

Multiple system atrophy (MSA) is a sporadic neurodegenerative disease clinically marked by autonomic failure and variable degrees of parkinsonism and cerebellar ataxias. The average age of onset is in the sixth decade of life with a five-year survival, although fulminant and more benign courses have been described (1). The pathologic hallmark of MSA is abundant α-synuclein-positive protein aggregates within oligodendrocytes (glial cytoplasmic inclusions, GCI) and neurons (neuronal cytoplasmic inclusions, NCI, and neuronal nuclear inclusions, NNI). The density of GCIs has been correlated with disease duration and severity of neuronal loss (2). Here, we present an unusual case of MSA clinically characterized by rapid decline followed by a 20-year disease duration in a severely affected state. At autopsy, the brain showed severe gliosis and neuronal loss in a pattern characteristic of MSA. Additionally, there were atypical Pick body-like and ring-shaped α-synuclein inclusions, most prominent in limbic regions, consistent with a recently described subtype of MSA designated “atypical MSA” (3). Four different α-synuclein antibodies highlighted only scant α-synuclein-positive pathology; however, Gallyas and Campbell-Switzer silver stains identified additional GCIs most prominent in subcortical white matter. Interestingly, many of the α-synuclein-positive limbic inclusions were Gallyas-negative, while many of the Gallyas-positive inclusions were α-synuclein-negative (4). This observation of markedly disparate α-synuclein immunohistochemical versus Gallyas reactivity of MSA inclusions is unusual and could support the possibility that MSA inclusions have variable compositions, something which has been recently suggested (5).
A 45-year-old Vietnamese woman presented with left upper extremity stiffness, light-headedness on standing, constipation, and urinary incontinence. Upon physical examination, orthostatic hypotension and parkinsonism were present in the absence of cerebellar signs, meeting clinical criteria for MSA-Parkinsonism (MSA-P) (6). There was no family history of parkinsonism or other neurodegenerative disease. Levodopa failed to significantly improve her parkinsonism. She started falling two years after diagnosis and was wheelchair-bound one year later. Seven years into her disease course, the patient developed stridor and vocal cord paralysis necessitating tracheostomy and dysphagia necessitating gastrostomy tube. Brain MRI, performed eleven years post-diagnosis, showed severe atrophy of the pons, cerebellum, basal ganglia, and frontal lobes, with severe hydrocephalus ex vacuo (Figure 1a). T2 hyperintensities of the pontine crossing fibres (hot-cross bun sign) and gliosis of the posterior putamen were observed, both characteristic of MSA (Figure 1b) (7). Genetic testing was not performed. Over the next 10 years, she suffered from anarthria and severe rigidity, resulting in complete dependence on caregivers. She died at age 66, twenty-one years after her initial diagnosis of MSA-P and her brain was donated (informed consent obtained).
Figure 1. Radiologic, gross anatomic, and histologic neurodegenerative features of a brain with longstanding atypical MSA.
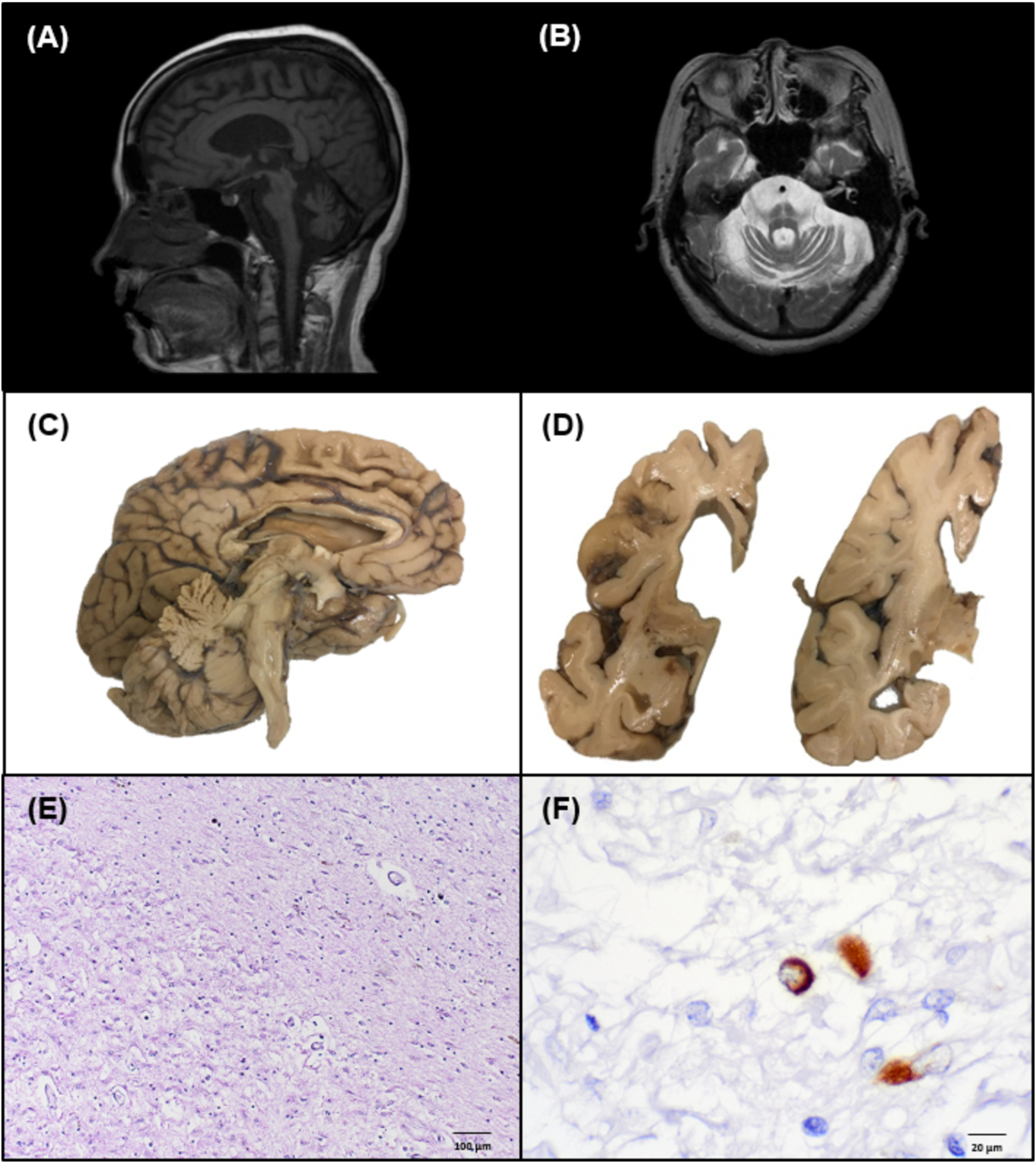
(A) Midsagittal T1-weighted MRI showing olivopontocerebellar atrophy. (B) Axial T1-weighted MRI showing cerebellar and pontine atrophy with increased T2 signal characteristic of the “hot cross bun sign” within the pons. (C) Mid-sagittal view of left hemibrain exhibiting olivopontocerebellar atrophy, medial frontal lobe atrophy, and cingulate gyrus atrophy. (D) Coronal view of left hemibrain highlighting basal ganglia, hippocampal atrophy, and hydrocephalus ex vacuo. (E) H&E stained section of the striatum demonstrating severe neuron loss, rarefaction, and gliosis. (F) Rare glial cytoplasmic inclusion identified on α-synuclein staining (81A IHC).
At autopsy, the formalin-fixed brain weighed 852 grams and showed severe frontal atrophy, moderate temporal and parietal atrophy, and severe hydrocephalus ex vacuo (Figure 1c). There was severe atrophy of the midbrain, pons, and cerebellum. Coronal sections showed severe atrophy of subcortical white matter and thinning of the corpus callosum. The caudate, putamen, and globus pallidus were severely atrophic with relative sparing of the thalamus (Figure 1d). There was moderate atrophy of the amygdala and severe atrophy of the hippocampus with relative sparing of the entorhinal cortex. The substantia nigra and locus coeruleus were completely depigmented. The cerebellar white matter was severely atrophic.
Haematoxylin and eosin (H&E)-stained neocortical sections exhibited moderate neuronal loss and vacuolation of cortical layer 2. There was marked white matter gliosis and rarefaction most notable in the frontal, temporal, and inferior parietal lobes. The hippocampus exhibited moderate gliosis and neuronal loss in CA1 and CA4, though no overt hippocampal sclerosis. Severe neuronal dropout was seen in the caudate, putamen, and globus pallidus with extensive rarefaction, mineralization, and gliosis especially in the globus pallidus (Figure 1e). The substantia nigra and locus coeruleus were devoid of neuromelanin-bearing neurons. The cerebellum demonstrated severe white matter rarefaction.
Immunohistochemistry for β-amyloid (ab69, 1:1,400, Dr. Edward Koo) showed sparse neuritic and diffuse plaques within the frontal and entorhinal cortices, diffuse plaques in the caudate, putamen, globus pallidus, midbrain, and focal staining in the cerebellum (Thal phase 5) (8). There was widespread severe amyloid angiopathy. Campbell-Switzer silver stain (performed by Dr Switzer’s lab, NeuroScience Associates) showed sparse neuritic plaques (CERAD A). Tau immunohistochemistry (PHF-1, 1:600, Dr Peter Davies) confirmed sparse neuritic plaques and frequent neurofibrillary tangles (NFTs) in the transentorhinal cortex and cornu ammonis (CA) regions with rare NFTs in the temporal cortex (Braak stage II/VI). Tau immunohistochemistry specific for 3R or 4R species (05–803,1:750, Millipore; 05–804, 1:200, Millipore) showed both 3R and 4R-positivity in entorhinal cortex, consistent with AD-type pathology. Overall, AD neuropathologic change was low (A3 B1 C1) (8). No pathological TDP-43 or TAF-15 positive inclusions were present (10782–2-AP, 1:12,000, Proteintech; TAFII68, 1:600, Bethyl).
α-synuclein immunohistochemistry (81A,1:15,000, Biolegend) highlighted scant (<10/slide) GCIs in the pons, midbrain, cingulate gyrus, hippocampus and basal ganglia (Figure 1f). More abundant but still rare NCIs and needle-like NNIs were also observed in these regions (Figure 2a). Scattered ring-shaped inclusions in neurons and oligodendroglia in upper cortical layers were observed, most prominent in frontal and inferior parietal regions. No astroglial α-synuclein immunoreactivity was observed in the glia limitans. There was a relative abundance of α-synuclein-positive inclusions in limbic regions compared to brainstem, cerebellum, or cortex. Immunohistochemistry for total tau (PHF-1) and α-synuclein each stained Pick body-like neuronal cytoplasmic inclusions in the posterior amygdala (Figure 2b,c) (3,9). Interestingly, these structures were also variably positive for 3R and 4R tau, as well as Campbell-Switzer silver stain. α-synuclein and tau also highlighted NFT-like inclusions in CA regions of hippocampus (Figure 2d) and ring-shaped α-synuclein-positive inclusions were seen in the dentate gyrus (Figure 2e) (10). Due to the limited identification of α-synuclein-positive inclusions, immunohistochemistry was repeated with three additional α-synuclein antibodies (MJF-R13, 1:20,000, Abcam; SYN303, 1:1,000, Biolegend; LB509, 1:200, Invitrogen), in parallel to a typical MSA control case. The typical MSA case showed abundant GCIs as well as NCIs and NNIs (not shown), confirming the scant α-synuclein-immunoreactive GCIs, NCIs, and NNIs in this case (Figure 2h). Staining with Gallyas silver stain highlighted additional GCIs and NCIs in subcortical white matter (Figure 2h). while much of the limbic α-synuclein-positive pathology was not demonstrated on Gallyas stain (Figure 2f). In a more typical case of MSA, the Gallyas method demonstrated a large number of the characteristic GCIs (not shown). Campbell-Switzer staining also showed GCIs and NCIs as above but to a greater degree in cingulate gyrus and subcortical white matter (Figure 2j) and more frequently highlighted ring-like inclusions in the hippocampus (Figure 2g).
Figure 2. Immunohistochemical and histochemical characterization of limbic-predominant pathology in brain with longstanding MSA.
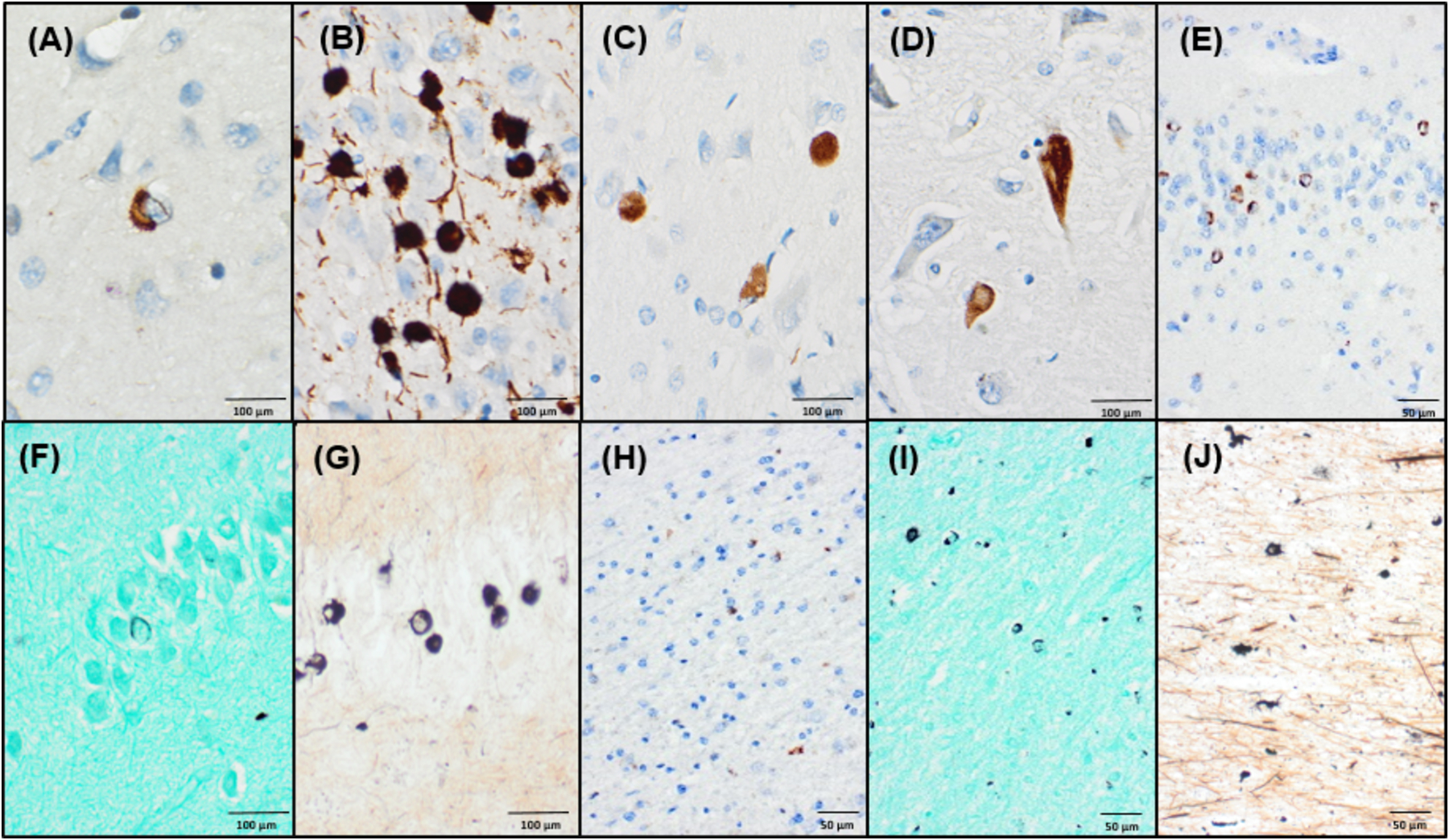
(A) α-synuclein (81A) positive neuronal cytoplasmic inclusion with a perinuclear skein in cingulate gyrus. (B) Tau (PHF-1) positive Pick-body like neuronal cytoplasmic inclusions seen in the posterior amygdala. (C) Many Pick-body like inclusions are also reactive for α-synuclein (81A). (D) α-synuclein (81A) positive neurofibrillary tangle in the CA1 region of the hippocampus. (E) Abundant α-synuclein (LB509) positive ring-like neuronal cytoplasmic inclusions are seen in the hippocampal dentate fascia. Only sparse ring-like inclusions are detected on Gallyas silver stain in the dentate fascia (F), compared to more abundant agyrophilic inclusions on Campbell-Switzer staining (G). (H-J) In frontal subcortical white matter, only sparse GCIs are detected by α-synuclein (LB509) IHC (H), but Gallyas staining (I) and Campbell-Switzer staining (J) more frequently highlight GCI pathology.
Overall, this case of clinical MSA-P of long duration showed macroscopic pathologic features of MSA, including both striatonigral degeneration and olivopontocerebellar atrophy, but additionally showed frontotemporal and limbic atrophy. Microscopically, there were GCIs, NCIs, and NNIs pathognomonic for MSA with a limbic predominance of α-synuclein-positive pathology that included Pick body-like, NFT-like, and ring-shaped α-synuclein positive NCIs in the hippocampus (1). Pathologically, this case is most consistent with a rare subtype of MSA Aoki et al. called “atypical MSA” or “FTLD-synuclein” (3). In 2015, these authors reported four patients with clinical features of frontotemporal dementia and pathological features including: 1) varying degrees of frontal and temporal atrophy associated with striatonigral degeneration and/or olivopontocerebellar atrophy, 2) varying degrees of α-synuclein-reactive GCIs from abundant to uncharacteristically sparse, 3) Pick body-like, NFT-like, and ring-shaped α-synuclein-reactive NCIs in limbic and cortical regions, and 4) co-localization of α-synuclein and tau in subset of neurons with NCIs (3). Our case shows these pathologic features. Similar NCI and hippocampal features have been documented in a very recent separate study of MSA cases of long duration that had severe hippocampal involvement, although many of the Pick body-like structures in our case were immunoreactive for PHF-1, 3R, and 4R tau, which was not observed in this report (11). Additionally, in our case α-synuclein-immunoreactive pathology was incongruously sparse given the longstanding disease duration, confirmed using four different α-synuclein antibodies against three epitopes. Interestingly, Gallyas silver staining highlighted additional GCI and NCIs but did not stain some of the α-synuclein-reactive limbic pathology, including much of the ring-shaped and NFT-like pathology.
This case also adds to a small but growing body of literature characterizing the clinical course and pathology of atypical MSA. The cases reported by Aoki et al. had prominent FTD features and an older age of onset (average of 69), although they cited a handful of previously published cases that had more typical ages of onset and who had been diagnosed with MSA clinically (3). This case met clinical criteria for MSA-P on initial examination at age 45 without evidence for FTD. Our patient had an unusual clinical course with a rapid decline, but with multiple medical interventions and extensive nursing care that likely prolonged her life. Further, she was anarthric for much of her clinical course, potentially confounding the reported absence of FTD symptoms.
This case raises interesting questions regarding α-synuclein species in MSA. First, the difference in α-synuclein immunohistochemical reactivity and Gallyas silver staining is intriguing, particularly given the anatomic distribution of lesions, in which most of the limbic pathology was strongly immunoreactive to α-synuclein but did not stain with Gallyas. Gallyas silver staining is known to react with the α-synuclein-positive inclusions of MSA but not with α-synuclein-positive Lewy bodies, while Campbell-Switzer silver staining reacts with α-synuclein positive GCIs in MSA as well as Lewy bodies (4). In this case, cingulate GCIs and ring-shaped structures in the hippocampus more frequently stained with Campbell-Switzer than Gallyas silver stain, raising the possibility that the conformation of the α-synuclein inclusions in limbic regions was closer to that of Lewy bodies than typical GCIs. Several groups have demonstrated differences in α-synuclein conformations, leading to different ‘strain’ like properties in Lewy body disease compared to MSA (12,13). These observations may not necessarily preclude the presence of multiple strains between MSA cases or within the same MSA case. The presence of sparse α-synuclein-positive inclusions despite severe disease of long duration could suggest a complex relationship between α-synuclein aggregation and disease pathogenesis in MSA. It is possible that in very longstanding disease, synuclein-positive inclusions may be partially cleared by cellular or immune-mediated mechanisms, as has been suggested in other synucleinopathies (14–16). Alternatively, it remains possible that the more abundant white matter inclusions identified on Gallyas and Campbell-Switzer stains consist of α-synuclein in a conformation that is less immunoreactive to the antibodies used, as it has been suggested that various synuclein conformers differentially expose various synuclein epitopes (17). These possibilities are speculative but warrant further study as they have ramifications for disease mechanisms and development of α-synuclein-directed biomarkers in MSA and other synucleinopathies.
Key Points.
This case describes a patient with MSA-P with an initial typical clinical decline but a 21-year survival, the majority of which was in a severely debilitated state.
Neuropathological assessment showed severe frontotemporal and limbic atrophy, widespread severe gliosis with scant α-synuclein positive glial cytoplasmic inclusions.
Unusual features including tau and α-synuclein positive Pick body-like inclusions and α-synuclein positive ring-like inclusions in the hippocampus and limbic regions were present and consistent with a pathological diagnosis of ‘atypical MSA’ or ‘FTLD-synuclein’
Campbell Switzer silver staining highlighted more abundant glial cytoplasmic inclusions than were demonstrated by multiple α-synuclein antibodies and Gallyas silver staining, suggesting the possible presence of unique α-synuclein conformations.
This report broadens the phenotypes associated with ‘atypical MSA’ or ‘FTLD-synuclein’ pathology.
Footnotes
Ethical Statement: All samples and patient information was collected according to the ethical guidelines of the UCSD ADRC in accordance with NIH.
References
- 1.Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: A review of 203 pathologically proven cases. Mov Disord [Internet]. 1997. [cited 2021 Mar 9];12(2):133–47. Available from: https://pubmed.ncbi.nlm.nih.gov/9087971/ [DOI] [PubMed] [Google Scholar]
- 2.Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations. Brain [Internet]. 2004. Dec [cited 2021 May 16];127(12):2657–71. Available from: https://pubmed.ncbi.nlm.nih.gov/15509623/ [DOI] [PubMed] [Google Scholar]
- 3.Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with α-synuclein. Acta Neuropathol [Internet]. 2015. Jul 17 [cited 2021 May 14];130(1):93–105. Available from: https://pubmed.ncbi.nlm.nih.gov/25962793/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uchihara T Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol [Internet]. 2007. May [cited 2021 Jul 18];113(5):483. Available from: /pmc/articles/PMC1868652/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hass EW, Sorrentino ZA, Lloyd GM, McFarland NR, Prokop S, Giasson BI. Robust α-synuclein pathology in select brainstem neuronal populations is a potential instigator of multiple system atrophy. Acta Neuropathol Commun 2021 91 [Internet]. 2021. May 3 [cited 2021 Jul 18];9(1):1–14. Available from: https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-021-01173-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massey LA, Micallef C, Paviour DC, O’Sullivan SS, Ling H, Williams DR, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord [Internet]. 2012/04/11. 2012;27(14):1754–62. Available from: http://onlinelibrary.wiley.com/doi/10.1002/mds.24968/abstract [DOI] [PubMed] [Google Scholar]
- 8.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2011/11/22. 2012;123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, et al. Co-localization of α-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration. Acta Neuropathol [Internet]. 2001. [cited 2021 May 18];101(3):285–93. Available from: https://pubmed.ncbi.nlm.nih.gov/11307630/ [DOI] [PubMed] [Google Scholar]
- 10.Homma T, Mochizuki Y, Komori T, Isozaki E. Frequent globular neuronal cytoplasmic inclusions in the medial temporal region as a possible characteristic feature in multiple system atrophy with dementia. Neuropathology [Internet]. 2016. Oct 1 [cited 2021 May 14];36(5):421–31. Available from: https://pubmed.ncbi.nlm.nih.gov/26970514/ [DOI] [PubMed] [Google Scholar]
- 11.Ando T, Riku Y, Akagi A, Miyahara H, Hirano M, Ikeda T, et al. Multiple system atrophy variant with severe hippocampal pathology. Brain Pathol [Internet]. 2021. [cited 2021 Aug 18];00:e13002. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/bpa.13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature [Internet]. 2018. May 24 [cited 2021 Jun 5];557(7706):558–63. Available from: https://www.nature.com/articles/s41586-018-0104-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature [Internet]. 2020. Sep 17 [cited 2021 Aug 18];585(7825):464–9. Available from: https://pubmed.ncbi.nlm.nih.gov/32461689/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009. Sep 1;35(3):385–98. [DOI] [PubMed] [Google Scholar]
- 15.Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun [Internet]. 2020. Dec 1 [cited 2021 May 14];11(1):1–14. Available from: 10.1038/s41467-020-15119-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018;557(7706):558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorrentino ZA, Goodwin MS, Riffe CJ, Dhillon J-KS, Xia Y, Gorion K-M, et al. Unique α-synuclein pathology within the amygdala in Lewy body dementia: implications for disease initiation and progression. Acta Neuropathol Commun. 2019;7(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]