

Abstract
The peptidoglycan (PG) cell wall is an extracytoplasmic glycopeptide polymeric structure that protects bacteria from osmotic lysis and determines cellular shape. Since the cell wall surrounds the cytoplasmic membrane, bacteria must add new material to the PG matrix during cell elongation and division. The lipid-linked precursor for PG biogenesis, Lipid II, is synthesized in the inner leaflet of the cytoplasmic membrane and subsequently translocated across the bilayer so that the PG building block can be polymerized and crosslinked by complex multiprotein machines. This review focuses on major discoveries that have significantly changed our understanding of PG biogenesis in the last decade. Particularly, we highlight progress made towards understanding the translocation of Lipid II across the cytoplasmic membrane by the MurJ flippase, as well as the recent discovery of a novel class of PG polymerases, the SEDS (shape, elongation, division and sporulation) glycosyltransferases RodA and FtsW. Since PG biogenesis is an effective target of antibiotics, these recent developments may lead to the discovery of much-needed new classes of antibiotics to fight bacterial resistance.
Graphical Abstract
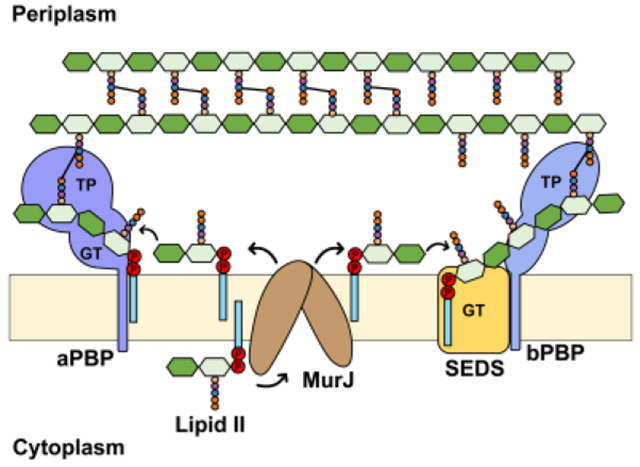
1. Introduction
Most bacteria build a peptidoglycan (PG) cell wall outside their cytoplasmic membrane. The rigid PG structure, or sacculus, surrounds the membrane and protects these unicellular microorganisms from osmotic lysis that would otherwise be caused by the high osmotic pressure in their cytoplasm.1,2 Indeed, the PG cell wall is so strong that it can be isolated from other cellular components by subjecting cells to extensive boiling in the presence of detergents.3 The PG cell wall can also serve as a structure onto which other envelope components such as proteins and polysaccharides can be attached. The essential role of PG in bacterial physiology has been exploited by many antibiotics that target PG biogenesis factors.4,5
The chemical composition of the PG structure is rather simple and highly conserved, but the three-dimensional structure is not only complex and diverse among bacteria but also responsible for the cell shape that is faithfully maintained through generations. From rod, to coccus, to spiral, and even star to name a few, bacterial cell shape is diverse but characteristic of each species.6,7 The PG cell wall is a polymeric mesh built in situ by enzymes that utilize a building block that is conserved across bacteria, except for some minor chemical alterations in some species. The conserved structure of the PG precursor is a disaccharide-pentapeptide composed of N-acetyl glucosamine-N-acetyl muramic acid (GlcNAc-MurNAc) and a stem pentapeptide. To build the mesh-like structure, glycosyltransferases polymerize the disaccharide into glycan chains that are covalently crosslinked via their stem peptides by transpeptidases.8 How and when these reactions occur dictate the final design of the PG structure and therefore cell shape. Not surprisingly, the foundational framework of the PG structure is directed by cytoskeletal elements that determine where the expansion, driven by synthesis or controlled lysis, of the PG sacculus and the building of a division septum occur during the cell cycle. How cells coordinate and accomplish these architectural activities using cytoskeletal and PG-biogenesis factors has fascinated scientists for decades and continues to motivate a significant amount research even today. Also impressive is the fact that when cells grow, they continuously add newly synthesized PG material to the pre-existing cell wall structure to accommodate the change in cell size without compromising cell integrity.
Many bacterial species have been historically classified into two groups based on their reaction towards the Gram stain: Gram-negative (e.g. Escherichia coli) and Gram-positive (e.g. Bacillus subtilis) bacteria. The differential staining is reflective of significant differences in the architecture of their cell envelope.9 Gram-negative cell envelopes consist of a cytoplasmic (or inner) membrane and an outer membrane, which are separated by an aqueous periplasm that contains a thin PG cell wall that is a few nanometers thick (Figure 1). By contrast, Gram-positive bacteria have only a cytoplasmic membrane fenced by a much thicker PG cell wall, ranging from 30 to 100 nm in thickness, into which teichoic acids and proteins are often embedded (Figure 1). Because of their thicker PG cell wall, Gram-positive bacteria retain the crystal violet primary stain during the Gram stain procedure, while Gram negatives lose it in the decolorization step.10,11
Figure 1. Architecture of the Gram-negative and Gram-positive cell envelopes.
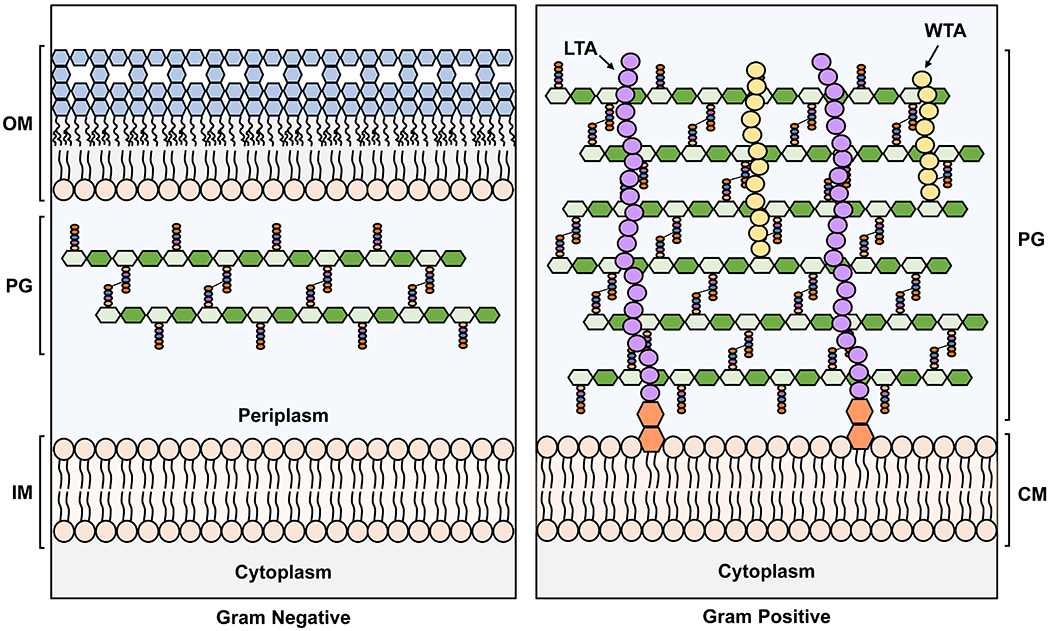
The Gram-negative inner membrane (IM) and the Gram-positive cytoplasmic membrane (CM) are phospholipid bilayers. In contrast, the outer membrane (OM) of Gram-negative bacteria is asymmetrical with phospholipids in the inner leaflet and lipopolysaccharide (LPS, with sugars in blue) in the outer leaflet. IM: inner membrane, PG: peptidoglycan, OM: outer membrane, LTA: lipoteichoic acid, WTA: wall teichoic acid. In the PG cartoon structure, the glycan strands are represented as polymers of hexagons, with N-acetylglucosamine (GlcNAc) in dark green and N-acetylmuramic acid (MurNAc) in light green, while the stem pentapeptides are shown as light orange, blue, purple and dark orange spheres. For more details on the structure of the PG building block, refer to Figure 2 and Section 2.1.
The PG disaccharide-pentapeptide precursor is made as a lipid-linked molecule known as Lipid II, which is synthesized via a biosynthetic pathway that starts from nucleotide-sugars and amino acids in the cytoplasm and then transitions to the inner leaflet of the cytoplasmic membrane when an isoprenoid lipid is added as a membrane anchor (Figure 2). Tethering the PG precursor to the membrane via a lipid prevents the diffusion of an otherwise soluble precursor and, as we will describe in this review, allows the cell to precisely control the site of PG polymerization. Once Lipid II synthesis is completed in the cytoplasmic side of the membrane, it is transported or flipped across the lipid bilayer and used by PG synthases to build new PG material. This overall biosynthetic strategy is not unique to PG biogenesis, as it is widely used by cells in the three domains of life for the biogenesis of diverse glycoconjugates such as surface-exposed polysaccharides and glycosylation of proteins.12,13,14
Figure 2. Chemical and cartoon structures of Lipid II.
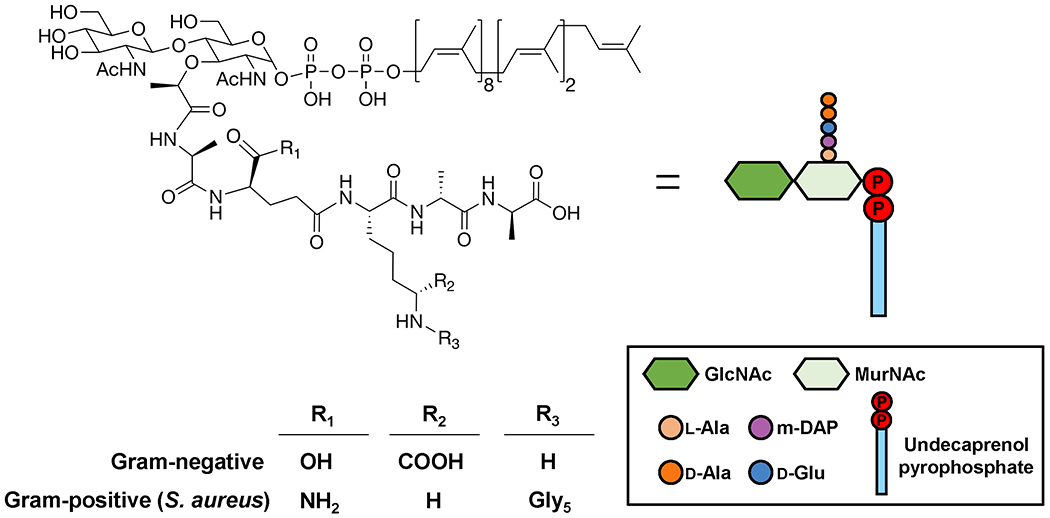
Functional groups specific to Gram-positive and Gram-negative bacteria are summarized in the table. The same cartoon representation for Lipid II is used in subsequent figures.
Despite having studied PG for decades, we continue to discover new proteins associated with cell wall synthesis and reveal the elusive role of others, while also investigating how bacteria utilize dynamic and transient multi-protein complexes to build their PG sacculus.15 Numerous exciting developments have been made in the past decade, largely from studies of the rod-shaped model organisms Escherichia coli and Bacillus subtilis. A combination of genetic, biochemical, structural, and microscopy approaches have advanced our knowledge about PG biogenesis and challenged several old assumptions. In this review, we will mainly discuss two discoveries that have fundamentally changed our view of PG biogenesis with respect to the translocation of Lipid II across the cytoplasmic membrane and PG polymerization. Specifically, we will review recent advances made in the past decade towards our understanding of the physiological function and mechanism of the Lipid II flippase MurJ, and a newly discovered class of PG glycosyltransferases, the SEDS (shape, elongation, division and sporulation) proteins RodA and FtsW. Although most of the work we discuss was carried out in the Gram-negative bacterium E. coli and to a lesser extent the Gram-positive B. subtilis, these proteins seem to be conserved among known PG producers.
2. Lipid II: Precursor for Peptidoglycan biogenesis
2.1. Structure of Lipid II
In the late 1960s and early 1970s, Strominger, Sweeley, and collaborators discovered that bacteria synthesize the monomeric unit precursor of PG, a disaccharide pentapeptide, on a lipid carrier.16–18 The lipid-linked precursor, called Lipid II, is an amphiphilic lipid consisting of a hydrophobic undecaprenyl (C55) tail with a pyrophosphate linkage to a β-1,4-linked heterodisaccharide of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc), and a pentapeptide branching off the 3’ carbon of MurNAc (Figure 2).16–18 As we describe below, Lipid II is synthesized in the inner leaflet of the cytoplasmic membrane and, once translocated across the lipid bilayer, its disaccharide unit is utilized to form a linear heteropolymer, or glycan strand, which is cross-linked to adjacent glycan strands via the peptide side-chains to build the mature PG cell wall structure. During glycan strand synthesis, the lipid carrier is released as undecaprenyl pyrophosphate (Und-PP), which is subsequently recycled. It is estimated that each lipid carrier undergoes a cycle of Lipid II synthesis and utilization and Und-PP recycling every 90 seconds.19
Notably, the C55 isoprenoid component of Lipid II is longer than the typical C14-C18 acyl chains of phospholipids present in bacterial membranes.20 Molecular dynamics simulations show that the isoprenoid chain on Lipid II is flexible and highly dynamic. With its pyrophosphate serving as an anchor at the water-membrane interface, the C55 isoprenoid is modeled to sample a large conformational space within the hydrophobic core of the membrane by acquiring extended and bent conformations.21,22 Accordingly, the highly dynamic Lipid II is predicted to disturb the surrounding lipids.21,22 As we discuss in section 3, Lipid II is translocated across the cytoplasmic membrane by transporters (i. e., flippases) during PG biogenesis. It is unknown whether the flexibility of Lipid II and its effect on the membrane play a role in this translocation, and by extension in the transport of phosphorylated forms of undecaprenol.
While the undecaprenyl-pyrophosphoryl-MurNAc-GlcNAc moiety of Lipid II is largely invariant between different organisms, the stem pentapeptide portion can vary widely. In general, Gram-negative bacteria synthesize the peptide l-Ala-γ-d-Glu-mDAP-d-Ala-d-Ala, where mDAP is a non-proteinogenic amino acid called meso-diaminopimelic acid; Gram-positive bacteria, on the other hand, synthesize l-Ala-γ-d-Gln-l-Lys-d-Ala-d-Ala, where the γ-d-Glu residue found in Gram-negative bacteria is amidated and the mDAP residue is replaced with l-Lys.23–25 The conversion of γ-d-Glu residue to d-Gln occurs at the membrane after Lipid II synthesis.25 Other amino acids can also exist at the first and third positions and, in some cases, they can vary depending on growth conditions.26,27 The reader is directed to an excellent review for a discussion of Lipid II variabilities in Gram-positive bacteria.28 The third amino acid (mDAP or l-Lys) of the stem peptide is particularly important when building the PG structure because it is responsible for forming cross-links to stem peptides on adjacent glycan strands by acting as a nucleophilic agent towards acyl-enzyme intermediates in the transpeptidation step of PG biogenesis. Most cross-links connect the third residue of one peptide with the fourth (most frequently) or third residue of another stem peptide. Notably, in some Gram-positive bacteria such as Staphylococcus aureus, the l-Lys residue is modified intracellularly with a peptide “bridge” (usually 2-5 amino acids depending on the bacterial species), resulting in longer distances between cross-linked stem peptides, which may become more frequent due to the flexibility of the type of linkage (Figure 2).23
The structural diversity of the stem peptide can also be a source of resistance to antibiotics targeting Lipid II. Lipid II is usually considered a very good antibiotic target because it is essential for viability but not abundant in cells (~1,000 molecules per E. coli cell) and there are few ways its structure can be altered to confer resistance while preserving function.29,30 Nevertheless, resistance resulting from alterations to Lipid II structure can still occur. For example, the glycopeptide antibiotic vancomycin acts by binding to the d-Ala-d-Ala terminus, preventing cross-link formation.31 Vancomycin-resistant enterococci replace d-Ala with either d-lactate or d-serine, reducing the H-bonding ability of vancomycin while preserving the ability of Lipid II to be used in polymerization reactions to build the PG matrix.32–34
Researchers have taken advantage of the fact that certain alterations to Lipid II are permitted by cells in order to develop chemical probes that can serve as tools to study PG biosynthesis. Reagents such as those that lead to metabolically labeled MurNAc or stem peptides have revolutionized the PG biogenesis field by providing means to monitor PG biogenesis in real time in cells via imaging.35,36 We refer the reader elsewhere for excellent reviews on this newly developing field. 37–39
2.2. Biosynthesis of the Lipid Carrier Undecaprenyl Phosphate.
Undecaprenyl phosphate (Und-P) is a C55 polyisoprenoid lipid that plays a central role in bacterial physiology owing to its utilization in the biogenesis of bacterial envelope glycopolymers such as PG, the O antigen of lipopolysaccharides (commonly known as LPS), wall teichoic acids, capsular polysaccharides, and enterobacterial common antigen.40–42 In all these polysaccharide biogenesis pathways, Und-P is attached to hydrophilic sugar moieties to form lipid-linked intermediates that are synthesized in the inner leaflet of the cytoplasmic membrane and then translocated to the outer leaflet.40–42 Und-P is produced by dephosphorylation of Und-PP in both de novo synthesis and recycling pathways (Figure 3). In the de novo synthesis pathway, the prenyl transferase Und-PP synthase (UppS) catalyzes the sequential condensation of isopentyl pyrophosphate (C5-PP) with farnesyl pyrophosphate (C15-PP) units to generate Und-PP. On the other hand, in the recycling pathway, Und-PP is released by pathway-specific transferases such as the PG glycosyltransferases during glycopolymers polymerization.40,42,43 In both cases, the cellular pool of Und-P is generated from the dephosphorylation of Und-PP by pyrophosphatases such as the membrane-integral pyrophosphatase BacA and members of the phosphatidic acid phosphatase (PAP2) super-family of phosphatases (PgpB, YbjG, and YeiU/LpxT). In E. coli, BacA is responsible for the majority of the Und-PP phosphatase activity, with smaller contributions by the PAP2 enzymes PgpB, YbjG, and LpxT (Figure 3).44,45 Interestingly, although undecaprenol (Und-OH) is absent in Gram-negative bacteria, it can be present in Gram-positive bacteria such as S. aureus and B. subtillis, where it is phosphorylated to form Und-P by an enzyme homologous to the diacylglycerol kinase of E. coli, DgkA.17,46
Figure 3: Schematic of the PG cell wall biogenesis pathway in E. coli.
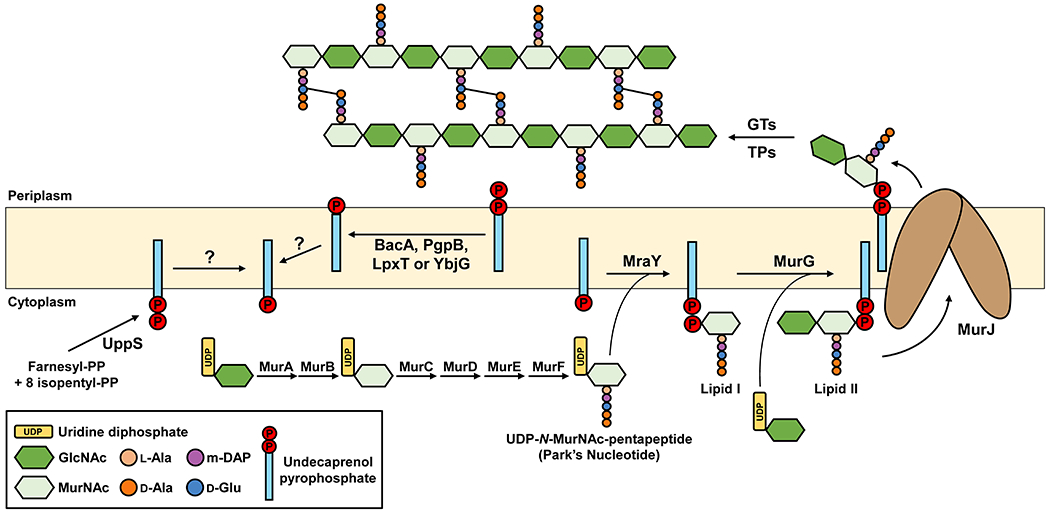
PG precursor synthesis starts in the cytoplasm where nucleotide-linked precursors UDP-N-acetylglucosamine (UDP-GlcNAc), UDP-N-acetylmuramic acid (UDP-MurNAc) and UDP-N-MurNAc-pentapeptide are synthesized. The latter precursor is linked to Und-P by MraY to generate Lipid I. Then, MurG utilizes Lipid I and UDP-N-GlcNAc to synthesize Lipid II. MurJ translocates Lipid II across the IM so that glycosyltransferases (GTs) can polymerize the disaccharide-pentapeptide into glycan chains and transpeptidases (TPs) catalyze cross-linking between stem peptides. After that, recycling of Und-P takes place by dephosphorylation of Und-PP by one of three enzymes (BacA, PgpB, YbjG, and LpxT) on the periplasmic side of the IM and subsequent flipping by an elusive flippase. A significant amount of undecaprenol (Und-OH) is present in Gram-positive bacteria where DgkA catalyzes the phosphorylation of Und-OH to Und-P.
A question that has been debated for years is how pyrophosphatases access both de novo synthesized Und-PP present in the inner leaflet of the cytoplasmic membrane and Und-PP released from transferase activity in the outer leaflet of the cytoplasmic membrane, given that active sites of the Und-PP phosphatases are thought to be present on the periplasmic side of the membrane.42,47–49 Moreover, Und-P enters polysaccharide biogenesis pathways from the inner leaflet of the cytoplasmic membrane; therefore, once Und-P is made by the pyrophosphatases, it must somehow be translocated across the lipid bilayer (Figure 3). It is possible that an unidentified flippase translocates Und-P to the inner leaflet of the cytoplasmic membrane after being produced in the outer leaflet by pyrophosphatases. It is also possible that de novo synthesized Und-PP undergoes a “flip-flop” mechanism of dephosphorylation in which Und-PP is first flipped towards the outside side of the membrane, dephosphorylated, and then flipped back to the cytoplasmic side of the membrane for utilization in polysaccharide biogenesis pathways.42 Moreover, one or more Und-PP pyrophosphatases might be capable of flipping Und-PP and/or Und-P. In support of pyrophosphatases functioning also as flippases, the structure of E. coli BacA displays an inverted repeat topology, a common feature of transporters that use an alternating-access mechanism of transport. In addition, although BacA’s active-site cavity opens to the periplasm, its active site is located mid-line with respect to the membrane. Based on these observations, and some structural similarities with MurJ, the Lipid II flippase that is described in detail in sections below, it has been recently speculated that BacA might function as both Und-PP phosphatase and Und-P(P) flippase.42,50,51 Although this model of BacA having dual function is undoubtedly novel and exciting, further studies are required to test it. For more information, the reader is directed to recent comprehensive reviews on the current knowledge of Und-P synthesis, its metabolism and recycling.40,42,46
2.3. Biosynthesis of Lipid II
Lipid II is synthesized in a linear sequence of reactions starting from fructose-6-phosphate in the cytoplasm to the completion of the lipid intermediate in the cytoplasmic side of the membrane (Figures 3 and 4). Over the past 50 years, this pathway has been characterized in detail through the efforts of many researchers. In the early years, biochemical and analytical analyses led to the identification of precursors and products of each step.52 Once the genes encoding the biosynthetic enzymes of this pathway were identified, they were found to be essential. Biochemical reconstitution and crystallization studies of the enzymes facilitated a better understanding of their mechanism of function and the development of inhibitors that could be used as antibiotics.52–54 The literature on the Lipid II biogenesis pathway has been reviewed extensively elsewhere. 30,41,52,55,56
Figure 4. Stepwise synthesis of Lipid II in E. coli.
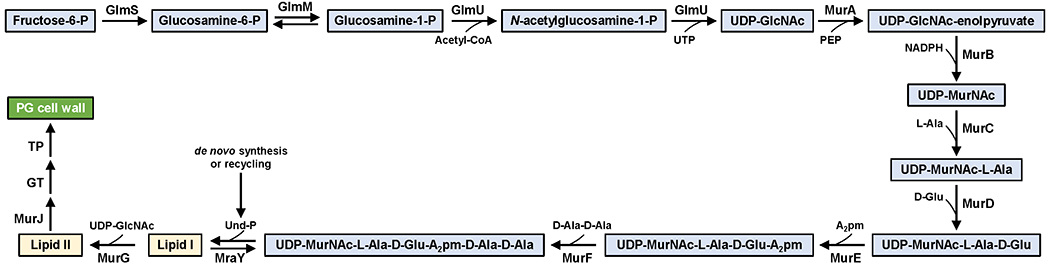
UTP: uridine triphosphate, PEP: phosphoenol pyruvate, NADPH: reduced nicotinamide adenine dinucleotide phosphate, A2pm: diaminopimelic acid, Ala: alanine, Glu: glutamic acid, GlcNAc: N-acetylglucosamine, MurNAc: N-acetylmuramic acid. Cytoplasmic intermediates are shown in blue boxes, while membrane-bound intermediates are shown in cream boxes.
Lipid II synthesis depends on the synthesis of both Und-P in the membrane (described in section 2.2) and nucleotide-sugar precursors in the cytoplasm (Figures 3 and 4). The nucleotide-sugar precursors UDP-GlcNAc and UDP-MurNAc-pentapeptide (also known as Park’s nucleotide) are synthesized by the Glm enzymes (GlmSMU) and the Mur enzymes (MurABCDEF enzymes), respectively, with MurA catalyzing the first committed step in the synthesis of PG.56 The integral membrane protein MraY catalyzes the first membrane-bound step by transferring the phospho-MurNAc-pentapeptide moiety of UDP-MurNAc-pentapeptide to Und-P to produce uridine-monophosphate (UMP) and Lipid I (undecaprenyl-pyrophosphoryl-MurNAc-pentapeptide). MurG then transfers the GlcNAc moiety from UDP-GlcNAc to Lipid I to yield Lipid II (Und-PP-MurNAc-(pentapeptide)-GlcNAc).41,55 A key finding made by Bupp and van Heijenoort was that MurG is associated with the cytoplasmic side of the membrane, indicating that fully synthesized Lipid II needed to be translocated across the cytoplasmic membrane for glycosyltransferases to make new glycan chains in the periplasm (Figure 3).57 At the time, nothing was known about Lipid II transport, but the past decade has witnessed significant discoveries in the transport and utilization of Lipid II by flippases and PG synthases, respectively, which are the focus of the next sections of this review.
3. Flipping Lipid II Across the Cytoplasmic Membrane
The demonstration by Bupp and van Heijenoort that Lipid II is synthesized in the cytoplasmic side of the membrane led to a highly debated question in PG biogenesis: How does Lipid II translocation across the membrane occur?57 Lipid II molecules present on either side of the membrane are chemically and structurally identical and thus thermodynamically equivalent (assuming no significant differences in charges between the cytoplasmic and periplasmic sides). Unassisted flip-flopping of Lipid II was not thought possible since it would require that the hydrophilic PP-GlcNAc-MurNAc-pentapeptide moiety traverse the hydrophobic interior of the membrane, a process which has a very high energy barrier and thus is kinetically disfavored. Bupp and van Heijenoort therefore proposed that one or more proteins would have to be responsible for the transport of Lipid II across the cytoplasmic membrane. Indeed, in vitro translocation assays using liposomes and fluorescently labeled Lipid II by Dam et al. later showed that flipping cannot occur spontaneously.57, 58
After much debate about the identity of the Lipid II transporter, the integral membrane protein MurJ was discovered as the main bacterial Lipid II flippase.59–62 Like other flippases of Und-P-linked glycoconjugates, MurJ is thought to reduce the energy barrier of translocation by providing a central aqueous cavity that protects the hydrophilic portion of Lipid II as it traverses the membrane, while maintaining the isoprenoid C55 tail in the hydrophobic core of the membrane. Based on an estimated translocation rate of ~5.4×104 Lipid II molecules per minute and ~78 copies of MurJ per E. coli cell in minimal medium, one expects that each MurJ protein can potentially translocate nearly 700 Lipid II molecules/min. 19,63
In the next sections, we describe how transporters of Lipid II were identified and the great progress towards our understanding of Lipid II translocation made over the last decade. We also highlight the fundamental questions that remain unanswered, many of which greatly depend on the yet-to-be-developed in vitro reconstitution of Lipid II flippase activity and future analyses of transport intermediates through biochemical and structural studies.
3.1. Identification of Lipid II Flippases
Identification of transporters of bacterial lipid-linked glycoconjugates is often driven by the location of their respective genes in the chromosome. In those instances, the flippase-encoding gene is within a locus containing the biosynthetic genes of the oligo/polysaccharide. As these transporters are integral membrane proteins, their transmembrane helix segments can be easily predicted bioinformatically from their gene sequence. Thus, being in the right gene neighborhood and encoding a membrane protein of unknown function often results in the designation of being the putative flippase for the lipid-linked oligo/polysaccharide. This designation can be strengthened by homology to known transporters. For example, the flippase for the lipid-linked wall-teichoic acid intermediate TarGH, belongs to the ABC (ATP-binding cassette) superfamily of transporters, which are easily identified through primary sequence conservation.64,65 However, it is important to recognize that these predictions need to be tested experimentally as they could be misleading. In fact, this prediction strategy led to the reasonable suggestion that FtsW could be a Lipid II flippase because, in many bacteria, this integral membrane protein is encoded in the same operon as mraY, murG, and murCDEF, the genes for enzymes required for the synthesis of Lipid II.66 However, as we describe in section 3.1.3, experimental evidence does not support that FtsW functions as a Lipid II transporter in cells.
3.1.1. Identification of MurJ
MurJ was the first Lipid II flippase to be identified. Since it is not encoded within a PG-related genomic locus, its identification as a Lipid II flippase candidate relied on different strategies when it was independently identified in two studies published in 2008. Using a reductionist bioinformatics approach, N. Ruiz searched for Lipid II flippase candidates by conducting a comparative conservation analysis between the inner membrane proteins of E. coli and the small proteome (<600 proteins) of related endosymbionts.60 This bioinformatics strategy, which had been proven to be a successful tool for the discovery of other proteins essential for cell envelope biogenesis, identified MurJ (previously known as MviN) as the only Lipid II flippase candidate in E. coli.67 In agreement with that prediction, Ruiz showed that MurJ is essential for growth and that depleting its cellular levels inhibits PG biosynthesis, leading to an accumulation of nucleotide and lipid-linked PG precursors. Similar in vivo evidence was independently obtained by Inoue et al., who uncovered that murJ is essential for PG biogenesis when systematically constructing large chromosomal deletions in E. coli.61 The authors observed that strains containing a temperature-sensitive allele of murJ lysed after being shifted to the nonpermissive temperature and displayed a two-fold increase in levels of lipid-linked PG intermediates.61 Neither study specified if the increase of lipid-linked precursors corresponded to Lipid I, Lipid II, or both. Nevertheless, a key piece of evidence that significantly strengthened the proposal that MurJ was a Lipid II flippase was the fact that it belongs to the MOP (Multidrug/Oligosaccharidyl-lipid/Polysaccharide) exporter superfamily of proteins, which include multidrug exporters of the MATE (Multidrug and Toxic compound Extrusion) family as well as Wzx proteins, which flip Und-PP-oligosaccharides across bacterial cytoplasmic membranes. Within the MOP exporter superfamily, MurJ is the founding member of the MVF family.59,61,68 The MviN (mouse virulence factor N) reference stems from a misnomer for MurJ resulting from erroneous mapping of a mutation affecting virulence to murJ instead of a nearby gene involved in motility.60
Addressing whether the loss of MurJ leads to the accumulation of Lipid II in the inner leaflet of the membrane, as it would be expected from the loss of Lipid II translocation, was crucial. Answering this question required the development of new methodology to overcome two technical barriers: 1) how to distinguish Lipid II located in the inner leaflet vs. the outer leaflet of the cytoplasmic membrane; and 2) how to quickly eliminate MurJ function in cells instead of relying on the slow depletion of MurJ protein, which makes distinguishing primary vs. secondary effects difficult. Both of these challenges were overcome by Sham et al., who developed an in vivo Lipid II flippase assay and a chemical genetic strategy to quickly and specifically inhibit MurJ function in E. coli cells (Figure 5).62 To differentiate the topological location of Lipid II with respect to the membrane, this Lipid II flippase assay makes use of the colicin M (ColM) toxin.69 When exogenously added to cells, ColM crosses the outer membrane to enter the periplasm where it specifically hydrolyzes periplasmic Lipid II into Und-OH and PP-disaccharide pentapeptide. Several lines of evidence indicate that ColM acts in the periplasmic side of the cytoplasmic membrane, not being able to target newly synthesized Lipid II that is yet-to-be flipped across the membrane.70 In addition, PP-disaccharide pentapeptide was shown to be further processed presumably by periplasmic carboxypeptidases into PP-disaccharide-tetrapeptide.62 Consequently, ColM specifically converts periplasmic (i.e. flipped) Lipid II into a molecule, PP-disaccharide-tetrapeptide, that is chemically different, and therefore can be separated and distinguished, from cytoplasmic Lipid II.62 To specifically inhibit MurJ function with fast kinetics, Sham et al. engineered functional MurJ variants containing single cysteine substitutions in their periplasmic-exposed portion (e.g. MurJA29C) that when chemically modified with the thiol-labelling reagent MTSES (2-Sulfonatoethyl methanethiosulfonate) induce rapid cell lysis, implying that PG synthesis is inhibited because of the loss of MurJ function (Figure 5). We now think, based on structural information, that these modifications (e.g. at position A29C of MurJ) leave MurJ unable to adopt all the structural conformations that are required for transport (see sections 3.2).71 With these methods in hand, Sham et al. monitored Lipid II translocation in cells before and after the specific fast inhibition of MurJ.62 They found that loss of MurJ function causes the simultaneous accumulation of cytoplasmic Lipid II and reduction in the levels of the PP-disaccharide-tetrapeptide derived from the soluble ColM product, which could be rescued upon compromising the structure of the cytoplasmic membrane. These critical results, together with the body of evidence we have highlighted in this section, led to the conclusion that MurJ is the Lipid II flippase in E. coli.
Figure 5. ColM-based in vivo flippase assay to measure translocation of Lipid II in E. coli cells.
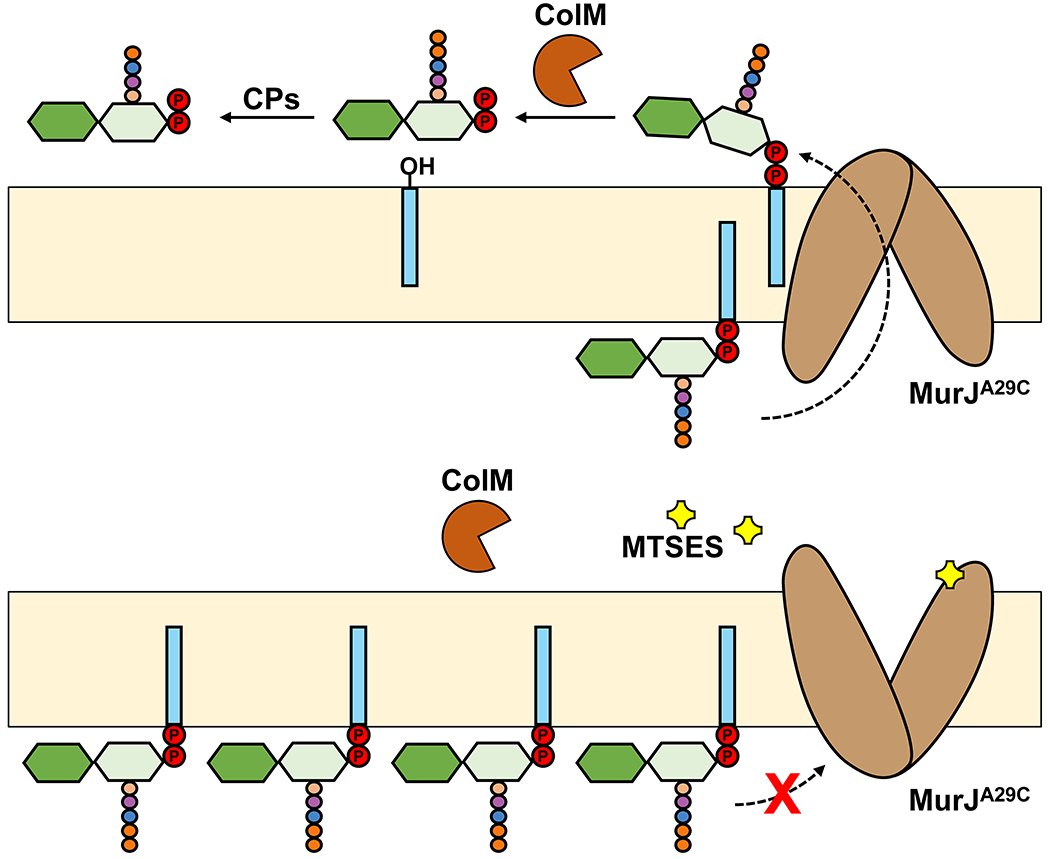
(a) Purified ColM toxin is added to actively growing E. coli cells producing MurJA29C, a functional variant that can be inactivated when the engineered cysteine at position 29 is modified with MTSES. ColM enters the periplasm and cleaves periplasmic Lipid II into membrane-bound undecaprenol and soluble PP-disaccharide-pentapeptide, which is further converted by periplasmic carboxypeptidases (CPs) into PP-disaccharide-tetrapeptide. Adding 3H-meso-diaminopimelic acid specifically labels PG and its DAP-containing precursors (not shown). When adding ColM, labeled cytoplasmic Lipid II and ColM-dependent PP-disaccharide-tetrapeptide can be separated and measured. Their ratio reflects the extent of Lipid II translocation across the cytoplasmic membrane. (b) Adding the cysteine-reactive compound MTSES causes inhibition of MurJ function, leading to an increase in the levels of Lipid II and the loss of ColM-dependent PP-disaccharide-tetrapeptide species.
3.1.2. Identification of Amj
Is MurJ conserved among PG-producing bacteria? Mohamed and Valvano found that the MurJ homolog in the Gram-negative Burkholderia cenocepacia is not only indispensable for PG biosynthesis and growth but can also substitute for the native MurJ in E. coli and vice versa, demonstrating that the two proteins are functionally equivalent.72 Essentiality for growth and PG biogenesis of the MurJ homolog were also demonstrated in Mycobacterium tuberculosis, although curiously this homolog contains additional non-essential domains that are thought to be involved in regulation of PG synthesis (see section 3.4). More recently, García-Heredia et al. also demonstrated that depletion of MurJ results in an accumulation of PG precursors in Mycobacterium smegmatis.73 Thus, MurJ is conserved across distant bacteria. However, the search for and functional characterization of MurJ homologs in Gram-positive bacteria was more difficult. A traditional BLAST homology search focused on targeting Gram-positive bacteria using the E. coli MurJ as query only revealed candidates with low homology levels, with YtgP being at the top of the list.74,75 The possibility of YtgP being the Gram-positive functional homolog of MurJ gained some traction following the demonstration that Streptococcus pyogenes YtgP complements E. coli strains depleted of their endogenous MurJ, and that YtgP is essential in Staphylococcus aureus and Streptococcus pneumoniae.76–78 Paradoxically, YtgP was shown to be not essential in B. subtilis despite being able to substitute for MurJ in E. coli, calling into question MurJ’s role in the essential process of Lipid II translocation across the cytoplasmic membrane.79,80 This dispute was solved by Meeske et al., who showed that ytgP is not essential in B. subtilis because this organism encodes a protein, AmJ, that is functionally redundant with YtgP (MurJ).81 B. subtilis encodes for 10 members of the MOP exporter superfamily including YtgP; Meeske et al. demonstrated that a mutant strain lacking all of them grows similarly to the wild type. The authors reasoned that given that YtgP/MurJ was essential in other bacteria, the lack of essentiality in B. subtilis likely stemmed from functional redundancy with a yet-to-be identified non-MOP protein(s). To identify this putative protein, Meeske et al. screened for transposon insertions that would cause synthetic lethality with a ∆ytgP allele, a strategy that resulted in the discovery of ydaH (renamed to amj, for “alternate to MurJ”). The authors confirmed that the simultaneous loss of Amj and YtgP in B. subtilis results in aberrant cell morphologies and extensive cell lysis, consistent with impaired cell wall synthesis. Importantly, they demonstrated that Amj and MurJ are functionally redundant for Lipid II translocation by showing that Amj can substitute for MurJ in E. coli cells using the aforementioned ColM-based flippase assay.81
Interestingly, Amj lacks sequence similarity with members of the MOP exporter superfamily including MurJ, or members of the ABC transporter superfamily, suggesting that it belongs to a new class of transporters of Und-PP-linked sugars.81 Alternatively, Amj could be a transporter of an unknown substrate that can promiscuously transport Lipid II. In fact, there is precedent for relaxed substrate specificity among transporters of Und-PP-linked oligosaccharides belonging to the MOP (i.e. Wzx) and ABC transporter superfamilies.81 An example relevant to substrate promiscuity involving Lipid II was illustrated by Elhenawy et al., who showed that Wzk, the ABC transporter that transports the lipopolysaccharide (LPS) O-antigen-Und-PP precursor in Helicobacter pylori, can substitute for MurJ in both H. pylori and E. coli.82
Collectively, the studies discussed in this section reveal the power of homology-driven studies as well as their most obvious limitations. They can be used to identify orthologs (i.e. genes that have a same function in diverse species and evolved from a common ancestral gene) among distantly related species, but they cannot inform on non-homologous, functionally redundant proteins. They also showed that lack of essentiality of a protein should not be used as evidence that the protein cannot perform an essential function, since paralogs can encode proteins that can substitute for each other. 74,80,83,84
3.1.3. Are FtsW and RodA Lipid II Flippases?
As mentioned earlier, the SEDS protein FtsW (and by extension its homolog RodA) was proposed to function as a Lipid II flippase based on its predicted membrane topology and its corresponding gene being located in a locus encoding for enzymes involved in Lipid II synthesis.66,85 In vitro reconstitution of FtsW into proteoliposomes seemed to support the Lipid II flippase function, but, paradoxically, the in vitro assay also led to the conclusion that FtsW could flip Lipid II even when its last 6 transmembrane helices were removed.66,86 Although FtsW’s structure is yet to be elucidated, the structure of its close homolog RodA is available. It is hard to envision how removing 6 of FtsW’s 10 transmembrane helices would result in a functional or even stable protein.87,88 As described in the next paragraph, there are concerns about the interpretation of the results obtained with the truncated proteins in the in vitro flippase assay.
As we discuss in section 4, RodA and FtsW were recently reported to function as PG glycosyltransferases that utilize Lipid II to synthesize PG glycan strands.89–91 Some researchers have speculated that these proteins could still function as both glycosyltransferases and flippases.92 Although this dual-function model seems formally possible, it is only based on the assumption that the in vitro FtsW reconstitution assay is truly reporting Lipid II flippase activity. As mentioned earlier, it is difficult to reconcile that extensively truncated FtsW variants can be functional flippases.86 In addition, the in vitro FtsW reconstitution assay also reported FtsW’s ability to translocate various phospholipids, but could not detect flippase activity when MurJ was incorporated into liposomes.66,86 Together, these findings call into question the specificity and interpretation of data obtained from the in vitro flippase assay. An alternative explanation for the reported results is that the in vitro assay was reporting on the integrity of proteoliposomes instead of Lipid II and phospholipid translocation. This is so because the assay relies on quenching of labeled substrates (Lipid II or phospholipids) with chemicals that cannot cross the proteoliposome; consequently, defects in liposome integrity (e. g. caused by misfolded proteins) could lead to misinterpretation.66,86 Moreover, at present, there is no in vivo experimental evidence supporting that FtsW functions as a Lipid II flippase. Instead, depleting E. coli cells of FtsW (even in the absence of RodA) was shown to not inhibit Lipid II translocation in the ColM assay described in section 3.1.1.62 Recent genetic evidence from B. subtilis also argues against a dual glycosyltransferase/flippase function for SEDS proteins. This bacterium can survive with RodA and FtsW as the only glycosyltransferases (see section 4.2.1), yet it still requires MurJ/Amj flippase activity.81,93 Thus, we believe that there is no convincing evidence supporting that FtsW flips Lipid II.
3.2. Structure of the MurJ flippase
MATE transporters are the best characterized members of the MOP exporter superfamily, which includes MurJ.68 Although MATE transporters do not share a high level of sequence identity, they share a common structure: 12 transmembrane helices (TM1-12) arranged into two bundles or lobes (TM1-6 and TM7-12) that form a V-shaped solvent-exposed central substrate-binding cavity (Figure 6a). Using in silico structural predictions and in vivo topological studies, Butler et al. showed that MurJ has 14 transmembrane domains and proposed that TM1-12 assemble into a structure similar to that reported for MATE transporters.94 This and a follow-up study also demonstrated that the predicted central solvent-exposed cavity includes residues whose charge is essential for MurJ function. These findings led the authors to propose that these charged residues in MurJ might interact with the hydrophilic portion of Lipid II and that the MurJ’s cavity might undergo conformational changes via an alternating-access mechanism of function during the transport cycle.94,95 Pushing structural predictions and structure-function analyses further, Butler et al. proposed that despite the low level of primary sequence homology between MurJ from E. coli and YtgP from S. pyogenes, these two proteins are structurally similar and demonstrated that both proteins contain 3 positively charged residues that are essential for function and similarly positioned in their predicted structures. When these studies were published in 2013 and 2014, the debate regarding whether MurJ or FtsW were Lipid II flippases was at its peak.59 Although these studies only provided very low-level-resolution structural information and structural predictions, they were empowered by extensive in vivo structure-function analyses and became key evidence supporting MurJ as a Lipid II flippase.
Figure 6. Crystal structures of PfMATE and MurJ.
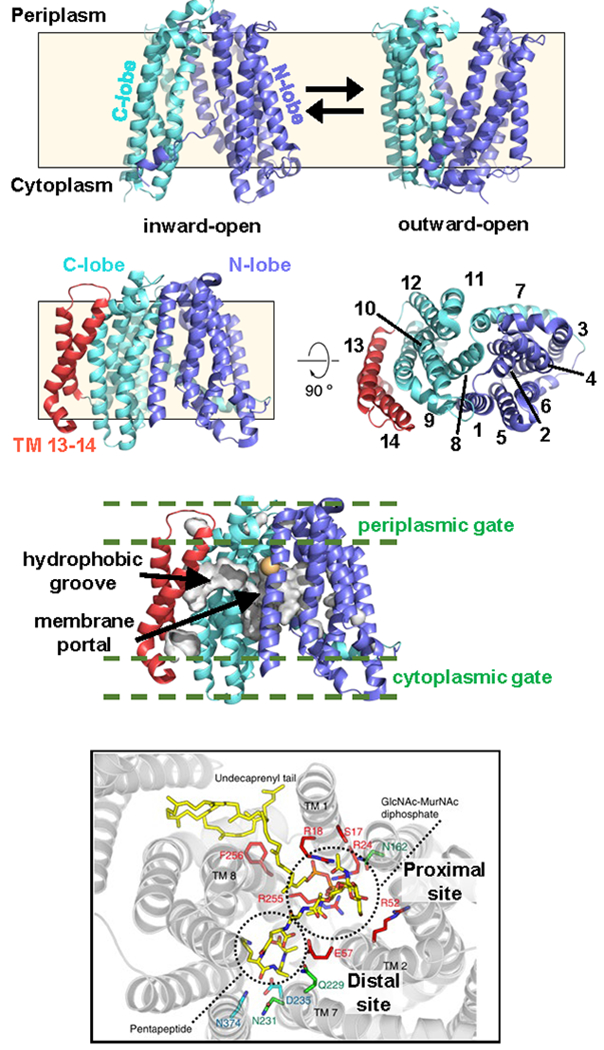
(a) Inward- and outward-open structures of PfMATE (PDB ID: 6FHZ and 6GWH, respectively). The N- and C-lobes are color-coded blue and cyan, respectively. The approximate location of the membrane is indicated with a cream-colored box. (b) Inward-open structure of MurJTa (PDB ID: 5T77) as shown from the membrane plane (left) and the periplasm (right). Transmembrane helix numbers (1-14) are indicated. The color scheme for the N- and C- lobes is the same as in panel (a), and TM13-14 are shown in red. (c) Surface representation of the hydrophobic groove extending from the central cavity into the TM13-14 pocket via the portal region located between TM1 and TM8. Regions corresponding to the cytoplasmic and periplasmic gates are also marked. Residue corresponding to F22 in E. coli MurJ is shown as gold sphere (see Section 3.3.1) (d) Docking model of Lipid II and MurJTa. This panel was reproduced with permission from reference 71. Copyright 2016, Nature Publishing Group. The proximal (containing essential arginines) and distal (negatively charged) sites discussed in the text are labeled.
The much awaited first high-resolution structure of MurJ was revealed in 2017. Kuk et al. successfully obtained a 2Å-resolution structure of MurJ from the hyperthermophilic bacterium Thermosipho africanus by lipidic cubic phase (LCP) crystallography.71 The MurJ structure revealed 14 transmembrane helicesarranged into two lobes called the N-lobe (TM1-6) and C-lobe (TM7-12), respectively (Figure 6b). These lobes are related by two-fold pseudo-rotational symmetry and form a V-shaped central cavity, as reported in MATE transporters.96,97 Interestingly, this MurJ structure was the first of a MOP exporter with its V-shaped central cavity captured in the inward-open conformation. Unlike other known MOP transporters, MurJ has two additional transmembrane helices (TM13-14) that form a ~20 Å-long, curved hydrophobic groove that lines the outside of the C-lobe and leads into a membrane portal region between TMs 1 and 8 that opens into the putative substrate-binding cavity (Figure 6b and 6c). Based on results from docking Lipid II into the MurJ structure, Kuk et al. proposed that the groove formed by TM13-14 associates with the undecaprenyl tail of Lipid II while the pyrophosphate-disaccharide-pentapeptide moiety associates with the central cavity (Figure 6d).71 This groove region has not yet been fully functionally characterized, and the proposed association between TM13-14 and the undecaprenyl tail of Lipid II is yet to be directly demonstrated by biochemical or structural data.
A prominent feature of this MurJ structure is the large internal cavity mainly lined by TMs 1, 2, 7 and 8 that is proposed to bind and shield the substrate during translocation (Figure 6b).71,94 The MurJ cavity is ~20 Å wide and opens to the cytoplasm, reaching ~20 Å up into the membrane. It contains multiple positively charged residues, including three essential arginine residues (R18, R24, R255 in T. africanus; R18, R24, R270 in E. coli) located near the aforementioned portal region between TM 1 and 8.94,95,98 These observations are consistent with a model in which the cavity interacts with Lipid II, which is large (~1,900 Da) and negatively charged. The cavity can be further subdivided into a proximal and a distal site with respect to the hydrophobic groove formed by TM13-14 (Figure 6d).71 The proximal site contains the three essential arginines, while the distal site has an overall negative charge. Docking data showed that the proximal and distal sites are likely to respectively bind to the PP-MurNAc-GlcNAc and pentapeptide moieties of Lipid II (Figure 6d).71 Based on these structural features and previous work on MATE transporters, Kuk et al. proposed that the MurJ cavity undergoes transitions between inward-open and outward-open states during the transport cycle. Furthermore, the inward-facing structure also led to a model explaining the fast inhibition of MurJ used in the in vivo flippase assay that was accomplished by chemically modifying specific engineered cysteine residues (e. g. A29C) located in the periplasmic region of MurJ with the sulfhydryl-reactive reagent MTSES (described in section 3.1.1). Kuk et al. proposed that these modifications cause steric hindrance between residues located in the periplasmic region of the N- and C-lobes and prevents MurJ from acquiring the inward-open state.71
Despite a low overall sequence identity (~28%) between T. africanus MurJ (MurJTa) and E. coli MurJ (MurJEc), the inward-open structure of MurJEc later solved by Zheng et al. revealed that the two proteins share the main structural features discussed above.99 Zheng et al. presented additional important insight regarding MurJ. By employing large-scale mutagenesis, the authors identified key residues in MurJ, although it is unclear whether changes affected activity or protein levels, since expression was not monitored.99 They also used evolutionary coupling analysis to predict additional conformational states, since this type of covariance technique can computationally identify pairs of residues that coevolve based on spatial proximity and interactions in the three-dimensional protein structure.100–102 These analyses predicted conserved contacts that are under evolutionary selection between the N- and C-lobes on both the cytoplasmic and periplasmic sides of MurJ, suggesting that, as hypothesized for MOP exporters, the MurJ cavity attains both inward- and outward- open states. A thin “periplasmic gate” containing the conserved D39-S263 interaction was predicted to stabilize the inward-open state, while several interactions on the cytoplasmic side were predicted to stabilize a “cytoplasmic gate” in the outward-open state. Collectively, these data led the authors to suggest that MurJ functions using a rocker-switch alternating-access mode of transport in which MurJ switches conformations that result in the cavity opening to one side of the membrane or the other to load or unload Lipid II. In a seminal 2019 study, Kuk et al. remarkably solved structures of MurJTa in inward-closed, inward-occluded, and the long-sought-after outward-open conformation, providing further support for an alternating-access mechanism of flipping and the importance of these periplasmic and cytoplasmic gates (Figure 7a).98 The structural snapshots obtained by Kuk et al. are discussed in section 3.3.3 together with the model for the mechanism of transport.
Figure 7. Proposed flipping mechanism and membrane potential-driven resetting of MurJ.
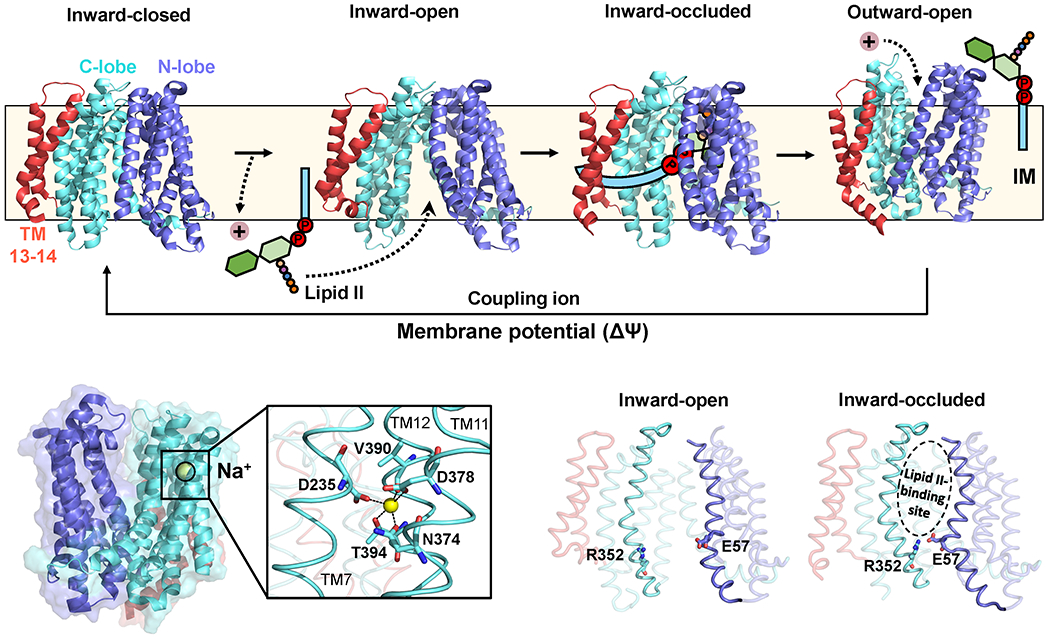
(a) Model showing proposed steps in the transport cycle. Structures were obtained from the PDB (inward-closed: 6NC6; inward-open: 6NC7; inward-occluded: 6NC8; outward-open: 6NC9). (b) A sodium ion (yellow sphere) is found in the outward-open structure, coordinated by TMs 7, 11 and 12. The coordinating residues are shown in the close-up. (c) The E57-R352 ion pair forms the thin gate. The Lipid II-binding pocket is indicated on the inward-occluded structure.
3.3. Mechanism of Lipid II Translocation by MurJ
To formulate a model for the mechanism of function of a transporter, at a minimum, one must consider the following fundamental issues: how the transporter interacts with its substrate; the type of conformations the transporter undergoes to complete each transport cycle; and, the source of energy driving transport. Addressing these questions experimentally is difficult because of how inherently dynamic and fast transport is in cells. It requires combining complementary biochemical, structural, and genetic approaches that can probe transport in vivo and in vitro, and analyze structural and substrate-transporter intermediates. In this section, we highlight the work that has advanced our understanding of how MurJ may interact with Lipid II and undergo conformational changes to translocate its substrate across the membrane, as well as questions that need to be addressed in future work.
3.3.1. Lipid II-MurJ Interactions
As described above, structural and in vivo structure-function analyses proposed that the central cavity mainly formed by TM1, 2, 7 and 8 interacts with the hydrophilic portion of Lipid II and undergoes conformational changes via an alternating-access mechanism of function to translocate Lipid II. Although we still lack experimental evidence demonstrating direct contacts between Lipid II and the central cavity of MurJ, recent studies have shown that MurJ binds Lipid II in vitro and in living cells.103–106
The first direct evidence that Lipid II and MurJ interact in cells was recently demonstrated by Rubino and Mollo et al. using site-specific cross-linking.103 In this study, we adapted a strategy previously developed to capture and detect transport intermediates of LPS (substrate) as they travel through the Lpt system (transporter).107,108 The strategy relies on genetically introducing single substitutions in the transporter so that a native residue is replaced with an unnatural amino acid that can be cross-linked to the substrate upon exposure to UV light if they are within ~2Å of each other.109 In order to detect Lipid II-MurJ cross-linked species in protein extracts from cells exposed to UV light, we developed an immunoblot-like method. In this method, the 5th amino acid on the stem peptide of Lipid II (d-Ala) is enzymatically exchanged with d-Lys-biotin in the MurJ:Lipid II cross-linked product; biotin can then bind to streptavidin conjugated with horse-radish peroxidase, allowing detection of the modified crosslinked product by chemiluminescence.110 Using this technique, we detected on-pathway intermediates of MurJ:Lipid II in living cells, providing evidence that MurJ and Lipid II molecules interact and that MurJ is directly responsible for Lipid II transport.103 The position on MurJEc that was crosslinked to Lipid II (residue F22 in TM1, Figure 6c) is localized at the lateral gate between TM1 and 8 that opens into the central cavity. Interestingly, the observed crosslinking of Lipid II was not significantly reduced in MurJ variants with substitutions in the conserved arginine residues located in the MurJ cavity (residues R18, R24, R270 in MurJEc) that are essential to support growth.94,95,103 As a result, we proposed that binding of Lipid II (likely via its pyrophosphate) to these essential arginines is subsequent to the initial association event, and leads to a conformational change in MurJ that is needed to proceed through the Lipid II transport cycle.71,94,95,98,103 This in vivo crosslinking technique should be further applied to reveal additional sites on MurJ that interact with Lipid II so that the translocation pathway can be elucidated.
The first studies to probe substrate specificity and quantify Lipid II binding to MurJ were carried out by Bolla et al. using purified, detergent-solubilized MurJ and native mass spectrometry (MS). The authors observed 1:1 high-affinity binding (with a dissociation constant Kd ~3 μM) of Lipid II to MurJ.106 This binding could be abrogated in the previously mentioned MurJA29C variant (see sections 3.1.1 and 3.2) upon modification with MTSES, which presumably prevents access to the inward-open state(s).62,71 To further investigate the binding mechanism, the authors used two antibiotics that specifically bind to different regions of Lipid II. They found that ramoplanin, which binds to the MurNAc and pyrophosphate moieties of Lipid II, dissociates Lipid II from MurJ, whereas vancomycin, which forms hydrogen bonds with the terminal mDAP/Lys-d-Ala-d-Ala segment of the pentapeptide, still binds to form a stable ternary complex with MurJ:Lipid II.31,111 Given these results and their inability to detect binding of MurJ to Und-P, Bolla et al. proposed that the pentapeptide stem and the undecaprenyl chain are not critical for binding to MurJ, whereas the pyrophosphate, MurNAc and GlcNAc moieties of Lipid II are essential for recognition.106 However, we note that their study did not specifically probe interactions of the GlcNAc component of Lipid II, so this putative interaction remains to be demonstrated. This is an important question to address because requiring binding to both MurNAc and GlcNAc would allow MurJ to differentiate between Lipid II and its precursor Lipid I. It is also possible that the first two amino acids of the stem peptide (l-Ala and d-Glu) may be important in the recognition of Lipid II by MurJ since, in S. aureus, Lipid II molecules that only contain the first two or three amino acids (owing to the absence or inhibition of specific Mur ligases) are translocated and incorporated into the bacterial peptidoglycan.112,113 Thus, the binding studies could not rule out contributions that the undecaprenyl chain, pentapeptide chain, and GlcNAc moieties might make to the transport cycle. They also did not reveal the region(s) or conformation(s) of MurJ mediating the reported binding between Lipid II and MurJ. In a follow-up study, however, Bolla et al. used a MS-based method to differentiate non-specific from specific lipid binding to MurJ to show that Lipid II, when bound to MurJ, does not readily exchange with detergent, presumably because it is sequestered inside the cavity.105
There are many questions still unanswered about the binding and specific recognition of Lipid II by MurJ. For example, how does MurJ differentiate between Lipid I and Lipid II? Can MurJ flip other substrates? To our knowledge, there is no evidence that MurJ can translocate substrates other than Lipid II. However, Bolla et al. reported that purified MurJ associates with the phospholipid cardiolipin to interfere with Lipid II binding, leading them to propose a regulatory role for cardiolipin in MurJ flippase activity.106 The physiological relevance of this interaction remains unknown. Requirements for substrate specificity are also not well understood in the related Wzx flippases of Und-PP-oligosaccharides of the MOP exporter superfamily. To address this question, Sham et al. performed a genetic selection that would allow for a Wzx flippase to substitute for MurJ in E. coli. Specifically, the authors focused on WzxC, the flippase of the capsule precursor Und-PP-colanic acid and found some WzxC variants that could translocate both their native substrate and Lipid II. Based on the positioning of the substitutions in a structural model of WzxC, they hypothesized that the alterations destabilize the inward-open conformation of the transporter.114 Accordingly, Sham et al. suggested that when a MOP transporter recognizes and loads its specific substrate in the inward-open state, it is destabilized, causing transition to the outward-open conformation and, therefore, substrate translocation. Studies like this one by Sham et al. are important not only for advancing our understanding of the nature of substrate specificity of MOP exporters but also to better understand how cross-specificity among MOP exporters could lead to resistance to inhibitors of MurJ.
3.3.2. Energetics Driving Transport in MurJ
MOP exporters are thought to share a common V-shaped structure that facilitates transport through an alternating-access mechanism. Accordingly, the directional transport of their substrates must include the transition of the inward-open substrate-bound structure to the outward-open state, the release of the substrate, and the subsequent resetting of the substrate-free transporter to the inward-open state. It is critical to understand the energy source driving these steps.
The MATE transporter family is proving to be a valuable case study when investigating MurJ. MATE exporters are secondary active antiporters in which the uphill export of amphipathic drugs is coupled to the import of an ion. Through this coupling, the cell uses the electrochemical gradient of the counter ion across the membrane (i.e. higher concentration of the ion in the periplasm than the cytoplasm) to power drug export. Several MATE transporters have been structurally and biochemically characterized in the past decade (>20 structures reported thus far; and their ion-coupling mechanisms are diverse).115–124 NorM from Vibrio cholerae (known as NorM-VC) was the first MATE transporter to be crystallized and is one of the best characterized MOP transporters.123 Like its homolog from Neisseria gonorrhoeae (NorM-NG), NorM-VC is a Na+-coupled transporter. Free energy from the binding of the substrate (a drug) to the inward-open conformation is thought to promote the transition to the outward-open conformation through an occluded state intermediate.124
A different mechanism of extrusion was proposed for PfMATE from Pyrococcus furiosus, a H+-coupled transporter. Two structures have been reported at two different pHs: a structure showing a bent TM1 helix at an acidic pH, and a structure with a straight TM1 helix at a basic pH.116 The protonation state of residue D41 was proposed to be responsible for this structural difference. Because the substrate-binding site does not overlap with this acidic site, it was proposed that protonation of the critical aspartate (D41) by extracellular protons causes TM1 bending, the collapse of the N-lobe cavity and concomitant extrusion of the substrate into the periplasm, and eventually the return to the inward-open state. However, Ficici et al. recently revisited available crystal structures of the PfMATE exporter and identified a putative Na+-binding site in the N-lobe of PfMATE that appears to be highly conserved across bacterial MATE exporters.125 They concluded that the Na+ ion had been previously wrongly assigned as a water molecule (H2O and Na+ have similar electron densities at low resolution). This novel site would imply that substrate release can also be induced by competition with Na+. The authors argued that the Na+-binding site is also weakly specific against H+, explaining why MATE exporters containing this motif, including PfMATE, have been thought to be H+-driven. These results are provocative as they suggest that MATE transporters might be much more accommodating with their counterion coupling than previously appreciated and that different experimental conditions may favor one ion over the other. Furthermore, Ficici et al. also proposed that some transporters might still contain additional H+-binding sites elsewhere in the protein.125 This proposal might explain why NorM-VC had been previously reported to simultaneously couple transport to both Na+ and H+ ions.126 These studies offer guidance when studying MurJ, but also illustrate the complexity of studying the energetics and mechanistic details of these transporters.
Not having the ability to study MurJ’s activity in an in vitro reconstitution has greatly limited our ability to study the energy source powering this transporter. Nevertheless, guided by the knowledge learned from MATE transporters, we, together with collaborators, recently showed that MurJ couples the membrane potential across the bacterial cytoplasmic membrane to Lipid II export in Rubino et al.127 The proton motive force or PMF, has two components: the membrane potential or Δψ (negative inside the cytoplasm relative to the periplasm), which is a measure of charge differences across the membrane; and the pH difference or ΔpH (alkaline inside the cytoplasm relative to the periplasm), which measures the proton concentration difference across the membrane.127,128 When the membrane potential (ΔΨ) was dissipated in live E. coli cells with the protonophore TCS (3,3′,4′,5-tetrachlorosalicylanilide), Lipid II pools increased by over an order of magnitude presumably because their translocation was inhibited. Treatment with valinomycin (a K+ ionophore that disrupts ΔΨ but not ΔpH) also led to Lipid II accumulation, whereas nigericin (an H+/K+ antiporter that disrupts ΔpH but not ΔΨ) failed to do so. These results led us to propose that the energy source of Lipid II transport by MurJ in E. coli is likely an ion other than H+.
Insights into the identity of a possible counterion used by MurJ came from the structural studies described in Section 3.2. The first inward-open structure of T. africanus MurJ (MurJTA) reported by Kuk et al. revealed a chloride ion bound to the proximal site near the essential arginines R24 and R255 (R24 and R270 in MurJEC).71 Because the protein was crystallized under high concentrations of Cl−, the authors suggested that the ion might have been non-specifically bound. However, they later found that omission of Cl− from the purification and crystallization buffers led to solving structures of MurJ in a range of different conformations.98 We think that the fact that Cl− restricts the available conformational landscape accessible to the protein by locking it in an inward-open conformation could argue in favor of a role of Cl− in the transport cycle of MurJ. Specifically, Cl− ions could be imported into the cytoplasm, driving the switch from outward-open to inward-open conformation. Indirect support in favor of such a hypothesis comes from Zakrzewska et al., who observed spontaneous binding of a Cl− ion to the inward-open structure of PfMATE by molecular dynamics simulations.119 Notably, this is the only inward-open structure obtained for a MATE exporter, and structural alignments revealed that the Cl− ion in MurJ is bound to an analogous location on PfMATE. These findings suggest that Cl− transport might be a conserved feature of MurJ and at least some MATE exporters.
The 2019 structural study by Kuk et al. discussed in the next section demonstrated the presence of a possible Na+-binding site in the C-lobe of MurJTA.71,98 The Na+ ion is kept in place by a trigonal bipyramidal coordination sphere formed by the side chains of residues D235, N374, D378, and T394, as well as the backbone carbonyl of V390 (Figure 7b). The placement of residue D235 at the hinge region of TM7 suggests that Na+ binding might induce the conformational changes in TM7 that were observed in the inward-to-outward transition. However, the relevance of this binding site in transport is yet to be demonstrated and difficult to assess since disrupting it leads to the loss of protein expression or degradation.
Lastly, we should note that the previously described study by Bolla et al. reported that Lipid II binding to purified MurJ measured by native MS is not sensitive to Na+ concentration but is affected by changes in H+ concentration with optimal binding at pH 7.0, consistent with the pH in the cytoplasm of E. coli (pH 7.2-7.8) where initial Lipid II binding to MurJ would take place.106 However, it is important to clarify that this assay does not assess the driving force for Lipid II transport by MurJ. Together, these studies and the sometimes seemingly conflicting conclusions we have discussed here highlight the need of developing an in vitro reconstitution system for MurJ that can be used to experimentally determine which, if any, of these ions—singly or in combination—drive specific conformational changes in MurJ during the transport cycle.
3.3.3. Alternating-Access Mechanism of MurJ
As discussed, the alternating-access model of Lipid II transport by MurJ was proposed early on once the structural similarities between MurJ and MATE transporters became evident.71,94,95,98,99,114 However, only recently have the biochemical and structural studies discussed in this section provided direct support of this model.
To probe the conformational changes of MurJ in growing E. coli cells, we developed a method using structure-guided cysteine cross-linking and proteolysis-coupled gel analysis.129,130 This method required introducing a cysteine residue into either the periplasmic or the cytoplasmic side of each of the lobes (N-lobe and C-lobe) of MurJEC. Structural studies predicted that, in each pair, residues would be near each other only in one of the predicted inward-open or outward-open conformations. Homo-bifunctional cross-linkers of various lengths were then added to cells to test if the specific cysteine pairs could be crosslinked. To clearly differentiate cross-linked vs. uncross-linked proteins via immunoblotting, a functional variant of MurJ containing a thrombin protease site in the loop connecting the N and C lobes was engineered. In this modified variant, the N and C lobes can be separated by gel electrophoresis after thrombin cleavage in uncross-linked MurJ but remain together in crosslinked species. Using this method, we demonstrated that, in vivo, MurJ adopts both inward- and outward-open conformations. This study also showed that MurJ relaxes into an inactive, outward-open state in cells treated with ionophores that collapse the membrane potential.129,130
Based on these results and those of others, we proposed the following model for Lipid II transport by MurJ (Figure 7a).129 Newly synthesized Lipid II localized in the cytoplasmic leaflet of the membrane binds to MurJ in the inward-open conformation. The undecaprenyl tail of Lipid II likely associates with the hydrophobic groove formed by TM13-14 while the pyrophosphate-disaccharide-pentapeptide moiety interacts with MurJ’s hydrophilic cavity.71,98,99 After engaging with Lipid II, the cavity undergoes the transition from inward-open to outward-open, similar to the proposed mechanism for some members of the MOP exporter superfamily.96,114 During this conformational change, the C55 lipid tail of Lipid II either remains associated with the hydrophobic groove (or other parts of MurJ) or is free in the hydrophobic core of the membrane, where its flexibility and ability to affect its surrounding lipid environment might aid in substrate translocation.21,22 Either way, the lateral gate between TM1 and TM8 that opens into the cavity would allow the unhindered flipping of Lipid II.71,95,98,99 Once in the outward-open conformation, MurJ releases Lipid II so that it can be used to build new PG cell wall. Based on both its dependence on membrane potential (∆ψ) and similarity with MATE exporters, MurJ is likely to bind to a cation in the outward-open conformation to release Lipid II and/or undergo the transition from the outward-open to the inward-open state during the Lipid II transport cycle.96 However, as discussed in section 3.3.2, alternative models such as voltage-driven or anion-coupled exchanger mechanisms cannot be ruled out.129
A major contribution to our understanding of MurJ and support in favor of the alternating-access model was the 2019 study by Kuk et al. reporting a series of structural snapshots of the MurJ conformational cycle.98 Since the inward-open conformation of MurJTA appeared to be stabilized by a Cl− ion (section 3.3.2), the authors strategically omitted the ion in purification and crystallization conditions, which successfully resulted in structures in the inward-closed, inward-open, inward-occluded and outward-facing conformations.71,98 As we describe below, these structures not only support the alternating-access mechanism but provide insight into molecular details driving the conformational cycle.
The different structural conformations obtained by Kuk et al. highlight the importance of H-bond interactions between N- and C-lobe residues in the periplasmic gate region in stabilizing the inward-facing state that allows loading of Lipid II.98 These structures also show that the size of the membrane portal that is proposed to serve as the entry gate for the hydrophilic portion of Lipid II into the central cavity of MurJ is regulated by rearrangements of TM 1 and TM8 and the cytoplasmic loop between TM4 and TM5. Examination of the inward-occluded conformation (Figure 7a), which presumably corresponds to a substrate-bound state that precedes the outward-open state, reveals a rigid-body rotation of the C-lobe towards the N-lobe by ~15° relative to the preceding inward-facing state; in addition, inward bending of TM2 towards the central cavity places residue E57 (located N-lobe) in proximity to residue R352 in TM10 (C-lobe), forming a thin gate that was proposed to sequester Lipid II inside the cavity prior to transition to the outward-open state (Figure 7c). The structures also revealed that the cavity in the outward-open structure is much shallower and narrower than in the inward-open structure. This difference in cavity size is similar to the one observed in MATE transporters and thought to be responsible for ejecting the substrate into the periplasmic space.115–118,131 The inward-to-outward open transition itself appears to be driven by the straightening of TM7 (the only helix bridging the N- and C-lobes) coupled to the lowering of the cytoplasmic loop connecting TM6 and TM7. TM1 also straightens en route to the outward-facing state, closing the lateral gate into the cavity.
An interesting result described in this study was the analysis of electron densities in the various structures along with tentative assignment of a Na+-binding site in the C-lobe (Figure 7b). The Na+ site in MurJ is analogous to the one in PfMATE but located in the C-lobe instead of the N-lobe as reported for MATE exporters. Furthermore, Kuk et al. postulated that Na+ binding to residue D235 is coupled to the aforementioned conformational changes in TM7. However, as described earlier, the significance of this putative Na+-binding site for MurJ function or structure could not be determined because mutations altering most of the coordination sphere residues led to significant or complete loss of expression, suggesting that the Na+-binding site might serve a structural role. Unfortunately, this problem prevents ruling out whether this Na+-binding site also has a functional role.
Lastly, by comparing the different crystal structures to one another, Kuk et al. identified R24-D25-R255 (residues R24-D25-R270 in MurJEC) as the putative substrate-binding triad responsible for coordinating the pyrophosphate group on Lipid II. 98 The triad is in close proximity to unmodeled electron density which cannot be reliably assigned but might originate from Lipid II that was added into the LCP for crystallization. Higher occupancy will be required in future structures to reliably place Lipid II inside the cavity. In addition, it would be very valuable to obtain equivalent structures of the transport cycle of the E. coli MurJ protein since we have already accumulated extensive structure-function data that could help interpret and validate such structures.94,95,99
3.4. Regulation of Lipid II Flippases
PG biosynthesis must be tightly regulated in time and space to balance cell growth with division and generate the appropriate cell shape and size.92 PG biosynthesis must also be coordinated with the growth of other envelope components such as the cytoplasmic and outer membranes in Gram-negative bacteria, but we know very little about this envelope-scale level of regulation.92,132 Ultimately, PG regulation must integrate both internal (i.e. metabolic) and extracellular cues. Given the complexity of PG biogenesis, there are multiple steps that could be subject to regulation, including Lipid II translocation across the membrane. There are three main mechanisms so far described that cells use to regulate Lipid II transport: regulating the production, the activity, and localization of Lipid II transporters. Although MurJEC is the best characterized Lipid II flippase in vivo, most of what we know about the regulation of Lipid II flippases comes from other orthologs and even Amj.
3.4.1. Regulation of Flippase Production
As described in Section 3.1.2, B. subtilis encodes Amj, a membrane protein not homologous to MurJ that can transport Lipid II and substitute for MurJBS protein (YtgP).81 One obvious question that this discovery raised is whether Amj is a ‘true’ Lipid II flippase or, rather, a promiscuous flippase that can translocate Lipid II in addition to its native (unknown) substrate. We favor the former model based on a significant finding reported by Meeske et al.81 The authors showed that amj is expressed under the control of the envelope stress-response sigma factor σM, which can be activated by antibiotics targeting PG biogenesis, providing a mechanism for regulating Lipid II flipping possibly in response to environmental cues.133,134 Meeske et al. suggested that the soil-dwelling B. subtilis bacterium might be exposed to MurJBS inhibitors produced by other microorganisms in the soil, and that such inhibition of MurJBS would activate the σM-dependent production of Amj, a Lipid II flippase that would likely be resistant to the inhibitor given its lack of homology to MurJBS.
Transcriptional regulation connecting cell wall stress to MurJ production was also recently discovered in Vibrio cholerae.135 In this bacterium, which lacks Amj, the two-component signal transduction system VxrAB increases murJ transcription upon β-lactam-induced cell wall stress.135,136 Whether upregulation of the expression of Lipid II flippases like the examples discussed here is widespread among bacteria is unknown.
3.4.2. Regulation of Flippase Activity
The first indication that MurJ could be subject to any type of regulation came from Mycobacterium tuberculosis. In this bacterium, MurJMtb is unique in that its conserved TM1-14 MOP superfamily domain is connected to three additional components: a pseudo-kinase domain, a transmembrane helix, and an extracellular domain found in some carbohydrate-binding proteins.137 While the conserved MOP superfamily domain is essential for viability, these additional C-terminal domains are not. The extracellular domain remains uncharacterized, but Gee et al. demonstrated that T947 in the pseudo-kinase domain of MurJMtb is phosphorylated by the essential Ser-Thr kinase PknB, which senses and becomes active in the presence of excess uncross-linked PG.137 Upon phosphorylation, MurJMtb binds to a forkhead-associated domain-containing protein called FhaA, which they proposed downregulates the activity of MurJMtb.137 However, this regulation was suggested from indirect evidence showing that overproduction of PknB inhibits PG synthesis and is lethal in M. tuberculosis, and that depletion of FhaA, while not lethal, leads to morphological effects. Yet, it is puzzling that the authors also showed that depletion of FhaA does not alter the levels of cytoplasmic nucleotide PG precursors, which have been shown to increase upon loss of MurJ function.60,62,127,137,138
In addition, it was recently shown that PknB also phosphorylates a protein called CwlM, which is essential for PG biogenesis139 and can associate with either FhaA or MurJMtb depending on its phosphorylation state. When phosphorylated, CwlM associates with FhaA; when unphosphorylated, CwlM binds to MurJMtb.139 These physical interactions have been proposed to lead to the downregulation or activation of MurJ activity, respectively. Thus, according to the model proposed by Turapov et al., the kinase activity of PknB affects MurJMtb activity in two manners: it removes the MurJMtb activator CwlM and stimulates association of FhaA with MurJMtb to downregulate Lipid II transport.139 Although this is an attractive model, and the physical interactions of CwlM and FhaA with MurJMtb and their regulation through PknB have been experimentally determined, there is no experimental evidence demonstrating that either CwlM or FhaA affect the activity of MurJMtb. Moreover, it appears as if FhaA also affects other steps of PG biosynthesis.140 It is therefore important that efforts are focused on testing the functional connection between MurJMtb and these factors, as well as whether this proposed regulation could also affect the localization of MurJMtb described in section 3.4.3. It is also yet to be determined if analogous systems regulating the activity of Lipid II flippases (MurJ or Amj) exist in other bacteria.
3.4.3. Regulating the Localization of Lipid II Translocation
Given that the PG cell wall is a rigid three-dimensional structure, it is critical that insertion of new PG material occurs at the right location. This is accomplished by directing PG biosynthetic complexes to specific sites in the cell, which vary according to cell shape and the state of the cell cycle.92 In rod-shaped bacteria like E. coli and B. subtilis, there are two modes of PG synthesis with respect to the cell cycle: elongation and septal PG synthesis. During cell elongation, PG-building blocks are added into the preexisting PG structure to accommodate the increase in cell size along the long axis of the cell by a protein complex called the elongasome (Figure 8).141 On the other hand, during cell division, the mother cell builds a septum containing a PG wall that will separate both daughter cells. The septum is built from entirely new material by means of a multiprotein complex termed the divisome (Figure 8).142 The cell division and elongation machineries contain multiple proteins and are characteristic of rod-shaped bacteria and functionally independent (Figure 8 and Section 4).143 In contrast, spherical cells (i.e. cocci) such as S. aureus rely primarily on the divisome and lack most components of the elongasome.144
Figure 8. Key components for PG biogenesis in the elongasome and mature divisome of E. coli.
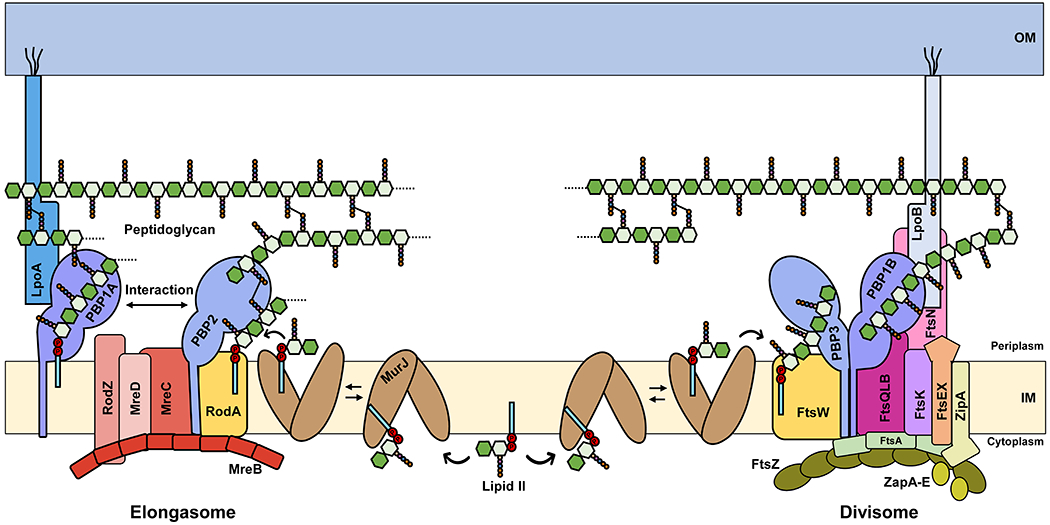
Lipid II is synthesized in the inner leaflet of the cytoplasmic or inner membrane (IM) and colocalizes with both the elongasome (left) and divisome (right) complexes. The exact molecular architecture of the elongasome and divisome, as well as how they insert new material into the pre-existing PG layer, remain unknown. LpoA and LpoB are lipoproteins that are anchored to the outer membrane (OM). For simplicity, PG hydrolases that are known to associate with each complex and to be important for their correct activity are not shown.
Liu et al. conducted the first localization studies of MurJ in E. coli.145 Using fluorescent protein fusions, they showed that MurJ localizes in the lateral membrane and is recruited to the septum with divisome proteins alongside MraY and MurG, the enzymes that synthesize Lipid I and Lipid II, respectively. The authors also found that MurJ septal localization is dependent on the synthesis of Lipid II and the presence of a mature and active divisome capable of synthesizing septal PG. However, MurJ flippase activity is not required for its septal localization. These results led the authors to conclude that the localization of MurJ to the divisome is mediated by affinity towards its substrate, Lipid II, rather than protein-protein interactions.145 Whether a similar type of recruitment occurs at the elongasome is yet to be determined.
Monteiro and Pereira et al. examined the localization of PG biogenesis proteins in coccoid S. aureus using fluorescent protein fusions and found that septal PG synthesis requires the recruitment of MurJ to the septum by some divisomal proteins.146 Interestingly, the authors found that MurJ recruitment coincides with a turning point in cytokinesis. Prior to MurJ recruitment, early cytokinesis is characterized by slow membrane invagination driven by treadmilling of the tubulin homolog FtsZ, which recruits divisomal proteins (Figure 8); once MurJ arrives at the septum, cytokinesis becomes faster, independent of FtsZ treadmilling, and driven by PG synthesis activity. Monteiro and Pereira et al. proposed that MurJ directs PG synthesis to the septum because through its own septal localization, it exclusively localizes flipped Lipid II to the septum, which in turn recruits PG synthases.146
While many rod-shaped bacteria such as E. coli and B. subtilis elongate by dispersing insertion of newly synthesized PG along the cell cylinder through the action of the elongasome, others such as members of the Alphaproteobacteria and Actinobacteria grow using a polar mode of growth by inserting new PG material at their poles.147–149 Among polar-growing bacteria, the localization of MurJ has been characterized in Mycobacteria. In M. smegmatis, Morita et al. showed that the cytoplasmic (or plasma) membrane is organized into distinct domains that can be separated using sucrose density gradient fractionation of cellular lysates.150 These domains include the intracellular membrane domain (IMD), which is not associated with the PG cell wall, and the conventional plasma membrane (PM-CW), which is tightly associated with the cell wall.150 Lipidomics and proteomics analyses revealed that the lipid and protein content of the IMD and PM-CW are different.150,151 Taking advantage of this difference in protein content, Hayashi et al. modified proteins specific to each domain with fluorescent tags and visualized them using live-imaging microscopy to find that the IMD forms distinct patches in the polar region where cell elongation occurs.151 In a following-up study, Garcia-Heredia et al. demonstrated that the Lipid II synthase MurG localizes to the IMD where it makes the PG precursor; Lipid II then moves to the PM-CW where MurJ and penicillin-binding proteins (PbpA/PbpB) reside to flip the precursor and use it to build new PG wall, respectively.138 The authors proposed that this functional partitioning of the membrane and PG biogenesis factors leads to directionally organized and more efficient PG synthesis in rod-shaped bacteria.138,152 Interestingly, Garcia-Heredia et al. also found that the protein DivIVA is involved in a feedback mechanism between PG wall synthesis in the PM-CW and the partitioning of the membrane into domains.138
Although the localization of MurJ is just beginning to be characterized, a common theme is emerging that highlights a role for the localized delivery of flipped Lipid II by MurJ in driving PG cell wall synthesis at specific cellular locations.
3.5. Inhibitors of MurJ
Since the discovery of penicillin, targeting PG biogenesis has proven to be an excellent strategy in the development of antibiotics. Being essential for PG biogenesis in important pathogens and absent in humans, MurJ represents a potentially ideal target for much-desired novel antibiotics.60,61,72,138,153 A few different types of MurJ inhibitors have been reported to date but none of them has entered clinical development.153–156 These inhibitors fall into two major classes according to their source: small molecules derived from large synthetic chemical libraries and naturally occurring inhibitors.
The first evidence that MurJ could be a novel antibiotic target came before MurJ was proposed to be a Lipid II flippase when scientists at Pfizer discovered that overexpression of and mutations in the S. aureus SA1575 gene conferred resistance to a novel type of anthranilic acid that inhibits PG synthesis.157 Mott et al. proposed that the compounds were inhibiting SA1575, a membrane protein of unknown function at the time, which we now know is MurJSA.157 Shortly thereafter, Huber et al. at Merck & Co. identified two novel MurJ inhibitors in a screen of a synthetic chemical library for compounds that restored β-lactam activity against methicillin-resistant S. aureus (MRSA).153 The resulting two structurally-related indole compounds, 3-1-methyl-2-pyridin-4-yl-1H-indole (DMPI) and 2-(2-Chlorophenyl)-3-[1-(2,3-dimethylbenzyl)piperidin-4-yl]-5-fluoro-1H-indole (CDFI), were shown to target MurJSA based on independent genetic approaches to identify genes with altered sensitivity.153 Unfortunately, these compounds are not effective in Gram-negative bacteria, likely because of their inability to penetrate the outer membrane or their susceptibility to efflux pumps; however, they were instrumental in studies by Monteiro and Pereira et al. described in section 3.4.3 on the localization of MurJSA and PG synthases in S. aureus.146,153
Recently, while investigating the mechanism of action of the lysis protein LysM of the E. coli levivirus M, Chamakura et al. discovered the first known natural inhibitor of MurJ. Phage M is a small RNA virus that induces lysis of its host after replication.154 Chamakura et al. demonstrated that LysM, a 37-residue hydrophobic protein, causes host lysis of E. coli cells by inhibiting the translocation Lipid II across the cytoplasmic membrane presumably by targeting MurJ.154,158 Although its mode of inhibition is yet to be determined, it is interesting that single-residue substitutions in TM2 and TM7 of MurJ confer resistance to LysM; recall that the transition in MurJ’s cavity from inward-open to outward-open has been proposed to be driven by the straightening of TM7. Whether LysM binds to the TM7 region to interfere with the transport cycle awaits future structural analysis of the MurJ:LysM complex.
In an independent study, Chu et al. discovered that humimycin antibiotics target MurJ.156 The authors conducted a bioinformatics search for natural products by identifying putative metabolite gene clusters found in the human bacterial microbiome. They then chemically synthesized the predicted products and tested for potential antimicrobial activity.156 Applying this approach, Chu et al. discovered the humimycin antibiotics, which are naturally produced by Rhodococcus species. Humimycins inhibit MurJ and potentiate β-lactam activity against some Gram-positive pathogens, including MRSA.156 In their initial characterization of humimycin, Chu et al. showed that most humimycin-resistant mutations they obtained change residues located in the cytoplasmic side of MurJ’s N-lobe.155,156 Interestingly, after optimizing humimycin, resistance to the more potent derivative humimycin 17s has so far not been detected.155 Although their mechanism of inhibition is currently unknown, it was proposed that humimycins might stall MurJ in an inward-open conformation by disrupting the closure of the cytoplasmic gate that is required for its transition to the outward-open conformation (Figure 7a).71,98,155
Together, these studies demonstrate that MurJ can be targeted by small molecules and even peptides. However, in all these examples, resistant MurJ variants could be readily isolated in the laboratory. Thus, whether MurJ inhibitors will be effectively developed for clinical use is yet to be determined.
4. Building the PG Cell Wall Structure: Glycosyltransferases and Transpeptidases
Once Lipid II is flipped across the cytoplasmic membrane, glycosyltransferases polymerize its GlcNAc-MurNAc-pentapeptide moiety into glycan strands that are cross-linked into the growing PG cell wall by transpeptidases (Figure 9). The activities and regulation of glycosyltransferases and transpeptidases are tightly coupled to one another to allow for a controlled expansion of the PG cell wall.92 These enzymes are targeted by many widely used antibiotics, underscoring their essential function in bacterial cells and the advantage that they offer as antibacterial agents localized outside of the cytoplasm. In this section, we will limit our discussion to recent developments on glycosyltransferases and transpeptidases that have called us to change our view of PG biogenesis. Specifically, we will describe the paradigm change involving the role of bifunctional penicillin-binding proteins (aPBPs) and SEDS/bPBP pairs, and discuss in detail recent studies of RodA/PBP2 and FtsW/PBP3, the SEDS/bPBP pairs that drive PG synthesis in the elongasome and divisome, respectively.
Figure 9. Overview of polymerization and cross-linking steps of PG biogenesis.
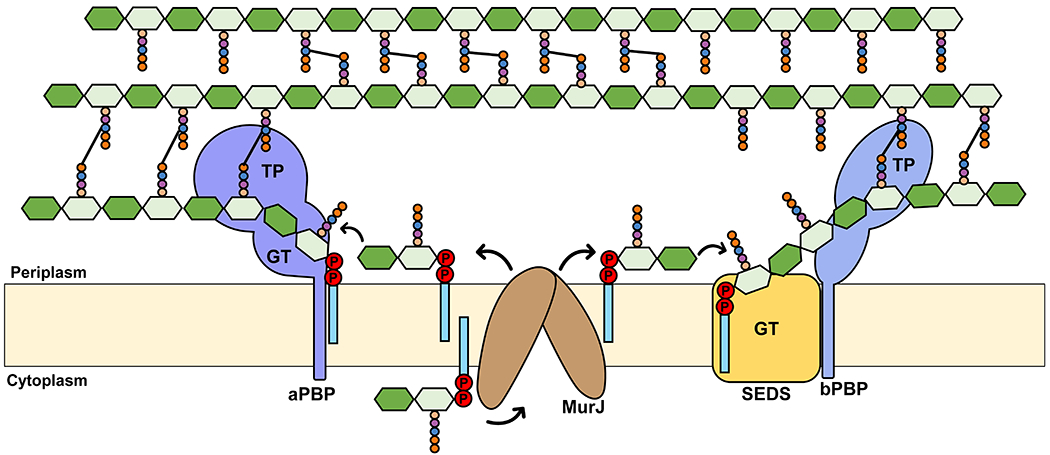
Newly synthesized Lipid II is flipped across the cytoplasmic membrane by MurJ. Glycan strand synthesis (i. e. polymerization) is catalyzed by either bifunctional aPBPs, MGTs (not shown), or SEDS proteins (RodA or FtsW) through their glycosyltransferase (GT) domain. Crosslinking of stem peptides is catalyzed by the transpeptidase (TP) domain of aPBPs and bPBPs. SEDS proteins function in conjunction with their cognate bPBP.
As described earlier, rod-shaped bacteria have two modes of PG synthesis with respect to the cell cycle: elongation catalyzed by the elongasome and septal PG synthesis catalyzed by the divisome (Figure 8). The elongasome is organized by the cytoplasmic actin-like protein MreB, which oligomerizes into filaments that localize to the lateral wall of bacteria to direct the movement of PG biosynthetic complexes.159,160 In E. coli, MreB interacts with the Rod complex, which includes the RodA-PBP2 synthase, RodZ, MreC, and MreD. RodZ physically links cytoplasmic MreB to the Rod complex.15 High-resolution microscopy has revealed that MreB organizes the insertion of new PG cell wall during elongation by localizing in discrete patches that create tracks perpendicular to the long axis of the cell that guide PG synthases.161,162 In Section 4.2, we will discuss how recent studies have shown that, as the complex moves around the cell, RodA synthesizes PG glycan strands that are crosslinked by PBP2. Incorporation of this new material into the pre-existing PG wall requires the concerted recruitment and activity of PG synthases and hydrolases. Many of the molecular details about the regulation of the localization and activity of the elongasome remains unknown.
The composition of the divisome is more complex than that of the elongasome. During cell division, at least twenty proteins are recruited in a sequential fashion to the septum to assemble the divisome (Figure 8). First, the tubulin homolog FtsZ is anchored to the cytoplasmic membrane at the location of the future septum by FtsA and ZipA. FtsZ polymerizes to build a dynamic Z-ring that undergoes treadmilling and recruits other divisome proteins by a diffusion-and-capture mechanism.163,164 The arrival of early-stage divisome proteins (e.g. Zap proteins and FtsEX) at the Z-ring forms the proto-ring, which then recruits the late-stage divisome proteins FtsK, FtsQ, FtsL, FtsB, FtsW, FtsI (also known as PBP3), PBP1b, FtsN, hydrolases, and regulatory proteins. FtsN marks the completion of the assembly of the core divisome and the start of septal PG synthesis.15,142 In Section 4.2, we will discuss recent work describing FtsW/PBP3 as the divisomal PG synthase.
4.1. High-Molecular Weight Penicillin-Binding Proteins (HMW-PBPs)
PBPs are among the most well-studied PG biogenesis factors. They first came into the spotlight because they are targets of some of the oldest and most successful antibiotics, the β-lactams, which include penicillin.165 Bacteria use varying numbers of PBPs (e.g. 13 in E. coli) for the synthesis, degradation, and modification of PG.165 Based on their size and functional domains, PBPs are conventionally divided into high- and low-molecular weight (HMW, LMW) PBPs. Unfortunately, the traditional nomenclature of PBPs (e.g. PBP2, PBP3) is confusing, as their numbers derive from their migration pattern on protein gels, which often does not correlate among different species.165 PBPs have been extensively reviewed elsewhere, and readers are directed to comprehensive reviews on structural, functional, and mechanistic details of PBPs and their inhibitors.165–168
HMW-PBPs are glycosyltransferases and/or transpeptidases that build the PG cell wall, while LMW-PBPs are primarily involved in its remodeling, recycling, and maintenance.165,166,169 The general structure of HMW-PBPs consists of an N-terminal cytoplasmic tail, a transmembrane helix, and two extracytoplasmic (or periplasmic) domains that are connected by a β-rich linker domain.165,166 Based on the structure and the enzymatic activity of their periplasmic domains, HMW-PBPs are further assigned to two classes: class A PBPs (aPBPs), which carry both glycosyltransferase and transpeptidase activities, and class B PBPs (bPBPs), which only have transpeptidase activity. The first periplasmic domain of aPBPs possesses glycosyltransferase activity and is responsible for synthesizing long glycan chains. In contrast, the equivalent domain of bPBPs lacks glycosyltransferase activity and is thought to mediate protein-protein interactions within PG biosynthetic complexes.165,170 Both aPBPs and bPBPs carry a C-terminal periplasmic domain that binds penicillin and provides the transpeptidase activity necessary to cross-link stem peptides of adjacent PG glycan chains. Although the LMW (or class C) PBPs are mainly composed of a transpeptidase domain, this domain generally acts as either a carboxypeptidase that removes the terminal residues from cross-linked stem peptides or as an endopeptidase that cleaves amide bonds between amino acids in stem peptides.166,167,171
E. coli expresses three bifunctional aPBPs (PBP1a, PBP1b, and PBP1c), belonging to the well-characterized GT51 family.165 The function of PBP1c is not entirely understood and will not be discussed further. In contrast, we know that PBP1a and PBP1b are required for the bulk of PG synthesis.91 In E. coli, both PBP1a and PBP1b have a non-catalytic domain (called ODD in PBP1A and UB2H in PBP1B) connecting their glycosyltransferase and transpeptidase domains.172,173 This linker domain functions as a binding site for LpoA and LpoB, which are outer membrane-anchored lipoproteins that activate PBP1a and PBP1b, respectively (Figure 8). Notably, Gram-positive bacteria do not appear to encode the LpoA and LpoB proteins. Binding of the lipoprotein activators to the respective ODD or UB2H domains induces conformational changes that increase both catalytic activities of the aPBPs.173–177 Early on, PBP1a and PBP1b were classified as enzymatically redundant, although, as discussed below, it was thought that PBP1a was functionally specialized to be the main PG synthase during elongation, while PBP1b was specialized in septal PG synthesis during division. In support of having redundant activities, cells lacking either PBP1a (or LpoA) or PBP1b (or LpoB) alone are viable under certain standard laboratory conditions, but cells lacking both are not.178,179 More recently, Mueller et al. demonstrated that the viability of single mutants lacking either PBP1a or PBP1b can be compromised in a pH-dependent manner as a result of pH affecting the activity of these seemingly redundant enzymes.180 It was always puzzling that, in addition to the aPBPs, E. coli possesses two monofunctional bPBPs whose respective transpeptidase activity is also essential for viability: PBP2, which is necessary for cell elongation, and PBP3, which is required for septum formation.165 Indeed, it was recognized decades ago that treatment of rod-shaped bacteria with the PBP2 inhibitor mecillinam resulted in the production of round cells, while PBP3 (FtsI) thermosensitive mutants formed filamentous cells under non-permissive conditions.181,182 Similarly, defects in RodA and FtsW were shown to alter morphology of E. coli by producing round and filamentous cells, respectively.183,184 Recent discoveries described below explain this paradox.
Traditionally, it was thought that PBP1a mediated PG synthesis in the elongasome, whereas PBP1b was responsible for PG synthesis in the divisome, but that the two enzymes could still somehow substitute for each other.185,186 This paradigm was mainly based on reported physical interactions between the aPBPs and protein members of cytoskeletal complexes (Figure 8), as well as on the synthetic lethality caused by the combined loss of PBP1a and PBP1b. However, as we describe in the next section, this paradigm in PG synthesis has been recently revised owing to the discovery that SEDS and their partner bPBPs (section 4.2) are the main PG synthases in the cell-cycle-specific cytoskeletal complexes that are organized by MreB and FtsZ to direct elongation and cell division, respectively (Figure 8).88,91 The precise role of aPBPs is now unclear, but it is important to note that despite functioning outside of the elongasome and divisome, they are still responsible for ~80% of the total PG synthesis in E. coli.91 A model we favor to explain the role of SEDS/bPBPs and aPBPs in PG biogenesis was proposed by Cho et al. and states that while SEDS/bPBPs pairs build the foundational structure of the PG cell wall, the aPBPs complete (or fill in) the remaining PG matrix during growth. Ultimately, both roles would be critical for properly building a strong PG sacculus.91 In agreement with this model, Vigouroux et al. recently showed that aPBPs do not contribute to determining cell shape but instead ensure the mechanical stability of the PG cell wall, which also led them to propose that PBP1b can sense and repair cell wall defects.187 A role for PBP1b in repairing the PG structure also agrees with findings by More et al. showing that PBP1b is required to prevent lysis of E. coli cells that have a defective outer membrane because of a compromised assembly of LPS at the cell surface.188 We remind readers that the Gram-negative envelope contains two load-bearing structures, the PG cell wall and the outer membrane, both contributing to cell integrity and prevention of osmotic rupture.189,190
It is worth mentioning here that not all bacterial species conform to the general model described in the next section in which SEDS/bPBP pairs build the foundational structure of the PG cell wall while aPBPs fill in the gaps and repair the PG structure. For example, members of the Rhizobiales, which includes the plant pathogen Agrobacterium tumefaciens, possess an FtsW-PBP3 pair used during cell division, but lack a Rod complex and, as such, must use a different cell wall synthase(s) to drive polar growth during cell elongation.191,192 Recently, Brown and co-workers used a combination of microscopy, genetic and biochemical analyses to demonstrate that in A. tumefaciens, PBP1a, a class-A PBP, is the main PG synthase that drives elongation at the poles.192 In fact, when PBP1a activity is lost in A. tumefaciens and other Rhizobiales, cells lose their rod shape and fail to grow. Similar findings on PBP1a essentiality for elongation has also been reported in Mycobacteria. This essential role of PBP1a contrasts what is known for most other rod-shaped bacteria and reminds us that bacteria can modify and reorganize their PG synthesis machinery and its regulation to accommodate different modes of synthesis and growth.192
Future work needs to focus on investigating the role of aPBPs and further testing the models presented above. We also note that many bacteria encode monofunctional murein glycosyltransferases (MGTs) whose function in PG cell wall biogenesis is unclear. The reader can refer elsewhere for a more in-depth discussion on the possible role of aPBPs in the biogenesis of PG in both Gram-negative and Gram-positive bacteria. 92,193–195
4.2. Shape, Elongation, Division, and Sporulation (SEDS) Protein Family
4.2.1. Discovery of Glycosyltransferase Activity of RodA and FtsW
For decades, aPBPs and MGTs were thought to be the only PG glycosyltransferases. However, recent landmark studies from various research groups discovered that the SEDS proteins RodA and FtsW serve as the primary glycosyltransferases of the elongasome and divisome, respectively (Figure 9).89–91,196 Early work by Ishino et al. hinted at the presence of an unidentified family of glycosyltransferases in bacteria.197 However, these findings were overlooked until McPherson and Popham showed, over a decade ago, that B. subtilis mutant cells lacking all known glycosyltransferases were viable and rod-shaped, and therefore could make PG.93 The paradigm about aPBPs being the only PG synthases was further challenged when Liechti et al. revealed that chlamydial species, which were previously thought to be wall-less, synthesize functional PG even though they lack aPBPs and MGTs, signifying that an unidentified glycosyltransferase was involved in synthesizing PG.198
Ishino et al. provided the first hint that eventually led to the identification of the proteins responsible for the missing glycosyltransferase activity 30 years later.197 They found that membranes of E. coli cells overexpressing the SEDS protein RodA and the bPBP PBP2 together, but not separately, could synthesize PG. Using PBP2-specific antibiotics as well as thermosensitive mutants of RodA and PBP2, they further showed that RodA was required for chain elongation, while the transpeptidase PBP2 was, as expected, needed for cross-linking of PG. Surprisingly, they rejected the possibility of RodA being a glycosyltransferase and instead inferred that PBP2 itself had glycosyltransferase activity that was somehow stimulated by RodA.15,197 These data were not considered for decades in models for PG biogenesis. In addition, the proposal that SEDS proteins functioned as Lipid II flippases (as discussed in section 3.1 and elsewhere) likely further diverted attention from the possibility that they might function as glycosyltransferases.59,66,85
Recent seminal studies from various labs have provided paradigm-shifting information regarding the roles of the SEDS proteins. Using homology and structural predictions, Meeske et al. identified similarities between SEDS proteins and O-antigen ligases and polymerases, which are glycosyltransferases that use Und-PP-linked-O-antigen and Und-PP-linked oligosaccharides to complete the periplasmic steps of LPS synthesis in Gram-negative bacteria.89,199 The homology to known glycosyltransferases of Und-PP-saccharides suggested that RodA/FtsW might function as PG glycosyltransferases that utilize Lipid II. Indeed, Meeske et al. demonstrated that purified B. subtilis RodA possesses glycosyltransferase activity in vitro.89 In an independent study, Emami et al. used a candidate-gene approach to find that overexpression of RodA suppresses the growth defect of B. subtilis mutants lacking all aPBPs, which was also independently observed by Meeske et al.89,196 Additionally, Cho et al. developed a novel assay to monitor PG polymerase activity in E. coli cells, which, together with localization studies of PG synthases, showed that elongasome-dependent PG synthesis still occurs when all aPBP activity is lost.91 Based on their results, Cho et al. concluded that cell wall synthesis is facilitated by two discrete polymerase systems functioning semi-autonomously: SEDS-family proteins, which work within the elongasome and divisome machines, and aPBP enzymes, which operate outside of these cytoskeletal complexes.91 More recently, Straume et al. reported that in the oval-shaped Gram-positive bacterium S. pneumoniae, aPBPs also function autonomously from the SEDS/bPBPs PG synthases present in the elongasome and divisome (RodA/PBP2b and FtsW/PBP2x, respectively), suggesting that they are not involved in primary PG synthesis.200 In addition, Reichmann and colleagues have shown that, in S. aureus, there are two SEDS/bPBP complexes (RodA-PBP3 and FtsW-PBP1) with different roles in PG synthesis and whose activities must be coordinated to maintain the characteristic coccoid shape of these bacteria. 201 The authors proposed that while the RodA-PBP3 complex mediates PG synthesis along the cell sidewall, the FtsW-PBP1 pair incorporates PG at the septum.
Taguchi et al. later provided important insight into the role of the physical interaction between SEDS proteins and their bPBP partners. They reported that divisome-associated FtsW orthologs from several organisms only have glycosyltransferase activity in complex with their cognate bPBPs.90,202 These results echoed those mentioned earlier obtained by Ishino et al. in 1986 showing that the combined overexpression of both RodA and PBP2 (bPBP) is needed for PG synthesis in E. coli membranes. Thus, the two-protein SEDS-bPBP complex represents a functionally integrated PG machine that recapitulates the bifunctionality of aPBPs.89–91,196 ( In the next section, we describe the structural characterization of RodA-PBP2 complexes that provides information on how these proteins may work as a unit, although it is notable that purified B. subtilis RodA was reported to have PG polymerization activity in vitro in the absence of its bPBP partner.89 87,88
The discovery of the glycosyltransferase activity of the SEDS proteins was a groundbreaking development in our understanding of how bacterial PG is synthesized during cell division and growth. Currently, only a few mechanistic details about SEDS activity are known, such as the requirement for divalent cations and insensitivity towards the antibiotic moenomycin, which targets aPBPs and MTGs, pointing towards a novel catalytic mechanism distinct from the aPBP-mediated PG polymerization.90,202 Elucidating the catalytic mechanism and possible regulation of these proteins will be critical towards understanding a reaction that is fundamental for bacterial growth and a potential target for novel antibiotics.
4.2.2. Structural Insights into the Function of SEDS/bPBP Complexes
In 2018, Sjodt et al. presented the first structure of a SEDS family protein, RodA from Thermus thermophilus.87 The structure was solved using LCP and refined to 2.9 Å resolution. Given the absence of prior similar structural data, the traditional mechanism of molecular replacement was not possible, so phase calculation was aided instead by evolutionary covariance (which was also used to model the outward-open MurJ structure, see section 3.2). The resulting structure revealed a unique fold consisting of 10 transmembrane helices positioned largely perpendicular to the plane of the membrane (Figure 10a). Large extracellular loops (ECLs) known to be involved in the protein’s catalytic activity were ordered in the crystal structure, except for the largest loop, ECL4, located between TM7 and TM8.87,89 The structure contained a lipid molecule (monoolein) nestled into a hydrophobic groove between TM2 and TM3, which led the authors to postulate might represent a binding site for the undecaprenyl moiety of Lipid II (Figure 10a). A large aqueous channel proximal to this groove contains a strictly conserved and essential Glu-Lys salt bridge whose function in transglycosylation remains to be determined.
Figure 10. Structure and function of the RodA-PBP2 complex.
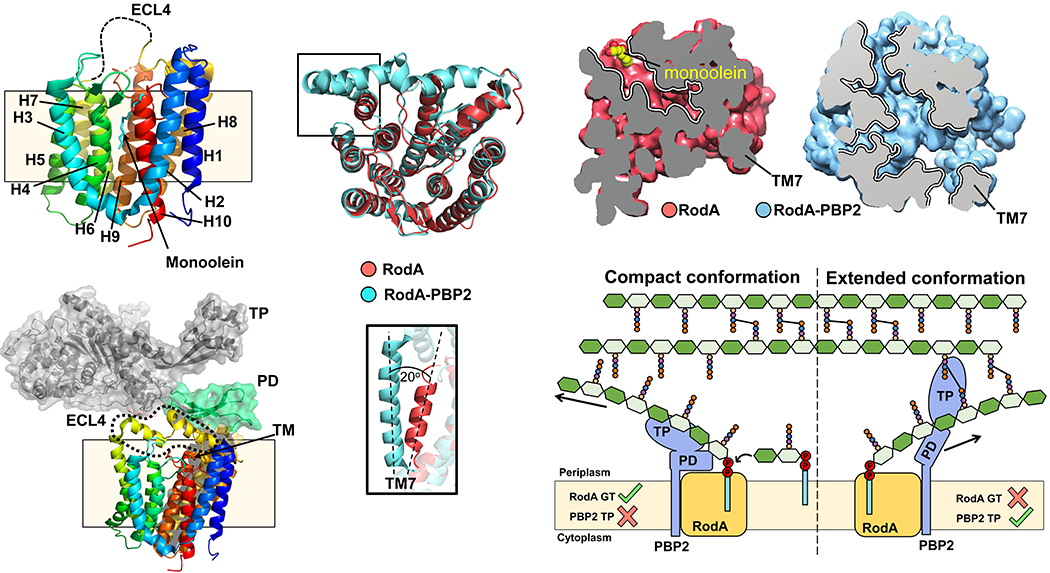
(a) Structure of RodA (top, PDB ID: 6BAR) and the RodA-PBP2 complex (bottom, PDB ID: 6PL5), both from T. thermophilus. For RodA, helix numbers are indicated (H1-10), along with the location of the monoolein molecule. ECL4, shown as a dashed loop, is not resolved. For RodA-PBP2, the resolved ECL4 is highlighted, along with the various domains of PBP2 (TM: transmembrane anchor; PD: pedestal domain; TP: transpeptidase domain). (b) Alignment of RodA with or without PBP2. The bottom inset shows tilting of TM7 by ~20° in the presence of PBP2. (c) Surface slices showing enlargement of the central cavity in RodA-PBP2. The location of TM7 is highlighted. (d) Proposed model for glycosyltransferase and transpeptidase activity of RodA-PBP2 in its ‘compact’ and ‘extended’ conformations.
Two years later, Sjodt et al. solved the structure of the same T. thermophilus RodA protein in complex with its cognate bPBP, PBP2 (Figure 10a and 10b).88 The complex observes a 1:1 stoichiometry, with the single transmembrane helix of PBP2 packed against TM8-9 of RodA. In the structure, the so-called pedestal domain of PBP2 is located directly on top of the ECLs of RodA, while the transpeptidase domain is right above the membrane ~80 Å from the core structure of RodA. Unlike in the structure obtained in the absence of PBP2, ECL4 of RodA is almost completely ordered in the complex as a result of its interaction with PBP2’s pedestal domain.
Since bPPBs induce the transglycosylase activity of their SEDS partners, Sjodt et al. tested for a possible role of the pedestal domain of PBP2 in regulating RodA’s function.87 Using an in vitro transglycosylase activity assay, they demonstrated that, indeed, the pedestal domain plays a significant role in the allosteric activation of RodA. The authors proposed a possible mechanism for this activation by comparing the structures of RodA alone and in association with PBP2. In the complex, RodA’s TM7 is tilted by as much as 20° away from the core of the protein (Figure 10b), creating a membrane-accessible cavity that is smaller and not accessible in the structure of RodA alone (Figure 10c). The authors postulated that the tilt of TM7 results from the interaction of the pedestal domain with ECL4, which connects TM7 and TM8. Because the cavity is large enough to accommodate Lipid II, the authors also proposed that the tilt of TM7 creates a putative substrate entry/exit site in RodA. The fact that residues in this cavity are highly conserved is in line with its proposed role, but additional experimental evidence is needed to support this model.
Finally, Sjodt et al. also proposed that this allosteric regulation might ultimately coordinate the transglycosylase and transpeptidase activities of this two-protein complex (Figure 10d).87 They speculated that the RodA-PBP2 complex is dynamic in cells, adopting a compact and an extended conformation. In the compact complex, the pedestal domain of PBP2 is in close contact with RodA, presumably stimulating its transglycosylase activity; however, PBP2’s catalytic domain would be too far to reach the PG structure for its transpeptidase domain to introduce cross-links. In contrast, in the extended conformation, the pedestal domain pulls away from RodA, which would prevent activation of RodA’s transglycosylase activity but allow PBP2 to reach the PG matrix and crosslink the newly synthesized glycan strand to preexisting glycan chains. This is an attractive model that needs to be thoroughly tested.
SEDS/bPBP complexes are part of the elongasome (RodA/PBP2) and the divisome (FtsW/PBP3) (Figure 8). We believe that to understand the function of these PG synthases, one must consider how their activities are regulated in vivo so that the cell can grow and divide while maintaining its characteristic shape. Protein interaction studies have led to the proposed architecture of these PG synthesis complexes, but it is poorly understood how dynamic some of the observed (and possibly yet-to-be identified) interactions are. Since the activities of components of these complexes have been often inferred by studying single proteins in isolation or as part of incomplete complexes in vitro, we caution readers to keep this in mind when revisiting the relevant literature.
4.3. Inhibitors of Glycosyltransferases and Transpeptidases
Although there is still a lot more to learn about how cells orchestrate the function of transglycosylases and transpeptidases to build their PG cell wall, these activities are clearly essential for viability of bacteria. PG biosynthesis depends primarily on the combined activities of the SEDS proteins and their cognate bPBPs, and to some extent on the action of aPBPs and MGTs. While no small-molecule inhibitors of SEDS proteins have been discovered to date, the transpeptidase activity of the PBPs is inhibited by the well-known class of antibiotics called the β-lactams. These antibiotics have been covered in much detail elsewhere,166,203 but, in short, they work by mimicking the natural d-Ala-d-Ala terminal of the stem peptide and acylating the nucleophilic serine in the active site of the transpeptidase. Structures of β-lactams are varied and encompass several sub-classes. Figure 11a provides the structure of an acylated transpeptidase along with the structure of the antibiotic subsequent to nucleophilic attack.204 The only natural product known to inhibit the active site of aPBP’s and MGT’s glycosyltransferase activity is moenomycin, produced by Streptomyces bacteria and active against a broad spectrum of Gram-positive bacteria.205,206 Used in the farming industry for decades, moenomycin acts as a mimic of the growing glycan chain to enter and compete for the donor site (Figure 11b), with very few known instances of resistance to date. 207,208 Several analogs have been designed and shown to have potent antibacterial activity, but thus far none have made it to the clinic, hindered in large part by adverse pharmacodynamics related to the necessity of a lipid tail to achieve the full antibacterial activity.209,210 That being said, the ability to carry out in vitro polymerization reactions with purified Lipid II holds great promise for the discovery of new inhibitors of the aPBPs as well as the SEDS proteins.89,90,110,202,211,212
Figure 11. Inhibitors of glycosyltransferases and transpeptidases.
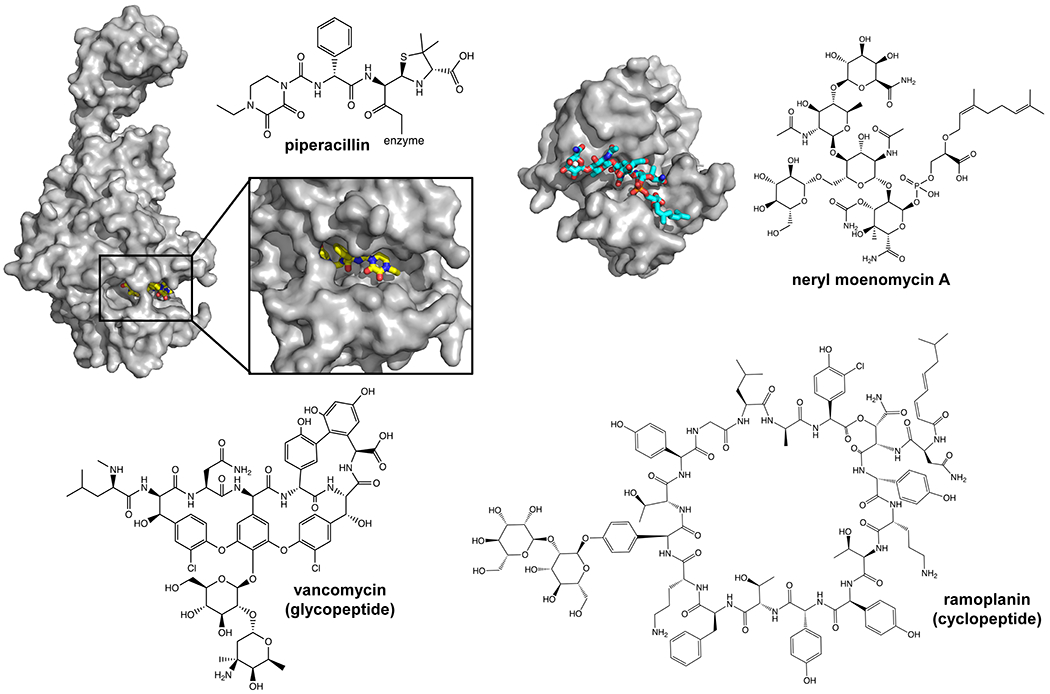
(a) Crystal structure of PBP3 from P. aeruginosa in complex with piperacillin (PDB ID: 4KQO). The chemical structure of the covalent adduct between the catalytic serine and piperacillin is shown. (b) Crystal structure of the glycosyltransferase domain from A. aeolicus and neryl moenomycin A (chemical structure shown on the right). (c) Structures of a representative glycopeptide antibiotic (vancomycin, left) and a cyclic peptide (ramoplanin, right).
Lastly, we should mention that there are several other compounds that inhibit the periplasmic steps of PG synthesis. They do not directly inhibit transglycosylases and transpeptidases, but instead prevent PG cell wall synthesis by binding to and sequestering Lipid II. These antimicrobials include the well-known glycopeptides (vancomycin, Figure 11c, left), which, as stated above, can also prevent transpeptidation reactions by binding to the d-Ala- d-Ala terminus in Lipid II, as well as the (poly)cyclic antibiotics (nisin, ramoplanin; Figure 11c, right). 213,31
5. Conclusions
In this review, we focused most of our attention on two classes of PG biogenesis factors that have been the subject of recent developments that have redrawn the textbook picture of PG biogenesis: the Lipid II flippase MurJ, and the SEDS transglycosylases.
Both in vivo and crystallographic studies have shed light on the function and structure of MurJ, leading to a model of alternating-access mechanism of flipping Lipid II. Despite these advances, fundamental details about its mechanism of function remain unclear. Among them is our lack of understanding of the energetics of transport and the interactions between Lipid II and MurJ during the transport cycle. A barrier that needs to be surpassed to address fundamental questions like these is the lack of an in vitro reconstitution assay. This technical advance would not only allow for better biochemical characterization of MurJ but also aid in the identification and characterization of small-molecule inhibitors that may be novel antibiotics. In addition to the biochemical reconstitution, we also need to expand in vivo work on this flippase to better understand how cells may regulate MurJ and coordinate its function with other PG biogenesis factors. The less-characterized Amj transporter also needs to be investigated as it will expand our understanding of how cells transport polyisoprenoid-linked sugars across membranes, an essential process in all domains of life. It is possible that its functional characterization might lead to the discovery of similar transporters that cannot be simply identified through standard homology searches.
Impressive progress has also been recently accomplished on SEDS/bPBP proteins. The ability to monitor polymerization activity of SEDS proteins in vitro has been crucial in these discoveries. Undoubtedly, future studies will focus on understanding the mechanistic details of how the activity of these proteins is coordinated with that of their partner bPBPs and the cytoskeletal proteins that direct them to build the PG wall. For this, reconstitution and structural studies should be expanded to complexes that include the main components of the elongasome and divisome machineries – a challenging goal. The discovery of SEDS/bPBP as the main PG synthases in the elongasome and divisome has called for a change in our view of PG biogenesis. We need to probe further not only how SEDS/bPBPs function in cells, but also the role of the bifunctional aPBPs in building the PG sacculus.
With antibiotic resistance set to become one of the main challenges facing humanity in the 21st century,214 the proteins we have focused on in this review represent promising novel antibiotic targets present in most bacteria. We hope that the growing knowledge about their function and experimental tools available will ultimately lead us to the discovery of new classes of antibiotics.
Acknowledgements
This work was supported by the National Institutes of Health awards R01-AI081059 and R01-AI153358 to D.K. and R01-GM100951 to N.R.
Biographies
Biographies
Sujeet Kumar completed his undergraduate in B.tech Biotechnology from Vellore Institute of Technology (VIT) in Vellore, India. He received his Ph.D in Biological Sciences in 2016 at Louisiana State University under the supervision of Prof. William Doerrler. His PhD thesis was focused on discovering the roles of members of the conserved DedA/Tvp38 membrane protein family in Escherichia coli drug resistance and alkaline pH tolerance. Presently, he is a postdoctoral scholar in the laboratory of Prof. Natividad Ruiz at the Ohio State University. His research interest is understanding the peptidoglycan biogenesis in Gram-negative bacteria.
Aurelio Mollo completed his undergraduate studies at Haverford College (Haverford, PA). He majored in chemistry with a concentration in biochemistry and worked under the supervision of Prof. Louise Charkoudian. For his thesis work, he reconstituted the activity of ComJ, a P450 enzyme which is responsible for carrying out an essential biaryl cross-linking step during the complestatin biosynthetic pathway. He then moved to Harvard University (Cambridge, MA) for his PhD studies, where he is currently working in the lab of Prof. Dan Kahne. His research is focused on studying intermediate states of transport of the Lipid II flippase MurJ.
Daniel Kahne is the Higgins Professor of Chemistry and Chemical Biology at Harvard University. His laboratory strives to develop new approaches to treat resistant bacterial infections. To do this, the Kahne Lab focuses on the protein machines that assemble the outer membrane that protects Gram-negative bacteria from toxic molecules. He was an undergraduate at Cornell University where he majored in Art History (and Chemistry). He earned his Ph.D. and did postdoctoral work in chemistry at Columbia University and was a professor at Princeton for sixteen years before moving to Harvard in 2004.
Natividad Ruiz is a Professor of Microbiology at The Ohio State University in Columbus, Ohio. Her laboratory studies the biogenesis of the cell envelope in Gram-negative bacteria, mainly focusing on the transport of envelope components across cellular compartments. She conducted her undergraduate studies at the University of Kansas and received a Ph.D. from Washington University in St. Louis.
Footnotes
The authors declare no competing financial interest.
References
- (1).Dik DA; Fisher JF; Mobashery S Cell-Wall recycling of the Gram-Negative bacteria and the nexus to antibiotic resistance. Chem. Rev 2018, 118, 5952–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Garde S; Chodisetti PK; Reddy M Peptidoglycan: Structure, synthesis, and regulation. EcoSal Plus. 2021, 9, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mett H; Bracha R; Mirelman D Soluble nascent peptidoglycan in growing Escherichia coli cells. J. Biol. Chem 1980, 255, 9884–9890. [PubMed] [Google Scholar]
- (4).Sarkar P; Yarlagadda V; Ghosh C; Haldar J A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Med. chem. comm 2017, 8, 516–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bush K Antimicrobial agents targeting bacterial cell walls and cell membranes. Rev. Sci. Tech 2012, 31, 43–56. [DOI] [PubMed] [Google Scholar]
- (6).Young KD The selective value of bacterial shape. Microbiol. Mol. Biol. Rev 2006, 70, 660–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Caccamo PD; Brun YV The molecular basis of noncanonical bacterial morphology. Trends. Microbiol 2018, 26, 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Vollmer W; Blanot D; de Pedro MA Peptidoglycan structure and architecture. FEMS Microbiol. Rev 2008, 32, 149–167. [DOI] [PubMed] [Google Scholar]
- (9).Coico R Gram staining. Curr. Protoc. Microbiol 2005, Appendix 3, Appendix 3C. [DOI] [PubMed] [Google Scholar]
- (10).Silhavy TJ; Kahne D; Walker S The bacterial cell envelope. Cold. Spring. Harb. Perspect. Biol 2010, 2, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Booth S; Lewis RJ Structural basis for the coordination of cell division with the synthesis of the bacterial cell envelope. Protein. Sci 2019, 28, 2042–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Caffalette CA; Kuklewicz J; Spellmon N; Zimmer J Biosynthesis and export of bacterial glycolipids. Annu. Rev. Biochem 2020, 89, 741–768. [DOI] [PubMed] [Google Scholar]
- (13).Eichler J; Guan Z Lipid sugar carriers at the extremes: The phosphodolichols Archaea use in N-glycosylation. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 2017, 1862, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Allen KN; Imperiali B Structural and mechanistic themes in glycoconjugate biosynthesis at membrane interfaces. Curr. Opin. Struct. Biol 2019, 59, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhao H; Patel V; Helmann JD; Dorr T Don’t let sleeping dogmas lie: new views of peptidoglycan synthesis and its regulation. Mol. Microbiol 2017, 106, 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Higashi Y; Strominger JL; Sweeley CC Structure of a lipid intermediate in cell wall peptidoglycan synthesis: a derivative of a C55 isoprenoid alcohol. Proc. Natl. Acad. Sci. U S A 1967, 57, 1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Higashi Y; Strominger JL; Sweeley CC Biosynthesis of the peptidoglycan of bacterial cell walls. XXI. Isolation of free C55-isoprenoid alcohol and of lipid intermediates in peptidoglycan synthesis from Staphylococcus aureus. J. Biol. Chem 1970, 245, 3697–3702. [PubMed] [Google Scholar]
- (18).Umbreit JN; Strominger JL Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J. Bacteriol 1972, 112, 1306–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Piepenbreier H; Diehl A; Fritz G Minimal exposure of Lipid II cycle intermediates triggers cell wall antibiotic resistance. Nat. Commun 2019, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Parsons JB; Rock CO Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid. Res 2013, 52, 249–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mulholland S; Turpin ER; Bonev BB; Hirst JD Docking and molecular dynamics simulations of the ternary complex nisin2:Lipid II. Sci. Rep 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Witzke S; Petersen M; Carpenter TS; Khalid S Molecular dynamics simulations reveal the conformational flexibility of Lipid II and its loose association with the defensin plectasin in the Staphylococcus aureus membrane. Biochemistry. 2016, 55, 3303–3314. [DOI] [PubMed] [Google Scholar]
- (23).Schneider T; Senn MM; Berger-Bachi B; Tossi A; Sahl HG; Wiedemann I In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol 2004, 53, 675–685. [DOI] [PubMed] [Google Scholar]
- (24).Morlot C; Straume D; Peters K; Hegnar OA; Simon N; Villard AM; Contreras-Martel C; Leisico F; Breukink E; Gravier-Pelletier C et al. Structure of the essential peptidoglycan amidotransferase MurT/GatD complex from Streptococcus pneumoniae. Nat. Commun 2018, 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Figueiredo TA; Sobral RG; Ludovice AM; Almeida JM; Bui NK; Vollmer W; de Lencastre H; Tomasz A Identification of genetic determinants and enzymes involved with the amidation of glutamic acid residues in the peptidoglycan of Staphylococcus aureus. PLoS. Pathog 2012, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mahapatra S; Crick DC; Brennan PJ Comparison of the UDP-N-acetylmuramate:L-alanine ligase enzymes from Mycobacterium tuberculosis and Mycobacterium leprae. J. Bacteriol 2000, 182, 6827–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schleifer KH; Kandler O Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev 1972, 36, 407–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Munch D; Sahl HG Structural variations of the cell wall precursor lipid II in Gram-positive bacteria - Impact on binding and efficacy of antimicrobial peptides. Biochim. Biophys. Acta 2015, 1848, 3062–3071. [DOI] [PubMed] [Google Scholar]
- (29).Muller A; Klockner A; Schneider T Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep 2017, 34, 909–932. [DOI] [PubMed] [Google Scholar]
- (30).Liu Y; Breukink E The membrane steps of bacterial cell wall synthesis as antibiotic targets. Antibiotics (Basel). 2016, 5, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Breukink E; de Kruijff B Lipid II as a target for antibiotics. Nature Reviews Drug Discovery. 2006, 5, 321–323. [DOI] [PubMed] [Google Scholar]
- (32).Marshall CG; Broadhead G; Leskiw BK; Wright GD d-Ala-d-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc. Natl. Acad. Sci. U S A 1997, 94, 6480–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bugg TD; Wright GD; Dutka-Malen S; Arthur M; Courvalin P; Walsh CT Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991, 30, 10408–10415. [DOI] [PubMed] [Google Scholar]
- (34).Arthur M; Depardieu F; Reynolds P; Courvalin P Quantitative analysis of the metabolism of soluble cytoplasmic peptidoglycan precursors of glycopeptide-resistant enterococci. Mol. Microbiol 1996, 21, 33–44. [DOI] [PubMed] [Google Scholar]
- (35).Liang H; DeMeester KE; Hou CW; Parent MA; Caplan JL; Grimes CL Metabolic labelling of the carbohydrate core in bacterial peptidoglycan and its applications. Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hsu YP; Hall E; Booher G; Murphy B; Radkov AD; Yablonowski J; Mulcahey C; Alvarez L; Cava F; Brun YV et al. Fluorogenic D-amino acids enable real-time monitoring of peptidoglycan biosynthesis and high-throughput transpeptidation assays. Nat. Chem 2019, 11, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Radkov AD; Hsu YP; Booher G; VanNieuwenhze MS Imaging bacterial cell wall biosynthesis. Annu. Rev. Biochem 2018, 87, 991–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Brown AR; Gordon RA; Hyland SN; Siegrist MS; Grimes CL Chemical biology tools for examining the bacterial cell wall. Cell Chem. Biol 2020, 27, 1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hernandez SB; Cava F New approaches and techniques for bacterial cell wall analysis. Curr. Opin. Microbiol 2021, 60, 88–95. [DOI] [PubMed] [Google Scholar]
- (40).Manat G; Roure S; Auger R; Bouhss A; Barreteau H; Mengin-Lecreulx D; Touze T Deciphering the metabolism of undecaprenyl-phosphate: The bacterial cell-wall unit carrier at the membrane frontier. Microb. Drug. Resist 2014, 20, 199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bouhss A; Trunkfield AE; Bugg TD; Mengin-Lecreulx D The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS. Microbiol. Rev 2008, 32, 208–233. [DOI] [PubMed] [Google Scholar]
- (42).Workman SD; Strynadka NCJ A slippery scaffold: Synthesis and recycling of the bacterial cell wall carrier lipid. J. Mol. Biol 2020, 432, 4964–4982. [DOI] [PubMed] [Google Scholar]
- (43).Pazos M; Peters K Peptidoglycan. Subcell. Biochem 2019, 92, 127–168. [DOI] [PubMed] [Google Scholar]
- (44).El Ghachi M; Bouhss A; Blanot D; Mengin-Lecreulx D The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J. Biol. Chem 2004, 279, 30106–30113. [DOI] [PubMed] [Google Scholar]
- (45).El Ghachi M; Derbise A; Bouhss A; Mengin-Lecreulx D Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J. Biol. Chem 2005, 280, 18689–18695. [DOI] [PubMed] [Google Scholar]
- (46).Kawakami N; Fujisaki S Undecaprenyl phosphate metabolism in Gram-negative and Gram-positive bacteria. Biosci. Biotechnol. Biochem 2018, 82, 940–946. [DOI] [PubMed] [Google Scholar]
- (47).Tatar LD; Marolda CL; Polischuk AN; van Leeuwen D; Valvano MA An Escherichia coli undecaprenyl-pyrophosphate phosphatase implicated in undecaprenyl phosphate recycling. Microbiology. 2007, 153, 2518–2529. [DOI] [PubMed] [Google Scholar]
- (48).Touze T; Blanot D; Mengin-Lecreulx D Substrate specificity and membrane topology of Escherichia coli PgpB, an undecaprenyl pyrophosphate phosphatase. J. Biol. Chem 2008, 283, 16573–16583. [DOI] [PubMed] [Google Scholar]
- (49).Manat G; El Ghachi M; Auger R; Baouche K; Olatunji S; Kerff F; Touze T; Mengin-Lecreulx D; Bouhss A Membrane topology and Biochemical characterization of the Escherichia coli BacA Undecaprenyl-Pyrophosphate Phosphatase. PLoS One. 2015, 10, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).El Ghachi M; Howe N; Huang CY; Olieric V; Warshamanage R; Touze T; Weichert D; Stansfeld PJ; Wang M; Kerff F et al. Crystal structure of undecaprenyl-pyrophosphate phosphatase and its role in peptidoglycan biosynthesis. Nat. Commun 2018, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Workman SD; Worrall LJ; Strynadka NCJ Crystal structure of an intramembranal phosphatase central to bacterial cell-wall peptidoglycan biosynthesis and lipid recycling. Nat. Commun 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).van Heijenoort J Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev 2007, 71, 620–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).El Zoeiby A; Sanschagrin F; Levesque RC Structure and function of the Mur enzymes: development of novel inhibitors. Mol. Microbiol 2003, 47, 1–12. [DOI] [PubMed] [Google Scholar]
- (54).Lovering AL; Safadi SS; Strynadka NC Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem 2012, 81, 451–478. [DOI] [PubMed] [Google Scholar]
- (55).Teo AC; Roper DI Core steps of membrane-bound peptidoglycan biosynthesis: Recent advances, insight and opportunities. Antibiotics (Basel). 2015, 4, 495–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Barreteau H; Kovac A; Boniface A; Sova M; Gobec S; Blanot D Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev 2008, 32, 168–207. [DOI] [PubMed] [Google Scholar]
- (57).Bupp K; van Heijenoort J The final step of peptidoglycan subunit assembly in Escherichia coli occurs in the cytoplasm. J. Bacteriol 1993, 175, 1841–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).van Dam V; Sijbrandi R; Kol M; Swiezewska E; de Kruijff B; Breukink E Transmembrane transport of peptidoglycan precursors across model and bacterial membranes. Mol. Microbiol 2007, 64, 1105–1114. [DOI] [PubMed] [Google Scholar]
- (59).Ruiz N Lipid flippases for bacterial peptidoglycan biosynthesis. Lipid. Insights 2015, 8, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ruiz N Bioinformatics identification of MurJ (MviN) as the peptidoglycan Lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. U S A 2008, 105, 15553–15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Inoue A; Murata Y; Takahashi H; Tsuji N; Fujisaki S; Kato J Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J. Bacteriol 2008, 190, 7298–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Sham LT; Butler EK; Lebar MD; Kahne D; Bernhardt TG; Ruiz N Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014, 345, 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li GW; Burkhardt D; Gross C; Weissman JS Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014, 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Thomas C; Aller SG; Beis K; Carpenter EP; Chang G; Chen L; Dassa E; Dean M; Duong Van Hoa F; Ekiert D et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Brown S; Zhang YH; Walker S A revised pathway proposed for Staphylococcus aureus wall teichoic acid biosynthesis based on in vitro reconstitution of the intracellular steps. Chem. Biol 2008, 15, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Mohammadi T; van Dam V; Sijbrandi R; Vernet T; Zapun A; Bouhss A; Diepeveen-de Bruin M; Nguyen-Disteche M; de Kruijff B; Breukink E Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO. J 2011, 30, 1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ruiz N; Gronenberg LS; Kahne D; Silhavy TJ Identification of two inner-membrane proteins required for the transport of Lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U S A 2008, 105, 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hvorup RN; Winnen B; Chang AB; Jiang Y; Zhou XF; Saier MH Jr. The Multidrug/Oligosaccharidyl-lipid/Polysaccharide (MOP) exporter superfamily. Eur. J. Biochem 2003, 270, 799–813. [DOI] [PubMed] [Google Scholar]
- (69).Touze T; Barreteau H; El Ghachi M; Bouhss A; Barneoud-Arnoulet A; Patin D; Sacco E; Blanot D; Arthur M; Duche D et al. Colicin M, a peptidoglycan Lipid-II-degrading enzyme: Potential use for antibacterial means? Biochem. Soc. Trans 2012, 40, 1522–1527. [DOI] [PubMed] [Google Scholar]
- (70).Touze T; Barreteau H; El Ghachi M; Bouhss A; Barneoud-Arnoulet A; Patin D; Sacco E; Blanot D; Arthur M; Duche D et al. Colicin M, a peptidoglycan Lipid-II-degrading enzyme: potential use for antibacterial means? Biochem Soc Trans. 2012, 40, 1522–1527. [DOI] [PubMed] [Google Scholar]
- (71).Kuk AC; Mashalidis EH; Lee SY Crystal structure of the MOP flippase MurJ in an inward-facing conformation. Nat. Struct. Mol. Biol 2017, 24, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Mohamed YF; Valvano MA A Burkholderia cenocepacia MurJ (MviN) homolog is essential for cell wall peptidoglycan synthesis and bacterial viability. Glycobiology. 2014, 24, 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Garcia-Heredia A; Pohane AA; Melzer ES; Carr CR; Fiolek TJ; Rundell SR; Lim HC; Wagner JC; Morita YS; Swarts BM et al. Peptidoglycan precursor synthesis along the sidewall of pole-growing Mycobacteria. Elife. 2018, 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ruiz N Streptococcus pyogenes YtgP (Spy_0390) complements Escherichia coli strains depleted of the putative peptidoglycan flippase MurJ. Antimicrob. Agents. Chemother 2009, 53, 3604–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Altschul SF; Gish W; Miller W; Myers EW; Lipman DJ Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- (76).Thanassi JA; Hartman-Neumann SL; Dougherty TJ; Dougherty BA; Pucci MJ Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 2002, 30, 3152–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Zalacain M; Biswas S; Ingraham KA; Ambrad J; Bryant A; Chalker AF; Iordanescu S; Fan J; Fan F; Lunsford RD et al. A global approach to identify novel broad-spectrum antibacterial targets among proteins of unknown function. J. Mol. Microbiol. Biotechnol 2003, 6, 109–126. [DOI] [PubMed] [Google Scholar]
- (78).Ruiz N Streptococcus pyogenes YtgP (Spy_0390) complements Escherichia coli strains depleted of the putative peptidoglycan flippase MurJ. Antimicrob Agents Chemother. 2009, 53, 3604–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Vasudevan P; McElligott J; Attkisson C; Betteken M; Popham DL Homologues of the Bacillus subtilis SpoVB protein are involved in cell wall metabolism. J. Bacteriol 2009, 191, 6012–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Fay A; Dworkin J Bacillus subtilis homologs of MviN (MurJ), the putative Escherichia coli Lipid II flippase, are not essential for growth. J. Bacteriol 2009, 191, 6020–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Meeske AJ; Sham LT; Kimsey H; Koo BM; Gross CA; Bernhardt TG; Rudner DZ MurJ and a novel Lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc. Natl. Acad. Sci. U S A 2015, 112, 6437–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Elhenawy W; Davis RM; Fero J; Salama NR; Felman MF; Ruiz N The O-Antigen flippase Wzk can substitute for MurJ in peptidoglycan synthesis in Helicobacter pylori and Escherichia coli. PLoS One. 2016, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Ruiz N; Davis RM; Kumar S YhdP, TamB, and YdbH are redundant but essential for growth and lipid homeostasis of the Gram-Negative outer membrane. mBio. 2021, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Doerrler WT; Sikdar R; Kumar S; Boughner LA New functions for the ancient DedA membrane protein family. J. Bacteriol 2013, 195, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Holtje JV Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev 1998, 62, 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Mohammadi T; Sijbrandi R; Lutters M; Verheul J; Martin NI; den Blaauwen T; de Kruijff B; Breukink E Specificity of the transport of Lipid II by FtsW in Escherichia coli. J. Biol. Chem 2014, 289, 14707–14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Sjodt M; Brock K; Dobihal G; Rohs PDA; Green AG; Hopf TA; Meeske AJ; Srisuknimit V; Kahne D; Walker S et al. Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature. 2018, 556, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Sjodt M; Rohs PDA; Gilman MSA; Erlandson SC; Zheng S; Green AG; Brock KP; Taguchi A; Kahne D; Walker S et al. Structural coordination of polymerization and crosslinking by a SEDS-bPBP peptidoglycan synthase complex. Nat. Microbiol 2020, 5, 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Meeske AJ; Riley EP; Robins WP; Uehara T; Mekalanos JJ; Kahne D; Walker S; Kruse AC; Bernhardt TG; Rudner DZ SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016, 537, 634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Taguchi A; Welsh MA; Marmont LS; Lee W; Sjodt M; Kruse AC; Kahne D; Bernhardt TG; Walker S FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat. Microbiol 2019, 4, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Cho H; Wivagg CN; Kapoor M; Barry Z; Rohs PDA; Suh H; Marto JA; Garner EC; Bernhardt TG Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol 2016, 1, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Egan AJF; Errington J; Vollmer W Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol 2020, 18, 446–460. [DOI] [PubMed] [Google Scholar]
- (93).McPherson DC; Popham DL Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J. Bacteriol 2003, 185, 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Butler EK; Davis RM; Bari V; Nicholson PA; Ruiz N Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J. Bacteriol 2013, 195, 4639–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Butler EK; Tan WB; Joseph H; Ruiz N Charge requirements of Lipid II flippase activity in Escherichia coli. J. Bacteriol 2014, 196, 4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Kusakizako T; Miyauchi H; Ishitani R; Nureki O Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim. Biophys. Acta. Biomembr 2019, 1862, 1–14. [DOI] [PubMed] [Google Scholar]
- (97).Claxton DP; Jagessar KL; McHaourab HS Principles of alternating access in Multidrug and Toxin Extrusion (MATE) transporters. J. Mol. Biol 2021, 433, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Kuk ACY; Hao A; Guan Z; Lee SY Visualizing conformation transitions of the Lipid II flippase MurJ. Nat. Commun 2019, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Zheng S; Sham LT; Rubino FA; Brock KP; Robins WP; Mekalanos JJ; Marks DS; Bernhardt TG; Kruse AC Structure and mutagenic analysis of the Lipid II flippase MurJ from Escherichia coli. Proc. Natl. Acad. Sci. U S A 2018, 115, 6709–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Marks DS; Colwell LJ; Sheridan R; Hopf TA; Pagnani A; Zecchina R; Sander C Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011, 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Hopf TA; Colwell LJ; Sheridan R; Rost B; Sander C; Marks DS Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012, 149, 1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hopf TA; Scharfe CP; Rodrigues JP; Green AG; Kohlbacher O; Sander C; Bonvin AM; Marks DS Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife. 2014, 3, 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Rubino FA; Mollo A; Kumar S; Butler EK; Ruiz N; Walker S; Kahne DE Detection of transport intermediates in the peptidoglycan flippase MurJ identifies residues essential for conformational cycling. J. Am. Chem. Soc 2020, 142, 5482–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Boes A; Olatunji S; Mohammadi T; Breukink E; Terrak M Fluorescence anisotropy assays for high throughput screening of compounds binding to lipid II, PBP1b, FtsW and MurJ. Sci. Rep 2020, 10, 6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Bolla JR; Corey RA; Sahin C; Gault J; Hummer A; Hopper JTS; Lane DP; Drew D; Allison TM; Stansfeld PJ et al. A Mass-Spectrometry-based Approach to distinguish annular and specific lipid binding to membrane proteins. Angew. Chem. Int. Ed. Engl 2020, 59, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Bolla JR; Sauer JB; Wu D; Mehmood S; Allison TM; Robinson CV Direct observation of the influence of cardiolipin and antibiotics on Lipid II binding to MurJ. Nat. Chem 2018, 10, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Okuda S; Freinkman E; Kahne D Cytoplasmic ATP hydrolysis powers transport of Lipopolysaccharide across the periplasm in E. coli. Science. 2012, 338, 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Owens TW; Taylor RJ; Pahil KS; Bertani BR; Ruiz N; Kruse AC; Kahne D Structural basis of unidirectional export of Lipopolysaccharide to the cell surface. Nature. 2019, 567, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Chin JW; Martin AB; King DS; Wang L; Schultz PG Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U S A 2002, 99, 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Qiao Y; Srisuknimit V; Rubino F; Schaefer K; Ruiz N; Walker S; Kahne D Lipid II overproduction allows direct assay of transpeptidase inhibition by beta-lactams. Nat. Chem. Biol 2017, 13, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Hu Y; Helm JS; Chen L; Ye XY; Walker S Ramoplanin inhibits bacterial transglycosylases by binding as a dimer to Lipid II. J Am Chem Soc. 2003, 125, 8736–8737. [DOI] [PubMed] [Google Scholar]
- (112).Sobral RG; Ludovice AM; de Lencastre H; Tomasz A Role of murF in cell wall biosynthesis: Isolation and characterization of a murF conditional mutant of Staphylococcus aureus. J. Bacteriol 2006, 188, 2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Ornelas-Soares A; de Lencastre H; de Jonge BL; Tomasz A Reduced methicillin resistance in a new Staphylococcus aureus transposon mutant that incorporates muramyl dipeptides into the cell wall peptidoglycan. J. Biol. Chem 1994, 269, 27246–27250. [PubMed] [Google Scholar]
- (114).Sham LT; Zheng S; Yakhnina AA; Kruse AC; Bernhardt TG Loss of specificity variants of WzxC suggest that substrate recognition is coupled with transporter opening in MOP-family flippases. Mol. Microbiol 2018, 109, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Tanaka Y; Iwaki S; Tsukazaki T Crystal structure of a plant Multidrug and Toxic Compound Extrusion family protein. Structure. 2017, 25, 1455–1460. [DOI] [PubMed] [Google Scholar]
- (116).Tanaka Y; Hipolito CJ; Maturana AD; Ito K; Kuroda T; Higuchi T; Katoh T; Kato HE; Hattori M; Kumazaki K et al. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature. 2013, 496, 247–251. [DOI] [PubMed] [Google Scholar]
- (117).Miyauchi H; Moriyama S; Kusakizako T; Kumazaki K; Nakane T; Yamashita K; Hirata K; Dohmae N; Nishizawa T; Ito K et al. Structural basis for xenobiotic extrusion by eukaryotic MATE transporter. Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Kusakizako T; Claxton DP; Tanaka Y; Maturana AD; Kuroda T; Ishitani R; McHaourab HS; Nureki O Structural basis of H(+)-dependent conformational change in a bacterial MATE transporter. Structure. 2019, 27, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Zakrzewska S; Mehdipour AR; Malviya VN; Nonaka T; Koepke J; Muenke C; Hausner W; Hummer G; Safarian S; Michel H Inward-facing conformation of a multidrug resistance MATE family transporter. Proc. Natl. Acad. Sci. U S A 2019, 116, 12275–12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Mousa JJ; Yang Y; Tomkovich S; Shima A; Newsome RC; Tripathi P; Oswald E; Bruner SD; Jobin C MATE transport of the E. coli-derived genotoxin colibactin. Nat. Microbiol 2016, 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Radchenko M; Symersky J; Nie R; Lu M Structural basis for the blockade of MATE multidrug efflux pumps. Nat. Commun 2015, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Lu M; Radchenko M; Symersky J; Nie R; Guo Y Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat. Struct. Mol. Biol 2013, 20, 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).He X; Szewczyk P; Karyakin A; Evin M; Hong WX; Zhang Q; Chang G Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010, 467, 991–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Lu M; Symersky J; Radchenko M; Koide A; Guo Y; Nie R; Koide S Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc. Natl. Acad. Sci. U S A 2013, 110, 2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Ficici E; Zhou W; Castellano S; Faraldo-Gomez JD Broadly conserved Na(+)-binding site in the N-lobe of prokaryotic multidrug MATE transporters. Proc. Natl. Acad. Sci. U S A 2018, 115, E6172–E6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Jin Y; Nair A; van Veen HW Multidrug transport protein NorM from Vibrio cholerae simultaneously couples to sodium- and proton-motive force. J. Biol. Chem 2014, 289, 14624–14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Rubino FA; Kumar S; Ruiz N; Walker S; Kahne DE Membrane potential is required for MurJ function. J. Am. Chem. Soc 2018, 140, 4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Krulwich TA; Sachs G; Padan E Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol 2011, 9, 330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Kumar S; Rubino FA; Mendoza AG; Ruiz N The bacterial Lipid II flippase MurJ functions by an alternating-access mechanism. J. Biol. Chem 2019, 294, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Kumar S; Ruiz N Probing conformational states of a target protein in Escherichia coli cells by in vivo cysteine cross-linking coupled with proteolytic gel analysis. Bio-protocol. 2019, 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Nishima W; Mizukami W; Tanaka Y; Ishitani R; Nureki O; Sugita Y Mechanisms for two-Step proton transfer reactions in the outward-facing form of MATE transporter. Biophys. J 2016, 110, 1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Gray AN; Egan AJ; Van’t Veer IL; Verheul J; Colavin A; Koumoutsi A; Biboy J; Altelaar AF; Damen MJ; Huang KC et al. Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. Elife. 2015, 4, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Eiamphungporn W; Helmann JD The Bacillus subtilis sigma(M) regulon and its contribution to cell envelope stress responses. Mol. Microbiol 2008, 67, 830–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Asai K Anti-sigma factor-mediated cell surface stress responses in Bacillus subtilis. Genes. Genet. Syst 2018, 92, 223–234. [DOI] [PubMed] [Google Scholar]
- (135).Shin JH; Choe D; Ransegnola B; Hong HR; Onyekwere I; Cross T; Shi Q; Cho BK; Westblade LF; Brito IL et al. A multifaceted cellular damage repair and prevention pathway promotes high-level tolerance to beta-lactam antibiotics. EMBO. Rep 2021, 22, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Teschler JK; Cheng AT; Yildiz FH The Two-component signal transduction system VxrAB positively regulates Vibrio cholerae biofilm formation. J. Bacteriol 2017, 199, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Gee CL; Papavinasasundaram KG; Blair SR; Baer CE; Falick AM; King DS; Griffin JE; Venghatakrishnan H; Zukauskas A; Wei JR et al. A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal 2012, 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Garcia-Heredia A; Kado T; Sein CE; Puffal J; Osman SH; Judd J; Gray TA; Morita YS; Siegrist MS Membrane-partitioned cell wall synthesis in Mycobacteria. Elife. 2021, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Turapov O; Forti F; Kadhim B; Ghisotti D; Sassine J; Straatman-Iwanowska A; Bottrill AR; Moynihan PJ; Wallis R; Barthe P et al. Two faces of CwlM, an essential PknB substrate, in Mycobacterium tuberculosis. Cell. Rep 2018, 25, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Viswanathan G; Yadav S; Joshi SV; Raghunand TR Insights into the function of FhaA, a cell division-associated protein in Mycobacteria. FEMS Microbiol. Lett 2017, 364, 1–9. [DOI] [PubMed] [Google Scholar]
- (141).Szwedziak P; Lowe J Do the divisome and elongasome share a common evolutionary past? Curr. Opin. Microbiol 2013, 16, 745–751. [DOI] [PubMed] [Google Scholar]
- (142).den Blaauwen T; Hamoen LW; Levin PA The divisome at 25: The road ahead. Curr. Opin. Microbiol 2017, 36, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Young KD Bacterial shape: Two-dimensional questions and possibilities. Annu. Rev. Microbiol 2010, 64, 223–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Pinho MG; Kjos M; Veening JW How to get (a)round: Mechanisms controlling growth and division of coccoid bacteria. Nat. Rev. Microbiol 2013, 11, 601–614. [DOI] [PubMed] [Google Scholar]
- (145).Liu X; Meiresonne NY; Bouhss A; den Blaauwen T FtsW activity and Lipid II synthesis are required for recruitment of MurJ to midcell during cell division in Escherichia coli. Mol. Microbiol 2018, 109, 855–884. [DOI] [PubMed] [Google Scholar]
- (146).Monteiro JM; Pereira AR; Reichmann NT; Saraiva BM; Fernandes PB; Veiga H; Tavares AC; Santos M; Ferreira MT; Macario V et al. Peptidoglycan synthesis drives an FtsZ-treadmilling-independent step of cytokinesis. Nature. 2018, 554, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Daniel RA; Errington J Control of cell morphogenesis in bacteria: Two distinct ways to make a rod-shaped cell. Cell. 2003, 113, 767–776. [DOI] [PubMed] [Google Scholar]
- (148).Baranowski C; Rego EH; Rubin EJ The dream of a Mycobacterium. Microbiol. Spectr 2019, 7, 1–14. [DOI] [PubMed] [Google Scholar]
- (149).Figueroa-Cuilan WM; Brown PJB Cell wall biogenesis during elongation and division in the plant pathogen Agrobacterium tumefaciens. Curr. Top. Microbiol. Immunol 2018, 418, 87–110. [DOI] [PubMed] [Google Scholar]
- (150).Morita YS; Velasquez R; Taig E; Waller RF; Patterson JH; Tull D; Williams SJ; Billman-Jacobe H; McConville MJ Compartmentalization of lipid biosynthesis in mycobacteria. J. Biol. Chem 2005, 280, 21645–21652. [DOI] [PubMed] [Google Scholar]
- (151).Hayashi JM; Luo CY; Mayfield JA; Hsu T; Fukuda T; Walfield AL; Giffen SR; Leszyk JD; Baer CE; Bennion OT et al. Spatially distinct and metabolically active membrane domain in Mycobacteria. Proc. Natl. Acad. Sci. U S A 2016, 113, 5400–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Puffal J; Garcia-Heredia A; Rahlwes KC; Siegrist MS; Morita YS Spatial control of cell envelope biosynthesis in Mycobacteria. Pathog. Dis 2018, 76, 1–15. [DOI] [PubMed] [Google Scholar]
- (153).Huber J; Donald RG; Lee SH; Jarantow LW; Salvatore MJ; Meng X; Painter R; Onishi RH; Occi J; Dorso K et al. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem. Biol 2009, 16, 837–848. [DOI] [PubMed] [Google Scholar]
- (154).Chamakura KR; Sham LT; Davis RM; Min L; Cho H; Ruiz N; Bernhardt TG; Young R A viral protein antibiotic inhibits Lipid II flippase activity. Nat. Microbiol 2017, 2, 1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Chu J; Vila-Farres X; Inoyama D; Gallardo-Macias R; Jaskowski M; Satish S; Freundlich JS; Brady SF Human microbiome inspired antibiotics with improved Beta-Lactam synergy against MDR Staphylococcus aureus. ACS. Infect. Dis 2018, 4, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Chu J; Vila-Farres X; Inoyama D; Ternei M; Cohen LJ; Gordon EA; Reddy BV; Charlop-Powers Z; Zebroski HA; Gallardo-Macias R et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol 2016, 12, 1004–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Mott JE; Shaw BA; Smith JF; Bonin PD; Romero DL; Marotti KR; Miller AA Resistance mapping and mode of action of a novel class of antibacterial anthranilic acids: Evidence for disruption of cell wall biosynthesis. J. Antimicrob. Chemother 2008, 62, 720–729. [DOI] [PubMed] [Google Scholar]
- (158).Chamakura K; Young R Phage single-gene lysis: Finding the weak spot in the bacterial cell wall. J. Biol. Chem 2019, 294, 3350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Shi H; Bratton BP; Gitai Z; Huang KC How to build a bacterial cell: MreB as the foreman of E. coli construction. Cell. 2018, 172, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Carballido-Lopez R The bacterial actin-like cytoskeleton. Microbiol. Mol. Biol. Rev 2006, 70, 888–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Dominguez-Escobar J; Chastanet A; Crevenna AH; Fromion V; Wedlich-Soldner R; Carballido-Lopez R Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science. 2011, 333, 225–228. [DOI] [PubMed] [Google Scholar]
- (162).Garner EC; Bernard R; Wang W; Zhuang X; Rudner DZ; Mitchison T Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science. 2011, 333, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Bisson-Filho AW; Hsu YP; Squyres GR; Kuru E; Wu F; Jukes C; Sun Y; Dekker C; Holden S; VanNieuwenhze MS et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science. 2017, 355, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Yang X; Lyu Z; Miguel A; McQuillen R; Huang KC; Xiao J GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science. 2017, 355, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Sauvage E; Kerff F; Terrak M; Ayala JA; Charlier P The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS. Microbiol. Rev 2008, 32, 234–258. [DOI] [PubMed] [Google Scholar]
- (166).Sauvage E; Terrak M Glycosyltransferases and transpeptidases/Penicillin-Binding Proteins: Valuable targets for new antibacterials. Antibiotics (Basel). 2016, 5, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Miyachiro MM; Contreras-Martel C; Dessen A Penicillin-Binding Proteins (PBPs) and bacterial cell wall elongation complexes. Subcell. Biochem 2019, 93, 273–289. [DOI] [PubMed] [Google Scholar]
- (168).Straume D; Piechowiak KW; Kjos M; Havarstein LS Class A PBPs: It is time to rethink traditional paradigms. Mol. Microbiol 2021, 116, 41–52. [DOI] [PubMed] [Google Scholar]
- (169).van Heijenoort J Peptidoglycan hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev 2011, 75, 636–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Sauvage E; Derouaux A; Fraipont C; Joris M; Herman R; Rocaboy M; Schloesser M; Dumas J; Kerff F; Nguyen-Disteche M et al. Crystal structure of penicillin-binding protein 3 (PBP3) from Escherichia coli. PLoS One. 2014, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Cochrane SA; Lohans CT Breaking down the cell wall: Strategies for antibiotic discovery targeting bacterial transpeptidases. Eur. J. Med. Chem 2020, 194, 1–15. [DOI] [PubMed] [Google Scholar]
- (172).Typas A; Banzhaf M; van den Berg van Saparoea B; Verheul J; Biboy J; Nichols RJ; Zietek M; Beilharz K; Kannenberg K; von Rechenberg M et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010, 143, 1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Paradis-Bleau C; Markovski M; Uehara T; Lupoli TJ; Walker S; Kahne DE; Bernhardt TG Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010, 143, 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Egan AJ; Jean NL; Koumoutsi A; Bougault CM; Biboy J; Sassine J; Solovyova AS; Breukink E; Typas A; Vollmer W et al. Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc. Natl. Acad. Sci. U S A 2014, 111, 8197–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Lupoli TJ; Lebar MD; Markovski M; Bernhardt T; Kahne D; Walker S Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J. Am. Chem. Soc 2014, 136, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Caveney NA; Egan AJF; Ayala I; Laguri C; Robb CS; Breukink E; Vollmer W; Strynadka NCJ; Simorre JP Structure of the peptidoglycan synthase activator LpoP in Pseudomonas aeruginosa. Structure. 2020, 28, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Sardis MF; Bohrhunter JL; Greene NG; Bernhardt TG The LpoA activator is required to stimulate the peptidoglycan polymerase activity of its cognate cell wall synthase PBP1a. Proc. Natl. Acad. Sci. U S A 2021, 118, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Kato J; Suzuki H; Hirota Y Dispensability of either penicillin-binding protein-1a or −1b involved in the essential process for cell elongation in Escherichia coli. Mol. Gen. Genet 1985, 200, 272–277. [DOI] [PubMed] [Google Scholar]
- (179).Yousif SY; Broome-Smith JK; Spratt BG Lysis of Escherichia coli by beta-lactam antibiotics: Deletion analysis of the role of penicillin-binding proteins 1A and 1B. J. Gen. Microbiol 1985, 131, 2839–2845. [DOI] [PubMed] [Google Scholar]
- (180).Mueller EA; Egan AJ; Breukink E; Vollmer W; Levin PA Plasticity of Escherichia coli cell wall metabolism promotes fitness and antibiotic resistance across environmental conditions. Elife. 2019, 8, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Spratt BG The mechanism of action of Mecillinam. J. Antimicrob. Chemother 1977, 3 Suppl B, 13–19. [DOI] [PubMed] [Google Scholar]
- (182).Curtis NA; Eisenstadt RL; Turner KA; White AJ Inhibition of penicillin-binding protein 3 of Escherichia coli K-12. Effects upon growth, viability and outer membrane barrier function. J. Antimicrob. Chemother 1985, 16, 287–296. [DOI] [PubMed] [Google Scholar]
- (183).Matsuzawa H; Hayakawa K; Sato T; Imahori K Characterization and genetic analysis of a mutant of Escherichia coli K-12 with rounded morphology. J. Bacteriol 1973, 115, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Khattar MM; Begg KJ; Donachie WD Identification of FtsW and characterization of a new ftsW division mutant of Escherichia coli. J. Bacteriol 1994, 176, 7140–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Denome SA; Elf PK; Henderson TA; Nelson DE; Young KD Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: Viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol 1999, 181, 3981–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Garcia del Portillo F; de Pedro MA Differential effect of mutational impairment of penicillin-binding proteins 1A and 1B on Escherichia coli strains harboring thermosensitive mutations in the cell division genes ftsA, ftsQ, ftsZ, and pbpB. J. Bacteriol 1990, 172, 5863–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Vigouroux A; Cordier B; Aristov A; Alvarez L; Ozbaykal G; Chaze T; Oldewurtel ER; Matondo M; Cava F; Bikard D et al. Class-A penicillin binding proteins do not contribute to cell shape but repair cell-wall defects. Elife. 2020, 9, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).More N; Martorana AM; Biboy J; Otten C; Winkle M; Serrano CKG; Monton Silva A; Atkinson L; Yau H; Breukink E et al. Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. mBio. 2019, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Berry J; Rajaure M; Pang T; Young R The spanin complex is essential for lambda lysis. J. Bacteriol 2012, 194, 5667–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Rojas ER; Billings G; Odermatt PD; Auer GK; Zhu L; Miguel A; Chang F; Weibel DB; Theriot JA; Huang KC The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature. 2018, 559, 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Brown PJ; de Pedro MA; Kysela DT; Van der Henst C; Kim J; De Bolle X; Fuqua C; Brun YV Polar growth in the Alphaproteobacterial order Rhizobiales. Proc. Natl. Acad. Sci. U S A 2012, 109, 1697–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Williams MA; Aliashkevich A; Krol E; Kuru E; Bouchier JM; Rittichier J; Brun YV; VanNieuwenhze MS; Becker A; Cava F et al. Unipolar peptidoglycan synthesis in the Rhizobiales requires an essential class A Penicillin-Binding Protein. mBio. 2021, 12, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Daitch AK; Goley ED Uncovering unappreciated activities and niche functions of bacterial cell wall enzymes. Curr. Biol 2020, 30, R1170–R1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Pazos M; Vollmer W Regulation and function of class A Penicillin-binding proteins. Curr. Opin. Microbiol 2021, 60, 80–87. [DOI] [PubMed] [Google Scholar]
- (195).Rohs PDA; Bernhardt TG Growth and division of the peptidoglycan matrix. Annu. Rev. Microbiol 2021, 315–336. [DOI] [PubMed] [Google Scholar]
- (196).Emami K; Guyet A; Kawai Y; Devi J; Wu LJ; Allenby N; Daniel RA; Errington J RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat. Microbiol 2017, 2, 16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Ishino F; Park W; Tomioka S; Tamaki S; Takase I; Kunugita K; Matsuzawa H; Asoh S; Ohta T; Spratt BG et al. Peptidoglycan synthetic activities in membranes of Escherichia coli caused by overproduction of penicillin-binding protein 2 and RodA protein. J. Biol. Chem 1986, 261, 7024–7031. [PubMed] [Google Scholar]
- (198).Liechti GW; Kuru E; Hall E; Kalinda A; Brun YV; VanNieuwenhze M; Maurelli AT A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature. 2014, 506, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Bertani B; Ruiz N Function and biogenesis of Lipopolysaccharides. EcoSal Plus. 2018, 8, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Straume D; Piechowiak KW; Olsen S; Stamsas GA; Berg KH; Kjos M; Heggenhougen MV; Alcorlo M; Hermoso JA; Havarstein LS Class A PBPs have a distinct and unique role in the construction of the pneumococcal cell wall. Proc. Natl. Acad. Sci. U S A 2020, 117, 6129–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Reichmann NT; Tavares AC; Saraiva BM; Jousselin A; Reed P; Pereira AR; Monteiro JM; Sobral RG; VanNieuwenhze MS; Fernandes F et al. SEDS-bPBP pairs direct lateral and septal peptidoglycan synthesis in Staphylococcus aureus. Nat. Microbiol 2019, 4, 1368–1377. [DOI] [PubMed] [Google Scholar]
- (202).Taguchi A; Kahne D; Walker S Chemical tools to characterize peptidoglycan synthases. Curr. Opin. Chem. Biol 2019, 53, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Fernandes R; Amador P; Prudêncio C β-Lactams: chemical structure, mode of action and mechanisms of resistance. Reviews in Medical Microbiology. 2013, 24, 7–17. [Google Scholar]
- (204).van Berkel SS; Nettleship JE; Leung IK; Brem J; Choi H; Stuart DI; Claridge TD; McDonough MA; Owens RJ; Ren J et al. Binding of (5S)-penicilloic acid to penicillin binding protein 3. ACS. Chem. Biol 2013, 8, 2112–2116. [DOI] [PubMed] [Google Scholar]
- (205).Goldman RC; Gange D Inhibition of transglycosylation involved in bacterial peptidoglycan synthesis. Curr. Med. Chem 2000, 7, 801–820. [DOI] [PubMed] [Google Scholar]
- (206).Halliday J; McKeveney D; Muldoon C; Rajaratnam P; Meutermans W Targeting the forgotten transglycosylases. Biochem. Pharmacol 2006, 71, 957–967. [DOI] [PubMed] [Google Scholar]
- (207).Yuan Y; Fuse S; Ostash B; Sliz P; Kahne D; Walker S Structural analysis of the contacts anchoring moenomycin to peptidoglycan glycosyltransferases and implications for antibiotic design. ACS. Chem. Biol 2008, 3, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Rebets Y; Lupoli T; Qiao Y; Schirner K; Villet R; Hooper D; Kahne D; Walker S Moenomycin resistance mutations in Staphylococcus aureus reduce peptidoglycan chain length and cause aberrant cell division. ACS. Chem. Biol 2014, 9, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Zuegg J; Muldoon C; Adamson G; McKeveney D; Le Thanh G; Premraj R; Becker B; Cheng M; Elliott AG; Huang JX et al. Carbohydrate scaffolds as glycosyltransferase inhibitors with in vivo antibacterial activity. Nat. Commun 2015, 6, 7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Derouaux A; Sauvage E; Terrak M Peptidoglycan glycosyltransferase substrate mimics as templates for the design of new antibacterial drugs. Front. Immunol 2013, 4, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Qiao Y; Lebar MD; Schirner K; Schaefer K; Tsukamoto H; Kahne D; Walker S Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J. Am. Chem. Soc 2014, 136, 14678–14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Welsh MA; Taguchi A; Schaefer K; Van Tyne D; Lebreton F; Gilmore MS; Kahne D; Walker S Identification of a functionally unique family of Penicillin-Binding Proteins. J. Am. Chem. Soc 2017, 139, 17727–17730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Blaskovich MAT; Hansford KA; Butler MS; Jia Z; Mark AE; Cooper MA Developments in Glycopeptide Antibiotics. ACS. Infect. Dis 2018, 4, 715–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Fair RJ; Tor Y Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem 2014, 6, 25–64. [DOI] [PMC free article] [PubMed] [Google Scholar]