

Abstract
Two α-amylase-producing strains of Aspergillus oryzae, a wild-type strain and a recombinant containing additional copies of the α-amylase gene, were characterized with respect to enzyme activities, localization of enzymes to the mitochondria or cytosol, macromolecular composition, and metabolic fluxes through the central metabolism during glucose-limited chemostat cultivations. Citrate synthase and isocitrate dehydrogenase (NAD) activities were found only in the mitochondria, glucose-6-phosphate dehydrogenase and glutamate dehydrogenase (NADP) activities were found only in the cytosol, and isocitrate dehydrogenase (NADP), glutamate oxaloacetate transaminase, malate dehydrogenase, and glutamate dehydrogenase (NAD) activities were found in both the mitochondria and the cytosol. The measured biomass components and ash could account for 95% (wt/wt) of the biomass. The protein and RNA contents increased linearly with increasing specific growth rate, but the carbohydrate and chitin contents decreased. A metabolic model consisting of 69 fluxes and 59 intracellular metabolites was used to calculate the metabolic fluxes through the central metabolism at several specific growth rates, with ammonia or nitrate as the nitrogen source. The flux through the pentose phosphate pathway increased with increasing specific growth rate. The fluxes through the pentose phosphate pathway were 15 to 26% higher for the recombinant strain than for the wild-type strain.
In the production of industrial enzymes, filamentous fungi are attractive as cell factories because they can secrete large amounts of protein into the medium. Presently, many important industrial enzymes such as lipases, cellulase, and amylases are produced by fermentation of filamentous fungi, and there also has been a report on the use of Aspergillus oryzae for production of mammalian proteins (14). Despite the industrial importance of these organisms, relatively little is known about their basic physiology. For example, for A. oryzae, information about which pathways are present and active under various growth conditions is limited. Such information is quite important for the optimization of a fermentation process, especially if the fluxes in wild-type cells and overproducing cells are compared, since it quantifies the drain of precursors from the central carbon metabolism to protein biosynthesis and the requirements for cofactors like NADPH for protein synthesis.
Metabolic fluxes may be calculated from measurements of fluxes into and out of the cell and metabolite mass balances (18, 28, 36, 43, 47). A more robust estimate of the metabolic fluxes is obtained from experiments with 13C-enriched glucose in which either the fractional enrichment of 13C in cellular metabolites or the isotopomer distributions of the metabolites are quantified by using nuclear magnetic resonance (31). However, this technique is technically complex, and the simple approach based on metabolite balancing is often preferred.
Our first objective was to compare a wild-type strain and an α-amylase-overproducing strain of A. oryzae with respect to metabolic fluxes and enzyme activities. In order to calculate the metabolic fluxes, we developed a metabolic flux model. Our second objective was to investigate the influence of different nitrogen sources on the metabolic fluxes and enzyme activities. The working hypothesis tested was whether the metabolic fluxes in the two strains are the same and whether the NADPH supply influences the α-amylase productivity.
MATERIALS AND METHODS
Symbols.
The following symbols are used for the metabolic flux analysis: G (J by Q), a stoichiometric matrix; F (= 10), degrees of freedom; gji (no dimension), stoichiometric coefficient for compound i in reaction j; J (= 69), number of reactions; Q (= 59), number of intracellular metabolites; Xi (moles gram [dry weight]−1), concentration of the intracellular compound i; μ (hour−1), specific growth rate; and vj (moles gram [dry weight]−1 hour−1), rate of reaction j.
Microorganism.
Two strains of A. oryzae were used. A1560 is a wild-type strain, and CF 1.1 is a recombinant strain containing additional copies of the α-amylase gene (11, 46). Both strains were provided by Novo Nordisk A/S.
Media.
The chemically defined medium for continuous cultivation with ammonia as a nitrogen source contained (per liter) 4 g of glucose · H2O, 2.5 g of (NH4)2SO4, 0.75 g of KH2PO4, 1 g of MgSO4 · 7H2O, 1 g of NaCl, 0.1 g of CaCl2 · 2H2O, 0.25 ml of antifoam (Struktol SB2121; Schill & Seilacher, Hamburg, Germany), 7.2 mg of ZnSO4 · 7H2O, 1.3 mg of CuSO4 · 5H2O, 0.3 mg of NiCl2 · 6H2O, and 6.9 mg of FeSO4 · 7H2O. When nitrate was used as the nitrogen source, (NH4)2SO4 was replaced with 3.2 g of NaNO3 per liter and 2.7 g of Na2SO4 per liter.
Cultivation conditions.
The cultivations were carried out as constant-mass chemostats in an MBR bioreactor (11). The temperature was 30°C, and the pH was kept at 6.0 by adding 4 M NaOH or 2 M H2SO4. The headspace pressure was 1.5 × 105 Pa, and the aeration was 0.4 to 0.9 liter liter−1 min−1, depending on the specific growth rate. The cultures were considered to be in physiological steady state when the biomass concentration, the CO2 evolution rate, and the extracellular α-amylase activity were stable for at least three residence times.
Sampling.
For biomass measurements, the samples were filtered, washed with 0.9% (wt/vol) NaCl, and dried at 105°C for 24 to 48 h. For determination of the macromolecular composition of the biomass, the sample was filtered, washed with 0.9% (wt/vol) NaCl, and freeze-dried. After the freeze-drying, the cells were crushed in a mortar and kept dry in a desiccator. For determination of in vitro enzyme activities, the sample was filtered, washed with cold 0.9% (wt/vol) NaCl, and kept at −80°C until assayed (not longer than 3 months).
Analysis of extracellular α-amylase.
Extracellular α-amylase activity was measured by a flow injection analysis system based on monitoring of the decolorization of an iodine-starch complex (10).
Analysis of carbohydrates.
Freeze-dried biomass was disrupted in distilled water with a mixer mill (Retsch MM2) for 2 h at 4°C with 0.75- to 1.0-mm-diameter glass beads. The sample was analyzed by the phenol method (19). The reported values for carbohydrate content have been corrected for the contributions from RNA and DNA.
Analysis of DNA.
DNA was measured by the diphenylamine reagent method with salmon testis DNA as a standard (19). The biomass used had been freeze-dried.
Analysis of RNA.
RNA was determined by the Schmidt-Tannhauser method (3). An extinction coefficient of 10,800 M−1 cm−1 and an average molecular mass of nucleotides of 340 g mol−1 was used. The biomass used had been freeze-dried.
Analysis of protein, free amino acids, and chitin.
Freeze-dried biomass was disrupted in distilled water in a mixer mill (Retsch MM2) for 45 min at 4°C with 0.75- to 1.0-mm-diameter glass beads. Samples were hydrolyzed with HCl (6 M) for 6, 24, and 72 h at 110°C. For analysis of free amino acids, the biomass was boiled in water for 15 min. The extract was centrifuged at 10,000 × g, and the supernatant was analyzed by high-pressure liquid chromatography (HPLC) for amino acid composition (2). Glutamine could not be distinguished from glutamic acid, and aspargine could not be distinguished from aspartic acid; therefore, the molar fractions of glutamine/glutamic acid and aspargine/aspartic acid were assumed to be 1. The nitrogen content originating from protein and chitin was calculated by subtracting the nitrogen content in RNA, DNA, and free amino acids from the total nitrogen content measured by the Kjeldahl method. The ratio of the amounts of chitin and protein was determined by HPLC analysis.
Analysis of glycerol and mannitol.
Freeze-dried biomass was suspended in 0.5 mM H2SO4 and boiled in water for 15 min. The cells were removed by centrifugation for 10 min at 5,000 × g, and the supernatant was analyzed by HPLC with an HPX-87H Aminex ion-exclusion column (36). The column was eluted at 60°C with 5 mM H2SO4 at a flow rate of 0.6 ml min−1. The detector was a Waters 410 refractive index detector.
Analysis of lipids.
Lipids were extracted with chloroform and methanol and determined gravimetrically (16).
Analysis of ash.
Freeze-dried biomass was heated at 600°C for 15 h, and the ash content was measured by weighing the remains.
Preparation of cell extracts for measurement of total enzyme activities.
A frozen sample was crushed in a mortar under liquid nitrogen. Thereafter, 0.1 g (wet weight) of mycelium was suspended in 1.5 ml of 0.1 M sodium phosphate (pH 7.4)–5 mM MgCl2–1 mM EDTA. To the suspension was added 0.15 ml of 26 mM dithiothreitol, and the suspension was crushed in a mixer mill (Retsch MM2) with 0.75- to 1.0-mm-diameter glass beads for 15 min at 4°C.
Preparation of cell extracts for separation of mitochondria and cytosol.
We prepared cell extracts by nitrogen cavitation (26) from cells grown on glucose and ammonia. The mycelia were filtered and washed with 0.9% (wt/vol) NaCl. Subsequently, 0.6 g (dry weight) of mycelia was suspended in 50 ml of 0.2 M sodium phosphate (pH 6.4) containing 0.6 M mannitol and 50 mg of Novozyme 234 (Sigma, St. Louis, Mo.). The suspension was incubated at room temperature (22 to 25°C) for 30 min with stirring. The mycelia were filtered through gauze and washed with 50 ml of ice-cold sodium phosphate-mannitol buffer. From this point on, the mycelium-cell extract was kept at 4°C. The mycelium was suspended in 50 ml of 0.1 M phosphate (pH 7.4) containing 0.25 M sucrose, 1 mM EDTA, 1 mM MgCl2, 1 mM CaCl2, 0.1 mM dithiothreitol, 8 μM phenylmethylsulfonyl fluoride, and 0.02 to 0.2% (wt/vol) bovine serum albumin (the cavitation buffer). Nitrogen pressure (106 Pa) was applied to the suspension for 30 min and then quickly released. The broken mycelium was filtered through two layers of gauze, and the filtrate was centrifuged at 700 × g for 10 min to remove cell debris. A mitochondrial pellet was obtained by centrifuging the supernatant at 45,000 × g for 45 min. The pellet was washed with the cavitation buffer and then resuspended in 4 ml of the cavitation buffer. The 45,000 × g supernatant represented the cytosolic fraction. Since the amount of extracted protein was rather low, two cavitation experiments were carried out in parallel, one experiment with 0.02% (wt/vol) bovine serum albumin in the cavitation buffer and the other with 0.2% (wt/vol) bovine serum albumin in the cavitation buffer. These experiments were used for measurement of the amount of extracted protein and the enzyme activities, respectively. Measured protein concentrations were corrected for the bovine serum albumin in the cavitation buffer.
Analysis of in vitro enzyme activities.
Enzyme assays were performed at 30°C with an HP8453 UV-visible light spectrophotometer. In all cases enzyme activities were proportional to the amount of cell extract added. Enzyme activities are expressed as micromoles minute−1 milligram−1. Protein was determined by the Bradford method (7) with bovine serum albumin as the standard. All specific enzyme activities were reduced by a factor of 2, since bovine serum albumin gives twice as high a signal as most other proteins in the Bradford assay. The enzyme activities were measured by using the reaction mixtures listed in Table 1.
TABLE 1.
Enzymatic assays
| Enzyme | Reaction mixturea | Reference |
|---|---|---|
| Citrate synthase (EC 4.1.3.7) | 180 mM Tris (pH 7.4), 3 mM MgCl2, 2.3 mM oxaloacetate, 0.2 mM acetyl-CoA, 0.1 mM DTNB, 0.04% Triton X-100 | 44 (modified) |
| Isocitrate dehydrogenase (NAD+) (EC 1.1.1.41) | 170 mM Tris (pH 7.4), 0.94 mM NAD+, 1.25 mM AMP, 9 mM MgCl2, 1.25 mM isocitrate, 0.03% Triton X-100 | 15 (modified) |
| Isocitrate dehydrogenase (NADP+) (EC 1.1.1.42) | 82 mM phosphate (pH 7.4), 8 mM MgCl2, 1.6 mM EDTA, 0.3 mM NADP+, 0.8 mM isocitrate, 0.02% Triton X-100 | 38 (modified) |
| Isocitrate lyase (EC 4.1.3.1) | 70 mM phosphate (pH 7.4), 6 mM MgCl2, 1.4 mM EDTA, 3.4 mM phenylhydrazine hydrochloride, 2 mM isocitrate, 0.03% Triton X-100 | 1 (modified) |
| Glucose-6-phosphate dehydrogenase (EC 1.1.1.49) | 40 mM Tris (pH 8.2), 0.64 mM NADP+, 5 mM MgCl2, 4.84 mM glucose-6-phosphate, 0.03% Triton X-100 | 50 (modified) |
| Glutamate dehydrogenase [NAD(P)+] (EC 1.4.1.2, EC 1.4.1.3) | 35 mM Tris (pH 7.4), 1.3 mM tricarbalylate, 0.6 mM MgCl2, 0.2 mM NAD(P)H, 6 mM α-ketoglutarate, 43 mM (NH4)2SO4, 0.02% Triton X-100 | |
| Glutamate oxaloacetate transaminase (EC 2.6.1.1) | 100 mM phosphate (pH 7.4), 4 mM MgCl2, 20 mM aspartate, 12 mM α-ketoglutarate, 0.15 mM NADH, 7 U of malate dehydrogenase, 0.03% Triton X-100 | 6 |
| Malic enzyme (EC 1.1.1.40) | 86 mM phosphate (pH 7.4), 4 mM MgCl2, 1.7 mM EDTA, 2 mM DTT, 0.25 mM NADP+, 10 mM malate, 0.02% Triton X-100 | 1 |
| Malate dehydrogenase (EC 1.1.1.37) | 215 mM Tris (pH 7.4), 8 mM tricarbalylate, 11 mM MgCl2, 2 mM oxaloacetate, 0.24 mM NADH, 0.02% Triton X-100 | 37 (modified) |
| Mannitol dehydrogenase (EC 1.1.1.138) | 55 mM phosphate (pH 7.4), 2.8 mM MgCl2, 1.1 mM EDTA, 170 mM mannitol, 0.3 mM NADP+, and 0.01% Triton X-100 | 22 (modified) |
Acetyl-CoA, acetyl coenzyme A; DTNB, 5,5-dithiobis(2-nitrobenzoic acid); DTT, dithiothreitol.
Isoelectric focusing and gel staining.
Isoelectric focusing was carried out on polyacrylamide gels (Ampholine PAGplate pI 4-6.5 and pI 3-10; Pharmacia Biotech, Uppsala, Sweden) under the conditions recommended by the manufacturer. The gels were washed with distilled water and stained for isocitrate dehydrogenase (NADP) by overlaying with a 0.8% (wt/vol) agarose gel containing 0.3 mg of thiazolyl blue tetrazolium bromide ml−1, 0.05 mg of N-methylphenazonium methyl sulfate ml−1, 0.5 mg of isocitrate ml−1, and 0.5 mg of NADP+ ml−1. For staining for total protein, the gels were fixed in trichloroacetic acid and stained with Coomassie brilliant blue.
Metabolic flux analysis.
Calculations of metabolic fluxes by metabolite balancing are based on a metabolic model normally represented as a stoichiometric matrix, G. In this matrix the rows represent reactions and the columns represents compounds. Thus, the stoichiometric coefficient for the compound i participating in reaction j is positioned as element gji in the stoichiometric matrix. With the rates (or fluxes) of reaction j given by vj, the mass balance for the intracellular compound i becomes:
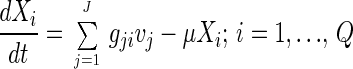 |
1 |
where Xi is the intracellular concentration of compound i and μ is the specific growth rate of the biomass. The last term accounts for dilution due to the cell growth and is generally negligible for intracellular metabolites. Furthermore, a pseudo-steady-state assumption can often be used for intracellular metabolites, and equation 1 then reduces to:
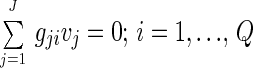 |
2 |
Equation 2 represents a set of algebraic equations (one for each intracellular metabolite) that relate the intracellular fluxes. If some of the fluxes can be measured, e.g., the flux of glucose into the cell or the flux of precursors incorporated into the biomass, then it may be possible to calculate the nonmeasured fluxes by using the algebraic equations represented by equation 2. If the total number of reactions included in the stoichiometric model is J and the number of intracellular metabolites in the pseudo-steady-state is Q, then the number of degrees of freedom is F = J − Q; i.e., F fluxes must be measured.
RESULTS
Enzyme activity measurements.
We analyzed several key enzymes to determine if these reactions were active and what the cofactor requirements were.
The glutamate dehydrogenase NADP+-dependent isoenzyme specific activities were 7 to 37 and 120 to 280 times higher than the NAD+-dependent isoenzyme specific activities with ammonia and nitrate, respectively, as the nitrogen source in a minimal medium. Only the reaction catalyzed by the NADP+-dependent isoenzyme was included in the metabolic model.
We found no activity of isocitrate lyase and malic enzyme.
Intracellular localization of enzymes.
Since intracellular compartmentation plays an important role in how the metabolic fluxes are distributed, eight enzymes were localized to the mitochondrial and cytosolic fractions (Table 2). The mitochondrial matrix citrate synthase is localized almost entirely in the mitochondrial fraction (80% of total activity). The activity found in the cytosolic fraction is probably from the breakage of some mitochondria during the separation.
TABLE 2.
Distribution of eight enzymes between the mitochondria and the cytosol
| Enzyme | % of total activity in:
|
|
|---|---|---|
| Mitochondria | Cytosol | |
| Citrate synthasea | 80 | 20 |
| Glucose-6-phosphate dehydrogenase | 1 | 99 |
| Isocitrate dehydrogenase (NADP) | 38 | 62 |
| Isocitrate dehydrogenase (NAD) | 100 | 0 |
| Glutamate oxaloacetate transaminase | 41 | 59 |
| Glutamate dehydrogenase (NADP) | 2 | 98 |
| Glutamate dehydrogenase (NAD) | 18 | 82 |
| Malate dehydrogenase | 14 | 86 |
The preparations showed latent activities of higher than 75% for citrate synthase in the mitochondrial fraction. Extent of latency = 100 · [1 − (activity without detergent/activity with detergent)]. The detergent was Triton X-100.
Since the cytosolic fraction contains 62% of the total isocitrate dehydrogenase (NADP) activity, compared to 20% for citrate synthase, the cytosolic activity of isocitrate dehydrogenase (NADP) cannot be explained simply by leakage from the mitochondria. After isoelectric focusing and staining for isocitrate dehydrogenase (NADP), only one band, with a pI of 5.3, was detected on the gel (data not shown). Presumably this single band results from identical pIs for the cytosolic and mitochondrial isocitrate dehydrogenases (NADP).
Biomass composition and α-amylase productivities.
The contents of the main biomass components were determined at different specific growth rates (Table 3). The measured biomass components could account for approximately 85% (wt/wt) of the biomass. The biomass contains approximately 10% (wt/wt) ash. The remaining 5% (wt/wt) can be accounted for by metabolites. For the flux analysis the entire biomass must be accounted for, so we normalized the measured composition to 100% (wt/wt).
TABLE 3.
Cellular compositions of A. oryzae wild-type and recombinant strains
| Component | % (wt/wt)a for the following strain, nitrogen source, and specific growth rate (h−1):
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A1560
|
CF 1.1
|
|||||||||
| Ammonia, 0.025 | Ammonia, 0.05 | Nitrate, 0.079 | Ammonia, 0.10 | Ammonia, 0.14 | Ammonia, 0.17 | Ammonia, 0.05 | Ammonia, 0.094 | Nitrate, 0.094 | Ammonia, 0.15 | |
| Proteinsb | 35 | 36 | 30 | 40 | 39 | 42 | 36 | 37 | 36 | 42 |
| Carbohydratesc | 43 | 34 | 34 | 28 | 29 | 28 | 32 | 31 | 33 | 23 |
| RNA | 3.0 | 3.5 | 3.9 | 5.3 | 5.4 | 5.5 | 4.6 | 4.9 | 5.3 | 6.7 |
| DNA | 1.0 | 0.8 | NDd | 0.8 | 0.8 | 0.9 | ND | ND | ND | ND |
| Lipids | 5.6 | 6.3 | ND | 6.8 | 5.1 | 5.1e | ND | ND | ND | ND |
| Mannitol | 2.4 | 3.8 | 8.8 | 3.3 | 2.8 | 2.1 | 8.8 | 5.5 | 4.8 | 4.0 |
| Glycerol | 0.5 | 0.4 | 0.2 | 0.7 | 0.3 | 0 | 0 | 0 | 0.2 | 0.3 |
| Otherf | 9.4 | 15 | 16 | 15 | 18 | 16 | 12 | 15 | 14 | 18 |
The standard deviations of the contents of protein, carbohydrate, lipid, mannitol, and RNA were less than 10%, and those of the contents of chitin, DNA, and glycerol were less than 17% based on three chemostat cultivations at 0.05 h−1 and four cultivations at 0.1 h−1 with the wild-type strain. All other data are based on one chemostat cultivation.
Free amino acids plus protein.
Glucans plus glycogen plus chitin.
ND, not determined. A value of 6.2% was used for lipid, and 0.8% was used for DNA.
The lipid content was not determined for this specific growth rate. A value of 5.1% was used to calculate the other.
This fraction contains ash and metabolites.
Table 4 shows the specific glucose uptake rates and the specific α-amylase productivities obtained for the different cultivations. We noticed that the specific α-amylase productivities were lower with nitrate as the nitrogen source than with ammonia as the nitrogen source. The nonlinear increase in specific α-amylase productivity at above a specific growth rate of 0.14 h−1 is due to glucose repression of the α-amylase productivity (11). From Table 4 it is seen that the α-amylase yields (millimoles of C [mole of glucose C]−1) are in the range of 5 to 29 for the wild-type strain and in the range of 44 to 88 for the recombinant strain.
TABLE 4.
Specific glucose uptake rates, specific α-amylase productivities, and α-amylase yields in the cultivations
| Parameter (unit) | Valuea for the following strain, nitrogen source, and specific growth rate (h−1)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A1560
|
CF 1.1
|
|||||||||
| Ammonia, 0.025 | Ammonia, 0.05 | Nitrate, 0.079 | Ammonia, 0.10 | Ammonia, 0.14 | Ammonia, 0.17 | Ammonia, 0.05 | Ammonia, 0.094 | Nitrate, 0.094 | Ammonia, 0.15 | |
| Specific glucose uptake rate (mmol of C g [dry wt]−1 h−1) | 1.8 | 3.6 | 5.8 | 6.7 | 8.3 | 10 | 3.3 | 6.2 | 8.3 | 8.2 |
| Specific α-amylase productivity (mmol of C g [dry wt]−1 h−1) | 0.053 | 0.075 | 0.035 | 0.12 | 0.14 | 0.051 | 0.26 | 0.55 | 0.40 | 0.36 |
| α-Amylase yield (mmol of C [mol of glucose C]−1) | 29 | 21 | 6.0 | 18 | 17 | 5.1 | 79 | 88 | 48 | 44 |
The data for the wild-type strain grown on ammonia are taken from reference 11.
Metabolic model.
We developed a stoichiometric model based on literature information (4, 5, 8, 9, 12, 13, 17, 20, 21, 25, 27, 29, 30, 32, 35, 38–42, 45, 48–50), the enzymatic activities measured in this study, and the measured localizations of various enzymes. The metabolic model contains 69 reactions and 59 intracellular metabolites. Since each intracellular metabolite gives a mass balance, 10 fluxes must be specified to calculate the reaction rates. These 10 fluxes are determined from measurements of the eight biomass components, glucose, and α-amylase. When the stoichiometry for production of a biomass component is known, the flux to this component is calculated by using the productivity (millimoles gram [dry weight]−1 hour−1) of the component, i.e., the specific growth rate (hour−1) times the fraction (grams gram [dry weight]−1) of the component in the biomass divided by the molar weight (grams millimole−1). To determine the stoichiometry for macromolecular synthesis, the compositions of the compounds must be known. In our analysis the composition of the proteins was taken as the average of eight measured protein compositions from the recombinant strain grown at specific growth rates of 0.05 to 0.15 h−1. The protein composition and content includes intracellular α-amylase, which accounts for less than 2% of the total cellular protein in the wild-type strain. In eucaryotes most of the central carbon metabolism takes place either in the cytosol or in the mitochondria, and the metabolic model includes the major metabolic reactions occurring in these two compartments. This compartmentalization is necessary to obtain reliable balances of all components that do not pass the mitochondrial membrane. NADH and NADPH cannot pass the mitochondrial membrane, and separate balances are needed for these cofactors in each compartment. Since several biosynthetic reactions in the mitochondria require NADPH, this cofactor must be synthesized in this compartment, presumably by the mitochondrial NADP+-linked isocitrate dehydrogenase. Due to the compartmentalization of isocitrate dehydrogenase (NADP), malate dehydrogenase and glutamate oxaloacetate transaminase are included twice in the model, since these enzymes are present in both the cytosolic and the mitochondrial compartments. The reactions catalyzed by glucose-6-phosphate dehydrogenase and glutamate dehydrogenase (NADP) are included only once, since they are present only in the cytosol, as are citrate synthase and isocitrate dehydrogenase (NAD), which are present only in the mitochondria.
Flux calculations.
Fluxes were calculated for four different scenarios (Table 5). We used the flux through the pentose phosphate pathway as an indicator for the change in fluxes. The flux through the pentose phosphate pathway is approximately constant in all four cases. We interpreted this result to mean that these pathways do not significantly alter the requirement for cytosolic NADPH. Therefore, we concluded that the assumptions regarding the biochemistry specified in Table 5 do not have much influence on the calculated flux distribution through the pentose phosphate pathway and the Embden-Meyerhof-Parnas pathway, and all further calculations were based on case a (see Table 5 and Appendix). If nitrate is the nitrogen source instead of ammonia, the calculated flux through the pentose phosphate pathway increases due to the large requirement for cytosolic NADPH in the reduction of nitrate to ammonia.
TABLE 5.
Relative fluxes through the pentose phosphate pathway with different assumptions regarding the biochemistry for growth of the recombinant strain at a specific growth rate of 0.094 h−1 with ammonia or nitrate as the nitrogen source
| Casea | Active cytosolic isocitrate dehydrogenase (NADP) | α-Ketoglutarate passes the mitochondrial membrane | Transport of oxaloacetate | Flux through pentose phosphate pathway (mol/100 mol of glucose-6-phosphate formed) with:
|
|
|---|---|---|---|---|---|
| Ammonia | Nitrate | ||||
| a | Yes | No | Malate | 40 | 125 |
| b | Yes | No | Aspartate | 46 | 130 |
| c | No | Yes | Malate | 50 | 132 |
| d | No | Yes | Aspartate | 50 | 132 |
For cases a and c, a surplus of oxaloacetate in the cytosol is converted to malate, which then enters the mitochondria, where it is converted back into oxaloacetate. For cases b and d, a surplus of oxaloacetate in the cytosol is converted to aspartate, which then enters the mitochondria, where it is converted back into oxaloacetate. For cases a and b, α-ketoglutarate needed in the cytosol is supplied by transport of isocitrate from the mitochondria to the cytosol and subsequent oxidation to α-ketoglutarate catalyzed by isocitrate dehydrogenase (NADP). For cases c and d, α-ketoglutarate needed in the cytosol is transported directly across the mitochondrial membrane.
The calculated fluxes (Fig. 1) through the pentose phosphate pathway in A. oryzae are 35% (wild-type strain) and 40% (recombinant strain) of the total glucose uptake and are determined by the overall NADPH requirement in the cytosol. The glucose uptake going through the pentose phosphate pathway ranges from 38 to 47% for the recombinant strain and from 29 to 37% for the wild-type strain (Fig. 2). The differences are due to increased specific α-amylase productivity in the recombinant strain. The fluxes catalyzed by isocitrate dehydrogenase (NAD) are 44 and 24% of the glucose uptake for the wild-type strain and the recombinant strain, respectively. Also, the flux through the anaplerotic reaction catalyzed by pyruvate carboxylase is higher in the recombinant strain than in the wild-type strain. These differences are caused by the difference in α-amylase productivity. For both strains the percentage of the glucose uptake going through the pentose phosphate pathway increases with increasing specific growth rate due to an increasing demand for cytosolic NADPH and increasing protein content.
FIG. 1.

Flux distributions of A. oryzae wild-type and recombinant strains during growth on glucose and ammonia at specific growth rates of 0.10 and 0.094 h−1, respectively. For each reaction, the numbers to the left are for the wild-type strain and the numbers to the right (italic) are for the recombinant strain. All fluxes are normalized with respect to the glucose uptake rate. Solid lines represent chemical reactions, and dashed lines represent transport across the mitochondrial membrane. It was assumed that cytosolic isocitrate dehydrogenase (NADP) is active, that α-ketoglutarate does not pass the mitochondrial membrane, and that oxaloacetate is transported from the cytosol to the mitochondria as malate. P, phosphate; acetyl-CoA, acetyl coenzyme A.
FIG. 2.

Relative fluxes through the pentose phosphate pathway at various specific growth rates for the wild-type strain (⧫) and the recombinant strain (□). The standard deviations at 0.05 and 0.10 h−1 are 6 and 3%, respectively, for the wild-type strain. All other data points are based on one chemostat cultivation. P, phosphate.
Comparison of in vitro enzyme activities with calculated fluxes.
We compared the measured in vitro activities with the calculated fluxes (Table 6). Since the measured enzyme activities are maximum activities in vitro, the determined activities are measures of only the amount of enzyme present in the cell and not the in vivo activity.
TABLE 6.
In vitro enzyme activities compared with calculated fluxes
| Enzyme and parametera | Value for the following strain, nitrogen source, and specific growth rate (h−1)
|
||||||
|---|---|---|---|---|---|---|---|
| CF 1.1
|
A1560
|
||||||
| Ammonia
|
Nitrate, 0.094 | Ammonia
|
Nitrate, 0.079 | ||||
| 0.05 | 0.094 | 0.15 | 0.05 | 0.15b | |||
| Glutamate dehydrogenase (NADP) | |||||||
| Activity | 0.10 | 0.12 | 0.33 ± 0.04 | 0.84 ± 0.08 | 0.12 ± 0.02 | 0.28 ± 0.005 | 1.81 ± 0.13 |
| Calculated flux | 0.015 | 0.029 | 0.043 | 0.027 | 0.013 | 0.034 | 0.019 |
| Glutamate oxaloacetate transaminase | |||||||
| Activity | 0.44 | 0.32 | 0.40 ± 0.04 | 0.50 ± 0.01 | 0.50 ± 0.02 | 0.50 ± 0.08 | 0.45 ± 0.015 |
| Calculated flux | 0.004 | 0.008 | 0.011 | 0.007 | 0.003 | 0.009 | 0.005 |
| Isocitrate dehydrogenase (NADP)c | |||||||
| Activity | 0.07 | 0.05 | 0.07 ± 0.005 | 0.06 ± 0.01 | 0.10 ± 0.01 | 0.08 ± 0.015 | 0.08 ± 0.01 |
| Calculated flux | 0.008 | 0.016 | 0.024 | 0.015 | 0.007 | 0.019 | 0.011 |
| Isocitrate dehydrogenase (NAD) | |||||||
| Activity | 0.011 | 0.007 | 0.011 | 0.019 | 0.014 | 0.017 | |
| Calculated flux | 0.009 | 0.013 | 0.013 | 0.030 | 0.019 | 0.017 | 0.025 |
All activities and fluxes are expressed as micromoles minute−1 milligram−1. Values are means and standard deviations.
The enzyme activities for the wild-type strain were measured at a specific growth rate of 0.15 h−1, whereas the fluxes were calculated for the wild type growing at a specific growth rate of 0.14 h−1.
The measured and calculated isocitrate dehydrogenase (NADP) activities and fluxes include both the mitochondrial and the cytosolic fractions.
DISCUSSION
Enzyme measurements.
We found the maximum specific activities of glutamate dehydrogenase (NADP) to be similar in the two strains and to increase with increasing flux catalyzed by this enzyme when ammonia was used as the nitrogen source. This is similar to results for Saccharomyces cerevisiae (36). When we used nitrate as the nitrogen source, the maximum specific activity was approximately 32 to 100 times higher than the calculated flux (it was 4 to 8 times higher than the calculated flux for ammonia as the nitrogen source). This effect might be due to a lower intracellular ammonia concentration during growth on nitrate compared to that on ammonia, which would require a high level of glutamate dehydrogenase (NADP) in the cell in order to maintain a certain flux to glutamate.
We did not find any changes in the maximum specific activity of glutamate oxaloacetate transaminase or isocitrate dehydrogenase (NADP) with the flux catalyzed by these enzymes, nor did we observe changes when the nitrogen source was changed or between the two strains. This result differs from that for Aspergillus nidulans grown in shake flasks, where the maximum specific activity of isocitrate dehydrogenase (NADP) increases by a factor of 25 when nitrate instead of ammonia is the nitrogen source (45). The high activity of isocitrate dehydrogenase (NADP) indicates that the in vivo activity (flux) is determined by the requirement for NADPH in the mitochondria and the requirement for α-ketoglutarate in the cytosol.
We found the maximum specific activities of isocitrate dehydrogenase (NAD) to be of the same magnitude as the calculated fluxes. For growth with ammonia as the nitrogen source, we observed decreasing maximum specific activities with increasing specific growth rates for both strains. The calculated fluxes and the maximum specific activities were higher for the wild-type strain than for the recombinant strain. The enzyme measurements are consistent with the calculated fluxes with respect to both the order of magnitude and the trend in the case of isocitrate dehydrogenase (NAD). We found calculated fluxes higher than the measured maximum specific activities for growth on nitrate. This difference could indicate that the metabolic model does not describe growth on nitrate as accurately as it does growth on ammonia. However, the activities correspond to activities found in Aspergillus niger (26).
We did not detect any isocitrate lyase and malic enzyme activity, which is similar to results for A. nidulans (33, 38, 45). We did not observe any significant difference in the activity of mannitol dehydrogenase during growth on ammonia or nitrate, which is similar to results for A. nidulans (45). This finding indicates the absence of the proposed NADPH-generating mannitol cycle (23, 24).
Except for glutamate dehydrogenase (NAD), which we found in both the mitochondria and the cytosol, all of the determined distributions of various enzymes between the mitochondria and the cytosol correspond to results obtained for A. nidulans (38). We found isocitrate dehydrogenase (NADP) in both fractions. However, we could detect only one band, with a pI of 5.3, during isoelectric focusing. This result is similar to findings for A. niger, where isocitrate dehydrogenase (NADP) was found in both the mitochondrial and the cytosolic fractions but only a single protein with a pI of 5.9 was identified (34). Isocitrate dehydrogenase (NADP) was detected in both fractions in A. nidulans, and two bands showing isocitrate dehydrogenase (NADP) activity were detected (38).
Biomass composition.
We found that the protein and RNA contents increased with specific growth rate. Both the wild type and the recombinant strain have approximately the same levels of intracellular protein and RNA, even though the recombinant strain produces approximately 24% more total protein (intracellular protein plus extracellular α-amylase) than the wild-type strain.
Carbohydrates and amino carbohydrates (chitin) decreased with increasing specific growth rate, while DNA, lipid, and glycerol levels remained relatively constant. Mannitol also decreased as a function of the specific growth rate. The decrease in the contents of these components can be explained by the increase in the contents of protein and RNA. It is difficult to explain the rather high mannitol content for the wild-type strain grown at a specific growth rate of 0.079 h−1 with nitrate as the nitrogen source. For the recombinant strain there is no significant difference in the mannitol contents for growth with ammonia and nitrate as nitrogen sources.
We conclude that the increased α-amylase productivity in the recombinant strain compared to the wild-type strain does not have any significant influence on the biomass composition.
Flux analysis.
The major branch points in the central metabolism where the fluxes are affected by changes in nitrogen source, specific growth rate, and α-amylase productivity are at the glucose-6-phosphate node and the flux catalyzed by isocitrate dehydrogenase (NAD). We calculated that the fraction of the glucose uptake diverted through the pentose phosphate pathway increased with increasing specific growth rate and α-amylase productivity. This effect is caused by the high NADPH demand for synthesis of proteins. Furthermore, at the pyruvate node we calculated an increased flux catalyzed by pyruvate carboxylase and a decreased tricarboxylic acid cycle flux in the recombinant strain compared with the wild-type strain. These changes are caused by the increased requirement for amino acid biosynthesis in the recombinant strain compared with the wild-type strain.
Growth with nitrate as the nitrogen source also leads to a high flux through the pentose phosphate pathway due to the demand for NADPH for reduction of nitrate to ammonia. We calculated the flux through the pentose phosphate pathway to be more than 100% of the glucose uptake rate, which means that fructose-6-phosphate is converted back to glucose-6-phosphate, which is possible since this reaction is known to be reversible. The low α-amylase productivities that we obtained with nitrate as the nitrogen source could be explained by a limited availability of amino acids due to a shortage of NADPH. When we used yeast extract (2 g liter−1) as an additional nitrogen and carbon source, the α-amylase productivity doubled compared to that when ammonia was the sole nitrogen source, which can be explained by a higher availability of amino acids. We calculated that the fluxes catalyzed by isocitrate dehydrogenase (NAD) were higher for growth on nitrate than for growth on ammonia. We observed low biomass and α-amylase yields during growth on nitrate, which results in low fluxes catalyzed by isocitrate dehydrogenase (NADP) in both the cytosol and the mitochondria, which divert a higher flux through isocitrate dehydrogenase (NAD).
We did not find large differences in the calculated flux distributions between the wild-type strain and the recombinant strain. This result may be a consequence of the almost identical biomass compositions of the two strains and the fact that protein production influences primarily the pentose phosphate pathway and the reaction catalyzed by isocitrate dehydrogenase (NAD). Assuming a very high α-amylase yield (0.50 mol of C mol of glucose C−1) and a lower biomass yield (0.10 mol of C mol of glucose C−1), the flux through the pentose phosphate pathway is 51% of the glucose uptake rate and the flux catalyzed by isocitrate dehydrogenase (NAD) is 25% of the glucose uptake rate. Thus, even at very high α-amylase yields, the flux distribution does not change dramatically. However, even though the relative fluxes do not change, the actual fluxes (millimoles gram [dry weight]−1 hour−1) increase significantly in such a high-yielding strain due to a high specific glucose uptake rate.
Based on the results that we obtained for the three nitrogen sources, we conclude that amino acid availability might be a limiting factor in the production of α-amylase. During growth with ammonia as the nitrogen source, the Embden-Meyerhof-Parnas and pentose phosphate pathways do not seem to be limited in the production of α-amylase due to rigid nodes, i.e., branch points over which the flux distribution is fixed to a certain range.
Appendix
Metabolic model for growth and α-amylase formation in A. oryzae with ammonia as the nitrogen source. Abbreviations: ex., extracellular; NADPHcyt, cytosolic NADPH; P, phosphate; acetyl-CoAmit, mitochondrial acetyl coenzyme A; FADH2, reduced flavin adenine dinucleotide; THF, tetrahydrofolate; PRPP, 5’-phosphoribosyl-1-pyrophosphate.
Case a
- 1.
H2S − SO42− (ex.) − 4 ATP − 4 NADPHcyt = 0
- 2.
Glucose-6-P − glucose (ex.) − 2 ATP = 0
- 3.
Fructose-6-P − glucose-6-P = 0
- 4.
Glyceraldehyde-3-P − dihydroxyacetone-P = 0
- 5.
Glyceraldehyde-3-P + dihydroxyacetone-P − fructose-6-P − ATP = 0
- 6.
3-Phosphoglycerate + NADHcyt + ATP − glyceraldehyde-3-P = 0
- 7.
Phosphoenolpyruvate − 3-phosphoglycerate = 0
- 8.
Pyruvate + ATP − phosphoenolpyruvate = 0
- 9.
Ribose-5-P + 2 NADPHcyt + CO2 − glucose-6-P = 0
- 10.
Erythrose-4-P + fructose-6-P − 2 ribose-5-P = 0
- 11.
Glyceraldehyde-3-P + fructose-6-P − erythrose-4-P − ribose-5-P = 0
- 12.
Acetyl-CoAmit + NADHmit + CO2 − pyruvate = 0
- 13.
Oxaloacetatemit − pyruvate − CO2 − ATP = 0
- 14.
Citrate − acetyl-CoAmit − oxaloacetatemit = 0
- 15.
Oxaloacetatecyt + acetyl-CoAcyt − citrate − ATP = 0
- 16.
α-Ketoglutaratecyt + NADPHcyt + CO2 − citrate = 0
- 17.
α-Ketoglutaratemit + NADPHmit + CO2 − citrate = 0
- 18.
α-Ketoglutaratemit + NADHmit + CO2 − citrate = 0
- 19.
Succinyl-CoA + CO2 + NADHmit − α-ketoglutaratemit = 0
- 20.
Malate + ATP + FADH2 − succinyl-CoA = 0
- 21.
Oxaloacetatemit + NADHmit − malate = 0
- 22.
Oxaloacetatecyt + NADHcyt − malate = 0
- 23.
2.6 ATP − NADHmit − 0.5 O2 = 0
- 24.
1.6 ATP − FADH2 − 0.5 O2 = 0
- 25.
1.6 ATP − NADHcyt − 0.5 O2 = 0
- 26.
Glutamate − α-ketoglutaratecyt − NADPHcyt − NH4+ (ex.) − ATP = 0
- 27.
Glutamine − glutamate − NH4+ (ex.) − 2 ATP = 0
- 28.
Proline − glutamate − 2 NADPHcyt − ATP = 0
- 29.
Arginine + α-ketoglutaratecyt + malate − aspartate − glutamate − glutamine − CO2 − NH4+ − NADPHmit − 5 ATP = 0
- 30.
Lysine + 2 α-ketoglutaratecyt + NADHcyt − acetyl-CoAmit − 2 glutamate − α-ketoglutaratemit − 2 ATP − 2 NADPHcyt = 0
- 31.
Aspartate + 0.59 α-ketoglutaratecyt + 0.41 α-ketoglutaratemit − glutamate − 0.59 oxaloacetatecyt − 0.41 oxaloacetatemit = 0
- 32.
Aspargine + glutamate − aspartate − glutamine − 2 ATP = 0
- 33.
Homoserine − aspartate − 2 NADPHcyt − ATP = 0
- 34.
Threonine − homoserine − ATP = 0
- 35.
Isoleucine + α-ketoglutaratecyt + NH4+ (ex.) + CO2 + ATP − glutamate − pyruvate − threonine − NADPHmit = 0
- 36.
Homocysteine − homoserine − H2S − 2 ATP = 0
- 37.
Methionine − homocysteine − NADHcyt − N5,N10-methylene-THF = 0
- 38.
S-Adenosylmethionine − methionine − ATP = 0
- 39.
Homocysteine − S-adenosylcysteine − 2 ATP
- 40.
Serine + α-ketoglutaratecyt + NADHcyt − 3-phosphoglycerate − glutamate = 0
- 41.
Glycine + N5,N10-methylene-THF − serine = 0
- 42.
Cysteine + succinyl-CoA + NADHcyt − homocysteine − serine = 0
- 43.
Alanine + α-ketoglutaratecyt − pyruvate − glutamate = 0
- 44.
Ketoisovalerate + CO2 − 2 pyruvate − NADPHmit = 0
- 45.
Valine + α-ketoglutaratemit − ketoisovalerate − glutamate = 0
- 46.
Leucine + α-ketoglutaratemit + CO2 + NADHcyt − ketoisovalerate − acetyl-CoAmit − glutamate = 0
- 47.
PRPP − ribose-5-P − 2 ATP = 0
- 48.
Histidine + 5′-aminoimidazole-4-carboxamide-ribonucleotide + α-ketoglutaratecyt + 2 NADHcyt − PRPP − ATP − glutamine = 0
- 49.
Chorismate − 2 phosphoenolpyruvate − erythrose-4-P − ATP − NADPHcyt = 0
- 50.
Tyrosine + α-ketoglutaratecyt + NADHcyt − chorismate − glutamate = 0
- 51.
Phenylalanine + α-ketoglutaratecyt − chorismate − glutamate = 0
- 52.
Tryptophan + pyruvate + glutamate + glyceraldehyde-3-P + CO2 − PRPP − serine − chorismate − glutamine = 0
- 53.
Protein − 0.095 alanine − 0.044 arginine − 0.046 aspargine − 0.046 aspartate − 0.011 cysteine − 0.080 glutamate − 0.080 glutamine − 0.094 glycine − 0.020 histidine − 0.045 isoleucine − 0.069 leucine − 0.057 lysine − 0.014 methionine − 0.031 phenylalanine − 0.047 proline − 0.066 serine − 0.048 threonine − 0.018 tryptophan − 0.028 tyrosine − 0.064 valine − 4 ATP = 0
- 54.
α-Amylase − 0.078 alanine − 0.021 arginine − 0.054 aspargine − 0.086 aspartate − 0.019 cysteine − 0.025 glutamate − 0.040 glutamine − 0.086 glycine − 0.015 histidine − 0.059 isoleucine − 0.071 leucine − 0.042 lysine − 0.019 methionine − 0.029 phenylalanine − 0.042 proline − 0.075 serine − 0.082 threonine − 0.021 tryptophan − 0.073 tyrosine − 0.065 valine − 4 ATP = 0
- 55.
Lipid + 3.8 CO2 + 1.1 S-adenosylcysteine − 18.12 acetyl-CoAcyt − 18 ATP − 0.623 dihydroxyacetone-P − 0.623 FADH2 − 0.3 glucose-6-P − 1.7 NADHcyt − 22.4 NADPHcyt − 1.1 S-adenosylmethionine − 0.4 serine − 2.6 O2 = 0
- 56.
UTP + glutamate + NADHcyt − aspartate − glutamine − PRPP − 4 ATP = 0
- 57.
CTP + glutamate − ATP − glutamine − UTP = 0
- 58.
5′-Aminoimidazole-4-carboxamide-ribonucleotide + 2 glutamate + malate + NADPHcyt − aspartate − 4 ATP − 2 glutamine − glycine − PRPP − N5,N10-methylene-THF = 0
- 59.
IMP + NADPHcyt − N5,N10-methylene-THF − 5′-aminoimidazole-4-carboxamide-ribonucleotide = 0
- 60.
Adenosine triphosphate + malate − aspartate − 3 ATP − IMP = 0
- 61.
GTP + glutamate + NADHcyt − 4 ATP − glutamine − IMP = 0
- 62.
RNA − 0.256 adenosine triphosphate − 0.196 CTP − 0.286 GTP − 0.262 UTP − ATP = 0
- 63.
DNA − 0.242 adenosine triphosphate − 0.258 CTP − 0.258 GTP − 0.242 UTP − 1.242 NADPHcyt − 0.242 N5,N10-methylene-THF − ATP = 0
- 64.
Carbohydrate − glucose-6-P − ATP = 0
- 65.
Chitin + glutamate − acetyl-CoAcyt − glucose-6-P − glutamine − 2 ATP = 0
- 66.
Mannitol − fructose-6-P − NADHcyt = 0
- 67.
Glycerol − dihydroxyacetone-P − FADH2 = 0
- 68.
−ATP = 0
- 69.
N5,N10-Methylene-THF + CO2 + NH4+ (ex.) + ATP + NADHmit − glycine = 0
Case b
Same as case a except for reaction 16, which is changed to
16. α-Ketoglutaratecyt − α-ketoglutaratemit = 0
Case c
Same as case a except for reactions 22 and 31, which are changed to
22. Glutamate + oxaloacetatecyt − aspartate − α-ketoglutaratecyt = 0
31. Glutamate + oxaloacetatemit − aspartate − α-ketoglutaratemit = 0
Case d
Same as case b except for reactions 22 and 31, which are changed to
22. Glutamate + oxaloacetatecyt − aspartate − α-ketoglutaratecyt = 0
31. Glutamate + oxaloacetatemit − aspartate − α-ketoglutaratemit = 0
The only change from cases a, b, c, and d with nitrate as the nitrogen source is the addition of one extra reaction:
70. NH4+ − NO3− − 4 NADPHcyt = 0
REFERENCES
- 1.Armitt S, McCullough W, Roberts C F. Analysis of acetate non-utilizing (acu) mutants in Aspergillus nidulans. J Gen Microbiol. 1976;92:263–282. doi: 10.1099/00221287-92-2-263. [DOI] [PubMed] [Google Scholar]
- 2.Barkholt V, Jensen A L. Amino acid analysis: determination of cysteine plus half-cystine in proteins after hydrochloric acid hydrolysis with a disulfide compound as additive. Anal Biochem. 1989;177:318–322. doi: 10.1016/0003-2697(89)90059-6. [DOI] [PubMed] [Google Scholar]
- 3.Benthin S, Nielsen J, Villadsen J. A simple and reliable method for the determination of cellular RNA content. Biotechnol Tech. 1991;5:39–42. [Google Scholar]
- 4.Bercovitz A, Peleg Y, Battat E, Rokem J S, Goldberg I. Localization of pyruvate carboxylase in organic acid-producing Aspergillus strains. Appl Environ Microbiol. 1990;56:1594–1597. doi: 10.1128/aem.56.6.1594-1597.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blumenthal H J. Reserve carbohydrates in fungi. In: Smith J E, Berry D R, editors. The filamentous fungi. II. London, United Kingdom: Edward Arnold; 1976. pp. 292–307. [Google Scholar]
- 6.Boehringer GmbH. Analysis kit for determination of glutamate oxaloacetate transaminase activity. Boehringer, Mannheim, Germany.
- 7.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Brown C E, Romano A H. Evidence against necessary phosphorylation during hexose transport in Aspergillus nidulans. J Bacteriol. 1969;100:1198–1203. doi: 10.1128/jb.100.3.1198-1203.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cabib E. The synthesis and degradation of chitin. In: Meister A, editor. Advances in enzymology. New York, N.Y: John Wiley & Sons; 1987. pp. 59–99. [DOI] [PubMed] [Google Scholar]
- 10.Carlsen M, Marcher J, Nielsen J. An improved FIA-system for measuring α-amylase in cultivation media. Biotechnol Tech. 1994;8:479–482. [Google Scholar]
- 11.Carlsen M, Nielsen J, Villadsen J. Growth and α-amylase production by Aspergillus oryzae during continuous cultivations. J Biotechnol. 1996;45:81–93. [Google Scholar]
- 12.Cartwright C P, Rose A H, Calderbank J, Keenan M H J. Solute transport. In: Rose A H, Harrison J S, editors. The yeasts. Vol. 3. London, United Kingdom: Academic Press; 1989. pp. 5–56. [Google Scholar]
- 13.Chattopadhyay P, Banerjee S K, Sen K. Lipid profiles of Aspergillus niger and its fatty acid auxotroph, UFA2. Can J Microbiol. 1985;31:352–355. doi: 10.1139/m85-067. [DOI] [PubMed] [Google Scholar]
- 14.Christensen T, Woeldike H, Boel E, Mortensen S B, Hjortshoej K, Thim L, Hansen M T. High level expression of recombinant genes in Aspergillus oryzae. Bio/Technology. 1988;6:1419–1422. [Google Scholar]
- 15.Cook R A, Sanwal B D. Isocitrate dehydrogenase (NAD-specific) from Neurospora crassa. Methods Enzymol. 1969;13:42–47. [Google Scholar]
- 16.Folch J, Lees M, Stanley G H S. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 17.Harding R W, Caroline D F, Wagner R P. The pyruvate dehydrogenase complex from the mitochondrial fraction of Neurospora crassa. Arch Biochem Biophys. 1970;138:653–661. doi: 10.1016/0003-9861(70)90393-0. [DOI] [PubMed] [Google Scholar]
- 18.Henriksen C M, Christensen L H, Nielsen J, Villadsen J. Growth energetics and metabolic fluxes in continuous cultures of Penicillium chrysogenum. J Biotechnol. 1996;45:149–164. doi: 10.1016/0168-1656(95)00164-6. [DOI] [PubMed] [Google Scholar]
- 19.Herbert D, Phipps P J, Strange R E. Chemical analysis of microbial cells. Methods Microbiol. 1971;5B:209–344. [Google Scholar]
- 20.Hersbach G J M, van den Beek C P, Dijk P W M. The penicillins: properties, biosynthesis and fermentation. In: Vandamme E J, editor. Biotechnology of industrial antibiotics. New York, N.Y: Marcel Dekker; 1984. pp. 66–145. [Google Scholar]
- 21.Hondmann D H A, Visser J. Carbon metabolism. In: Martinelli S D, Kinghorn J R, editors. Aspergillus: 50 years on. Progress in industrial microbiology. Vol. 29. Amsterdam, The Netherlands: Elsevier; 1994. pp. 61–139. [PubMed] [Google Scholar]
- 22.Horikoshi K, Iida S, Ikeda Y. Mannitol and mannitol dehydrogenase in conidia of Aspergillus oryzae. J Bacteriol. 1965;89:326–330. doi: 10.1128/jb.89.2.326-330.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hult K, Gatenbeck S. Production of NADPH in the mannitol cycle and its relation to polyketide formation in Alternaria alternata. Eur J Biochem. 1978;88:607–612. doi: 10.1111/j.1432-1033.1978.tb12487.x. [DOI] [PubMed] [Google Scholar]
- 24.Hult K, Veide A, Gatenbeck S. The distribution of the NADPH regenerating mannitol cycle among fungal species. Arch Microbiol. 1980;128:253–255. doi: 10.1007/BF00406168. [DOI] [PubMed] [Google Scholar]
- 25.Hunter K, Rose A H. Yeast lipids and membranes. In: Rose A H, Harrison J S, editors. The yeasts. Physiology and biochemistry of yeasts. II. London, United Kingdom: Academic Press; 1971. pp. 211–270. [Google Scholar]
- 26.Jaklitsch W M, Kubicek C P, Scrutton M C. Intracellular location of enzymes involved in citrate production by Aspergillus niger. Can J Microbiol. 1991;37:823–827. doi: 10.1139/m91-142. [DOI] [PubMed] [Google Scholar]
- 27.Jones E W, Fink G R. Regulation of amino acid and nucleotide biosynthesis in yeast. In: Strathern J N, Jones E W, Broach J R, editors. The molecular biology of the yeast Saccharomyces. Metabolism and gene expression. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1982. pp. 181–299. [Google Scholar]
- 28.Jørgensen H, Nielsen J, Villadsen J. Metabolic flux distributions in Penicillium chrysogenum during fed-batch cultivations. Biotechnol Bioeng. 1995;46:117–131. doi: 10.1002/bit.260460205. [DOI] [PubMed] [Google Scholar]
- 29.Lara M, Blanco L, Campomanes M, Calva E, Palacios R, Mora J. Physiology of ammonium assimilation in Neurospora crassa. J Bacteriol. 1982;150:105–112. doi: 10.1128/jb.150.1.105-112.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mark C G, Romano A H. Properties of the hexose transport systems of Aspergillus nidulans. Biochim Biophys Acta. 1971;249:216–226. doi: 10.1016/0005-2736(71)90098-8. [DOI] [PubMed] [Google Scholar]
- 31.Marx A, de Graaf A A, Wiechert W, Eggeling L, Sahm H. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol Bioeng. 1996;49:111–129. doi: 10.1002/(SICI)1097-0290(19960120)49:2<111::AID-BIT1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 32.McCorkindale N J. The biosynthesis of terpenes and steroids. In: Smith J E, Berry D R, editors. The filamentous fungi. II. London, United Kingdom: Edward Arnold; 1976. pp. 369–422. [Google Scholar]
- 33.McCullough W, Shanks A. Properties of genes involved in the control of isocitrate lyase production in Aspergillus nidulans. J Gen Microbiol. 1993;139:509–511. doi: 10.1099/00221287-139-3-509. [DOI] [PubMed] [Google Scholar]
- 34.Meixner-Monori B, Kubicek C P, Harrer W, Schreferl G, Rohr M. NADP-specific isocitrate dehydrogenase from the citric acid accumulating fungus Aspergillus niger. Biochem J. 1986;236:549–557. doi: 10.1042/bj2360549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen J. Physiological engineering aspects of Penicillium chrysogenum. Singapore: World Scientific Publishing; 1997. [Google Scholar]
- 36.Nissen T L, Schulze U, Nielsen J, Villadsen J. Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology. 1997;143:203–218. doi: 10.1099/00221287-143-1-203. [DOI] [PubMed] [Google Scholar]
- 37.Ochoa S. Malic dehydrogenase from pig heart. Methods Enzymol. 1955;1:735–745. [Google Scholar]
- 38.Osmani S A, Scrutton M C. The sub-cellular localisation of pyruvate carboxylase and of some other enzymes in Aspergillus nidulans. Eur J Biochem. 1983;133:551–560. doi: 10.1111/j.1432-1033.1983.tb07499.x. [DOI] [PubMed] [Google Scholar]
- 39.Paszewski A, Grabski J. Enzymatic lesions in methionine mutants of Aspergillus nidulans: role and regulation of an alternative pathway for cysteine and methionine synthesis. J Bacteriol. 1975;124:893–904. doi: 10.1128/jb.124.2.893-904.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pateman J A, Kinghorn J R. Nitrogen metabolism. In: Smith J E, Berry D R, editors. The filamentous fungi. II. London, United Kingdom: Edward Arnold; 1976. pp. 159–237. [Google Scholar]
- 41.Pronk J T, Steensma H Y, van Dijken J P. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast. 1996;12:1607–1633. doi: 10.1002/(sici)1097-0061(199612)12:16<1607::aid-yea70>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberger R F. The cell wall. In: Smith J E, Berry D R, editors. The filamentous fungi. II. London, United Kingdom: Edward Arnold; 1976. pp. 328–344. [Google Scholar]
- 43.Sauer U, Hatzimanikatis V, Hohmann H P, Manneberg M, van Loon A P G M, Bailey J E. Physiology and metabolic fluxes of wild-type and riboflavin-producing Bacillus subtilis. Appl Environ Microbiol. 1996;62:3687–3696. doi: 10.1128/aem.62.10.3687-3696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwitzguebel J P, Möller I M, Palmer J M. Changes in density of mitochondria and glyoxysomes from Neurospora crassa: a re-evaluation using silica sol gradient centrifugation. J Gen Microbiol. 1981;126:289–295. [Google Scholar]
- 45.Singh M, Scrutton N S, Scrutton M C. NADPH generation in Aspergillus nidulans: is the mannitol cycle involved? J Gen Microbiol. 1988;134:643–654. doi: 10.1099/00221287-134-3-643. [DOI] [PubMed] [Google Scholar]
- 46.Spohr A, Carlsen M, Nielsen J, Villadsen J. α-Amylase production in recombinant Aspergillus oryzae during fed-batch and continuous cultivations. J Ferment Bioeng. 1998;86:49–56. [Google Scholar]
- 47.Stephanopolus G, Vallino J J. Network rigidity and metabolic engineering in metabolite overproduction. Science. 1991;252:1675–1681. doi: 10.1126/science.1904627. [DOI] [PubMed] [Google Scholar]
- 48.Tuite M F. Protein synthesis. In: Rose A H, Harrison J S, editors. The yeasts. London, United Kingdom: Academic Press; 1989. pp. 161–204. [Google Scholar]
- 49.Walker P, Woodbine M. The biosynthesis of fatty acids. In: Smith J E, Berry D R, editors. The filamentous fungi. II. London, United Kingdom: Edward Arnold; 1976. pp. 137–158. [Google Scholar]
- 50.Wennekes L M J, Goosen T, van den Broek P J M, van den Broek H W J. Purification and characterization of glucose-6-phosphate dehydrogenase from Aspergillus nidulans. J Gen Microbiol. 1993;139:2793–2800. doi: 10.1099/00221287-139-11-2793. [DOI] [PubMed] [Google Scholar]