

Abstract
β-Xylosidase secreted by the shoyu koji mold, Aspergillus oryzae, is the key enzyme responsible for browning of soy sauce. To investigate the role of β-xylosidase in the brown color formation, a major β-xylosidase, XylA, and its encoding gene were characterized. β-Xylosidase XylA was purified to homogeneity from culture filtrates of A. oryzae KBN616. The optimum pH and temperature of the enzyme were found to be 4.0 and 60°C, respectively, and the molecular mass was estimated to be 110 kDa based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The xylA gene comprises 2,397 bp with no introns and encodes a protein consisting of 798 amino acids (86,475 Da) with 14 potential N-glycosylation sites. The deduced amino acid sequence shows high similarity to Aspergillus nidulans XlnD (70%), Aspergillus niger XlnD (64%), and Trichoderma reesei BxII (63%). The xylA gene was overexpressed under control of the strong and constitutive A. oryzae TEF1 promoter. One of the A. oryzae transformants produced approximately 13 times more of the enzyme than did the host strain. The partial-length antisense xylA gene expressed under control of the A. oryzae TEF1 promoter decreased the β-xylosidase level in A. oryzae to about 20% of that of the host strain.
The brown color characteristic of the traditional Japanese fermented food soy sauce, or shoyu, is one of the important parameters of its quality; the lighter brown is preferable. The brown color is formed by the aminocarbonyl reaction between the reducing sugars and amino acids in soy sauce mash. Of the reducing sugars, pentose, especially xylose, has been considered to be mainly responsible for the brown color formation (4). Xylose in soy sauce mash is formed as a result of xylan hydrolysis in soybean and wheat with the xylanolytic enzymes produced by the shoyu koji mold, Aspergillus oryzae. The major xylanolytic enzyme complex includes endo-(1,4)-β-xylanases (EC 3.2.1.8), the so called xylanases, which hydrolyze the polysaccharide backbone, and β-xylosidases (EC 3.2.1.37), which hydrolyze xylo-oligosaccharides to xylose. The use of A. oryzae mutants producing low levels of the xylanolytic enzymes, especially β-xylosidase, has been reported to prevent browning of soy sauce (14). Therefore, among the xylanolytic enzymes secreted by A. oryzae, β-xylosidase is the key enzyme responsible for browning of soy sauce.
Numerous studies on fungal β-xylosidases have been done, but only three fungal β-xylosidase genes have been isolated, from Aspergillus nidulans (16), Aspergillus niger (18), and Trichoderma reesei (13). When grown on a shoyu koji composed of soybean and wheat, A. oryzae secretes three β-xylosidases (14). However, little is known about the enzymatic properties of β-xylosidases and the molecular mechanisms controlling their gene expression in A. oryzae. Therefore, we purified a major β-xylosidase (XylA) from an industrial shoyu koji mold strain, A. oryzae KBN616, and characterized its enzymatic properties. The β-xylosidase gene (xylA) was cloned and sequenced. In order to assess the role of β-xylosidase in the formation of the dark brown color during soy sauce brewing, the sense or antisense xylA gene was expressed in A. oryzae under control of the promoter of the A. oryzae TEF1 gene (10). Expression of the antisense gene reduced the β-xylosidase level significantly.
MATERIALS AND METHODS
General procedures.
Recombinant DNA techniques, double-strand DNA sequencing, A. oryzae cultivation, and genomic DNA isolation were carried out by standard procedures (8, 17). Total RNA was extracted from mycelia grown for 3 days in xylan-peptone (XP) medium (2% birchwood xylan, 1% polypeptone, 0.5% KH2PO4, 0.5% KCl, 0.1% NaNO3, 0.05% MgSO4 · 7H2O) by the method of Cathala et al. (3). Reverse transcription-PCR was performed with total RNA by using the Access RT-PCR system (Promega, Madison, Wis.).
Purification of the A. oryzae β-xylosidase.
A. oryzae KBN616 was grown in XP medium for 10 days. After removal of mycelia through filtration, 250 ml of the culture filtrate was diluted 10 times with 10 mM potassium phosphate buffer, pH 6.0. Protein was adsorbed to a STREAMLINE DEAE (Pharmacia Biotech, Uppsala, Sweden) column (2.6 by 12.0 cm) equilibrated with the same buffer and eluted by pulse elution with the same buffer containing 1.0 M NaCl. The β-xylosidase-containing fractions were diluted 10 times with the same buffer and loaded on an HR16/20 Fast Flow Q-Sepharose anion-exchange column (Pharmacia Biotech) followed by elution with a linear gradient of 0 to 0.4 M NaCl. Fractions containing β-xylosidase activity were pooled, dialyzed against 10 mM potassium phosphate buffer, pH 6.0, and rechromatographed on an HR16/20 Fast Flow Q-Sepharose anion-exchange column under the same conditions. The active pool was then dialyzed against 10 mM potassium phosphate buffer, pH 6.8, and loaded on a Gigapite (Seikagakukogyo, Tokyo, Japan) hydroxyapatite column (1.0 by 7.0 cm) equilibrated in the same buffer. Elution was performed with a linear gradient of 10 to 300 mM potassium phosphate buffer, pH 6.8. Finally, β-xylosidase was purified by using a HiLoad 26/60 Superdex 200-pg gel column (Pharmacia Biotech).
Enzyme assay.
β-Xylosidase activity was assayed as described by Ooi et al. (15). α-Galactosidase, α-l-arabinofuranosidase, α-l-arabinopyranosidase, β-l-arabinopyranosidase, β-galactosidase, β-glucosidase, and β-mannosidase activities were determined as described by Margolles-Clark et al. (13). Xylanase activity was assayed as described by Bailey (1).
N-terminal and internal amino acid sequencing.
The purified enzyme was blotted onto a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Richmond, Calif.), and its N-terminal amino acid sequence was determined with an Applied Biosystems 477A-120A sequencer according to the manufacturer’s instructions (Applied Biosystems, Foster City, Calif.). The purified enzyme was digested with V8 protease, and the resulting peptides were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Three major peptides, designated P1, P2, and P3, were subjected to amino acid sequencing as described above.
Amplification of β-xylosidase genomic DNA sequences by PCR.
A genomic DNA fragment encoding a portion of the β-xylosidase gene was amplified by PCR with chromosomal DNA of A. oryzae KBN616 as a template. Two oligonucleotide primers were synthesized based on the amino acid sequences: a sense primer [5′-GC(T/C)GA(T/C)CT(T/C)AT(T/C)AT(T/C)TT(T/C)GC(T/C)GG-3′] corresponding to peptide P2 (ADLIIFAG) and an antisense primer [5′-GT (A/G)TA(A/G)AA(A/G)AG(A/G)CC(A/G)TG(A/G)CC(A/G)AA-3′] corre-sponding to peptide P3 (FGHGLFYT). The amplified 419-bp fragment designated BXF1 was cloned on pUC118 and sequenced.
For amplification of the 5′ and 3′ regions of BXF1, cassette ligation-mediated PCR (CLM-PCR) was performed with an LA PCR in vitro cloning kit (Takara Shuzo, Kyoto, Japan) as described by Isegawa et al. (6). Four specific primers, I-1 (5′-CAGCCGGATACTGCGTCGTAACTAGCCGAG-3′), I-2 (5′-TAGGTCCGCGAGCTTGGTTATTAGGGAGAG-3′), II-1 (5′-CTCGGCTAGTTACGACGCAGTATCCGGCTG-3′), and II-2 (5′-CTCTCCCTAATAACCAAGCTCGCGGACCTA-3′), were synthesized on the basis of BXF1. The 5′ region was amplified from a Sau3AI cassette-ligated genomic DNA with two sets of primers (C1/I-1 in the primary amplification and C2/I-2 in the second round of PCR). The 3′ region was also amplified, in a similar manner, with two sets of primers (C1/II-1 in the primary amplification and C2/II-2 in the second round of PCR). The amplified fragments, a 2.5-kb fragment for the 5′ region and a 1.5-kb fragment for the 3′ region, were cloned and sequenced.
Expression of the A. oryzae xylA gene under control of the A. oryzae TEF1 promoter.
To express the xylA gene under control of the A. oryzae TEF1-α gene (TEF1) promoter (10), the expression plasmid vector pTFBX200 was constructed as follows. An EcoT22I cleavage site was introduced just before the ATG codon of the xylA gene by PCR in order to ligate the TEF1 promoter fragment precisely next to the xylA coding region. The 2.9-kb EcoT22I xylA DNA fragment was cloned into the EcoT22I site on pTF100 (10) to create plasmid pTFBX100, which contained the xylA gene under control of the TEF1 promoter. The 3.7-kb PstI-XbaI fusion gene fragment excised from pTFBX100 was cloned into the PstI and XbaI sites on pND300 containing the A. oryzae niaD gene on pUC119 (9) to create plasmid pTFBX200. Then, pTFBX200 was introduced to the niaD-deficient A. oryzae strain KBN616-39 by the method described previously (9).
Expression of the partial antisense A. oryzae xylA gene under control of the A. oryzae TEF1 promoter.
To express the partial antisense xylA gene by use of the A. oryzae TEF1 promoter, a plasmid vector, pASBX200, was constructed as follows. A 192-bp KpnI fragment from xylA at nucleotides −51 to 141 was cloned in the antisense orientation into the KpnI site on pTF100 to create plasmid pASBX100, which expressed the antisense RNA for the xylA gene under control of the A. oryzae TEF1 promoter. The 1.0-kb PstI-EcoRI fusion fragment excised from pASBX100 was treated with Klenow fragment and cloned into the SmaI site on pND300 to create plasmid pASBX200. Then, pASBX200 was introduced to the niaD-deficient A. oryzae strain KBN616-39 as described above.
Nucleotide sequence accession number.
The sequence of the xylA gene has been deposited in the DDBJ, EMBL, and GenBank nucleotide sequence databases under accession no. AB013851.
RESULTS AND DISCUSSION
Purification and characterization of the A. oryzae β-xylosidase.
A. oryzae produced three β-xylosidases with different molecular masses, and a major enzyme was purified 65.6-fold with recovery of 11% of the initial activity from culture supernatants of A. oryzae KBN616, as described in Materials and Methods. The purified enzyme showed a single protein band with an apparent molecular mass of 110 kDa on analysis by SDS-PAGE (Fig. 1). The molecular mass of 110 kDa is on the same order of magnitude as reported before for β-xylosidases of other Aspergillus spp. (90 to 122 kDa [11, 12, 15, 18]).
FIG. 1.
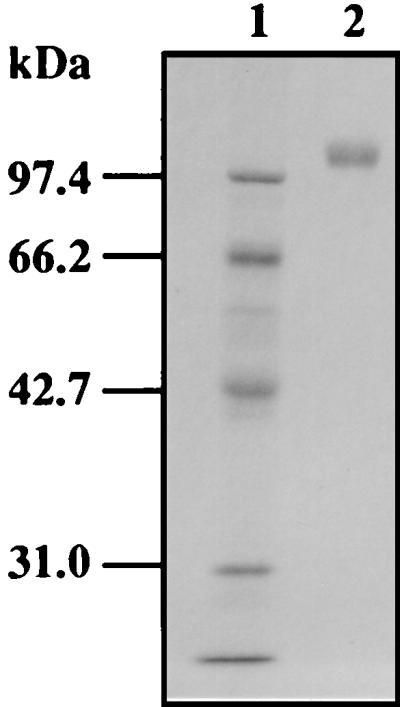
SDS-PAGE of purified β-xylosidase XylA. XylA was purified as described in Materials and Methods, subjected to SDS-PAGE on a 10% gel, and stained with Coomassie brilliant blue. Lane 1, molecular size markers (rabbit muscle phoshorylase b, 97.4 kDa; bovine serum albumin, 66.2 kDa; hen egg white ovalbumin, 42.7 kDa; bovine carbonic anhydrase, 31.0 kDa); lane 2, purified XylA.
The enzymatic features of β-xylosidase—pH and temperature optima and pH and thermal stabilities—were determined with the purified protein. The pH optimum was determined by incubation for 10 min at 40°C in 50 mM sodium acetate buffers of various pHs (3.0 to 7.0). The temperature optimum was determined by incubation at various temperatures (35 to 65°C) in 50 mM sodium acetate buffer of the optimal pH. The enzyme has a pH optimum of 4.0 and temperature optimum of 60°C. The pH optimum is similar to that of the Aspergillus awamori β-xylosidase (12). The thermal and pH stabilities were determined by incubation of the enzyme at various temperatures (35 to 65°C) for 10 min and at various pHs (3.0 to 7.0) for 20 h at 30°C, respectively. The enzyme was stable in the wide pH range of 3.0 to 7.0 and at temperatures of up to 45°C; it was inactivated gradually above 45°C. The pH stability was similar to that of the Aspergillus aculeatus β-xylosidase (15).
The hydrolytic properties of β-xylosidase toward several p-nitrophenyl substrates were determined. Besides xylosidase activity, the enzyme exhibited clearly α-l-arabinofuranosidase and β-l-arabinopyranosidase activities (Table 1). Substrate ambiguity of β-xylosidase has been reported for β-xylosidases of fungal origins such as A. niger and T. reesei (13).
TABLE 1.
Specific activities of purified XylA toward p-nitrophenyl substratesa
| p-Nitrophenyl substrateb | Sp act (U/mg) |
|---|---|
| βXylNp | 76.0 |
| αGalNp | 0.2 |
| αArafNp | 8.8 |
| αArapNp | 21.2 |
| βArapNp | 0.2 |
| βGalNp | 0.0 |
| βGluNp | 0.8 |
| βManNp | 0.0 |
Activities were measured as described in Materials and Methods.
βXylNp, p-nitrophenyl-β-d-xylopyranoside; αGalNp, p-nitrophenyl-α-galactopyranoside; αArafNp, p-nitrophenyl-α-l-arabinofuranoside; αArapNp, p-nitrophenyl-α-l-arabinopyranoside; βArapNp, p-nitrophenyl-β-l-arabinopyranoside; βGalNp, p-nitrophenyl-β-d-galactopyranoside; βGluNp, p-nitrophenyl-β-d-glucopyranoside; βManNp, p-nitrophenyl-β-d-mannopyranoside.
Amino acid sequencing of β-xylosidase and amplification of a partial β-xylosidase gene.
N-terminal amino acid sequencing of the purified enzyme failed, indicating that the N terminus might be blocked. V8 protease digestion of the purified enzyme, followed by SDS-PAGE, yielded three major peptides: P1, P2, and P3. Their N-terminal amino acid sequences were SFHDQFVSRQDL, ADLIIFAGGIDNTLETEAQD, and FGHGLFYT, respectively. Upon comparison of these sequences to those of other β-xylosidases, the amino acid sequences of the P1, P2, and P3 peptides showed high homologies to those of A. niger XlnD and T. reesei BxlI. Therefore, two oligonucleotide mixtures were designed based on the N-terminal amino acid sequence from residues 1 to 8 of the P2 peptide and the P3 peptide amino acid sequence. The amplified 419-bp fragment, designated BXF1, was sequenced and found to contain an open reading frame encoding 139 amino acids bearing high homology to the sequences of A. niger XlnD and T. reesei BxlI.
Characterization of the A. oryzae xylA gene.
For amplification of the 5′ and 3′ regions of BXF1, four specific primers, termed I-1, I-2, II-1, and II-2, were synthesized on the basis of the nucleotide sequence of BXF1. By CLM-PCR with a Sau3AI cassette-ligated genomic DNA as the template, a 2.5-kb fragment for the 5′ region, designated BXF2, and a 1.5-kb fragment for the 3′ region, designated BXF3, were amplified as described in Materials and Methods. BXF2 has the nucleotide sequence of the 5′ region of BXF1 and the ATG start codon of the xylA gene. BXF3 has the nucleotide sequence of the 3′ region of BXF1 and the TAG stop codon of the xylA gene. Based on the sequences of BXF1, BXF2, and BXF3, the entire nucleotide sequence of the xylA gene was determined (Fig. 2).
FIG. 2.

Restriction map of the xylA gene. The sequencing strategy for the xylA gene is represented below the restriction map by arrows. Solid and hatched boxes indicate the coding regions of the xylA gene and the BXF1 fragment amplified by PCR, respectively.
The coding region consists of 2,397 bp and contains no introns by sequence comparisons of cDNA and genomic DNA. The coding sequence comprises 798 amino acids, which should include amino acids of a signal peptide since β-xylosidase was extracellularly produced by A. oryzae. The region of the N-terminal 20 amino acid residues was found to be highly hydrophobic, and the N terminus of the purified enzyme was blocked, as described above. Based on the properties of signal peptide cleavage sites proposed by von Heijne (20), the secretory precursor could be processed at a specific cleavage site between Gly-20 and Gln-21. The mature protein would thus be 778 amino acids long, with a calculated molecular mass of 84,657 Da. Fourteen potential N-glycosylation sites were found, and some of them appeared to be glycosylated, since the molecular mass of the purified enzyme was found to be 110 kDa, as judged by SDS-PAGE (Fig. 1). The amino acid sequences determined chemically for the internal peptides P1, P2, and P3, generated by V8 protease digestion, were found in residues 354 to 365, 500 to 519, and 637 to 644 of the deduced amino acid sequence, respectively (Fig. 3).
FIG. 3.

Nucleotide and deduced amino acid sequences of the xylA gene. Numbers on the right refer to the nucleotide sequence (negative numbers refer to nucleotides upstream from the xylA ATG) and the amino acid sequence. The TATA and CCAAT sequences are double underlined, and CreA consensus binding sites are underlined. The GGCTAAA sequence is boxed. An asterisk (∗) marks the translation stop codon. A putative termination sequence [TAG…TA(T)GT…TTT] is underlined. The amino acids determined by sequencing of the P1, P2, and P3 peptides are also underlined, in the derived amino acid sequence. #, potential N-glycosylation site.
In the 5′ noncoding region of the xylA gene, a potential TATA box at −74 and a CCAAT sequence at −433 were found. Three CreA (a negatively acting regulatory protein mediating carbon catabolite repression in A. nidulans) consensus binding sites were present at −201, −247, and −787 and were suggested to participate in repression of the gene in response to glucose. The GGCTAAA sequence, which has been shown to be the binding site for XlnR, a transcriptional activator of the xylanolytic system in A. niger (19), was found at −456. In common with some other fungal genes, the typical polyadenylation signal, AATAAA, was not present within the 3′ noncoding region. However, the sequence defined by Zaret and Sherman (21) to be involved in transcription termination in Saccharomyces cerevisiae, TAG…TA(T)GT…TTT, was present at nucleotides 2395 to 2415 and 2517 to 2685 of the xylA gene.
Comparison of the amino acid sequence of A. oryzae XylA with those of other β-xylosidases.
Many of the β-xylosidases known so far have been classified by hydrophobic cluster analysis into three distinct groups: families 39, 43, and 52 (5). Although XylA shows no amino acid similarity to these families of β-xylosidases, it exhibits significant similarity to A. nidulans XlnD (70%), A. niger XlnD (64%), and T. reesei BxlI (63%), which have high sequence similarity to family 3 β-glucosidases. Several highly conserved sequences found among XylA, A. nidulans XlnD, A. niger XlnD, and T. reesei BxlI might be involved in the catalytic reaction, binding of the substrate, or both. One putative active-site Asp residue which might take part in the catalytic activity, as determined for the Aspergillus wentii β-glucosidase A3 (2), was also conserved in XylA (Asp-310).
Overexpression of the A. oryzae β-xylosidase gene.
In order to obtain high-level expression of the xylA gene in A. oryzae, A. oryzae KBN616-39 (niaD) was transformed with pTFBX200, which contained the xylA gene under control of the A. oryzae TEF1 promoter. Of 100 transformants screened on plates containing 2% glucose and 0.5 mM 4-MUX (4-methylumbelliferyl-β-d-xyloside), 95 transformants showed β-xylosidase activity after 24 h of growth, whereas the reference strain showed no β-xylosidase activity in the presence of glucose because of carbon catabolite repression. Two highly XylA-producing strains selected by the plate assay produced extracellularly about 18 U of β-xylosidase per ml (about 250 mg/liter) when grown in glucose-peptone medium for 10 days. A 110-kDa XylA was detected as a prominent band on an SDS-polyacrylamide gel stained with Coomassie brilliant blue (Fig. 4, lanes 4 and 5). In XP medium, these transformants produced 23 U of the enzyme per ml, about 13 times more than the reference strain. Southern blot analysis revealed that multiple copies of plasmid pTFBX200 were integrated into chromosomal DNAs of these transformants (Fig. 5).
FIG. 4.

SDS-PAGE of XylA overexpressed by A. oryzae transformants. One milliliter of culture filtrate was precipitated with 10% trichloroacetic acid, solubilized in a small volume, and subjected to SDS-PAGE on a 10% gel. Molecular size markers (lane 1) are as described in the legend to Fig. 1. Lane 2, purified XylA; lane 3, culture supernatant of A. oryzae KBN616-39; lanes 4 and 5, culture supernatant of A. oryzae carrying pTFBX200.
FIG. 5.

Southern blot analysis of chromosomal DNAs isolated from A. oryzae KBN616 and two representative transformants. DNA (about 5 μg) was digested with PstI and processed for Southern blot hybridization. Hybridization was done with the AlkPhos Direct system (Amersham, Little Chalfont, United Kingdom) with a 300-bp fragment (+1 to +300, with the translation start site at +1) as a probe. Lane 1, A. oryzae KBN616; lanes 2 and 3, transformants.
Antisense inhibition of A. oryzae β-xylosidase gene expression.
In order to inhibit the expression of the xylA gene in A. oryzae, the partial-length antisense xylA gene was expressed under control of the A. oryzae TEF1 promoter by introducing pASBX200 into A. oryzae KBN616-39. Of 84 transformants screened on plates containing 2% xylan and 0.5 mM 4-MUX, two transformants, designated ASBX13 and ASBX33, showed barely detectable levels of β-xylosidase activity after 24 h of growth. Quantitative assays revealed low β-xylosidase activities in ASBX13 and ASBX33 (99 and 115 mU/ml), indicating that 75 to 80% inhibition was achieved (Table 2). Southern blot analysis of ASBX13 and ASBX33 revealed that the antisense construct was integrated into their genomes (Fig. 6). Since the A. oryzae TEF1 gene promoter is a strong constitutive promoter, as described previously (10), a high level of the partial-length antisense RNA could be accumulated in the transformed cells, leading to a reduction in sense xylA mRNA levels, probably as a consequence of degradation of sense-antisense complexes (7, 22). However, xylanase activity in these transformants remained unaltered (Table 2). This indicates that the antisense gene specifically inhibited xylA gene expression.
TABLE 2.
Antisense inhibition of β-xylosidase expression in A. oryzae carrying the antisense construct pASBX200a
| Strain | β-Xylosidase activity (mU/ml) | Xylanase activity (U/ml) |
|---|---|---|
| KBN616-39 | 463 (100) | 15.4 (100) |
| ASBX13 | 99 (21) | 15.1 (98) |
| ASBX33 | 115 (25) | 15.7 (101) |
Activities were measured in culture filtrate after growth on 2% birchwood xylan at 30°C for 5 days. Values in parentheses indicate relative enzyme activities, in percentages.
FIG. 6.

Southern blot analysis of chromosomal DNAs isolated from A. oryzae KBN616 and two transformants, ASBX13 and ASBX33. DNA (about 5 μg) was digested with EcoRI and PstI and processed for Southern blot hybridization as described in the legend to Fig. 5. Lane 1, A. oryzae KBN616; lane 2, transformant ASBX13; lane 3, transformant ASBX33.
We are now trying to use the high- and low-level XylA-producing transformants in soy sauce brewing and to evaluate the effects of expression of the xylA gene on brown color formation.
ACKNOWLEDGMENT
We thank S. Karita of the Center for Molecular Biology and Genetics, Mie University, for determination of the amino acid sequences of the three peptides.
REFERENCES
- 1.Bailey M J. A note on the use of dinitrosalicylic acid for determining the products of enzymatic reactions. Appl Microbiol Biotechnol. 1988;29:494–496. [Google Scholar]
- 2.Bause E, Legler G. Isolation and structure of a tryptic glycopeptide from the active site of β-glucosidase A3 from Aspergillus wentii. Biochim Biophys Acta. 1980;626:459–465. doi: 10.1016/0005-2795(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 3.Cathala G, Savouret J F, Mendez B, West B L, Karin M, Martial J A, Baxter J D. A method for isolation of intact, translationally active ribonucleic acid. DNA. 1983;2:329–335. doi: 10.1089/dna.1983.2.329. [DOI] [PubMed] [Google Scholar]
- 4.Hashiba H. Oxidative browning of soy sauce (II) J Jpn Soy Sauce Res Inst. 1981;11:189–199. . (In Japanese.) [Google Scholar]
- 5.Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1993;193:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isegawa Y, Sheng J, Sokawa Y, Yamanishi K, Nakagomi O, Ueda S. Selective amplification of cDNA sequence from total RNA by cassette-ligation mediated polymerase chain reaction (PCR): application to sequencing 6.5 kb genome segment of hantavirus strain B-1. Mol Cell Probes. 1992;6:467–475. doi: 10.1016/0890-8508(92)90043-w. [DOI] [PubMed] [Google Scholar]
- 7.Judelson H S, Dudler R, Pieterse C M J, Unkles S E, Michelmore R W. Expression and antisense inhibition of transgenes in Phytophthora infestans is modulated by choice of promoter and position effects. Gene. 1993;133:63–69. doi: 10.1016/0378-1119(93)90225-r. [DOI] [PubMed] [Google Scholar]
- 8.Kitamoto N, Go M, Shibayama T, Kimura T, Kito Y, Ohmiya K, Tsukagoshi N. Molecular cloning, purification and characterization of two endo-1,4-β-glucanases from Aspergillus oryzae KBN616. Appl Microbiol Biotechnol. 1996;46:538–544. doi: 10.1007/s002530050857. [DOI] [PubMed] [Google Scholar]
- 9.Kitamoto N, Kimura T, Kito Y, Ohmiya K, Tsukagoshi N. The nitrate reductase gene from a shoyu koji mold, Aspergillus oryzae KBN616. Biosci Biotechnol Biochem. 1995;59:1795–1797. doi: 10.1271/bbb.59.1795. [DOI] [PubMed] [Google Scholar]
- 10.Kitamoto N, Matsui J, Kawai Y, Kato A, Yoshino S, Ohmiya K, Tsukagoshi N. Utilization of the TEF1-α gene (TEF1) promoter for expression of polygalacturonase genes, pgaA and pgaB, in Aspergillus oryzae. Appl Microbiol Biotechnol. 1998;50:85–92. doi: 10.1007/s002530051260. [DOI] [PubMed] [Google Scholar]
- 11.Kitpreechavanich V, Hayashi M, Nagai S. Purification and characterization of extracellular β-xylosidase and β-glucosidase from Aspergillus fumigatus. Agric Biol Chem. 1986;50:1703–1711. [Google Scholar]
- 12.Kurakake M, Osada S, Komaki T. Transglycosylation of β-xylosidase from Aspergillus awamori K4. Biosci Biotechnol Biochem. 1997;61:2010–2014. doi: 10.1271/bbb.61.2010. [DOI] [PubMed] [Google Scholar]
- 13.Margolles-Clark E, Tenkanen M, Nakari-Setälä T, Penttilä M. Cloning of genes encoding α-l-arabinofuranosidase and β-xylosidase from Trichoderma reesei by expression in Saccharomyces cerevisiae. Appl Environ Microbiol. 1996;62:3840–3846. doi: 10.1128/aem.62.10.3840-3846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakata, Y., M. Morishita, T. Hashimoto, and Y. Tsuji. Unpublished data.
- 15.Ooi T, Fujimoto H, Wang S-L, Takizawa T, Hidaka H, Ogura S, Murao S, Arai M. Purification and some properties of Aspergillus aculeatus β-xylosidase. Oyo Toshitsu Kagaku. 1995;42:45–48. [Google Scholar]
- 16.Pérez-González J A, van Peij N N M E, Bezoen A, MacCabe A P, Ramón D, de Graaff L H. Molecular cloning and transcriptional regulation of the Aspergillus nidulans xlnD gene encoding a β-xylosidase. Appl Environ Microbiol. 1998;64:1412–1419. doi: 10.1128/aem.64.4.1412-1419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 18.van Peij N N M E, Brinkmann J, Vrsanská M, Visser J, de Graaff L H. β-Xylosidase activity, encoded by xlnD, is essential for complete hydrolysis of xylan by Aspergillus niger but not for induction of the xylanolytic enzyme spectrum. Eur J Biochem. 1997;245:164–173. doi: 10.1111/j.1432-1033.1997.00164.x. [DOI] [PubMed] [Google Scholar]
- 19.van Peij N N M E, Visser J, de Graaff L H. Isolation and analysis of xlnR, encoding a transcriptional activator coordinating xylanolytic expression in Aspergillus niger. Mol Microbiol. 1998;27:131–142. doi: 10.1046/j.1365-2958.1998.00666.x. [DOI] [PubMed] [Google Scholar]
- 20.von Heijne G. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaret K S, Sherman F. DNA sequence required for efficient transcription termination in yeast. Cell. 1982;28:563–573. doi: 10.1016/0092-8674(82)90211-2. [DOI] [PubMed] [Google Scholar]
- 22.Zheng X F, Kobayashi Y, Takeuchi M. Construction of a low-serine-type-carboxypeptidase-producing mutant of Aspergillus oryzae by the expression of antisense RNA and its use as a host for heterologous protein secretion. Appl Microbiol Biotechnol. 1998;49:39–44. doi: 10.1007/s002530051134. [DOI] [PubMed] [Google Scholar]