

Summary
Antiandrogen treatment resistance is a major clinical concern in castration-resistant prostate cancer (CRPC) treatment. Using xenografts of VCaP cells we showed that growth of antiandrogen resistant CRPC tumors were characterized by a higher intratumor dihydrotestosterone (DHT) concentration than that of treatment responsive tumors. Furthermore, the slow tumor growth after adrenalectomy was associated with a low intratumor DHT concentration. Reactivation of androgen signaling in enzalutamide-resistant tumors was further shown by the expression of several androgen-dependent genes. The data indicate that intratumor DHT concentration and expression of several androgen-dependent genes in CRPC lesions is an indication of enzalutamide treatment resistance and an indication of the need for further androgen blockade. The presence of an androgen synthesis, independent of CYP17A1 activity, has been shown to exist in prostate cancer cells, and thus, novel androgen synthesis inhibitors are needed for the treatment of enzalutamide-resistant CRPC tumors that do not respond to abiraterone.
Subject areas: Endocrinology, Cancer
Graphical abstract
Highlights
-
•
Enzalutamide treatment response is transient in VCaP prostate cancer xenografts
-
•
AR-signaling pathway remains active in enzalutamide-resistant VCaP tumors
-
•
Antiandrogen resistance is associated with upregulation of intratumor DHT
-
•
Adrenalectomy is more efficient than antiandrogen in the VCaP CRPC model
Endocrinology; Cancer
Introduction
Prostate cancer (PCa) remains the second most frequent cancer and the fifth leading cause of cancer death in men. It was recently estimated that there were almost 1.3 million new cases of PCa and over 350,000 PCa-associated deaths worldwide in 2018 (Bray et al., 2018). Androgens and androgen receptors (AR) are primary regulators of normal prostate development, growth, and function. Furthermore, most PCa cases are hormone dependent, and the androgen-signaling axis has a central role in disease initiation and progression (Dehm and Tindall, 2006; Yuan et al., 2014).
Most patients with metastatic PCa are treated with androgen-deprivation therapy (ADT) using LHRH agonists, antagonists or orchiectomy as a first-line hormonal therapy (Horwich et al., 2013; Parker et al., 2015). ADT can be combined with antiandrogens that block binding of any potential remaining androgens to the androgen receptor (AR). The ADT treatment response is usually effective, but during treatment, most tumors stop responding and progress to castration-resistant PCa (CRPC) (Antonarakis et al., 2007). Despite androgen ablation, CRPC still expresses AR and is androgen dependent (Gregory et al., 2001; Visakorpi et al., 1995). Therefore, along with cytotoxic agents (e.g., docetaxel), AR-targeting therapeutics confer treatment response even during the late stage of the disease.
CRPC is defined as a rising serum prostate-specific antigen (PSA) concentration, despite a low level of serum testosterone (T) (Cornford et al., 2017). It is known that therapies leading to suppression of androgen action may lead to the development of novel AR-related resistance mechanisms. These mechanisms include 1) overexpression of AR (Chen et al., 2004; Koivisto et al., 1997; Linja et al., 2001), causing sensitization of cancer cells to low levels of androgens (Waltering et al., 2009), 2) somatic point mutations in the AR that reduce the steroid specificity of the receptor (Bohl et al., 2005; Korpal et al., 2013; Sun et al., 2006), 3) alternative AR gene splicing, including the expression of ligand-independent, constitutively active, AR splice variants (Antonarakis et al., 2017; Sun et al., 2010; Watson et al., 2010), and 4) induction of intratumoral androgen [T and dihydrotestosterone (DHT)] biosynthesis de novo or from circulating precursors derived from the adrenal gland (Cai et al., 2011; Locke et al., 2008)). Accordingly, second-line hormonal therapies with antiandrogens (e.g., enzalutamide) and/or with androgen synthesis inhibitors (such as abiraterone) are widely used treatment options in advanced PCa. Enzalutamide (Enza) blocks the binding of androgens to AR and prevents AR nuclear translocation and its binding to DNA (Scher et al., 2010). Enza binds to AR with an 8-fold higher affinity than first-generation antiandrogens, such as bicalutamide (Shaw et al., 2018; Tran et al., 2009). Despite these beneficial properties, Enza improves overall survival after chemotherapy for only 52% of the patients, by an average of 4.8 months (Scher et al., 2012), and almost all Enza-treated patients develop drug resistance that leads to the development of lethal disease.
Studies focused on mechanisms of acquired Enza resistance have suggested that the resistance mechanisms are similar to those involved in the development of CRPC. Mechanisms suggested to be involved include, e.g., mutations (Joseph et al., 2013; Korpal et al., 2013) and splice variants of the AR (Antonarakis et al., 2017; Li et al., 2013; Scher et al., 2016), signaling pathways that bypass androgen signaling (Arora et al., 2013; Isikbay et al., 2014), increased intratumoral androgen biosynthesis (Efstathiou et al., 2015; Liu et al., 2015), a switch from adenocarcinoma to neuroendocrine PCa (Beltran et al., 2016; Ku et al., 2017; Mu et al., 2017) and the contributions of the factors in the tumor microenvironment such as inflammatory cytokines (Handle et al., 2016); there are also data showing that glucocorticoid receptor (GR) could recapitulate, at least partially, the AR action in CRPC settings (Prekovic et al., 2018). However, proof for the mechanisms is still largely lacking.
Tumor xenografts are classical preclinical models for cancer research, and have provided valuable information about CRPC growth regulation. One of the widely used models is based on VCaP cells that originate from a vertebral metastasis of PCa (Korenchuk et al., 2001). VCaP tumor xenografts present with a good take-rate in intact immunodeficient mice, respond to castration with a reduced growth rate, and exhibit regrowth after castration, and growth after castration is associated with AR overexpression, upregulation of AR splice variants and activation of intratumoral androgen biosynthesis (Huhtaniemi et al., 2018; Knuuttila et al., 2014, 2018), which are also classical features in clinical CRPC. Moreover, we and others recently showed that, in contrast to the previous hypothesis, mouse adrenals produce biologically relevant levels of active androgens, as well as precursors of androgen synthesis, thus contributing to CRPC growth in the VCaP model (Huhtaniemi et al., 2018; Mostaghel et al., 2019).
Similar to clinical CRPC, castration-resistant VCaP tumors initially respond to Enza treatment. Importantly, we have shown that the treatment response was associated with reduced intratumoral androgen concentrations (Knuuttila et al., 2018), thus potentially representing a novel mechanism of action of antiandrogens in clinical CRPC. In the present study, we show that, similar to many CRPC patients, the Enza response in castration-resistant VCaP tumors is transient and that the treatment resistance in our preclinical model is associated with markedly higher intratumoral T and DHT concentration and reactivation of the AR compared to that during the treatment response. Furthermore, after removing adrenal precursors by adrenalectomy, a similar upregulation in intratumoral androgens was not observed, and accordingly, adrenalectomy resulted in a long-term and stable treatment response. These findings strongly suggest that intratumoral ligand availability is key in the progression of treatment resistance.
Results
VCaP xenografts gain resistance to enzalutamide treatment in vivo
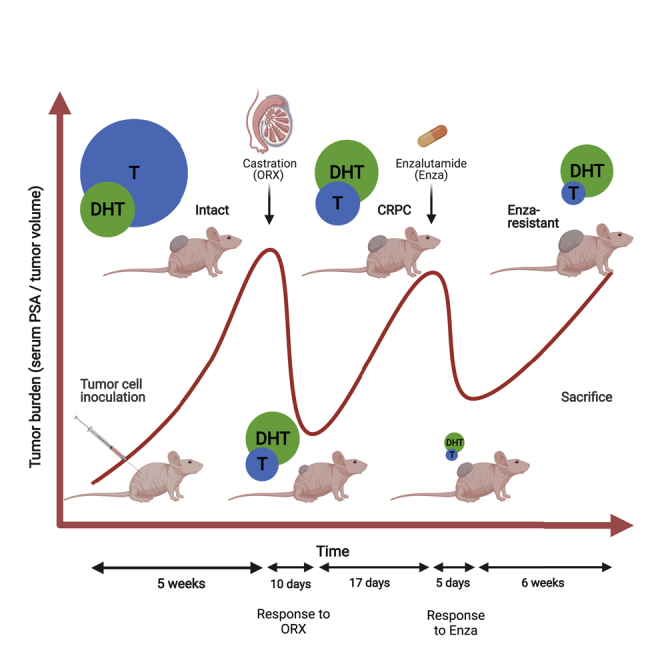
To study antiandrogen (enzalutamide) resistance in the VCaP xenograft model, animals with VCaP tumors were allocated to three study groups: orchiectomized (ORX) mice, orchiectomized and Enza-treated (ORX + Enza) mice, and orchiectomized and adrenalectomized (ORX + ADX) mice (Figure 1A). These three groups were further divided into subgroups I and II with varying treatment times (Figure 1A). Similar to results often observed in patients, the present study shows that Enza response in VCaP xenografts in nude mice after ORX is transient and effective for approximately 3 weeks, as measured by tumor growth (Figure 1B). Thereafter, the tumor growth increased to a level higher than that observed in the untreated (ORX) mice. The doubling time for the untreated control tumors was 68 days. During the Enza response, the growth was markedly slower, with a doubling time of 311 days. However, the growth was reactivated after 3 weeks of treatment, showing a doubling time of 37 days in tumors escaping the Enza response. Serum PSA measurements showed a similar pattern, with a more obvious, but shorter, Enza treatment response for 5-10 days, while after 14 days of Enza treatment, the measured PSA level was at the level observed before the treatment (Figure 1C). In our recent study, we showed that mouse adrenals contribute to castration-resistant growth of VCaP xenografts in mice (Huhtaniemi et al., 2018). We thus compared the long-term effects of adrenalectomy and antiandrogen treatment on tumor growth in mice after ORX. Importantly, the long-term treatment response to ADX was stronger than that to Enza, resulting in a significantly smaller (p < 0.005) tumor volume at the end of the study in ORX + ADX mice than in Enza-treated mice. The doubling time of the tumor volume was 94 days over the whole treatment period, and no evidence of induced tumor growth was observed after long-term treatment with ADX (Figure 1B). Identical data, indicating a stronger treatment response with ADX compared to Enza, were obtained by following the serum PSA concentration (Figure 1C). This resulted in significantly lower serum PSA concentrations and smaller tumor volumes at the end of the study in ADX mice than in Enza-treated mice. Notably, at the end of the experiment, two out of the 13 mice from the ADX group had a serum PSA concentration below the detection level and when palpated, the tumors were barely detectable.
Figure 1.

VCaP xenografts gain resistance toward enzalutamide treatment in vivo
Study flow of the VCaP xenograft model (created with BioRender.com) (A) and tumor volume and (B) serum PSA concentration (C) in nude mice after inoculation of VCaP prostate cancer cells subcutaneously. Mice were castrated (ORX) or castrated and adrenalectomized (ORX + ADX) 5 weeks after inoculation of the tumor cells, and enzalutamide (Enza) treatment (ORX + Enza) started 3 weeks after castration. Although the Enza response was transient (shaded area), ADX presented tumor growth inhibition throughout the study period. This resulted in a smaller tumor volume and lower serum PSA at the end of the study in the ORX + ADX mice compared to those treated with ORX + Enza. The data shown represent the mean ± SEM. Two-way ANOVA multiple comparisons were used to test the differences in tumors grown in castrated mice (ORX I and ORX II, n = 17 + 17), tumors grown in castrated mice treated with Enza (ORX + Enza I and ORX + Enza II, n = 14 + 14) and those grown in ORX + ADX (ORX + ADX I and ORX + ADX II, n = 15 + 13) mice. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Androgen receptor signaling is restored in enzalutamide-resistant VCaP xenografts
To understand the potential androgen action in Enza-resistant tumors, we compared AR expression in the different treatment groups and noted that AR-FL mRNA and protein (Figures 2A and 2B) and the mRNAs for the main AR splice variants, V1 and V7 (Figures 2C and 2D), were expressed very similarly in the tumors of the different treatment groups, except for the upregulation of all AR mRNA forms in the tumors after a long exposure to ORX + ADX (ORX + ADX II). To characterize whether common AR mutations would account for maintained AR signaling in Enza-treated tumors (Enza I and Enza II), we analyzed the tumor samples for the common mutations T878A, F877L, and L702H found in CRPC patients. However, none of the samples carried any of these AR mutations. This is in line with a previously published in vitro Enza-resistant VCaP model (Kregel et al., 2016). Immunohistochemical staining showed that AR localized to the cytoplasm during the Enza treatment response (ORX + Enza I) and in the tumors of the ORX + ADX mice, while more nuclear staining was observed in the untreated tumors (ORX II) and those that showed the Enza resistance (ORX + Enza II) (Figure 2E). In addition, in the RNA-sequencing (RNA-seq) data, the full-length AR was highly expressed in both Enza-responsive (ORX + Enza I) and Enza-resistant (ORX + Enza II) tumors. In Enza-resistant tumors, AR had the fourth highest expression level of the entire transcriptome, with mean expression levels of 7,895 (Enza I) and 7,105 (Enza II) CPM (counts-per-million). This further suggests a central role for AR in the mechanism of Enza resistance.
Figure 2.

Androgen receptor signaling is restored in the enzalutamide resistant VCaP xenografts
Androgen receptor signaling is restored in the enzalutamide resistant VCaP xenografts Expression of full-length androgen receptor mRNA (AR-FL) (A), AR protein (B), and AR mRNA splice variants V1 (C) and V7 (D), as well as AR immunohistochemistry (E), were performed in VCaP prostate cancer cells inoculated subcutaneously into nude mice. Mice were orchiectomized (ORX) or orchiectomized and adrenalectomized (ORX + ADX) 5 weeks after inoculation of the tumor cells, and enzalutamide (Enza) treatment started 3 weeks after castration (ORX + Enza group). The following groups of mice were analyzed: ORX I (orchiectomized mice, sacrificed 10 days after ORX), ORX II (orchiectomized mice, sacrificed 76 days after ORX), ORX + Enza I (sacrificed 34 days after ORX and 5 days after initiating Enza treatment), ORX + Enza II (sacrificed 76 days after ORX and 47 days after initiating Enza treatment), ORX + ADX I (sacrificed 10 days after ORX and ADX) and ORX + ADX II (sacrificed 76 days after ORX and ADX).
(A, C, and D) AR mRNAs increased in VCaP tumors in ORX + ADX II mice, as measured by quantitative RT–PCR.
(B) Western blot analysis of AR protein in the VCaP tumors indicates that the induction of mRNA expression in ORX + ADX mice also translates to slightly higher AR protein expression. ImageJ software was used to compare and quantify the intensity of the stained protein fragments in the Western blot. The RT–qPCR results were normalized to L19 expression, and AR signals in the Western blot were normalized to the ꞵ-actin.
(E) Representative immunohistochemical staining for AR in the tumors. The staining shows cytoplasmic localization for AR during the Enza response (ORX + Enza I), whereas the staining was mostly nuclear in the Enza-resistant tumors (ORX + Enza II). In ORX + ADX, the AR is localized mostly in the cytoplasm. The Kruskal-Wallis with Dunn post hoc test was used for statistical testing of the data in (A–D). Data are expressed as median and range with Tukey box and whisker plots (A–D). (A, C, and D): ORX I n = 10, ORX II n = 10, ORX + Enza I n = 9, ORX + Enza II n = 9, ORX + ADX I n = 8, ORX + ADX II n = 8. (B): n = 4 in every group. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Scale bars = 50 μm (E).
Increased intratumoral DHT concentration is associated with tumor growth and enzalutamide resistance of VCaP xenografts
Because the data indicated that AR also plays a pivotal role in the late stage of PCa progression, we measured the concentrations of the classical sex steroids of the tumors, adrenal glands and serum. Similar to our recently published observation (Knuuttila et al., 2018), only low levels of T and DHT were observed in the tumors at the time of the Enza treatment response (5 days after initializing Enza treatment, ORX + Enza I, Figure 3A). However, in those tumors that had escaped the transient treatment response (collected approximately 40 days after initiation of the Enza treatment, ORX + Enza II), the T and DHT concentrations were restored to the levels observed in the untreated ORX tumors (ORX I and ORX II, Figure 3A). Thus, 38-fold and 17-fold higher T and DHT concentrations, respectively, were observed in the Enza-resistant (ORX + Enza II) tumors than in the Enza-responsive (Enza I) tumors. Such concentrations are likely able to induce AR-dependent growth even in the presence of Enza. The average DHT concentration in the Enza-resistant tumors of 2,045 pg/g (range: 71 to 4,324 pg/g), was 5-fold higher than the mean T concentration of 445 pg/g (range: 11 to 1,036 pg/g). Notably, in our previous study (Huhtaniemi et al., 2018), the mean concentrations of T and DHT in intact mice were 9,000 pg/g and 3,902 pg/g, respectively. Thus, the DHT was only 2-fold higher in the intact mice than in the Enza resistant tumors in the present study. The tumor concentrations of progesterone (P4) and androstenedione (A-dione) were 139 pg/g (range: 15 to 1,061 pg/g) and 66 pg/g (range: 42 to 115 pg/g), respectively. Thus, such P4 and A-diones were likely not able to contribute to AR activation in the presence of much higher concentrations of T and DHT with higher affinity toward AR. However, their role as precursors for local DHT production should be studied further. In conclusion, our data indicate a major role for DHT as a growth-promoting androgen in the Enza-resistant VCaP tumors in castrated mice. In contrast to Enza treatment, after ORX + ADX, there was no significant difference in the intratumoral T or DHT between the two time points measured, and the DHT concentration measured 10 weeks after performing ADX was 3-fold lower than that observed in Enza-resistant tumors (ORX + ADX II, Figure 3A). Thus, the treatment response of the VCaP xenografts treated with both Enza and ADX was associated with a low intratumoral DHT concentration. This response was only transient in the Enza-treated mice, whereas a more stable suppression of intratumoral DHT was obtained after ADX treatment.
Figure 3.

Increased intratumoral DHT concentration is associated with tumor growth and enzalutamide resistance of VCaP xenografts
Concentrations of progesterone (P4), androstenedione (A-dione), testosterone (T) and dihydrotestosterone (DHT) in tumors (A), adrenal glands (B) and serum (C) in orchiectomized (ORX) and orchiectomized and enzalutamide-treated (ORX + Enza) or orchiectomized and adrenalectomized (ORX + ADX) mice. Groups are denoted as follows: ORX I (orchiectomized mice, sacrificed 10 days after ORX), ORX II (orchiectomized mice, sacrificed 76 days after ORX), ORX + Enza I (sacrificed 34 days after ORX and 5 days after initiating Enza treatment), ORX + Enza II (sacrificed 76 days after ORX and 47 days after initiating Enza treatment), ORX + ADX I (sacrificed 10 days after ORX and ADX) and ORX + ADX II (sacrificed 76 days after ORX and ADX). Enza treatment and ADX significantly altered the steroid profiles compared to ORX alone in the tumors and serum.
(A) Intratumoral T and DHT concentrations were reduced in Enza-responsive tumors (ORX + Enza I) but were restored to the level of tumors in ORX mice in the Enza-resistant tumors (ORX + Enza II). The reduced tumor growth after ADX treatment (Figure 1) is associated with reduced DHT concentration in ORX + ADX II compared with ORX alone (ORX II), whereas this difference is not observed between ORX II and ORX + Enza II or in T concentrations between the mice after ORX + ADX treatment compared to ORX + Enza II.
(B) The adrenal glands produced significant amounts of the measured hormones in ORX mice, and the levels were increased further during long-term treatment with Enza (ORX + Enza II).
(C) Circulating P4 was markedly reduced by ADX, whereas other treatments did not show significant changes. Long-term ADX (ORX + ADX II) also resulted in significantly lower serum T and DHT levels than those in the Enza-treated mice (ORX + Enza II). Kruskal-Wallis with Dunn’s post hoc tests was used for statistical analyses. Data are expressed as the median and range with Tukey box and whisker plots (A–C). n = 17 in ORX I and ORX II, n = 14 in ORX + Enza I and II, n = 15 in ORX + ADX I and n = 11 in ORX + ADX II (A–C). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
By measuring the intra-adrenal levels of the key androgens (A-dione, T and DHT) and P4, we showed that all of the above-mentioned hormones were markedly produced by the adrenals in ORX mice. Of the steroids measured, the P4 concentration was considerably higher compared to the androgens (Figure 3B), and the large drop in serum P4 after ADX treatment (Figure 3C) is in line with this and our former (Huhtaniemi et al., 2018) data, indicating that circulating P4 in male mice is partially of adrenal origin. Production of active androgens in the mouse adrenals is also indicated by the high adrenal/serum ratio of the steroids (Figure 4A). Enza treatment in ORX mice did not affect adrenal P4 production (Figure 3B) or the serum levels (Figure 3C), whereas its concentration in the tumors showed a tendency for lower levels during Enza treatment (Figure 3A). This, together with the low tumor-to-serum ratio (mean 0.19, range 0.07–0.46) in mice with Enza-responsive tumors (ORX + Enza I) and in mice with Enza-resistant tumors (mean 0.35, range 0.12–0.83), indicates the activation of P4 metabolism in the Enza-treated tumors (Figure 4B). In all the other treatment groups, the tumor-to-serum ratio was close to 1, suggesting that the P4 metabolism in those tumors was minimal. The activation of steroid synthesis using P4 as a precursor is also suggested by the increased intratumoral T and DHT levels during the Enza treatment (Figure 3A). Notably, the production of A-dione, T and DHT was higher in the adrenals of mice after long-term Enza treatment than that measured during the treatment response, potentially also contributing to the concentration of DHT in the tumors (Figure 3B). The mechanisms for the enhanced treatment response in the castration-resistant VCaP xenografts after blocking hormone synthesis in the adrenals as compared to blocking androgen action at the receptor level in the tumors should be studied further.
Figure 4.

Active androgens are produced in adrenal glands and progesterone metabolism is activated in the Enza-treated tumors
The high adrenal-to-serum steroid ratios indicate the production of active androgens in the mouse adrenal gland (A), and the low tumor-to-serum ratios in mice treated with Enza (ORX + Enza I and ORX + Enza II) indicate the activation of progesterone (P4) metabolism in tumors treated by Enza, while in other treatment groups, the ratio was close to 1 (B). Kruskal-Wallis with Dunn’s post hoc test was used for statistical analyses. Data are expressed as the median and range with Tukey box and whisker plots (A–C). n = 17 in ORX I and ORX II, n = 14 in ORX + Enza I and II, n = 15 in ORX + ADX I and n = 11 in ORX + ADX II (A–C). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Enzalutamide resistance is associated with enhanced androgen action, as revealed by RNA-seq
By analyzing the gene expression between the ORX + Enza I and ORX + Enza II groups by RNA-seq, we identified 292 genes with significantly (FC > 1.5, FDR >0.05) altered expression between the two groups (Figure 5A). Of those, 230 genes were upregulated in ORX + Enza II, and 62 genes were downregulated (see Table S1). Thus, there was a clear bias toward induced gene expression in the Enza-resistant tumors. Both the principal component analysis (PCA) (data not shown) and hierarchical clustering of all significantly changed genes showed that the Enza II samples were scattered into three main clusters, A, B and C (Figure 5B). The heterogeneity in the ORX + Enza II group was obvious, as among all the genes with > 3-fold altered expression, only AKAP12 (A-kinase anchoring protein 12), MAP1B (microtubule-associated protein 1B) and THBS1 (thrombospondin 1) were shared with all three clusters and thus considered among the most constantly altered genes (Figure 5C).
Figure 5.

Enzalutamide resistance is associated with enhanced androgen action as revealed by RNA-seq
The differential gene expression in the tumors between the ORX + Enza I and ORX + Enza II mice by RNA-seq.
(A) By using FC > 1.5 and FDR <0.05, we identified 292 genes with altered expression between the two groups, presented by a volcano plot. Thresholds used in filtering are marked in the plot with dashed lines, upregulated genes (230 genes) are colored red, and downregulated genes (62 genes) are colored green.
(B) Heatmap of hierarchical clustering of the differentially expressed genes in the tumors of the ORX + Enza I and ORX + Enza II mice define three separate clusters for the Enza II mice (A–C). Each row represents one differentially expressed gene, and each column represents one sample.
(C) RNA-seq data of AKAP12, MAP1B and THBS1 representing transcripts with those constantly altered between ORX + Enza I and ORX + Enza II tumors.
(D) Hierarchical clustering of the set of AR-associated genes provides a clear separation of the ORX + Enza I and ORX + Enza II tumors, indicating altered androgen action during the progression of Enza resistance. The heatmaps are based on the differentially expressed AR-related genes (FDR <0.05 and FC >1.5).
(E) RNA-seq data of well-characterized AR-regulated genes (KLK3, FKBP5, ELL2 and NOV) in ORX + Enza I and ORX + Enza II tumors further support the enhanced androgen action in Enza-resistant tumors compared to those with Enza response.
(F) Hierarchical clustering of the RNA-seq data of Enza responsive (ORX + Enza I) and Enza-resistant (ORX + Enza II) tumors using the leading edge genes among enzymes of the CYP, SDR and AKR families potentially involved in steroid metabolism. The leading edge determined which 68 subsets of genes contributed the most to the enrichment signal of a given set of 124 genes. The color gradients (B, D, and F) represents the intensity of gene expression in tumors, with blue indicating low expression in tumor and red indicating high gene expression during ORX + Enza I and ORX+Enza II treatments.
When analyzing the expression of AR-associated genes in all samples of the ORX + Enza I and ORX + Enza II groups, 253 of the 536 tested genes were revealed to be expressed in both groups. In hierarchical clustering using AR-associated genes, a clear separation was observed between the ORX + Enza I and ORX + Enza II mice (Figure 5D), indicating a difference in androgen action between the Enza-responsive and Enza-resistant tumors. Of these genes, 30 genes were upregulated and 2 genes were downregulated in the Enza-resistant (ORX + Enza II) tumors compared to the Enza-responsive tumors (ORX + Enza I) (Table 1). The genes included, among others, several well-characterized androgen-regulated genes, such as KLK3, LOX, ELL2, FKBP5, TMPRSS2 and PMEPA; downregulation of NOV expression, which was a recently defined androgen-suppressed gene, was also observed (Figure 5E). Thus, it was evident that androgen action was higher in Enza-resistant tumors than in those collected during the Enza-responsive stage. The overall variation in the expression levels across the androgen-dependent genes was markedly higher in the Enza-resistant tumors than in the tumors during the Enza response, further indicating increased heterogeneity among the tumors growing after long-term Enza treatment.
Table 1.
Significantly changed androgen receptor regulated genes in the enzalutamide treated VCaP tumors
| ID | mean RPKM Enza I | mean RPKM Enza II | Fold change | FDR |
|---|---|---|---|---|
| TNFAIP3 | 4.8 | 22.5 | 4.1 | 0.000 |
| KLK3 | 58.1 | 195.0 | 3.3 | 0.000 |
| MAP1B | 7.9 | 26.8 | 3.1 | 0.000 |
| LOX | 9.8 | 26.7 | 2.6 | 0.000 |
| STEAP4 | 8.3 | 21.4 | 2.4 | 0.000 |
| NR3C2 | 12.4 | 29.7 | 2.3 | 0.000 |
| ELL2 | 102.6 | 214.4 | 2.1 | 0.000 |
| FKBP5 | 140.8 | 286.1 | 2.0 | 0.000 |
| ANXA2 | 3.0 | 6.9 | 2.0 | 0.008 |
| PFKFB2 | 22.7 | 43.0 | 1.9 | 0.000 |
| SLC45A3 | 154.3 | 286.8 | 1.9 | 0.000 |
| RASD1 | 2.0 | 4.6 | 1.9 | 0.007 |
| MBOAT2 | 27.1 | 49.3 | 1.8 | 0.000 |
| TMPRSS2 | 328.5 | 582.4 | 1.8 | 0.000 |
| ABHD2 | 573.7 | 1006.7 | 1.8 | 0.009 |
| ZBTB16 | 18.8 | 33.4 | 1.7 | 0.016 |
| NFKBIA | 41.1 | 71.2 | 1.7 | 0.036 |
| BMPR1B | 114.2 | 195.0 | 1.7 | 0.008 |
| LDLR | 6.8 | 12.2 | 1.7 | 0.023 |
| SPOCK1 | 346.3 | 584.1 | 1.7 | 0.008 |
| PMEPA1 | 557.1 | 937.3 | 1.7 | 0.009 |
| DNAJB1 | 103.8 | 168.5 | 1.6 | 0.040 |
| SLC2A3 | 1.5 | 3.0 | 1.6 | 0.011 |
| ATP1A1 | 390.8 | 623.3 | 1.6 | 0.027 |
| SOCS2 | 19.3 | 31.3 | 1.6 | 0.007 |
| ACSL3 | 111.6 | 176.3 | 1.6 | 0.012 |
| C1orf21 | 32.6 | 51.3 | 1.6 | 0.023 |
| ALDH1A3 | 78.0 | 119.5 | 1.5 | 0.023 |
| TUBA3D | 5.9 | 9.5 | 1.5 | 0.037 |
| PIK3AP1 | 9.0 | 14.1 | 1.5 | 0.049 |
| GLI3 | 3.1 | 1.4 | −1.7 | 0.000 |
| NOV | 25.9 | 8.1 | −3.0 | 0.000 |
Using list of AR-associated genes containing known androgen-regulated genes as well as AR interacting proteins, 30 genes were upregulated and 2 genes downregulated in the Enza-resistant (ORX + Enza II, n = 10) tumors compared to the Enza responsive tumors (ORX + Enza I, n = 10). (FDR <0.05 and FC > 1.5).
To define the route of DHT synthesis within the tumors, we also studied the expression of steroidogenic enzymes and other proteins potentially contributing to the increased intratumoral DHT in the Enza-resistant tumors. Hierarchical clustering analysis with the leading genes among all SDR, AKR and CYP enzymes (Figure 5F) separated the Enza-responsive and Enza-resistant tumors into different clusters, indicating altered steroid metabolism machinery in these two groups of tumors. However, of the 124 genes analyzed by RNA-seq, only 8 were found to be differentially expressed with a fold change higher than 1.5, including CYP1B1, SLCO2A1, CYP4F62P, SULT1C4, CYP4F8, CYP4F30P, FASN and RDH11. Hematoxylin-eosin staining of the tumors showed that the stromal contribution in the tumors is minimal. Furthermore, RT–qPCR data revealed that classical steroidogenic enzymes, such as CYP11A1, CYP17A1, HSD3B1 and -2, HSD17B3, and SRD5A1 and -2, were not markedly expressed in the mouse stromal compartment. Our data therefore did not reveal the route for the accumulation of DHT in the Enza-resistant tumors. However, RNA-seq data indicated that various HSD17B enzymes with variable catalytic efficacy to convert A-dione to T were expressed, as were HSD3B7 and SR5A1 and SDR5A3 (Figure 5F). Because the AR level was not altered between the treatment groups, the increased ligand concentration produced by a nonconventional pathway is likely the key determinant for the enhanced androgen action in the Enza-resistant tumors.
We also performed pathway analyses of differentially expressed genes between the Enza I and Enza II tumors using enrichment analysis of the GO terms of biological processes. The analysis revealed terms such as inflammatory response, various terms for angiogenesis and blood vessel development and morphogenesis, and cell adhesion and motility, all with p < 1x 10−10 (see Table S2). Similar conclusions could be drawn using a PubMed search of the 10 genes with the highest upregulation and downregulation in Enza II tumors compared to Enza I, identified by RNA-seq (see Table S3). Of those genes, the majority were associated with androgen action, whereas they were also strongly associated with proliferation/apoptosis, invasiveness, cell migration, EMT and neuroendocrine development. Thus, the resistance mechanism involves multiple pathways promoting tumor progression.
Discussion
VCaP cells, containing endogenous AR gene amplification and overexpression, are sensitive to low levels of androgens (Waltering et al., 2009) and thus represent an excellent model to identify mechanisms leading to antiandrogen therapy resistance. Our study indicates that in castrated mice, Enza treatment response is only transient in VCaP xenografts, whereas ADX treatment resulted in more pronounced long-term growth inhibition. Importantly, we showed that in the Enza-resistant tumors, the AR-signaling pathway is active and thus remains a potential pathway for targeted therapy in the Enza-resistant patients. Our results further indicate that activation of AR signaling during antiandrogen therapy resistance in this preclinical model is associated with upregulation of the intratumoral DHT concentration, which is a potential marker for antiandrogen therapy resistance.
Our results are in agreement with those observed in earlier studies showing elevated A-dione and T levels in the bone marrow aspirates of patients with bone metastases treated with Enza (Efstathiou et al., 2015), with the exception that in our model, DHT was the main active androgen in the resistant tumors. It has been shown that although castration reduces circulating T levels by 90–95% (Labrie et al., 1980, 1985; Moghissi et al., 1984), intraprostatic DHT is typically reduced by only 50% after medical or surgical castration, and there is no decrease in A-dione concentrations in the prostate after castration (Bélanger et al., 1989; Miyamoto et al., 1998). Furthermore, an increase in intratumoral androgen biosynthesis is reported in Enza-resistant cells in vitro (Liu et al., 2015). However, to our knowledge, the present study is the first to report that enhanced intratumoral levels of active androgens, especially DHT, might be a key feature in a set of Enza-resistant CRPC tumors in humans.
To date, serum PSA, coded by the KLK3 gene (Riegman et al., 1991), is the best characterized marker to follow PCa progression and is used as a diagnostic tool clinically to detect early-stage disease, to track tumor burden and to monitor the efficacy of treatments (Nash and Melezinek, 2000; Ryan et al., 2006). KLK3 is well known to be androgen dependent, and the possibility of using PSA to follow-up clinical tumor progression after androgen-deprivation therapy and other treatments also speaks for the androgen dependency of the majority of advanced PCa cases. Our data are in line with these observations, and KLK3 was one of the genes with the highest upregulation in the Enza-resistant tumors compared to the Enza-responsive tumors, likely increasing the sensitivity of detecting Enza-resistant tumors by serum PSA. A similar increase in KLK3 gene expression was also observed in Enza-resistant VCaP cells in vitro (Kregel et al., 2016). The increased expression of several other classical AR-regulated genes further indicates that the Enza-resistant tumors in our model are at least partially AR-driven. Nanomolar intratumoral DHT concentrations (approximately 300 pg/g) are sufficient to drive the expression of AR-dependent genes, and after medical or surgical castration even lower levels are sufficient to activate AR (Gregory et al., 2001) because of increased AR expression during disease progression. In this study, the nuclear localization of AR was increased in Enza-resistant tumors compared to responsive tumors and those grown in mice after ORX + ADX. These data are in agreement with other studies (Gregory et al., 1998, 2001; Kregel et al., 2016) and further support the activation of androgen signaling in Enza-resistant VCaP tumors.
Notably, in addition to increasing intratumoral DHT concentrations, long-term Enza treatment was also found to increase the intra-adrenal androgen concentrations, suggesting adrenal androgen synthesis induction in castrated mice by antiandrogen treatment. Adrenalectomy resulted in a significantly lower intratumoral DHT concentration compared to the Enza-resistant tumors. Thus, DHT in CRPC tumors is evidently affected by adrenal-derived factors, inducing de novo steroidogenesis in tumors (synthesized from cholesterol) (Locke et al., 2008) or providing active androgens and/or precursors for DHT synthesis in tumors (Cai et al., 2011). Our data, together with previous studies (Barnard et al., 2020; Huhtaniemi et al., 2018; Mostaghel et al., 2019), evidently reveal a marked role for adrenals in promoting the growth of CRPC in rodents. This is novel, while the role of rodent models has been largely criticized because of the lack of DHEA and DHEA-S production, which is suggested to be the main source of intratumoral androgen production in clinical CRPC tumors (Cai et al., 2011). From a clinical perspective, the value of identifying increased intratumoral DHT as a result of antiandrogen treatment and defining its role as one mechanism of antiandrogen resistance suggests that a combination of an antiandrogen (e.g., Enza) with a CYP17 or CYP11A1 inhibitor would be of benefit to achieve substantial inhibition of androgen action in a set of CRPC tumors. Several clinical studies have been carried out with a combination of Enza and abiraterone, but unfortunately, those have failed to show a benefit for the combination regimen (Efstathiou et al., 2020; Morris et al., 2019). Enza is a potent inducer of CYP3A4 (Gibbons et al., 2015; Narayanan et al., 2016), and CYP3A4, in turn, is the major route for abiraterone metabolism (Bernard et al., 2015). Thus, Enza stimulates the degradation of abiraterone, and to minimize the risk of treatment failure, it is recommended to avoid CYP3A4 inducers when a therapeutic alternative is available (Bernard et al., 2015). Furthermore, a CYP17A1-independent DHT synthesis route has been shown to be present in PCa cells (de Mello Martins et al., 2017).
In this study, the reactivation of androgen action was associated with multiple biological processes, as indicated by the gene expression changes between Enza I and Enza II tumors. Simultaneously, according to direct markers of androgenic activity, pathways and processes such as inflammation, angiogenesis, invasiveness, EMT and neuroendocrine development were induced to increase the progression of tumorigenesis. Nephroblastoma overexpressed (NOV, former CCN3) is a marker of active androgen action (Knuuttila et al., 2018; Wu et al., 2014). It has been shown to promote cell differentiation (Chen et al., 2017) and to exhibit both tumor-suppressive and tumorigenic roles, depending on the cellular context (Perbal, 2008). In PCa NOV has been proposed to function as a tumor suppressor (Fong et al., 2017; Wu et al., 2014), and thus, its reduced expression is consistent with the increased proliferation in the Enza-resistant tumors compared to Enza responsive tumors. In addition to being a direct target of ligand-activated AR (Wu et al., 2014), NOV is an inhibitor of the P1K3/AKT/mTOR pathway (Huang et al., 2019). P1K3/AKT/mTOR signaling regulates, e.g., cell survival, growth and proliferation, metabolism, angiogenesis, and differentiation of stem cell-like properties (Bitting and Armstrong, 2013). Therefore, the suppression of NOV could be one of the effects by which the increased intratumoral DHT drives Enza resistance. Similarly, it has been demonstrated that PLOD2, which is among the most upregulated genes in the Enza-resistant tumors (fold change 4.41, FDR <0.005), mediates resistance by promoting the stemness P1K3/AKT/mTOR pathway, as evidenced in glioma cells (Song et al., 2017) and laryngeal cancer cells (Sheng et al., 2019). Interestingly, there are data suggesting that NOV activates ICAM-1 expression in prostate cancer cells (Chen et al., 2012), while in our data, the genes coding for these two proteins behaved in opposite fashion, ICAM-1 was highly upregulated in several of the Enza-resistant tumors, although there was high variation in the tumors. The potential roles of MAP1B and THBS1, which are upregulated in Enza-resistant tumors, remain to be investigated with respect to promoting the growth of tumors. To date, studies have indicated a tumor suppressive function for THBS1, which is likely not a driver of the Enza-resistant growth. However, the increased expression of MAP1B is of interest, as it has been shown that MAP1B overexpression in urothelial carcinoma is associated with adverse clinical features and predicts poor prognosis (Chien et al., 2020), and the expression of microtubule-associated proteins (MAPs) has also been associated with chemotherapy resistance in cancer (Bhat and Setaluri, 2007; Chien et al., 2020), the mechanisms of which remain to be solved.
In addition to the putative effects of adrenal-derived androgens and their precursors as growth-promoting factors in Enza-resistant tumors, there is a possibility that the lack of adrenal medulla-derived noradrenaline and/or adrenalin is the reason for the adrenalectomy response in our VCaP model. In fact, the increased expression of β2-adrenergic receptor (ADRB2) by 1.6-fold (FDR 0.02), as assessed by RNA-seq, in the Enza-resistant xenografts (ORX + Enza II) compared to Enza-responsive xenografts (ORX + Enza I) suggests activation of β-adrenergic signaling as a putative driver of Enza resistance in tumors with AR dependency. In line with these data, previous studies have shown ADRB2 to be an AR-regulated gene (Braadland et al., 2015; Guthrie et al., 1990; Massie et al., 2007) and indicated that ADRB2 is associated with luminal differentiation (Braadland et al., 2019). Interestingly, downregulation of ADRB2 has been associated with poor prognosis and treatment resistance caused by treatment-induced neuroendocrine differentiation (tNEPC) (Braadland et al., 2015, 2019). A marked upregulation of genes, including AKAP12, CCL2 and NPY, connected to adrenergic signaling in the Enza-resistant tumors in our study suggests a novel role of adrenergic signaling in androgen-driven resistance.
The expression of AKAP12, one of the three genes that was uniformly increased in heterogeneous Enza-resistant tumors, is reported to be associated with recruitment, desensitization and recycling of ADRB2, e.g., in cardiac myocytes. Furthermore, C-C motif chemokine ligand 2 (CCL2) was one of the most upregulated transcripts in the Enza-resistant tumors (fold change above 5). Although CCL2 has been found to be linked to ADRB2 by promoting the production of ADRB2 in astrocytes (Gutiérrez et al., 2018), it is also known as one of the main chemokines involved in PCa establishment in the bone marrow (Lu et al., 2009; Zhang et al., 2010). In line with the potential roles of several mechanisms, including both AR- and ADRB2-signaling, in Enza resistance, the transcriptomic data of Enza-resistant tumors were separated into three separate clusters. These clusters shared a number of transcripts that were differentially expressed between the Enza-responsive and Enza-resistant tumors but showed markedly more cluster-specific alterations. In summary, our data show that the occurrence of Enza resistance coincides with an increase in intratumoral DHT and reactivation in androgen signaling, and thus measuring the intratumoral DHT concentration in CRPC tumors may offer a tool to define the appearance of Enza resistance in a set of patients, benefiting from therapies suppressing androgen reactivation by another mode of action.
Limitations of the study
It is unfortunate that the PCa cells mimicking the local steroid synthesis often present in clinical CRPC is currently limited to VCaP cells only: as such, the current study should be confirmed when further cell lines become available. Currently, only the four most commonly used PCa cell lines, LNCaP, 22Rv, MDA PCa2b and VCaP, express AR and secrete PSA (Shi et al., 2019). The VCaP cell line is known to be the only one that expresses high levels of only wild-type AR and secretes PSA (Van Bokhoven et al., 2003), responds well to androgen ablation and exhibits classical features of CRPC, as we have proven.
From a clinical perspective, our results obtained with the preclinical model emphasize the possibility that elevated intratumor DHT could also indicate an antiandrogen resistant stage in clinical CRPC tumors. Thus, intratumor DHT could be used as a biomarker of the need for more intensive treatment to reduce the tumor androgen levels to nil or by using other modes of action. It is important to note that clinical studies that failed to show benefit for the combination regimen of antiandrogen and abiraterone included an unselected group of patients, whereas more personalized patient selection would be necessary because of the heterogeneous androgen concentrations shown in the CRPC specimens in some of the clinical studies (Efstathiou et al., 2020). Furthermore, the DHT synthesis pathway bypassing CYP17A1 exists (de Mello Martins et al., 2017).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-AR [EPR1535(2)] | Abcam | ab133273 |
| HRP-linked anti-rabbit IgG | Cell Signaling Technology | #7074 |
| AR (N-20) | Santa Cruz Biotechnology | sc-816 |
| Anti-rabbit antibody conjugated with polymer-HRP | Dako | K4002 |
| Chemicals, peptides, and recombinant proteins | ||
| BD Matrigel Matrix High Concentration | BD Biosciences | Cat#354248 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat#74034 |
| TruSeq Stranded mRNA LT Sample Prep Kit | Illumina | Cat#20020594 |
| Deposited data | ||
| NCBI’s Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147541F | Edgar et al. (2002) | GSE147541 |
| Experimental models: Cell lines | ||
| VCaP | ATCC | CRL-2876 |
| Experimental models: Organisms/strains | ||
| Athymic Nude male mice (Hsd:Athymic Nude-Foxn1nu | Envigo, France | Order code: 069 |
| Oligonucleotides | ||
| In the Table S4 | ||
| Software and algorithms | ||
| Study groups allocations | Tero Aittokallio (Laajala et al., 2016) | http://rvivo.tcdm.fi/ |
| FastQC tool | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR v2.5.0c | Dobin et al. (2013) | |
| Subread tool version 1.6.2 | Liao et al., 2014 | |
| R version 3.2.2 | R Development Core Team (2016) | |
| Other | ||
| Reagents for PSA time-resolved fluorometric assay | Kim Petterson (Lovgren et al., 1996) | N/A |
| Steroid profiling with GC-MS/MS | Claes Ohlsson (Nilsson et al., 2015) | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matti Poutanen (matti.poutanen@utu.fi), University of Turku, Turku, Finland.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell culture
Tested and authenticated VCaP cells were obtained from the American Type Cell Culture (ATCC, Manassas, VA, USA), and the cells were confirmed to be free of any harmful rodent pathogens before initiation of the study (Surrey Diagnostics, Surrey, United Kingdom). All cell culture reagents were purchased from Gibco, Thermo Fisher Scientific (Waltham, MA, USA), except HyClone fetal bovine serum was obtained from GE Healthcare, (Marlborough, MA, USA).
Generation of castration-resistant VCaP xenografts
The castration-resistant VCaP xenografts were generated, as previously reported (Huhtaniemi et al., 2018). Shortly, 180 Athymic Nude male mice (Hsd:Athymic Nude-Foxn1nu, Envigo, Gannat, France), weighing between 20 and 30 g, were used at 4-6 weeks of age. The mice were housed in individually ventilated cages (IVC, Techniplast, Buguggiate, Italy; one mouse/cage), under controlled conditions of light (12 h light/12 h dark), temperature (22°C ± 2°C) and humidity (55% ± 15%) in specific pathogen-free conditions at the Central Animal Laboratory, University of Turku, Finland. The mice were given irradiated soy-free natural-ingredient feed [RM3 (E), Special Diets Services, Essex, UK] and autoclaved tap water ad libitum. The cells were suspended in the culture medium described above at a density of 26.6 x106 cells/mL. Thereafter, high protein concentration Matrigel (BD Biosciences, Bedford, MA, USA) was added (1:1), and a 150 μL aliquot of this suspension (2 million cells per mice) was inoculated subcutaneously (s.c.) to the right flank of each mouse by using a 25 G needle.
Development and growth of the VCaP tumors were monitored by measuring the tumor volume twice a week, and by detecting the serum concentration of PSA every 10 days. The volume of the tumors were calculated according to following formula: W2 x L/2 (W = shorter diameter, L = longer diameter of the tumor). For PSA analysis, blood was collected from saphenous vein, and the PSA was measured with an in-house time-resolved fluorometric assay (Lovgren et al., 1996).
Tumors were grown for 5 weeks, until the mean volume of the tumors reached approximately 500 mm3 (range 211-1554 mm3), and the mean serum PSA value was 12 μg/L (range 1.4–53.8 μg/L). Mice were then allocated to study groups using our published algorithm (Laajala et al., 2016), the groups being orchiectomized (ORX; n = 34) mice, orchiectomized and Enza treated (ORX + Enza; n = 28) mice, and orchiectomized and adrenalectomized (ORX + ADX; n = 28) mice. Orchiectomy and adrenalectomy were carried out under the isoflurane (2-3% Isofluran, Baxter S.A., Lessines, Belgium) induced anesthesia. For pain relief, mice were injected s.c. with buprenorphine (0.1 mg/kg, Temgesic® 0.3 mg/mL, Reckitt Benckiser Healthcare, Hull, United Kingdom) and carpofen 5 mg/kg (Vet Rimadyl ® 50 mg/mL, Pfizer SA, Louvain-La-Neuve, Belgium) before and after the operations. The Enza treatment was started 31 days after ORX, and thereafter, 20 mg/kg of the drug was administered orally (p.o.) via gavage once daily, for 5 days or for 46 days. The mice were sacrificed via cervical dislocation at different time points (Figure 1A). The following group of mice were allocated again and analyzed: ORX I (orchiectomized mice, sacrificed 10 days after ORX), ORX II (orchiectomized mice, sacrificed 76 days after ORX), ORX + Enza I (sacrificed 34 days after ORX and 5 days after initiating Enza treatment), ORX + Enza II (sacrificed 76 days after ORX and 47 days after initiating Enza treatment), ORX + ADX I (sacrificed 10 days after ORX and ADX) and ORX + ADX II (sacrificed 76 days after ORX and ADX). Tumor, adrenal gland and serum samples were stored at −80°C after initial freezing in liquid nitrogen, and specimens for histology and immunohistochemistry were fixed in 10% formalin for 24 h prior to paraffin embedding.
This study has been performed according to the guidelines for the welfare and use of animals in cancer research(Workman et al., 2010), and following the EU legislation related to the use of animals for scientific purposes. National Animal Experiment Board of Finland authorized the animal studies with the license ESAVI/4199/04.10.07/2014 that were performed according to the instructions given by the Institutional Animal Care and Use Committees of the University of Turku.
Method details
Measurement of intratumoral, intra-adrenal and serum steroid concentrations
Tumors and adrenal glands were homogenized in sterile water using a TissueLyzer LT homogenizer (Qiagen, Venlo, The Netherlands), and intratumoral, intra-adrenal and serum concentrations of progesterone (P4), androstenedione (A-dione), T and DHT were measured using a previously described method applying GC-MS/MS (Nilsson et al., 2015). Using mouse serum, the lower limit of quantitation (LLOQ) for P4, A-dione, T and DHT with the assay are 74 pg/mL, 12 pg/mL, 8 pg/mL, 2.5 pg/mL, respectively. Results under the LLOQ were calculated to be half of each lower limit of quantitation value to avoid overestimation of low values in the analysis. To make intratumoral steroid concentrations comparable to those in the serum, 1 g of tumor was considered equivalent to 1 mL of serum.
Quantitative RT–qPCR analyses and RNA-sequencing
For RT–qPCR and RNA-sequencing (RNA-seq), total RNA was extracted from tumors by homogenizing the specimens to TRIsure (Invitrogen, Carlsbad, CA, USA), and using RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. The expression levels of full length AR (AR-FL) and AR splice variants V1 and V7 were analyzed by real-time RT–qPCR using the primers presented at Table S4. Possible contribution of mouse stromal cells into intratumoral androgen synthesis was studied by analyzing the expression of mouse steroidogenic enzymes from tumor samples using RT–qPCR (see Table S4).
RNA-seq from tumors of ORX mice treated for 5 days (n = 10) or 46 days (n = 10) with Enza, were carried out at the Finnish Functional Genomics Center (University of Turku, Åbo Akademi University and Biocenter Finland). The samples were treated according to Illumina TruSeq® Stranded mRNA Sample Preparation Guide, and sequenced with Illumina HiSeq 2500 instrument (Illumina, San Diego, CA, USA), using Hiseq v2 Rapid sequencing chemistry and 50 bp single-end read length.
The quality of the sequenced reads was confirmed using the FastQC tool (Babraham Bioinformatics, Babraham Institute, Cambridge, UK). STAR v2.5.0c (Dobin et al., 2013) was used to align the reads to the mouse reference genome mm10, available at University of California, Santa Cruz Genome Bioinformatics Group (Illumina iGenomes website, San Diego, CA). The number of uniquely mapped reads associated with each gene, according to RefSeq gene annotation, was counted using the Subread tool version 2.6.1b (Liao et al., 2014). The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE147541 (NCBI’s Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147541). The downstream analysis of the data was performed using R version 3.2.2 (R Development Core Team, 2016) and its corresponding Bioconductor module. The read counts were normalized for library size using the Trimmed Mean of M-values (TMM) method implemented in the R-package named as edgeR (Robinson et al., 2010). R package ROTS (Suomi et al., 2017) was used for performing the statistical testing. False discovery rate (FDR) < 0.05 and absolute fold change (FC) > 1.5 was required to consider the gene to be differentially expressed between the groups analyzed. The hierarchical clustering of the normalized expression values of differentially expressed AR-regulated genes was performed using Euclidean distance and Ward’s method, implemented in the R package pheatmap (Kolde, 2015).
For analyzing the role of AR signaling in our model, we generated a list of AR-associated genes containing known androgen-regulated genes as well as AR interacting proteins using common up-regulated genes in VCaP and LNCaP upon DHT treatment (Asangani et al., 2014), Database Resources of the National Center for Biotechnology Information (Coordinators, 2016), androgen pathway product analysis list provided by SwitchGear Genomics (Menlo Park, CA) and Regulators of Androgen Action Resource (RAAR) database (Depriest et al., 2016).
In order to detect whether AR mutations T878A, F877L and L702H would contribute the Enza resistance, we amplified AR exon four and beginning of exon 8 from cDNA samples via PCR (primers: AR exon 4 SE: ACAGGAGGAAGGAGAGGCTT; AR exon 4 As: CCCACTTGACCACGTGTACA; AR exon 7 SE: ACATCCTGCTCAAGACGCTT; AR exon 8 As: TGGGTGTGGAAATAGATGGGC). Amplified products were run on a 2% agarose gel and DNA containing gel pieces were cut out. The DNA was extracted from the gel and purified with NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. The DNA samples were prepared for sequencing with the Mix2Seq kit (Eurofins Genomics, Konstanz, Germany) and sequencing was performed at Eurofins Genomics Sequencing Europe using cycle sequencing on ABI 3730XL machines (Applied Biosystems, Foster City, CA).
Immunoblotting
Tumor samples were homogenized using a TissueLyzer LT and stainless steel beads (Qiagen, Hilden, Germany) in radioimmunoprecipitation assay (RIPA)-lysis buffer containing the following ingredients: 150 mmol/L Tris-HCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mmol/L EDTA, 1 mmol/L SDS, 100 mmol/L sodium orthovanadate (Sigma-Aldrich, St.Louis, MO, USA), and cOmplete Mini protease inhibitor (Roche Diagnostics, Mannheim, Germany). Samples (30 μg) were centrifuged at 8000 g for 10 min at 4°C, and total protein concentrations in the supernatant were measured with a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). The samples were loaded onto an 10% Mini-PROTEAN TGX Precast Protein SDS-PAGE Gel (Bio-Rad, Hercules, USA) and separated under reducing conditions, followed by transfer onto an Amersham Hybond P 0.45 PVDF blotting membrane (GE Healthcare Life Sciences, Chicago, IL, USA).
The membranes of tumor samples were probed with rabbit monoclonal anti-AR antibody (Abcam, Cambridge, UK) with 1:2000 dilution followed by HRP-linked anti-rabbit IgG antibody (dilution 1:5000, Cell Signaling Technology, Danvers, MA, USA). The membranes were visualized using Cy5 and Cy3 detection and a Typhoon laser scanner (GE Healthcare) and imaged with an ImageQuant LAS 4000 camera system (GE Healthcare). ImageJ software version 1.51K (NIH, Bethesda, MD) was used to compare and quantify the intensity of bands on scanned images of membranes.
Immunohistochemistry
Formalin-fixed and paraffin-embedded tumor samples were cut to sections prior to deparafinization and rehydration. The sections were exposed to the antigen retrieval in a steamer in 10 mM sodium citrate buffer (Citric acid monohydrate Sigma-Aldrich, St Louis, USA) and Tri-sodium citrate dehydrate (Merck, Darmstadt, Germany) for 30 min. The sections were then incubated in a humidified chamber overnight with the primary antibody against the N-terminus of the AR (N-20: sc-816, dilution 1:250, Santa Cruz Biotechnology, Dallas, Texas, USA) at 4 °C. Endogenous peroxidase activity was blocked by applying 1% H2O2 for 20 min at room temperature and the section were then incubated for 30 min with anti-rabbit antibody conjugated with polymer-HRP (Dako, Glostrup, Denmark), washed and visualized with Envision+ System-HRP DAB staining (Dako). The sections were counterstained with hematoxylin, mounted and digitized using a Pannoramic 250 slide scanner (3DHISTECH, Budapest, Hungary).
Quantification and statistical analysis
The statistical tests were chosen depending on the results of the preliminary Shapiro-Wilk tests of data normality. Non-parametric Mann-Whitney, Kruskal-Wallis and Dunn’s multiple comparison tests were applied in RT–qPCR comparisons on single gene level, and to test the differences in the steroid concentrations in ORX, ORX + ADX and ORX + Enza treated mice. These univariate statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA). p < 0.05 was considered statistically significant.
Acknowledgments
Contributions from nonauthors:
Kim Petterson (University of Turku, Finland) kindly provided reagents for the PSA assay. The authors would like to thank the personnel of Turku Center for Disease Modeling (www.TCDM.fi) and the Histology Core Facility at the Institute of Biomedicine, University of Turku. The Turku Center for Disease Modeling is part of the Biocenter Finland Model Organisms Infrastructure.
Funding sources:
The Academy of Finland, Cancer Society of Finland, Drug Research Doctoral Programme, Finnish Cultural Foundation, Orion Pharma, Sigrid Jusélius Foundation, Swedish government, under the Agreement for Medical Education and Research, and the Swedish Research Council and University of Turku.
Author contributions
Conceptualization, R.H., P.S., R.O., E.A., M.H.T., P.K., S.M., M.V.J.M., and M.P.; Methodology, R.H., M.K., P.K., S.M., and M.P.; Software, T.D.L. and T.A.; Validation, R.H. and M.P.; Formal Analysis, R.H., A.M., T.D.L., and T.A.; Investigation, R.H.; Resources, P.S., R.O., E.A., T.A., and M.P.; Data Curation, R.H. and M.P.; Writing – Original Draft, R.H., P.S., and M.P.; Writing – Review & Editing, R.H., P.S., R.O., M.K., A.M., E.A., T.D.L., T.A., M.H.T., P.K., S.M., M.V.J.M., and M.P.; Visualization, R.H. and A.M.; Supervision, P.K., S.M., and M.P.; Project Administration, R.H. and M.P.; Funding Acquisition, R.H., E.A., and M.P.
Declaration of interests
R.O. and E.A. are employees and M.V.J.M. and P.K. are former employees of Orion Corporation, Orion Pharma. M.P. is a consultant of Forendo Pharma. The other authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104287.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the STAR Methods section.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Antonarakis E.S., Blackford A.L., Garrett-Mayer E., Eisenberger M.A. Survival in men with nonmetastatic prostate cancer treated with hormone therapy: a quantitative systematic review. J. Clin. Oncol. 2007;25:4998–5008. doi: 10.1200/JCO.2007.11.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis E.S., Lu C., Luber B., Wang H., Chen Y., Zhu Y., Silberstein J.L., Taylor M.N., Maughan B.L., Denmeade S.R., et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J. Clin. Oncol. 2017;35:2149–2156. doi: 10.1200/jco.2016.70.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora V.K., Schenkein E., Murali R., Subudhi S.K., Wongvipat J., Balbas M.D., Shah N., Cai L., Efstathiou E., Logothetis C., et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cellule. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I.A., Dommeti V.L., Wang X., Malik R., Cieslik M., Yang R., Escara-Wilke J., Wilder-Romans K., Dhanireddy S., Engelke C., et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard M., Mostaghel E.A., Auchus R.J., Storbeck K.H. The role of adrenal derived androgens in castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2020;197:105506. doi: 10.1016/j.jsbmb.2019.105506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger B., Bélanger A., Labrie F., Dupont A., Cusan L., Monfette G. Comparison of residual C-19 steroids in plasma and prostatic tissue of human, rat and Guinea pig after castration: unique importance of extratesticular androgens in men. J. Steroid. Biochem. 1989;32:695–698. doi: 10.1016/0022-4731(89)90514-1. [DOI] [PubMed] [Google Scholar]
- Beltran H., Prandi D., Mosquera J.M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B.V.S.K., Varambally S., et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A., Vaccaro N., Acharya M., Jiao J., Monbaliu J., De Vries R., Stieltjes H., Yu M., Tran N., Chien C. Impact on abiraterone pharmacokinetics and safety: open-label drug-drug interaction studies with ketoconazole and rifampicin. Clin. Pharmacol. Drug Dev. 2015;4:63–73. doi: 10.1002/cpdd.132. [DOI] [PubMed] [Google Scholar]
- Bhat K.M.R., Setaluri V. Microtubule-associated proteins as targets in cancer chemotherapy. Clin. Cancer. Res. 2007;13:2849–2854. doi: 10.1158/1078-0432.CCR-06-3040. [DOI] [PubMed] [Google Scholar]
- Bitting R.L., Armstrong A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer. 2013;20:R83–R99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- Bohl C.E., Gao W., Miller D.D., Bell C.E., Dalton J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. U S A. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braadland P.R., Ramberg H., Grytli H.H., Taskén K.A. β-Adrenergic receptor signaling in prostate cancer. Front. Oncol. 2015;4:375. doi: 10.3389/fonc.2014.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braadland P.R., Ramberg H., Grytli H.H., Urbanucci A., Nielsen H.K., Guldvik I.J., Engedal A., Ketola K., Wang W., Svindland A., et al. The β2-adrenergic receptor is a molecular switch for neuroendocrine transdifferentiation of prostate cancer cells. Mol. Cancer Res. 2019;17:2154–2168. doi: 10.1158/1541-7786.MCR-18-0605. [DOI] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer. J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Cai C., Chen S., Ng P., Bubley G.J., Nelson P.S., Mostaghel E.A., Marck B., Matsumoto A.M., Simon N.I., Wang H., et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer. Res. 2011;71:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.D., Welsbie D.S., Tran C., Baek S.H., Chen R., Vessella R., Rosenfeld M.G., Sawyers C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen P.C., Lin T.H., Cheng H.C., Tang C.H. CCN3 increases cell motility and ICAM-1 expression in prostate cancer cells. Carcinogenesis. 2012;33:937–945. doi: 10.1093/carcin/bgs108. [DOI] [PubMed] [Google Scholar]
- Chen P.C., Tai H.C., Lin T.H., Wang S.W., Lin C.Y., Chao C.C., Yu H.J., Tsai Y.C., Lai Y.W., Lin C.W., Tang C.H. CCN3 promotes epithelial-mesenchymal transition in prostate cancer via FAK/Akt/HIF-1α-induced twist expression. Oncotarget. 2017;8:74506–74518. doi: 10.18632/oncotarget.20171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien T.M., Chan T.C., Huang S.K.H., Yeh B.W., Li W.M., Huang C.N., Li C.C., Wu W.J., Li C.F. Role of microtubule-associated protein 1b in urothelial carcinoma: overexpression predicts poor prognosis. Cancers (Basel) 2020;12:630. doi: 10.3390/cancers12030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coordinators N.R. Database resources of the national center for biotechnology information. Nucleic. Acids. Res. 2016;44:D7–D19. doi: 10.1093/nar/gkv1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornford P., Bellmunt J., Bolla M., Briers E., De Santis M., Gross T., Henry A.M., Joniau S., Lam T.B., Mason M.D., et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur. Urol. 2017;71:630–642. doi: 10.1016/j.eururo.2016.08.002. [DOI] [PubMed] [Google Scholar]
- de Mello Martins A.G.G., Allegretta G., Unteregger G., Haupenthal J., Eberhard J., Hoffmann M., van der Zee J.A., Junker K., Stöckle M., Müller R., et al. CYP17A1-independent production of the neurosteroid-derived 5α-pregnan-3β,6α-diol-20-one in androgen-responsive prostate cancer cell lines under serum starvation and inhibition by Abiraterone. J. Steroid Biochem. Mol. Biol. 2017;174:183–191. doi: 10.1016/j.jsbmb.2017.09.006. [DOI] [PubMed] [Google Scholar]
- Dehm S.M., Tindall D.J. Molecular regulation of androgen action in prostate cancer. J. Cell. Biochem. 2006;99:333–344. doi: 10.1002/jcb.20794. [DOI] [PubMed] [Google Scholar]
- Depriest A.D., Fiandalo M.V., Schlanger S., Heemers F., Mohler J.L., Liu S., Heemers H.V. Regulators of androgen action resource: a one-stop shop for the comprehensive study of androgen receptor action. Database. 2016;2016:bav125. doi: 10.1093/database/bav125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou E., Titus M., Wen S., Hoang A., Karlou M., Ashe R., Tu S.M., Aparicio A., Troncoso P., Mohler J., Logothetis C.J. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur. Urol. 2015;67:53–60. doi: 10.1016/j.eururo.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou E., Titus M., Wen S., Troncoso P., Hoang A., Corn P., Prokhorova I., Araujo J., Dmuchowski C., Melhem-Bertrandt A., et al. Enzalutamide in combination with abiraterone acetate in bone metastatic castration-resistant prostate cancer patients. Eur. Urol. Oncol. 2020;3:119–127. doi: 10.1016/j.euo.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong K.W., Zhao J.C., Kim J., Li S., Yang Y.A., Song B., Rittie L., Hu M., Yang X., Perbal B., Yu J. Polycomb-mediated disruption of an androgen receptor feedback loop drives castration-resistant prostate cancer. Cancer Res. 2017;77:412–422. doi: 10.1158/0008-5472.CAN-16-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons J.A., de Vries M., Krauwinkel W., Ohtsu Y., Noukens J., van der Walt J.S., Mol R., Mordenti J., Ouatas T. Pharmacokinetic drug interaction studies with enzalutamide. Clin. Pharm. 2015;54:1057–1069. doi: 10.1007/s40262-015-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory C.W., Hamil K.G., Kim D., Hall S.H., Pretlow T.G., Mohler J.L., French F.S. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–5724. [PubMed] [Google Scholar]
- Gregory C.W., Johnson R.T., Mohler J.L., French F.S., Wilson E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- Guthrie P.D., Freeman M.R., Liao S., Chung L.W.K. Regulation of gene expression in rat Prostate by androgen and β-adrenergic receptor pathways. Mol. Endocrinol. 1990;4:1343–1353. doi: 10.1210/mend-4-9-1343. [DOI] [PubMed] [Google Scholar]
- Gutiérrez I.L., González-Prieto M., García-Bueno B., Caso J.R., Feinstein D.L., Madrigal J.L.M. CCL2 induces the production of β2 adrenergic receptors and modifies astrocytic responses to noradrenaline. Mol. Neurobiol. 2018;55:7872–7885. doi: 10.1007/s12035-018-0960-9. [DOI] [PubMed] [Google Scholar]
- Handle F., Erb H.H.H., Luef B., Hoefer J., Dietrich D., Parson W., Kristiansen G., Santer F.R., Culig Z. SOCS3 modulates the response to enzalutamide and is regulated by androgen receptor signaling and CpG methylation in prostate cancer cells. Mol. Cancer Res. 2016;14:574–585. doi: 10.1158/1541-7786.MCR-15-0495. [DOI] [PubMed] [Google Scholar]
- Horwich A., Hugosson J., de reijke T., Wiegel T., Fizazi K., Kataja V., Parker C., Bellmunt J., Berthold D., Bill-axelson A., et al. Prostate cancer: ESMO consensus conference guidelines 2012. Ann. Oncol. 2013;24:1141–1162. doi: 10.1093/annonc/mds624. [DOI] [PubMed] [Google Scholar]
- Huang X., Ni B., Mao Z., Xi Y., Chu X., Zhang R., Ma X., You H. NOV/CCN3 induces cartilage protection by inhibiting PI3K/AKT/mTOR pathway. J. Cell. Mol. Med. 2019;23:7525–7534. doi: 10.1111/jcmm.14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhtaniemi R., Oksala R., Knuuttila M., Mehmood A., Aho E., Laajala T.D., Nicorici D., Aittokallio T., Laiho A., Elo L., et al. Adrenals contribute to growth of castration-resistant VCaP prostate cancer xenografts. Am. J. Pathol. 2018;188:2890–2901. doi: 10.1016/j.ajpath.2018.07.029. [DOI] [PubMed] [Google Scholar]
- Isikbay M., Otto K., Kregel S., Kach J., Cai Y., Vander Griend D.J., Conzen S.D., Szmulewitz R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer. 2014;5:72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J.D., Lu N., Qian J., Sensintaffar J., Shao G., Brigham D., Moon M., Maneval E.C., Chen I., Darimont B., Hager J.H. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Knuuttila M., Mehmood A., Huhtaniemi R., Yatkin E., Häkkinen M.R., Oksala R., Laajala T.D., Ryberg H., Handelsman D.J., Aittokallio T., et al. Antiandrogens reduce intratumoral androgen concentrations and induce androgen receptor expression in castration-resistant prostate cancer xenografts. Am. J. Pathol. 2018;188:216–228. doi: 10.1016/j.ajpath.2017.08.036. [DOI] [PubMed] [Google Scholar]
- Knuuttila M., Yatkin E., Kallio J., Savolainen S., Laajala T.D., Aittokallio T., Oksala R., Häkkinen M., Keski-Rahkonen P., Auriola S., et al. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am. J. Pathol. 2014;184:2163–2173. doi: 10.1016/j.ajpath.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T., Kallioniemi O.P. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- Kolde R. 2015. Pheatmap: pretty heatmaps [WWW Document]. R Packag. Version 1.0.8. https://cran.r-project.org/web/packages/pheatmap/index.html. [Google Scholar]
- Korenchuk S., Lehr J.E., MClean L., Lee Y.G., Whitney S., Vessella R., Lin D.L., Pienta K.J. VCaP, a cell-based model system of human prostate cancer. In Vivo (Brooklyn) 2001;15:163–168. [PubMed] [Google Scholar]
- Korpal M., Korn J.M., Gao X., Rakiec D.P., Ruddy D.A., Doshi S., Yuan J., Kovats S.G., Kim S., Cooke V.G., et al. An F876l mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (Enzalutamide) Cancer Discov. 2013;3:1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- Kregel S., Chen J.L., Tom W., Krishnan V., Kach J., Brechka H., Fessenden T.B., Isikbay M., Paner G.P., Szmulewitz R.Z., Vander Griend D.J. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016;7:26259–26274. doi: 10.18632/oncotarget.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku S.Y., Rosario S., Wang Y., Mu P., Seshadri M., Goodrich Z.W., Goodrich M.M., Labbé D.P., Gomez E.C., Wang J., et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laajala T.D., Jumppanen M., Huhtaniemi R., Fey V., Kaur A., Knuuttila M., Aho E., Oksala R., Westermarck J., Mäkelä S., et al. Optimized design and analysis of preclinical intervention studies in vivo. Sci. Rep. 2016;6:30723. doi: 10.1038/srep30723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie F., Cusan L., Séguin C., Bélanger A., Pelletier G., Reeves J., Kelly P.A., Lemay A., Raynaud J.P., GOURDEAU Y. Antifertility effects of LHRH agonists in the male. J. Androl. 1980;1:209–228. doi: 10.1002/j.1939-4640.1980.tb00034.x. [DOI] [PubMed] [Google Scholar]
- Labrie F., Dupont A., Belanger A., Giguere M., Lacoursiere Y., Emond J., Monfette G., Bergeron V. Combination therapy with flutamide and castration (LHRH agonist or orchiectomy) in advanced prostate cancer: a marked improvement in response and survival. J. Steroid. Biochem. 1985;23:833–841. doi: 10.1016/S0022-4731(85)80024-8. [DOI] [PubMed] [Google Scholar]
- Li Y., Chan S.C., Brand L.J., Hwang T.H., Silverstein K.A.T., Dehm S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Linja M.J., Savinainen K.J., Saramaki O.R., Tammela T.L.J., Vessella R.L., Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- Liu C., Lou W., Zhu Y., Yang J.C., Nadiminty N., Gaikwad N.W., Evans C.P., Gao A.C. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke J.A., Guns E.S., Lubik A.A., Adomat H.H., Hendy S.C., Wood C.A., Ettinger S.L., Gleave M.E., Nelson C.C. Androgen Levels Increase by Intratumoral De Novo Steroidogenesis during Progression of Castration-Resistant Prostate Cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- Lovgren T., Merio L., Mitrunen K., Makinen M.L., Makela M., Blomberg K., Palenius T., Pettersson K. One-step all-in-one dry reagent immunoassays with fluorescent europium chelate label and time-resolved fluorometry. Clin. Chem. 1996;42:1196–1201. doi: 10.1093/clinchem/42.8.1196. [DOI] [PubMed] [Google Scholar]
- Lu Y., Chen Q., Corey E., Xie W., Fan J., Mizokami A., Zhang J. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis. 2009;26:161–169. doi: 10.1007/s10585-008-9226-7. [DOI] [PubMed] [Google Scholar]
- Massie C.E., Adryan B., Barbosa-Morais N.L., Lynch A.G., Tran M.G., Neal D.E., Mills I.G. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO. Rep. 2007;8:871–878. doi: 10.1038/sj.embor.7401046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto H., Yeh S., Lardy H., Messing E., Chang C. δ5-Androstenediol is a natural hormone with androgenic activity in human prostate cancer cells. Proc. Natl. Acad. Sci. U S A. 1998;95:11083–11088. doi: 10.1073/pnas.95.19.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghissi E., Ablan F., Horton R. Origin of plasma androstanediol glucuronide in men. J. Clin. Endocrinol. Metab. 1984;59:417–421. doi: 10.1210/jcem-59-3-417. [DOI] [PubMed] [Google Scholar]
- Morris M.J., Heller G., Bryce A.H., Armstrong A.J., Beltran H., Hahn O.M., McGary E.C., Mehan P.T., Goldkorn A., Roth B.J., et al. Alliance A031201: a phase III trial of enzalutamide (ENZ) versus enzalutamide, abiraterone, and prednisone (ENZ/AAP) for metastatic castration resistant prostate cancer (mCRPC) J. Clin. Oncol. 2019;37:5008. doi: 10.1200/jco.2019.37.15_suppl.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaghel E.A., Zhang A., Hernandez S., Marck B.T., Zhang X., Tamae D., Biehl H.E., Tretiakova M., Bartlett J., Burns J., et al. Contribution of adrenal glands to intratumor androgens and growth of castration-resistant prostate cancer. Clin. Cancer Res. 2019;25:426–439. doi: 10.1158/1078-0432.CCR-18-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P., Zhang Z., Benelli M., Karthaus W.R., Hoover E., Chen C.C., Wongvipat J., Ku S.Y., Gao D., Cao Z., et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R., Hoffmann M., Kumar G., Surapaneni S. Application of a “Fit for Purpose” PBPK model to investigate the CYP3A4 induction potential of enzalutamide. Drug Metab. Lett. 2016;10:172–179. doi: 10.2174/1872312810666160729124745. [DOI] [PubMed] [Google Scholar]
- Nash A.F., Melezinek I. The role of prostate specific antigen measurement in the detection and management of prostate cancer. Endocr. Relat. Cancer. 2000;7:37–51. doi: 10.1677/erc.0.0070037. [DOI] [PubMed] [Google Scholar]
- Nilsson M.E., Vandenput L., Tivesten Å., Norlén A.-K., Lagerquist M.K., Windahl S.H., Börjesson A.E., Farman H.H., Poutanen M., Benrick A., et al. Measurement of a comprehensive sex steroid profile in rodent serum by high-sensitive gas chromatography-tandem mass spectrometry. Endocrinology. 2015;156:2492–2502. doi: 10.1210/en.2014-1890. [DOI] [PubMed] [Google Scholar]
- Parker C., Gillessen S., Heidenreich A., Horwich A. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015;26:v69–v77. doi: 10.1093/annonc/mdv222. [DOI] [PubMed] [Google Scholar]
- Perbal B. CCN3: doctor jekyll and mister hyde. J. Cell Commun. Signal. 2008;2:3–7. doi: 10.1007/s12079-008-0028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekovic S., van den Broeck T., Linder S., van Royen M.E., Houtsmuller A.B., Handle F., Joniau S., Zwart W., Claessens F. Molecular underpinnings of enzalutamide resistance. Endocr. Relat. Cancer. 2018;25:R545–R557. doi: 10.1530/ERC-17-0136. [DOI] [PubMed] [Google Scholar]
- R Development Core Team R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. Vienna Austria 0. 2016. [DOI]
- Riegman P.H.J., Vlietstra R.J., van der Korput J.A.G.M., Brinkmann A.O., Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol. Endocrinol. 1991;5:1921–1930. doi: 10.1210/mend-5-12-1921. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C.J., Smith A., Lal P., Satagopan J., Reuter V., Scardino P., Gerald W., Scher H.I. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: an early predictor of relapse or incomplete androgen suppression. Urology. 2006;68:834–839. doi: 10.1016/j.urology.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Scher H.I., Fizazi K., Saad F., Taplin M.-E., Sternberg C.N., Miller K., de Wit R., Mulders P., Chi K.N., Shore N.D., et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Scher H.I., Anand A., Rathkopf D., Shelkey J., Morris M.J., Danila D.C., Larson S., Humm J., Fleisher M., Sawyers C.L., et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher H.I., Lu D., Schreiber N.A., Louw J., Graf R.P., Vargas H.A., Johnson A., Jendrisak A., Bambury R., Danila D., et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016;2:1441–1449. doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J., Leveridge M., Norling C., Karén J., Molina D.M., O’Neill D., Dowling J.E., Davey P., Cowan S., Dabrowski M., et al. Determining direct binders of the androgen receptor using a high-throughput cellular thermal shift assay. Sci. Rep. 2018;8:163. doi: 10.1038/s41598-017-18650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X., Li Y., Li Y., Liu W., Lu Z., Zhan J., Xu M., Chen L., Luo X., Cai G., Zhang S. PLOD2 contributes to drug resistance in laryngeal cancer by promoting cancer stem cell-like characteristics. BMC. Cancer. 2019;19:840. doi: 10.1186/s12885-019-6029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Chen X., Tan D. Development of patient-derived xenograft models of prostate cancer for maintaining tumor heterogeneity. Transl. Androl. Urol. 2019;8:519–528. doi: 10.21037/tau.2019.08.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Zheng S., Wang J., Long H., Fang L., Wang G., Li Z., Que T., Liu Y., Li Y., et al. Hypoxia-induced PLOD2 promotes proliferation, migration and invasion via PI3K/Akt signaling in glioma. Oncotarget. 2017;8:41947–41962. doi: 10.18632/oncotarget.16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Shi Y., Xu L.L., Nageswararao C., Davis L.D., Segawa T., Dobi A., McLeod D.G., Srivastava S. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006;25:3905–3913. doi: 10.1038/sj.onc.1209424. [DOI] [PubMed] [Google Scholar]
- Sun S., Sprenger C.C.T., Vessella R.L., Haugk K., Soriano K., Mostaghel E.A., Page S.T., Coleman I.M., Nguyen H.M., Sun H., et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomi T., Seyednasrollah F., Jaakkola M.K., Faux T., Elo L.L. ROTS: an R package for reproducibility-optimized statistical testing. PLoS Comput. Biol. 2017;13:e1005562. doi: 10.1371/journal.pcbi.1005562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C., Ouk S., Clegg N.J., Chen Y., Watson P.A., Arora V., Wongvipat J., Smith-Jones P.M., Yoo D., Kwon A., et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bokhoven A., Varella-Garcia M., Korch C., Johannes W.U., Smith E.E., Miller H.L., Nordeen S.K., Miller G.J., Lucia M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- Visakorpi T., Hyytinen E., Koivisto P., Tanner M., Keinänen R., Palmberg C., Palotie A., Tammela T., Isola J., Kallioniemi O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Waltering K.K., Helenius M.A., Sahu B., Manni V., Linja M.J., Jänne O.A., Visakorpi T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141–8149. doi: 10.1158/0008-5472.CAN-09-0919. [DOI] [PubMed] [Google Scholar]
- Watson P.A., Chen Y.F., Balbas M.D., Wongvipat J., Socci N.D., Viale A., Kim K., Sawyers C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P., Aboagye E.O., Balkwill F., Balmain A., Bruder G., Chaplin D.J., Double J.A., Everitt J., Farningham D.A.H., Glennie M.J., et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Runkle C., Jin H.J., Yu J., Li J., Yang X., Kuzel T., Lee C., Yu J. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene. 2014;33:504–513. doi: 10.1038/onc.2012.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Cai C., Chen S., Chen S., Yu Z., Balk S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33:2815–2825. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lu Y., Pienta K.J. Multiple roles of chemokine (C-C Motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer. Inst. 2010;102:522–528. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the STAR Methods section.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.