

SUMMARY
Dopamine (DA)-releasing neurons in the substantia nigra pars compacta (SNcDA) inhibit target cells in the striatum through postsynaptic activation of γ-aminobutyric acid (GABA) receptors. However, the molecular mechanisms responsible for GABAergic signaling remain unclear, as SNcDA neurons lack enzymes typically required to produce GABA or package it into synaptic vesicles. Here, we show that aldehyde dehydrogenase 1a1 (Aldh1a1), an enzyme proposed to function as a GABA synthetic enzyme in SNcDA neurons, does not produce GABA for synaptic transmission. Instead, we demonstrate that SNcDA axons obtain GABA exclusively through presynaptic uptake using the membrane GABA transporter Gat1 (encoded by Slc6a1). GABA is then packaged for vesicular release using the vesicular monoamine transporter Vmat2. Our data therefore show that presynaptic transmitter recycling can substitute for de novo GABA synthesis and that Vmat2 contributes to vesicular GABA transport, expanding the range of molecular mechanisms available to neurons to support inhibitory synaptic communication.
In brief
Melani and Tritsch demonstrate that inhibitory co-transmission from midbrain dopaminergic neurons does not depend on cell-autonomous GABA synthesis but instead on presynaptic import from the extracellular space through the membrane transporter Gat1 and that GABA loading into synaptic vesicles relies on the vesicular monoamine transporter Vmat2.
Graphical Abstract

INTRODUCTION
A detailed appreciation for how neurons modify the activity of surrounding cells through the release of transmitters is key to our understanding of the brain. Because neurons rely primarily on the release of a single classical neurotransmitter to transmit signals across synapses, it is typically sufficient to manipulate the activity of presynaptic cells to evaluate their postsynaptic contribution. However, this approach is insufficient for neurons that release multiple classical transmitters. Chief among them are midbrain neurons of the substantia nigra pars compacta (SNc). These cells are best known for their ability to release dopamine (DA), but they can also directly affect the discharge of target cells in striatum through monosynaptic glutamatergic and GABAergic signaling (Hnasko et al., 2010; Tecuapetla et al., 2010; Tritsch et al., 2012; Trudeau et al., 2014). The molecular mechanisms that enable glutamate co-release from DA-releasing SNc (SNcDA) neurons are well established (Eskenazi et al., 2021). By contrast, much less is known about inhibitory co-transmission (Tritsch et al., 2016).
An important contributor to this gap in knowledge is that nigrostriatal DAergic neurons do not possess the molecular machinery thought to be essential for GABAergic synaptic transmission; they do not express the GABA synthetic enzymes Gad65 and Gad67 or the vesicular GABA transporter Vgat (Poulin et al., 2014; Saunders et al., 2018; Tritsch et al., 2012, 2014). Instead, inhibitory co-transmission from SNcDA neurons was proposed to rely on the vesicular monoamine transporter Vmat2 (Tritsch et al., 2012), which is not known to transport GABA (Yelin and Schuldiner, 1995). Several endogenous ligands activate GABAA receptors (R) (Belelli and Lambert, 2005; Johnston, 1996); it is therefore conceivable that the transmitter released by SNcDA neurons is not GABA but a ligand that functions as a GABAAR agonist and serves as a substrate for Vmat2. Another possibility is that GABA is the transmitter but that the mechanisms involved remain to be defined. Both scenarios point to an incomplete understanding of molecular pathways that neurons can leverage to inhibit postsynaptic targets.
We previously suggested that SNcDA neurons import GABA from the extracellular milieu, as they contain mRNA for the membrane GABA transporter Gat1 and pharmacological inhibition of GABA uptake impairs GABAergic co-transmission (Tritsch et al., 2014). However, a recent study found that Gat1 does not distribute to the axon of SNcDA neurons and that Gat1 inhibition non-selectively depresses vesicular release from SNcDA axons (Roberts et al., 2020). In addition, SNcDA neurons were proposed to synthetize GABA de novo using the mitochondrial enzyme Aldh1a1 (Kim et al., 2015), which a subpopulation of SNcDA neurons expresses abundantly (Liu et al., 2014; McCaffery and Dräger, 1994; Poulin et al., 2014). Here, we show that SNcDA neurons do not produce GABA using Aldh1a1 but rely instead on Gat1-mediated uptake to obtain GABA for Vmat2-dependent vesicular release. Our data therefore demonstrate that membrane GABA transporters and vesicular monoamine transporters can respectively substitute for Gad-dependent GABA synthesis and Vgat-mediated vesicular loading, expanding the range of molecular mechanisms available to neurons to support inhibitory transmission.
RESULTS
Aldh1a1 does not directly contribute to inhibitory co-transmission from SNcDA neurons
To examine how Adlh1a1 contributes to inhibitory transmission from SNcDA neurons, we obtained Aldh1a1 germline knockout mice (Aldh1a1−/−; Fan et al., 2003). Using immunofluorescence, we confirmed that Aldh1a1 is no longer detected in the brain of knockouts, including in SNcDA neurons (Figures 1A and 1B). We next bred Aldh1a1−/− mice to DatIRES-Cre knockin mice (Bäckman et al., 2006) to enable adeno-associated virus (AAV)-mediated expression of channelrhodopsin 2 (ChR2) in SNcDA neurons (Figures S1A and S1B). We obtained acute brain slices from these mice, stimulated SNcDA axons using 1-ms blue light flashes, and recorded GABAAR inhibitory postsynaptic currents (IPSCs) from striatal projection neurons (SPNs) using whole-cell voltage clamp (Figure 1C). We limited recordings to dorsal striatum where Aldh1a1+ SNcDA axons project most densely (Figures S2A and S2B). The amplitude of optogenetically evoked IPSCs (oIPSCs) in Aldh1a1−/− mice was reduced compared with controls, but not statistically different (Figure 1D). The synaptic latency (Figure 1D) and rise time of oIPSCs (Figure 1D), as well as their sensitivity to the GABAAR antagonist SR95531 (Figure S1C), were also comparable across conditions, indicating that genetic deletion of Aldh1a1 does not strongly impair the ability of SNcDA neurons to inhibit SPNs.
Figure 1. Aldh1a1 does not contribute to GABA co-release from DA neurons.
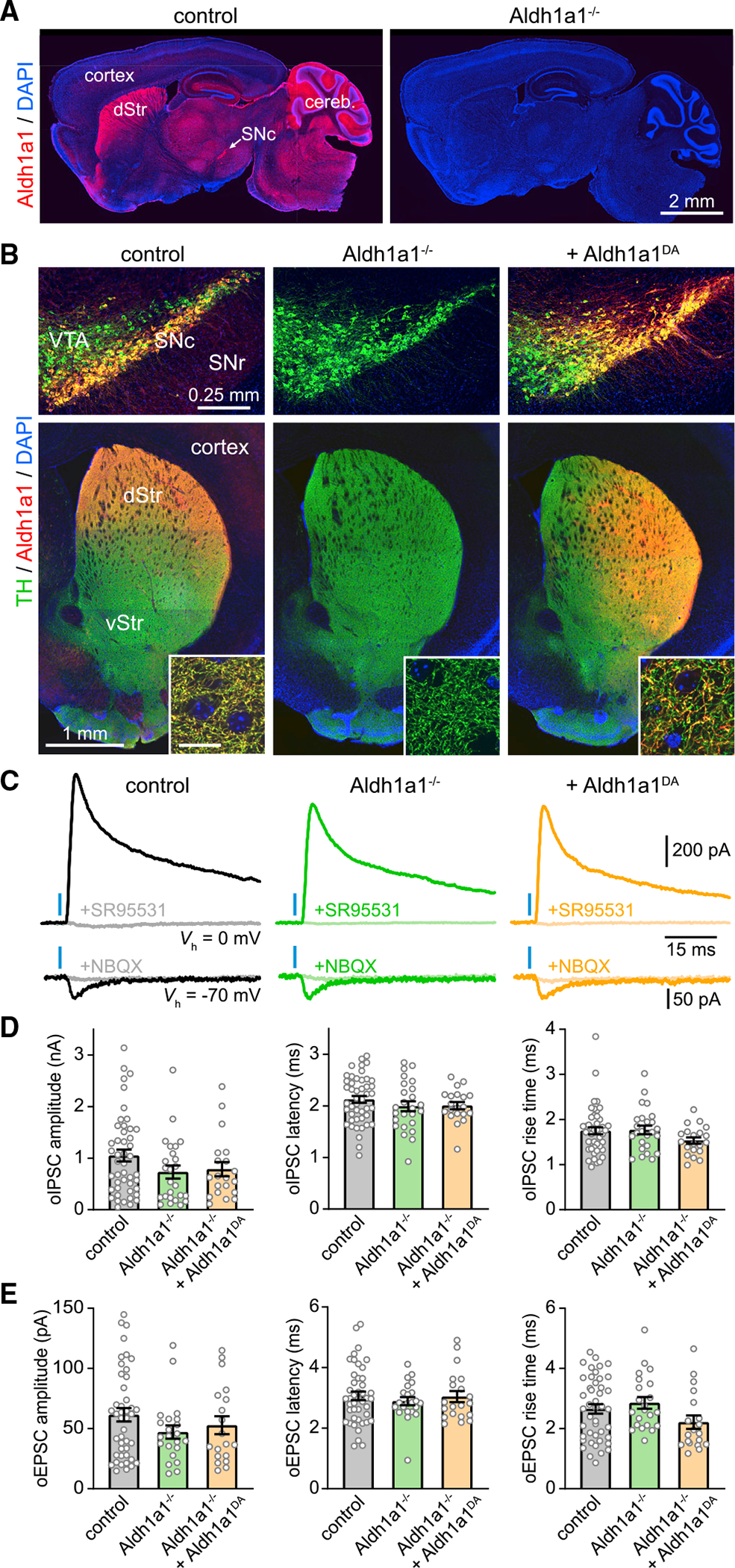
(A) Sagittal sections from control (left) and Aldh1a1−/− (right) mice immunolabeled for Aldh1a1 (red) and DAPI nuclear stain (blue). dStr, dorsal striatum; Cereb, cerebellum.
(B) Coronal brain sections from a control DatIRES-Cre/+ mouse (left), a DatIRES-Cre/+;Aldh1a1−/− mouse (middle), and a DatIRES-Cre/+;Aldh1a1−/− mouse in which Aldh1a1 is selectively restored in DA neurons (right) immunolabeled for TH (green), Aldh1a1 (red), and DAPI (blue). Top: confocal image of SNc, ventral tegmental area (VTA), and substantia nigra pars reticulata (SNr). Bottom: epifluorescence image of striatum. vStr, ventral striatum. Inset: confocal detail in dorsal striatum (scale, 10 μm).
(C) Postsynaptic responses recorded from SPNs voltage-clamped (Vh) at 0 (top) or −70 mV (bottom) upon stimulation of SNcDA axons (blue bar) before and after application of the GABAAR antagonist SR95531 (10 μM) or the ionotropic glutamate receptor blockers NBQX (10 μM).
(D) Mean amplitude (left), synaptic latency (middle), and 10% to 90% rise time (right) of oIPSCs recorded from SPNs in slices from control (n = 46), Aldh1a1−/− (n = 25), and Aldh1a1−/− mice expressing Aldh1a1 in SNcDA neurons (n = 20; amplitude: p = 0.16; latency: p = 0.41; rise time: p = 0.17; Kruskal-Wallis). Individual values shown along with population mean ± SEM.
(E) Same as (D) for oEPSCs (amplitude: p = 0.338; latency: p = 0.94; rise time: p = 0.07; Kruskal-Wallis).
Because Aldh1a1 expression is not exclusive to SNc (Figures 1A and S2A), it is unclear whether the reduction in inhibitory co-transmission in Aldh1a1−/− mice results from the specific loss of Aldh1a1 in SNcDA neurons. If Aldh1a1 functions as a GABA synthetic enzyme, we reasoned that selectively restoring its expression in SNcDA neurons ought to increase inhibitory co-transmission in Aldh1a1−/− mice. We therefore generated an AAV expressing Cre-dependent Aldh1a1, which we co-injected along with Cre-dependent ChR2 in the midbrain of DatIRES-Cre/+;Aldh1a1−/− mice (Figures 1B and S1A). This manipulation reestablished Aldh1a1 protein expression in DA neurons (Figure 1B) but failed to increase the amplitude of oIPSCs in SPNs (Figure 1D).
To assess whether differences in ChR2 expression hindered our ability to reveal a role for Aldh1a1, we recorded optogenetically evoked excitatory postsynaptic currents (oEPSCs) from the same SPNs. oEPSCs reflect glutamate co-release (Trudeau et al., 2014) and therefore serve as an independent metric of stimulation strength. We did not observe differences in the prevalence, amplitude, and kinetics of oEPSCs across conditions (Figures 1E and S1D), indicating that we recruited similar numbers of SNcDA axons. We nevertheless carried out an additional experiment to minimize variability in ChR2 expression: we expressed it genetically in all DAergic neurons by breeding a conditional allele of ChR2 (Madisen et al., 2012) into DatIRES-Cre/+;Aldh1a1−/− mice. Here, too, we failed to detect any difference in the amplitude of oIPSCs or oEPSCs in Aldh1a1+/+ and Aldh1a1−/− mice (Figures S1E–S1G and S2C). Collectively, these results fail to support an important, cell-autonomous role for Aldh1a1 in producing GABA for inhibitory co-transmission from SNcDA axons.
Selective deletion of Gat1 from SNcDA neurons abolishes GABAergic co-transmission
SNcDA neurons in mice do not express the canonical GABA synthetic enzymes Gad65 and Gad67 (Poulin et al., 2014; Tritsch et al., 2014; Saunders et al., 2018). If Aldh1a1 does not produce GABA either, how do SNcDA neurons inhibit SPNs? We previously proposed that SNcDA neurons acquire GABA using Gat1 (Tritsch et al., 2014). To test this directly, we generated mice in which Gat1 can be conditionally knocked out (cKO) by Cre recombinase (Gat1fl mice; Figures 2A and S3A–S3D). To validate our strategy, we first deleted Gat1 constitutively by breeding Gat1fl mice to a line expressing germline Cre (Gat1 cKOgermline). As shown in Figure 2B, Gat1 protein is not detected in the brain of Gat1 cKOgermline mice, confirming our ability to ablate Gat1 in a Cre-dependent fashion.
Figure 2. GABAergic co-transmission from SNcDA neurons is abolished in Gat1 cKODA mice.
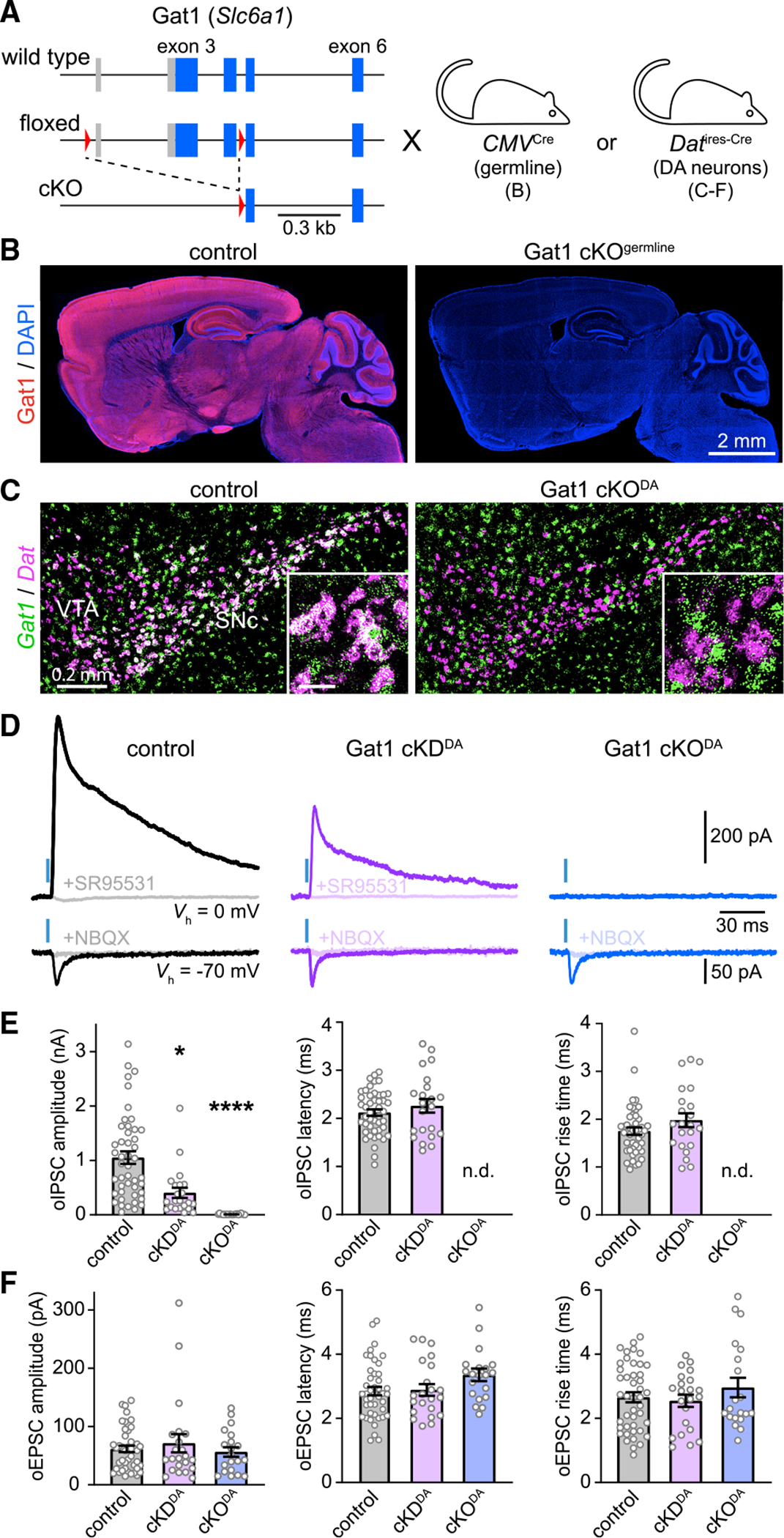
(A) Schematic of Gat1 locus (top), floxed allele before (middle) and after (bottom) Cre-mediated excision in CMVCre or DatIRES-Cre mice. Boxes: exons with protein-coding regions in blue; red triangles: LoxP sites.
(B) Sagittal brain sections from control (left) and Gat1 cKOgermline mice (right) immunolabeled for Gat1 (red) and DAPI (blue).
(C) Fluorescence in situ hybridization for Gat1 (green) and Dat (magenta; labels DA neurons) in coronal brain sections from control (DatIRES-Cre/+;Gat1+/+; left) and Gat1 cKODA (DatIRES-Cre/+;Gat1fl/fl; right) mice. Colocalization appears white. Inset: detail (scale, 50 μm).
(D) Example oIPSCs (top) and oEPSCs (bottom) in SPNs held at 0 and −70 mV, respectively, in DatIRES-Cre/+ mice with no (control; black), one (cKDDA; purple), or both (cKODA; blue) alleles of Gat1 conditionally deleted from SNcDA neurons before and after application of SR95531 (10 μM) or NBQX (10 μM).
(E) Meanamplitude(left), synaptic latency (middle), and 10% to 90% rise time (right) of oIPSCs recorded from SPNs in control (n = 46), cKDDA (n = 22), and cKODA (n = 22) slices (amplitude: p = 2.94 × 10−13; Kruskal-Wallis; *p = 0.012, ***p = 1.01 × 10−13 versus control, Dunn’s multiple comparison; latency: p = 0.65; rise time: p = 0.18 between control and Gat1 cKDDA; Mann-Whitney). nd, not detected. Individual values shown along with population mean ± SEM.
(F) Same as (E) for oEPSCs (amplitude: p = 0.85; latency: p = 0.18; rise time: p = 0.76; Kruskal-Wallis).
We next crossed Gat1fl and DatIRES-Cre mice to generate offspring in which Gat1 is selectively deleted from DAergic cells (DatIRES-Cre/+;Gat1fl/fl or Gat1 cKODA mice; Figures 2A, 2C, and S3A). Using in situ hybridization, we confirmed that Gat1 mRNA is no longer detected in SNcDA neurons of Gat1 cKODA mice yet is intact in surrounding inhibitory neurons within the substantia nigra (Figures 2C, S3E, and S3F). To assess inhibitory co-transmission, we virally expressed ChR2 in SNcDA neurons and recorded light-evoked postsynaptic currents in SPNs. Although oIPSCs are readily observed in control slices, they were entirely absent in Gat1 cKODA mice (Figures 2D–2E and S3G). Importantly, the same SPNs showed oEPSCs that were similar to controls in prevalence, amplitude, and kinetics (Figures 2D, 2F, and S3H), confirming our ability to stimulate SNcDA neurons. Moreover, in mice in which Gat1 is conditionally knocked down in DAergic neurons (DatIRES-Cre/+;Gat1fl/+ or Gat1 cKDDA mice), oIPSCs were 60% smaller than controls but were otherwise comparable in prevalence and kinetics (Figures 2D, 2E, and S3G). These data suggest that Gat1 expression in SNcDA neurons modulates the strength of inhibitory co-transmission onto SPNs in a dose-dependent fashion, with complete removal of Gat1 abolishing GABAergic signaling entirely.
Deleting Gat1 from SNcDA neurons does not impair basic nigrostriatal physiology
Our inability to evoke oIPSCs in Gat1 cKODA mice may reflect deleterious effects of our manipulation on nigrostriatal function. To test this, we first determined whether Gat1 loss affects the development of SNcDA neurons using immunofluorescence for the DA synthetic enzyme tyrosine hydroxylase (TH). Figures S4A–S4C show that the number of TH+ cell bodies in SNc and the density of TH+ axons in striatum are indistinguishable in control and Gat1 cKODA mice. We also did not observe any difference in TH or Aldh1a1 levels or in the cellular distribution of Aldh1a1 in SNcDA neurons (Figures S4D and S4E), indicating that deleting Gat1 from DA neurons does not grossly impair their development.
Gat1 plays an important role in limiting the buildup of extracellular GABA. Recent studies show that elevating extracellular GABA significantly dampens the excitability of SNcDA axons through activation of presynaptic GABAARs and GABABRs (Brodnik et al., 2019; Lopes et al., 2019; Roberts et al., 2020; Kramer et al., 2020). Deleting Gat1 from SNcDA neurons may therefore depress their ability to release transmitters indirectly by causing ambient GABA levels to rise. To test this, we estimated extracellular GABA levels in striatum by measuring the amplitude of tonic GABAAR currents in SPNs using the GABAAR blocker picrotoxin. Picrotoxin-mediated shifts in holding current were not different in control and Gat1 cKODA slices (Figures S4F and S4G), suggesting that ambient GABA levels are comparable between conditions. We also found no differences in the membrane resistance or capacitance of SPNs (Figures S3I and S3J) or in the amplitude and frequency of spontaneous IPSCs (Figures S4H and S4I), indicating that the membrane properties of SPNs and their capacity to detect GABAergic currents are not affected in Gat1 cKODA mice.
Last, we assessed the integrity of the vesicular release machinery of SNcDA neurons in Gat1 cKODA mice. Although glutamate release from SNcDA neurons appears intact (Figure 2F), it is anatomically and molecularly distinct from DA release (Fortin et al., 2019; Silm et al., 2019; Zhang et al., 2015). By contrast, DAergic and GABAergic transmission both depend on Vmat2 (Tritsch et al., 2012). We therefore compared the magnitude of DA transients evoked by water rewards in control and Gat1 cKODA mice using fiber photometry of the DA sensor GRAB-DA2m in striatum (Figure S4J; Sun et al., 2020). As shown in Figures S4K and S4L, reward-evoked DA transients were not smaller in Gat1 cKODA mice compared with controls, indicating that activity-dependent DA exocytosis is not compromised in SNcDA neurons lacking Gat1. Together, these results point to Gat1 cKODA mice having a selective deficit in the ability of SNcDA neurons to liberate the transmitter that inhibits SPNs.
Inhibitory co-transmission relies on Gat1-mediated import of GABA
Gat1 normally transports GABA from the synaptic cleft into the cytosol (Scimemi, 2014). In SNcDA neurons, Gat1 may therefore serve as a source of GABA for vesicular loading. Alternatively, Gat1 may operate in reverse to mediate non-vesicular GABA efflux (Wu et al., 2007; Bertram et al., 2011), leaving the source of GABA in SNcDA neurons to be identified. Gat1 may also be part of a molecular complex that controls inhibitory signaling from SNcDA neurons independently of its transporter function (Deken et al., 2000; Ryan et al., 2021). To distinguish these possibilities, we selectively expressed constructs in SNcDA neurons to restore inhibitory transmission in Gat1 cKODA mice.
We first made a Cre-dependent AAV encoding Gat1 tagged with influenza hemagglutinin (HA), which we injected in the midbrain of adult Gat1 cKODA mice along with ChR2. Immunostaining for HA confirmed our ability to reintroduce Gat1 specifically in SNcDA neurons (Figure 3A). We observed Gat1-HA throughout the axons of SNcDA neurons in dorsal striatum (Figure S5A), consistent with Gat1’s usual presynaptic distribution (Uchigashima et al., 2016). Importantly, expressing Gat1-HA in SNcDA neurons of Gat1 cKODA mice fully restored GABAergic signaling in SPNs (Figures 3C and 3H–3J). Together, these data show that SNcDA neurons remain capable of inhibiting SPNs in adult Gat1 cKODA mice if provided with Gat1.
Figure 3. GABA co-release from SNcDA axons requires Gat1-mediated GABA uptake.
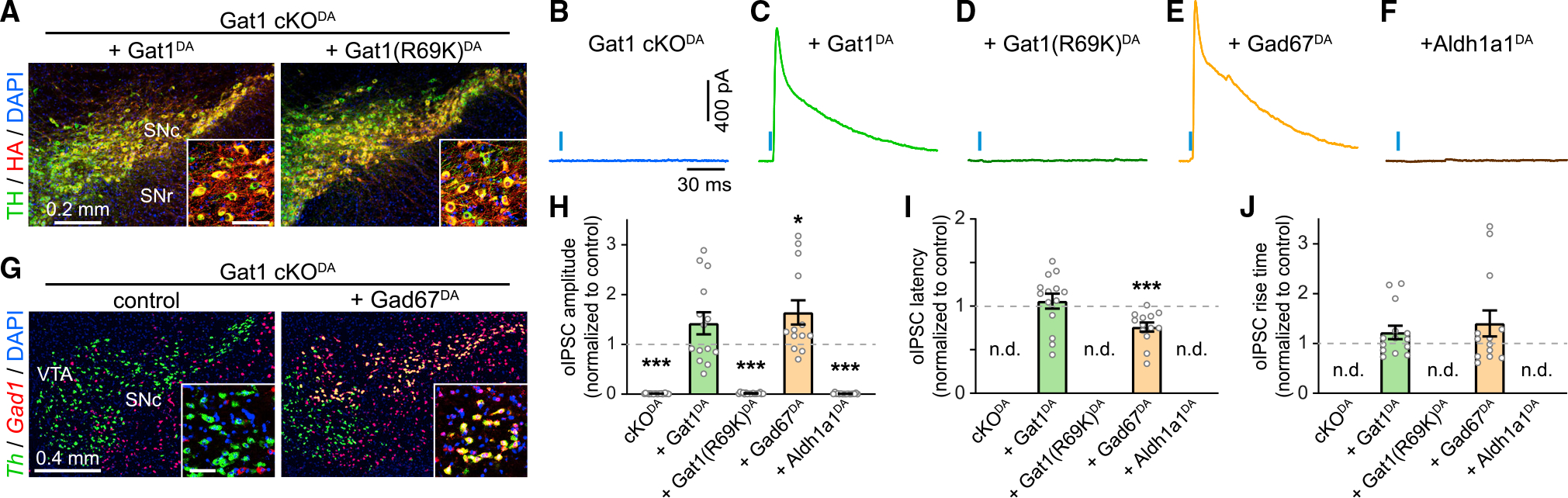
(A) Confocal images of SNc in Gat1 cKODA mice expressing Cre-dependent HA-tagged Gat1 (left) and Gat1(R69K) (right) immunolabeled for TH (green), HA (red), and DAPI (blue). Inset: detailed view (scale, 50 μm).
(B–F) oIPSCs recorded from SPNs (Vh = 0 mV) upon stimulation of SNcDA axons (blue bar) in Gat1 cKODA mice (B) exogenously expressing Gat1 (C), Gat1(R69K) (D), Gad67 (E), or Aldh1a1 (F).
(G) Fluorescence in situ hybridization for Gad67 (red) and TH (green) in Gat1 cKODA mice with (right) or without (left) Gad67 in DA neurons. Inset: detail (scale, 50 μm).
(H) oIPSC amplitude in SPNs of Gat1 cKODA mice virally expressing ChR2 in SNcDA neurons (n = 22, p = 1 × 10−15), or ChR2 + Gat1 (n = 14; p = 0.07), ChR2 + Gat1(R69K) (n = 18; p = 1 × 10−15), ChR2 + Gad67 (n = 13; p = 0.018), or ChR2 + Aldh1a1 (n = 17; p = 2 × 10−15) normalized to control. All p values versus control, Mann-Whitney. nd, not detected. Individual values shown along with population mean ± SEM.
(I) Same as (H) for latency (Gat1DA: p = 0.32; Gad67DA: p = 0.0007 versus control, Mann-Whitney).
(J) Same as (I) for rise time (Gat1DA: p = 0.18; Gad67DA: p = 0.39 versus control, Mann-Whitney).
To distinguish whether the dependence on Gat1 stems from its ability to transport GABA across the plasma membrane or interact with other molecules, we expressed Gat1-HA with a single amino acid mutation (R69K) that abolishes GABA transport but not membrane expression (Dayan-Alon and Kanner, 2019; Pantanowitz et al., 1993) along with ChR2 in SNcDA neurons of Gat1 cKODA mice. Despite strong expression of Gat1(R69K)-HA in SNcDA cell bodies and axons (Figures 3A and S5A) and successful activation of SNcDA axons in striatum (Figures S5B–S5E), we were unable to evoke oIPSCs in SPNs (Figures 3D and 3H). This suggests that SNcDA neurons depend on Gat1’s transport function to inhibit SPNs.
If Gat1 acts to import GABA for vesicular release, a strong prediction is that other means of supplying cytosolic GABA to SNcDA axons of Gat1 cKODA mice should also reinstate inhibitory transmission. To test this, we transduced SNcDA neurons of Gat1 cKODA mice with ChR2 and the cytosolic GABA synthetic enzyme Gad67. Consistent with our prediction, expressing Gad67 in SNcDA neurons lacking Gat1 rescued inhibitory co-transmission (Figures 3E and 3H–3J). We obtained similar results with Gad65 (Figures S5F and S5G), but not with Aldh1a1 (Figures 3F and 3H). Interestingly, oIPSCs evoked from SNcDA neurons expressing Gad65 or Gad67 were significantly less prone to rundown with successive stimulation compared with Gat1-expressing controls (Figure S5H). Together, these experiments show that Gat1 contributes to inhibitory co-transmission by importing GABA into the cytoplasm of SNcDA neurons for vesicular loading.
Synaptic release of GABA from SNcDA neurons depends on Vmat2
We previously suggested using pharmacology that GABAergic transmission from SNcDA neurons requires Vmat2 (Tritsch et al., 2012). However, GABA does not resemble other Vmat2 substrates, raising the possibility that GABA is loaded into synaptic vesicles using another transporter that is sensitive to Vmat2 antagonists. To rule out this possibility, we measured oIPSCs in slices in which ChR2 is expressed genetically in DAergic axons (Madisen et al., 2012) and Vmat2 is genetically knocked down (DatIRES-Cre/+:Ai32+/−;Vmat2fl/+ or Vmat2 cKDDA mice; Figure 4A), as deletion of Vmat2 results in post-natal death (Isingrini et al., 2016). DA release is highly sensitive to Vmat2 levels (Fon et al., 1997). If GABA is similarly dependent on Vmat2 for vesicular transport, we hypothesized that the amplitude of oIPSCs should be depressed in Vmat2 cKDDA mice. Indeed, oIPSCs were 50% smaller compared with controls (Figures 4B and 4C) but otherwise comparable in synaptic latency and kinetics (Figure S6A). By contrast, oEPSCs were unchanged (Figures 4B, 4C, and S6B), indicating that knocking down Vmat2 does not impair the release of transmitters that do not depend on Vmat2 for vesicular loading.
Figure 4. Vmat2 is required for vesicular transport of GABA.

(A) Strategy to generate mice in which the expression of ChR2 and Vmat2 in DA neurons is controlled genetically. cKDDA mice have one allele of Vmat2 conditionally deleted.
(B) Example oIPSCs (top) and oEPSCs (bottom) recorded from SPNs at 0 and −70 mV, respectively, in DatIRES-Cre/+;Rosa26Ai32/−;Vmat2+/+ (control; black) and in DatIRES-Cre/+: Rosa26Ai32/−; Vmat2fl/+ (Vmat2 cKDDA; red) mice before and after bath application of SR95531 (10 μM) or NBQX (10 μM).
(C) Mean amplitude of oIPSCs (left) and oEPSCs (right) recorded from SPNs in control (n = 29) Vmat2 cKDDA (n = 18) mice (p = 0.008, Mann-Whitney). Individual values shown along with population mean ± SEM.
(D) Strategy to conditionally knock out Vmat2 from SNcDA neurons unilaterally in the adult nervous system.
(E) Confocal image of SNc in a DatIRES-Flpo/+;Vmat2fl/fl mouse transduced with AAVs encoding Flp-dependent Cre and immunolabeled for TH (green) and Cre (red) showing specific expression of Cre in SNcDA neurons.
(F) Example oIPSCs (top) and oEPSCs (bottom) recorded from SPNs in DatIRES-Flpo/+;Vmat2+/+ (black) and Vmat2 cKODA (purple) mice before and after application of SR95531 (10 μM) or NBQX (10 μM).
(G) Same as (C) for control (n = 6) and Vmat2 cKODA (n = 7) SPNs (oIPSCs: p = 0.0012; oEPSCs: p = 0.53, Mann-Whitney).
(H) oIPSCs recorded from SPNs upon stimulation of Gad67-expressing SNcDA axons (blue bar) in Gat1 cKODA mice with (red) or without (yellow) reserpine.
(I) Amplitude of oIPSCs in Gat1 cKODA mice expressing Gad67 in SNcDA neurons without (n = 13) or with reserpine (n = 9; p = 4.02 × 10−6, Mann-Whitney). Individual values shown along with population mean ± SEM.
To further implicate Vmat2, we bred the Vmat2fl allele in knockin mice expressing Flp recombinase specifically in DAergic neurons (DatIRES-Flpo). Adult mice with no (control; DatIRES-Flpo/+;Vmat2+/+) or both alleles of Vmat2 floxed (cKODA: DatIRES-Flpo/+;Vmat2fl/fl) were injected unilaterally with two AAVs in SNc; one encoding Flp-dependent Cre and the other Cre-dependent ChR2 (Figure 4D) to restrict Vmat2 deletion and ChR2 expression to SNcDA neurons in the mature nervous system (Figure 4E). Vmat2 cKODA mice showed strong ipsiversive rotational bias consistent with severe unilateral loss of DA release 16 weeks after viral transduction (Isingrini et al., 2017). We obtained slices from these mice and recorded oIPSCs and oEPSCs from SPNs in dorsal striatum (Figure 4F). ChR2 stimulation reliably evoked oEPSCs of comparable amplitude and kinetics in control and Vmat2 cKODA mice (Figures 4G and S6C) but failed to evoke oIPSCs in Vmat2 cKODA slices (Figures 4F and 4G), demonstrating that Vmat2 is essential for inhibitory co-transmission from SNcDA neurons.
To confirm that GABA itself can be a substrate for Vmat2-mediated vesicular loading, we compared oIPSCs recorded in Gat1 cKODA mice expressing Gad67 in SNcDA neurons in the presence or absence of the Vmat2 antagonist reserpine. We selected these mice, as the inhibitory transmitter released by SNcDA neurons is unquestionably GABA. Compared with untreated slices, reserpine strongly depressed the amplitude of oIPSCs, but not oEPSCs (Figures 4H, 4I, and S6D), providing strong evidence that Vmat2 supports vesicular transport of GABA.
DISCUSSION
In addition to releasing DA, SNcDA neurons exert a rapid and potent inhibitory influence on downstream striatal neurons through synaptic co-release of an inhibitory transmitter, the identity of which has remained speculative (Tritsch et al., 2016). Here, we provide strong evidence that the neurotransmitter that SNcDA neurons co-release is GABA, that Vmat2 is required for vesicular packaging and release of GABA, and that SNcDA neurons sustain inhibitory transmission without producing GABA, relying instead on presynaptic uptake through the membrane GABA transporter Gat1.
The chemical identity of a neurotransmitter is traditionally established by determining if a synapse possesses the means to (1) acquire said transmitter, typically by expressing synthetic enzymes, (2) package it into synaptic vesicles, (3) detect it postsynaptically using specific receptors, and (4) terminate its synaptic actions, most commonly through membrane uptake or enzymatic degradation. SNcDA neuron stimulation evokes nanoampere-sized GABAAR IPSCs in SPNs (Kim et al., 2015; Nelson et al., 2014; Tritsch et al., 2012), the decay kinetics of which are shaped by GABA transporters (Tritsch et al., 2014), suggesting that the released transmitter is GABA. However, this assertion was called into question, as mouse SNcDA neurons lack molecules considered essential for GABA release and rely on a vesicular transporter not known to transport GABA. Here, we show that inhibitory co-transmission from SNcDA neurons is abolished upon selective deletion of Gat1 from DA neurons. We demonstrate that this effect is not secondary to developmental or experimental confounds, as SNcDA neurons remain structurally and functionally competent to co-release DA, glutamate, and even GABA (if GABA is provided to DA neurons through cell-type-specific expression of Gad65 or Gad67). Importantly, we restore inhibitory co-transmission from SNcDA neurons in adult mice by selectively introducing Gat1, Gad67, or Gad65 in SNcDA neurons, but not a GABA transport-deficient Gat1 point-mutant. This indicates that Gat1’s transporter function is required for SNcDA neurons to inhibit SPNs and that Gad65 and Gad67 can functionally replace Gat1. Last, we provide fresh genetic evidence that Vmat2 supports the vesicular transport of GABA. When combined with previous reports showing that GABA can be observed in close association with Vmat2+ synaptic vesicles in striatal DA terminals (Stensrud et al., 2014) and that Vgat can substitute for Vmat2 to sustain inhibitory transmission from SNcDA neurons (Tritsch et al., 2012), our results establish that GABA possesses all the necessary attributes to be the transmitter that SNcDA neurons release along with DA and glutamate.
Our results provide important mechanistic insights into GABAergic transmission. First, our study describes a mammalian GABAergic synapse lacking the ability to produce GABA. In mice, GABA is almost exclusively synthetized de novo from glutamate by Gad65 and Gad67, with Gad67 accounting for most of the GABA produced (Asada et al., 1996, 1997; Tian et al., 1999). Gad65 and Gad67 are therefore considered critical elements for conferring neurons a GABAergic identity. Our results call for an expansion of this list, as SNcDA neurons rely exclusively on Gat1-mediated uptake as their source of GABA. This mechanism explains why GABA co-release is not detected in primary DA neuron cultures not supplemented with extracellular GABA (Sulzer et al., 1998) and raises the interesting possibility that GABA co-release may vary at individual synapses with extracellular GABA levels and Gat1 transporter activity. More importantly, our findings suggest that the function of GABA transporters extends beyond terminating GABA’s actions in the synaptic cleft to include sustaining vesicular filling. Such contribution has proven difficult to dissect at Gad-containing synapses (Bragina et al., 2008; Jensen et al., 2003; Wang et al., 2013).
Second, they reveal that, in the absence of Gat1, SNcDA neurons do not possess GABA for synaptic release. This is at odds with a report claiming that Aldh1a1 produces GABA de novo in SNcDA neurons (Kim et al., 2015). However, the manipulations employed were not strictly limited to SNcDA neurons. Our experiments show that GABA co-release is not significantly diminished in Aldh1a1−/− mice and that overexpressing Aldh1a1 in adult SNcDA neurons fails to elevate GABA co-release. Although we did not directly measure Aldh1a1’s enzymatic activity, our genetic loss- and gain-of-function manipulations call into question its ability to produce GABA. Indeed, deleting Gat1 from DA neurons abolishes GABA co-release without altering Aldh1a1 expression and overexpressing Gad67 or Gad65 in SNcDA neurons of Gat cKODA mice restores inhibitory co-transmission onto SPNs, but overexpressing Aldha1 does not. Interfering with Aldh1a1 may modify GABA co-transmission indirectly; Aldh1a1 is widely expressed throughout the brain of humans and mice (Adam et al., 2012; Saunders et al., 2018; Zhang et al., 2014), where it controls numerous biological processes that may impact GABAergic signaling. For instance, Aldh1a1 is one of three enzymes responsible for converting retinal into retinoic acid, an important developmental morphogen and transcriptional regulator (Rhinn and Dollé, 2012). Interfering with Aldh1a1 disrupts the development of DA axons and mu opioid receptorrich patches in striatum (Jacobs et al., 2007; Sgobio et al., 2017; Pan et al., 2019). In DA neurons, loss of Aldh1a1 also leads to the accumulation of toxic DA metabolites and α-synuclein aggregates that may affect GABA release (Anderson et al., 2011; Goldstein et al., 2013; Liu et al., 2014). Last, aldehyde dehydrogenases in astrocytes modulate synaptic transmission by altering extracellular GABA homeostasis and neuronal excitability (Kwak et al., 2020; Jin et al., 2021; Melani and Tritsch, 2021).
Last, although Vgat is typically considered essential for packaging GABA into vesicles, Vgat knockout mice are incompletely deficient in synaptic GABA release (Wojcik et al., 2006), suggesting that other vesicular GABA transporters exist. Our data provide fresh evidence that Vmat2 contributes to vesicular packaging of GABA along with DA. First, we show that inhibitory co-transmission is completely blocked when Vmat2 is conditionally deleted from SNcDA neurons. Second, we report that reserpine blocks the release of GABA produced by Gad67 in Gat1 cKODA mice. Together, our data redefine the minimal molecular machinery necessary to impart neurons with a GABAergic phenotype and pave the way for studies dissecting the specific contribution of GABA co-release from SNcDA neurons to behavior.
Limitations of the study
Although our study calls into question the ability of Aldh1a1 to synthesize GABA in SNcDA neurons, it does not directly rule out the possibility that Aldh1a1 serves in that capacity in other cells. Our study does not establish whether Vmat2 transports GABA into synaptic vesicles directly or indirectly via interactions with other vesicular proteins and whether DA and GABA are copackaged into the same vesicles or released from distinct presynaptic specializations.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nicolas X. Tritsch (nicolas.tritsch@nyulangone.org).
Materials availability
Plasmids for viral vectors generated in this study (AAV8.DIO.Aldh1a1, AAV8.DIO.Gat1, AAV8.DIO.Gat1(R69K), AAV8.DIO.Gad65 and AAV8.DIO.Gad67) are deposited to Addgene.
Gat1fl mice generated in this study were deposited at Jackson Laboratories (strain # 037219 - B6.Cg-Slc6a1<tm1.1Ntrch>/J).
Data and code availability
All data reported in this paper lead contact are available at https://github.com/TritschLab/Melani-Tritsch-2022.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult mice (8–28 weeks old) of both sexes were used in this study in accordance with protocols approved by the New York University Langone Health Institutional Animal Care and Use Committee (protocol #170123). Mice were housed in groups under a reverse 12 h light-dark cycle (dark from 10 a.m. to 10 p.m.) with ad libitum access to food and water. The following transgenic mice were obtained from the Jackson Laboratory: knock-in mice bearing an internal ribosome entry site (IRES)-linked Cre recombinase gene downstream of the gene Slc6a3, which encodes the plasma membrane DA transporter Dat (referred to as DatIRES–Cre mice; strain # 006660), CMVCre mice (strain # 006054), mice expressing the channelrhodopsin-2/EYFP fusion protein upon exposure to Cre recombinase (referred to as Rosa26Ai32 mice; strain # 033673), and knock-in mice bearing an IRES-FLPo cassette downstream of Slc6a3, (referred to as DatIRES-Flpo mice, strain # 035436). Mice in which the gene encoding Aldh1a1 is conventionally knocked out (B6.129-Aldh1a1tm1Gdu/J; referred to as Aldh1a1−/− mice) were kindly donated by Dr. Huaibin Cai (NIH). Mice with a conditional allele of Vmat2 (referred to as Vmat2fl) were kindly donated by Dr. Bruno Giros (McGill University). Transgenic lines were maintained on a C57Bl6/J background (Jackson Laboratory strain # 000664) and bred to one another to generate experimental animals, which were genotyped both in house by the NYU Genotyping Core and by Transnetyx.
METHOD DETAILS
Generation of conditional Gat1 knockout mice
Gat1fl mice were generated by genOway by homologous recombination in mouse embryonic stem (ES) cells. LoxP sites flank exons 2, 3 and 4 of Slc6a1 (Figures 2A and S3A), which contain the ATG initiation codon, the N-terminal intracellular tail, 2 sodium binding sites essential for GABA transport and 3 of 12 transmembrane domains (Bendahan and Kanner, 1993; Pantanowitz et al., 1993). The targeting vector contained a cassette encoding diphtheria toxin and a neomycin resistance cassette flanked by FRT sites for negative and positive selection of ES cell clones, respectively. Homologous recombination at the intended locus were confirmed by PCR screening and Southern blotting of ES cell clones’ genomic DNA (Figure S3B). ES cell clones positive for the knock-in (ki) allele were injected into blastocysts to obtain chimeric males, which were bred to Flp deleter mice to excise the Neomycin selection cassette and generate heterozygous mice carrying the floxed allele of Gat1 (referred to as Gat1fl mice). Crossing these mice with lines expressing Cre recombinase enabled the excision of the loxP-flanked region, resulting in a Gat1 conditional knockout allele (cKO): CMVCre mice mediate germline recombination to knock Gat1 out throughout the body, while DatIRES-Cre mice delete Gat1 embryonically from Dat-expressing DA neurons only. The following primers were designed and validated by GenOway for the specific detection Gat1fl and Gat1KO alleles: GAT1-FloxF and GAT1-FloxR yielding 170 bp wild-type and 276 bp conditional knock-out bands (Figure S3C), and GAT1-Flox14F, GAT1-Flox18F and GAT1-Flox15R yielding 211 bp wild-type and 818 bp constitutive knock-out bands (Figure S3D).
Adeno-associated viruses (AAVs)
Cre-dependent AAV viruses were used to selectively transduce midbrain DA neurons with channelrhodopsin 2 (ChR2) and express constructs to rescue GABA co-release in DatIRES-Cre/+;Aldh1a1−/− and DatIRES-Cre/+;Gat1fl/fl (Gat1 cKODA) mice. AAV8.EF1a.DIO.hChR2(H134R)-mCherry was obtained from Addgene (# 20297) and AAV9.hSyn.GRAB_DA2 m was provided by Dr. Yulong Li (Peking University). Flp-dependent AAV virus AAV8.EF1a.fDIO-Cre (Addgene # 121675) was used to selectively express Cre recombinase in DA neurons of DatIRES-Flpo/+ mice. AAV8.EF1a.DIO.Aldh1a1, AAV8. EF1a.DIO.Gat1, AAV8. EF1a.DIO.Gad65 and AAV8. EF1a.DIO.Gad67 were generated by replacing the coding sequence of ChR2-mCherry in Addgene’s plasmid #20297 with the protein-coding sequence for Aldh1a1 (NM_013467.3), Gat1 (NM_178703.4), Gat1(R69K), Gad65 (NM_008078.2) or Gad67 (NM_008077.5), respectively, using Asc1 and Nhe1 restriction sites (Genscript). Three copies of the influenza hemagglutinin (HA) coding sequence were inserted at the C-terminus of all constructs except for Gad65 and Gad67 to facilitate immunohistochemical identification. Plasmids were subsequently packaged into AAVs (serotype 8) by the Boston Children Hospital viral core, stored in undiluted aliquots at −80°C and diluted 2–10X in sterile 0.9% saline immediately prior to intracranial injection. All viral constructs created in this study were deposited at Addgene.
Stereotaxic surgery
For stereotaxic viral injections, mice were anesthetized with isoflurane, placed in an animal stereotaxic frame (David Kopf Instruments) and injected (rate: 100 nL/min) in the right SNc of DatIRES-Cre mice with 1 μL of a mix of AAVs (Table S1) encoding Cre-dependent ChR2 (1 × 1012 GC/mL) and either Cre-dependent Aldh1a1 (2.4 × 1010 GC/mL), Gat1 (7.5 × 1011 GC/mL), Gat1(R69K) (5.7 × 1011 GC/mL), Gad65 (1.1 × 1012), Gad67 (3.2 × 1012) or saline. DatIRES-Flpo/+ mice were injected into SNc with 1 μL of a mix of AAVs encoding Flp-dependent Cre recombinase (4.2 × 1011) and Cre-dependent ChR2 (1 × 1012 GC/mL). The optimal dilution of each AAV was empirically determined to enable strong expression while avoiding toxicity. Working dilutions of AAVs were prepared on ice and loaded into a pulled glass injection micropipette (~100 μm tip; Wiretrol II; Drummond) connected to a syringe pump (Legato 111; KD scientific) fitted with a Hamilton syringe (Gastight 1701N) via PE tubing filled with mineral oil immediately prior to intracranial injection. Injection coordinates for SNc were (from bregma): AP −3.12 mm, ML +1.6 mm and DV −4.2 mm (from pia). For fiber photometry, 250 nL of AAV9-hSyn-GRAB_
DA2m (5.3 × 1012 GC/mL) was injected 2.2 mm below pia into the right dorsal striatum (from bregma: AP, +0.5 mm; ML, +2.0 mm). Mice were allowed to recover for at least 3 weeks prior to electrophysiology. DatIRES-Flpo/+;Vmat2fl/fl mice were kept for 16 weeks following transduction with Flp-dependent Cre AAV to ensure complete depletion of Vmat2 proteins in SNcDA neurons (Isingrini et al., 2017).
Slice electrophysiology
Acute brain slices and whole-cell voltage-clamp recordings from SPNs were obtained blind to experimental conditions using standard methods, as described previously (Tritsch et al., 2012, 2014). Briefly, mice were anesthetized and perfused with ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4 and 11 glucose (295 mOsm·kg−1). Sagittal slices of striatum (275-μm thick) were subsequently obtained in cold choline-based cutting solution (in mM: 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 25 glucose, 11.6 ascorbic acid, and 3.1 pyruvic acid) using a Leica VT1200 S vibratome. Following 15 min recovery in ACSF at 34°C, slices were kept at room temperature (20–22°C) until use. All solutions were constantly bubbled with 95% O2/5% CO2. For recording, slices were transferred to a Plexiglas chamber mounted on an upright microscope (SliceScope Pro 6000; Scientifica) and imaged through a 40X water-immersion objective (Olympus) with infrared light (780 nm) and differential interference contrast (DIC) optics using a complementary metal-oxide-semiconductor (CMOS) camera (ORCA-spark; Hamamatsu). Slices were held down by a flattened U-shaped platinum wire strung with individual strands of synthetic dental floss fibers and were continually superfused (2–2.5 mL/min) with ACSF at 33–34°C by passing it through a feedback-controlled in-line heater (SH-27B; Warner Instruments) before entering the chamber. Whole-cell voltage-clamp recordings were established from SPNs in dorsal striatum identified visually and electrophysiologically. For oIPSC/oEPSC recordings, patch pipettes (2–3 MΩ) were filled with a cesium-based low-chloride internal solution containing (in mM): 135 CsMeSO3, 10 HEPES, 1 EGTA, 3.3 QX-314 (Cl salt), 4 Mg-ATP, 0.3 Na-GTP, 8 Na2-phosphocreatine (pH 7.3 adjusted with CsOH; 295 mOsm·kg−1). Under these conditions, GABAAR currents can be isolated as outward currents when SPNs are held at 0 mV, while glutamatergic currents can be isolated as inward currents at ECl = −70 mV. Inward tonic GABAergic currents and sIPSC in Figure S4 were recorded at −70 mV using an internal solution containing (in mM) 125 CsCl, 10 TEA-Cl, 10 HEPES, 0.1 Cs-EGTA, 3.3 QX-314 (Cl–salt), 4 Mg-ATP, 0.3 Na-GTP, 8 Na2-Phosphocreatine (pH 7.3 adjusted with CsOH; 295 mOsm) in the presence of NBQX, R-CPP, and CGP55845. For all voltage-clamp experiments, errors due to the voltage drop across the series resistance (<20 MΩ) were left uncompensated. Membrane potentials were corrected for a ~7 mV liquid junction potential. To activate ChR2-expressing SNcDA axons, 1 ms-long full-field pulses of light from a 473-nm LED (p300 CoolLED) were delivered through the objective (10 mW.mm−2 under the objective) at 30 s intervals.
Pharmacological reagents
Drugs (all from Tocris) were applied by bath perfusion: SR95531 (10 μM), picrotoxin (100 μM), CGP55845 (5 μM), 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(f)quinoxaline (NBQX; 10 μM), R,S-3-(2-carboxypiperazin-4-yl)propyl-1- phosphonic acid (R-CPP; 10 μM). To inhibit monoaminergic vesicular transport and deplete transmitter-filled vesicles, mice were injected intraperitoneally with the irreversible Vmat inhibitor reserpine (5 mg.kg−1) 24 h and 2 h before slicing. Brain sections from these animals were prepared as described above, but were incubated and recorded in ACSF containing 1 μM reserpine.
Fiber photometry
Upon stereotaxic injection of AAV9.hSyn.GRAB-DA2m in dorso-lateral striatum, a 400 μm optic fiber (FP400URT, Thorlabs) housed in a 1.25 mm ceramic ferrule (CFLC440, Thorlabs) was implanted 0.2 mm above the injection site using C&B metabond (Parkel). Following 2 weeks of recovery in their home cage, mice were habituated for a minimum of 5 days to locomote while head-fixed on a cylindrical wheel placed into a dark soundproof chamber and were water restricted to incite consumption of water rewards delivered from a spout at semi-random 8 to 12 s intervals. Licking was monitored by a capacitive touch sensor (AT42QT1010; Sparkfun). Extracellular DA levels in striatum were imaged by providing 470 nm excitation light from a LED (M470F3; Thorlabs) coupled to an optical fiber (400 μm, 0.48 NA; Doric) to a fluorescence mini-cube (FMC3_E(460–490)_F(500–550)_S; Doric) connected to the mouse via a fiber optic patch cord (400 μm, 0.48 NA; Doric) and zirconia sleeve. Emission light (500–550 nm) was collected through the same patch cord and mini-cube by a femtowatt photoreceiver (2151; Newport) fitted with a FC adapter (FOA_2151_FC; Doric) via a 600 mm 0.48NA fiber optic (Doric).
Immunohistochemistry
Mice were deeply anesthetized with isoflurane and perfused transcardially with 4% paraformaldehyde in 0.1 M sodium phosphate buffer. Brains were post-fixed for 1–3 days, sectioned coronally (50 μm in thickness) using a vibratome (Leica; VT1000S) and processed for immunofluorescence staining for tyrosine hydroxylase (Millipore; AB152, 1:2000), Gat1 (Cell Signaling; 37342, 1:200), Aldh1a1 (R&D Systems; AF5869, 1:200), HA (Abcam; ab18181, 1:500) or Cre (Synaptic Systems; 257 004, 1:500). After primary antibodies incubation (4°C, overnight), the following secondary antibodies (all from Thermo Fisher Scientific) were applied for 2 h at room temperature: Goat anti-mouse IgG Alexa Fluor 568 (A11004), Goat anti-mouse IgG Alexa Fluor 488 (A11029), Goat anti-rabbit IgG Alexa Fluor 488 (A11034), Goat anti-rabbit IgG Alexa Fluor 568 (A11036), Donkey anti-goat IgG Alexa Fluor 568 (A11057), Donkey anti-rabbit IgG Alexa Fluor 488 (A21206). Brain sections were mounted on Superfrost slides and coverslipped with DAPI Fluoromount-G (SouthernBiotech; 0100-20).
In situ hybridization
Mice were deeply anesthetized with isoflurane, the brain was rapidly extracted and frozen on dry ice in plastic cryomolds containing OCT compound (Fisher Scientific; 4585). Coronal sections (16 mm in thickness) were obtained using a Leica CM3050S cryostat, immediately mounted onto Superfrost Plus glass slides (Fisher Scientific; 1255015) and kept at −80°C until use. RNAscope assay was performed following Advanced Cell Diagnostic (ACD) manual Fluorescent Multiplex Kit. Briefly, slides were fixed in ice-cold 4% PFA for 15 min, then dehydrated in 50%, 70% and 100% ethanol before incubation with protease IV at room temperature for 30 min. The following target probes were used: Slc6a3/Dat (31544-C2), Th/TH (317621), Gad1/Gad67 (400951-C3), Gad2/Gad65 (439371-C2). For Slc6a1/Gat1, a custom probe was designed by ACD targeting 243–718 bp of NM_178703.4. After the last wash step, sections were incubated in DAPI at room temperature for 30 s before coverslipping with Fluoromount-G (SouthernBiotech; 0100-01).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data acquisition
Membrane currents were amplified and low-pass filtered at 2 kHz using a Multiclamp 700B amplifier (Molecular Devices), digitized at 10 kHz using National Instruments acquisition boards (BNC 2110) and a custom version of ScanImage (Pologruto et al., 2003 https://github.com/bernardosabatinilab/SabalabSoftware_Nov2009) written in MATLAB (Mathworks). Electrophysiology data were analyzed offline using Igor Pro 6.02A (Wavemetrics). In figures, voltage-clamp traces represent the averaged waveform of 3–5 consecutive acquisitions. Single waveforms aligned to optogenetic light stimulus onset were used to measure postsynaptic current peak amplitude, latency (from light onset to current onset), and 10–90% rise time. Only oIPSCs and oEPSCs larger than 11 pA in amplitude were considered to compute prevalence, latency and rise time. For sIPSCs, the detection threshold was set to 20 pA to facilitate event detection in the presence of large and noisy tonic GABA currents. For photometry, signals were continuously digitized at 2 kHz using a National Instruments acquisition board (USB-6343) and Wavesurfer software (Janelia), low-pass filtered and down-sampled to 30 Hz, and aligned to the first lick within 500 ms of water delivery for analysis in MATLAB. For in situ hybridization and immunofluorescence analyses, brain sections were first digitized with an Olympus VS120 slide-scanning microscope. High-resolution images of regions of interest were subsequently acquired with a Zeiss LSM 800 confocal microscope. Confocal images in figures represent maximum intensity projections of 3 μm-thick confocal stacks. To estimate the number of DA neurons in control and Gat1 cKODA mice (Figure S4), we used Cellpose (Stringer et al., 2021) to automatically segment the cell bodies of TH+ neurons in confocal images of ventral midbrain. We report the mean number of TH+ cells counted per mouse across the 3 evenly-spaced coronal sections of SNc (−2.9, −3.1, −3.3 mm AP, from bregma). We calculated the mean fluorescence intensity of TH and Aldh1a1 in DA neurons in ImageJ (Schindelin et al., 2012) using the masks to obtained with CellPose. The fraction of TH+ cells that are Aldh1a1+ was estimated by counting the fraction of TH+ cell masks where Aldh1a1 fluorescence intensity exceeded a threshold common to all sections. DA axon density was calculated as the mean percentage of dorsal striatal neuropil (excluding neuronal somata and corticofugal axon bundles) covered by TH+ axons across 4 coronal sections (0.75, 0.65, 0.55, 0.45 mm AP, from bregma) per mouse.
Statistical analysis
Data (reported in text and figures as mean ± SEM) were compared using Prism 9 (GraphPad) with the following non-parametric statistical tests (as indicated in the text): Mann-Whitney for comparisons between test and control groups, and Kruskal–Wallis analysis of variance (ANOVA) followed by Dunn’s Multiple Comparison Test for multiple group comparisons. Two-way repeated measures ANOVA were used in experiment characterizing the time course of synaptic transmission rundown (Figure S5H). For electrophysiology experiments, n-values represent the number of recorded cells. In most cases, a single cell was recorded per slice, with each animal contributing at most 5 individual recordings per condition. For immunohistochemistry, n represents the number of mice analyzed (Figures S4B–S4D) or the total number of TH+ cells quantified (Figure S4E). For fiber photometry recordings, n represents the number of imaging sessions performed, with a maximum of 2 imaging sessions per mouse. Exact p-values are provided in text and figure legends, and statistical significance in figures is presented as *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit policlonal anti-Tyrosine Hydroxylase | Millipore | Cat#: AB152; RRID:AB_390204 |
| Mouse monoclonal anti-HA | Abcam | Cat#: ab18181; RRID:AB_444303 |
| Goat ployclonal anti-Aldh1a1 | R&D Systems | Cat#: AF5869; RRID:AB_2044597 |
| Rabbit monoclonal anti-Gat1 | Cell Signaling | Cat#: 37342; RRID: AB_2905573 |
| Guinea pig polyclonal anti-Cre-recombinase | Synaptic Systems | Cat#: 257 004; RRID: AB_2782969 |
| Goat anti-mouse IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat#: A11029; RRID:AB_2534088 |
| Goat anti-mouse IgG Alexa Fluor 568 | Thermo Fisher Scientific | Cat#: A11004; RRID:AB_2534072 |
| Goat anti-rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat#: A11034; RRID:AB_2576217 |
| Goat anti-rabbit IgG Alexa Fluor 568 | Thermo Fisher Scientific | Cat#: A11036; RRID:AB_10563566 |
| Donkey anti-goat IgG Alexa Fluor 568 | Thermo Fisher Scientific | Cat#: A11057; RRID:AB_2534104 |
| Donkey anti-rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat#: A21206; RRID:AB_2535792 |
| Goat anti-guinea pig IgG Alexa Fluor 568 | Abcam | Cat#: ab175714; RRID:AB_2864763 |
| GAT1-Flox14F: TGTGCTGTCCTTCTGGCTGAACATCTACT | This manuscript | N/A |
| GAT1-Flox18F: TGGAGATAGTCGTTCAGGACAAGCCTACC | This manuscript | N/A |
| GAT1-Flox15R: AGCATGGCTGGTCAGGGAAGACACTATAG | This manuscript | N/A |
| Lab ID: GAT1-FloxF: TTCCTGAGTCCCAACATGCCTCG | This manuscript | N/A |
| GAT1-FloxR: AGTGACAGTGGTGAGTCCATGCAGC | This manuscript | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pAAV-EF1a-double fioxed-hChR2(H134R)-mCherry-WPRE-HGHpA | Addgene | Cat#: 20297-AAV8; RRID:Addgene_20297 |
| pAAV-hSyn-DA2m | Sun et al., 2020 | Kindly provided by Dr. Yulong Li |
| pAAV-EF1a-fDIO-Cre | Addgene | Cat#: 121675-AAV8; RRID:Addgene_121675 |
| pAAV-EF1a-double floxed-Aldh1a1-HA | This manuscript | Cat#: 184633; RRID:Addgene_184633 |
| pAAV-EF1a-double floxed-Gat1-HA | This manuscript | Cat#: 184635; RRID:Addgene_184635 |
| pAAV-EF1a-double floxed-Gat1(R69K)-HA | This manuscript | Cat#: 184636; RRID:Addgene_184631 |
| pAAV-EF1a-double floxed-Gad65 | This manuscript | Cat#: 184636; RRID:Addgene_184631 |
| pAAV-EF1a-double floxed-Gad67 | This manuscript | Cat#: 184632; RRID:Addgene_184632 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| SR95531 | Tocris | Cat#: 1262; CAS#: 104104-50-9 |
| CGP55845 hydrochloride | Tocris | Cat#: 1248; CAS#: 149184-22-5 |
| NBQX disodium salt | Tocris | Cat#: 1044; CAS#: 479347-86-9 |
| R-CPP | Tocris | Cat#: 0247; CAS#: 126453-07-4 |
| Reserpine | Tocris | Cat#: 2742; CAS#: 50-55-5 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mm-Slc6a1-O1 | Advanced Cell Diagnostics | Cat#: 1086931-C3 |
| Mm-Gad1 | Advanced Cell Diagnostics | Cat#: 400951-C3 |
| Mm-Gad2 | Advanced Cell Diagnostics | Cat#: 439371-C2 |
| Mm-Slc6a3 | Advanced Cell Diagnostics | Cat#: 31544-C2 |
| Mm-Th | Advanced Cell Diagnostics | Cat#: 317621 |
| Protease IV | Advanced Cell Diagnostics | Cat#: 322340 |
| Fluorescent Multiplex Detection Reagents: AMP 1, AMP2, AMP 3, AMP 4, DAPI | Advanced Cell Diagnostics | Cat#: 320851 |
|
| ||
| Deposited data | ||
|
| ||
| Data for figures | This manuscript | https://github.com/TritschLab/Melani-Tritsch-2022 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| B6.C-Tg(CMV-cre)1Cgn/J | Jackson Laboratory (Schwenk et al., 1995) | Strain #: 006054; RRID:IMSR_JAX:006054 |
| B6.SJL-Slc6a3tm1.1(cre)Bkmn/J | Jackson Laboratory (Bäckman et al., 2006) | Strain #: 006660; RRID:IMSR_JAX:006660 |
| B6.Cg-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J | Jackson Laboratory (Madisen et al., 2012) | Strain # 024109; RRID:IMSR_JAX:024109 |
| B6N(Cg)-Slc6a3tm1.1(flpo)Fuyu/J | Jackson Laboratory | Strain # 033673; RRID:IMSR_JAX:033673 |
| B6.129-Aldh1a1tm1Gdu/J | Fan et al., 2003 | Kindly provided by Dr. Huaibin Cai |
| VMAT2fl/fl | Isingrini et al., 2016 | Kindly provided by Dr. Bruno Giros |
| Gat1−/− | This manuscript | N/A |
| Gat1fl - B6.Cg-Slc6a1tm1.1Ntrch/J | This manuscript | Strain #: 037219 |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji ImageJ | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| Cellpose | Stringer et al., 2021 | https://www.cellpose.org/ |
| GraphPad Prism 9.0 | GraphPad | RRID:SCR_002798 |
| Custom MATLAB code | Pologruto et al., 2003 | RRID:SCR_014307 |
| Igor Pro 6.02A | Wavemetrics | RRID:SCR_000325 |
| OlyVIA 2.8 | Olympus | https://www.olympus-lifescience.com/en/discovery/image-sharing-made-easy-meet-olyvia/ |
| ZEN 2.6 | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
|
| ||
| Other | ||
|
| ||
| Fluoromount-G | SouthernBiotech | Cat#: 0100-01 |
| DAPI Fluoromount-G | SouthernBiotech | Cat#: 0100-20 |
| O.C.T compound embedding medium | Fisher Scientific | Cat#: 4585 |
Highlights.
SNcDA neurons release GABA through unconventional mechanisms
Aldh1a1 is neither necessary nor sufficient to provide GABA in SNcDA neurons
Selective deletion of Gat1 from SNcDA neurons abolishes GABA co-release
Selective deletion of Vmat2 from SNcDA neurons abolishes GABA co-release
ACKNOWLEDGMENTS
We thank Adam Carter, Margaret Rice, Dick Tsien, and members of the Tritsch laboratory for comments on the manuscript; Yulong Li for the GRAB-DA2m DA sensor; and Huaibin Cai and Bruno Giros for kindly providing the Aldh1a1−/− and VMAT2f/f mice, respectively. We acknowledge the New York University Medical Center Rodent Genetic Engineering Laboratory for rederivation, the Genotyping Core Laboratory for mouse genotyping, the Department of Comparative Medicine for animal care and maintenance, and the Neuroscience Institute’s imaging facilities for microscope availability. R.M. is supported by a Marlene and Paolo Fresco Postdoctoral Fellowship. N.X.T. is an Alfred P. Sloan Research Fellow in Neuroscience and is supported by grants from the Dana, Whitehall and Feldstein Medical Foundations, as well as from the National Institutes of Health (DP2NS105553).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110716.
REFERENCES
- Adam SA, Schnell O, Pöschl J, Eigenbrod S, Kretzschmar HA, Tonn J-C, and Schüller U (2012). ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients. Brain Pathol. 22, 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DW, Schray RC, Duester G, and Schneider JS (2011). Functional significance of aldehyde dehydrogenase ALDH1A1 to the nigrostriatal dopamine system. Brain Res. 1408, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, et al. (1996). Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem. Biophys. Res. Commun. 229, 891–895. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, and Obata K (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U S A 94, 6496–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. [DOI] [PubMed] [Google Scholar]
- Belelli D, and Lambert JJ (2005). Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat. Rev. Neurosci. 6, 565–575. [DOI] [PubMed] [Google Scholar]
- Bendahan A, and Kanner BI (1993). Identification of domains of a cloned rat brain GABA transporter which are not required for its functional expression. FEBS Lett. 318, 41–44. [DOI] [PubMed] [Google Scholar]
- Bertram S, Cherubino F, Bossi E, Castagna M, and Peres A (2011). GABA reverse transport by the neuronal cotransporter GAT1: influence of internal chloride depletion. Am. J. Physiol. Cell Physiol. 301, C1064–C1073. [DOI] [PubMed] [Google Scholar]
- Bragina L, Marchionni I, Omrani A, Cozzi A, Pellegrini-Giampietro DE, Cherubini E, and Conti F (2008). GAT-1 regulates both tonic and phasic GABA(A) receptor-mediated inhibition in the cerebral cortex. J. Neurochem. 105, 1781–1793. [DOI] [PubMed] [Google Scholar]
- Brodnik ZD, Batra A, Oleson EB, and España RA (2019). Local GABAA receptor-mediated suppression of dopamine release within the nucleus accumbens. ACS Chem. Neurosci. 10, 1978–1985. [DOI] [PubMed] [Google Scholar]
- Dayan-Alon O, and Kanner BI (2019). Internal gate mutants of the GABA transporter GAT1 are capable of substrate exchange. Neuropharmacology 161, 107534. [DOI] [PubMed] [Google Scholar]
- Deken SL, Beckman ML, Boos L, and Quick MW (2000). Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat. Neurosci. 3, 998–1003. [DOI] [PubMed] [Google Scholar]
- Eskenazi D, Malave L, Mingote S, Yetnikoff L, Ztaou S, Velicu V, Rayport S, and Chuhma N (2021). Dopamine neurons that cotransmit glutamate, from synapses to circuits to behavior. Front. Neural Circuits 15, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe S-I, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, and Duester G (2003). Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell Biol. 23, 4637–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun B-C, Killeen N, Sulzer D, and Edwards RH (1997). Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron 19, 1271–1283. [DOI] [PubMed] [Google Scholar]
- Fortin GM, Ducrot C, Giguére N, Kouwenhoven WM, Bourque M-J, Pacelli C, Varaschin RK, Brill M, Singh S, Wiseman PW, et al. (2019). Segregation of dopamine and glutamate release sites in dopamine neuron axons: regulation by striatal target cells. FASEB J. 33, 400–417. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ, and Sharabi Y (2013). Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 126, 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, and Edwards RH (2010). Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron 65, 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isingrini E, Guinaudie C, C Perret L, Rainer Q, Moquin L, Gratton A, and Giros B (2017). Genetic elimination of dopamine vesicular stocks in the nigrostriatal pathway replicates Parkinson’s disease motor symptoms without neuronal degeneration in adult mice. Sci. Rep. 7, 12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isingrini E, Perret L, Rainer Q, Sagueby S, Moquin L, Gratton A, and Giros B (2016). Selective genetic disruption of dopaminergic, serotonergic and noradrenergic neurotransmission: insights into motor, emotional and addictive behaviour. J. Psychiatry Neurosci. 41, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs FMJ, Smits SM, Noorlander CW, von Oerthel L, van der Linden AJA, Burbach JPH, and Smidt MP (2007). Retinoic acid counteracts developmental defects in the substantia nigra caused by Pitx3 deficiency. Development 134, 2673–2684. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu C-S, Sokolova I, Lester HA, and Mody I (2003). GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol. 90, 2690–2701. [DOI] [PubMed] [Google Scholar]
- Jin S, Cao Q, Yang F, Zhu H, Xu S, Chen Q, Wang Z, Lin Y, Cinar R, Pawlosky RJ, et al. (2021). Brain ethanol metabolism by astrocytic ALDH2 drives the behavioural effects of ethanol intoxication. Nat. Metab 3, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston GA (1996). GABAA receptor pharmacology. Pharmacol. Ther. 69, 173–198. [DOI] [PubMed] [Google Scholar]
- Kim J-I, Ganesan S, Luo SX, Wu Y-W, Park E, Huang EJ, Chen L, and Ding JB (2015). Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PF, Twedell EL, Shin JH, Zhang R, and Khaliq ZM (2020). Axonal mechanisms mediating g-aminobutyric acid receptor type A (GABA-A) inhibition of striatal dopamine release. eLife 9, e55729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Koh W, Kim S, Song K, Shin J-I, Lee JM, Lee EH, Bae JY, Ha GE, Oh J-E, et al. (2020). Astrocytes control sensory acuity via tonic inhibition in the thalamus. Neuron 108, 691–706.e10. [DOI] [PubMed] [Google Scholar]
- Liu G, Yu J, Ding J, Xie C, Sun L, Rudenko I, Zheng W, Sastry N, Luo J, Rudow G, et al. (2014). Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Invest 124, 3032–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes EF, Roberts BM, Siddorn RE, Clements MA, and Cragg SJ (2019). Inhibition of nigrostriatal dopamine release by striatal GABAA and GABAB receptors. J. Neurosci. 39, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo J, Berenyi A, Fujisawa S, Hsu Y-WA, Garcia AJ, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffery P, and Dräger UC (1994). High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc. Natl. Acad. Sci. U S A 91, 7772–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani R, and Tritsch NX (2021). How alcohol affects motor control: not your usual suspects. Nat. Metab. 3, 293–294. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, and Kreitzer AC (2014). Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron 82, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Yu J, Sun L, Xie C, Chang L, Wu J, Hawes S, Saez-Atienzar S, Zheng W, Kung J, et al. (2019). ALDH1A1 regulates postsynaptic m-opioid receptor expression in dorsal striatal projection neurons and mitigates dyskinesia through transsynaptic retinoic acid signaling. Sci. Rep. 9, 3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantanowitz S, Bendahan A, and Kanner BI (1993). Only one of the charged amino acids located in the transmembrane alpha-helices of the gamma-aminobutyric acid transporter (subtype A) is essential for its activity. J. Biol. Chem. 268, 3222–3225. [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, and Svoboda K (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin J-F, Zou J, Drouin-Ouellet J, Kim K-YA, Cicchetti F, and Awatramani RB (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep. 9, 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, and Dollé P (2012). Retinoic acid signalling during development. Developement 139, 843–858. [DOI] [PubMed] [Google Scholar]
- Roberts BM, Doig NM, Brimblecombe KR, Lopes EF, Siddorn RE, Threlfell S, Connor-Robson N, Bengoa-Vergniory N, Pasternack N, Wade-Martins R, et al. (2020). GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 11, 4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RM, Ingram SL, and Scimemi A (2021). Regulation of glutamate, GABA and dopamine transporter uptake, surface mobility and expression. Front. Cell. Neurosci. 15, 670346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, and Rajewsky K (1995). A cre -transgenic mouse strain for the ubiquitous deletion of loxP -flanked gene segments including deletion in germ cells. Nucleic Acids Research 23 (24), 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A (2014). Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci 8, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgobio C, Wu J, Zheng W, Chen X, Pan J, Salinas AG, Davis MI, Lovinger DM, and Cai H (2017). Aldehyde dehydrogenase 1-positive nigrostriatal dopaminergic fibers exhibit distinct projection pattern and dopamine release dynamics at mouse dorsal striatum. Sci. Rep. 7, 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silm K, Yang J, Marcott PF, Asensio CS, Eriksen J, Guthrie DA, Newman AH, Ford CP, and Edwards RH (2019). Synaptic vesicle recycling pathway determines neurotransmitter content and release properties. Neuron 102, 786–800.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stensrud MJ, Puchades M, and Gundersen V (2014). GABA is localized in dopaminergic synaptic vesicles in the rodent striatum. Brain Struct. Funct. 219, 1901–1912. [DOI] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, and Pachitariu M (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, and Rayport S (1998). Dopamine neurons make glutamatergic synapses in vitro. J. Neurosci. 18, 4588–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zhou J, Dai B, Qian T, Zeng J, Li X, Zhuo Y, Zhang Y, Wang Y, Qian C, et al. (2020). Next-generation GRAB sensors for monitoring dopaminergic activity in vivo. Nat. Methods 17, 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, et al. (2010). Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J. Neurosci. 30, 7105–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, and Nicoll R (1999). The role of the synthetic enzyme GAD65 in the control of neuronal g-aminobutyric acid release. Proc. Natl. Acad. Sci. U S A 96, 12911–12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, and Sabatini BL (2012). Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 490, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Granger AJ, and Sabatini BL (2016). Mechanisms and functions of GABA co-release. Nat. Rev. Neurosci. 17, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Oh W-J, Gu C, and Sabatini BL (2014). Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife 3, e01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau L-E, Hnasko TS, Wallén-Mackenzie A, Morales M, Rayport S, and Sulzer D (2014). The multilingual nature of dopamine neurons. Prog. Brain Res. 211, 141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Ohtsuka T, Kobayashi K, and Watanabe M (2016). Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc. Natl. Acad. Sci. U S A 113, 4206–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tu P, Bonet L, Aubrey KR, and Supplisson S (2013). Cytosolic transmitter concentration regulates vesicle cycling at hippocampal GABAergic terminals. Neuron 80, 143–158. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, and Rhee J-S (2006). A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50, 575–587. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Díez-Sampedro A, and Richerson GB (2007). Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelin R, and Schuldiner S (1995). The pharmacological profile of the vesicular monoamine transporter resembles that of multidrug transporters. FEBS Lett. 377, 201–207. [DOI] [PubMed] [Google Scholar]
- Zhang S, Qi J, Li X, Wang H-L, Britt JP, Hoffman AF, Bonci A, Lupica CR, and Morales M (2015). Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat. Neurosci. 18, 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper lead contact are available at https://github.com/TritschLab/Melani-Tritsch-2022.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.