

Abstract
Calcium is an important second messenger regulating a bioenergetic response to the workloads triggered by neuronal activation. In embryonic mouse cortical neurons using glucose as only fuel, activation by NMDA elicits a strong workload (ATP demand)-dependent on Na+ and Ca2+ entry, and stimulates glucose uptake, glycolysis, pyruvate and lactate production, and oxidative phosphorylation (OXPHOS) in a Ca2+-dependent way. We find that Ca2+ upregulation of glycolysis, pyruvate levels, and respiration, but not glucose uptake, all depend on Aralar/AGC1/Slc25a12, the mitochondrial aspartate-glutamate carrier, component of the malate-aspartate shuttle (MAS). MAS activation increases glycolysis, pyruvate production, and respiration, a process inhibited in the presence of BAPTA-AM, suggesting that the Ca2+ binding motifs in Aralar may be involved in the activation. Mitochondrial calcium uniporter (MCU) silencing had no effect, indicating that none of these processes required MCU-dependent mitochondrial Ca2+ uptake. The neuronal respiratory response to carbachol was also dependent on Aralar, but not on MCU. We find that mouse cortical neurons are endowed with a constitutive ER-to-mitochondria Ca2+ flow maintaining basal cell bioenergetics in which ryanodine receptors, RyR2, rather than InsP3R, are responsible for Ca2+ release, and in which MCU does not participate. The results reveal that, in neurons using glucose, MCU does not participate in OXPHOS regulation under basal or stimulated conditions, while Aralar-MAS appears as the major Ca2+-dependent pathway tuning simultaneously glycolysis and OXPHOS to neuronal activation.
SIGNIFICANCE STATEMENT Neuronal activation increases cell workload to restore ion gradients altered by activation. Ca2+ is involved in matching increased workload with ATP production, but the mechanisms are still unknown. We find that glycolysis, pyruvate production, and neuronal respiration are stimulated on neuronal activation in a Ca2+-dependent way, independently of effects of Ca2+ as workload inducer. Mitochondrial calcium uniporter (MCU) does not play a relevant role in Ca2+ stimulated pyruvate production and oxygen consumption as both are unchanged in MCU silenced neurons. However, Ca2+ stimulation is blunt in the absence of Aralar, a Ca2+-binding mitochondrial carrier component of Malate-Aspartate Shuttle (MAS). The results suggest that Ca2+-regulated Aralar-MAS activation upregulates glycolysis and pyruvate production, which fuels mitochondrial respiration, through regulation of cytosolic NAD+/NADH ratio.
Keywords: Aralar/AGC1/Slc25a12, calcium regulation, glycolysis, malate aspartate shuttle, mitochondrial calcium uniporter, neuronal metabolism
Introduction
The mammalian brain is a prominent consumer of glucose and oxygen in the resting state. In the adult brain, neurons have the highest energy demand (Attwell and Laughlin, 2001). The restoration of the ion movements generated by postsynaptic currents and the action potentials are major neuronal ATP consumers. Neurons respond to these workloads (increases in ATP demand) through a stimulation of ATP production, which is provided through increases in glycolysis and oxidative phosphorylation (OXPHOS) (Hall et al., 2012; Mergenthaler et al., 2013; Connolly et al., 2014; Rangaraju et al., 2014; Ashrafi and Ryan, 2017; Dienel, 2019). The mitochondrial response to workloads is immediate (Llorente-Folch et al., 2013, 2015; Connolly et al., 2014); and in the case of small workloads, it takes place without a detectable decrease in cytosolic ATP levels (Baeza-Lehnert et al., 2019), in dendrites (Gerkau et al., 2019), and the presynapse (Rangaraju et al., 2014).
It has been known for a long time that the rate of respiration depends not only on ATP demand but also on Ca2+ (Territo et al., 2000; Denton, 2009; Glancy and Balaban, 2012; Llorente-Folch et al., 2015), providing a feedforward system to adjust OXPHOS to energy demand. In neurons, this regulatory role of Ca2+ was shown to be independent of its effect in ATP demand, at least under certain stimulation conditions (Llorente-Folch et al., 2013). Ca2+ may upregulate respiration thanks to its entry in mitochondria through the mitochondrial calcium uniporter complex (MCUC) (de Stefani et al., 2016), activating pyruvate dehydrogenase, phosphatase 1 (Fecher et al., 2019), NAD+-linked isocitrate dehydrogenase, and α-KGDH (Denton, 2009; Armstrong et al., 2014). However, the finding that a global MCU deletion in outbred CD1 mice did not cause gross dysfunction was at odds with the functions attributed to MCU in the control of bioenergetics (Pan et al., 2013; Wang et al., 2020, Kosmach et al., 2021).
Another way in which Ca2+ may operate to increase coupled respiration is through an increase in the activity of mitochondrial metabolite carriers regulated by Ca2+, with Ca2+-binding motifs facing the intermembrane space (Satrústegui et al., 2007). For example, the aspartate-glutamate exchanger Aralar/AGC1/Slc25a12, a component of the malate aspartate shuttle (MAS), is regulated by Ca2+, with half-maximal effects at ∼300 nm (Pardo et al., 2006; Contreras et al., 2007). In brain mitochondria (Gellerich et al., 2013) and neurons using glucose, Ca2+ activation of MAS drives pyruvate away from lactate and into mitochondria (Llorente-Folch et al., 2013).
In addition to its role during the response to workloads, Ca2+ has shown to be important in the regulation of basal respiration in many cell types (Cárdenas et al., 2010; Mallilankaraman et al., 2012; Filadi et al., 2018; Tomar et al., 2019). A constitutive transfer of Ca2+ from ER-to-mitochondria was required to maintain basal respiration and ATP/AMP levels. It involved inositol-3P receptors (IP3Rs) on the ER, the MCU in mitochondria, and additional proteins involved in contacts between ER and mitochondria (Rossi et al., 2019). It is unknown whether this pathway is also functional in neurons, and its specific components in ER and mitochondria, including MCU.
In this work, we have evaluated the contribution of these two systems (MCU and Aralar-MAS) as Ca2+-regulation mechanisms to activate mitochondrial respiration in response to workloads and in the regulation of basal respiration in neurons using glucose as fuel. A second fundamental question in this work is the influence of these Ca2+ regulation mechanisms in glycolysis itself.
We report that while Mcu-KD blocks the increases in mitochondrial Ca2+ elicited by carbachol (Cch) and NMDA, it is dispensable for regulating neuronal respiration using glucose in every condition studied: basal respiration, Cch-stimulated respiration, and NMDA-stimulated respiration. We find that neurons respond to NMDA by a rapid increase in cytosolic pyruvate and lactate production, which is blocked in the absence of Ca2+. Remarkably, the Ca2+-dependent increase in pyruvate, but not lactate, is blunted in Aralar-KO neurons, revealing a dependence of Aralar-MAS in the provision of cytosolic pyruvate for mitochondrial respiration. Moreover, we show that NMDA triggers a Ca2+-dependent increase in glycolysis, which also depends on Aralar. Thus, in neurons using glucose, neuronal activity is met with a Ca2+-dependent mechanism, which boosts simultaneously glycolysis and respiration by activating Aralar-MAS, as metabolic link between glycolysis and OXPHOS. It is possible that the Ca2+-binding motifs of Aralar N-terminus are involved in this activation, but additional/alternative calcium sensors cannot be excluded as the disruption of Aralar removes both domains of the protein, calcium sensor, and mitochondrial carrier.
Materials and Methods
Animals
Mice with targeted disruption of Aralar/AGC1/Slc25a12 (Aralar-KO) were described previously (Jalil et al., 2005). Aralar-KO mice on a mixed SVJ129xC57BL/6 background were used. WT and Aralar-KO E16 embryos of either sex for preparing neuronal cultures were obtained by breeding of heterozygous Aralar-KO animals.
All animal procedures were performed in compliance with animal protocols approved by institutional ethical committee (Center of Molecular Biology Severo Ochoa) and Autónoma University (CEEA-CBMSO-23/159), and in accordance with Spanish regulations (BOE 67/8509-12, 1988) and European regulations (EU directive 86/609, EU decree 2001-486), reporting followed the ARRIVE Guidelines. All efforts were made to minimize the number of animals used and their suffering.
Drugs
Cells were treated as indicated with cytochalasin B (CytB), 2,4-dinitrophenol (DNP), antimycin A (A), oligomycin (Olig), rotenone (R), caffeine, ionomycin, Cch, TTX, dantrolene, NMDA, MK-801, and CNQX all purchased from Sigma-Aldrich, also with AR-C155858 and (S)-3,5-dihydroxyphenylglycine hydrate (DHPG) from Tocris Bioscience, and ryanodine from Calbiochem. Sodium lactate, sodium pyruvate, and sodium iodoacetate (IAA) were from Sigma-Aldrich and used in the conditions indicated. 3-Desmethyl Xestospongin B (3-dmXeB) was produced by chemical synthesis and dissolved in methanol. For in vitro experiments, compounds dissolved in DMSO or ethanol, both solvents were never higher than 0.001%. Corresponding vehicle solutions were used in control experiments. Doses and concentrations of the different drugs were chosen on the basis of previous published data or preliminary experiments.
Primary neuronal culture
Neuronal cultures were obtained from E15-E16 mouse embryos, as described previously (Pardo et al., 2006; Ramos et al., 2011). Cerebral cortices were enzymatically dissociated in PBS containing 1% BSA, 0.4 mg/ml papain (Roche), and 6 mm glucose. Dissociated cells were plated on poly-L-lysine and laminin-coated glass coverslips in medium containing 20% horse serum. After 3 h, medium was completely replaced by serum-free Neurobasal medium supplemented with 2% B27, 1% Glutamax (all from Invitrogen), and 100 mg/ml penicillin-streptomycin. Neurons were maintained in these conditions until DIV 8-10 for experimental analysis. Neurons represented >80% of the total cell population (Pardo et al., 2006).
Constructs
For Mcu knockdown (KD) using shRNA, we used recombinant adeno-associated virus (rAAV) vectors with a U6 promoter driving shRNA expression that coexpress also mCherry under control of CaMKII promoter to identify infected neurons, pAAV.shRNA.CaMKI.mCherry, kindly provided by Hilmar Bading and Giles E. Hardingham (Qiu et al., 2013). The Mcu hairpin and the scrambled one, used as control, have been described previously (Qiu et al., 2013). To generate shRNA RyR2 constructs, two target sequences, shRyR2-1 and shRyR2-2, used previously (More et al., 2018 and Mu et al., 2014, respectively) were selected. RyR2-sh1, targeting sequences 5′-AGGAAGCAATGGTGGACAAGTTGGCTGAA-3′ and RyR2-sh2, 5′-CGTCCACATACTATTACTC-3′ were annealed and subcloned into the pAAV.shRNA.CaMKI.mCherry vector using standard molecular cloning techniques as described previously (Rueda et al., 2015). All constructs were verified by sequencing.
rAAV obtention
Viral particles (chimeric rAAVs containing the capsid proteins of both AAV1 and AAV2 at equal ratios) were produced for Scr, RyR2-KD, and Mcu-KD. rAAVs were purified and titrated, as described previously (McClure et al., 2011). Briefly, human HEK293 cells were cotransfected with the pAAV.shRNA.CaMKI.mCherry plasmid, the serotype-specific AAV helper plasmids, pH 21 and pRV-1, encoding rep and cap genes of AAV1 and AAV2, respectively, and the adenovirus helper plasmid (pFΔ6) by standard CaPO4 transfection. Cells were harvested 48 h after transfection, treated with benzonase (Sigma-Aldrich) to reduce the viscosity, and cell debris were cleared by centrifugation for 15 min. The rAAV particles were purified using HiTrap heparin affinity columns (Sigma-Aldrich) and concentrated using Amicon Ultra centrifugal filters (EMD Millipore), and solutions were pooled and sterile filtered. Primary cortical neurons were infected with 107 rAAV particles/μl at DIV 3. Infection efficiencies were determined at DIV 9 by analyzing the fluorescence of mCherry, they ranged from 80% to 90% of the viable neurons. rAAV infection decreased viable neurons. In infected neurons, oxygen consumption and calcium and fluorescence measurements were performed at DIV 9-10.
Transfections
Primary cortical neurons, seeded on chambered cover glass 4-well plates (Lab-Tek) at 1.5 × 105 cells/well density, were transfected at DIV 7-8 using calcium phosphate protocol with 1 µg of plasmids coding fluorescent sensors for the measurement of cytosolic glucose [FLII12Pglu-700μδ6; Addgene #28002 (Takanaga et al., 2008)], pyruvate [Pyronic; Addgene #51308 (San Martín et al., 2014)], lactate [Laconic; Addgene #44238 (San Martín et al., 2013)], cytosolic ATP [cyt GO-ATeam1; (Rueda et al., 2015)], ATP/ADP cytosolic ratio [Perceval-HR; Addgene #49083 (Tantama et al., 2013)], and for mitochondrial Ca2+ determinations [4mtD3cpv; Addgene #58184 (Palmer et al., 2006)]. For the transfection of rAAV-transduced cells, neurons were seeded at higher density (2 × 105 cells/well), and transfections with fluorescent sensors were performed using Lipofectamine 2000 (Invitrogen) following supplied instructions. FRET measurements were usually performed 24 or 48 h after transfection.
Mitochondrial respiration measurements
Cellular oxygen consumption rate (OCR) was measured using Seahorse XF24 Extracellular Flux Analyzer (Agilent) as previously described (Llorente-Folch et al., 2013; Rueda et al., 2015; Juaristi et al., 2019a). At DIV 9-10, cells were equilibrated with bicarbonate-free low-buffered DMEM (without pyruvate, L-lactate, glucose, glutamine, or calcium) supplemented with 2.5 mm glucose and 2 mm CaCl2 for 1 h before XF assay. When required,1 μm BAPTA-AM (Thermo Fisher Scientific) was added to the incubation medium. OCR was determined under basal conditions and after the sequential injections of medium or NMDA at the concentrations indicated and 6 μm oligomycin, 0.5 mm DNP, and 1 μm rotenone/1 μm antimycin. Values of basal, ATP-linked, nonmitochondrial and mitochondrial uncoupled respiration were determined (Qian and Van Houten, 2010; Brand and Nicholls, 2011). Protein from each well was extracted with 0.1% NP-40 PBS solution and quantified with BCA protein assay kit (Thermo Fisher Scientific), and data were expressed as a function of protein concentration. Nonmitochondrial OCR was subtracted from OCR values to obtain basal values. OCR/ extracellular acidification rate (ECAR) ratio values were calculated under basal conditions of OCR and ECAR for each well.
Cytosolic calcium, sodium, and pH imaging
Primary cortical neurons were seeded on glass coverslips on 24-well plates at 2.0 × 105, or 1.5 × 105 for Cch stimulations, cells/well. At DIV 8-9, neurons were incubated during 30 min at 37°C in HEPES-buffered control salted solution (HCSS; 137 mm NaCl, 1.25 mm MgSO4, 10 mm HEPES, 3 mm KCl, 2 mm NaHCO3, and 1% BSA, pH 7.4) supplemented with 5 μm fura-2 AM (Thermo Fisher Scientific), 50 μm pluronic acid F-127 (Thermo Fisher Scientific), and 2.5 mm glucose without calcium (plus 100 μm EGTA) for Ca2+ imaging, and with 0.12 μm BCECF-AM (Thermo Fisher Scientific), 50 μm pluronic acid F-127, 2.5 mm glucose, and 2 mm CaCl2 for pH imaging. Single-cell measurements of cytosolic Na+ were performed as previously described previously (Llorente-Folch et al., 2013; Juaristi et al., 2019a) using sodium-binding benzofuran isophthalate-AM (SBFI-AM, Thermo Fisher Scientific) as Na+ indicator. After loading, neurons were washed in HCSS with 2.5 mm glucose and 2 mm CaCl2 during 20 min at 37°C, and then imaging was performed at 37°C as described previously (Juaristi et al., 2019a) with a Neofluar 40×/0.75 NA objective in an Axiovert 75 M microscope (Carl Zeiss). Acute additions were made as a bolus at the times indicated. ROIs were selected covering single-cell soma. Image acquisition was performed with Aquacosmos 2.5 software (Hamamatsu).
FRET experiments
For pyronic, laconic, FLII12Pglu-700μδ6 experiments, cells were pre-incubated 1 h in HCSS (+Ca2+) at 37°C; for Ca2+-free assays, cells were washed once in HCSS (-Ca2+) prior imaging. Experiments were performed in HCSS supplemented with 2.5 mm glucose and 2 mm CaCl2 or 100 μm EGTA and the additions indicated. In pyronic and laconic experiments, 2 mm pyruvate or lactate was added at the end of the experiment to verify that the probe was not saturated. Neurons were excited at 430 nm (CFP), and emission was collected with a dual pass dichroic filter CFP-YFP (440-500 nm and 510-610 nm). Exposure time was 200 ms. Images were collected every 5 s using a filter wheel λ 10-3, Sutter Instruments (Chroma) and recorded by a Hamamatsu C9100-02 camera mounted on an Axiovert 200 M inverted microscope equipped with a 40×/1.3 oil Plan-Neofluar objective.
ROIs containing only somas were selected on each cell and analyzed using MetaMorph (Universal Imaging) and ImageJ (National Institutes of Health, https://fiji.sc/) software. Background was subtracted from YFP and CFP images; and subsequently, the ratio between CFP and YFP emissions was calculated. Rates of fluorescence increase in response to NMDA stimulation were calculated as the lineal increase in fluorescence ratio (ΔF/min) during the first 30 s after addition. Maximal fluorescence ratio increase (ΔF max) and fluorescence ratio at the end of the recording (ΔF endpoint) after NMDA stimulation were calculated. Mitochondrial calcium was analyzed on neurons expressing the 4mt-D3cpv calcium sensor as described previously (Juaristi et al., 2019a). Variations in cytosolic ATP levels and ATP/ADP ratio using cytosolic FRET probes cyt GO-ATeam1 and Perceval-HR, respectively, were determined as in Rueda et al. (2015).
Adenine nucleotide quantification
Adenine nucleotide total levels were determined by HPLC (de Korte et al., 1995). Neuronal cultures were treated with 660 mm HClO4 and 10 mm theophylline immediately frozen and kept them overnight at −80°C. Neurons were scrapped and homogenized on ice, then centrifuged at 16,000 × g 15 min 4°C. The supernatants were neutralized using 2.8 m K2CO3, kept on ice 10 min, and then at −80°C for at least 2 h to allow precipitation of HClO4. Extracts were centrifuged, and 50 µl of the supernatants was injected for adenine nucleotide determination. To determine the identity of each peak and quantify the amount of the species of interest, standard curves were constructed by plotting peak heights versus known concentrations of ATP, ADP, and AMP.
qRT-PCR
RNA from primary cortical neurons was isolated using Trizol reagent (TRIzol; Sigma-Aldrich) following the manufacturer's instructions. Retrotranscription reactions were performed using the iScript cDNA Synthesis kit (Bio-Rad PN170-8891) following the manufacturer's instructions. Thermal conditions consisted of the following steps: 5 min × 25°C, 20 min × 46°C, and 1 min × 95°C. cDNA was amplified with Fast SYBR MasterMix probe (Thermo Fisher Scientific) in the ABI Prism 7900HT sequence detection system (Thermo Fisher Scientific) at the Genomics and Massive Sequencing Facility. The primers used for amplifying the target genes were as follows: mouse RyR2 (5′-GAATTCATCATGGATACTCTACC-3′; 5′-GTCATGCACATTATCTTCTGCAT-3′). Mouse β-actin (5′-CTAAGGCCAACCGTGAAAAG-3′; 5′-ACCAGAGGCATACAGGGACA-3′) and mouse GAPDH (5′-TGCGACTTCAACAGCAACTC-3′; 5′-GGATAGGGCCTCTCTTGCTC-3′) were used as housekeeping genes. ValidPrime expression (Tataa Biocenter, A106S10) was checked in all the samples, and necessary corrections were done as described by Laurell et al. (2012). The relative expression of genes was calculated using the 2-ΔΔCt Livak comparative method (Schmittgen and Livak, 2008).
Western blotting and antibodies
To examine protein levels, total protein extracts were prepared in RIPA buffer (125 mm NaCl, 25 mm Tris-Cl, pH 7.4, 1 mm EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and Complete EDTA-free protease inhibitor, Roche); 22.5 µg (for RyR2 analysis) or 40 µg (for MCU and AMPK analysis) of total proteins was loaded. Proteins were separated by SDS-PAGE electrophoresis, and transferred onto PVDF or nitrocellulose membranes (for RyR2 or other analysis, respectively) by wet electrophoretic transfer. Blots were blocked 1 h at room temperature with 5% nonfat dry milk in TBS-Tween (0.5 m Tris, 1.5 m NaCl, 0.01% Tween) solution and incubated at 4°C with primary antibodies. For phospho-antibodies, 5% BSA in TBS-Tween was used as blocking solution. Secondary antibodies were incubated 1 h at room temperature. The following antibodies were used: anti-MCU (1:1000, Sigma-Aldrich), anti-RYR2 (1:500, Thermo Fisher Scientific), anti-GAPDH (1:1000, Merk), anti-AMPKα (1:1000, Cell Signaling Technology), anti-p-AMPK T172 (1:1000, Cell Signaling Technology), and anti-β-F1ATPase (1:5000, kindly provided by Jose M. Cuezva). Secondary HRP-conjugated antibodies were purchased from Bio-Rad and used at 1:5000 dilution. The signal was detected by chemiluminescence with detection solution (75 mm Tris, pH 8.8, 2.5 mm luminol, 0.40 mm p-coumaric acid, 0.01% H2O2). The quantification of the bands was performed with ImageJ (National Institutes of Health) software. We used 661W cells (expressing RyR2) as positive control.
Quantification and statistical analysis
Statistical details of experiments can be found in the figure legends. All results are representative of at least three independent experiments unless otherwise specified and are presented as mean ± SEM. Significance was calculated by ANOVA (one-way or two-way), post hoc Bonferroni test, or t test. All statistical tests were plotted either with Sigma Plot 11.0 software or GraphPad Prism version 6 or 7 software.
Results
Control of basal neuronal respiration by ER calcium: the role of ER Ca2+ stores and MCU
In 2010, a constitutive transfer of Ca2+ from ER to mitochondria was shown to be required to maintain basal respiration and ATP/AMP levels, and to block macroautophagy via an AMPK-dependent pathway (Cárdenas et al., 2010; Mallilankaraman et al., 2012).
We have examined whether this constitutive Ca2+ transfer pathway plays a similar role in neurons, and the role of MCU in this process. As neurons express two types of Ca2+ release channels in the ER, IP3R, and ryanodine receptors (RyRs) (Zalk et al., 2007; Galeotti et al., 2008; Egorova and Bezprozvanny, 2018), we have analyzed the role of both. To study the role of ER efflux via IP3R, we used a membrane-permeable selective blocker of all types of IP3R, analog of Xestospongin B (XeB), a macrocyclic bis-1-oxaquinolizidine alkaloid originally extracted from the marine sponge Xestospongia exigua (Gafni et al., 1997; Jaimovich et al., 2005). This analog 3-dmXeB has been produced by chemical synthesis and exerts similar actions as XeB on IP3R (Podunavac et al., 2021).
Figure 1A shows that 60 min preincubation of cortical neurons with 5 μm 3-dmXeB substantially reduces the Ca2+ transient induced by 200 μm DHPG, an agonist of Group I metabotropic glutamate receptors (Niswender and Conn, 2010) consistent with the ability of XeB to block IP3R (Jaimovich et al., 2005; Cárdenas et al., 2010); 10 μm 3-dmXeB was also inhibitory but induced cellular damage and was not used any further. 3-dmXeB did not modify the basal respiration rate or OCR/ECAR ratio in the same cortical neurons, as determined with a Seahorse extracellular flux analyzer (Fig. 1B–D), indicating that Ca2+ efflux from the ER through IP3R does not play a relevant role in the control of basal respiration.
Figure 1.
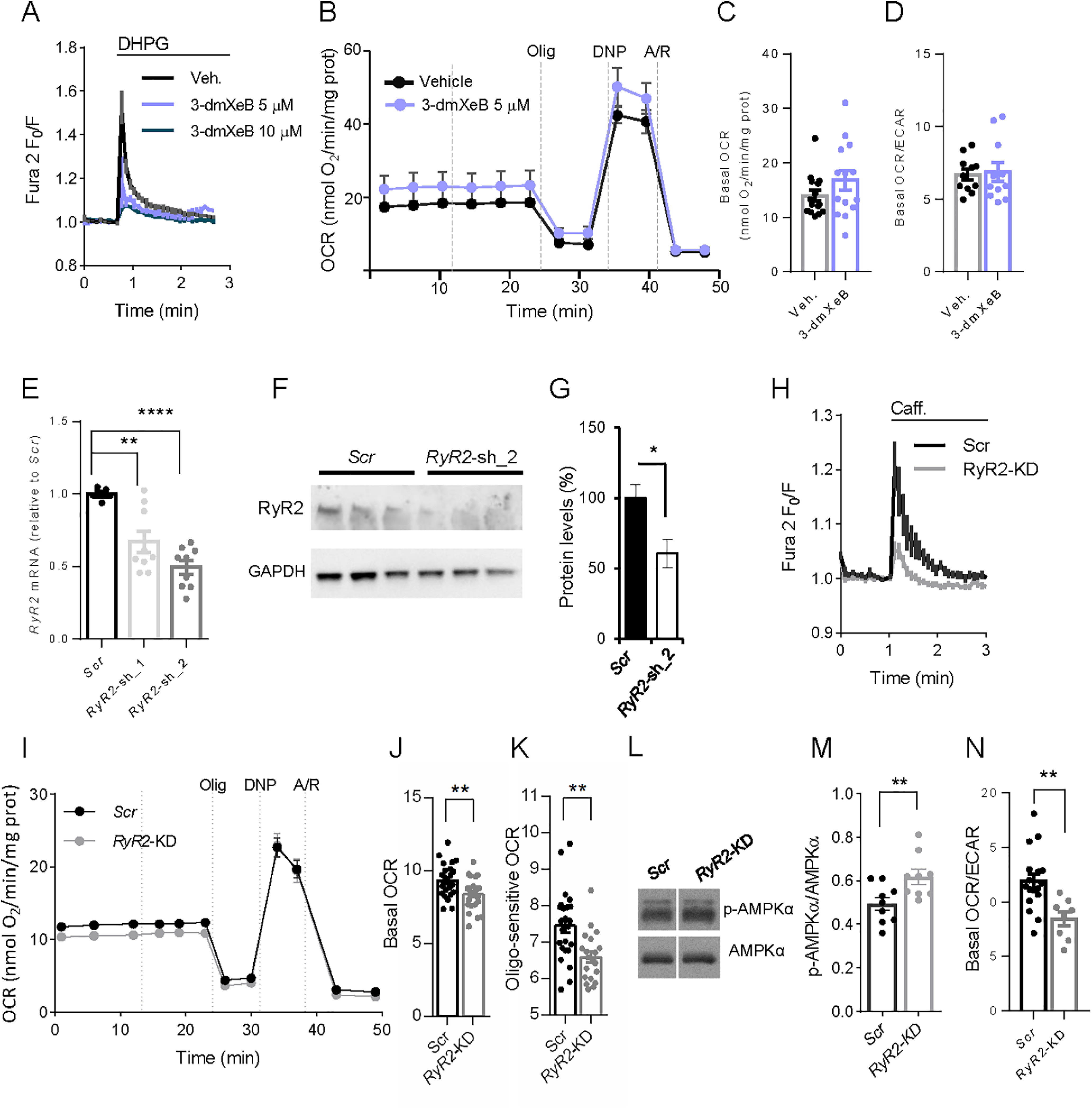
RyRs control neuronal mitochondrial respiration. A, 5 and 10 μm 3-dmXeB substantially reduces the Ca2+ transients induced by 200 μm DHPG, an agonist of Group I metabotropic glutamate receptors in cortical neurons. Neuronal cultures were preincubated with 3-dmXeB 60 min before the experiment was started. B, C, Incubation with 5 μm 3-dmXeB does not change basal respiration in cortical neurons. 3-dmXeB was added to the culture 60 min before the experiment was started. Mitochondrial function was determined through sequential addition of 6 μm oligomycin (Olig), 0.5 mm DNP, and 1 μm/1 μm antimycin A/rotenone (A/R) at the indicated time points. Data are mean ± SEM; n = 7. C, Basal respiration corrected for nonmitochondrial respiration. Dots represent individual data. Error bars indicate mean ± SEM; n = 14 or 15. D, Basal OCR/ECAR ratio values. Data are mean ± SEM; n = 8-17. E, qPCR analysis of the cDNA obtained from neuronal cultures transduced with rAAV containing two different RyR2-directed (RyR2-KD1 or RyR2-KD) or Scrambled (Scr) shRNA using specific oligos; values were normalized to RyR2 mRNA in Scr neurons. Data are mean ± SEM, 5-9 wells (dots) from two independent experiments. One-way ANOVA: **p ≤ 0.01, ****p ≤ 0.0001, post hoc Bonferroni test. F, Western blot analysis of RyR levels in neurons transduced with rAAV containing RyR2-directed (RyR2-KD) or Scrambled shRNA (Scr). GAPDH was used as loading control. G, Quantitative analysis from Western blot of RyR2/GAPDH. Data are mean ± SEM; n = 3. *p = 0.049 (two-tailed t test). H, RyR2 silencing decreases caffeine-induced [Ca2+]i signal, which is blocked by ryanodine or dantrolene (see Extended Data Fig. 1-1A,B). I–K, RyR2 silencing decreases basal and oligomycin-sensitive OCR levels in neurons. I, Data are mean ± SEM; n = 7 or 8. J, Data are mean ± SEM; n = 14-16. **p = 0.0037. K, Data are mean ± SEM; n = 20-24. **p = 0.0017 (t test). L, Representative Western blot analysis of p-AMPK and AMPK levels. M, Quantitative analysis from Western blot of p-AMPK/AMPK levels. Dots represent individual data. Error bars indicate mean ± SEM; n = 9. **p = 0.0069 (t test). N, RyR2 silencing decreases basal OCR/ECAR ratio values. Data are mean ± SEM; n = 8-17, from 3 independent experiments and does not change ATP, ADP, or AMP levels (see Extended Data Fig. 1-1C–G). **p = 0.0041 (two-tailed t test).
Caffeine-discharge ER-Calcium and Adenine nucleotide content in RyR2-KD neurons. A-B: 15 mM Caffeine (Caff.) discharges ER calcium by activating RyR in cortical neurons at 9 DIV. Inhibition of the response to caffeine by preincubation with 50 µM ryanodine (Ryan.) (A) or 10 µM dantrolene (Dandr.) (B) during 15 or 30 min. C-G: Adenine nucleotide content, measured by HPLC in neurons transduced with rAAV containing RyR2-directed (RyR-KD) or Scrambled shRNA (Scr). Mean ± SEM (bars), 5 independent experiments (dots). fura-2 AM Download Figure 1-1, TIF file (299.2KB, tif) .
RyRs, but not MCU, are involved in the maintenance of basal respiration in cortical neurons
In neurons, ER Ca2+ mobilization via activation of RyR is involved in calcium-induced calcium release of CICR (Shmigol et al., 1995; Solovyova et al., 2002; Patel et al., 2009). As observed (see Extended Data Fig. 1-1A), 15 mm caffeine can discharge the ER Ca2+ store, as shown previously (Solovyova et al., 2002), resulting in an increase in cytosolic Ca2+. Preincubation with RyR inhibitors, such as ryanodine (50 μm; Extended Data Fig. 1-1A,B) (Solovyova et al., 2002) or dantrolene (10 μm; Extended Data Fig. 1-1A,B) (Liu et al., 2009; Oulès et al., 2012) was able to block the release of Ca2+ caused by caffeine, confirming that these compounds block RyR in cortical neurons.
To study the role of RyR in providing a constitutive Ca2+ flow regulating respiration in cortical neurons, we have silenced RyR2, the major RyR isoform in brain cortex and hippocampus (Zalk et al., 2007; Galeotti et al., 2008), using selected sequences used previously (Mu et al., 2014; More et al., 2018). Neurons were infected with two rAAV-containing vectors encoding RyR2-directed small hairpin RNA (shRNA-1 and shRNA2), or nontargeted control sequence (Scrambled, Scr). Silencing was verified by qPCR (Fig. 1E) and Western blot analysis, indicating a decrease in RyR2 protein levels of ∼50% with shRNA2. (Fig. 1F,G). RyR2 silencing with shRNA2, RyR2-KD, resulted in a marked decrease in caffeine-induced Ca2+ release (Fig. 1H).
A significant decrease in basal respiration was observed in the RyR2-silenced neurons (RyR2-KD, Fig. 1I,J), which affected oligomycin-sensitive respiration (Fig. 1K) supporting a constitutive flow of ER Ca2+ to mitochondria involving, at least partially, RyR2 (Fig. 1I).
Failure to maintain ER to mitochondria Ca2+ flux causes an increase in autophagy triggered by activation of AMPK (Cárdenas et al., 2010; Mallilankaraman et al., 2012). Indeed, AMPK phosphorylation status increased in RyR2-KD neurons (Fig. 1L,M). This was accompanied by an increase in ECAR and a decrease in the OCR/ECAR ratio (Fig. 1N), suggesting a higher glycolytic flux in response to AMPK activation. Indeed, when evaluating the adenine nucleotide levels present in these cells, RyR2 silencing did not lead to any significant changes (Extended Data Fig. 1-1C–G) in agreement with the robust energetic stability in neurons (Baeza-Lehnert et al., 2019).
We have next studied the involvement of the MCUC in this constitutive pathway. To this end, we silenced Mcu, the Ca2+ transporter component of MCUC (de Stefani et al., 2016). Neurons were transduced with rAAV containing Mcu-directed shRNA, or nontargeted control sequence (Scrambled, Scr) (Qiu et al., 2013). Analysis by Western blot shows a 65 ± 10% decrease in MCU protein level (Fig. 2A,B). Figure 2C shows that the basal FRET ratio of the mitochondrial Ca2+ probe 4mtD3cpv in Mcu-KD neurons is very similar to that of Scr neurons. Indeed, unchanged resting Ca2+-mit levels were observed for other tissue-specific MCU deletions, as muscle- (Gherardi et al., 2019), cardiomyocyte- (Kwong et al., 2015), or neuronal-specific Mcu-KD (Qiu et al., 2013; Ashrafi et al., 2020). Silencing of Mcu had no effect on basal respiration with glucose as the main substrate compared with neurons transduced with adenovirus carrying Scrambled sequences (Fig. 2D,E). Similarly, neuron-specific KD of MCU did not cause changes in basal respiration in cortical neurons (Nichols et al., 2018). As MCU is clearly able to take up Ca2+ in mitochondria (see below), we conclude that it is not required as part of the ER to mitochondria Ca2+ flow maintaining basal respiration in neurons using glucose.
Figure 2.
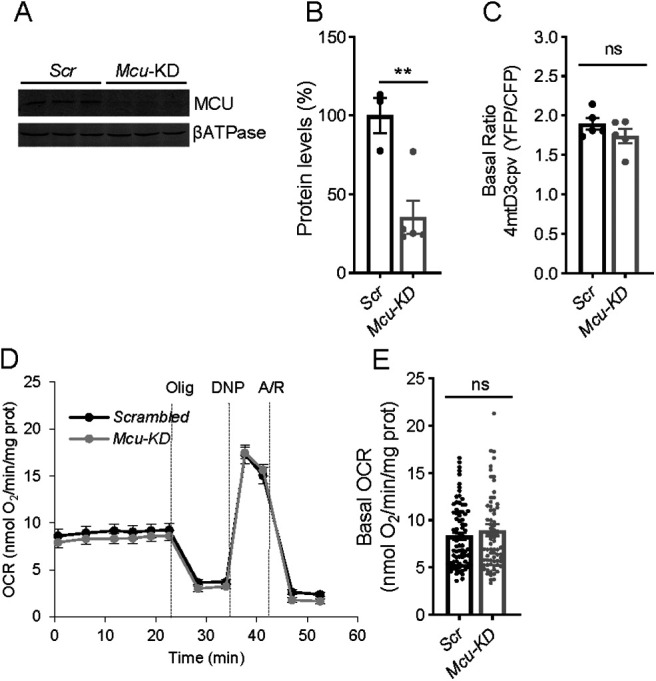
Basal mitochondrial Ca2+ and respiration do not change in Mcu-KD cortical neurons. A, Representative Western blot analysis of MCU levels in neurons transduced with rAAV-containing MCU-directed (Mcu-KD) or Scrambled shRNA (Scr). βATPase was used as loading control. B, Quantitative analysis from Western blot of MCU/ βATPase. Dots represent individual data. Error bars indicate mean ± SEM; n = 3-5. **p = 0.0072 (t test). C, Basal ratio of 4mtD3cpv probe reflecting basal mitochondrial Ca2+ levels in Scr and Mcu-KD neurons. Dots represent individual data. Error bars indicate mean ± SEM; n = 19-21 neurons per condition from 5 independent platings. p = 0.2335 (t test). D, E, Basal respiration in Scrambled and Mcu-KD neurons expressed as OCR (nmol O2/min/mg protein). Dots represent individual data. Error bars indicate mean ± SEM; n = 26 per condition from 5 independent platings. p = 0.4193 (t test).
Therefore, our results show that a pathway for Ca2+ flow from ER to mitochondria that controls basal respiration exists in cortical neurons, but it is not dependent on IP3R on the ER or Ca2+ entry in mitochondria along MCU.
The neuronal respiratory response to activation of acetylcholine receptors by Cch depends on Aralar-MAS but not MCU
Having shown that ER to mitochondria calcium flow via IP3R does not control neuronal respiration under basal conditions, we have investigated its role in neuronal stimulation by Cch (Delmas et al., 2002, 2004). The spontaneous Ca2+ activity of the neuronal network is synaptically driven (Bacci et al., 1999; Opitz et al., 2002; Young et al., 2005), as it is blocked by TTX and ionotropic glutamate receptor inhibitors (Extended Data Fig. 3-1A,B), and is enhanced by Cch with an increase in the amplitude of the Ca2+ transients (Fig. 3A,B). The Cch-induced workload was fully Ca2+-dependent with no changes in Na+-cyt (Fig. 3C) and accordingly, caused a stimulation of neuronal respiration only in the presence of extracellular Ca2+ (Fig. 3D,E).
Figure 3.
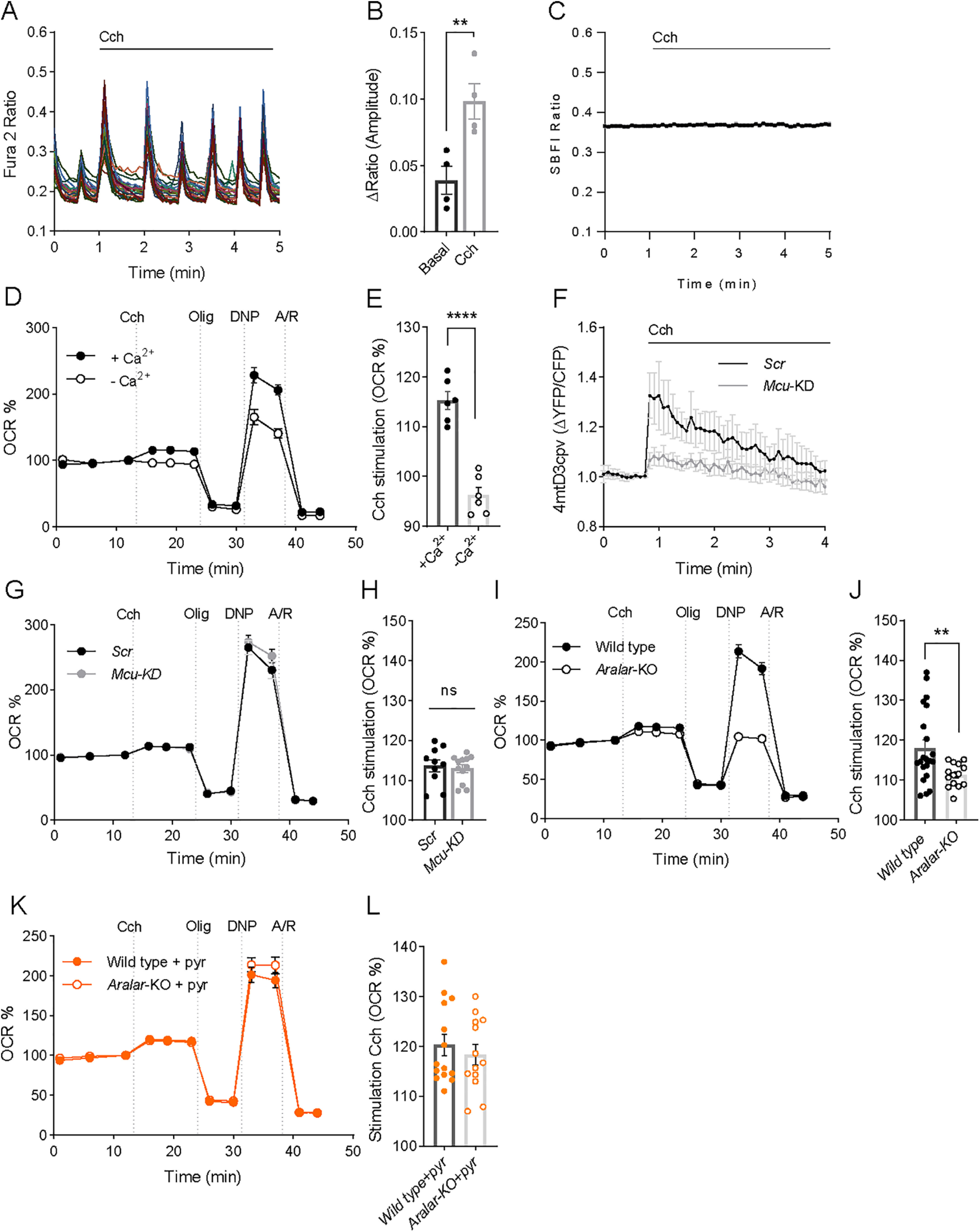
Cch stimulation of mitochondrial respiration depends on calcium and the ARALAR-MAS pathway. A, fura-2 AM [Ca2+]i signals in neurons in HCSS medium containing 2 mm CaCl2, on addition of 250 μm Cch where indicated. The figure shows a representative experiment; each trace corresponds to a single neuron from the same recording field. Spontaneous Ca2+ oscillations are blocked by inhibitors of ionotropic glutamate receptors (Extended Data Fig. 3-1A,B). B, Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM comparing basal spontaneous Ca2+ oscillations to Cch-enhanced Ca2+ oscillations. Data were obtained from 4 independent experiments. Means were compared using one-tailed t test (p = 0.0064). C, SBFI [Na+]i signals in neurons on addition of 250 μm Cch where indicated. Data are mean ± SEM: 3 experiments, n = 96. D, Stimulation by 250 μm Cch of OCR expressed as percentage of basal OCR in the absence of Ca2+ (–Ca2+) or presence of 2 mm Ca2+ (+Ca2+) in the incubation medium. E, Quantification of percentage of respiratory stimulation (OCR % 3 min after Cch addition). Data were obtained from 6 independent experiments (n = 6). Means were compared using one-tailed t test (p ≤ 0.0001). The effect of Cch on Ca2+ oscillations was the same in MCU-KD or in Aralar-KO neurons (Extended Data Fig. 3-1C–H). F, 4mtD3cpv mitochondrial Ca2+ signals in Scrambled or Mcu-KD neurons on addition of 250 μm Cch where indicated. Data are normalized to the initial values and are expressed as mean ± SEM. Data were obtained from 3 independent experiments (n = 8-11 cells). G, Cch (250 μm) stimulation of OCR expressed as percentage of basal OCR in Scrambled and Mcu-KD neurons. H, Percentage of respiratory stimulation (OCR % 3 min after Cch addition) in neurons infected with Scr and Mcu rAAVs. Data were obtained from 3 independent experiments (n = 10-12). I, Cch (250 μm) stimulation of OCR expressed as percentage of basal OCR in WT and Aralar-KO neurons. J, Percentage of respiratory stimulation (OCR % 3 min after Cch addition) in WT and Aralar-KO neurons. Data were obtained from 6 independent experiments (n = 14-21). Means were compared using one-tailed t test (p = 0.0063). K, Cch (250 μm) stimulation of OCR in WT and Aralar-KO neurons in the presence of 2 mm pyruvate. L, Percentage of respiratory stimulation (OCR % 3 min after Cch addition). Data were obtained from 3 independent experiments (n = 11). Means were compared using one-tailed t test (p = 0.53).
Cch induced cytosolic Ca2+ signals. Effects of MCU-KD and Aralar-KO. A. Mouse cortical neurons plated at high density and cultured for 8-9 days showed spontaneous Ca2+ activity, consisting of repetitive Ca2+ transients occurring at the same time in all neurons. These Ca2+ transients were blocked by treatment with the AMPA/Kainate receptor antagonist CNQX (10 μm), the NMDAR antagonist MK-801 (10 μm), or the Na+ channel blocker TTX (1 μm) (results not shown). B. Cch addition in the presence of TTX, which blocks other responses except those due to Ca2+ mobilization by IP3R, resulted in a weak response. C-D: fura-2 AM [Ca2+]i signals in Scr (C) or Mcu-KD (D) neurons in HCSS medium containing 2 mM CaCl2 and 2.5 mM glucose upon addition of 250 μm Cch where indicated. Representative experiments, each trace corresponds to a single neuron from the same recording field. E: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM. Data were obtained from 3 independent experiments. F-G: fura-2 AM [Ca2+]i signals in WT (E) and Aralar-KO (G) neurons in HCSS medium containing 2 mM CaCl2 and 2.5 mM glucose upon addition of 250 μm Cch where indicated. Representative experiments, each trace corresponds to a single neuron from the same recording field. H: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM. Data were obtained from 5 independent experiments. Download Figure 3-1, TIF file (405.7KB, tif) .
To study the impact of MCU signaling on the Cch-induced increase in respiration, we silenced Mcu and studied its effect on Cch-stimulated respiration. Downregulation of MCU protein resulted in drastic block in mitochondrial Ca2+ signals, as Cch-driven mitochondrial Ca2+ rise was dramatically decreased in Mcu-silenced neurons (Fig. 3F), while the increase in cytosolic Ca2+ transients evoked by Cch were the same in both Scrambled (Scr) or Mcu-KD neurons. No differences were found in amplitude or number of peaks after Cch addition (Extended Data Fig. 3-1C–E).
As described, basal respiration rates were similar in Scrambled and Mcu silenced neurons (Fig. 2D,E). Strikingly, although Cch-induced increase in matrix Ca2+ did not occur under Mcu silencing conditions (Fig. 3F), the stimulation of mitochondrial respiration by Cch was not affected: 114 ± 2% in Scrambled and 113 ± 1% (p = 0.71, t test) in Mcu-silenced neurons over basal levels, respectively (Fig. 3G,H).
Having shown that MCU is not required for Cch stimulation of respiration, we investigated the role of Aralar. We first verified that cytosolic Ca2+ signals induced by Cch in cortical neurons derived from WT and Aralar-KO mice were the same (Cch enhanced Ca2+ oscillations to the same level; Extended Data Fig. 3-1F–H). Next, we studied Cch stimulation of respiration in the two genotypes finding that Cch stimulation of respiration is reduced by ∼40% in the absence of Aralar (WT neurons: 118 ± 1% and Aralar-KO neurons: 111 ± 1% increase over basal levels, p = 0.0063, t test; Fig. 3I,J). It is likely that the remaining stimulation of respiration is due exclusively to Ca2+-stimulated workload (i.e., Ca2+-induced ATP consumption), not to Ca2+ signaling. Although much smaller than that dependent on Na+ entry, Ca2+-stimulated workload plays also a role in neuronal energy consumption (Llorente-Folch et al., 2013; Díaz-García et al., 2021). The lack of Aralar also caused a pronounced decrease in DNP-stimulated respiration (Fig. 3I).
Activation by extramitochondrial Ca2+ of Aralar-MAS results in an increase in NADH production in neuronal mitochondria, which is blocked in Aralar-KO neurons, and the presence of exogenous pyruvate in Aralar-KO neurons overcomes this block (Pardo et al., 2006). It has been proposed that Ca2+ activation of Aralar functions as a “gas pedal” to increase pyruvate formation (Gellerich et al., 2009, 2012, 2013). Indeed, Cch-stimulated respiration in Aralar-KO neurons was fully restored in the presence of 2 mm pyruvate (Fig. 3K,L), and this also restored DNP-stimulated respiration (Fig. 3G). Therefore, we conclude that the lack of Aralar results in a limitation in substrate (pyruvate) supply to mitochondria, which prevents full Cch-induced stimulation of OCR.
Role of MCU and Aralar-MAS in Ca2+-dependent stimulation of neuronal respiration triggered by NMDA
Activation of NMDARs leads to Na+ and Ca2+ entry in cortical neurons (Rueda et al., 2015). In order to restore the electrochemical gradients, Na+/K+-ATPases, plasma membrane and ER Ca2+ ATPases, and Na+/Ca2+ exchangers are activated, leading to stimulated ATP use and to the upregulation of mitochondrial respiration to meet the energetic demands. In the absence of extracellular Ca2+, 100 μm NMDA evoked a smaller increase in respiration than in the presence of calcium (Rueda et al., 2015). As the responses to NMDA were much larger than those elicited by Cch, we have switched to NMDA as agonist to analyze in more detail the role played by Aralar and MCU and other metabolic pathways in the responses.
The Ca2+ dependence of the OCR response in cortical neurons is also observed at lower NMDA concentrations, 25 μm NMDA (Fig. 4A,B) larger responses in the presence than in the absence of Ca2+. The response to NMDA entails increased respiratory substrate utilization, which probably limits the maintenance of the OCR beyond the initial response (Fig. 4A). In addition, the maximal respiratory response was lower than in nonstimulated neurons (compare the responses to the uncoupler in Fig. 1B with those in Fig. 4A). Thus, after correction for nonmitochondrial respiration, uncoupler-stimulated respiration was 280.7 ± 9.6% or 83.2 ± 5.9% of basal respiration in unstimulated or NMDA stimulated neurons, respectively (p < 0.001), indicating a limitation in substrate supply to maintain the response to the uncoupler after stimulation by NMDA. The limitation of substrate supply after glutamate stimulation of neuronal OCR has been confirmed experimentally, by showing that the addition of other biofuels at the time of uncoupler addition restores maximal OCR to control values (Laird et al., 2013). Thus, in agreement with the larger NMDA-OCR stimulation in the presence than absence of Ca2+, maximal respiration was larger in Ca2+-free media, in which the drain of substrates during NMDA stimulation is lower (Fig. 4A).
Figure 4.
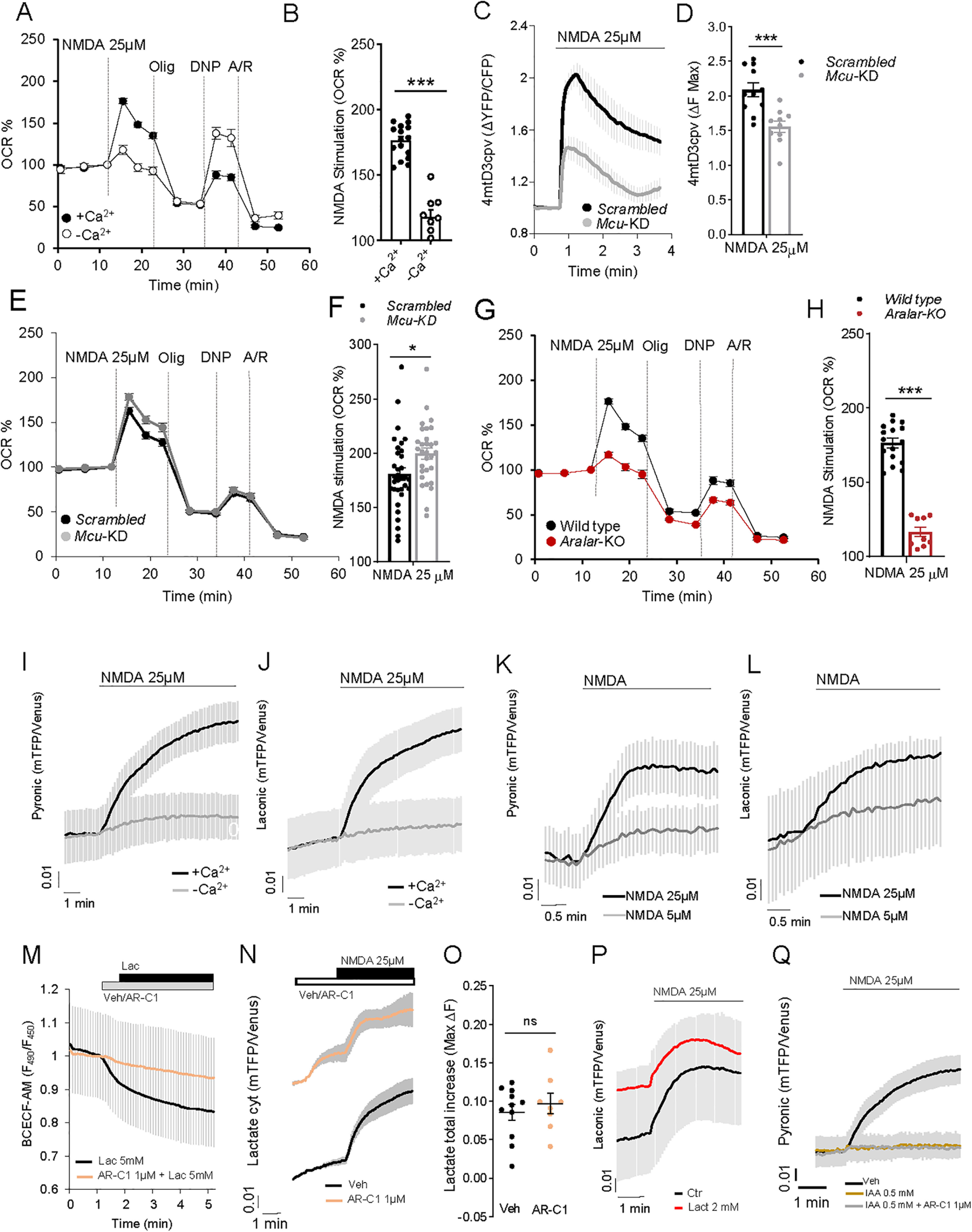
NMDA stimulation of OCR and cytosolic pyruvate and lactate formation. A, 25 μm NMDA-stimulated respiration in cortical neurons incubated in the presence of 2 mm Ca2+ (+Ca2+) or without calcium (–Ca2+; plus 100 μm EGTA), expressed as percentage of basal values (OCR %). B, NMDA-stimulated OCR (%). Data are mean ± SEM (bars); n = 9-16 (dots) from two different experiments. One-way ANOVA: ***p ≤ 0.001, post hoc Bonferroni test. C, 25 and 5 μm (Extended Data Fig. 4-1A,B) NMDA-induced changes in mitochondrial Ca2+ (Ca2+-mit) in Scrambled (Scr) and Mcu-silenced (Mcu-KD) neurons transfected with 4mtD3cpv probe. D, Quantification of maximum Ca2+-mit increment (ΔF max) after NMDA stimulation. Data are mean ± SEM (bars); n = 10 or 11 from 3 independent experiments. ***p = 0.0006 (t test). NMDA-induced cytosolic Ca2+ signals were the same in Scrambled and MCU-KD neurons (Extended Data Fig. 4-2A–D). E, 25 and 5 μm (Extended Data Fig. 4-1C,D) NMDA-stimulated respiration in Scrambled and Mcu-KD neurons, expressed as percentage of basal values (OCR %). F, Quantification of NMDA-stimulated OCR (%). Data are mean ± SEM (bars); n = 30-33, 5 independent experiments. *p = 0.0173 (t test). G, 25 and 5 μm (Extended Data Fig. 4-1E,F) NMDA-stimulated respiration in WT and Aralar-KO neurons, expressed as percentage of basal values (OCR %). H, Quantification of NMDA-stimulated OCR (%). Data are mean ± SEM (bars); n = 9-16 (dots) from 2 independent experiments. ****p < 0.0001 (t test). Cytosolic and mitochondrial Ca2+ signals were the same in WT and Aralar-KO neurons (Extended Data Fig. 4-2E–L). I, J, Effect of NMDA in the presence (+Ca2+) or absence (–Ca2+) of calcium on cytosolic pyruvate (Pyr) and lactate (Lac) in neurons transfected with pyronic (I) and laconic (J) probes. K, L, Dose-dependent effect of 5 and 25 μm NMDA induction of Pyr (K) and Lac (L) production. Data are mean ± SEM; n = 5-15 neurons per condition from 2-6 independent experiments. M, Intracellular pH variation determined in BCECF-AM-loaded neurons after 5 mm lactate (Lac) acute addition with or without 1 min pre-incubation with 1 μm MCT1/2 inhibitor AR-C155858 (AR-C1). Data are mean ± SEM; n = 6 per condition from 2 independent experiments. N, Lactate changes induced by 25 μm NMDA in neurons to which 1 μm AR-C1 or medium (Veh) was pre-added. O, Quantification of maximum fluorescence change (ΔF max) in Lac after Veh/1 μm AR-C1 and 25 μm NMDA. P, Lactate changes induced by 25 μm NMDA in neurons in the presence (Lact) or absence (Ctr) of extracellular 2 mm lactate. Q, Pyruvate changes induced by 25 μm NMDA in neurons pre-incubated 30 min with HCSS (Veh), 0.5 mm IAA, or IAA + 1 μm AR-C1 (IAA + AR-C1). Data are mean ± SEM; n = 8-20 per condition from 4-8 independent experiments. The 4mtD3cpv, pyronic, laconic, and BCECF-AM experiments were performed in HCSS, 2.5 mm glucose, and 2 mm Ca2+ or 100 μm EGTA (–Ca2+).
Changes in mitochondrial Ca2+ and OCR upon stimulation with 5 μm NMDA. A: Changes in mitochondrial Ca2+ (Ca2+-mit) in Scrambled (Scr) and Mcu-silenced (Mcu-KD) neurons transfected with 4mtD3cpv probe, after 5 μm NMDA stimulation in 2.5 mM glucose and 2 mM Ca2+ HCSS; B: Quantification of maximum Ca2+-mit increment (ΔF max) after NMDA stimulation. Data are mean ± SEM (bars), n = 8-9 (dots) from three independent experiments; T- test *p = 0.0365. C: 5 μm NMDA-stimulated respiration in Scrambled and Mcu-KD neurons maintained in 2.5 mM glucose and 2 mM Ca2+ DMEM, expressed as percentage of basal values (OCR %); D: Quantification of NMDA-stimulated OCR (%); mean ± SEM (bars), n = 14-15 (dots) from 5 independent experiments; T test *p = 0.0339. E: 5 μm NMDA-stimulated respiration in wild type and Aralar-KO neurons maintained in 2.5 mM glucose and 2 mM Ca2+ DMEM, expressed as percentage of basal values (OCR %); F: Quantification of NMDA-stimulated OCR (%); mean ± SEM (bars), n = 4-6 (dots) from 2 independent experiments; T test ***p = 0.0007. Download Figure 4-1, TIF file (311.5KB, tif) .
NMDA-induced Cytosolic and mitochondrial Ca2+ signals in Scr, Mcu-KD, Wild type and Aralar-KO neurons. A-D: Changes in cytosolic Ca2+ (Ca2+-cyt) after 5 (A, C) and 25 μm NMDA (B, D) stimulation in Scr (A, B) or Mcu-KD (C, D) neurons. E-I: Changes in cytosolic Ca2+ (Ca2+-cyt), after 5 (E, G) and 25 μm (F, H) NMDA in wild type (E, I) and Aralar-KO (G, H) Fura2-AM loaded neurons. Individual cell recordings (gray) and average (black) are shown. I: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM (bars) comparing 5 and 25 μm NMDA stimulus; two-way ANOVA, **p ≤ 0.01, ***p ≤ 0.001, post hoc Bonferroni test. Recordings from at least 50 cells/condition from 4-7 wells (dots) and two independent experiments. J-L: Changes in mitochondrial Ca2+ (Ca2+-mit), in wild type (J) and Aralar-KO neurons (K) transfected with 4mtD3cpv probe, after 5 and 25 μm NMDA stimulation; L: maximum Ca2+-mit increment (ΔF max) after NMDA stimulation. Mean ± SEM (bars), 5-10 cells (dots) from two independent cultures; two-way ANOVA, *p ≤ 0.05, post hoc Bonferroni test. All cells were maintained in 2.5 mM glucose and 2 mM Ca2+ HCSS. Download Figure 4-2, TIF file (351.2KB, tif) .
To study the contribution of MCU and Aralar-MAS as Ca2+ regulation mechanisms in NMDA stimulation of respiration, we started by silencing Mcu in cortical neurons, as described above. NMDA-stimulated increase in matrix Ca2+ was clearly smaller in Mcu-KD than Scrambled neurons both with 25 μm NMDA (Fig. 4C,D) and 5 μm NMDA (see Extended Data Fig. 4-1A,B). However, Mcu silencing did not block completely NMDA-stimulated Ca2+ entry in mitochondria as reported previously (Qiu et al., 2013) in agreement with the incomplete block of Ca2+ uptake in isolated brain mitochondria from global Mcu-KO mice (Hamilton et al., 2018; Szibor et al., 2020). This diminished Ca2+ transport into mitochondria was not translated into an increase in cytosolic Ca2+ in Mcu-silenced neurons; rather, 5 and 25 μm NMDA caused a similar increase in cytosolic Ca2+ in Mcu-silenced and Scrambled neurons (Extended Data Fig. 4-2A–D).
Figure 5.

NMDA-induced changes in cytosolic pyruvate and lactate, in WT and Aralar-KO primary neuronal cultures. A, D, Cortical neurons from WT and Aralar-KO mice transfected with pyronic (A) and laconic (D) probes were stimulated with 25 μm NMDA in 2.5 mm glucose HCSS, in the presence of 2 mm Ca2+ (+Ca2+) or 100 μm EGTA (–Ca2+). FRET changes reporting cytosolic pyruvate or lactate levels are shown. B, E, Quantification of maximum fluorescence change (ΔF max) in pyruvate (B) and Lac (E) after NMDA stimulation in the presence of Ca2+. C, F, Quantification of velocity of increase of pyruvate (C) and Lac (F) (ΔF/min) in the presence of Ca2+ as the increment of fluorescence ratio during the first 30 s after stimulation. n = 10-22 per condition from 5-8 independent experiments for all pyruvate and Lac assays; 5 mm pyruvate or Lac was added 3 min after NMDA as a control. Data are mean ± SEM. *p = 0.0133 (t test). In the absence of Ca2+, pyruvate and Lac output was the same in Aralar-KO and control neurons (A,D). MCU-KD or Scrambled neurons had similar increases in pyronic or laconic fluorescence on NMDA exposure (Extended Data Fig. 5-1).
Effects of NMDA stimulation on cytosolic pyruvate and lactate production in Mcu-silenced neurons. Cytosolic pyruvate (Pyr) (A, C) and lactate (Lac) (B, D) levels of Scr and Mcu-KD neurons transfected with Pyronic and Laconic probes, respectively, upon neuronal stimulation with 5 μm (A, B) or 25 μm NMDA (C, D). Neurons were incubated in 2.5 mM glucose and 2 mM Ca2+ HCSS. Data are mean ± SEM, n = 7-15 neurons per condition from 2 independent cultures. Download Figure 5-1, TIF file (293.6KB, tif) .
Remarkably, 5 μm (Extended Data Fig. 4-1C,D) and 25 μm (Fig. 4E,F) NMDA-stimulated OCR was not decreased by Mcu-silencing as expected if matrix Ca2+ played a role in stimulation of respiration; indeed, the stimulation was slightly larger compared with Scrambled neurons. The lack of inhibition of NMDA-stimulated respiration by Mcu silencing cannot be because of incomplete inhibition of Ca2+ entry in mitochondria, as that would result in small inhibition or no inhibition of NMDA-stimulated OCR, but not in an even larger response. Indeed, Mcu-KD in cultured neurons was effective in preventing glutamate excitotoxicity (Qiu et al., 2013). Therefore, the results clearly indicate that cortical neurons using glucose do not require MCU and matrix Ca2+ to upregulate respiration, in response to NMDA-induced increase in workload.
To explore the role of Aralar, we have studied the respiratory response to 5 and 25 μm NMDA. Stimulation of respiration by 5 μm (Extended Data Fig. 4-1E,F) and 25 μm (Fig. 4G,H) NMDA was strikingly blunted in Aralar-KO compared with WT, as found in response to 50 μm glutamate (Llorente-Folch et al., 2016), whereas the absence of Aralar did not change the NMDA-dependent rise in cytosolic (Extended Data Fig. 4-2E–I) and mitochondrial Ca2+ (Extended Data Fig. 4-2J–L) at either of the two NMDA concentrations. Therefore, the presence of Aralar-MAS and not MCU has a critical role in Ca2+ regulation of respiration in response to NMDA.
Activation of NMDARs increases cytosolic pyruvate and lactate production in a Ca2+-dependent way
The effect of the lack of Aralar on Cch-stimulated respiration is rescued by exogenous pyruvate (Fig. 3K,L) (Llorente-Folch et al., 2013, 2016). Therefore, we have analyzed the impact of the presence or absence of Aralar in cytosolic pyruvate and lactate levels on NMDA stimulation.
To this end, cortical neurons were transfected with pyronic or laconic FRET nanosensors (San Martín et al., 2013, 2014) to register pyruvate or lactate levels, respectively, as described previously (Juaristi et al., 2019a). The addition of 25 μm NMDA to neurons in the presence of Ca2 resulted in a prominent increase in both cytosolic pyruvate and lactate levels. The increase in both metabolites was drastically reduced in a Ca2+ free medium (Fig. 4I,J). NMDA-induced pyruvate and lactate production was dependent on NMDA concentration (Fig. 4K,L).
We next addressed the origin of lactate and pyruvate. Although purity of primary neuronal cultures is high, glial contamination can occur; and the surrounding astrocytes could provide lactate taken up by neurons.
In order to elucidate the origin of these metabolites, bilateral transport across plasma membrane was blocked through pharmacological inhibition of neuronal monocarboxylate transporter 2 (MCT2) with AR-C155858 (AR-C1), a potent inhibitor of both MCT1 and MCT2 (Ovens et al., 2010). MCT2 catalyzes the cotransport of a monocarboxylate (i.e., pyruvate, lactate, β-hydroxybutyrate) with a proton (Alvarez et al., 2003). To demonstrate the inhibitory effect of AR-C1 in our conditions, we have used the acidification associated with MCT2 activity as readout. Acute addition of 5 mm lactate (Lac) caused the expected intracellular acidification, as indicated from the drop of F490/F450 fluorescence ratio in BCECF-AM loaded neurons (Fig. 4M). This was prevented by 1 min preincubation with 1 μm AR-C1 revealing the effectiveness of AR-C1 as MCT2 blocker. Acute inhibition of neuronal MCT2 with 1 μm AR-C1 leads to an accumulation of lactate per se, that further increases with 25 μm NMDA stimulation (Fig. 4N); thus, the total increase in cytosolic lactate after AR-C1 + NMDA is similar to that caused by NMDA alone (Fig. 4N,O). These results show that lactate arises from neuronal synthesis rather than by capture from the external media, as shown in activated brain neurons (Díaz-García et al., 2017). It should be noted that neuronal cultures have a relatively large lactate production even in the basal state (Juaristi et al., 2019a, 2019b), possibly larger than that of brain neurons, likely limited by astrocyte lactate buildup in the extracellular space. However, regardless of the actual basal state, NMDA clearly stimulates lactate production even in the presence of 2 mm lactate in the media (Fig. 4P).
In the case of pyruvate, 1 μm AR-C1 also increased rather than decreased pyruvate accumulation (results not shown), indicating an internal origin of pyruvate. This origin is glycolytic, since 30 min pre-incubation with 0.5 mm IAA, an inhibitor of GAPDH, prevented neuronal generation of pyruvate (Fig. 4Q), and partially inhibited lactate production (results not shown), confirming its glycolytic origin. Together, our results show that neurons stimulate glycolytic production of pyruvate and lactate on 25 μm NMDA action in a Ca2+-dependent manner.
NMDA-induced pyruvate production drops in the absence of Aralar-MAS
NMDA (25 μm)-induced pyruvate production decreased in Aralar-KO neurons with the maximum pyruvate increase reduced by 44% compared with WT (Fig. 5A,B; 0.059 ± 0.007 and 0.033 ± 0.006 for WT and Aralar-KO). Pyruvate production in Ca2+-free media was largely abolished in Aralar-KO, and WT neurons (Fig. 5A–C). However, NMDA-induced lactate production was not affected by the absence of Aralar (Fig. 5D), as reflected in the same maximum increase (Fig. 5E; 0.093 ± 0.002 and 0.086 ± 0.002 for WT and Aralar-KO), and in the same velocity of lactate production (Fig. 5F; 0.063 ± 0.002 and 0.046 ± 0.001 for WT and Aralar-KO). Ca2+ dependence of lactate production on 25 μm NMDA stimulation was also observed in Aralar-KO neurons (Fig. 5D–F).
It should be noted that, following the application of a smaller workload, short theta bursts (40 pulses in 11 s) changes in lactate and pyruvate were not detected in hippocampal neurons (Baeza-Lehnert et al., 2019) probably because of the small dynamic range of the probe, especially around basal metabolite levels (San Martín et al., 2014; Juaristi et al., 2019a). This may explain the variability in stimulation-induced increase in pyronic signal in WT and Aralar-KO cultures found in this study. However, studies in brain neurons found increases in lactate and pyruvate after electrical stimulation (Díaz-García et al., 2017, 2021; Díaz-García and Yellen, 2019) or by arousal-evoked cortical activity (Zuend et al., 2020).
Together, these results suggest that Aralar is required to stimulate mitochondrial respiration in response to an NMDA-induced workload in the presence of Ca2+ by providing pyruvate to mitochondria. The Ca2+-dependent stimulation of pyruvate generation is consistent with the following: (1) the stimulation of MAS by Ca2+ conferred by Aralar in isolated brain mitochondria (Pardo et al., 2006; Contreras et al., 2007); (2) the stimulation by Ca2+ of respiration on glutamate + malate of brain mitochondria (Gellerich et al., 2009, 2012, 2013; Szibor et al., 2020); and (3) the observed recovery of Aralar-KO neuronal respiration by external pyruvate administration during stimulation (Fig. 3L) (Pardo et al., 2006; Llorente-Folch et al., 2013). On the other side, Mcu-KD had no influence on NMDA-induced production of pyruvate and lactate (Extended Data Fig. 5-1A–D).
Activation of NMDARs activates glucose uptake and glycolysis in a Ca2+-dependent way
We wondered about the origin of glucose fueling this process and its stimulation by NMDA. To monitor cytosolic glucose, we have used the glucose FRET nanosensor FLII12Pglu-700μδ6 (Takanaga et al., 2008), which is pH-independent, thanks to the replacement of eYFP by citrine (Takanaga et al., 2008; Bittner et al., 2010). Indeed, NMDA induces a rapid cytosolic acidification (Fig. 6A, see comparison with that caused by acute addition of HCl to BCECF-loaded neurons) as shown previously (Wu et al., 1999; Rathje et al., 2013). However, the response to NMDA observed with FLII12Pglu-700μδ6 was an upward deflection of the FRET ratio consistent with an increase in glucose levels (Fig. 6B). In contrast, HCl addition did not cause any increase in FRET ratio, but rather a gradual decrease probably because of side effects and probe quenching. Therefore, the FLII12Pglu-700μδ6 fluorescence changes observed in our experiments are because of changes in cytosolic glucose levels.
Figure 6.
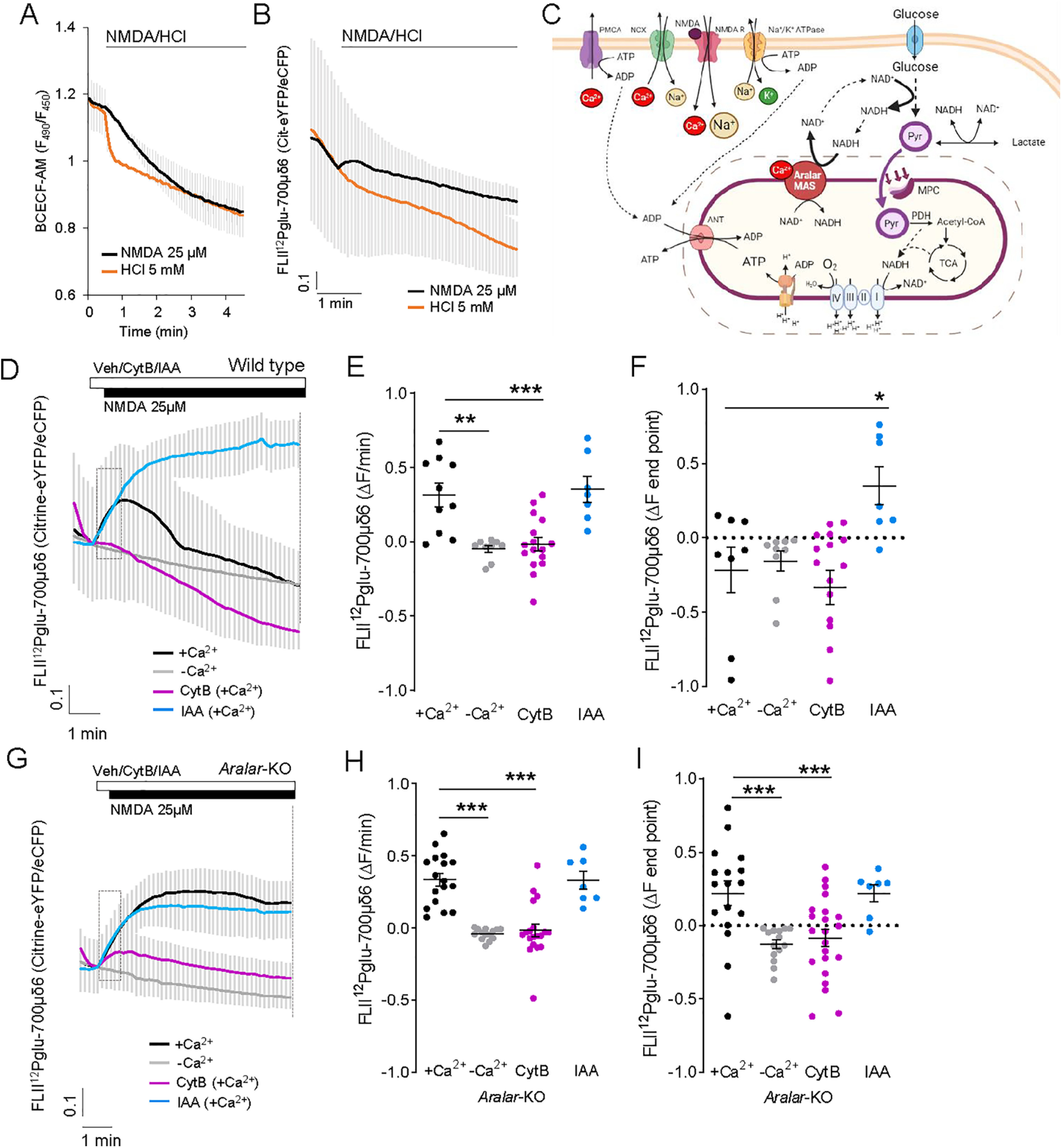
25 μm NMDA-induced changes in cytosolic glucose levels. A, B, Changes in cytosolic pH (A) or intracellular glucose (B) in BCECF-loaded or FLII12Pglu-700μδ6 transfected neurons after the acute addition of 25 μm NMDA or 5 mm HCl. Data are mean ± SEM; recordings from 5 or 6 cells per condition. C, Representative scheme Ca2+-regulated Aralar-MAS activation upregulates glycolysis and pyruvate production, which fuels mitochondrial respiration, through regulation of cytosolic NAD+/NADH ratio. Image created with www.BioRender.com. D, F, 25 μm NMDA induced changes in glucose levels in WT (D) and Aralar-KO (G) neurons, with or without calcium or with a 20 s pre-incubation with 50 μm CytB or 0.5 mm IAA. E, H, Velocity of increase of glucose levels during the first 30 s after stimulation (ΔF/min) in WT (E) and Aralar-KO (H) neurons. F, I, FRET ratio change between pre-addition of NMDA and the end of the recording (ΔF endpoint) in WT (F) and Aralar-KO (I) neurons. Data are mean ± SEM; recordings from 7 to 17 cells per condition from 3 to 5 independent experiments. One-way ANOVA: *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001, post hoc Bonferroni test. ΔF at the endpoint after NMDA addition is higher in Aralar-KO than in WT neurons. Two-way ANOVA: **p ≤ 0.005, post hoc Bonferroni test. Assays using BCECF and FLII12Pglu-700μδ6 probes were performed in 2.5 mm glucose and 2 mm Ca2+ (+Ca2+) or 100 μm EGTA (–Ca2+) HCSS.
Neuronal stimulation is expected to decrease intracellular glucose levels, because of activation of glycolysis to cope with ATP demand. However, the results depicted in Fig. 6D (black traces) show that FLII12Pglu-700μδ6 fluorescence ratio increases immediately after 25 μm NMDA stimulation. The rise in cytosolic glucose levels is maintained during a short period, and then steadily declines, suggesting an initial upregulation of glucose uptake or synthesis which is subsequently consumed.
To determine whether the increase in glucose obtained after NMDA exposure arises from uptake from the external medium, cortical neurons were treated with 50 μm CytB, an inhibitor of plasma membrane glucose transporters (Basketter and Widdas, 1978), 30 s before the addition of 25 μm NMDA (Fig. 6D, purple traces). NMDA-induced increases in citrine-eYFP/eCFP fluorescence ratio in terms of slope (Fig. 6E) and at the end of the experiment (ΔF endpoint; Fig. 6F) are completely abolished in the presence of CytB, indicating that the increase in intracellular glucose arises from uptake from extracellular medium.
The following results clarify that the subsequent steady decline in cytosolic glucose is because of glycolysis: (1) NMDA-stimulated glucose consumption still continues in the presence of CytB (Fig. 6D, purple traces); and (2) when 0.5 mm IAA was administered before NMDA addition (Fig. 6D, blue traces), glucose levels dramatically increased consistent with a clear inhibition of glucose consumption by glycolysis. By using mild electrical stimulation rather than bath application of NMDA, such an increase in cytosolic glucose (recorded with the same probe) and subsequent decrease was not observed (Baeza-Lehnert et al., 2019), possibly because increased uptake was balanced with increased glucose consumption.
Surprisingly, the NMDA-dependent rise in cytosolic glucose completely disappears in the absence of Ca2+, as shown by the block in the initial increase in glucose levels (Fig. 6D,E, gray traces), and the NMDA-dependent decay in cytosolic glucose is also slower (ΔF endpoint; Fig. 6F). Together, these results indicate that activation of NMDARs in cortical neurons upregulates glucose uptake from the external medium and its immediate consumption in glycolysis, both processes being dependent on Ca2+.
NMDA stimulation of glycolysis, but not glucose uptake, is strongly inhibited in Aralar-KO neurons
We next studied the involvement of Aralar in the upregulation of glucose uptake and glycolysis. Strikingly, NMDA-induced a much higher increase in glucose levels in Aralar-KO than in WT neurons (Fig. 6F,I; +Ca2+ in WT and Aralar-KO; p = 0.0009, unpaired t test), with no differences in velocity of increase between genotypes (Fig. 6E,H). NMDA-elevated glucose levels in Aralar-KO neurons were maintained throughout the recording interval instead of being immediately consumed as in WT neurons (Fig. 6G,I, black traces). NMDA-induced increase in glucose was blocked by incubation with 50 μm CytB (Fig. 6G,H, purple traces) and in Ca2+-free medium (Fig. 6G, gray traces), confirming that it was because of uptake from the external medium and was Ca2+-dependent also in Aralar-KO neurons. Thus, glucose uptake in neurons seems to be a Ca2+-dependent process, but independent from Aralar.
Unlike WT neurons (Fig. 6D), Aralar-KO neurons failed to consume cytosolic glucose in the absence or presence of CytB during the recording interval (Fig. 6G, compare black and purple traces). The very low cytosolic glucose consumption in Aralar-KO neurons was confirmed in incubations with 0.5 mm IAA, which caused only a small increase in glucose accumulation (Fig. 6I) compared with its effects in WT neurons (Fig. 6F). Together, the results reveal that (1) NMDA-induced glucose uptake is Ca2+-dependent in cortical neurons and is not affected by the lack of Aralar, and (2) NMDA-induced stimulation of glycolysis is also Ca2+-dependent and requires Aralar (Fig. 6C).
Basal glucose consumption and lactate production are the same in WT and Aralar-KO neurons (0.40 ± 0.08 and 0.43 ± 0.08 μmol glucose/mg/h, and 0.42 ± 0.05 and 0.44 ± 0.07 μmol lactate/mg/h, respectively) (Juaristi et al., 2017). In the presence of Ca2+, NMDA-induced stimulation of glycolysis is much smaller in Aralar-KO than in WT neurons, both in the absence (compare the glucose disappearance rate in WT and Aralar-KO in Fig. 6D,G, black traces) or presence of CytB (compare purple traces in Fig. 6D,G). This is followed by a smaller stimulation of pyruvate formation (Fig. 5A) and respiration (Fig. 4G,H), while lactate formation was not significantly altered (Fig. 5D). This entails that the fate of the small NMDA increase in glycolysis in Aralar-KO neurons is largely lactate, with a diminished pyruvate formation and respiration.
Ca2+ dependence of NMDA stimulation of glycolysis and OXPHOS: workload and/or signaling
The Ca2+ dependence of NMDA stimulation of glycolysis-pyruvate formation-respiration may be because of a feedback mechanism arising as a consequence of Ca2+-induced increased workload caused by NMDA exposure in the presence of Ca2+ and also to feedforward Ca2+ signaling on downstream targets. This dual role of Ca2+ is well known in the case of the muscle metabolic response to contraction (Balaban, 2009; Richter and Hargreaves, 2013; Hargreaves and Spriet, 2020). To tell apart these possibilities, we have measured the changes in intracellular Na+, Ca2+, ATP, and ATP/ADP ratio on addition of 25 μm NMDA to cortical neurons in the presence or absence of external calcium. As shown in Figure 7, the increase in cytosolic Ca2+ was abolished in Ca2+-free medium, and the increases in Na+ triggered by NMDA were the same in the presence or absence of Ca2+ in the external medium. Thus, the combined workload (Na+ + Ca2+ extrusion) is higher in the presence than in the absence of Ca2+. Accordingly, the fall in cytosolic ATP and in the ATP/ADP ratio is larger in the presence than in the absence of Ca2+ (Fig. 7). These results with 25 μm NMDA differ from those obtained with 100 μm NMDA (Rueda et al., 2015) and indicate that, in the presence of Ca2+, NMDA induces a larger workload than in its absence. We conclude that the larger fall in the ATP/ADP ratio could upregulate respiration, pyruvate supply, and glycolysis simply via increased ATP demand (i.e., via a feedback mechanism).
Figure 7.

Dependence of external calcium or BAPTA-AM of the changes in cytosolic Ca2+, Na+, ATP, and ATP/ADP ratio and OCR induced by 25 μm NMDA in neocortical neuronal cultures. A, E, Cytosolic Ca2+ measurements in fura-2-loaded neurons; B, F, cytosolic Na+ in SBFI-loaded neurons; C, G, cytosolic ATP measurement in GO-ATeam-transfected neurons; D, H, cytosolic ATP/ADP ratio in Perceval-transfected neurons. A–D, In the presence of Ca2+. E–H, In nominally Ca2+-free media. The results correspond to representative experiments. I, K, OCR response to 25 μm NMDA in wt and Aralar KO neurons incubated with Ca2+ and in presence or absence of 1 μm BAPTA-AM. Data are mean ± SEM; n = 4-9. J, Histogram represents the decrease (***p < 0.001, n = 9, unpaired t test) in NMDA-induced stimulation of respiration in wt neurons in the presence of BAPTA-AM. K, In Aralar-KO neurons, the decrease was not significant.
To investigate whether Ca2+ signaling could also play a role in the effects of Ca2+ in addition to that on workload, we have used BAPTA-AM, a rapid intracellular Ca2+ chelator. BAPTA loading blunts cytosolic Ca2+ signals (Llorente-Folch et al., 2013) and hence Ca2+ signaling, but does not reduce Ca2+ inflow through NMDARs (Legendre et al., 1993) or voltage-dependent Ca2+-channels (Adler et al., 1991), thus maintaining the Ca2+-dependent fraction of the global workload. In the presence of Ca2+, incubation with 1 μm BAPTA-AM diminishes the OCR response to NMDA in control neurons (Fig. 7I,J), suggesting that Ca2+ signaling, and not only Ca2+-dependent workload, contributes to upregulation of OCR. Interestingly, BAPTA-AM had no detectable effect in Aralar-KO neurons (Fig. 7K), suggesting that the absence of Aralar blocks the Ca2+ signaling mechanism present in control neurons. Together, the results suggest a major role of Ca2+-dependent workload in the metabolic response to NMDA (i.e., a feedback effect) and also a signaling role of calcium relying on Aralar-MAS in boosting this response. Although the Ca2+-binding N-terminus of Aralar is likely the Ca2+ target involved in NMDA-dependent MAS activation by Ca2+, we cannot exclude that other Ca2+ sensors play a role, as the disruption of Aralar removes both protein domains, Ca2+ sensing and mitochondrial carrier. A rigorous study of this possibility will require selective inactivation of the Ca2+-binding domain while maintaining an intact carrier domain.
Discussion
Constitutive Ca2+ flow from ER to mitochondria maintains basal respiration in neurons depends on RyR2, and not IP3R, and does not require MCU
The constitutive Ca2+ flow from ER to mitochondria has been shown to regulate basal respiration in many cell lines (Cárdenas et al., 2010; Filadi et al., 2018), hepatocytes (Tomar et al., 2019), and skeletal muscle (Diaz-Vegas et al., 2018). We find that such a pathway also operates in neurons, with RyR2, but not IP3R, as source of ER Ca2+. This would seem surprising since IP3R are in close apposition to mitochondria, and IP3R1 is abundant in brain neurons (Egorova and Bezprozvanny, 2018). However, the role of RyR in controlling basal respiration is not new; RyR and IP3R control basal respiration in skeletal muscle (Diaz-Vegas et al., 2018). Indeed, RyR are also close to mitochondria in neurons (Jakob et al., 2014), facilitate a rapid Ca2+ transfer from ER/SR to the matrix (Hajnóczky et al., 2002), participate in MAM formation in brain (Ma et al., 2017; Völgyi et al., 2018), and are positioned in MCU enriched nanodomains in heart mitochondria (de la Fuente et al., 2018), therefore in an adequate position to participate in a constitutive Ca2+ flow to mitochondria.
We find that Mcu-KD has no influence on neuronal basal respiration. It could be argued that this is because of compensatory adaptations to MCU deletion masking its true role. By using shRNAs to acutely silence Mcu in neurons, our approach is not expected to cause compensatory effects; therefore, the lack of effect of Mcu-KD in basal respiration (this work and Nichols et al., 2018) shows that MCU is not part of the ER to mitochondria Ca2+ flow regulating basal energetics in neurons using glucose. The Ca2+ sensor on mitochondria remains to be identified.
Stimulation of mitochondrial respiration by agonists: stimulation by Cch requires Aralar-MAS but not MCU
Our results clearly show that, in cortical neurons with spontaneous Ca2+ activity, Cch induces an increase in Ca2+ oscillations and an increase in neuronal respiration. Cch-induced Ca2+ oscillations reached mitochondria, resulting in an increase in matrix Ca2+. Although matrix Ca2+ signals were definitely reduced in Mcu-silenced neurons, Mcu-KD did not affect Cch-stimulated respiration.
In contrast, Cch stimulation of respiration was blunted in Aralar-KO neurons. This reflects a limitation in substrate supply to mitochondria, as pyruvate addition abolished the differences in Cch-stimulated OCR between Aralar-KO and WT neurons. As neither MCU nor Aralar deficiency altered Cch-induced cytosolic Ca2+ oscillations, these results suggest that the Aralar-MAS pathway, but not MCU-driven Ca2+ entry in mitochondria, is involved in boosting respiration by Cch-enhanced Ca2+ oscillations. The mechanism whereby Aralar-MAS regulates respiration in response to Cch becomes apparent from the analysis of the neuronal response to NMDA.
Stimulation of mitochondrial respiration by agonists: NMDA stimulation in cortical neurons causes Ca2+-dependent increases in glucose uptake, glycolysis, pyruvate and lactate formation, and respiration
We have shown that NMDA exposure results in a Ca2+-dependent stimulation of respiration, and Ca2+-dependent increases in cytosolic pyruvate and lactate, both of which arose from glycolysis.
NMDA also triggers a Ca2+-dependent disappearance of cytosolic glucose blocked by inhibitors of glycolysis, showing that neurons respond to NMDA with a Ca2+-dependent stimulation of glycolytic glucose utilization. Additionally, NMDA stimulates Ca2+-dependent glucose uptake.
It makes sense that Ca2+ activates at the same time respiration, substrate supply to mitochondria, glycolysis, and glucose uptake in response to an increase in workload. As neurotransmission is linked to Ca2+ signals, in addition to a direct feedback response to the increased workload, Ca2+ regulation of all these steps can be taken as a feedforward mechanism to increase glycolysis and mitochondrial function on initiation of the workload itself. But does this occur through a single mechanism or are there several Ca2+-dependent individual steps controlling the fate of glucose in neurons up to its combustion in mitochondria? Based on our results, we suggest two Ca2+-dependent steps: one regarding glucose uptake and another one involving Aralar-MAS driven glycolysis and OXPHOS regulation. Our results show that glucose uptake has a specific Ca2+ regulation system independent of glycolysis. An increase in glucose uptake on synaptic activity or NMDA stimulation has been observed in both cerebellar and cortical neuronal cultures (Ferreira et al., 2011; Connolly et al., 2014). It required GLUT3/Slc2a3 translocation to the plasma membrane, via activation of AMPK (Connolly et al., 2014) or nNOS and cGMP-dependent protein kinase (Ferreira et al., 2011). Ashrafi and Ryan (2017) have found that, similarly to muscle, activity at synapses triggers the rapid (seconds) translocation of GLUT4 to the plasma membrane, driven by AMPK. As CaMKKβ is involved in AMPK activation by depolarization in neurons (Kawashima et al., 2012), it is possible that these mechanisms provide Ca2+ sensitivity to the NMDA-dependent increase in glucose uptake found in this study.
Regarding the other steps beyond glucose uptake, in one possible scenario there may be at least two. One is Ca2+ regulation of glycolysis, and the other is Ca2+ activation of mitochondrial dehydrogenases (Denton, 2009) which may boost the activity of the tricarboxylic acid cycle, and the use of pyruvate when Ca2+ enters the matrix. Ca2+ stimulation of glycolysis may involve a Ca2+-dependent activation of PFKFB3 (Rider et al., 2004). PFKFB3 is activated by AMPK (Almeida et al., 2004), which in turn is phosphorylated by CAMKKβ in response to rise in cytosolic Ca2+ (Sanders et al., 2007; Xiao et al., 2011). However, expression of PFKFB3 in neurons is low because of continuous degradation (Herrero-Méndez et al., 2009), and the Yellen group has shown that AMPK is not involved in regulation of glycolysis in stimulated neurons (Díaz-García et al., 2021), suggesting that this mechanism does not operate in response to acute NMDA stimulation. On the other hand, Ca2+ activation of mitochondrial dehydrogenases requires MCU.
In another scenario, glycolysis and OXPHOS are Ca2+-modulated by the same mechanism, Aralar-MAS. The results from this study support this second possibility.
Ca2+ dependence of NMDA stimulation of glycolysis, pyruvate formation, and respiration depends on Aralar-MAS and not MCU
Glycolysis can only proceed if there is sufficient supply of NAD+. The oxidation of glucose requires NAD+ (2 molecules for each glucose) that becomes NADH in the GAPDH reaction. Therefore, to sustain glycolysis, NAD+ needs to be continuously regenerated either by lactate dehydrogenase with conversion of pyruvate to lactate, or by NADH shuttle systems of which MAS is prevalent in neurons (Juaristi et al., 2019a). MAS diverts pyruvate away from lactate and into OXPHOS and is regulated by cytosolic Ca2+ (Pardo et al., 2006; Contreras et al., 2007; Gellerich et al., 2012, 2013, 2020; Llorente-Folch et al., 2013, 2015; Rueda et al., 2014), providing a potential feedforward mechanism linking early Ca2+ signals arising from synaptic activity to both glycolysis and OXPHOS, as suggested previously (Ashrafi and Ryan, 2017).
The results from the present study indicate that, in neurons, using glucose NMDA triggers a substantial Ca2+-dependent workload met by a Ca2+-regulated response in which Ca2+ boosts glycolysis and OXPHOS by means of Ca2+ upregulation of Aralar-MAS. In contrast, Mcu-KD neurons were unaffected in terms of NMDA stimulation of respiration or pyruvate production. Although NMDA-induced Ca2+ entry in mitochondria was only partially blocked by MCU-KD, this indicates that MCU-induced Ca2+ entry in mitochondria and activation of mitochondrial dehydrogenases (McCormack and Denton, 1993) are largely dispensable in the stimulation of respiration of neurons using glucose as also clearly shown by Díaz-García et al. (2021) and Szibor et al. (2020).
In conclusion, it is important to emphasize that the reliance on Aralar-MAS as Ca2+ regulation switch connecting neuronal activity to metabolic response rather than MCU is limited to the use of glucose as substrate. Using a mixture of lactate and pyruvate instead of glucose, Ashrafi et al. (2020) have found that MCU is essential for synaptic vesicle endocytosis and the maintenance of ATP homeostasis after electrical stimulation in nerve terminals. By providing external pyruvate and not glucose, Aralar-MAS function is bypassed, allowing a role of MCU and possibly matrix Ca2+ activation of NAD+-linked isocitrate dehydrogenase, alpha-KGDH and pyruvate dehydrogenase in the control of respiration. It is likely that matrix pyruvate levels need to increase beyond those present in neurons using glucose to allow a functional role of Ca2+-dependent activation of mitochondrial dehydrogenases in upregulating neuronal respiration.
Footnotes
This work was supported by Spanish Ministry of Science, Innovation and Universities SAF2014-56929R to J.S. and B.P.; SAF2017-82560-R to A.d.A. and B.P.; Fundación Ramón Areces to J.S.; and Fundación Ramón Areces institutional grant to Centro de Biología Molecular Severo Ochoa (CBMSO). I.P.-L. and L.G.-M. received predoctoral fellowships from MINECO. P.G.-S. received a postdoctoral research contract from Comunidad de Madrid. The qPCR experimental development was provided by the Genomics and NGS Core Facility at the CBMSO. We thank Isabel Manso, Beatriz García, and Bárbara Sesé for technical support; and the unit of Optical and Confocal Microscopy from CBMSO.
The authors declare no competing financial interests.
References
- Adler EM, Augustine GJ, Duffy SN, Charlton MP (1991) Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci 11:1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Moncada S, Bolaños JP (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 6:45–51. 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- Alvarez G, Ramos M, Ruiz F, Satrústegui J, Bogonez E (2003) Pyruvate protection against β-amyloid-induced neuronal death: role of mitochondrial redox state. J Neurosci Res 73:260–269. 10.1002/jnr.10648 [DOI] [PubMed] [Google Scholar]
- Armstrong CT, Anderson JL, Denton RM (2014) Studies on the regulation of the human E1 subunit of the 2-oxoglutarate dehydrogenase complex, including the identification of a novel calcium-binding site. Biochem J 459:369–381. 10.1042/BJ20131664 [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Ryan TA (2017) Glucose metabolism in nerve terminals. Curr Opin Neurobiol 45:156–161. 10.1016/j.conb.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, de Juan-Sanz J, Farrell RJ, Ryan TA (2020) Molecular tuning of the axonal mitochondrial Ca2+ uniporter ensures metabolic flexibility of neurotransmission. Neuron 105:678–687. 10.1016/j.neuron.2019.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. 10.1097/00004647-200110000-00001 [DOI] [PubMed] [Google Scholar]
- Bacci A, Verderio C, Pravettoni E, Matteoli M (1999) Synaptic and intrinsic mechanisms shape synchronous oscillations in hippocampal neurons in culture. Eur J Neurosci 11:389–397. 10.1046/j.1460-9568.1999.00440.x [DOI] [PubMed] [Google Scholar]
- Baeza-Lehnert F, Saab AS, Gutiérrez R, Larenas V, Díaz E, Horn M, Vargas M, Hösli L, Stobart J, Hirrlinger J, Weber B, Barros LF (2019) Non-canonical control of neuronal energy status by the Na+ pump. Cell Metab 29:668–680. 10.1016/j.cmet.2018.11.005 [DOI] [PubMed] [Google Scholar]
- Balaban RS (2009) The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta 1787:1334–1341. 10.1016/j.bbabio.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basketter DA, Widdas WF (1978) Asymmetry of the hexose transfer system in human erythrocytes: comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J Physiol 278:389–401. 10.1113/jphysiol.1978.sp012311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner CX, Loaiza A, Ruminot I, Larenas V, Sotelo-Hitschfeld T, Gutiérrez R, Córdova A, Valdebenito R, Frommer WB, Barros LF (2010) High resolution measurement of the glycolytic rate. Front Neuroenergetics 2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435:297–312. 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283. 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly NM, Düssmann H, Anilkumar U, Huber HJ, Prehn JH (2014) Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J Neurosci 34:10192–10205. 10.1523/JNEUROSCI.3127-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrústegui J (2007) Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem 282:7098–7106. 10.1074/jbc.M610491200 [DOI] [PubMed] [Google Scholar]
- de Korte D, Haverkort WA, van Gennip AH, Roos D (1985) Nucleotide profiles of normal human blood cells determined by high-performance liquid chromatography. Anal Biochem 147:197–209. 10.1016/0003-2697(85)90028-4 [DOI] [PubMed] [Google Scholar]
- De La Fuente S, Lambert J, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS, Csordás G (2018) Spatial separation of mitochondrial calcium uptake and extrusion for energy-efficient mitochondrial calcium signaling in the heart. Cell Rep 24:3099–3107. 10.1016/j.celrep.2018.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R, Pozzan T (2016) Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85:161–192. 10.1146/annurev-biochem-060614-034216 [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA (2002) Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron 34:209–220. 10.1016/s0896-6273(02)00641-4 [DOI] [PubMed] [Google Scholar]
- Delmas P, Nauli SM, Li X, Coste B, Osorio N, Crest M, Brown DA, Zhou J (2004) Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB J 18:740–742. 10.1096/fj.03-0319fje [DOI] [PubMed] [Google Scholar]
- Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316. 10.1016/j.bbabio.2009.01.005 [DOI] [PubMed] [Google Scholar]
- Díaz-García CM, Yellen G (2019) Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res 97:883–889. 10.1002/jnr.24374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal Stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26:361–374.e4. 10.1016/j.cmet.2017.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García CM, Meyer DJ, Nathwani N, Rahman M, Martínez-François JR, Yellen G (2021) The distinct roles of calcium in rapid control of neuronal glycolysis and the tricarboxylic acid cycle. Elife 10:e64821. 10.7554/Elife.64821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Vegas AR, Cordova A, Valladares D, Llanos P, Hidalgo C, Gherardi G, De Stefani D, Mammucari C, Rizzuto R, Contreras-Ferrat A, Jaimovich E (2018) Mitochondrial calcium increase induced by RyR1 and IP3R channel activation after membrane depolarization regulates skeletal muscle metabolism. Front Physiol 9:791. 10.3389/fphys.2018.00791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA (2019) Brain glucose metabolism: integration of energetics with function. Physiol Rev 99:949–1045. 10.1152/physrev.00062.2017 [DOI] [PubMed] [Google Scholar]
- Egorova PA, Bezprozvanny IB (2018) Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J 285:3547–3565. 10.1111/febs.14366 [DOI] [PubMed] [Google Scholar]
- Fecher C, Trovò L, Müller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kühn R, Hartmann J, Karl RM, Konnerth A, Korn T, Wurst W, Merkler D, Lichtenthaler SF, Perocchi F, Misgeld T (2019) Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci 22:1731–1742. 10.1038/s41593-019-0479-z [DOI] [PubMed] [Google Scholar]
- Ferreira JM, Burnett AL, Rameau GA (2011) Activity-dependent regulation of surface glucose transporter-3. J Neurosci 31:1991–1999. 10.1523/JNEUROSCI.1850-09.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Leal NS, Schreiner B, Rossi A, Dentoni G, Pinho CM, Wiehager B, Cieri D, Calì T, Pizzo P, Ankarcrona M (2018) TOM70 sustains cell bioenergetics by promoting IP3R3-mediated ER to mitochondria Ca2+ transfer. Curr Biol 28:369–382. 10.1016/j.cub.2017.12.047 [DOI] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN (1997) Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 19:723–733. 10.1016/S0896-6273(00)80384-0 [DOI] [PubMed] [Google Scholar]
- Galeotti N, Quattrone A, Vivoli E, Norcini M, Bartolini A, Ghelardini C (2008) Different involvement of type 1, 2, and 3 ryanodine receptors in memory processes. Learn Mem 15:315–323. 10.1101/lm.929008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Arandarcikaite O, Jerzembek D, Vielhaber S, Seppet E, Striggow F (2009) Extramitochondrial Ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS One 4:e8181. 10.1371/journal.pone.0008181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Trumbekaite S, Korzeniewski B, Gaynutdinov T, Seppet E, Vielhaber S, Heinze HJ, Striggow F (2012) Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem J 443:747–755. 10.1042/BJ20110765 [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z, Vielhaber S (2013) The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life 65:180–190. 10.1002/iub.1131 [DOI] [PubMed] [Google Scholar]
- Gerkau NJ, Lerchundi R, Nelson JS, Lantermann M, Meyer J, Hirrlinger J, Rose CR (2019) Relation between activity-induced intracellular sodium transients and ATP dynamics in mouse hippocampal neurons. J Physiol 597:5687–5705. 10.1113/JP278658 [DOI] [PubMed] [Google Scholar]
- Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, Bonaldo P, De Stefani D, Rizzuto R, Mammucari C (2019) Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ 26:362–381. 10.1038/s41418-018-0191-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Balaban RS (2012) Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51:2959–2973. 10.1021/bi2018909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G, Csordás G, Yi M (2002) Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32:363–377. 10.1016/s0143416002001872 [DOI] [PubMed] [Google Scholar]
- Hall CN, Klein-Flügge MC, Howarth C, Attwell D (2012) Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci 32:8940–8951. 10.1523/JNEUROSCI.0026-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N (2018) Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem 293:15652–15663. 10.1074/jbc.RA118.002926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves M, Spriet LL (2020) Skeletal muscle energy metabolism during exercise. Nat Metab 2:817–828. 10.1038/s42255-020-0251-4 [DOI] [PubMed] [Google Scholar]
- Herrero-Méndez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752. 10.1038/ncb1881 [DOI] [PubMed] [Google Scholar]
- Jaimovich E, Mattei C, Liberona JL, Cárdenas C, Estrada M, Barbier J, Debitus C, Laurent D, Molgó J (2005) Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108-15) cells. FEBS Lett 579:2051–2057. 10.1016/j.febslet.2005.02.053 [DOI] [PubMed] [Google Scholar]
- Jakob R, Beutner G, Sharma VK, Duan Y, Gross RA, Hurst S, Jhun BS, O-Uchi J, Sheu SS (2014) Molecular and functional identification of a mitochondrial ryanodine receptor in neurons. Neurosci Lett 575:7–12. 10.1016/j.neulet.2014.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Marmol P, Horiuchi M, Shimotsu K, Nakagawa S, Okubo A, Sameshima M, Isashiki Y, Del Arco A, Kobayashi K, Satrústegui J, Saheki T (2005) Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem 280:31333–31339. 10.1074/jbc.M505286200 [DOI] [PubMed] [Google Scholar]
- Juaristi I, García-Martín ML, Rodrigues TB, Satrústegui J, Llorente-Folch I, Pardo B (2017) ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J Neurochem 142:132–139. 10.1111/jnc.14047 [DOI] [PubMed] [Google Scholar]
- Juaristi I, Llorente-Folch I, Satrústegui J, Del Arco A (2019a) Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 67:759–774. 10.1002/glia.23574 [DOI] [PubMed] [Google Scholar]
- Juaristi I, Contreras L, González-Sánchez P, Pérez-Liébana I, González-Moreno L, Pardo B, Del Arco A, Satrústegui J (2019b) The response to stimulation in neurons and astrocytes. Neurochem Res 44:2385–2391. 10.1007/s11064-019-02803-7 [DOI] [PubMed] [Google Scholar]
- Kawashima J, Alquier T, Tsuji Y, Peroni OD, Kahn BB (2012) Ca2+/calmodulin-dependent protein kinase kinase is not involved in hypothalamic AMP-activated protein kinase activation by neuroglucopenia. PLoS One 7:e36335. 10.1371/journal.pone.0036335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmach A, Roman B, Sun J, Femnou A, Zhang F, Liu C, Combs CA, Balaban RS, Murphy E (2021) Monitoring mitochondrial calcium and metabolism in the beating MCU-KO heart. Cell Rep 37:109846. 10.1016/j.celrep.2021.109846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD (2015) The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12:15–22. 10.1016/j.celrep.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Clerc P, Polster BM, Fiskum G (2013) Augmentation of normal and glutamate-impaired neuronal respiratory capacity by exogenous alternative biofuels. Transl Stroke Res 4:643–651. 10.1007/s12975-013-0275-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell H, Iacovoni JS, Abot A, Svec D, Maoret JJ, Arnal JF, Kubista M (2012) Correction of RT-qPCR data for genomic DNA-derived signals with ValidPrime. Nucleic Acids Res 40:e51. 10.1093/nar/gkr1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook GL (1993) Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci 13:674–684. 10.1523/JNEUROSCI.13-02-00674.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I (2009) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 29:9148–9162. 10.1523/JNEUROSCI.0660-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrústegui J (2013) Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 33:13957–13971. 10.1523/JNEUROSCI.0929-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrústegui J (2015) The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 593:3447–3462. 10.1113/JP270254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Pérez-Liébana I, Satrústegui J, Pardo B (2016) L-lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J Neurosci 36:4443–4456. 10.1523/JNEUROSCI.3691-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Chu W, Chai J, Shen C, Li D, Wang X (2017) ER stress and subsequent activated calpain play a pivotal role in skeletal muscle wasting after severe burn injury. PLoS One 12:e0186128. 10.1371/journal.pone.0186128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14:1336–1343. 10.1038/ncb2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure C, Cole KL, Wulff P, Klugmann M, Murray AJ (2011) Production and titering of recombinant adeno-associated viral vectors. J Vis Exp 27:e3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Denton RM (1993) Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev Neurosci 15:165–173. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. 10.1016/j.tins.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- More JY, Bruna BA, Lobos PE, Galaz JL, Figueroa PL, Namias S, Sánchez GL, Barrientos GC, Valdés JL, Paula-Lima AC, Hidalgo C, Adasme T (2018) Calcium release mediated by redox-sensitive RyR2 channels has a central role in hippocampal structural plasticity and spatial memory. Antioxid Redox Signal 29:1125–1146. 10.1089/ars.2017.7277 [DOI] [PubMed] [Google Scholar]
- Mu YH, Zhao WC, Duan P, Chen Y, Zhao WD, Wang Q, Tu HY, Zhang Q (2014) RyR2 modulates a Ca2+-activated K+ current in mouse cardiac myocytes. PLoS One 9:e94905. 10.1371/journal.pone.0094905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols M, Pavlov EV, Robertson GS (2018) Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis 9:606. 10.1038/s41419-018-0607-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz T, De Lima AD, Voigt T (2002) Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J Neurophysiol 88:2196–2206. 10.1152/jn.00316.2002 [DOI] [PubMed] [Google Scholar]
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M (2012) Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci 32:11820–11834. 10.1523/JNEUROSCI.0875-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP (2010) AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7-10. Biochem J 425:523–530. 10.1042/BJ20091515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY (2006) Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 13:521–530. 10.1016/j.chembiol.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15:1464–1472. 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrústegui J (2006) Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem 281:1039–1047. 10.1074/jbc.M507270200 [DOI] [PubMed] [Google Scholar]
- Patel JC, Witkovsky P, Avshalumov MV, Rice ME (2009) Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J Neurosci 29:6568–6579. 10.1523/JNEUROSCI.0181-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podunavac M, Mailyan AK, Jackson JJ, Lovy A, Farias P, Huerta H, Molgó J, Cárdenas C, Zakarian A (2021) Scalable total synthesis, IP3R inhibitory activity of desmethylxestospongin B, and effect on mitochondrial function and cancer cell survival. Angew Chem Int Ed Engl 60:11278–11282. 10.1002/anie.202102259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Van Houten B (2010) Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods 51:452–457. 10.1016/j.ymeth.2010.03.006 [DOI] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4:2034. 10.1038/ncomms3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos M, Pardo B, Llorente-Folch I, Saheki T, Del Arco A, Satrústegui J (2011) Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J Neurosci Res 89:2008–2017. 10.1002/jnr.22639 [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. 10.1016/j.cell.2013.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathje M, Fang H, Bachman JL, Anggono V, Gether U, Huganir RL, Madsen KL, Shmigol A (2013) AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc Natl Acad Sci USA 110:14426–14431. 10.1073/pnas.1312982110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Hargreaves M (2013) Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev 93:993–1017. 10.1152/physrev.00038.2012 [DOI] [PubMed] [Google Scholar]
- Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L (2004) 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 381:561–579. 10.1042/BJ20040752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Pizzo P, Filadi R (2019) Calcium, mitochondria and cell metabolism: a functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res 1866:1068–1078. 10.1016/j.bbamcr.2018.10.016 [DOI] [PubMed] [Google Scholar]
- Rueda CB, Llorente-Folch I, Amigo I, Contreras L, González-Sánchez P, Martínez-Valero P, Juaristi I, Pardo B, del Arco A, Satrústegui J (2014) Ca(2+) regulation of mitochondrial function in neurons. Biochim Biophys Acta 1837:1617–1624. 10.1016/j.bbabio.2014.04.010 [DOI] [PubMed] [Google Scholar]
- Rueda CB, Traba J, Amigo I, Llorente-Folch I, González-Sánchez P, Pardo B, Esteban JA, del Arco A, Satrústegui J (2015) Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J Neurosci 35:3566–3581. 10.1523/JNEUROSCI.2702-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, Barros LF (2013) A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One 8:e57712. 10.1371/journal.pone.0057712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Baeza-Lehnert F, Lerchundi R, Valdebenito R, Contreras-Baeza Y, Alegría K, Barros LF (2014) Imaging mitochondrial flux in single cells with a FRET sensor for pyruvate. PLoS One 9:e85780. 10.1371/journal.pone.0085780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403:139–148. 10.1042/BJ20061520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrústegui J, Pardo B, Del Arco A (2007) Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev 87:29–67. 10.1152/physrev.00005.2006 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Shmigol A, Verkhratsky A, Isenberg G (1995) Calcium-induced calcium release in rat sensory neurons. J Physiol 489:627–636. 10.1113/jphysiol.1995.sp021078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A (2002) Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J 21:622–630. 10.1093/emboj/21.4.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szibor M, Gizatullina Z, Gainutdinov T, Endres T, Debska-Vielhaber G, Kunz M, Karavasili N, Hallmann K, Schreiber F, Bamberger A, Schwarzer M, Doenst T, Heinze HJ, Lessmann V, Vielhaber S, Kunz WS, Gellerich FN (2020) Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J Biol Chem 295:4383–4397. 10.1074/jbc.RA119.011902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga H, Chaudhuri B, Frommer WB (2008) GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta 1778:1091–1099. 10.1016/j.bbamem.2007.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M, Martínez-François JR, Mongeon R, Yellen G (2013) Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4:2550. 10.1038/ncomms3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Territo PR, Mootha VK, French SA, Balaban RS (2000) Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol 278:C423–C435. 10.1152/ajpcell.2000.278.2.C423 [DOI] [PubMed] [Google Scholar]
- Tomar D, Jaña F, Dong Z, Quinn WJ, Jadiya P, Breves SL, Daw CC, Srikantan S, Shanmughapriya S, Nemani N, Carvalho E, Tripathi A, Worth AM, Zhang X, Razmpour R, Seelam A, Rhode S, Mehta AV, Murray M, Slade D, et al. (2019) Blockade of MCU-mediated Ca2+ uptake perturbs lipid metabolism via PP4-dependent AMPK dephosphorylation. Cell Rep 26:3709–3725. 10.1016/j.celrep.2019.02.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völgyi K, Badics K, Sialana FJ, Gulyássy P, Udvari EB, Kis V, Drahos L, Lubec G, Kékesi KA, Juhász G (2018) Early presymptomatic changes in the proteome of mitochondria-associated membrane in the APP/PS1 mouse model of Alzheimer's disease. Mol Neurobiol 55:7839–7857. 10.1007/s12035-018-0955-6 [DOI] [PubMed] [Google Scholar]
- Wang P, Fernandez-Sanz C, Wang W, Sheu SS (2020) Why don't mice lacking the mitochondrial Ca2+ uniporter experience an energy crisis? J Physiol 598:1307–1326. 10.1113/JP276636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ML, Chen JH, Chen WH, Chen YJ, Chu KC (1999) Novel role of the Ca(2+)-ATPase in NMDA-induced intracellular acidification. Am J Physiol 277:C717–C727. 10.1152/ajpcell.1999.277.4.C717 [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233. 10.1038/nature09932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KW, Billups D, Nelson CP, Johnston N, Willets JM, Schell MJ, Challiss RA, Nahorski SR (2005) Muscarinic acetylcholine receptor activation enhances hippocampal neuron excitability and potentiates synaptically evoked Ca(2+) signals via phosphatidylinositol 4,5-bisphosphate depletion. Mol Cell Neurosci 30:48–57. [DOI] [PubMed] [Google Scholar]
- Zalk R, Lehnart SE, Mark AR (2007) Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76:367–385. 10.1146/annurev.biochem.76.053105.094237 [DOI] [PubMed] [Google Scholar]
- Zuend M, Saab AS, Wyss MT, Ferrari KD, Hösli L, Looser ZJ, Stobart JL, Duran J, Guinovart JJ, Barros LF, Weber B (2020) Arousal-induced cortical activity triggers lactate release from astrocytes. Nat Metab 2:179–191. 10.1038/s42255-020-0170-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Caffeine-discharge ER-Calcium and Adenine nucleotide content in RyR2-KD neurons. A-B: 15 mM Caffeine (Caff.) discharges ER calcium by activating RyR in cortical neurons at 9 DIV. Inhibition of the response to caffeine by preincubation with 50 µM ryanodine (Ryan.) (A) or 10 µM dantrolene (Dandr.) (B) during 15 or 30 min. C-G: Adenine nucleotide content, measured by HPLC in neurons transduced with rAAV containing RyR2-directed (RyR-KD) or Scrambled shRNA (Scr). Mean ± SEM (bars), 5 independent experiments (dots). fura-2 AM Download Figure 1-1, TIF file (299.2KB, tif) .
Cch induced cytosolic Ca2+ signals. Effects of MCU-KD and Aralar-KO. A. Mouse cortical neurons plated at high density and cultured for 8-9 days showed spontaneous Ca2+ activity, consisting of repetitive Ca2+ transients occurring at the same time in all neurons. These Ca2+ transients were blocked by treatment with the AMPA/Kainate receptor antagonist CNQX (10 μm), the NMDAR antagonist MK-801 (10 μm), or the Na+ channel blocker TTX (1 μm) (results not shown). B. Cch addition in the presence of TTX, which blocks other responses except those due to Ca2+ mobilization by IP3R, resulted in a weak response. C-D: fura-2 AM [Ca2+]i signals in Scr (C) or Mcu-KD (D) neurons in HCSS medium containing 2 mM CaCl2 and 2.5 mM glucose upon addition of 250 μm Cch where indicated. Representative experiments, each trace corresponds to a single neuron from the same recording field. E: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM. Data were obtained from 3 independent experiments. F-G: fura-2 AM [Ca2+]i signals in WT (E) and Aralar-KO (G) neurons in HCSS medium containing 2 mM CaCl2 and 2.5 mM glucose upon addition of 250 μm Cch where indicated. Representative experiments, each trace corresponds to a single neuron from the same recording field. H: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM. Data were obtained from 5 independent experiments. Download Figure 3-1, TIF file (405.7KB, tif) .
Changes in mitochondrial Ca2+ and OCR upon stimulation with 5 μm NMDA. A: Changes in mitochondrial Ca2+ (Ca2+-mit) in Scrambled (Scr) and Mcu-silenced (Mcu-KD) neurons transfected with 4mtD3cpv probe, after 5 μm NMDA stimulation in 2.5 mM glucose and 2 mM Ca2+ HCSS; B: Quantification of maximum Ca2+-mit increment (ΔF max) after NMDA stimulation. Data are mean ± SEM (bars), n = 8-9 (dots) from three independent experiments; T- test *p = 0.0365. C: 5 μm NMDA-stimulated respiration in Scrambled and Mcu-KD neurons maintained in 2.5 mM glucose and 2 mM Ca2+ DMEM, expressed as percentage of basal values (OCR %); D: Quantification of NMDA-stimulated OCR (%); mean ± SEM (bars), n = 14-15 (dots) from 5 independent experiments; T test *p = 0.0339. E: 5 μm NMDA-stimulated respiration in wild type and Aralar-KO neurons maintained in 2.5 mM glucose and 2 mM Ca2+ DMEM, expressed as percentage of basal values (OCR %); F: Quantification of NMDA-stimulated OCR (%); mean ± SEM (bars), n = 4-6 (dots) from 2 independent experiments; T test ***p = 0.0007. Download Figure 4-1, TIF file (311.5KB, tif) .
NMDA-induced Cytosolic and mitochondrial Ca2+ signals in Scr, Mcu-KD, Wild type and Aralar-KO neurons. A-D: Changes in cytosolic Ca2+ (Ca2+-cyt) after 5 (A, C) and 25 μm NMDA (B, D) stimulation in Scr (A, B) or Mcu-KD (C, D) neurons. E-I: Changes in cytosolic Ca2+ (Ca2+-cyt), after 5 (E, G) and 25 μm (F, H) NMDA in wild type (E, I) and Aralar-KO (G, H) Fura2-AM loaded neurons. Individual cell recordings (gray) and average (black) are shown. I: Quantification of peak amplitude as ΔRatio (F340/F380) ± SEM (bars) comparing 5 and 25 μm NMDA stimulus; two-way ANOVA, **p ≤ 0.01, ***p ≤ 0.001, post hoc Bonferroni test. Recordings from at least 50 cells/condition from 4-7 wells (dots) and two independent experiments. J-L: Changes in mitochondrial Ca2+ (Ca2+-mit), in wild type (J) and Aralar-KO neurons (K) transfected with 4mtD3cpv probe, after 5 and 25 μm NMDA stimulation; L: maximum Ca2+-mit increment (ΔF max) after NMDA stimulation. Mean ± SEM (bars), 5-10 cells (dots) from two independent cultures; two-way ANOVA, *p ≤ 0.05, post hoc Bonferroni test. All cells were maintained in 2.5 mM glucose and 2 mM Ca2+ HCSS. Download Figure 4-2, TIF file (351.2KB, tif) .
Effects of NMDA stimulation on cytosolic pyruvate and lactate production in Mcu-silenced neurons. Cytosolic pyruvate (Pyr) (A, C) and lactate (Lac) (B, D) levels of Scr and Mcu-KD neurons transfected with Pyronic and Laconic probes, respectively, upon neuronal stimulation with 5 μm (A, B) or 25 μm NMDA (C, D). Neurons were incubated in 2.5 mM glucose and 2 mM Ca2+ HCSS. Data are mean ± SEM, n = 7-15 neurons per condition from 2 independent cultures. Download Figure 5-1, TIF file (293.6KB, tif) .