

CONSPECTUS
Protein aggregation is a biological phenomenon in which aberrantly processed or mutant proteins misfold and assemble into a variety of insoluble aggregates. Decades of studies have delineated the structure, interaction, and activity of proteins in either their natively folded structures or in insoluble aggregates such as amyloid fibrils. However, a variety of intermediate species exist between these two extreme states in the protein folding landscape. Herein, we collectively term these conformations as misfolded protein oligomers, including soluble oligomers and pre-amyloidal oligomers that are formed by unfolded or misfolded proteins. While extensive tools have been developed to study folded proteins or amyloid fibrils, research to understand the properties and activities of misfolded protein oligomers has been limited by the lack of methods to detect and interrogate these species in live cells.
In this Account, we describe our efforts in the development of chemical methods that allow for the characterization of the multi-step protein aggregation process, in particular the misfolded protein oligomers, in living cells. As the start of this journey, we attempted to develop a fluorogenic method, wherein the misfolded oligomers could turn on the fluorescence of chemical probes that are conjugated to the protein-of-interest (POI). To this end, we produced a series of destabilized HaloTag variants, formulating the primary component of the AgHalo sensor, which misfold and aggregate when cells are subjected to stress. When AgHalo is covalently conjugated with a solvatochromic fluorophore, misfolding of the AgHalo conjugation would activate fluorescence, resulting in the observation of misfolded oligomers. Following this work, we extended the scope of detection from AgHalo to any protein-of-interest via the AggTag method, wherein the POIs are genetically fused to self-labeling protein tags (HaloTag or SNAP-tag). Focusing on the molecular rotor-based fluorophores, we applied the modulated FP chromophore core as a prototype for the AggTag probes, to enable the fluorogenic detection of misfolded soluble oligomers of multiple proteins in live cells. Next, we further developed the AggTag method to distinguish insoluble aggregates from misfolded oligomers, using two classes of probes that differently activate fluorescence emission towards these two conformations. To enable this goal, we applied physical organic chemistry and computational chemistry to discover a new category of triode-like fluorophores, wherein the π orbitals of either an electron density regulator or the donor-acceptor linkages are used to control the rotational barriers of fluorophores at the excited states. This mechanism allows us to rationally design molecular rotor-based fluorophores that have desired responses to viscosity, thus extending the application of the AggTag method.
In summary, our work allows the field to directly monitor the intermediate misfolded oligomers and differentiate insoluble aggregates from this conformation in live cells, thus enabling the studies of many currently unanswered questions in protein aggregation. Future directions are to develop methods that enable quantitative analyses of the protein aggregation process. Further, new methods are needed to detect and quantify the formation and maturation of protein or RNA condensates that form membraneless organelles.
Graphical Abstract
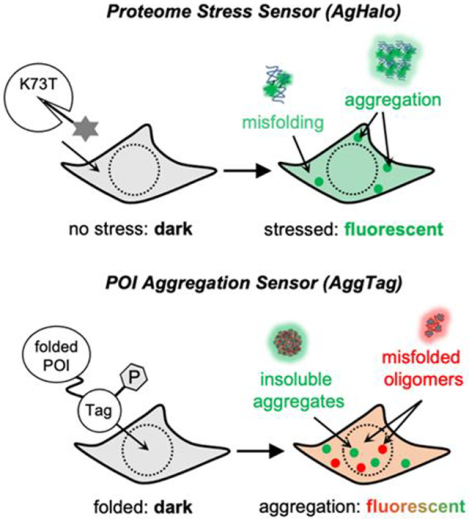
1. Introduction
Unfolded polypeptides emerging from the ribosome after translation need to properly fold into native three-dimensional structures to perform their physiological functions. The folding of the proteome within a cell is assisted by a set of highly conserved folding and quality control machineries, comprising macromolecular chaperones, chaperonins, ubiquitin-proteasome systems and autophagy5, 6. These machineries form a concerted proteostasis network to maintain an appropriate level of proteome homeostasis, which is reflected by a balance between the folded, misfolded and aggregated states of the cellular proteome7–10. However, endogenous and exogenous stress conditions (including genetic mutations, environmental perturbations, chemical toxins, and pathogen invasion) impair the integrity of proteostasis by shifting the free energy landscape of protein folding and/or inducing chemical or conformational changes in folded proteins11, 12.
Failure to maintain proteostasis during stress leads to global misfolding and aggregation of the endogenous proteome, resulting in a series of aberrant conformations that include misfolded proteins in the form of misfolded monomers, soluble oligomers, amorphous aggregates, and fibrils with ordered β-sheet structures. Extensive studies have connected cellular proteostasis to various biological processes including development, oncogenesis, metabolism and evolution. Stress-induced cellular protein aggregation has been associated with a growing number of human diseases, including neurodegenerative disorders, metabolic disorders, and some cancers13, 14. Protein aggregation is a multi-step process. Upon protein misfolding, misfolded monomers are produced and subsequently bind with one another to form misfolded oligomers (step 1, Fig 1A). Through a phase transition process, misfolded oligomers progress from their initial soluble oligomeric form into insoluble aggregates that exist in various forms, including amyloid-β fibrils, amorphous aggregates (step 2, Fig 1A). While extensive efforts have focused on either folded proteins or insoluble aggregates, a less studied species between these two states is the soluble misfolded oligomers intermediates. Evidence shows that the misfolded oligomers may play key roles in both cell physiology and pathology.15
Figure 1: The multi-step protein aggregation process.
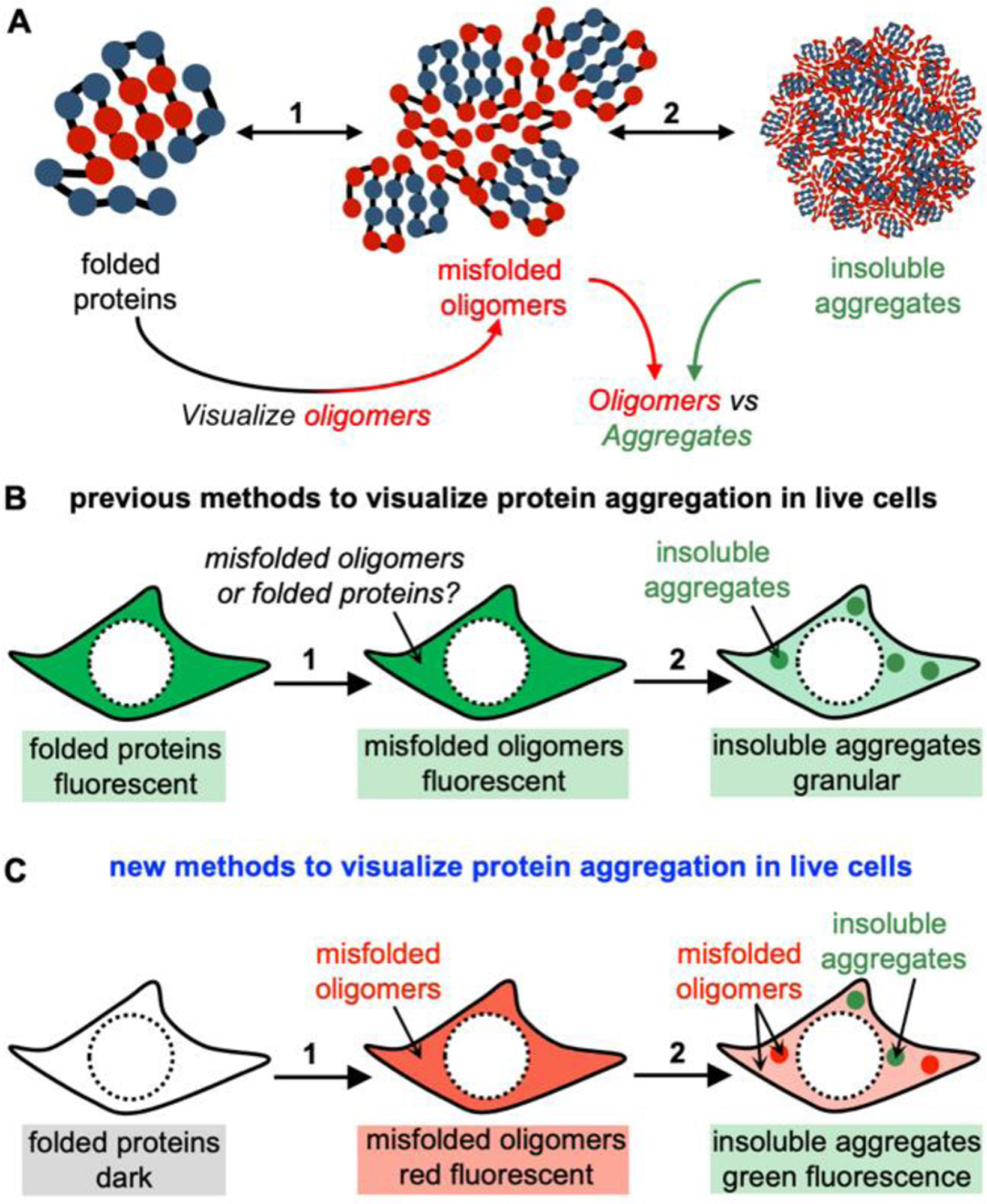
(A) Folded proteins misfold to form soluble oligomers, which proceed to accumulate and evolve into insoluble aggregates. (B) Previous methods employ FP-tagged or fluorescently labeled proteins to monitor protein aggregation. Events of aggregation are visualized by the transition from the diffused signal to the granular fluorescent structure. These methods, however, could not reveal misfolded oligomers in live cells. (C) The proposed new method to visualize protein aggregation in live cells. While folded proteins exhibit dark fluorescent signal, the misfolded oligomers emit red fluorescence in both diffuse and punctate structures. By contrast, the insoluble aggregates emit green fluorescence with primarily granular morphology.
Despite the emergence of a group of methods16–23, no simple and direct method is available to directly visualize the multi-step process of protein aggregation in live cells, in particular the misfolded oligomers. In the past, the aggregation of POIs has been mostly visualized by either the fusion of fluorescent protein (FP) tags or covalent labeling of fluorescent probes. In this technical scheme, diffused fluorescent signals represent folded and soluble POIs; whereas, punctate or granular fluorescent structures are indicative of protein aggregations (Fig 1B). These approaches are exemplified by destabilized client proteins that are fused to GFP16, 17, incorporation of fluorescent unnatural amino acids to thermally-labile proteins18, labeling FlAsH dye to proteins that are fused to a tetracysteine motif19, destabilized retroaldolase enzyme that is labeled with a fluorophore24, destabilized barnase enzyme22, GFP-fused heat-shock proteins25, and destabilized GFP26, 27. Despite the broad applications, the limitation remains - that is the fluorescence signal exists before and after protein misfolding or aggregation. Thus, these methods cannot report on soluble oligomers because these oligomers do not have granular structures. Furthermore, modern cell biology has revealed that RNAs and proteins can spontaneously undergo the liquid-liquid phase separation (LLPS) process to form biological condensates or membraneless organelles, which exhibit granular morphology both in vitro and in cells28–30. Most proteins involved in LLPS contain intrinsically disordered regions, making this process connected to protein aggregation. In many cases, the condensates are formed as liquid droplets; however, the material state can convert from the liquid to the solid states that are likely protein aggregates31–34. While the FP-fusion method can rely on the change of fluorescence to monitor when and how POIs go through LLPS to form biological condensates, this method cannot delineate the folding states of POIs (folded structures or soluble misfolded oligomers) in the liquid droplets, because both of which exhibit liquid-like characteristics. As a result, the traditional FP-fusion method is particularly unsuited to reveal the protein aggregation process or membraneless organelles because of their granular morphology.
Therefore, visualizing the multi-step process of protein aggregation, particularly the intermediate misfolded oligomers, has been increasingly recognized as an important field in the biomedical and biochemical communities. To this end, our group has developed a series of fluorescence-based imaging methodologies, which enable fluorogenic detection (turn-on fluorescence) of the entire protein aggregation process in live cells. This Account describes two goals that we had achieved (Fig 1C). First, we had developed chemical methods and fluorescent probes to detect previously invisible misfolded oligomers. Second, we also developed methods to distinguish insoluble aggregates from misfolded oligomers, using probes that only recognize insoluble aggregates and harbor distinct spectral properties from probes used in Step 1. Ultimately, a combination of these probes will allow live-cell imaging of misfolded oligomers using turn-on fluorescence, and differentiation between oligomers and insoluble aggregates using orthogonal fluorescent signals (Fig 1C). In a long term, these novel research tools will make positive and significant contributions to bridge the knowledge gaps in protein biochemistry and cell biology.
2. AgHalo: A protein-based fluorogenic sensor to quantify cellular proteome stress
We began with the development of a two-component proteome stress sensor that is composed of a metastable protein and a fluorogenic probe. The metastable protein only aggregates in the cells with deficient proteostasis; whereas the fluorogenic probe covalently conjugates with the protein and undergo changes of fluorescence (e.g. turn-on when the protein-probe conjugate misfolds and aggregates) (Fig 2B). Without proteome stress, the sensor remains folded. In the presence of proteome stress, the sensor misfolds and aggregates. During this process, the fluorogenic probe would exhibit changes in fluorescence intensity that can be quantified to reflect the extent of sensor aggregation, as a measure of proteome stress. We started with the HaloTag protein, an engineered dehalogenase that reacts with chloroalkane ligands to form a stable covalent enzyme-ligand conjugate35. HaloTag is an ideal platform because of its thermodynamic stability (ΔGfolding = −5.6 kcal/mol), established warhead for a bio-orthogonal reaction in various cells, and a rapid labeling kinetics (107 M−1 s−1). By leveraging the HaloTag technology, we bypass the hurdle to develop a new bioorthogonal labeling reaction and jump-start on the HaloTag-based proteome stress sensor, hereinafter named AgHalo.
Figure 2: The AgHalo-1 conjugate activates fluorescence when AgHalo misfolds in temperature or chemical stressed cells.
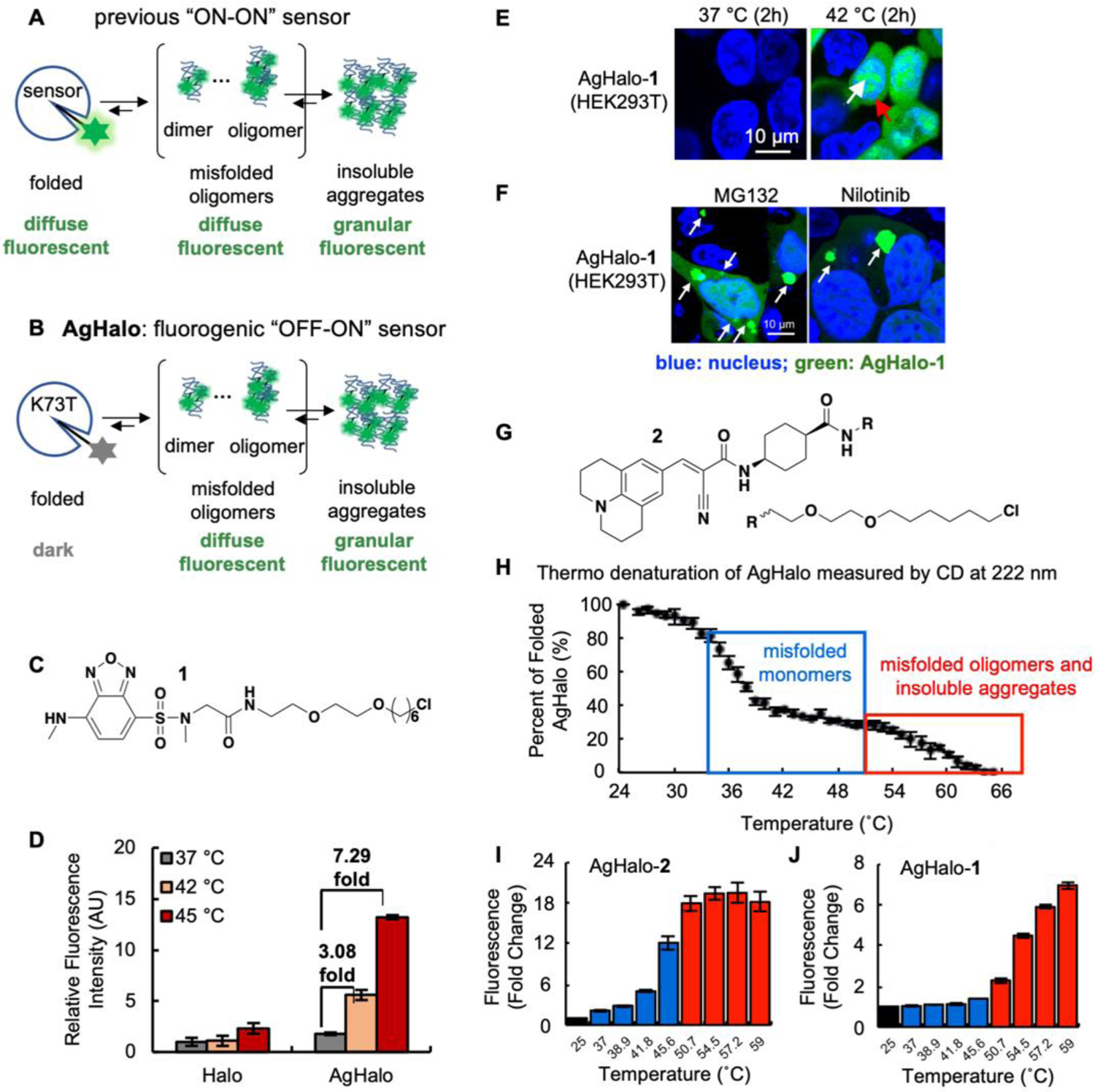
(A) Previous protein aggregation detection methods exhibit similar diffuse fluorescence signal in the folded states and the misfolded oligomeric states. (B) The proposed experimental goal wherein the AgHalo (K73T) conjugated with a fluorophore could activate fluorescence in misfolded oligomers and insoluble aggregates. (C) Chemical structure of 1. (D) The AgHalo-1 conjugate activates fluorescence in heat-shocked cells. (E–F) Fluorescence microscopy visualizes the misfolded oligomers of AgHalo-1 in cells treated with heat (E) or chemical stressors (F). (G) Chemical structure of 2. (H) Thermo denaturation of AgHalo results the formation of misfolded monomers (blue box), and misfolded oligomers or insoluble aggregates (red box). (I–J) Fluorescence response of molecular rotor-based probe 2 (I) and solvatochromic probe 1 (J) during thermo denaturation of AgHalo. (A–F) Reproduced with permission from Ref. 1. Copyright 2017 John Wiley & Sons, Inc. (G–I) Reproduced with permission from Ref. 41. Copyright 2018 American Chemical Society.
Using a screening strategy, we firstly identified a series of metastable HaloTag variants for the proteostasis sensor, as represented by K73T (ΔGfolding = −2.2 kcal/mol), which was used to study its aggregation in mammalian HEK293T cells treated with heat-shock.1 We found that K73T aggregated at elevated temperature using a fractionation experiment: 4%, 11%, and 40% of K73T was found in the insoluble fraction at 37 °C, 42 °C and 45 °C, respectively. Furthermore, treating cells at 37 °C with a proteasome inhibitor MG-132 that is known to cause proteome aggregation resulted in 23% of K73T in the insoluble fraction. These results suggest that K73T is suitable to report on cellular proteostasis deficiency via its aggregation.
To construct a turn-on fluorescence ligand for the proteome stress sensor, we explored solvatochromic fluorophores that exhibit fluorescence intensity increase when their adjacent surroundings transit from polar to non-polar microenvironment36, 37. For a folded protein, the hydrophobic residues are normally buried in its core; whereas the exterior surface is occupied with the hydrophilic residues, resulting in a primarily polar microenvironment (corresponding to dielectric constant of ~30). Protein misfolding disrupts the hydrophobic core and thus exposes the non-polar residues to the surface of misfolded proteins (corresponding to dielectric constant of ~3–6). Thus, solvatochromic fluorophores are expected to activate fluorescence when proteins transit from folded to misfolded conformations. Guided by this principle, we designed and synthesized a probe harboring a sulfonyl-benzoxadiazole (SBD) moiety (Fig 2C)38, 39, which is a well-established solvatochromic fluorophore that increases fluorescence intensity when local polarity decreases. An optimized sarcosine linker was installed on the original linker of HaloTag ligands to extend the SBD moiety out of HaloTag and position SBD in a hydrophilic environment5. The resulting ligand 1 had low fluorescence in PBS buffer (quantum yield: ϕ=0.002) and when conjugated to folded K73T (ϕ=0.02). When the K73T-1 conjugate aggregated at 59°C, we observed a blue shift of emission maximum from 600 nm to 545 nm and a 10-fold fluorescence increase (ϕ=0.22). Importantly, we demonstrated that soluble oligomers of the K73T-1 conjugate are sufficient to activate fluorescence. Thus, the AgHalo sensor bears the premise to visualize misfolded oligomers in live cells.
Next, we assessed whether the AgHalo sensor detects heat-induced proteome stress in live cells (Fig 2D). Fluorescence of the AgHalo sensor could not only report the presence of heat (42°C) stress in real time (Fig 2D), but also distinguish its severity by the different fold-of-change (7.29-fold at 45°C, 3.08-fold at 42°C; Fig 2D). Confocal fluorescence microscopy confirmed that fluorescence signal originated from both misfolded oligomers and insoluble aggregates formed by the AgHalo-1 aggregates, shown by the diffuse (red arrow) and punctate (white arrow) fluorescent structures, respectively (Fig 2E). We also studied drug-induced proteome stress, using MG132 that is known to cause proteome aggregation, and five common anti-cancer drugs which their side effect is to compromise cellular proteostasis that cannot be detected by common cytotoxicity assays. In the cases of MG132 and nilotinib, we also observed the activation of fluorescence in both diffuse and punctate structures, suggesting that the AgHalo-1 fluorescence reports on both the misfolded oligomers and insoluble aggregates (Fig 2F). AgHalo-1 has been employed in other research groups to resolve various biological questions. As an example, Wang et al. directed AgHalo into the endoplasmic reticulum (AgHaloER) to demonstrate that phosphorylation of protein disulfide isomerase (PDI) at the position of Ser357 provides a folding favorable ER proteostasis. In the PDI knock-out HepG2 cells, AgHaloER-1 emitted turn-on fluorescence and formed granular structures, suggesting aggregation of AgHalo. This fluorescent signal could be significantly reduced when the wild-type PDI was over-expressed, indicating rescue of AgHalo folding and inhibition of aggregation40.
In addition to the solvatochromic probe 1 that is sensitive to local polarity, we further devised a molecular rotor-based probe 2 that is sensitive to local viscosity (Fig 2G)41. Molecular rotors have been applied to detect insoluble protein aggregates and amyloid fibrils. Using the 9-(2-carboxy-2-cyanovinyl)julolidine (CCVJ) fluorophore as an example (probe 2), we demonstrated that the misfolded state of AgHalo is able to induce fluorescence increase by hindering rotation of probe 2. Interestingly, fluorescence is activated when the AgHalo-2 forms misfolded monomers (blue box; Fig 2H), resulting in a 12-fold increase (blue bars; Fig 2I). This signal further increased to ~20-fold when misfolded oligomers or insoluble aggregates form (red box; Fig 2H and red bars; Fig 2I). By contrast, the solvatochromic fluorophore probe 1, when conjugated with AgHalo, cannot detect misfolded monomers of AgHalo-1 (blue bars; Fig 2J), suggesting that the initial misfolded state of AgHalo may not change its dielectric constant sufficiently to turn on fluorescence of 1. This result inspired us to expand the molecular rotor-based fluorophores to the detection of protein misfolding in live cells.
3. Detecting misfolded oligomers in live cells using the AggTag method and chemically modified fluorescent protein chromophores
While the AgHalo system allowed the visualization of misfolded oligomers for AgHalo-1 or AgHalo-2 conjugates, it would be more significant to image the misfolded oligomers of any proteins-of-interest in live cells. Hence, we proposed the AggTag (Aggregation Tag) method, which can visualize misfolded oligomers and insoluble aggregates within the cellular milieu (Fig 3A)2. This method features small molecular AggTag probes (Fig 3A) that turn on their fluorescence only upon interaction with misfolded oligomers and insoluble aggregates, due to the increased surface hydrophobicity and/or elevated intermolecular rigidity of these species compared to folded proteins. When AggTag probes are covalently conjugated to a protein tag that is genetically-fused to folded POI, fluorescence of probes is quenched. Formation of misfolded oligomers and/or insoluble aggregates of the POI, however, inhibits the quenching and yields the turn-on fluorescence via intramolecular interactions with the probes. Unlike the conventional FP-fusion methods that track the location of late-stage aggregates via the appearance of fluorescent puncta, AggTag probes specifically fluoresce in response to changes in protein conformation and thus report on early events during protein aggregation.
Figure 3: Visualizing the misfolded oligomers using the AggTag method.
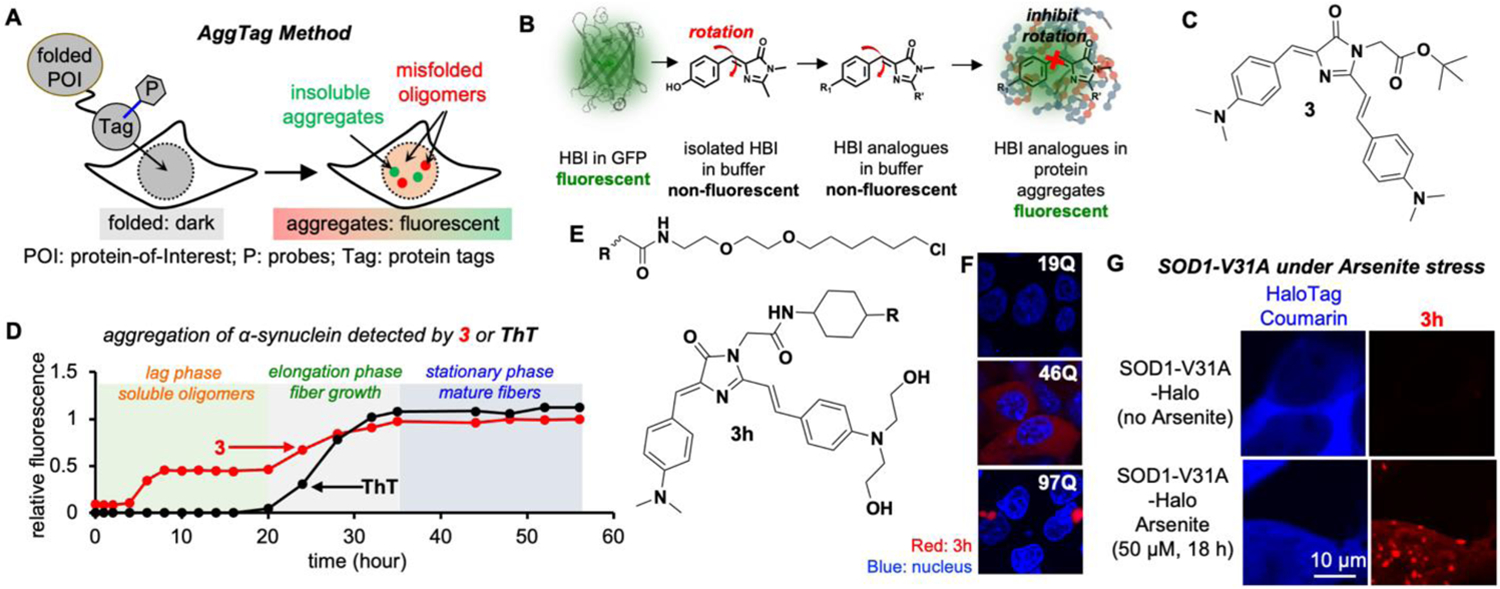
(A) Overview of the AggTag method. (B) Chemical modulation of the FP chromophore (HBI) to detect protein aggregation. (C) Chemical structure of 3. (D) 3 detects misfolded oligomers during α-synuclein aggregation. (E) Chemical structure of 3h. (F–G) Detecting misfolded oligomers of Htt (F) and SOD1-V31A (G). Reproduced with permission from Ref. 2. Copyright 2018 American Chemical Society.
To develop such probes, we studied the chromophore core of the green fluorescent protein, 4-hydroxybenzylidene-imidazolinone (HBI), which belongs to molecular-rotor based fluorophores (Fig 3B).42 Upon photoexcitation, HBI readily undergoes intramolecular rotation and enters low energy non-radiative twisted intramolecular charge transfer (TICT) state. Inhibition of the TICT pathway is often achieved by increasing environmental viscosity, hence suppressing the internal rotation and restoring fluorescence. Based on our experience with CCVJ, formation of misfolded protein oligomers and/or protein aggregates increases intermolecular rigidity and microviscosity. Thus, we anticipated that HBI analogues would exhibit turn-on fluorescence when their TICT is inhibited in the rigid environment within protein aggregates (Fig 3B). Structural variation of HBI was achieved by replacing the hydroxide group to a better electron donating group—N, N-dimethyl amino group and installing an extended π conjugation on the imidazole heterocycle. The final HBI derivative, probe 3 (Fig 3C), exhibited enhanced fluorescence when dissolved in glycerol, a viscous solvent mimicking compact microenvironment in misfolded oligomers, while remaining dark in non-viscous solvents. Probe 3 exhibits quantum yield (ϕ) value of 0.22 in glycerol, comparable to ϕ of the Kaede protein as 0.3343. The molar extinction coefficient (ε) of 3 is 39,049 M−1•cm−1, about half of mCherry (72,000 M−1•cm−1). Thus, the turn-on fluorescence and the brightness of probe 3 in glycerol make it appropriate for live cell imaging of misfolded oligomers.
To test 3, we chose α-synuclein (α-syn), whose aggregation is associated with Parkinson’s disease.44 Purified α-syn aggregates via a three-step process: formation of the soluble oligomers, growth of the amyloid fibers, and maturation of the fibers (Fig 3D). Thioflavin-T (ThT) monitors the growth and the maturation of the fibers, but not the misfolded oligomers (black curve in Fig 3D). Different from ThT, 3 exhibited fluorescence activation as early as 4 h, followed by further increase at 20 h until 36 h to plateau (red curve in Fig 3D). We further demonstrated that the AggTag approach could report POI aggregation in live cells, with a focus on the previously “invisible” misfolded oligomers. To this end, we genetically fused HaloTag to the POI, and synthesized probe 3h bearing a HaloTag reactive warhead and 2-hydroxyl group for enhanced solubility (Fig 3E). We tested whether 3h detects soluble oligomers in live cells, using the Huntingtin exon 1 protein (Htt-polyQ) that harbors varying lengths of poly-glutamine repeats45, 46. Htt-polyQ with short glutamine repeats (Htt-16Q-Halo) resulted in no fluorescence (top panel, Fig 3F). The moderate length Htt-Q46-Halo, by contrast, showed a diffused fluorescence in cytosol (mid panel, Fig 3F); whereas, Htt-polyQ with longer glutamine repeats (Htt-97Q-Halo) resulted in granular structures as turn-on fluorescence from 3h without any background (lower panel, Fig 3F). Similar results were found with a mutant of superoxide dismutase 1 (SOD1-V31A). Sodium arsenite (NaAsO2) induces the accumulation of reactive oxygen species (ROS), causing cellular oxidative stress. Using the coumarin fluorescence, we found that SOD1-V31A-Halo was primarily located in the cytosol, and the oxidative stressor NaAsO2 induced partial translocation of V31A to nucleus33, 47. While the coumarin fluorescence remained diffuse before and after stress (left panel, Fig 3G), 3h only exhibited both diffuse and punctate fluorescent structures in stressed cells (right panel, Fig 3G). Taken together, we have achieved observing the previously invisible misfolded protein oligomers from properly folded protein monomers in live cells using the fluorogenic AggTag approach.
4. AggFluor: a fluorogenic toolbox to visualize the multi-step process of protein aggregation
Protein aggregation is a multi-step process that involves various aggregation species with different conformational states. It is important to distinguish insoluble aggregates from misfolded oligomers, because not only do they have distinct functions in cells, but also are managed differently by cells. In particular, misfolded oligomers do not necessarily display as a diffusive structure, instead they can reside in granular structures that appear to be almost identical to granules formed by insoluble aggregates48, 49. In this case, probe 3h is unable to distinguish granules that contain misfolded oligomers or insoluble aggregates, because their fluorescence could be equally activated in both conformations. When considering the physicochemical features of these two conformations, the misfolded oligomers should be less viscous and exhibit greater intermolecular spaces than the insoluble aggregates. Thus, we proposed to develop two classes of molecular rotor-based probes, named AggFluor, to distinguish between these two conformations: the Class 1 probe activates fluorescence when incubated with both conformations, while the Class 2 probe only turns on fluorescence when incubated with insoluble aggregates3.
To achieve this goal, we revisited our previous fluorophores, to discover fluorophores that could remain dark upon binding to misfolded oligomers while activate fluorescence for insoluble aggregates. We used glycerol at 25 °C (972 cP) to mimic the viscous environment for misfolded oligomers, and glycerol frozen at −80 °C (3 × 1011 cP) to mimic insoluble aggregates. To our surprise, probe 4 (Fig 4A), which was originally rejected for the detection of misfolded oligomer due to its low quantum yield (Φ=0.027) in glycerol, showed an 11-fold fluorescence increase in frozen glycerol (Fig 4B). By contrast, probe 3, which was optimized for misfolded oligomers detection, showed only a 1.5-fold increase in frozen glycerol (Fig 4C). This side-by-side comparison revealed a significant viscosity sensitivity difference between probes 3 and 4. While probe 3 exhibited a notable emission signal in the medium viscosity environment, probe 4 requires a much more rigid local environment than probe 3 to be fluorescent.
Figure 4: Detecting the multi-step protein aggregation process in live cells.
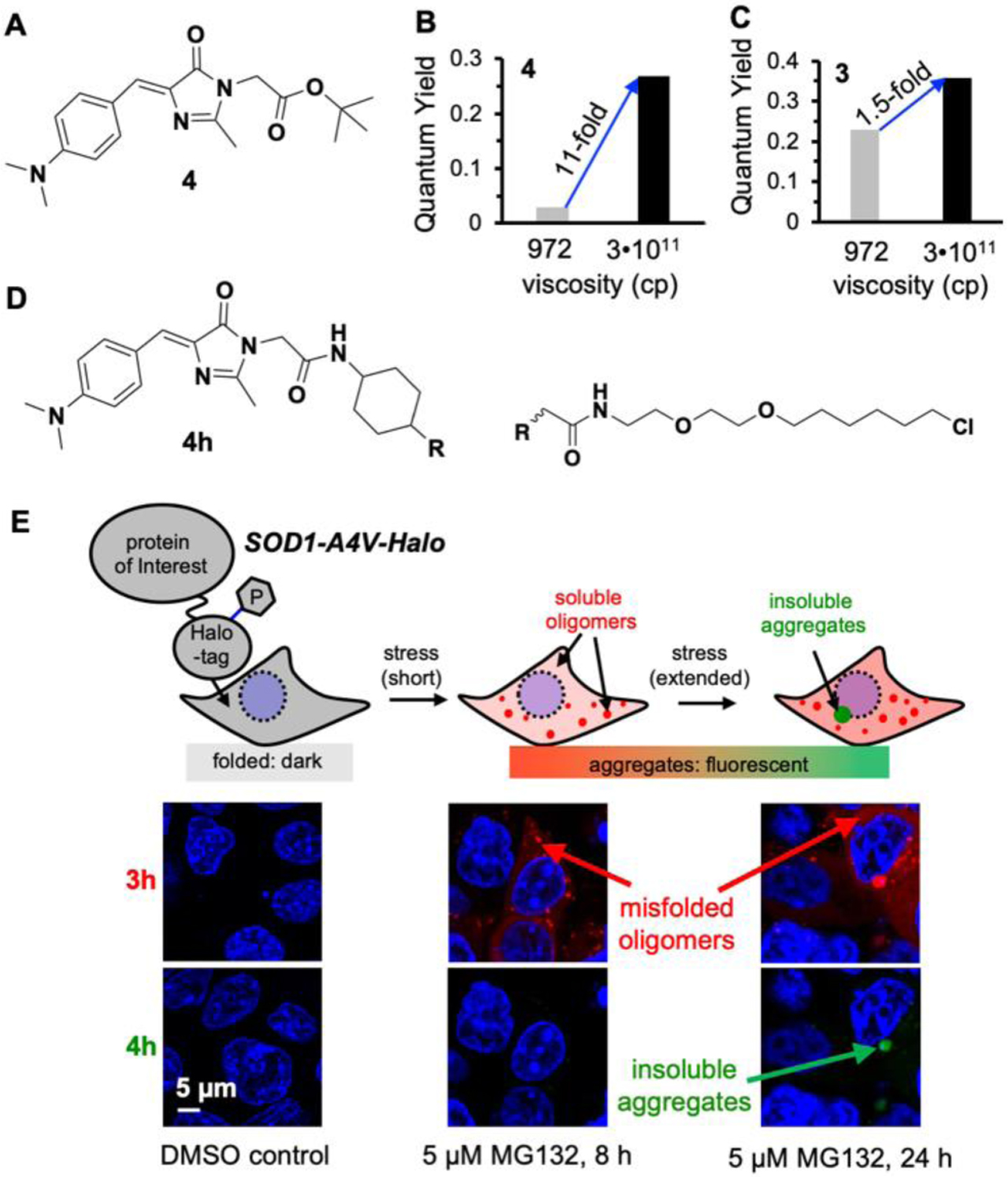
(A) Chemical structure of 4. (B–C) Fluorescence activation of 4 and 3 in glycerol at room temperature or frozen temperature. (D) Chemical structure of 4h. (E) Formation of misfolded oligomers and insoluble aggregates of SOD1-A4V in MG132 treated cells. Reproduced with permission from Ref. 3. Copyright 2020 American Chemical Society.
Based on these data, we combined the fluorescently orthogonal probe 3 (Ex/Em = 530/600 nm) and probe 4 (Ex/Em = 450/520 nm) to enable a two-color imaging platform, wherein these two probes bear distinct viscosity sensitivity to distinguish different aggregation species. The red fluorescence from probe 3 would arise from both the less compact misfolded oligomers and the highly compact insoluble aggregates. Whereas, the substantial turn-on of green fluorescence from probe 4 will distinguish insoluble aggregates from soluble oligomers. To test the two-color imaging strategy, we expressed SOD1(A4V)-Halo in live HEK 293T cells and co-labeled the protein with probes 3h and 4h (Fig 4D), derivatives of probes 3 and 4 with a Halo-Tag reactive warhead. In the absence of cellular stress, both probes emit minimal fluorescent background (Fig 4E). When cells were treated with 5 μM protease inhibitor MG132 for 8h (Fig 4E), we started to observe the red fluorescence signal from 3h in both diffusive and small granule forms. However, no significant green fluorescence signal was detected from 4h at this point. When MG132 stress persisted for 24h (Fig 4E), the red fluorescence from 3h continued to increase and started to develop perinuclear aggresome structures, wherein the green fluorescence signal from 4h was found at the core. Thus, this two-color imaging strategy has allowed us, for the first time, to directly test the multi-step protein aggregation process that was proposed a decade ago: In mammalian cells, aggregation of cytosolic proteins initiates from misfolding into oligomers as reflected by the diffused fluorescence of 3h; the oligomers continue to assemble into granular structures that only activate fluorescence from 3h but not 4h; these granular structures finally result in perinuclear aggresomes wherein the insoluble aggregates deposit to enhance fluorescence of 4h.
5. Rational control of excited state rotational barrier for molecular rotor-based fluorophores
Why did probes 3 and 4 respond so differently to viscosity with such a minor structural difference? Isomerization of the HBI derivatives, along either the phenolate (P) or imidazolinone (I) bonds, is the primary nonradiative decay process to reduce HBI’s fluorescence quantum yield. In particular, the synthetic HBI undergoes rotation along the I bond at excited state (Fig 5A), thus exhibiting dark fluorescence due to nonradiative decay via TICT50. We hypothesize that the correlation between rotational inhibition and environmental viscosity is primarily due to the excited state rotation barrier between the luminescent and TICT state (Fig 5B)4. Through a computational analysis, interesting differences were revealed for the potential energy surface (PES) of both probes at their ground state (S0) and the first singlet electronic excited state (S1): 3 exhibited a higher barrier (Ea is 0.36 eV for 3 and 0.08 eV for 4), a greater angle as the transition state of rotation (45° for 3 and 30° for 4), and a greater energy gap at the TICT state (ETICT is 1.76 eV for 3 and 1.38 eV for 4). The π electron of the 2’-stilbene-dimethylaminobenzyl (S-DMB) group in 3 overlapped with the acceptor to form π conjugation that elongated to the bond of rotation. Thus, the higher π orbital composition of 3 than 4 contributes to the higher rotational barrier of 3. We then named the S-DMB group as an electron-density regulator (EDR) and synthesized a series of HBI derivatives with varying π electron density of EDR. The resulting AggFluor probes (3–20) harbor varying rotational barriers and viscosity responses. While probes with higher rotational barriers (represented by 3) should activate fluorescence in microenvironment that have lower viscosities, probes with lower rotational barriers (represented by 4) would only be fluorescent with high viscosities.
Figure 5: Chemical control of the rotational barrier of molecular rotor-based fluorophores.
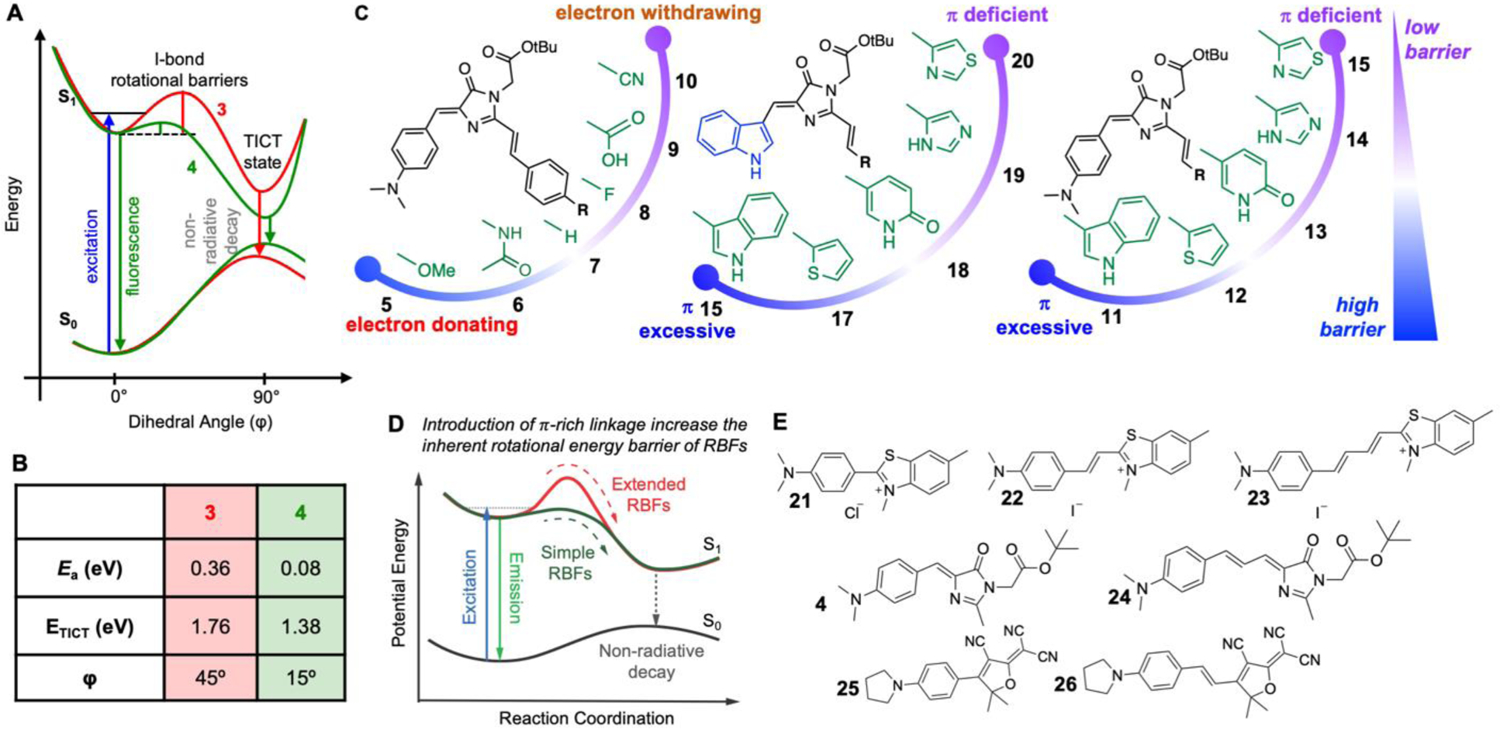
(A) Schematic diagram of potential energy surface of 3 and 4 at both ground and excite states. (B) Calculated height and position of rotational barrier for 3 and 4, as well as the energy gap between excited and ground states. (C) Chemical structures of 5–20. (D) The linkage between donor and acceptor controls the rotational barrier of rotor-based fluorophores. (E) Chemical structures of 21–26. (A–C) Reproduced with permission from Ref. 3. Copyright 2020 American Chemical Society. (D–E) Reproduced with permission from Ref. 4. Copyright 2021 John Wiley & Sons, Inc.
In addition to EDR, we recently reported another mechanism to tune the viscosity sensitivity of rotor-based fluorophores by installing π-rich alternating bridges between electron donor and electron acceptor groups. This structural modification forces excited-state fluorophore to undergo high energy barrier via the trans-cis isomerization, leading to reduced viscosity sensitivity. This strategy was applied to three reprehensive class of molecular rotor-based fluorophores, including benzothiazolium (21–23), HBI (4 and 25) and 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (DCDHF, 26–27). In all cases, we observed higher rotational barriers when the extended linkages incorporate increasing number of double bonds. Collectively, these methods significantly advance the capacity of the current imaging method, resulting an entire family of fluorophores with diverse viscosity sensitivity which could enable high temporal and spatial resolution, and multi-color imaging, to report biological processes including but not limited to protein aggregation. We envision similar strategies could be developed to modulate a wide variety of photoisomerizable fluorophores, and molecular rotors and aggregation-induced emission fluorophores, thereby expanding their chemical control and targeted applications.
6. Conclusion and perspective
Protein aggregation is a multi-step process that has been associated with a growing number of human diseases. Misfolded proteins form soluble oligomers that evolve into insoluble aggregates. While extensive efforts have focused on either folded proteins or insoluble aggregates, a less studied species between these two states is a series of intermediate conformations as soluble misfolded oligomers. In this Account, we have described how our recent work has chemically modified FP chromophores, a class of molecular rotor-based fluorophores, to enable the direct visualization of the previously invisible misfolded oligomers in live cells. Furthermore, we had discovered the principles to control the excited state rotational barriers of molecular rotor-based fluorophores, generating a series of AggFluor probes with varying sensitivities towards microviscosity. As a result, these probes allow us to simultaneously image misfolded oligomers and insoluble aggregates using orthogonal fluorescent signals, thus displaying the multi-step protein aggregation pathway in live cells.
The described fluorogenic probes provide powerful means to light up the misfolded oligomers and emit distinct fluorescent signals for the multiple conformations along the pathway of protein aggregation. However, such strategies could fall short when the microenvironment of protein aggregates needs to be quantitively analyzed. While the emission intensity of probes can be quantitively correlated with the polarity or viscosity when probes are measured in bulk solution, it is technically difficult to derive such a quantitative correlation from live cell imaging results. This is primarily due to the changes of fluorescence intensity could be triggered by local concentration changes instead of microenvironmental changes; thus, fluorogenic methods could introduce potential artifacts. By contrast, fluorescence lifetime or emission spectra is independent of fluorophore concentrations, hence providing more robust measurements of protein aggregation. In this regard, recent probes from aggregation-induced emission (AIE) family have demonstrated great potential to be used for concentration-independent imaging51. Functionalized AIE based probes that change fluorescence lifetime52 or emission spectra53 have been used to map cellular polarity and detect protein aggregation, respectively. In another work, a class of solvatochromic crystallization-induced emission fluorophores have been developed to exhibit both emission spectra shift that is dependent on polarity and intensity increase that is dependent on viscosity54. Using these probes, the authors found that polarity primarily decreases during protein misfolding and viscosity mainly increases when insoluble aggregates are formed. Furthermore, probes have been developed to covalently label protein aggregates based on their microenvironment to exhibit emission spectra changes55. More in-depth quantitative imaging analyses of protein aggregation would benefit from probes with appropriate bathochromic and/or lifetime changes in response to viscosity and/or polarity.
In addition to protein aggregation, a related process is the liquid-liquid phase separation (LLPS) of protein and RNA molecules to form liquid droplets or condensates. LLPS is a natural biological process in which the cellular components separate into a protein-rich phase and a protein-dilute phase. The phase separation produces several types of liquid droplets based on the driving forces behind the process. Forces that are known to drive phase separation are cation-π interactions, electrostatics, and hydrophobic effects. Thus, future work is needed to discern the driving force for LLPS under specific conditions, to quantify the physicochemical property of protein or RNA condensates, and to discern how these properties are connected to the biological function of membraneless organelles that are formed via LLPS.
Acknowledgments.
We thank support from the Burroughs Welcome Fund Career Award at the Scientific Interface 1013904 (X.Z.), Paul Berg Early Career Professorship (X.Z.), Lloyd and Dottie Huck Early Career Award (X.Z.), Sloan Research Fellowship FG-2018-10958 (X.Z.), PEW Biomedical Scholars Program 00033066 (X.Z.), and National Institute of Health R35 GM133484 (X.Z.).
Biographies
Songtao Ye was born in Qingdao, China in 1995. He received a B.E. in polymer science and engineering from Beijing University of Chemical Technology, an M.S. in polymer science from the University of Akron, and recently obtained his Ph.D. in chemistry from the Pennsylvania State University. His doctorate research focused on developing and employing environmental sensitive fluorophores for protein aggregation and liquid-liquid phase separation study under the supervision of Prof. Xin Zhang. In 2021, he becomes a postdoctoral fellow at Westlake University, China.
Chia-Heng Hsiung received his Ph.D. in Biochemistry, Microbiology and Molecular Biology from The Pennsylvania State University. His dissertation work utilizes the novel fluorescent probes developed in the Zhang group for live cell imaging to elucidate the physicochemical properties of biomolecular condensates. Chia-Heng received his B.S. degree from Saginaw Valley State University (Saginaw, MI) in 2011. He then worked as a research assistant at Central Michigan University, College of Medicine (Mt. Pleasant, MI) under Professor Edward McKee. In 2021, he becomes a postdoctoral fellow at Westlake University, China.
Yuqi Tang was born in Chongqing, China. She is a graduate student at Westlake University. She received her bachelor’s degree in chemistry from Beijing Normal University in 2020 and master’s degree in chemistry at Pennsylvania State University in 2021. Her current research focuses on the understanding of protein phase separation and related organizations.
Xin Zhang is the Professor of Chemistry and Cell Biology at the Westlake University, Hangzhou. Prior to joining the faculty at Westlake in 2021, Zhang was the Associate Professor of Chemistry and of Biochemistry and Molecular Biology at the Pennsylvania State University. Zhang was a postdoctoral fellow at the Scripps Research Institute and earned a doctoral degree at the California Institute of Technology.
Reference:
- 1.Liu Y; Fares M; Dunham NP; Gao Z; Miao K; Jiang X; Bollinger SS; Boal AK; Zhang X, AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew Chem Int Ed 2017, 56 (30), 8672–8676. [DOI] [PubMed] [Google Scholar]; This work describes the development of the AgHalo sensor whose misfolded oligomers can be visualized in live cells using fluorescence microscopy.
- 2.Liu Y; Wolstenholme CH; Carter GC; Liu H; Hu H; Grainger LS; Miao K; Fares M; Hoelzel CA; Yennawar HP; Ning G; Du M; Bai L; Li X; Zhang X, Modulation of Fluorescent Protein Chromophores To Detect Protein Aggregation with Turn-On Fluorescence. J Am Chem Soc 2018, 140 (24), 7381–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]; This reference describes the development of the AggTag method that can be applied to detect misfolded oligomers of any proteins in live cells. The chromophore of fluorescent protein is modified to serve as the AggTag probes that activate fluorescence with misfolded oligomers.
- 3.Wolstenholme CH; Hu H; Ye S; Funk BE; Jain D; Hsiung CH; Ning G; Liu Y; Li X; Zhang X, AggFluor: Fluorogenic Toolbox Enables Direct Visualization of the Multi-Step Protein Aggregation Process in Live Cells. J Am Chem Soc 2020, 142 (41), 17515–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]; This reference describes the development of the AggFluor probes that enable the visualization of the multi-step protein aggregation process in live cells. The concept of triode-like fluorophores is proposed to guide the chemical control of rotational barriers of molecular rotor-based fluorophores at the excited states.
- 4.Ye S; Zhang H; Fei J; Wolstenholme CH; Zhang X, A General Strategy to Control Viscosity Sensitivity of Molecular Rotor-Based Fluorophores. Angew Chem Int Ed 2021, 60 (3), 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes chemically modulating the linkage between electron donor and acceptor can serve as a general strategy to control rotational barriers of molecular rotor-based fluorophores.
- 5.Hartl FU, Molecular chaperones in cellular protein folding. Nature 1996, 381 (6583), 571–579. [DOI] [PubMed] [Google Scholar]
- 6.Bukau B; Weissman J; Horwich A, Molecular chaperones and protein quality control. Cell 2006, 125 (3), 443–451. [DOI] [PubMed] [Google Scholar]
- 7.Lindquist S, The heat-shock response. Annu Rev Biochem 1986, 55, 1151–1191. [DOI] [PubMed] [Google Scholar]
- 8.Lindquist S; Craig EA, The heat-shock proteins. Annu Rev Genet 1988, 22, 631–677. [DOI] [PubMed] [Google Scholar]
- 9.Morimoto RI, Transcription factors: positive and negative regulators of cell growth and disease. Curr Opin Cell Biol 1992, 4 (3), 480–487. [DOI] [PubMed] [Google Scholar]
- 10.Morimoto RI, Cells in stress: transcriptional activation of heat shock genes. Science 1993, 259 (5100), 1409–1410. [DOI] [PubMed] [Google Scholar]
- 11.Miyazaki Y; Chen LC; Chu BW; Swigut T; Wandless TJ, Distinct transcriptional responses elicited by unfolded nuclear or cytoplasmic protein in mammalian cells. Elife 2015, 4, e07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace EW; Kear-Scott JL; Pilipenko EV; Schwartz MH; Laskowski PR; Rojek AE; Katanski CD; Riback JA; Dion MF; Franks AM; Airoldi EM; Pan T; Budnik BA; Drummond DA, Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress. Cell 2015, 162 (6), 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balch WE; Morimoto RI; Dillin A; Kelly JW, Adapting proteostasis for disease intervention. Science 2008, 319 (5865), 916–919. [DOI] [PubMed] [Google Scholar]
- 14.Eisele YS; Monteiro C; Fearns C; Encalada SE; Wiseman RL; Powers ET; Kelly JW, Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov 2015, 14 (11), 759–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haass C; Selkoe DJ, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007, 8 (2), 101–112. [DOI] [PubMed] [Google Scholar]
- 16.Gupta R; Kasturi P; Bracher A; Loew C; Zheng M; Villella A; Garza D; Hartl FU; Raychaudhuri S, Firefly luciferase mutants as sensors of proteome stress. Nat Methods 2011, 8 (10), 879–884. [DOI] [PubMed] [Google Scholar]
- 17.Winkler J; Seybert A; Konig L; Pruggnaller S; Haselmann U; Sourjik V; Weiss M; Frangakis AS; Mogk A; Bukau B, Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J 2010, 29 (5), 910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh TY; Nillegoda NB; Tyedmers J; Bukau B; Mogk A; Kramer G, Monitoring Protein Misfolding by Site-Specific Labeling of Proteins In Vivo. Plos One 2014, 9 (6), e99395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ignatova Z; Gierasch LM, Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (2), 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramdzan YM; Polling S; Chia CPZ; Ng IHW; Ormsby AR; Croft NP; Purcell AW; Bogoyevitch MA; Ng DCH; Gleeson PA; Hatters DM, Tracking protein aggregation and mislocalization in cells with flow cytometry. Nature Methods 2012, 9 (5), 467–470. [DOI] [PubMed] [Google Scholar]
- 21.Nath S; Meuvis J; Hendrix J; Carl SA; Engelborghs Y, Early aggregation steps in alpha-synuclein as measured by FCS and FRET: evidence for a contagious conformational change. Biophys J 2010, 98 (7), 1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood RJ; Ormsby AR; Radwan M; Cox D; Sharma A; Vopel T; Ebbinghaus S; Oliveberg M; Reid GE; Dickson A; Hatters DM, A biosensor-based framework to measure latent proteostasis capacity. Nat Commun 2018, 9 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura A; Nagata K; Kinjo M, Conformational Analysis of Misfolded Protein Aggregation by FRET and Live-Cell Imaging Techniques. International Journal of Molecular Sciences 2015, 16 (3), 6076–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y; Zhang X; Chen W; Tan YL; Kelly JW, Fluorescence Turn-On Folding Sensor To Monitor Proteome Stress in Live Cells. J Am Chem Soc 2015, 137 (35), 11303–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marisa Pereira DT, Domingues Ana S., Varanda Ana S., Paulo Cristiana, Santos Manuel A. S., Soares Ana R., A fluorescence-based sensor assay that monitors general protein aggregation in human cells. Biotechnology Journal 2018, 13 (4), 1700676. [DOI] [PubMed] [Google Scholar]
- 26.Waldo GS; Standish BM; Berendzen J; Terwilliger TC, Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol 1999, 17 (7), 691–695. [DOI] [PubMed] [Google Scholar]
- 27.Wigley WC; Stidham RD; Smith NM; Hunt JF; Thomas PJ, Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat Biotechnol 2001, 19 (2), 131–136. [DOI] [PubMed] [Google Scholar]
- 28.Boeynaems S; Alberti S; Fawzi NL; Mittag T; Polymenidou M; Rousseau F; Schymkowitz J; Shorter J; Wolozin B; Van Den Bosch L; Tompa P; Fuxreiter M, Protein Phase Separation: A New Phase in Cell Biology. Trends in cell biology 2018, 28 (6), 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyman AA; Weber CA; Julicher F, Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 2014, 30, 39–58. [DOI] [PubMed] [Google Scholar]
- 30.Alberti S; Hyman AA, Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38 (10), 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Y; Protter DS; Rosen MK; Parker R, Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Molecular cell 2015, 60 (2), 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel A; Lee HO; Jawerth L; Maharana S; Jahnel M; Hein MY; Stoynov S; Mahamid J; Saha S; Franzmann TM; Pozniakovski A; Poser I; Maghelli N; Royer LA; Weigert M; Myers EW; Grill S; Drechsel D; Hyman AA; Alberti S, A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162 (5), 1066–1077. [DOI] [PubMed] [Google Scholar]
- 33.Mateju D; Franzmann TM; Patel A; Kopach A; Boczek EE; Maharana S; Lee HO; Carra S; Hyman AA; Alberti S, An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J 2017, 36 (12), 1669–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J; Choi JM; Holehouse AS; Lee HO; Zhang X; Jahnel M; Maharana S; Lemaitre R; Pozniakovsky A; Drechsel D; Poser I; Pappu RV; Alberti S; Hyman AA, A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174 (3), 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M; Simpson D; Mendez J; Zimmerman K; Otto P; Vidugiris G; Zhu J; Darzins A; Klaubert DH; Bulleit RF; Wood KV, HaloTag: a novel protein labeling technology for cell imaging and protein analysis. Acs Chemical Biology 2008, 3 (6), 373–382. [DOI] [PubMed] [Google Scholar]
- 36.Reichardt C, Solvatochromic Dyes as Solvent Polarity Indicators. Chemical Reviews 1994, 94 (8), 2319–2358. [Google Scholar]
- 37.Loving GS; Sainlos M; Imperiali B, Monitoring protein interactions and dynamics with solvatochromic fluorophores. Trends Biotechnol 2010, 28 (2), 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neto BAD; Carvalho PHPR; Correa JR, Benzothiadiazole Derivatives as Fluorescence Imaging Probes: Beyond Classical Scaffolds. Accounts of Chemical Research 2015, 48 (6), 1560–1569. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y; Miao K; Dunham NP; Liu H; Fares M; Boal AK; Li X; Zhang X, The Cation-pi Interaction Enables a Halo-Tag Fluorogenic Probe for Fast No-Wash Live Cell Imaging and Gel-Free Protein Quantification. Biochemistry 2017, 56 (11), 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu J; Li T; Liu Y; Wang X; Zhang J; Wang X; Shi G; Lou J; Wang L; Wang CC; Wang L, Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J 2020, 39 (10), e103841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fares M; Li Y; Liu Y; Miao K; Gao Z; Zhai Y; Zhang X, A Molecular Rotor-Based Halo-Tag Ligand Enables a Fluorogenic Proteome Stress Sensor to Detect Protein Misfolding in Mildly Stressed Proteome. Bioconjug Chem 2018, 29 (1), 215–224. [DOI] [PubMed] [Google Scholar]
- 42.Hong Y; Lam JW; Tang BZ, Aggregation-induced emission. Chem Soc Rev 2011, 40 (11), 5361–5388. [DOI] [PubMed] [Google Scholar]
- 43.Ando R; Hama H; Yamamoto-Hino M; Mizuno H; Miyawaki A, An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci U S A 2002, 99 (20), 12651–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lashuel HA; Overk CR; Oueslati A; Masliah E, The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013, 14 (1), 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gidalevitz T; Ben-Zvi A; Ho KH; Brignull HR; Morimoto RI, Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311 (5766), 1471–1474. [DOI] [PubMed] [Google Scholar]
- 46.DiFiglia M; Sapp E; Chase KO; Davies SW; Bates GP; Vonsattel JP; Aronin N, Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277 (5334), 1990–1993. [DOI] [PubMed] [Google Scholar]
- 47.Zhong Y; Wang J; Henderson MJ; Yang P; Hagen BM; Siddique T; Vogel BE; Deng HX; Fang S, Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife 2017, 6, e23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaganovich D; Kopito R; Frydman J, Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454 (7208), 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sontag EM; Vonk WIM; Frydman J, Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol 2014, 26, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin ME; Negri F; Olivucci M, Origin, nature, and fate of the fluorescent state of the green fluorescent protein chromophore at the CASPT2//CASSCF resolution. Journal of the American Chemical Society 2004, 126 (17), 5452–5464. [DOI] [PubMed] [Google Scholar]
- 51.Zhang S; Greening DW; Hong Y, Recent advances in bioanalytical methods to measure proteome stability in cells. Analyst 2021, 146 (7), 2097–2109. [DOI] [PubMed] [Google Scholar]
- 52.Sabouri S; Liu M; Zhang S; Yao B; Soleimaninejad H; Baxter AA; Armendariz‐Vidales G; Subedi P; Duan C; Lou X, Construction of a Highly Sensitive Thiol‐Reactive AIEgen‐Peptide Conjugate for Monitoring Protein Unfolding and Aggregation in Cells. Advanced Healthcare Materials 2021, 2101300. [DOI] [PubMed] [Google Scholar]
- 53.Owyong TC; Subedi P; Deng J; Hinde E; Paxman JJ; White JM; Chen W; Heras B; Wong WWH; Hong Y, A Molecular Chameleon for Mapping Subcellular Polarity in an Unfolded Proteome Environment. Angew Chem Int Ed 2020, 59 (25), 10129–10135. [DOI] [PubMed] [Google Scholar]
- 54.Wan W; Zeng L; Jin W; Chen X; Shen D; Huang Y; Wang M; Bai Y; Lyu H; Dong X; Gao Z; Wang L; Liu X; Liu Y, A Solvatochromic Fluorescent Probe Reveals Polarity Heterogeneity upon Protein Aggregation in Cells. Angew Chem Int Ed 2021, 60 (49), 25865–25871. [DOI] [PubMed] [Google Scholar]
- 55.Wan W; Huang Y; Xia Q; Bai Y; Chen Y; Jin W; Wang M; Shen D; Lyu H; Tang Y; Dong X; Gao Z; Zhao Q; Zhang L; Liu Y, Covalent Probes for Aggregated Protein Imaging via Michael Addition. Angew Chem Int Ed 2021, 60 (20), 11335–11343. [DOI] [PubMed] [Google Scholar]