

Abstract
DNA break lesions pose a serious threat to the integrity of the genome. Eukaryotic cells can remove these lesions using the homologous recombination (HR) pathway that guides the repair reaction by using a homologous DNA template. The budding yeast Saccharomyces cerevisiae is an excellent model system with which to study this repair mechanism and the resulting patterns of genomic change resulting from it. In this chapter, we describe an approach that utilizes whole genome sequencing (WGS) data to support the analysis of tracts of loss-of-heterozygosity (LOH) that can arise from mitotic recombination in the context of the entire diploid yeast genome. The workflow and the discussion in this chapter are intended to enable classically trained molecular biologists and geneticists with limited experience in computational methods to conceptually understand and execute the steps of the genome-wide LOH analysis, as well as to adapt and apply them to their own specific studies and experimental models.
1. INTRODUCTION
The budding yeast Saccharomyces cerevisiae has traditionally been, and continues to be, one of the premier model systems for the study of mechanisms of meiotic and mitotic recombination. Successful adoption of this model organism has been due in large part to the many tools available since the early 1980’s for the genetic manipulation and construction of sophisticated recombination reporters that produce selectable phenotypes. Assays using budding yeast allow for the quantitative measurement of recombination rates and the isolation of clones carrying recombinant sequences that can be characterized for insight into the molecular mechanisms of genetic rearrangement. These genetic systems continue to be developed and still drive innovation in this field (Klein 2001). However, such reporter assays can be limiting as they are typically only informative about the DNA sequences in the immediate vicinity of the reporter. Little to no information can be gleaned for recombination events that may occur elsewhere in the genome. This is because conventional laboratory strains are either haploid or homozygous diploid, and tend to lack genetic markers that can be followed outside of the reporter locus.
This limitation has been overcome in the last 15–20 years of study in this field, and unprecedented discoveries have been made regarding the distribution of recombination processes across the whole yeast genome (Gerton et al. 2000; Mancera et al. 2008; Pan et al. 2011; St Charles and Petes 2013; Laureau et al. 2016; McGinty et al. 2017; Zheng and Petes 2018). Our current ability to study recombination on a genome-wide scale can be attributed to the combination of two factors: The availability of fully characterized genome sequences from multiple S. cerevisiae strains that are evolutionarily diverged from the S288c reference strain (Cherry et al. 2012), and the dissemination and increasing affordability of methods for genome-wide analyses, including genotyping microarrays and whole genome sequencing (WGS).
Use of heterozygous hybrid strain backgrounds:
Allelic polymorphisms between two haploid parents give rise to structural and nucleotide-level heterozygosity in their respective hybrid diploid. In mitotically dividing cells, recombination-based repair of DNA double strand breaks (DSBs) using as template the allelic sequences on the homologous chromosome can lead to loss-of-heterozygosity (LOH) (Symington et al. 2014). As its name suggests, LOH constitutes the loss of genetic information resulting in homozygosis or hemizygosis of a region of the genome. Whole genome sequence analysis (WGS) of >1000 natural diverged S. cerevisiae isolates has revealed a broad spectrum of nucleotide variation between haploids, and heterozygosity among diploid strains ranging from ~337 heterozygous single nucleotide polymorphisms (referred to in this chapter as HetSNPs) in the laboratory strain Sigma1278b, to >40,000 HetSNPs in the clinical isolate YJM311 (Magwene et al. 2011; Strope et al. 2015; Peter et al. 2018a).
Natural heterozygosity can offer valuable insight into the life history of a strain. For example, our group has extensively characterized the genome of a wild isolate called JAY270, originally isolated from a bioethanol production facility in Brazil (Basso et al. 2008). JAY270 is only moderately heterozygous (~12,000 HetSNPs) (Argueso et al. 2009; Rodrigues Prause et al. 2018). These HetSNPs, however, are not evenly distributed across the genome (Fig. 1A). Rather, there are extended tracts of homozygosity (Fig. 1A), which span whole chromosomes (e.g., Chr1) and large sections of chromosome arms (Fig. 1A, distal left and right arms of Chr4). While we cannot directly infer the pattern and timing of recombination events that have shaped the genome of JAY270, it is tempting to speculate that the original heterozygosity of JAY270 has eroded over its life history due to LOH events resulting from mitotic recombination and meiotic recombination followed by mating between sibling spores. The JAY270 genome illustrates the premise that in the context of a heterozygous background, we can “see” the outcomes of inter-homolog recombination on a genome-wide scale. However, because heterozygous markers are absent in significant portions of the genome, this natural diploid only supports high-resolution characterization of new recombination in the genomic regions where it remains heterozygous.
Figure 1.
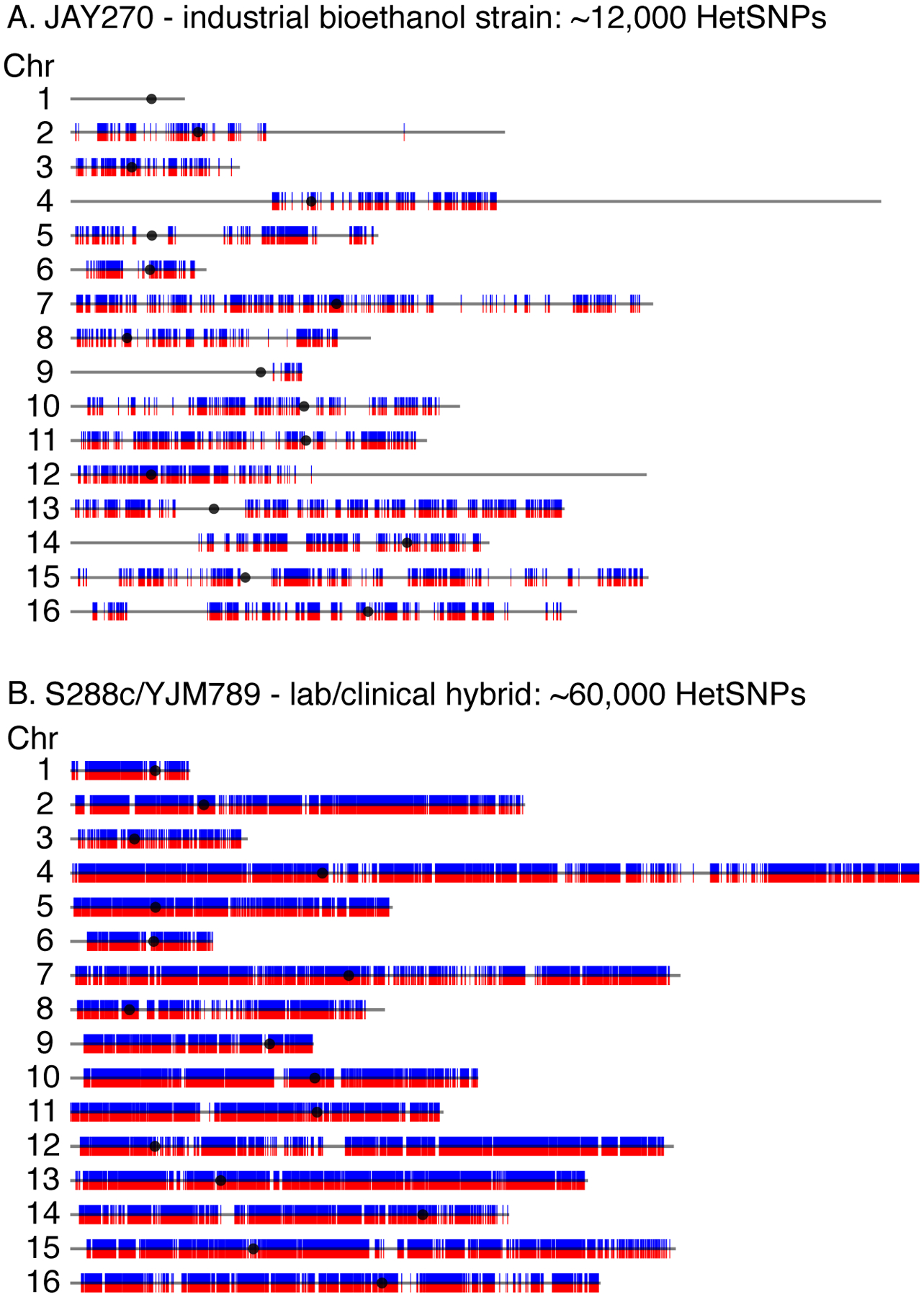
Levels and distribution of heterozygosity in natural and artificially created hybrid diploid S. cerevisiae strains. A) Linear representation of all nuclear chromosomes in the JAY270 diploid strain (Rodrigues Prause et al. 2018) displaying the specific positions of HetSNPs. Each red/blue line indicates a HetSNP between the two homologous chromosomes. Regions that lack HetSNP lines are entirely homozygous. Black circles indicate the positions of centromeres. JAY270 contains ~12,000 HetSNPs. B) Linear representation of each chromosome in the S288c/YJM789 hybrid diploid showing the high density and uniform distribution of its ~60,000 HetSNPs (Mancera et al. 2008).
For many years now, homologous recombination (HR) and specifically LOH events have been studied using hybrid diploid strains that are highly heterozygous because they are formed by mating two evolutionarily diverged haploid parents (Mancera et al. 2008; St Charles and Petes 2013; Dutta et al. 2017). These genetic backgrounds enable researchers to map recombination breakpoints and tracts of homozygosity with high resolution, and have been instrumental in expanding our understanding of HR-based DNA repair (Zheng and Petes 2018). Currently, our group uses a well-characterized hybrid diploid formed by mating the reference strain S288c (Mortimer and Johnston 1986; Goffeau et al. 1996) to YJM789, a clinical isolate (Tawfik et al. 1989; McCusker et al. 1994). The S288c/YJM789 diploid has ~60,000 HetSNPs distributed evenly throughout the genome (Fig. 1B, Wei et al. 2007), making it an excellent background for genome-wide characterization of recombination events.
Genome-wide assessment of recombination outcomes:
The ability to characterize the outcomes of recombination on a genome-wide basis has only been possible because of the introduction of single nucleotide polymorphism (SNP) genotyping microarrays and WGS technologies (Magwene et al. 2011; Zheng and Petes 2018; Peter et al. 2018b). SNP microarrays were used to produce the first high-resolution maps of both mitotic and meiotic recombination events in the yeast genome (Mancera et al. 2008; St Charles and Petes 2013). These arrays can map recombination events and other chromosome rearrangements to a resolution of ~1kb (Zheng and Petes 2018), depending on the array design. More recently, WGS has been used to map recombination events with higher resolution than is practical with SNP microarrays. When would one want to consider mitotic recombination events on a genome-wide scale? SNP microarrays and WGS analysis have been used to assess the global responses to DNA damaging agents such as ultra-violet radiation, as well as to identify recombination-prone regions (e.g., fragile sites) that are stimulated by replication stress (Lemoine et al. 2005; Lemoine et al. 2008; St Charles et al. 2012; Song et al. 2014; O’Connell et al. 2015; Zheng et al. 2016). In addition, our own group uses WGS analysis to characterize mitotic recombination events that co-occur in the genome simultaneously (Sampaio et al. 2017).
In this chapter, we describe a general procedure used by our group to detect genome-wide LOH associated with mitotic recombination in budding yeast. In the specific type of assay described here, we start with a parent diploid strain that contains well-characterized HetSNPs distributed across the genome, and at least one hemizygous counter-selectable marker. Selection for loss of the marker typically yields clones that contain an LOH tract that resulted from inter-homolog recombination in the chromosome arm where the marker was inserted. While LOH of the marker may also arise via other events such as point mutation, segmental deletion, and chromosome loss (see Notes), the spontaneous rates of mitotic recombination are substantially higher than the rates of those alternative events, so most of the clones recovered are by such selection regime are typically due to mitotic recombination. The procedure outlined below is able to distinguish between the causal mechanisms. Moreover, we can place these mutagenic events in the context of the entire genome to identify genome-wide mutational patterns surrounding the reporter regions and also any unselected co-occurring mutations that may be present in clones examined (Sampaio et al. 2017).
2. MATERIALS and METHODS
Isolation of yeast clones carrying LOH tracts:
Into the S288c/YJM789 hybrid strain background, we have inserted the counter-selectable URA3 gene onto a distal position in the right arm of the S288c chromosome IV (Chr4) (Fig. 2, red homolog). Both endogenous copies of the URA3 gene were mutated or fully deleted from the native locus on Chr5. When a DSB lesion occurs at a proximal position on the S288c Chr4 homolog containing the URA3 insertion, it may be repaired by HR using the allelic position on the YJM789 homolog as template and be resolved as a crossover (Fig. 2). In this scenario, depending on the segregation of recombinant chromatids in the subsequent mitosis, one of the resulting daughter cells will lose the URA3 insertion, become resistant to 5-fluoroorotic acid (5-FOA), and form a colony on selective media (Fig. 2, FOAr) ((Boeke et al. 1984; Boeke et al. 1987). Again, we note that this procedure may also yield at low frequency clones that became resistant to 5-FOA through mechanisms other that mitotic recombination (see Notes). The same general approach could also be pursued using other counter-selectable markers such as CAN1 and AmdS (Solis-Escalante et al. 2013).
Figure 2.
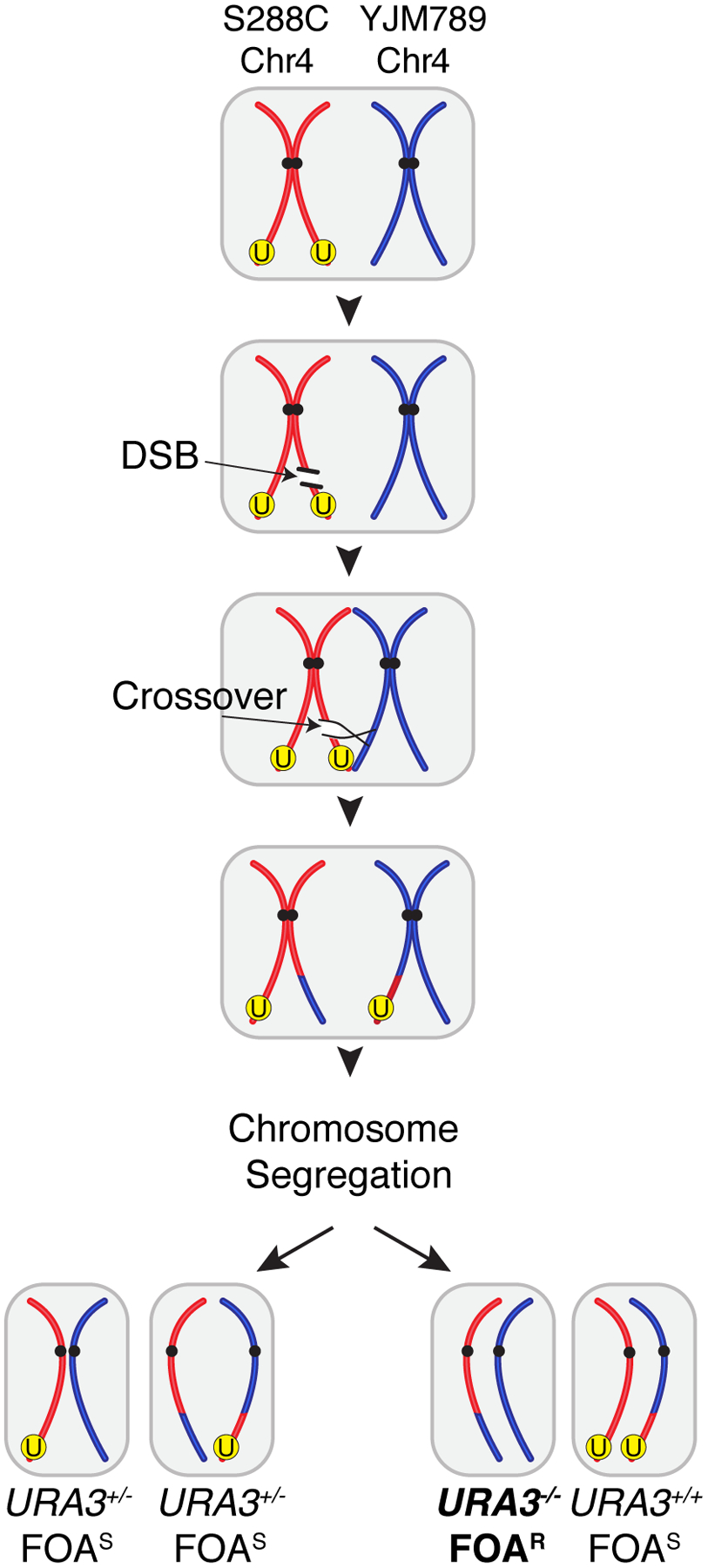
Inter-homolog allelic mitotic recombination leading to loss-of-heterozygosity (LOH) of a counter-selectable marker. The sequence of panels (from top to bottom) illustrates the steps of a DSB repair event by HR. Other possible scenarios such as gene conversion and break induced replication are not shown. A pair of duplicated homologous chromosomes represent the S288c/YJM789 hybrid background; S288c in red, YJM789 in blue. Black circles represent centromeres, a “U” labeled yellow circle represents the counter-selectable URA3 marker which has been inserted at a distal region of the chromosome. One of the chromatids in the S288c homolog sustains a DSB. Repair of the DSB occurs through HR using as template the allelic position from a chromatid in the YJM789 homolog. The recombination intermediate is resolved as a crossover resulting in a reciprocal exchange of genetic information. In the subsequent mitosis, two possible outcomes of segregation of the parental and recombinant chromatids produce genetically distinct sets of daughter cells. Left: one daughter cell receives two parental chromatids while the other receives two recombinant chromatids. Both daughter cells remain heterozygous for the URA3 marker insertion and are thus 5-FOA sensitive, although HetSNP phasing is altered in one of them. Right: each daughter cell receives one parental and one recombinant chromatid. One cell becomes homozygous for the S288c sequences at positions distal to the crossover site, including the URA3 marker insertion and remains sensitive to 5-FOA. The other daughter cell (bold) becomes homozygous for the YJM789 sequences, thus losing the URA3 marker and becoming resistant to 5-FOA.
To prepare a culture from which to select clones that have experienced LOH of URA3, we streak cells of the above strain to a rich YPD plate (1% yeast extract, 2% peptone, 2% dextrose, 2% bacteriological agar) at a low density that enables single colonies to grow in isolation. Colonies are grown for 2 days at which point they are individually picked up with a sterile toothpick and inoculated into a tube with 5 mL of YPD to expand the culture during overnight incubation. The following morning, each culture is appropriately diluted and plated on synthetic complete media (0.17% yeast nitrogen base without amino acids, 0.14% complete drop out mix, 0.5% ammonium sulfate, 2% dextrose, 2% bacteriological agar) supplemented with 1 g/L 5-FOA such that individual 5-FOA resistant colonies can be selected. Plates are incubated at 30C until colonies are visible (~2–3 days). To ensure independence between clones, a single 5-FOA resistant colony from each plated culture is then picked using a sterile toothpick and patched to a new 5-FOA plate in order to expand the clone under selective conditions. Next, cells from this patch are inoculated into a tube with 5 mL of YPD and grown overnight. From this saturated culture, an aliquot is frozen in glycerol, and remaining cell pellet is used for genomic DNA isolation and subsequent Illumina WGS. Further protocol details regarding the use of this type of assay for quantitative LOH rate determination have been described in our previous work (Conover et al. 2015; Cornelio et al. 2017; Rodrigues Prause et al. 2018).
Whole genome Illumina sequencing:
Genome sequencing involves two primary steps that are not described in this protocol: Preparation of sequencing libraries from genomic DNA, and processing of the libraries in a sequencer instrument to generate sequencing read data. These two steps can be outsourced completely to a core facility or a service provider, or carried out entirely in-house if the equipment is available. Most small labs do not have a next-generation sequencer, but the materials and equipment needed for library preparation are relatively simple and generally available. Thus, many users will prepare their own libraries, and then ship them out for sequencing at a core facility or private vendor. Preparing libraries in-house significantly decreases the overall sequencing costs. However, initially, outsourcing both library preparation and sequencing may produce more consistent sequencing data output until the user becomes confident enough to prepare their own high quality libraries. Protocols for genomic DNA isolation and library preparation of yeast genomes can be found at the following references (Wilkening et al. 2013; Baym et al. 2015). Libraries should then be sequenced using an Illumina sequencing platform to generate reads of approximately 150 bp in length. The reads do not necessarily need to be pair-ended for the analysis described here, although doing so can provide information with which to characterize structural genomic variation. Whether one chooses to generate single or paired end reads, at least for the analysis described here, will come down to the cost of library preparation kits and sequencing. In order to reliably call LOH at HetSNPs, we aim to produce an overall read coverage of between 60x and 120x. In our experience, coverage of 90x supports optimal downstream analysis, whereas coverage above 120x has diminishing returns for the type of LOH analysis described here. Multiplexing approaches appropriate for the specific sequencing instrument’s data generation capacity should be used in order to achieve these coverage targets while maximizing the number of LOH clones analyzed per sequencer lane and minimizing cost.
Computational analysis of sequencing data to identify LOH tracts:
Although the availability and affordability of WGS technologies has greatly increased, the computational analysis of sequencing data remains a limiting step and is often intimidating to classically trained geneticists, biochemists, and cell and molecular biologists. We admit sharing this hesitancy and consider ourselves to be in that generation of colleagues who are not yet fully versed in computational methods. With this in mind, we describe a generalized workflow, using primarily commercial bioinformatics software packages, that allows researchers with limited command line programming skills to process Illumina sequencing data in order to extract single nucleotide variation information and ultimately identify LOH tracts in yeast clones. We describe each step of the analysis in broad terms, as many of the specific actions will depend on the software used. We use the CLC Genomics Workbench proprietary software (Qiagen) in our laboratory due to its intuitive graphical user interface and broad sequencing-based applicability. However, comparable analyses can be achieved using other commercial packages and a number of open-source sequence read mappers such as BWA (https://sourceforge.net/projects/bio-bwa/)(Li and Durbin 2009, 2010) and Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)(Langmead and Salzberg 2012).
This workflow is designed to interrogate a specific list of genomic positions known to be heterozygous (HetSNPs) in the parent hybrid diploid strain from which a FOA-resistant LOH clone is derived. While the raw sequencing data also contains the information required to identify de novo point mutations, we do not discuss them here. In order to interrogate the specific sites mentioned above, the analysis requires that the user generate or have access to an accurate list of the reference genome coordinates and base variants of the majority of HetSNPs present in the parent diploid strain (in this protocol we refer to this as the HetSNP map). For instance, we use a refinement of the HetSNP map for the S288c/YJM789 hybrid diploid originally reported previously (Mancera et al. 2008). In the case of experiments using the JAY270 strain, we generated and reported the HetSNPs map ourselves (Rodrigues Prause et al. 2018). These HetSNP maps are then used to interrogate the heterozygous status of known positions in the genome and to filter out base variants detected at uncharacterized genomic positions.
To detect, analyze, and identify LOH tracts in selected clones, sequencing reads are processed in the following manner (for schematic, see Fig. 3):
Figure 3.
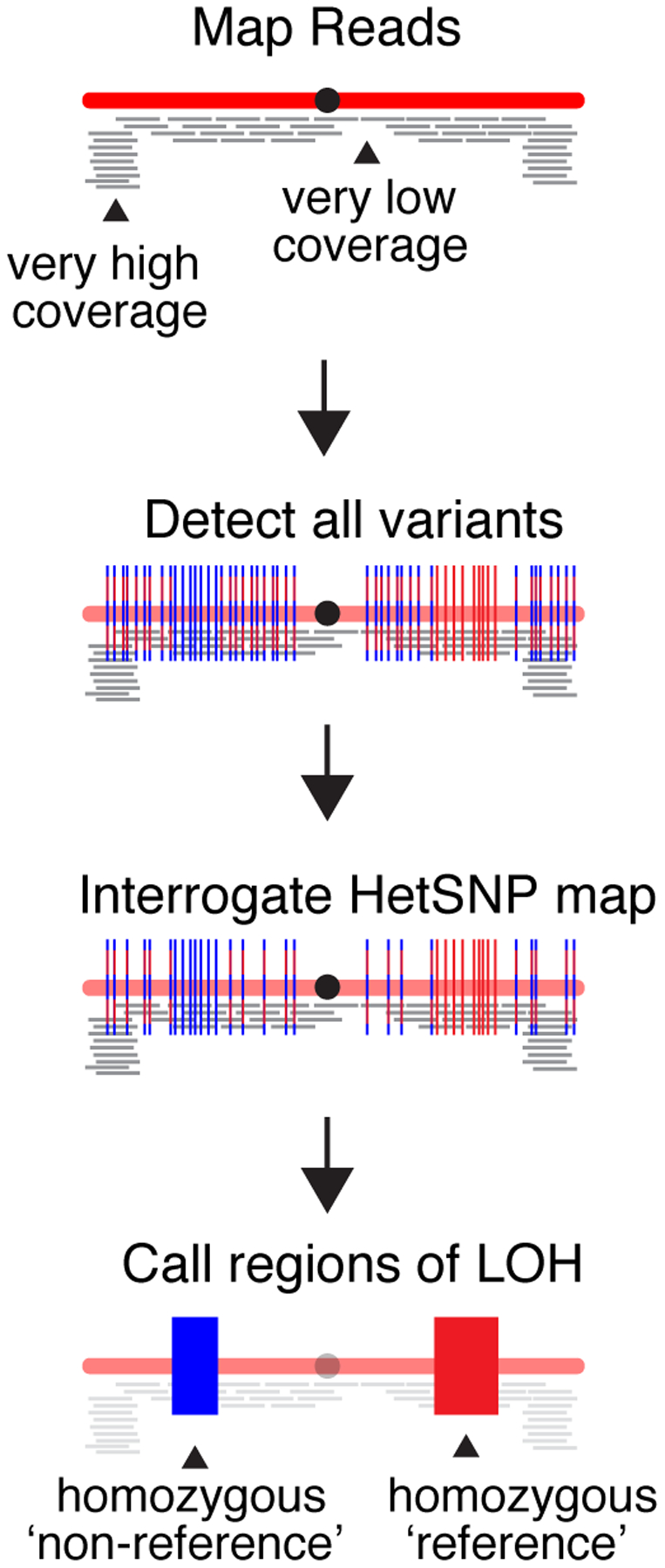
Summary of the approach used to identify tracts of LOH. Sequencing reads are mapped to the S288c reference genome (red) and regions that are excessively over- or under-represented are identified and excluded from further analysis. Next, sequence variants are detected with relaxed stringency. Variants that remained heterozygous are shown as red/blue dashed lines. Variants that have become homozygous are shown as either solid red or solid blue lines. Following low-stringency variant detection, specific allele frequency data are interrogated and retrieved for only HetSNP positions known to exist in the parent diploid. The remaining variants are filtered out and not considered further. Finally, LOH calls are made based on the allele frequencies at the HetSNP positions.
Step 1. Importing sequencing reads derived from a LOH clone:
Illumina reads are stored in FASTQ format, a file type that describes both the nucleotide sequence and the corresponding base calling quality scores. These files can be imported into any of the above read mapping programs. During import, information about the availability of paired ends is included. Paired end reads should be imported as such, even if the matching read information will not be used in the downstream steps.
Step 2. Mapping reads to the reference genome:
Once imported, sequencing reads from a LOH clone can be mapped to the reference genome. This process assigns each read to a corresponding location in the reference genome. Optional inclusion of paired-end read information may be taken into consideration at this step, although it will not influence the primary LOH analysis described here. The reference S. cerevisiae genome from the haploid strain S288c is available for download on the Saccharomyces Genome Database (SGD, https://www.yeastgenome.org)(Cherry et al. 2012). We use the most recent version (released 01-31-2015), although it is essential to keep the reference genome version specifically coordinated with the strain’s HetSNPs map (see Notes). Under the SEQUENCE tab on the SGD home page, choose DOWNLOAD. Next, choose the S288c_reference/ folder. Then, choose the genome_releases/ folder. Finally, download the S288c_reference_genome_Current_Release.tgz file. This file contains full chromosomal sequence assemblies as well as the associated annotations of the sequence features. The S288c reference genome contains 17 contigs, one for each of the 16 nuclear chromosomes as well as a single contig of the mitochondrial genome. The mitochondrial genome is not used for the LOH analysis described here.
Sequencing reads spanning repetitive regions may map to more than one genomic site. Due to this ambiguity, these reads should be ignored during the downstream analysis. In addition, some reads may fail to map to any portion of the S288c reference genome. This may be because they are unique to the non-reference parent of the hybrid strain (i.e., YJM789), because they are of poor quality, or because they originate from a contaminating source. These data are ignored in the analysis described here because the HetSNP map does not contain any bases present in the mitochondrial chromosome, or at repetitive elements, or within regions of structural variation.
Step 3. Identification of all sequence variants:
Once reads derived from a LOH clone have been mapped onto the reference genome, a software function is run to detect the presence of sequence variants between the reads and the reference genome. In our case, we use CLC Genomics Workbench to detect these variants, and we keep the following variables in mind and adjust detection parameters accordingly. In general, the parameters that we set for variant detection are quite liberal, in the sense that even minor frequency differences (resulting from sequencing errors or mis-mapped reads) will be tolerated and populated into a long variant detection output table (VDOT) containing all detected variants. This is not a concern because we ultimately filter this table to extract data only for the highly specific HetSNPs map. The use of such liberal parameters for variant detection ensures that positions in the HetSNP map that remained heterozygous in the LOH clone have a high probability of being detected and populated in the VDOT. Our liberal settings enable us to detect variants at these positions even in cases where coverage was relatively low, or if the measured allele frequency deviated too far from 50% due to stochasticity in the sequencing library. These are the parameters we typically use for variant detection:
Fixed ploidy SNP detection: This parameter enables variant detection with the assumption that the genome of the sequenced LOH clone is diploid. This is useful for initial automated prediction of LOH in CLC, but is not strictly necessary for the interrogation of HetSNP positions described below. Furthermore, and as we discuss below, the assumption that the experimentally-derived LOH clone is in fact diploid is not always correct. Copy number analysis should be conducted to validate and supplement the sequence-based genotype calls.
Discard positions with excessively high coverage: As stated above, some genomic segments are present at multiple copies in the genome or share high similarity to other sequences (e.g. transposable elements, rDNA, telomeric and subtelomeric sequences). Reads can and will often be erroneously mapped, resulting in artificially high coverage at these repetitive regions. Because it is difficult to confidently call variants in these regions, these over-represented sequences should be ignored from the subsequent analysis. To determine the upper coverage limit value, we first examine the read mapping file and evaluate the median coverage for the whole genome and for specific non-repetitive regions. We then set the high coverage cutoff at 5–8x the median coverage.
Set the minimum read coverage: Just like over-represented sequences, under-represented sequences can also challenge the confidence of variant detection. Thus, we typically exclude sequences/regions that are present at less than ~20% the median coverage.
Set the minimum variant count for detection at 1: When performing sequence analyses of clonal derivatives of heterozygous diploids (e.g., a clone that was selected for LOH), three classes of genotypes need to be identified for each HetSNP position in the parent strain: A/B, loci that remained heterozygous in the experimentally-derived LOH clone (frequency of non-reference nucleotide [i.e., YJM789] ~50%); B/B, loci that became homozygous for the non-reference nucleotide (frequency of non-reference nucleotide >95%); and A/A, loci that became homozygous for the reference nucleotide (frequency of non-reference nucleotide <5%). The first two genotype classes are easily detected directly because a large proportion (>50%) of reads differ specifically from the reference, and are rarely missed during the SNP variant detection. A/A tracts of homozygosity are more challenging to call because the majority of reads, and occasionally all of them, will match to the reference genome and will not be detected as a variant position, and thus will not be populated into the VDOT. In order to boost our ability to identify A/A tracts of homozygosity, we set an extremely relaxed stringency parameter for SNP variant detection (minimum variant count = 1). As discussed above, this parameter will allow many genomic positions to be included in the VDOT, including variant nucleotides that arose from amplification or base calling errors during sequencing. However, because we have a specific HetSNP map for all heterozygous positions present in the parental genome, we are able to recognize these artifacts and interrogate the genomic positions of interest to determine that they have become homozygous for the reference. For instance, consider a heterozygous genomic position in which the reference base is T and the non-reference base is A. If this site is heterozygous, then ~50% of the reads mapped to that position will contain T and the other 50% will contain A. However, if this site was mapped with similar coverage as the rest of the genome, and 97% of the reads contained T and 3% contained a C (which is neither the reference or the non-reference base), we would conclude that this position had become homozygous for the reference base. If instead of 97%, 100% of the reads matched the reference base at that site, it would not be included in the VDOT. While we can indirectly infer that this type of site had likely become homozygous by examining the genotypes at neighboring HetSNPs, we also take the following additional measures to ensure that we correctly call the homozygous reference region. First, we always conduct a simple visual inspection of the read mapping file at the HetSNP positions that were called homozygous reference by an indirect inference. Second, we can map the same sequencing reads to the non-reference genome (e.g., to YJM789 instead of S288c; see Notes). In doing so, genotype classes A/B and A/A are called directly, whereas genotype B/B is called through indirect inference. Combining and cross-referencing the analyses based on the two independent read mappings to each haploid parent reference genome removes any ambiguity and enhances the resolution of genotype calls to equal all positions on the HetSNP map.
Run the variant detection function: Once the above parameters are specified, the software will analyze the whole read mapping file and create a VDOT containing each detected polymorphic position, the chromosome and nucleotide coordinate of the reference base, the identity of the reference base itself, the identity of the variant bases detected at that position, and the total coverage and specific number of reads displaying each of the base variants. Note that this table also contains lines that show small insertions and deletion polymorphisms (indels) of +/− 1 or a few bases between the two haploid genomes that make up the hybrid diploid. In some cases, this information can be used later to refine the resolution of LOH tract breakpoints, but will not be used initially because the HetSNP map contains only single base variants.
Step 4. Interrogation of the VDOT to call genotypes at HetSNPs:
The variant detection procedure described above outputs a long VDOT containing a low stringency list of variants between the sequencing reads and the reference genome. This table can be exported as a Microsoft Excel spreadsheet, where it is most convenient to perform the final formatting and LOH tract identification using built-in Excel functions. First, we refine the VDOT to isolate only the genomic positions present in the HetSNP map for the parental strain. Specifically, we use Excel’s VLOOKUP function to retrieve only data from the VDOT rows corresponding to the coordinates in the HetSNP map specific to the hybrid diploid used. To identify VDOT rows containing known HetSNPs, we use a unique search term called a MarkerID that consists of the chromosome number, the underscore symbol, and the nucleotide coordinate within the reference chromosome (e.g., Chr03_157963; Chr14_75492). As discussed above, genomic positions that have become 100% homozygous for the reference base will not be detected, and will not be present in the VDOT. Thus, the VLOOKUP approach will not be able to identify a row specifying nucleotide information for that genomic position. Those positions are relatively uncommon when coverage is higher than 50–60x. The genotype calls in those cases are made initially through indirect inference, followed by independent positive validation as described above.
Once retrieved, the base count data for the HetSNP positions are used to calculate overall coverage and individual base frequencies in the reads. Those data are used to make the genotype calls at each HetSNP.
Positions that remained heterozygous (A/B) in the sequenced clone have 40–60% of reads that match the reference genome. Higher sequencing coverage will narrow this distribution closer to ~50%. Cases where the frequency of the reference base is close to 33% or 66% indicate a possible copy number gain from 2 copies in the parent strain to 3 copies in the sequenced clone (see below).
Positions that became homozygous for the non-reference base (B/B) or that simply lost the reference base through a deletion (B/-) have <5% of reads that match the reference base and >95% that match the non-reference base.
Position that became homozygous for the reference base (A/A) or that simply lost the non-reference base through deletion (A/-) have >95% of reads that match the reference genome. In cases where 100% of reads match the reference base, the call needs to be made by indirect inference.
Mitotic recombination often affects continuous segments of the genome, rather than individual bases independently. Because these events typically affect multiple neighboring HetSNPs within the same recombination tract, we are able to enhance the confidence of genotype calls by placing them in the context of a regional segment. When we make an indirect inference of the A/A genotype, it is helpful to assess the heterozygosity of HetSNPs surrounding that individual position. Ultimately, though, these regions should be reviewed by visually examining the mapped sequencing reads, and in some cases, confirmation of a genotype may require validation using PCR and Sanger sequencing.
Step 5. Copy number analysis:
The HetSNP analysis described above is not sufficient to differentiate between copy neutral LOH (e.g., crossover type; Fig. 2) and LOH caused by segmental deletion or chromosome loss. It is essential to also take into consideration the depth of read coverage at the regions of genotypic change in order to unambiguously distinguish LOH mechanisms. For example, in cases where the above HetSNP analysis identified genotypes A/A or A/-, the copy neutral A/A call would likely be made initially because the rate of mitotic recombination leading to copy neutral LOH is usually 10–100x higher than the rates of either segmental deletions or chromosome loss (Fig. 4B; (Symington et al. 2014; Klein et al. 2019). However, these two possible genotype alternatives can be distinguished through the analysis of the read depth coverage information. The example shown in Fig. 4B, illustrates how an initial interpretation of copy neutral gene conversion (A/A), can be refined to a segmental deletion call after copy number analysis has been integrated into the inference. In this example, CNV analysis shows a ~50% reduction in depth coverage coinciding with the LOH tract. Similarly, cases where LOH of an entire chromosome is identified using HetSNP analysis might be initially interpreted as a case of chromosome loss (Fig. 4C). However, CNV analysis may show that coverage for that chromosome is similar to the coverage for the whole diploid genome, indicating that there are in fact two copies of the remaining homolog. Thus, instead of calling LOH due to monosomy (single copy of a chromosome), the call is refined to uniparental disomy.
Figure 4.
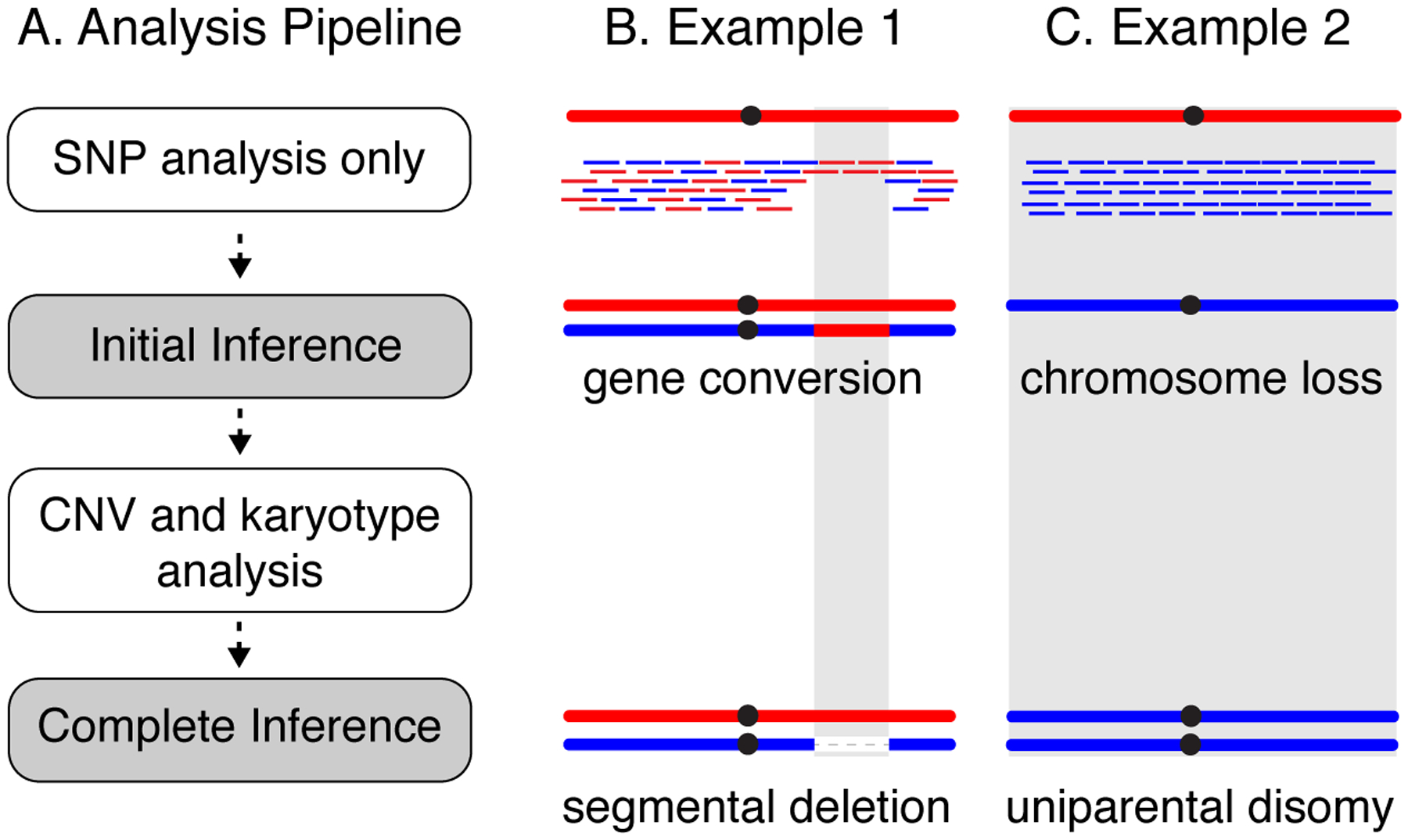
CNV analysis is used in conjunction with HetSNP analysis to refine inferences of genotype and genome structure. A) First, use HetSNP analysis to identify LOH tracts. This alone can lead to an incomplete inference of genotype and genome structure. Next, perform CNV and karyotype analysis. With these additional data, refined inferences of genotype and genome rearrangement mechanisms can be made. B) Example 1, HetSNP genotyping alone would suggest that this clone has become homozygous for a region of red homolog. However, CNV analysis detects a ~50% lower coverage in the region, indicating that that this LOH genotype is in fact caused by a segmental deletion on the blue homolog. C) Example 2, HetSNP genotyping would suggest that this clone has become monosomic due to loss of the red homolog. CNV analysis detects coverage similar to the median for rest of the genome, indicating that the clone is actually disomic for the blue chromosome (uniparental disomy).
Before the broad dissemination and cost reduction of next generation sequencing, genome-wide CNV analysis would be primarily obtained using microarray-based competitive genomic hybridization (aCGH). This technique, which is still useful and remains in use, exploits the relative annealing of genomic DNA from an experimental clone and from a reference strain to tiled arrays of oligonucleotides, each from a defined genomic position, to estimate the number of copies at which that sequence is present in the clone of interest (Argueso et al. 2008; Zhang et al. 2013). When WGS data is available, analogous relative copy number information can be extracted by examining trends in read depth coverage along the genome. For researchers like us with limited coding skills, proprietary software such as Nexus Copy Number (BioDiscovery) provide powerful, yet accessible genome-wide CNV determination and visualization using BAM read mapping files as input. When using this approach, the sequencing coverage of the parental diploid strain is used as a reference for normalization. Pairwise coverage comparison between the read depth in the experimental LOH clone and in the parental strain identifies under-, neutral, or over-represented gene dosage regions. The output of CNV calls from Nexus Copy Number includes specific coordinates where the copy number changes occurred, and that information is then compared to the HetSNP genotype calls to refine the assessment of genomic changes in the experimental clones (Fig. 4). Supplementation of these analyses with physical chromosome length measurement by pulse-field gel electrophoresis (PFGE) karyotyping can also be used to further characterize changes chromosome structure and aneuploidy.
4. NOTES and CONCLUSIONS
Alternative mechanisms of 5-FOA resistance:
Most of the clones resistant to 5-FOA isolated through the procedure described above loose the entire URA3 marker insertion through a mitotic recombination event leading to homozygosis at the corresponding region of the homolog lacking URA3. However, clones that became 5-FOA resistant through non-crossover mechanisms can also be recovered at low frequency. These can include segmental deletions and whole chromosome loss similar to the examples shown in Fig. 4, as well as point mutations that inactivate the URA3 gene without affecting heterozygosity or copy number in the region. Each of these possibilities can be specifically discerned through the combination of HetSNP genotyping and CNV analysis, supplemented by PFGE, PCR and Sanger sequencing.
Strain-specific HetSNP map refinement and reference genome version:
The analysis above relies on a map of HetSNPs between the two parent haploids used to create a hybrid diploid. These lists do not necessarily need to, and indeed should not, include every possible position where the two haploids differ. While such a complete HetSNP map would provide maximal resolution for the characterization of LOH tracts, it is best to use a conservative HetSNP map that contains only single base variants within unique DNA sequences. One should avoid positions that are vulnerable to read mapping errors, such as repetitive elements and homopolymer runs. Such refinement of the HetSNPs map might reduce resolution by ~10% of the actual variation between the haploids, but it substantially improves the reliability and reproducibility of the genotype calls. A good way to optimize a HetSNP map is to generate control sequencing reads directly from each of two the haploid parents as well as from the hybrid diploid, and then run those control data through multiple iterations of the procedure above. Haploid control reads should always return the expected single genotype calls and hybrid diploid control reads should always return heterozygosity for all positions in the HetSNP map. The positions that do not always conform to these strict expectations should be removed from the HetSNP map to improve the reliability of genotype calls made from reads derived from the experimental clones. Once this high confidence HetSNP map is developed, or if it is obtained from prior publications, it is essential to have the genomic coordinates in the map match the reference genome version that is used in the read mapping step. Each time the reference genome is updated (Cherry et al. 2012), there are usually minor changes in the coordinates for each nucleotide between versions. If the coordinates in the HetSNP map are from an older version of the reference genome, they can be corrected to match the latest genome release, or the corresponding older version of the reference genome should be downloaded and used for read mapping.
Mapping reads to the non-reference genome:
As mentioned above, a good way to resolve ambiguities that may emerge from indirect calling of regions of homozygosity for the reference genome base (e.g., the A/A genotype) is to use the orthogonal approach of mapping the sequencing reads to the non-reference genome. In doing so, the same positions (those that contain the reference base) will now be detected as variants relative to the non-reference genome. In principle, these two independent read mappings should be used. However, in practice, mapping reads to a non-reference genome is more challenging because the assembly of non-S288c genomes, including the YJM789 genome (Wei et al. 2007), is not as contiguous and complete. Instead, these diverged genomes are usually available for download as a collection of contigs that are much more numerous and smaller than the 16 nuclear chromosomes assembled for the S288c genome. Therefore, interpreting the mapping of reads to a non-reference genome requires the additional step of generating a good alignment between the reference and non-reference genome assemblies, and deriving a set of corresponding coordinates between the two. A reasonable, but effective alternative approach is to conduct non-reference read mappings primarily for specific genomic segments that are assembled into longer contigs of the non-reference genome, and for situations where the homozygous reference LOH call (A/A) cannot be independently validated by direct visualization of mapped reads to the S288c reference genome assembly.
Conclusions:
Here, we have provided a discussion of how the combination of traditional selectable marker-based genetic assays (such as LOH, (Klein et al. 2019)) with WGS sequencing and CNV analyses can provide genome-wide context and characterization of the qualitative spectrum of recombination events. The discussion and workflow included in this chapter are intended to enable classically trained molecular biologists and geneticists with limited experience in sequence analysis to conceptually understand the above approach, adapt and apply it to their own studies and experimental models, and ultimately advance our collective knowledge of the causes, consequences and mechanisms of genomic instability.
ACKNOWLEDGEMENTS
Mitotic recombination and general genome stability research in the Argueso laboratory was supported by NIH grant R35GM119788 to JLA.
Keywords and Abbreviations
- DSBs
Double-strand breaks
- HR
Homologous Recombination
- WGS
Whole Genome Sequencing
- LOH
Loss of Heterozygosity
- SNPs
Single Nucleotide Polymorphisms
- HetSNPs
Heterozygous Single Nucleotide Polymorphisms
- PFGE
Pulsed-Field Gel Electrophoresis
- aCGH
Microarray-based Comparative Genomic Hybridization
- VDOT
Variant Detection Output Table
REFERENCES
- Argueso JL, Carazzolle MF, Mieczkowski PA, Duarte FM, Netto OV, Missawa SK, Galzerani F, Costa GG, Vidal RO, Noronha MF, Dominska M, Andrietta MG, Andrietta SR, Cunha AF, Gomes LH, Tavares FC, Alcarde AR, Dietrich FS, McCusker JH, Petes TD, Pereira GA (2009) Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production. Genome Res 19 (12):2258–2270. doi: 10.1101/gr.091777.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA (2008) Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci U S A 105 (33):11845–11850. doi: 10.1073/pnas.0804529105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso LC, de Amorim HV, de Oliveira AJ, Lopes ML (2008) Yeast selection for fuel ethanol production in Brazil. FEMS Yeast Res 8 (7):1155–1163. doi: 10.1111/j.1567-1364.2008.00428.x [DOI] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R (2015) Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10 (5):e0128036. doi: 10.1371/journal.pone.0128036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5’-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Molecular \& general genetics: MGG 197 (2):345–346. doi:papers3://publication/uuid/042E2D03-B3E9-43FB-BD01-932C628D66CD [DOI] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods in Enzymology 154:164–175. doi:papers3://publication/uuid/82568CDB-07EC-4C65-A395-9EDED2E6F6DC [DOI] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ, Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M, Weng S, Wong ED (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40 (Database issue):D700–705. doi: 10.1093/nar/gkr1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover HN, Lujan SA, Chapman MJ, Cornelio DA, Sharif R, Williams JS, Clark AB, Camilo F, Kunkel TA, Argueso JL (2015) Stimulation of Chromosomal Rearrangements by Ribonucleotides. Genetics 201 (3):951–961. doi: 10.1534/genetics.115.181149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelio DA, Sedam HN, Ferrarezi JA, Sampaio NM, Argueso JL (2017) Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair (Amst) 52:110–114. doi: 10.1016/j.dnarep.2017.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Lin G, Pankajam AV, Chakraborty P, Bhat N, Steinmetz LM, Nishant KT (2017) Genome Dynamics of Hybrid Saccharomyces cerevisiae During Vegetative and Meiotic Divisions. G3 (Bethesda, Md) 7 (11):3669–3679. doi:papers3://publication/doi/10.1534/g3.117.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, Petes TD (2000) Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 97 (21):11383–11390. doi: 10.1073/pnas.97.21.11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science (New York, NY) 274 (5287):546-563-547. doi:papers3://publication/uuid/15DEC284-A24D-4364-83BF-6FB9DF29C064 [DOI] [PubMed] [Google Scholar]
- Klein HL (2001) Spontaneous chromosome loss in Saccharomyces cerevisiae is suppressed by DNA damage checkpoint functions. Genetics 159 (4):1501–1509. doi:papers3://publication/uuid/89DCC842-CEBF-4D33-B72E-56947DDEA06A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein HL, Bačinskaja G, Che J, Cheblal A, Elango R, Epshtein A, Fitzgerald DM, Gómez-González B, Khan SR, Kumar S, Leland BA, Marie L, Mei Q, Miné-Hattab J, Piotrowska A, Polleys EJ, Putnam CD, Radchenko EA, Saada AA, Sakofsky CJ, Shim EY, Stracy M, Xia J, Yan Z, Yin Y, Aguilera A, Argueso JL, Freudenreich CH, Gasser SM, Gordenin DA, Haber JE, Ira G, Jinks-Robertson S, King MC, Kolodner RD, Kuzminov A, Lambert SA, Lee SE, Miller KM, Mirkin SM, Petes TD, Rosenberg SM, Rothstein R, Symington LS, Zawadzki P, Kim N, Lisby M, Malkova A (2019) Guidelines for DNA recombination and repair studies: Cellular assays of DNA repair pathways. Microb Cell 6 (1):1–64. doi: 10.15698/mic2019.01.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9 (4):357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureau R, Loeillet S, Salinas F, Bergström A, Legoix-Né P, Liti G, Nicolas A (2016) Extensive Recombination of a Yeast Diploid Hybrid through Meiotic Reversion. PLoS Genet 12 (2):e1005781. doi: 10.1371/journal.pgen.1005781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Kokoska RJ, Petes TD (2008) Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol Cell Biol 28 (17):5359–5368. doi: 10.1128/MCB.02084-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120 (5):587–598. doi: 10.1016/j.cell.2004.12.039 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14):1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5):589–595. doi: 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene PM, Kayıkçı Ö, Granek JA, Reininga JM, Scholl Z, Murray D (2011) Outcrossing, mitotic recombination, and life-history trade-offs shape genome evolution in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 108 (5):1987–1992. doi: 10.1073/pnas.1012544108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454 (7203):479–485. doi: 10.1038/nature07135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker JH, Clemons KV, Stevens DA, Davis RW (1994) Genetic characterization of pathogenic Saccharomyces cerevisiae isolates. Genetics 136 (4):1261–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RJ, Rubinstein RG, Neil AJ, Dominska M, Kiktev D, Petes TD, Mirkin SM (2017) Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair. Genome Res 27 (12):2072–2082. doi: 10.1101/gr.228148.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR (1986) Genealogy of principal strains of the yeast genetic stock center. Genetics 113 (1):35–43. doi:papers3://publication/uuid/4B4ECA76-CC7B-4EA0-93DB-38C49F09EBFC [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell K, Jinks-Robertson S, Petes TD (2015) Elevated Genome-Wide Instability in Yeast Mutants Lacking RNase H Activity. Genetics 201 (3):963–975. doi: 10.1534/genetics.115.182725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, Hochwagen A, Keeney S (2011) A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144 (5):719–731. doi: 10.1016/j.cell.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, Cruaud C, Labadie K, Aury J-M, Istace B, Lebrigand K, Barbry P, Engelen S, Lemainque A, Wincker P, Liti G, Schacherer J (2018a) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. doi:papers3://publication/uuid/A69F80AD-26ED-4982-9809-8783E47F09F0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue JX, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, Cruaud C, Labadie K, Aury JM, Istace B, Lebrigand K, Barbry P, Engelen S, Lemainque A, Wincker P, Liti G, Schacherer J (2018b) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556 (7701):339–344. doi: 10.1038/s41586-018-0030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues Prause A, Sampaio NMV, Gurol TM, Aguirre GM, Sedam HNC, Chapman MJ, Malc EP, Ajith VP, Chakraborty P, Tizei PA, Pereira GAG, Mieczkowski PA, Nishant KT, Argueso JL (2018) A Case Study of Genomic Instability in an Industrial Strain of Saccharomyces cerevisiae. G3: Genes|Genomes|Genetics. doi:papers3://publication/doi/10.1534/g3.118.200446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio NMV, Rodrigues Prause A, Ajith VP, Gurol TM, Chapman MJ, Malc EP, Chakraborty P, Duarte FM, Aguirre GM, Tizei PA, Pereira GAG, Mieczkowski PA, Nishant KT, Argueso JL (2017) Mitotic systemic genomic instability in yeast. bioRxiv:161869. doi:papers3://publication/doi/10.1101/161869 [Google Scholar]
- Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P (2013) amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 13 (1):126–139. doi: 10.1111/1567-1364.12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Dominska M, Greenwell PW, Petes TD (2014) Genome-wide high-resolution mapping of chromosome fragile sites in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 111 (21):E2210–2218. doi: 10.1073/pnas.1406847111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Charles J, Hazkani-Covo E, Yin Y, Andersen SL, Dietrich FS, Greenwell PW, Malc E, Mieczkowski P, Petes TD (2012) High-resolution genome-wide analysis of irradiated (UV and γ-rays) diploid yeast cells reveals a high frequency of genomic loss of heterozygosity (LOH) events. Genetics 190 (4):1267–1284. doi: 10.1534/genetics.111.137927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Charles J, Petes TD (2013) High-resolution mapping of spontaneous mitotic recombination hotspots on the 1.1 Mb arm of yeast chromosome IV. PLoS Genet 9 (4):e1003434. doi: 10.1371/journal.pgen.1003434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strope PK, Skelly DA, Kozmin SG, Mahadevan G, Stone EA, Magwene PM, Dietrich FS, McCusker JH (2015) The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res 25 (5):762–774. doi: 10.1101/gr.185538.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M (2014) Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198 (3):795–835. doi: 10.1534/genetics.114.166140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik OW, Papasian CJ, Dixon AY, Potter LM (1989) Saccharomyces cerevisiae pneumonia in a patient with acquired immune deficiency syndrome. J Clin Microbiol 27 (7):1689–1691. doi:papers3://publication/uuid/0EA423B4-8C95-43AB-A8C6-C04549FB3A38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, McCusker JH, Hyman RW, Jones T, Ning Y, Cao Z, Gu Z, Bruno D, Miranda M, Nguyen M, Wilhelmy J, Komp C, Tamse R, Wang X, Jia P, Luedi P, Oefner PJ, David L, Dietrich FS, Li Y, Davis RW, Steinmetz LM (2007) Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proceedings of the National Academy of Sciences of the United States of America 104 (31):12825–12830. doi:papers3://publication/doi/10.1073/pnas.0701291104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkening S, Tekkedil MM, Lin G, Fritsch ES, Wei W, Gagneur J, Lazinski DW, Camilli A, Steinmetz LM (2013) Genotyping 1000 yeast strains by next-generation sequencing. BMC Genomics 14:90. doi: 10.1186/1471-2164-14-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zeidler AFB, Song W, Puccia CM, Malc E, Greenwell PW, Mieczkowski PA, Petes TD, Argueso JL (2013) Gene copy-number variation in haploid and diploid strains of the yeast Saccharomyces cerevisiae. Genetics 193 (3):785–801. doi:papers3://publication/doi/10.1534/genetics.112.146522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng DQ, Petes TD (2018) Genome Instability Induced by Low Levels of Replicative DNA Polymerases in Yeast. Genes (Basel) 9 (11). doi: 10.3390/genes9110539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng DQ, Zhang K, Wu XC, Mieczkowski PA, Petes TD (2016) Global analysis of genomic instability caused by DNA replication stress in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 113 (50):E8114–E8121. doi: 10.1073/pnas.1618129113 [DOI] [PMC free article] [PubMed] [Google Scholar]