

Abstract
The contribution of the biochemical pathways nitrification, denitrification, and dissimilatory NO3− reduction to NH4+ (DNRA) to the accumulation of NO2− in freshwaters is governed by the species compositions of the bacterial populations resident in the sediments, available carbon (C) and nitrogen (N) substrates, and environmental conditions. Recent studies of major rivers in Northern Ireland have shown that high NO2− concentrations found in summer, under warm, slow-flowing conditions, arise from anaerobic NO3− reduction. Locally, agricultural pollutants entering rivers are important C and N sources, providing ideal substrates for the aquatic bacteria involved in cycling of N. In this study a range of organic C compounds commonly found in agricultural pollutants were provided as energy sources in 48-h incubation experiments to investigate if the chemical compositions of the pollutants affected which NO3− reduction pathway was followed and influenced subsequent NO2− accumulation. Carbon stored within the sediments was sufficient to support DNRA and denitrifier populations, and the resulting NO2− peak (80 μg of N liter−1 [approximate]) observed at 24 h was indicative of the simultaneous activities of both bacterial groups. The value of glycine as an energy source for denitrification or DNRA appeared to be limited, but glycine was an important source of additional N. Glucose was an efficient substrate for both the denitrification and DNRA pathways, with a NO2− peak of 160 μg of N liter−1 noted at 24 h. Addition of formate and acetate stimulated continuous NO2− production throughout the 48-h period, caused by partial inhibition of the denitrification pathway. The formate treatment resulted in a high NO2− accumulation (1,300 μg of N liter−1 [approximate]), and acetate treatment resulted in a low NO2− concentration (<100 μg of N liter−1).
Nitrogen present in freshwaters is derived from three key sources: (i) agriculture, (ii) domestic sewage and industrial effluents, and (iii) rainfall and dry deposition (11). In Northern Ireland, associated with large inputs of agriculturally derived N substrates into the aquatic environments, there have been reports of elevated nitrite (NO2−) concentrations (31) which are believed to be toxic to aquatic biota (12). Concentrations of NO2−, an intermediate in oxidative and reductive pathways mediated by bacteria (Fig. 1), regularly exceed the European Union guideline of 0.003 mg of N liter−1 for rivers supporting salmonid fish (9). The two main substrates for NO2− production are NH4+, through nitrification, and NO3−, via a number of NO3−-reductive pathways. Previous experiments in our laboratory have shown that NH4+ oxidation via nitrification, occurring only in the oxygen diffusion zone, is mainly responsible for the elevated NO2− levels observed in fast-flowing aerobic small streams (30). However, the high concentrations of NO2− found in larger rivers in summer under warm, slow-flowing conditions have recently been attributed to anaerobic NO3−-reducing processes (13). Our laboratory incubation experiments showed that maximal concentrations of NO2− were found in anaerobic sediments deeper than 6 cm and were associated with a high concentration of added metabolizable C where dissimilatory NO3− reduction to NH4+ (DNRA) was the predominant NO3−-reducing pathway.
FIG. 1.

Nitrogen transformation processes that involve NO2− production and consumption. Pathway 1, denitrification; pathway 2, DNRA; pathway 3, nitrification. (Reproduced with permission from Kelso et al. [13]).
The partitioning of NO3− between denitrification and fermentative DNRA is dependent on the activities of two distinct bacterial populations (21). The complexity of the functioning of these multispecies microbial communities is only beginning to be understood (5), but it is believed that the availability of organic matter is probably of prime importance in regulating the relative rates of DNRA and denitrification. Organic matter has the potential to mediate between DNRA and denitrification directly by providing a direct electron donor (C substrate) and indirectly by taking up oxygen, thus creating anoxic conditions (35). The ratio between available C, which acts as an electron donor, and NO3−, an electron acceptor, is important not only in influencing which NO3−-reducing pathway is followed (36) but also in determining what end products are produced (28). Denitrification is the dominant process in NO3−-rich sediments with a poor C supply; conversely, DNRA is the dominant process in environments rich in C which are preferentially colonized by fermentative bacteria (36). To ensure their survival, microbial communities must be very versatile, and this versatility is reflected in their ability to be able to metabolize a large range of C substrates which are available in the absence of oxygen (3). However, the chemical structure of the C source may have diverse effects on the biochemical reduction rates of NO3− and NO2− (18, 27).
The aim of this study was to investigate the effects of a range of C compounds commonly found in agricultural pollutants on the biochemical pathways of NO2− accumulation. Our previous anaerobic study (13) focused on NO2− accumulation in rivers where sediment, taken from a range of depths, was supplemented with C in the form of glucose under a high concentration of NO3− (13 mg of N liter−1) and negligible NH4+ concentrations. In the present study differentially 15N-labeled NH4NO3 and a range of C supplements were provided at environmental concentrations for sediment incubations in order to deduce which N pathway supported NO2− accumulation.
MATERIALS AND METHODS
Experimental design.
Approximately 20 liters of surface water was collected from the midstream section of a continuously monitored “unpolluted” station on the Upper Bann River, stored in polyethylene bottles, and returned immediately to the laboratory. The water was filtered through GF/C filters (pore size, 0.45 μm; Whatman International Ltd., Kent, United Kingdom) and refrigerated at 4°C. Sediment was collected in October 1997 from a sampling station on the Upper Bann River (Irish Grid reference, J192 429, described previously in the report of Kelso et al. [13]), which drains an agricultural catchment in the counties of Armagh and Down, Northern Ireland. Eleven sediment cores (5 cm [diameter] by 15 cm [depth]) were collected by pushing Plexiglas tubes into areas of sediment accumulation. The top end of the column was closed with a rubber stopper, and the column was then pulled out with the intact core. The stopper was removed, and a plunger was inserted into the bottom end to push the core out. The sediments from the cores were pooled, sieved (sieve pore size, <8 mm), thoroughly mixed, and left at room temperature for 48 h. One-ninth of a core (approximately one-third of the core size used in our previous study [13]) was added to approximately 180 ml of river water in 90 Kilner jars. Then one of two N treatment samples in 10-ml aliquots was added to the jars to give a N concentration of 6 mg of N liter−1, either as 15NH4NO3, where the NH4+ moiety was labeled at 40 atom% excess, or as NH415NO3, where the NO3− moiety was labeled at 40 atom% excess. In addition to the two differentially labeled substrates, five C treatment solutions (a distilled-water control, glucose, glycine, acetate, and formate) were added to obtain a final C concentration of 1.0 g of C liter−1. Acetate and formate were prepared by neutralizing the respective acids to pH 7 with KOH (22). During incubation there was approximately 5 cm of water above the sediment. Immediately after the addition of all substrates, a nylon lid with a gas-sampling septum was fitted to each jar with an O-ring to form a gastight seal. Each treatment was replicated three times, with replicates randomly distributed in incubators maintained at a temperature of 23°C. Analyses were made at time zero, with subsequent destructive sampling carried out at 6, 24, and 48 h.
Gas analyses.
At each sampling time prior to destructive sampling, two 12-ml gas samples were taken through the gas-sampling septum by using a 20-ml gastight syringe with a push-button valve into evacuated (<100 Pa) septum-capped Exetainers (Europa Scientific, Crewe, United Kingdom). The gas was analyzed to determine the 15N contents of N2O and N2 and the concentration of N2O by continuous-flow isotope-ratio mass spectrometry. Analyses were performed with a Europa Scientific model 20-20 stable-isotope analyzer interfaced to a Europa Scientific trace gas preparation system with a Gilson auto-sampler. The valve switching was automated so that the 15N contents of N2 and N2O could be determined from the same sample. The ion currents (I) at m/z 44, 45, and 46 enabled 45R(45I/44I) and 46R(46I/44I), where R is the ratio of I and the superscript number indicates the mass/charge ratio, to be calculated for N2O. The 15N content of the N2O was calculated from either the 45R, with equations 5 and 7, or the 46R, with equations 6 and 7 in the work of Stevens et al. (33). For N2, the ion currents at m/z 28, 29, and 30 were measured by isotope-ratio mass spectrometry (33). The differences between 29R(29I/28I) and 30R(30I/28I) for enriched and normal atmospheres enabled the flux of N2 to be calculated (19). The concentration of N2O was calculated as described by Stevens et al. (33) from measurements of 44I, 45I, and 46I. The flux of N2O was calculated from the change in N2O concentration with time. It was assumed that the concentration of N2O at the start of each flux measurement was 310 ppb. A further two 5-ml gas samples were transferred to helium-filled 10-ml crimp-capped septum vials to determine the concentrations of CO2 and CH4 in the headspace by using a Varian Genesis headspace auto-sampler interfaced to a Varian model 3800 chromatograph fitted with a 5-m by 2-mm Porapak gas QS column (80-100 mesh). Carbon dioxide was measured with a thermal conductivity detector, and CH4 was measured with a flame ionization detector.
Analysis of N fractions.
The pH of the liquid fraction was measured with the aid of an Orion expandable ion analyzer, model EA940. For determining the 15N contents of NH4+, NO2−, and NO3−, a sufficient amount of KCl was added to the solution to produce a 2 M KCl solution, which was subsequently filtered sequentially through GF/C and GF/F filters (Whatman International Ltd.). Concentrations of NO2− were determined spectrophotometrically (Hitachi spectrophotometer model V-2000) by the sulfanilamide-naphthylene ethylene diamine reaction (24). Concentrations of NO3− and NH4+ in the KCl solutions were determined by segmented-flow analysis (Technicon Random Access Automated Chemistry System 800+) (4). Particulate organic N (PON) was calculated by the methodology of Koike and Hattori (14) as follows: PON = (total 15N added originally) − (15N in NH4+ + NO2− + NO3− + N2O + N2). The 15N contents of NO2−, NO3−, and NH4+ were analyzed by methods based on their conversion to N2O (16, 32).
Calculation of simultaneous nitrification and NO3− reduction rates.
The equations employed were those developed by Koike and Hattori (15):
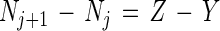 |
1 |
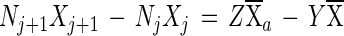 |
2 |
where j is time (6, 24, or 48 h); Nj is the NO3− plus NO2− concentration at time j (in milligrams of N per liter); Xj is the 15N content of NO3− plus NO2− in excess of the level in nature (natural abundance) at time j (atom percent excess); 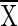 is the average 15N content of NO3− plus NO2− in excess of natural abundance between time j and time j plus 1 day (atoms percent excess);
is the average 15N content of NO3− plus NO2− in excess of natural abundance between time j and time j plus 1 day (atoms percent excess); 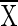 a is the average 15N content of NH4+ in excess of natural abundance between time j and time j plus 1 day (atoms percent excess); Z is the rate of nitrification per time interval; and Y is the rate of NO3− reduction per time interval. Equation 1 states that the change in NO2− and NO3− concentrations per time interval is equal to the amounts of NO2− and NO3− produced from nitrification minus the reduction in NO2− and NO3− brought about by NO3−-reducing processes in that same time interval, i.e., the nitrification rate minus the NO3− reduction rate per time period. Equation 2 states that the change in the 15N content of NO2− and NO3− per time interval is equal to the average 15N content of NH4+ nitrified minus the average of the combined 15N contents of NO2− and NO3− reduced, all within that time period. Natural abundance is assumed to be 0.37%, and the rates of nitrification and NO3− reduction are assumed to be constant during each time interval.
a is the average 15N content of NH4+ in excess of natural abundance between time j and time j plus 1 day (atoms percent excess); Z is the rate of nitrification per time interval; and Y is the rate of NO3− reduction per time interval. Equation 1 states that the change in NO2− and NO3− concentrations per time interval is equal to the amounts of NO2− and NO3− produced from nitrification minus the reduction in NO2− and NO3− brought about by NO3−-reducing processes in that same time interval, i.e., the nitrification rate minus the NO3− reduction rate per time period. Equation 2 states that the change in the 15N content of NO2− and NO3− per time interval is equal to the average 15N content of NH4+ nitrified minus the average of the combined 15N contents of NO2− and NO3− reduced, all within that time period. Natural abundance is assumed to be 0.37%, and the rates of nitrification and NO3− reduction are assumed to be constant during each time interval.
Statistical analyses.
To determine the significance of the effects of the different C treatments, the logarithms of concentration and enrichment data from the sediment studies were subjected to analysis of variance with Genstat software (10).
RESULTS
To deduce and quantify the mechanisms by which NO2− is produced, we employed the paired-incubation technique where the NH4+ and NO3− pools are differentially labeled with 15N and used the results in 15N pool dilution calculations (15). The rationale for the paired-incubation technique is that if NO3−-reductive pathways, e.g., denitrification and DNRA, are solely responsible for NO2− accumulation (i.e., all the accumulated NO2− came from the NO3− pool), then the 15N enrichment of the NO2− pool derived from NH415NO3 (Table 1) would be expected to match that of the NO3− pool once isotopic equilibrium had been established. In contrast, if nitrification was the only source of NO2− during 15NH4NO3 supplementation (Table 2) then the levels of enrichment of the NO2− and NH4+ pools would be similar.
TABLE 1.
Atom percent excess of N measured in nitrogen fractions in sediment enriched with NH415NO3a
| Carbon treatment | Nitrogen fraction | Mean atom% excess (SEM) (n = 3) at:
|
||
|---|---|---|---|---|
| 6 h | 24 h | 48 h | ||
| Control | NO3− | 37.12 (0.12) | 36.92 (0.09) | 36.16 (0.20) |
| NH4+ | 0.43 (0.26) | 0.29 (0.02) | 0.33 (0.08) | |
| NO2− | 28.68 (0.98) | 31.47 (1.36) | 28.09 (2.74) | |
| N2O | 37.85 (0.31) | 36.92 (0.10) | 36.04 (0.38) | |
| N2 | 35.97 (9.02) | 37.30 (0.33) | 36.97 (0.33) | |
| Glycine | NO3− | 37.25 (0.02) | 37.16 (0.03) | 36.50 (0.04) |
| NH4+ | 0.22 (0.03) | 0.20 (0.08) | 0.32 (0.03) | |
| NO2− | 25.53 (0.88) | 23.54 (0.57) | 26.37 (3.16) | |
| N2O | 37.54 (0.17) | 37.09 (0.09) | 36.57 (0.16) | |
| N2 | 43.63 (5.51) | 36.63 (0.00) | 36.63 (0.00) | |
| Acetate | NO3− | 37.39 (0.09) | 37.48 (0.04) | 37.17 (0.11) |
| NH4+ | 0.07 (0.01) | 0.19 (0.03) | 0.12 (0.01) | |
| NO2− | 18.77 (1.63) | 24.95 (0.32) | 32.92 (0.29) | |
| N2O | 34.36 (0.54) | 36.55 (0.08) | 36.16 (0.08) | |
| N2 | 0 | 0 | 0 | |
| Formate | NO3− | 37.48 (0.04) | 37.75 (0.06) | 37.08 (0.06) |
| NH4+ | 0.07 (0.02) | 0.13 (0.01) | 0.10 (0.01) | |
| NO2− | 25.45 (3.16) | 31.58 (2.05) | 36.37 (0.10) | |
| N2O | 35.92 (0.23) | 36.75 (0.11) | 36.80 (0.03) | |
| N2 | 0 | 0 | 0 | |
| Glucose | NO3− | 37.30 (0.06) | 37.01 (0.04) | 32.31 (1.55) |
| NH4+ | 0.16 (0.01) | 0.67 (0.07) | 2.03 (0.08) | |
| NO2− | 36.02 (1.04) | 34.48 (0.32) | 36.16 (0.21) | |
| N2O | 37.43 (0.48) | 37.14 (0.09) | 36.81 (0.23) | |
| N2 | 30.30 (1.86) | 37.63 (0.00) | 37.63 (0.00) | |
15N was measured in nitrogen fractions during incubation of sediment enriched with NH415NO3 and supplemented with different sources of carbon at 1.0 g of C liter−1.
TABLE 2.
Atom percent excess of N measured in nitrogen fractions in sediment enriched with 15NH4NO3a
| Carbon treatment | Nitrogen fraction | Mean atom% excess (SEM) (n = 3) at:
|
||
|---|---|---|---|---|
| 6 h | 24 h | 48 h | ||
| Control | NO3− | 0.11 (0.04) | 0.51 (0.16) | 0.90 (0.26) |
| NH4+ | 31.38 (0.30) | 29.52 (0.30) | 26.51 (0.35) | |
| NO2− | 2.24 (0.63) | 3.73 (0.50) | 4.13 (0.79) | |
| N2O | 0.57 (0.14) | 0.53 (0.05) | 0.57 (0.07) | |
| Glycine | NO3− | 0.05 (0.01) | 0.62 (0.20) | 0.21 (0.01) |
| NH4+ | 23.25 (0.77) | 19.13 (1.52) | 14.32 (0.23) | |
| NO2− | 0.35 (0.02) | 0.51 (0.05) | 0.25 (0.04) | |
| N2O | 0.18 (0.06) | 0.30 (0.02) | 0.26 (0.02) | |
| Acetate | NO3− | 0.07 (0.02) | 0.05 (0.01) | 0.08 (0.01) |
| NH4+ | 28.92 (0.14) | 28.18 (0.16) | 27.45 (0.21) | |
| NO2− | 0.00 (0.00) | 0.03 (0.01) | 0.01 (0.01) | |
| N2O | 0.03 (0.02) | 0.07 (0.01) | 0.06 (0.01) | |
| Formate | NO3− | 0.20 (0.09) | 0.02 (0.00) | 0.05 (0.01) |
| NH4+ | 28.87 (0.09) | 28.20 (0.34) | 27.66 (0.27) | |
| NO2− | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | |
| N2O | 0.02 (0.03) | 0.02 (0.00) | 0.00 (0.00) | |
| Glucose | NO3− | 0.18 (0.11) | 0.32 (0.17) | 1.12 (0.36) |
| NH4+ | 31.39 (0.35) | 28.33 (0.72) | 23.34 (2.45) | |
| NO2− | 1.03 (0.05) | 1.24 (0.21) | 0.59 (0.22) | |
| N2O | 0.41 (0.03) | 0.34 (0.02) | 0.30 (0.01) | |
15N was measured in nitrogen fractions during incubation of sediment enriched with 15NH4NO3 and supplemented with different sources of carbon at 1.0 g of C liter−1. NB, it is not possible to measure 15N enrichment of N2 under 15NH4NO3 conditions.
Control treatment.
Because organic C is normally present in freshwater sediment, additional organic C supplementation is not essential as an energy source to stimulate NO3−-reductive pathways (34). This was observed in the control treatment, where although no additional organic C was provided, 44.5% of the initial 15NO3− label was consumed during a 48-h period (Fig. 2) at an average rate of 1.1 mg of N liter−1 day−1 (Table 3). This consumption was reciprocated by an increase in NO2− concentrations, which showed a peak at 24 h with a concentration of 82 μg of N liter−1 (Fig. 3). The average 15N enrichment of the NO2− produced under NH415NO3-enriched conditions (29.4 atom% excess) was closer to that of NO3− (36.7 atom% excess) than to that of NH4+ (0.35 atom% excess) (Table 1), suggesting that the NO2− was predominantly of NO3− origin. Although the average rate of NO3− production via nitrification was substantially lower than the rate of NO3− utilization (Table 3), the 4 atom% excess enrichment of the NO2− pool detected under 15NH4NO3-supplemented conditions (Table 2) occurring in conjunction with dilution of the NO2− pool arising from NH415NO3 incubations (Table 1) is evidence that nitrification of NH4+ does contribute to the NO2− pool. Nitrous oxide can be produced by both NO3− reduction and NH4+ oxidation (23, 25); however, the absence of a statistical difference between the 15N atom percent excesses of NO3− and N2O produced from NH415NO3 (Table 1) indicates that only NO3−-reductive pathways were responsible for the 146 μg of N2O-N produced. Nitrogen gas, the terminal product of denitrification, was produced in concentrations similar to those of N2O, acting as a sink for 10% of the original 15NO3− label (Fig. 2). There is evidence for the DNRA pathway via NH4+ production, since 1% of NO3− was detected in NH4+ fractions (Fig. 2). Further, CH4 concentrations, a possible indicator of fermentative activity, increased by 40% from a base concentration of 3 ppm (base concentrations were similar in all treatments), while CO2 concentrations, indicative of fermentative and mineralization activity, doubled from a typical initial concentration of 2,000 ppm. It was not possible to directly determine whether the fate of NH4+ originating from the DRNA pathway was assimilated into PON. After 48 h the unexplained fraction, which we assume to be NH4+ assimilated into PON, represented as much as 22% of the original 15NO3− label (Fig. 2).
FIG. 2.

Fate of NO3− label during anaerobic incubation with NH415NO3 under the control conditions and with four carbon treatments. The remainder of the label was still present in the NO3− pool.
TABLE 3.
Rates of nitrification and NO3− reduction in sediment enriched with differentially 15N-labeled NH4NO3a
| Carbon treatment | mg of N produced liter−1 day−1 by:
|
|
|---|---|---|
| Nitrification | NO3− reduction | |
| Control | 0.06 | 1.10 |
| Glycine | 0.04 | 1.64 |
| Acetate | 0.01 | 0.30 |
| Formate | 0.00 | 0.00 |
| Glucose | 0.08 | 2.57 |
Rates of nitrification and NO3− reduction (averaged over 48 h) were measured during incubation with sediment enriched with differentially 15N-labeled NH4NO3 and supplemented with different carbon substrates at 1.0 g of C liter−1.
FIG. 3.

Concentrations of NO2− produced under the control conditions and with four carbon treatments. Error bars indicate the standard errors of the means (n = 6).
Glycine treatment.
The value of glycine as an additional energy source appeared to be limited, as the rate of NO3− reduction was similar to that of the control (Table 3). However, glycine was an important source of additional N via deamination and provided an NH4+ influx of 60 mg of N liter−1. A maximal NO2− accumulation of 74 μg of N liter−1 was reported at 24 h (Fig. 3). Under NH415NO3-supplemented conditions, the 15N enrichment of the NO2− pool (Table 1) indicated that there was an additional source of NO2− other than that formed from NO3− reduction. However a nitrification rate of 0.04 mg of N liter−1 day−1 (Table 3) in addition to a NO2− pool enrichment of 0.25 to 0.51 atom% excess reported after 15NH4NO3 treatments (Table 2) is inadequate to represent another large source of NO2−. The mole fraction of N2O [i.e., N2O/(N2O + N2)] remained constant at almost 50%, at concentrations resembling that of the control (Fig. 2). A substantial proportion (7.67%) of the initial 15NO3− label was present in the NH4+ pool (Fig. 2) and, together with almost a trebling in CO2 concentrations and a 20% increase in CH4, was strong evidence that DNRA was active. After 48 h, the unexplained fraction was equivalent to 22.9% of the initial 15NO3− label.
Acetate treatment.
The addition of acetate, for potential usage as an energy source, significantly retarded (P < 0.001) rates of NO3− utilization (Table 3), with 72% of the initial 15NO3− label still unprocessed after 48 h (Fig. 2). In contrast to what occurred with other treatments, NO2− accumulated at a constant rate to approximately 80 μg of N liter−1 at the end of the 48-h monitoring period (Fig. 3). Although nitrifying bacteria were inactive as evidenced by the low 15N enrichment of NO3− produced under 15NH4NO3-enriched conditions (0.08 atom% excess at most [Table 2]) and the almost negligible nitrification rates (Table 3) under NH415NO3-enriched conditions, the NO2− pool was not entirely derived from NO3, with the 15N enrichment of the NO2− 18.8 atom% excess initially increasing to 32.9 atom% excess at the termination of the investigation. The concentration of nitrous oxide produced was significantly lower than that of the control (P < 0.001), with only 56 μg of N2O-N detected in a 48-h period. None of the N2O was reduced further to N2. Only 0.35% of the initial 15NO3− label accumulated as NH4+, and 21.76% was calculated by difference to be present in the PON pool.
Formate treatment.
Apart from a significant accumulation of NO2−, concentrations of terminal products detected in formate-amended sediments were not significantly different from concentrations detected after the acetate treatment. However, NO2− accumulated in a linear fashion to a concentration of 1,343 μg of N liter−1 (Fig. 3), which contrasted with the small N2O concentrations (72 μg of N2O-N) arising from NO2− reduction and zero N2 production (Fig. 2). After 48 h, 11.8% of the initial 15NO3− label was detected in the PON pool. Although CO2 concentrations were twice the original levels, only 0.3% of the initial 15NO3− label was processed by DNRA into the NH4+ pool (Fig. 2). Nitrification rates were shown to be negligible (Table 3).
Glucose treatment.
In glucose-enriched sediments, NO3− reduction pathways were markedly stimulated to the extent that 5.3 mg of N liter−1 (90.6% of the 15NO3− label) was metabolized within 48 h at an average rate of 2.6 mg of N liter−1 day−1 (Table 3). At 24 h, NO2− concentrations showed a peak at 163 μg of N liter−1; thereafter, the NO2− concentration decreased to 53 μg of N liter−1 (Fig. 3). Throughout the investigation, the 15N enrichment of the NO2− pool under NH415NO3 conditions was approximately 36 atom% excess, which is very similar to the initial 37 atom% excess of the NO3− pool (Table 1), indicating that NO3− reduction accounted for NO2− production. The rapid NO2− depletion occurred in association with a trebling in CH4 efflux and a 10-fold increase in CO2 concentrations. Of the initial NO3− pool, 48% was transformed to N2 via complete denitrification. Fermentative DNRA activity as indicated by CO2, CH4, and NH4+ measurements was also significant, with 2.01% of the 15N label being detected in the NH4+ pool. Assimilation of NO3− into PON was 50% greater than that of the other treatments, incorporating 34.8% of the 15NO3− label. Nitrification was responsible for 3.5% of NO2− produced (Table 3). However, activity was retarded after 24 h of activity, when the enrichment of the NO2− pool derived from 15NH4NO3 declined from 1.24 to 0.59 atom% excess (Table 2), probably as a consequence of increasing anoxia.
DISCUSSION
Potential of carbon substrates to influence NO2− formation pathways.
Nitrite is a common intermediate in at least three different biochemical pathways that occur in freshwater sediments: nitrification, denitrification, and DNRA (Fig. 1). The relative contribution of these processes to the accumulation of NO2− is governed by the species compositions of the bacterial populations resident in the sediments, available C and N substrates, and the surrounding environmental conditions. In this study, NO3−-reducing processes deemed responsible for large NO2− concentrations were predominantly controlled by the C substrate present. Two dissimilar patterns of NO2− accumulation reflecting different formation pathways were observed (Fig. 3). The most common pattern, observed in the glucose, glycine, and control treatments, exhibited a NO2− peak at 24 h. The second pattern, observed with the acetate and formate treatments, exhibited continuous NO2− production that resulted in a high concentration of NO2− (1,300 μg of N liter−1 [approximate]) in the formate treatment and a low concentration of NO2− (<100 μg of N liter−1) in the acetate treatment.
Glucose, glycine, and control treatments.
Nitrite accumulation patterns detected in the glucose, glycine, and control treatments were indicative of the multistage DNRA and denitrification processes occurring simultaneously. Not all denitrifiers have the capability of completely reducing NO3− to N2, with the enzyme NO2− reductase commonly being absent (35). Rather than being limited by the genetic capability of the organism, reduction beyond the NO2− step is restricted more by the environmental conditions (29). For NO2− to accumulate in the environment, activity of the NO3− reductase enzyme, which reduces NO3− to NO2−, must function at a higher rate than the corresponding NO2− reductase. Often it is the environmental O2 concentrations that regulate the reduction of NO2− in denitrification, through stimulation of the enzyme NO2− reductase when suitable conditions are induced. This finding is in contrast to what occurs with the corresponding NO3− reductase, which is ever present and functional in natural environments (6, 8) and may elevate NO2− concentrations before sufficient synthesis of the NO2− reductase has occurred. The accumulation of NO2− is not entirely restricted to denitrification, since it is also believed to be a common trait of DNRA (7) due to either inhibitory effects of NO3− on the fermentative NO2− reductase enzyme (28) or repression of this enzyme (21).
Acetate and formate treatments.
Acetate and formate are capable of reducing NO3− only through the denitrification pathway (3). In Escherichia coli, it has been shown that formate-dependent NO2− reductase is active only when NO3− is scarce and NO2− is available (6). This can lead to substantial concentrations of NO2−, which under acidic conditions accumulates predominantly as the protonated species, nitrous acid (HNO2) (1, 2, 20). It has been suggested that toxic effects on the cell may be exerted by HNO2, which is capable of increasing the proton permeability of the cell membrane by shuttling protons between the two sides. To expedite the release of protons from a cell, a large proportion of energy (26), which rations the C essential to promote N-reducing processes, is required. Because acetate is a terminal product of C metabolism with a low redox potential (37), the low NO3− utilization rates resulting in reduced NO2− accumulations observed in the present study may have arisen from the inability of acetate to support further N transformations.
Environmental implications.
In Northern Ireland, agriculture has a major impact on the environment and has the potential to significantly elevate NO2− concentrations in freshwaters, either directly through leaching or indirectly by providing C and N substrates for sediment transformations. The application of slurry to grassland is a common agricultural practice. Acetic acid is the largest organic acid constituent of slurry (4.6 g liter−1) and has the potential to stimulate NO2− accumulation by denitrifying populations in freshwaters. On the other hand, silage effluent, the most common pollutant of rivers (17), is capable of supplying readily available C-rich substrates, e.g., glycine and glucose, which may support NO2− accumulation by both denitrifying and DNRA bacteria.
ACKNOWLEDGMENTS
We are very grateful to Jeff Cole, University of Birmingham, for helpful discussions. We also thank David Lennox and Tony Poland of the Department of Agriculture for Northern Ireland for assistance with statistical and chemical analyses, respectively.
This work was supported by the award of a studentship from the Department of Agriculture for Northern Ireland to B.H.L.K.
REFERENCES
- 1.Almeida J S, Júlio S M, Reis M A M, Carrondo M J T. Nitrite inhibition of denitrification by Pseudomonas fluorescens. Biotechnol Bioeng. 1995;46:194–201. doi: 10.1002/bit.260460303. [DOI] [PubMed] [Google Scholar]
- 2.Anthonisen A C, Loehr R C, Prakasam T B S, Srinath E G. Inhibition of nitrification by ammonia and nitrous acid. J Water Pollut Control Fed. 1976;48:835–852. [PubMed] [Google Scholar]
- 3.Beauchamp E G, Trevors J T, Paul J W. Carbon sources for bacterial denitrification. Adv Soil Sci. 1989;10:113–142. [Google Scholar]
- 4.Bran and Luebbe Analysing Technologies. Operator manual for Traacs Analyser 800 System. Brixworth, Northants, United Kingdom: Bran and Luebbe Analysing Technologies; 1989. [Google Scholar]
- 5.Coghlan A. Slime. New Sci. 1996;151:32–36. [Google Scholar]
- 6.Cole J. Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation. FEMS Microbiol Lett. 1996;136:1–11. doi: 10.1111/j.1574-6968.1996.tb08017.x. [DOI] [PubMed] [Google Scholar]
- 7.Cole J A. Assimilatory and dissimilatory reduction of nitrate to ammonia. In: Cole J A, Ferguson S J, editors. The nitrogen and sulphur cycles. Cambridge, United Kingdom: Cambridge University Press; 1988. pp. 281–329. [Google Scholar]
- 8.Dendooven L, Anderson J M. Dynamics of reduction enzymes involved in the denitrification process in pasture soil. Soil Biol Biochem. 1994;26:1501–1506. [Google Scholar]
- 9.European Economic Community. A directive of the quality of freshwater needing protection or improvement in order to support fish life. Off J Eur Comm. 1978;L222:34–54. [Google Scholar]
- 10.Genstat 5 Committee. Genstat 5 release 3 reference manual. Oxford, United Kingdom: Oxford Science Publications, Clarendon Press; 1993. [Google Scholar]
- 11.Heathwaite A L. Nitrogen cycling in surface waters and lakes. In: Burt T P, Heathwaite A L, Trudgill S T, editors. Nitrate: processes, patterns and management. Chichester, United Kingdom: John Wiley and Sons Ltd.; 1993. pp. 99–140. [Google Scholar]
- 12.Kelso, B. H. L., D. M. Glass, and R. V. Smith. Toxicity of nitrite to freshwater invertebrates. In W. S. Wilson, A. S. Ball, and R. H. Hinton (ed.), Managing risks of nitrates to humans and the environment, in press. Royal Society of Chemistry, London, United Kingdom.
- 13.Kelso B H L, Smith R V, Laughlin R J, Lennox S D. Dissimilatory nitrate reduction in anaerobic sediments leading to river nitrite accumulation. Appl Environ Microbiol. 1997;63:4679–4685. doi: 10.1128/aem.63.12.4679-4685.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koike I, Hattori A. Simultaneous determinations of nitrification and nitrate reduction in coastal sediments by a 15N dilution technique. Appl Environ Microbiol. 1978;35:853–857. doi: 10.1128/aem.35.5.853-857.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koike I, Hattori A. Denitrification and ammonia formation in anaerobic coastal sediments. Appl Environ Microbiol. 1978;35:278–282. doi: 10.1128/aem.35.2.278-282.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laughlin R J, Stevens R J, Zhuo S. Determining nitrogen-15 in ammonium by producing nitrous oxide. Soil Sci Soc Am J. 1997;61:462–465. [Google Scholar]
- 17.Lennox S D, Foy R H, Smith R V, Unsworth E F, Smyth D R. A comparison of agricultural water pollution incidents in Northern Ireland with those in England and Wales. Water Res. 1998;32:649–656. [Google Scholar]
- 18.Monteith H D, Bridle T R, Sutton P M. Industrial waste carbon sources for biological denitrification. Prog Water Technol. 1980;12:127–141. [Google Scholar]
- 19.Mulvaney R L, Boast C W. Equations for determination of N-15 labeled dinitrogen and nitrous oxide by mass-spectrometry. Soil Sci Soc Am J. 1986;50:360–363. [Google Scholar]
- 20.Parsonage D, Greenfield A J, Ferguson S J. The high affinity of Paracoccus denitrificans cells for nitrate as an electron acceptor. Analysis of possible mechanisms of nitrate and nitrite movement across the plasma membrane and the basis of inhibition by added nitrite of oxidase activity in permeabilized cells. Biochim Biophys Acta. 1985;807:81–95. [Google Scholar]
- 21.Paul J W, Beauchamp E G. Denitrification and fermentation in plant-residue-amended soil. Biol Fertil Soils. 1989;7:303–309. [Google Scholar]
- 22.Paul J W, Beauchamp E G, Trevors J T. Acetate, propionate, butyrate, glucose and sucrose as carbon sources for denitrifying bacteria in soil. Can J Microbiol. 1989;35:754–759. [Google Scholar]
- 23.Payne W J. Denitrification. New York, N.Y: John Wiley; 1981. [Google Scholar]
- 24.Rider B F, Mallon M G. Colorimetric determination of nitrites. Ind Eng Chem (Anal) 1946;18:96–99. [Google Scholar]
- 25.Ritchie G A F, Nicholas D J D. Identification of the sources of nitrous oxide produced by oxidative and reductive processes in Nitrosomonas europaea. Biochem J. 1972;126:1181–1191. doi: 10.1042/bj1261181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sijbesma W F H, Almeida J S, Reis M A M, Santos H. Uncoupling effect of nitrite during denitrification by Pseudomonas fluorescens: an in vivo 31P-NMR study. Biotechnol Bioeng. 1996;52:176–182. doi: 10.1002/(SICI)1097-0290(19961005)52:1<176::AID-BIT18>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 27.Skrinde J E, Bhagat S K. Industrial wastes as carbon sources in biological denitrification. J Water Pollut Control Fed. 1982;54:370–377. [Google Scholar]
- 28.Smith M S. Dissimilatory reduction of nitrite to ammonium and nitrous oxide by a soil Citrobacter sp. Appl Environ Microbiol. 1982;43:854–860. doi: 10.1128/aem.43.4.854-860.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith M S, Zimmerman K. Nitrous oxide production by nondenitrifying soil nitrate reducers. Soil Sci Soc Am J. 1981;45:865–871. [Google Scholar]
- 30.Smith R V, Burns L C, Doyle R M, Lennox S D, Kelso B H L, Foy R H, Stevens R J. Free ammonia inhibition of nitrification in river sediments leading to nitrite accumulation. J Environ Qual. 1997;26:1049–1055. [Google Scholar]
- 31.Smith R V, Foy R H, Lennox S D, Jordan C, Burns L C, Cooper J E, Stevens R J. Occurrence of nitrite in the Lough Neagh river system. J Environ Qual. 1995;24:952–959. [Google Scholar]
- 32.Stevens R J, Laughlin R J. Determining nitrogen-15 in nitrite or nitrate by producing nitrous oxide. Soil Sci Soc Am J. 1994;58:1108–1116. [Google Scholar]
- 33.Stevens R J, Laughlin R J, Atkins G J, Prosser S J. Automated determination of 15N-labeled dinitrogen and nitrous oxide by mass spectrometry. Soil Sci Soc Am J. 1993;57:981–988. [Google Scholar]
- 34.Terry R E, Nelson D W. Factors influencing nitrate transformations in sediments. J Environ Qual. 1975;4:549–554. [Google Scholar]
- 35.Tiedje J M. Ecology of denitrification and dissimilatory nitrate reduction to ammonia. In: Zehnder A J B, editor. Biology of anaerobic microorganisms. New York, N.Y: John Wiley and Sons; 1988. pp. 179–244. [Google Scholar]
- 36.Tiedje J M, Sexstone A J, Myrold D D, Robinson J A. Denitrification: ecological niches, competition and survival. Antonie Leeuwenhoek. 1982;48:569–583. doi: 10.1007/BF00399542. [DOI] [PubMed] [Google Scholar]
- 37.Zehnder A J B, Stumm W. Geochemistry and biogeochemistry of anaerobic habitats. In: Zehnder A J B, editor. Biology of anaerobic microorganisms. New York, N.Y: John Wiley and Sons; 1988. pp. 1–38. [Google Scholar]