

Abstract
Background:
Type 1 angiotensin (AT1) receptors are expressed on immune cells, and we previously found that bone marrow-derived type 1 angiotensin (AT1) receptors protect against Angiotensin (Ang) II-induced hypertension. CD11c is expressed on myeloid cells derived from the bone marrow, including dendritic cells (DCs) that activate T lymphocytes. Here we examined the role of AT1 receptors on CD11c+ cells in hypertension pathogenesis.
Methods:
Mice lacking the dominant murine AT1 receptor isoform, AT1a, on CD11c+ cells (DC AT1aR KO) and wild-type (WT) littermates were subjected to Ang II-induced hypertension. Blood pressures were measured by radiotelemetry.
Results:
DC AT1aR KO mice had exaggerated hypertensive responses to chronic Ang II infusion with enhanced renal accumulation of effector memory T cells and CD40+ DCs. CCL5 recruits T cells into injured tissues, and CCR7 facilitates DC and T cell interactions in the kidney lymph node to allow T cell activation. DCs from the hypertensive DC AT1aR KO kidneys expressed higher levels of CCL5 and CCR7. mRNA expressions for CCR7 and tumor necrosis factor-α were increased in CD4+ T cells from the renal lymph nodes of DC AT1aR KO mice. During the 2nd week of Ang II infusion when blood pressures between groups diverged, DC AT1aR KO mice excreted less sodium than WTs. Expressions for epithelial sodium channel subunits were increased in DC AT1aR KO kidneys.
Conclusions:
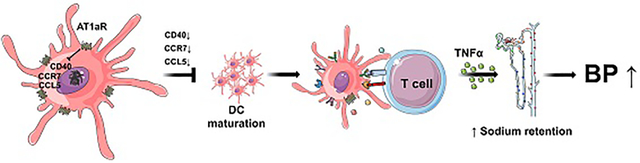
Following activation of the renin angiotensin system, AT1aR stimulation on DCs suppresses renal DC maturation and T cell activation with consequent protection from sodium retention and blood pressure elevation.
Keywords: Hypertension, AT1aR, Dendritic cells, T cells, sodium transporters
Graphical Abstract
Introduction
Hypertension is highly prevalent worldwide, impacting more than a billion people and causing catastrophic cardiovascular complications. Activation of the renin-angiotensin system (RAS) contributes to blood pressure elevation as evidenced by the capacity of type 1 angiotensin receptor (AT1R) blockers (ARBs) to reduce blood pressures in hypertensive patients1. The AT1R in the kidney plays a dominant role in blood pressure elevation2, 3. However, recent studies suggest that AT1R stimulation on hematopoietic cells may have beneficial effects in models of kidney damage4–7. In our previous studies, activating the dominant murine AT1R receptor isoform AT1A on bone marrow-derived cells afforded protection against angiotensin (Ang) II-induced hypertension8. However, deletion of AT1A on T lymphocytes9 or macrophages5 did not impact the hypertensive response. As dendritic cells (DCs) are another bone marrow-derived cell lineage critical for T cell activation, we hypothesized that stimulating AT1a receptors (AT1aRs) on DCs could limit blood pressure elevation.
As the most potent antigen presenting cells (APCs) in the body, DCs can have a profound effect on the hypertensive response10–13. In experimental models, DC-mediated T cell activation can raise blood pressure by promoting oxidative stress and sodium reabsorption in the kidney14–16. T cells mediate these effects through the elaboration of cytokines that influence renal epithelial function and even through direct contact with renal tubular cells17–22. Costimulatory molecules on DCs including CD40, CD80, and CD86 provide accessory signals during T cell activation, yielding effector memory T (Tem) cells marked by CD44hiCD62lo expression. These Tem cells are required to launch a full hypertensive response23, 24. In these same models, circulating activated CD4+CD69+ T cells are increased and contribute to blood pressure elevation25.
To investigate the role of the AT1aR on CD11c-expressing cells in hypertension, we employed a Cre-loxp strategy to generate mice lacking the AT1aR on CD11c+ cells (DC AT1aR KO). With this mouse model, we explored the role of the AT1aR on DCs in T cell activation and blood pressure regulation following RAS stimulation. We employed Ang II infusion plus unilateral nephrectomy to model hypertension in chronic kidney disease as in our earlier bone marrow chimera studies8.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Detailed Materials and Methods are presented in the Supplemental Material.
Results
Generation of mice with deletion of the AT1aR on CD11c+ myeloid cells
To examine the renal accumulation of CD11c+ cells in our hypertension model, we first bred a Cd11c-Cre mouse line with a double fluorescence reporter mouse (mT/mG), in which Cre expression is marked by GFP and the absence of Cre expression is marked by red fluorescent protein. We then infused these Cd11c+ mT/mG animals with vehicle (saline) or Ang II (500ng/kg/min) via an osmotic mini-pump (Figure 1A & Figure S1). This reporter strain revealed sparse accumulation of CD11c+ cells in the renal interstitium after 7 days of Ang II (Figure 1A & Figure S1). We then bred the Cd11c-Cre mouse line with an AT1aRflox/flox line as previously described9. To confirm CD11c+ cell-specific deletion of AT1aR in our DC AT1aR KO cohort, we labeled splenocytes with lymphocyte markers and sorted DCs (CD11c+MHCIIhi), B cells (CD19+) and T cells (CD3+), respectively. RNA from immune cells, kidney, and heart were extracted, and mRNA levels for Agtr1a were assessed via qPCR. Compared to WT littermates, DC AT1aR KO mice exhibited about 87% deletion of the AT1aR in CD11c+MHCIIhi cells but preserved AT1aR expression in other tissues examined (Figure 1B).
Figure 1. Validation of AT1aR mRNA deletion in CD11c+ myeloid cells.
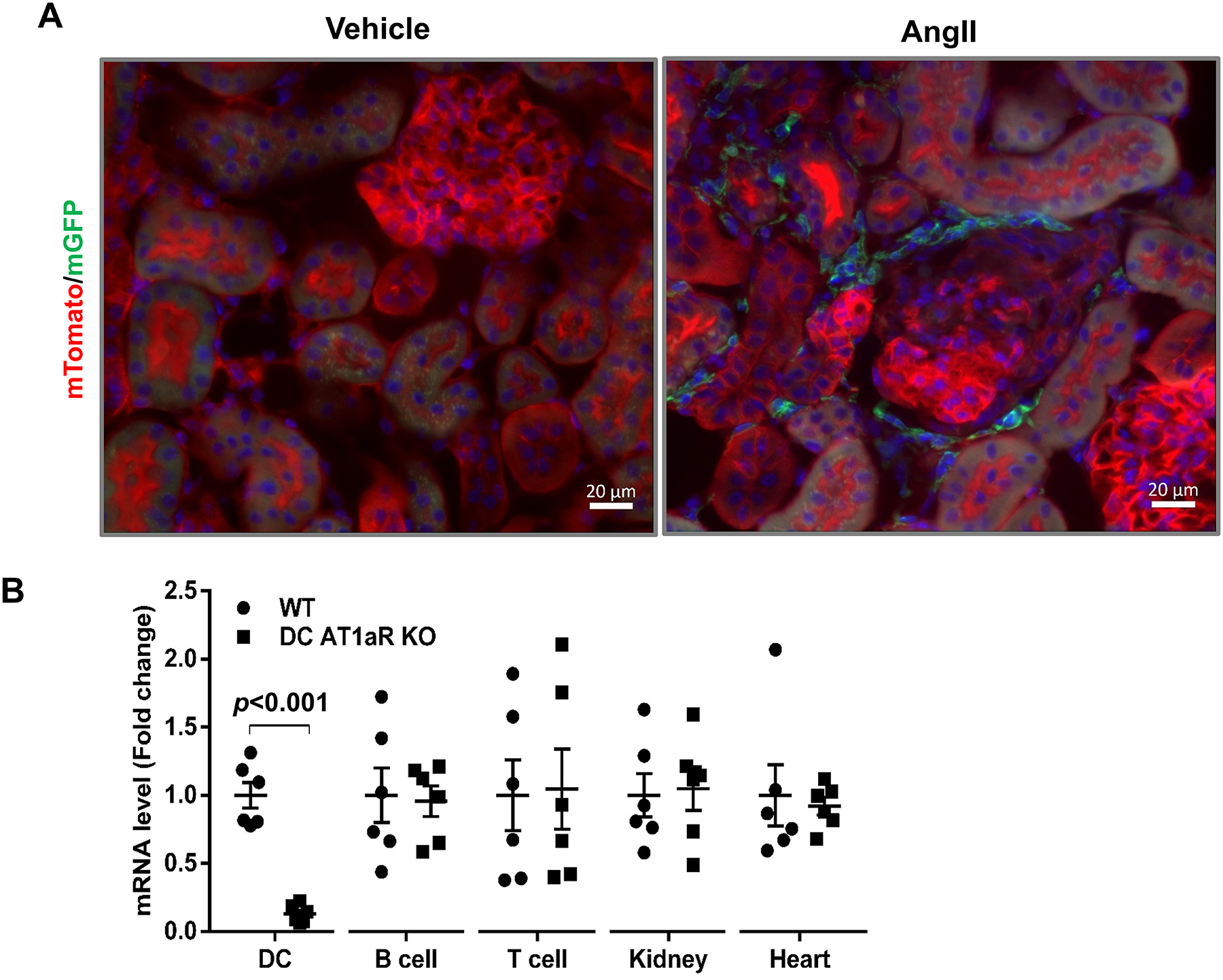
(A) Representative sections of kidneys from Cd11c-Cre+ mT/mG mice infused with Ang II or vehicle for 7 days. Cre expression marked by green fluorescence. Red: mTomato, green: mGFP. (B) mRNA levels of Agtr1a in multiple tissues of WT and DC AT1aR KO mice at baseline. “DC” labeled by CD11c+ MHCIIhi. N≥6 mice/group. Data are mean ± SE.
Deletion of AT1aR on CD11c+ cells exacerbates Ang II-dependent hypertension
DC AT1aR KO mice and WT controls underwent unilateral nephrectomy to enhance salt sensitivity, followed by implantation of pressure-sensing radiotelemetry catheters. Baseline blood pressures measured by telemetry were virtually identical in the WT and DC AT1aR KO mice (Figure 2A). To induce hypertension, mice were infused subcutaneously with Ang II (500ng/kg/min) via an osmotic mini-pump. During Ang II infusion, mean arterial blood pressures (MAPs) in the WT group rose approximately 30 mmHg and remained elevated throughout the infusion period. However, the elevation of blood pressure was exacerbated in the DC AT1aR KO cohort compared to WT controls (Figure 2A). This difference in blood pressures became evident during the 2nd and 3rd weeks of chronic Ang II infusion. Despite these blood pressure differences, after 4 weeks of Ang II-induced hypertension the groups had similar and mild levels of renal injury (Figure S2). In contrast to the Ang II model, WT and DC AT1aR KO mice had a similar chronic hypertensive response to L-NAME administration (Figure S3). These data suggest that deleting AT1a receptors on CD11c+ cells worsens the severity of hypertension independently of injury through mechanisms that specifically require RAS activation.
Figure 2. AT1aR stimulation on CD11c+ myeloid cells limits angiotensin (Ang) II-induced hypertension.
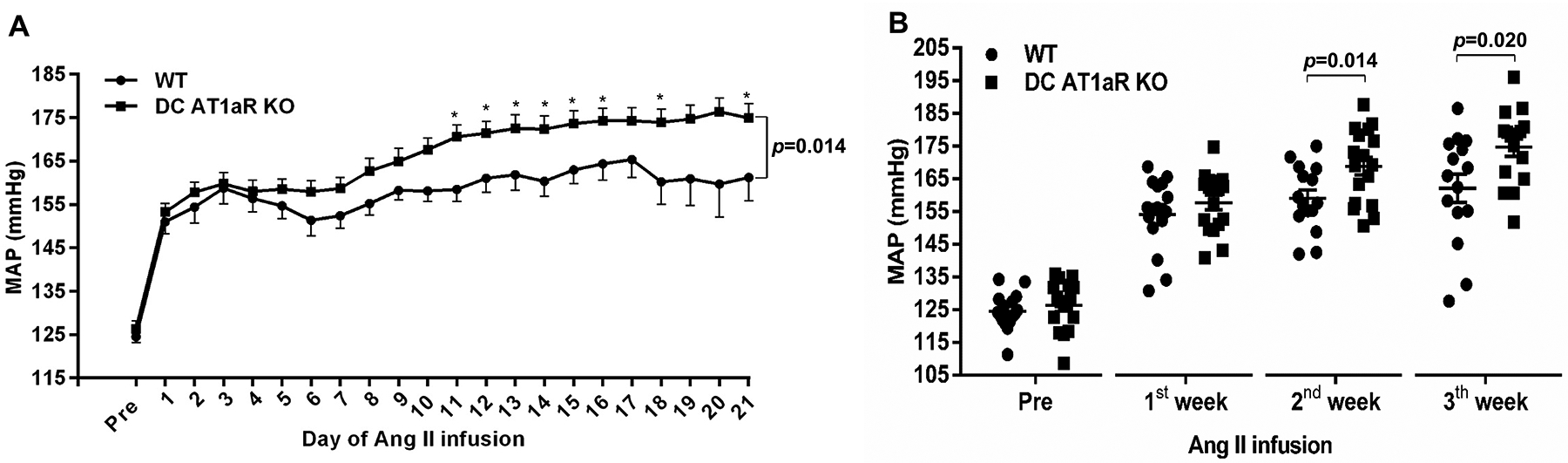
(A) Mean arterial pressures (MAP) were measured by radiotelemetry in WT (circles) and DC AT1aR KO (squares) mice at baseline (“pre”) and during chronic Ang II infusion. (B) Weekly average MAPs. N≥15 mice/group. Data are mean ± SE.
AT1aR deficiency on CD11c+ myeloid cells augments the accumulation of activated T cells in the kidney during hypertension
Ang II provokes T cell accumulation in the kidney, and cytokines produced by these cells have the capacity to influence blood pressure elevation9, 26. Tem cells can be recruited to the kidney to regulate blood pressure24, whereas activated T cells marked by cell surface expression of CD69 also contribute to RAS-mediated hypertension25. To assess the accumulation of these T cell populations in the hypertensive kidney, we performed flow cytometric analysis on single cell suspensions from the kidney after 4 weeks of Ang II. As illustrated by our gating strategy (Figure 3A), cells were stained with fluorescently labeled anti-CD45, anti-CD3, anti-CD4, Anti-CD8, Anti-CD62L, Anti-CD44, and Anti-CD69. We detected greater numbers of CD3+ T lymphocytes in the kidneys from our DC AT1aR KO mice compared to WT controls (Figure 3B). Moreover, the absolute number of CD4+ and CD8+ T cells was increased in the DC AT1aR KO kidneys (Figure 3C). To evaluate the intrarenal distribution of T cells, kidney sections of the DC AT1aR KO and WT kidneys were stained for the T cell marker CD3. In these sections, T lymphocytes were clustered around the blood vessels in the kidney (Figure 3D & Figure S4). The numbers of renal Tem cells marked by CD62LloCD44hi expression were markedly increased within the CD4+ T cell subset, but not in the CD8+ subset from the DC AT1aR KO kidneys (Figure 3E). Similarly, numbers of renal CD69+ T cells were significantly increased within the CD4+ T cell subset, but not in the CD8+ subset from the DC AT1aR KO cohort (Figure 3F). Thus, abrogating AT1aR signals in CD11c-expressing myeloid cells may accentuate blood pressure elevation by increasing the renal accumulation of activated CD4+ T cells.
Figure 3. AT1aR deficiency on CD11c+ cells enhances the accumulation of effector memory T cells in the kidney during hypertension.
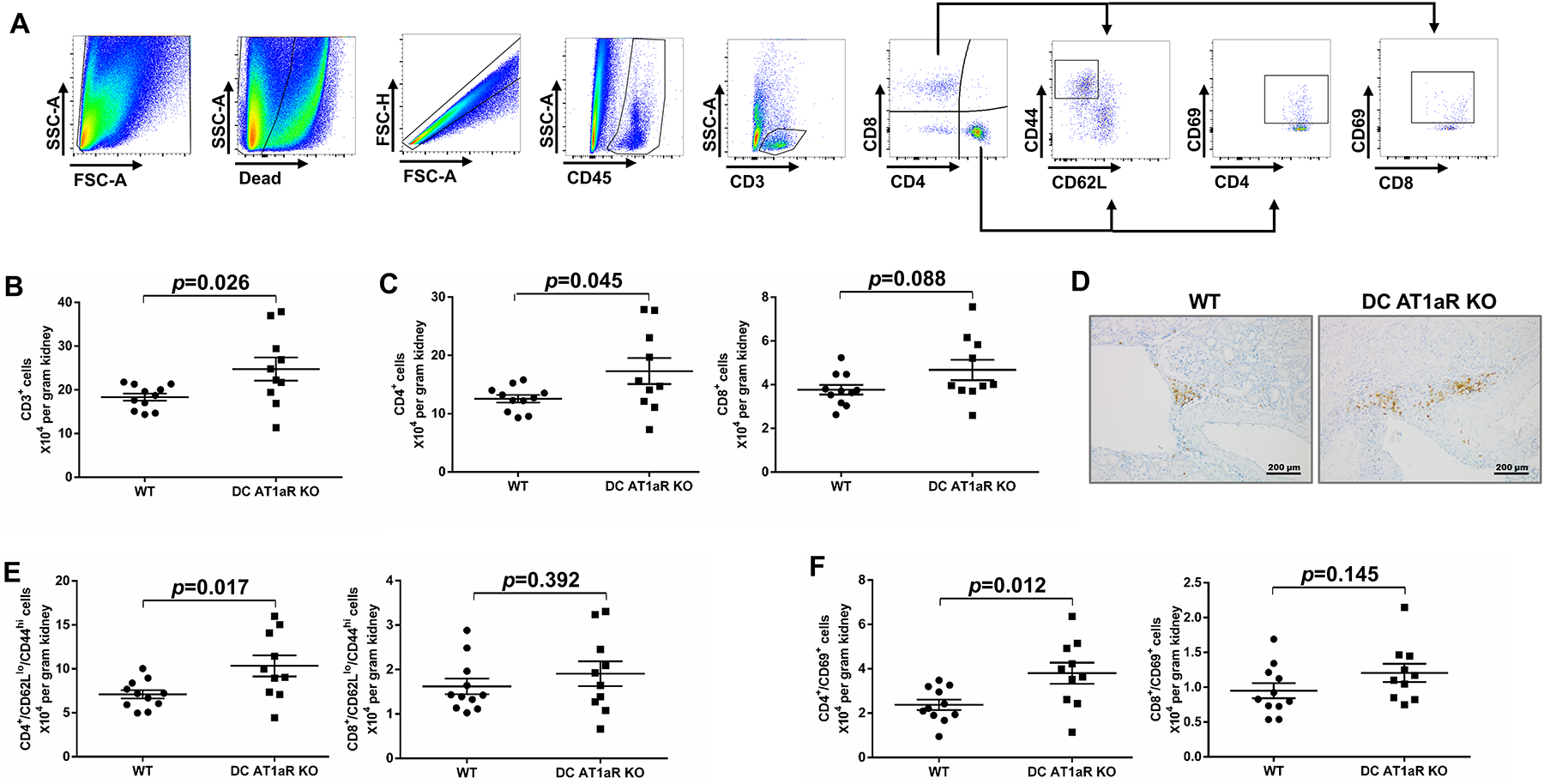
Flow cytometric analysis of single cell suspensions from the kidney harvested after 4 weeks of Ang II infusion. (A) Gating strategy for parsing T cell populations in the kidney. (B-C) Absolute numbers of (B) CD3+ T lymphocytes, (C) CD4+ T lymphocytes and CD8+ T lymphocytes. (D) Representative images of perivascuclar CD3 staining in kidney. (E) Absolute numbers of CD4+ CD44hiCD62Llo T lymphocytes and CD8+ CD44hiCD62Llo T lymphocytes. (F) Absolute numbers of CD4+ CD69+ T lymphocytes and CD8+ CD69+ T lymphocytes. N≥10 mice/group. Data are mean ± SE.
AT1aR deletion enhances renal DC activation during Ang II-dependent hypertension
CD40, CD80, and CD86 on the DCs provide accessory signals during DC-mediated activation of the T cell27. To determine if the AT1aR regulates DC activation in our hypertension model, we examined the expression of these co-stimulatory molecules on DCs isolated from the kidney after 28 days of Ang II infusion. We selected for CD45+ live hematopoietic cells that did not express markers for T cells (CD3), B cells (CD19), or NK cells (NK1.1), and then analyzed the expression of co-stimulatory molecules on the remaining cells co-expressing the DC markers CD11c and MHCII (Figure 4A). We found that the absolute number of CD11c+MHCIIhi DCs in the hypertensive kidney from DC AT1aR KO mice was numerically but not significantly higher than in WT mice (Figure 4B). The absolute numbers of CD40+ DCs were elevated in the DC AT1aR KO mice kidneys compared to the WT cohort whereas the numbers of CD80+ and CD86+ DCs were similar between two groups. (Figure 4C–E). Accordingly, AT1aR deletion on DCs upregulates their expression of the co-stimulatory molecule CD40, providing a possible mechanism through which the AT1aR on DCs limits renal T cell activation.
Figure 4. AT1aR suppresses renal dendritic cell (DC) activation during hypertension.
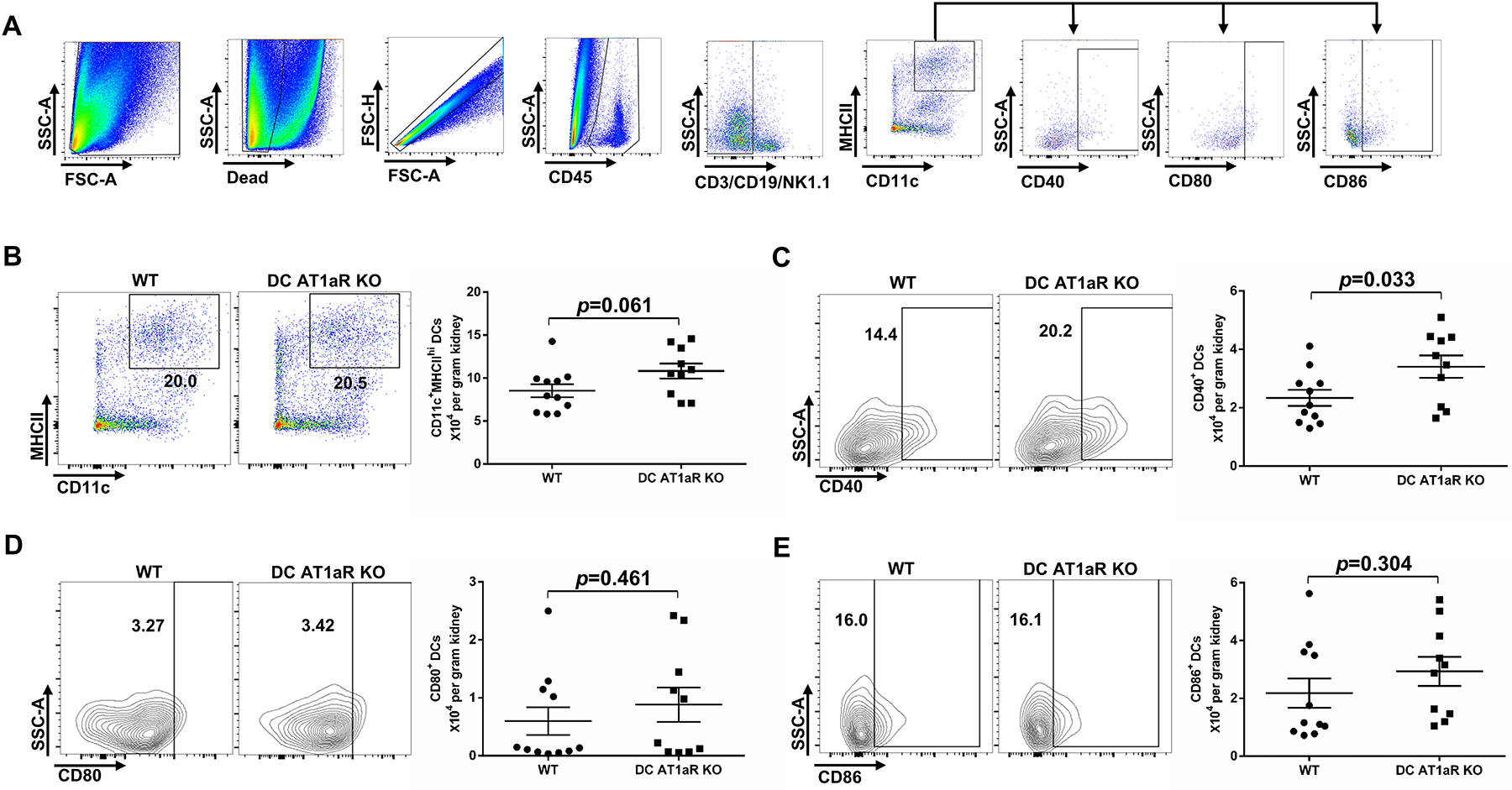
Flow cytometric assessment of co-stimulatory molecule expression on CD11c+MHCIIhi DCs isolated from the kidney after 4 weeks of Ang II infusion. (A) Gating strategy for parsing for CD40, CD80, or CD86 positivity on DCs in the kidney, based on “fluorescence minus one” strategy. (B) Representative flow plots and the absolute number of CD11c+MHCIIhi Cells from WT and DC AT1aR KO groups. (C-E) Representative flow plots and the absolute number of (C) CD40+, (D) CD80+, and (E) CD86+ DCs from WT and DC AT1aR KO mice. N≥10 mice/group. Data are mean ± SE.
Ablation of AT1aR on CD11c+ cells increases the expression of inflammatory mediators in DCs and T cells during hypertension
Expression of C-C chemokine receptor type 7 (CCR7) on DCs stimulates their maturation and migration into the kidney lymph node where they can activate T cells28–31. Chemokine ligand 5 (CCL5) recruits T cells into injured tissues, whereas expression of CCR7 on the T cell similarly allows its recruitment into the draining lymph node for interaction with DCs. Once activated, both DCs and T cells can elaborate TNF-α, a pro-hypertensive cytokine that can contribute to blood pressure elevation through diverse actions in the kidney32–35. We therefore measured the mRNA expression of these mediators in sorted DCs and T cells from Ang II-infused WT and DC AT1aR KO mice. We found that mRNA levels for CCR7 were increased in DCs from hypertensive kidneys (Figure 5A) and CD4+ T cells from renal lymph nodes (Figure 5B) in the DC AT1aR KO mice. The expression of CCL5 was also significantly higher in DCs from the DC AT1aR KOs compared to the WTs (Figure 5A), offering another potential mechanism underpinning the enhanced accumulation of activated T cells in the DC AT1aR KO hypertensive kidney. Given the importance of NF-κB activation to DC maturation and the coordinated regulation of NF-κB by AT1 receptors and the β-arrestin scaffolding signal proteins36, we measured gene expression levels for these proteins in splenic DCs, and found mRNA levels for β-arrestin 1 but not β-arrestin 2 to be blunted in the DC AT1aR KO cells (Figure S5). Finally, mRNA levels for TNF-α were significantly higher in CD4+ T cells from renal lymph nodes of the DC AT1aR KOs (Figure 5B). By contrast, we saw similar expression levels for CCR7 and TNF-α within CD8+ T cells from the WT and DC AT1aR KO renal lymph nodes (Figure 5C). These data suggest that preventing AT1aR activation in CD11c+ myeloid cells augments blood pressure elevation by permitting upregulated expression of mediators that drive renal accumulation and activation of CD4+ T cells.
Figure 5. Mice lacking AT1aR on CD11c+ cells have augmented expression of inflammatory mediators in renal mononuclear cells.
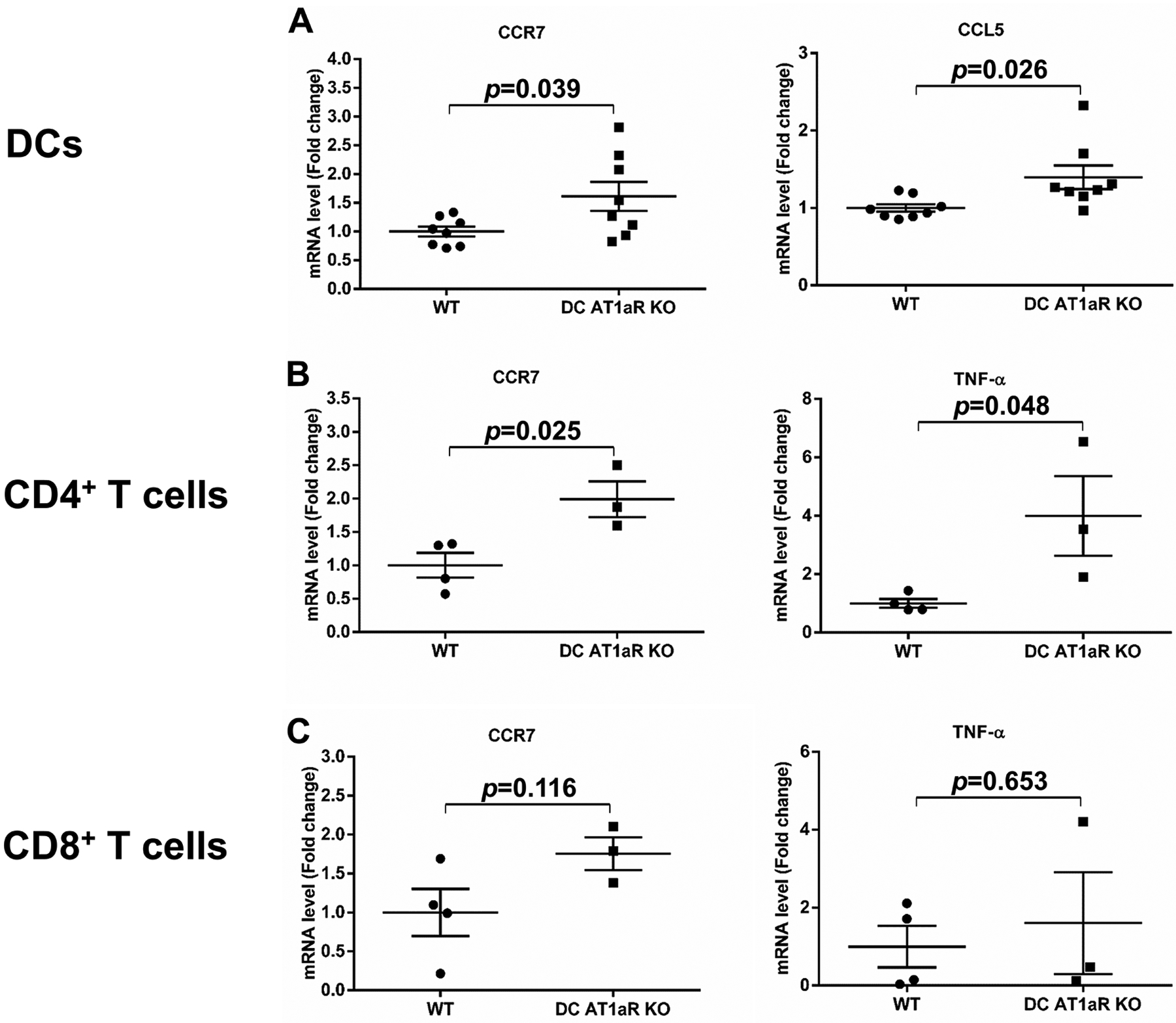
(A) Kidneys from hypertensive WT and DC AT1aR KO mice were harvested, and CD11c+MHCIIhi DCs were sorted for analysis of CCR7 and CCL5 mRNA expression. (B-C) Renal lymph nodes were harvested from WT and DC AT1aR KO mice at 4 weeks of Ang II, and CD4+ T cells and CD8+ T cells were isolated for RNA extraction. qPCR for mRNA levels of CCR7 and TNF-α in (B) CD4+ T cells and (C) CD8+ T cells. N≥3 mice/group. Data are mean ± SE.
AT1aR in CD11c+ myeloid cells facilitates diuresis and blunts renal sodium transporter expression
TNF-α can promote blood pressure elevation during RAS activation by modulating renal tubular function35, 37, 38. To assess the effects of the DC AT1aR on RAS-dependent sodium retention, uni-nephrectomized WT, and DC AT1aR KO mice were placed into metabolic cages beginning 5 days prior to the initiation of chronic Ang II infusion for quantitation of daily sodium ingestion and urinary sodium excretion. Food ingestion and body weights (Figure S6) remained similar in the two groups before and after the study. Prior to Ang II and during the first 5 days of Ang II infusion, the 2 groups had similar sodium excretion (Figure 6A). However, concomitant with the separation in blood pressure between the WT and DC AT1aR KO groups during the 2nd week of Ang II, the DC AT1aR KO cohort excreted less sodium compared the WTs (Days 6–10, Figure 6A). Consistent with this finding, mRNA levels for the epithelial sodium channel subunits, α-ENaC and β-ENaC, were significantly increased while γ-ENaC was numerically but not significantly upregulated in the DC AT1aR KO kidneys compared to WT controls (Figure 6B). By contrast, the levels of sodium-hydrogen antiporters 3 (NHE3) and sodium-potassium-chloride cotransporter 2 (NKCC2) were similar in the two groups (Figure 6B). To determine the protein level of sodium transporters in the kidney, we performed western blotting of kidney tissue and found that β-ENaC and cleaved-γ-ENaC were significantly increased in the DC AT1aR KOs, while the levels of α-ENaC, full-length γ-ENaC, NHE3, and NKCC2 were similar in the two groups (Figure 6C–D). Thus, AT1aR deletion on CD11c+ myeloid cells modulates sodium transporter expression and renal sodium retention, promoting blood pressure elevation.
Figure 6. AT1aR activation on CD11c+ cells attenuates renal sodium retention during hypertension.
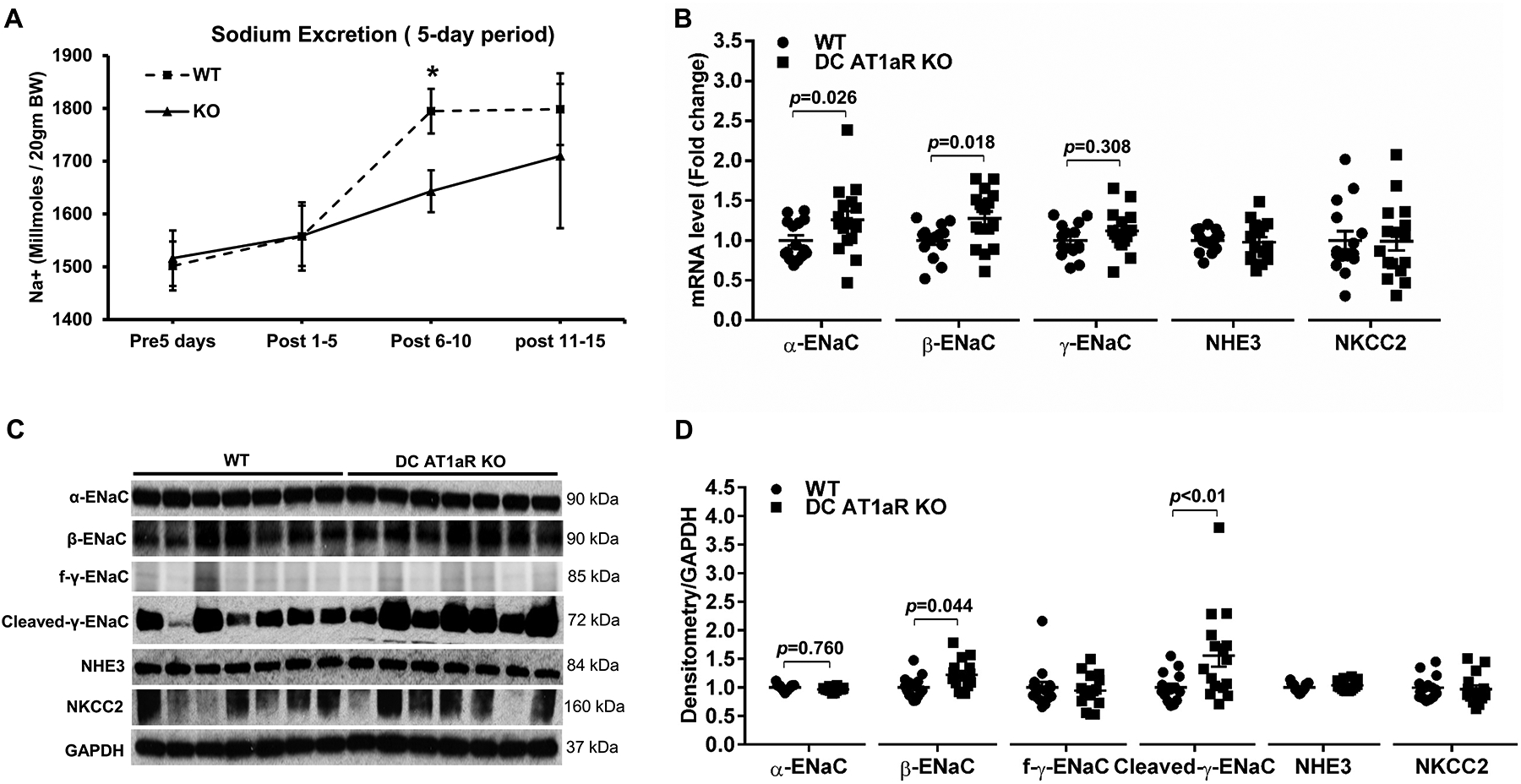
(A) Urinary sodium excretion at baseline and during Ang II infusion. N=6 mice/group. (B) mRNA levels for α-ENaC, β-ENaC, γ-ENaC, NHE3, and NKCC2 in the kidney after 4 weeks of Ang II. (C) kidney protein levels of the sodium transporters as determined by immunoblotting. (D) Densitometry of these blots. normalized to GAPDH. N≥15 mice/group. Data are mean ± SE. “KO” is DC AT1aR KO. Urinary sodium excretion is reported as Na+ (millmoles/ 20gm body weight). “f-y-ENaC” stands for full-length γ-ENaC.
Discussion
Activation of AT1 receptors in the kidney and the vasculature drives blood pressure elevation, and hypertensive renal injury that is responsive to treatment with ARBs1, 2, 39. However, recent studies from other groups and our own point to a paradoxical role for AT1 receptors in the immune system to ameliorate hypertension and its complications4, 5, 9, 40. For example, we have previously demonstrated that the AT1aR on bone marrow-derived cells protects against Ang II-induced hypertension8. To identify the population of immune cells that limit blood pressure elevation via AT1 receptor functions, we generated mouse lines with conditional deletion of AT1aR on T lymphocytes or macrophages, but did not see effects on blood pressure despite exacerbations in target organ injury5, 9. In the current study, we examined the actions of the AT1aR on CD11c+ myeloid cells in the pathogenesis of hypertension.
After validating specific deletion of the Agtr1a gene from CD11c+ cells in our mouse model, we found that baseline blood pressures in our DC AT1aR KOs were similar to WT controls, consistent with our previous studies using bone-marrow chimeras8, suggesting that the AT1aR on CD11c+ cells does not contribute to normal blood pressure homeostasis. The lack of effects of the CD11c+ cell AT1aR on blood pressure here may accrue from the relatively low frequency of CD11c+ cells in the kidney at baseline as detected with our fluorescent reporter mice. By contrast, with chronic Ang II infusion, the DC AT1aR KO animals had an augmented chronic hypertensive response, indicating that the AT1aR on CD11c+ cells limits RAS-mediated blood pressure elevation. This result corroborates our findings with AT1aR-deficient bone marrow chimeras8 and, together with the normal hypertensive response in conditional mutants lacking AT1aR’s on T cells or macrophages5, 9, suggests that AT1aR stimulation on dendritic cells (DCs) attenuates blood pressure elevation. Alternatively, a contribution of signaling via the alternate rodent AT1 receptor isoform AT1B or the AT2 receptor in the DC AT1aR KOs seems unlikely as earlier radioligand binding studies suggested that AT1a binding accounts for all Ang II binding to murine immune cells41, and we are unable to detect mRNA for the genes encoding AT1b or the AT2 receptor in splenic CD11c+ cells from our WT or DC AT1aR KO animals (data not shown). Our studies in the L-NAME hypertension model would indicate that RAS stimulation is required to exert the protective effects of the AT1aR on CD11c+ cells in limiting blood pressure elevation.
Inappropriate immune activation can trigger blood pressure elevation by acting on cardiovascular control centers, such as the heart, kidney, vasculature, adipose tissue, and nervous system42. T lymphocytes accumulate within the kidney and vascular wall during hypertension to augment the hypertensive response as established in several animal models33, 43. Consistent with the enhanced hypertension in the DC AT1aR KOs, we find an increased accumulation of both CD4+ and CD8+ T cells within the kidneys from the Ang II-infused DC AT1aR KO cohort. Interactions of T cells with DCs can promote differentiation of T cells to effector memory T (Tem) cells that express high levels of CD44 but low levels of CD62L. These Tem cells and also activated T cells as marked by expression of CD69 contribute to RAS-dependent hypertension24, 25. In the current studies, we find markedly increased numbers of CD4+ but not CD8+ Tem and CD69+ T cells in the DC AT1aR KO kidneys compared to WT controls. This correlation of renal CD4+ T cells with the severity of blood pressure may relate to our use of the salt-sensitive 129SvEv strain here as studies from others and our group using the C57BL/6 strain have implicated CD8+ rather than CD4+ T cells in hypertension pathogenesis22, 44, 45.
DCs activate T cells via the presentation of antigen in the cleft of a major histocompatibility complex. Upregulation of co-stimulatory molecule receptors, including CD80, CD86, and/or CD40 on the surface of the maturing DCs potentiates T cell activation46. In the hypertensive kidneys, we found only non-significantly increased numbers of DCs marked by CD11c and MHCII co-expression in the DC AT1aR KOs. However, upon evaluation of additional DC markers, we detected significantly increased numbers of CD40+ DCs but not CD80+ or CD86+ DCs in the DC AT1aR KO versus WT kidneys. These results suggest that the AT1aR on CD11c+ myeloid cells may constrain T cell activation by suppressing the expression of CD40 on renal DCs. Moreover, the AT1aR KO renal DCs expressed higher mRNA levels for CCR7, which should promote their interactions with T cells30, 31, and CCL5, which can promote the recruitment of T cells into injured tissues during hypertension47. In addition, we found blunted β-arrestin-1 expression in CD11c+ cells from the DC AT1aR KO mice, which can permit increased NF-κB signals36. Thus, the AT1aR on CD11c+ cells appears to regulate their expression of several factors that could impact T cell accumulation and activation during hypertension.
Following activation by DCs, T cells can modulate blood pressure through the elaboration of inflammatory cytokines, including TNF-α, to alter vascular and renal epithelial cell functions34, 35, 37, 38, 48. In our sorted CD4+ T cells, mRNA expression for TNF-α was increased in the DC AT1aR KO cohort leading us to examine sodium retention and epithelial cell transporter expression in our model. We found that the DC AT1aR KO cohort excreted less sodium during Ang II infusion as their blood pressures rose beyond those of the WTs despite ingesting similar amounts of food. Moreover, the DC AT1aR KO kidneys showed upregulated mRNA or protein expression of multiple sodium channels, including α-ENaC, β-ENaC, and cleaved-γ-ENaC, which should increase the capacity for tubular sodium reabsorption. Nevertheless, we did not detect differences between groups in NHE3 or NKCC2 mRNA expression in this study. Thus, the AT1aR on CD11c+ cells appears to impact renal sodium handling in the distal nephron. However, we cannot exclude the possibility that TNF in this system is acting to influence NKCC2 activity rather than expression in the thick ascending limb as has been reported21, 37.
In sum, we find that AT1aR activation on CD11c+ myeloid cells protects against hypertension and limits renal T cell accumulation and activation. This attenuation in T cell recruitment is associated with blunted inflammatory cytokine and ENaC expression with consequent protection from sodium retention and RAS-mediated blood pressure elevation.
Perspectives
Although the efficacy of ARBs in lowering blood pressure highlights the importance of AT1 receptor activation to the pathogenesis of hypertension1, ARB therapy does not completely prevent complications accruing from hypertension. We posit that some of this incomplete efficacy may accrue from divergent actions AT1 receptors in tissues such as the immune system that can modulate blood pressure and target organ damage. In a RAS-mediated hypertension model, we find that activating the AT1aR on CD11c-expressing myeloid cells protects against hypertension in part by limiting the accumulation of effector memory T cells in the kidney and constraining renal sodium retention. Exploring inflammatory pathways regulated by the AT1 receptor in dendritic cells may elucidate novel interventions for patients with hypertension that is resistant to current therapies.
Supplementary Material
Pathophysiological Novelty and Significance.
What is New?
The AT1aR on CD11c-expressing myeloid cells constrains the chronic hypertensive response to Ang II.
The AT1aR on CD11c+ myeloid cells limits renal dendritic cell (DC) maturation during Ang II-induced hypertension.
The AT1aR on CD11c-expressing myeloid cells restricts renal T cell activation and suppresses the expression of renal epithelial sodium channel subunits.
What Is Relevant?
CD11c is expressed on myeloid cells derived from the bone marrow, including dendritic cells (DCs), which are potent antigen-presenting cells that activate T lymphocytes. We investigate how AT1 receptors on CD11c+ cells are protective in hypertension pathogenesis, identifying novel actions of the myeloid AT1R during renin-angiotensin system (RAS) activation.
Clinical/Pathophysiological Implications
Following RAS activation, AT1aR stimulation on DCs suppresses renal DC maturation and T cell activation, constraining expression of inflammatory mediators, sodium retention, and blood pressure elevation.
Sources of Funding
This work was supported by NIH grants DK118019, HL128355; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893; VA Clinician Scientist Investigator Award; American Heart Association Grants 19POST34380480 and 18TPA34170047.
Nonstandard Abbreviations and Acronyms
- AT1R
type 1 angiotensin receptor
- Ang
angiotensin
- ARBs
angiotensin receptor blockers
- APCs
antigen presenting cells
- CCL5
C-C Motif chemokine ligand 5
- CCR7
C-C Motif chemokine receptor 7
- CD
cluster of differentiation
- DC
dendritic cell
- ENaC
epithelial sodium channel
- KO
knock out
- RAS
renin-angiotensin system
- L-NAME
L-NG-Nitro arginine methyl ester
- NHE3
sodium-hydrogen antiporter 3
- NKCC2
sodium-potassium-chloride cotransporter 2
- NK
nature killer
- Na+
sodium
- mT/mG
cell membrane-localized tdTomato / cell membrane-localized EGFP
- MHC II
major histocompatibility complex II
- SE
standard error
- TNF
tumor necrosis factor
Footnotes
Disclosures
None
References
- 1.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, Group LS. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (life): A randomised trial against atenolol. Lancet. 2002;359:995–1003 [DOI] [PubMed] [Google Scholar]
- 2.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim H-S, Smithies O, Le TH, Coffman TM. Angiotensin ii causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crowley SD, Zhang J, Herrera M, Griffiths R, Ruiz P, Coffman TM. Role of at₁ receptor-mediated salt retention in angiotensin ii-dependent hypertension. Am J Physiol Renal Physiol. 2011;301:F1124–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishida M, Fujinaka H, Matsusaka T, Price J, Kon V, Fogo AB, Davidson JM, Linton MF, Fazio S, Homma T, Yoshida H, Ichikawa I. Absence of angiotensin ii type 1 receptor in bone marrow-derived cells is detrimental in the evolution of renal fibrosis. J Clin Invest. 2002;110:1859–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, Lefler WS, Chen D, Crowley SD. Type 1 angiotensin receptors on macrophages ameliorate il-1 receptor-mediated kidney fibrosis. J Clin Invest. 2014;124:2198–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Rudemiller NP, Patel MB, Wei Q, Karlovich NS, Jeffs AD, Wu M, Sparks MA, Privratsky JR, Herrera M, Gurley SB, Nedospasov SA, Crowley SD. Competing actions of type 1 angiotensin ii receptors expressed on t lymphocytes and kidney epithelium during cisplatin-induced aki. J Am Soc Nephrol. 2016;27:2257–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Y, Rudemiller NP, Zhang J, Jeffs AD, Griffiths R, Lu X, Ren J, Privratsky J, Crowley SD. Stimulating type 1 angiotensin receptors on t lymphocytes attenuates renal fibrosis. Am J Pathol. 2019;189:981–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley SD, Song Y-S, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux P-L, Ruiz P. A role for angiotensin ii type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin ii-dependent hypertension. Hypertension. 2010;55:99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on t lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110:1604–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen S-c, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R, Cifuentes-Araneda F, Gonzalez AA, Amador CA, Pacheco R, Michea L. Myeloid cd11c+ antigen-presenting cells ablation prevents hypertension in response to angiotensin ii plus high-salt diet. Hypertension. 2018;71:709–718 [DOI] [PubMed] [Google Scholar]
- 13.Lu X, Rudemiller NP, Privratsky JR, Ren J, Wen Y, Griffiths R, Crowley SD. Classical dendritic cells mediate hypertension by promoting renal oxidative stress and fluid retention. Hypertension. 2020;75:131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P. Lymphocyte responses exacerbate angiotensin ii-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. 2012;59:673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299:R590–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17a regulates renal sodium transporters and renal injury in angiotensin ii-induced hypertension. Hypertension. 2016;68:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2016;311:F555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the nkcc2 co-transporter in the nephron. Cell Metab. 2016;23:360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, Hoover RS, He B, Mu S. Cd8(+) t cells stimulate na-cl co-transporter ncc in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun. 2017;8:14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of t-lymphocyte activation and vascular inflammation produced by angiotensin ii-induced hypertension. Circulation research. 2010;107:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller DN, Shagdarsuren E, Park J-K, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin ii-induced renal damage. The American journal of pathology. 2002;161:1679–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Gool SW, Vandenberghe P, de Boer M, Ceuppens JL. Cd80, cd86 and cd40 provide accessory signals in a multiple-step t-cell activation model. Immunol Rev. 1996;153:47–83 [DOI] [PubMed] [Google Scholar]
- 28.Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, Qin S, Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–2769 [DOI] [PubMed] [Google Scholar]
- 29.Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromley SK, Thomas SY, Luster AD. Chemokine receptor ccr7 guides t cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901 [DOI] [PubMed] [Google Scholar]
- 31.Wen Y, Rudemiller NP, Zhang J, Lu X, Ren J, Privratsky JR, Griffiths R, Zhang JJ, Hammer GE, Crowley SD. C-c motif chemokine receptor 7 exacerbates hypertension through effects on t lymphocyte trafficking. Hypertension. 2020;75:869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (c-reactive protein, interleukin-6, and tnf-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154 [DOI] [PubMed] [Google Scholar]
- 33.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin ii-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin ii-dependent hypertension. Hypertension. 2014;64:1275–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. Beta-arrestin inhibits nf-kappab activity by means of its interaction with the nf-kappab inhibitor ikappabalpha. Proc Natl Acad Sci U S A. 2004;101:8603–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via rho/rho kinase in the thick ascending limb. Hypertension. 2012;59:1145–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramseyer VD, Garvin JL. Tumor necrosis factor-alpha: Regulation of renal function and blood pressure. Am J Physiol Renal Physiol. 2013;304:F1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1a angiotensin ii receptor gene. Proc Natl Acad Sci U S A. 1995;92:3521–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato H, Ishida J, Nagano K, Honjo K, Sugaya T, Takeda N, Sugiyama F, Yagami K, Fujita T, Nangaku M, Fukamizu A. Deterioration of atherosclerosis in mice lacking angiotensin ii type 1a receptor in bone marrow-derived cells. Lab Invest. 2008;88:731–739 [DOI] [PubMed] [Google Scholar]
- 41.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM. Angiotensin ii regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104:1693–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coffman TM. Under pressure: The search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409 [DOI] [PubMed] [Google Scholar]
- 43.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. Cd247 modulates blood pressure by altering t-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu X, Rudemiller NP, Wen Y, Ren J, Hammer GE, Griffiths R, Privratsky JR, Yang B, Sparks MA, Crowley SD. A20 in myeloid cells protects against hypertension by inhibiting dendritic cell-mediated t-cell activation. Circ Res. 2019;125:1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Gool SW, Vandenberghe P, de Boer M, Ceuppens JL. Cd80, cd86 and cd40 provide accessory signals in a multiple-step t-cell activation model. Immunol Rev. 1996;153:47–83 [DOI] [PubMed] [Google Scholar]
- 47.Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, Sagan A, Wu J, Vinh A, Marvar PJ, Guzik B, Podolec J, Drummond G, Lob HE, Harrison DG, Guzik TJ. Role of chemokine rantes in the regulation of perivascular inflammation, t-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016;30:1987–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kroetsch JT, Levy AS, Zhang H, Aschar-Sobbi R, Lidington D, Offermanns S, Nedospasov SA, Backx PH, Heximer SP, Bolz SS. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat Commun. 2017;8:14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.