
Conflict Interest
The authors declared that no conflict interest exists.
References
- 1.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA and Weissman IL (2003). Biology of Hematopoietic Stem Cells and Progenitors: Implications for Clinical Application. Annu Rev Immunol, 21,759–806. [DOI] [PubMed] [Google Scholar]
- 2.Attar EC, and Scadden DT (2004). Regulation of hematopoietic stem cell growth. Leukemia, 18, 1760–8. [DOI] [PubMed] [Google Scholar]
- 3.Akala OO, and Clarke MF (2006). Hematopoietic stem cell self-renewal. Curr Opin Genet Dev,16,496–501. [DOI] [PubMed] [Google Scholar]
- 4.Zon LI (2008). Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature, 453,306–13. [DOI] [PubMed] [Google Scholar]
- 5.Walasek MA, van Os R, de Haan G (2012). Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci, 1266,138–50. [DOI] [PubMed] [Google Scholar]
- 6.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M (2004). Stem cells and cancer; the polycomb connection. Cell, 118, 409–18. [DOI] [PubMed] [Google Scholar]
- 7.Sparmann A, and van Lohuizen M (2006). Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer, 6, 846–856. [DOI] [PubMed] [Google Scholar]
- 8.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, and Cavalli G (2007). Genome Regulation by Polycomb and Trithorax Proteins. Cell, 128, 735–45. [DOI] [PubMed] [Google Scholar]
- 9.Pietersen AM, and van Lohuizen M (2008). Stem cell regulation by polycomb repressors: postponing commitment. Curr Opin Cell Biol, 20, 201–207. [DOI] [PubMed] [Google Scholar]
- 10.Bracken AP, and Helin K (2009). Polycomb Group Proteins: Navigators of Lineage Pathways Led Astray in Cancer. Nature Review of Cancer, 9, 773–84. [DOI] [PubMed] [Google Scholar]
- 11.Simon JA, and Kingston RE (2009). Mechanisms of Polycomb Gene Silencing: Knowns and Unknowns. Nat Rev Mol Cell Biol, 10, 697–708. [DOI] [PubMed] [Google Scholar]
- 12.Sauvageau M, and Sauvageau G (2010). Polycomb Group Proteins: Multi-Faceted Regulators of Somatic Stem Cells and Cancer. Cell Stem Cell, 7, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konuma T, Oguro H, Iwama A (2010). Role of the polycomb group proteins in hematopoietic stem cells. Dev Growth Differ, 52,505–16. [DOI] [PubMed] [Google Scholar]
- 14.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature, 423,302–5. [DOI] [PubMed] [Google Scholar]
- 15.Lessard J, Sauvageau G (2003). Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature, 423,255–60. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Liu F, Yu H, Zhao X, Sahsida G, Deblasio A, Chen Z, Lin HK, Di Giandomenico S, Elf SE, Yang YY, Miyata Y, Huang G, Menendez S, Mellinghoff I, Pandolfi PP, Hedvat CV and Nimer SD (2012). Akt Phosphorylates the Transcriptional Repressor Bmi1 to Block Its Association with Tumor Suppressing Ink4a-Arf locus. Science Signaling, 5, ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, Nakauchi H, Iwama A (2010). Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell, 6,279–86. [DOI] [PubMed] [Google Scholar]
- 18.Gao R, Chen S, Kobayashi M, Yu H, Young SK, Soltis A, Zhang Y, Wan Y, Vemula S, Fraenkel E, Cantor A, Xu Y, Yoder MC, Wek R, Ellis S, Kapur R, Zhu X and Liu Y (2015). Bmi1 promotes erythroid development through regulating ribosome biogenesis. Stem Cells, 33,925–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi M, Lin Y, Mishra A, Shelly C, Gao R, Reeh CW, Wang PZ, Xi R, Liu Y, Wenzel P, Ghosn E, Liu Y, Yoshimoto M (2020). Bmi1 Maintains the Self-Renewal Property of Innate-like B Lymphocytes. J Immunol, 204,3262–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature, 397, 164–168. [DOI] [PubMed] [Google Scholar]
- 21.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Mönch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K (2007). The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev, 21, 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H (2006). Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med, 203, 2247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reya T, Clevers H. (2005). Wnt signalling in stem cells and cancer. Nature. 434(7035):843–50. [DOI] [PubMed] [Google Scholar]
- 24.Staal FJ, Sen JM. (2008). The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur J Immunol, 38,1788–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staal FJ, Chhatta A, Mikkers H (2016). Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp Hematol, 44, 451–7. [DOI] [PubMed] [Google Scholar]
- 26.Luis TC, Naber BA, Roozen PP, Brugman MH, de Haas EF, Ghazvini M, Fibbe WE, van Dongen JJ, Fodde R, Staal FJ (2011). Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell, 9,345–56. [DOI] [PubMed] [Google Scholar]
- 27.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. (2003). A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature, 423,409–14. [DOI] [PubMed] [Google Scholar]
- 28.Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. (2005). Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol, 6,314–22. [DOI] [PubMed] [Google Scholar]
- 29.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. (2008). A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med, 205,2163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Fernald AA, Anastasi J, Le Beau MM, Qian Z. (2010). Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood,115,3481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Elf SE, Miyata Y, Sashida G, Liu YH, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A and Nimer SD (2009). p53 Regulates Hematopoietic Stem Cell Quiescence. Cell Stem Cell, 4, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Wang Q, Yu H, Capitano ML, Vemula S, Nabinger SC, Gao R, Yao C, Kobayashi M, Geng Z, Fahey A, Henley D, Liu SZ, Barajas S, Cai W, Wolf ER, Ramdas B, Cai Z, Gao H, Luo N, Sun Y, Wong TN, Link DC, Liu Y, Boswell HS, Mayo LD, Huang G, Kapur R, Yoder MC, Broxmeyer HE, Gao Z, Liu Y (2019). Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun, 10,5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu M, Mazor T, Huang H, Huang HT, Kathrein KL, Woo AJ, Chouinard CR, Labadorf A, Akie TE, Moran TB, Xie H, Zacharek S, Taniuchi I, Roeder RG, Kim CF, Zon LI, Fraenkel E, Cantor AB (2012). Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol Cell, 45, 330–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Liu D, Sun X, Yang K, Yao J, Cheng C, Wang C, Zheng J (2019). CDX2 inhibits the proliferation and tumor formation of colon cancer cells by suppressing Wnt/beta-catenin signaling via transactivation of GSK-3beta and Axin2 expression. Cell Death Dis, 10,26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J, Kuttler F, Malanchi I, Birchmeier W, Leutz A, Huelsken J, Held W (2008). Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood, 111,142–9. [DOI] [PubMed] [Google Scholar]
- 36.Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG, Leutz A (2006). Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol, 7,1037–47. [DOI] [PubMed] [Google Scholar]
- 37.Luis TC, Weerkamp F, Naber BA, Baert MR, de Haas EF, Nikolic T, Heuvelmans S, De Krijger RR, van Dongen JJ, Staal FJ (2009). Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood, 113, 546–54. [DOI] [PubMed] [Google Scholar]
- 38.Luis TC, Naber BA, Fibbe WE, van Dongen JJ, Staal FJ (2010). Wnt3a nonredundantly controls hematopoietic stem cell function and its deficiency results in complete absence of canonical Wnt signaling. Blood, 116, 496–7. [DOI] [PubMed] [Google Scholar]
- 39.Kabiri Z, Numata A, Kawasaki A, Tenen DG, Virshup DM (2015). Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood, 126,1086–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho JH, Dimri M, Dimri DP (2013). A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J Biol Chem, 288,3406–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu F, Zhou C, Zeng H, Liu Y, Li S (2018). BMI1 activates WNT signaling in colon cancer by negatively regulating the WNT antagonist IDAX. Biochem Biophys Res Commun, 496,468–474. [DOI] [PubMed] [Google Scholar]
- 42.Yu T, Chen X, Zhang W, Colon D, Shi J Napier D, Rychahou P, Lu W, Lee EY, Weiss HL, Evers BM, Liu C (2012). Regulation of the potential marker for intestinal cells, Bmi1, by β-catenin and the zinc finger protein KLF4: implications for colon cancer. J Biol Chem, 287,3760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]