

Abstract
Non‐alcoholic steatohepatitis (NASH) has emerged as a major challenge for public health because of high global prevalence and lack of evidence‐based therapies. Most animal models of NASH lack sufficient validation regarding disease progression and pharmacological treatment. The Gubra‐Amylin NASH (GAN) diet‐induced obese (DIO) mouse demonstrate clinical translatability with respect to disease etiology and hallmarks of NASH. This study aimed to evaluate disease progression and responsiveness to clinically effective interventions in GAN DIO‐NASH mice. Disease phenotyping was performed in male C57BL/6J mice fed the GAN diet high in fat, fructose, and cholesterol for 28–88 weeks. GAN DIO‐NASH mice with biopsy‐confirmed NASH and fibrosis received low‐caloric dietary intervention, semaglutide (30 nmol/kg/day, s.c.) or lanifibranor (30 mg/kg/day, p.o.) for 8 and 12 weeks, respectively. Within‐subject change in nonalcoholic fatty liver disease (NAFLD) Activity Score (NAS) and fibrosis stage was evaluated using automated deep learning‐based image analysis. GAN DIO‐NASH mice showed clear and reproducible progression in NASH, fibrosis stage, and tumor burden with high incidence of hepatocellular carcinoma. Consistent with clinical trial outcomes, semaglutide and lanifibranor improved NAS, whereas only lanifibranor induced regression in the fibrosis stage. Dietary intervention also demonstrated substantial benefits on metabolic outcomes and liver histology. Differential therapeutic efficacy of semaglutide, lanifibranor, and dietary intervention was supported by quantitative histology, RNA sequencing, and blood/liver biochemistry. In conclusion, the GAN DIO‐NASH mouse model recapitulates various histological stages of NASH and faithfully reproduces histological efficacy profiles of compounds in advanced clinical development for NASH. Collectively, these features highlight the utility of GAN DIO‐NASH mice in preclinical drug development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Given the lack of effective therapies for non‐alcoholic steatohepatitis (NASH), there is a need to establish animal models that better predict clinical outcomes. A clear limitation in preclinical drug discovery is the lack of translational animal models, which recapitulate the various stages of NASH, and faithfully reproduce the individual efficacy profile of current clinical interventions.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study addressed disease progression and validated treatment outcomes of dietary intervention and compounds in advanced clinical development in the GAN diet‐induced obese (DIO) and biopsy‐confirmed mouse model of NASH.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The GAN DIO‐NASH mouse model presents with metabolic, biochemical, and histological hallmarks of progressive NASH, characterized by deteriorating liver fibrosis and increasing hepatocellular carcinoma burden. The differential clinical histological efficacy profiles of low‐caloric dietary intervention, semaglutide, and lanifibranor were closely reproduced in the model.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This preclinical model will be instrumental for probing efficacy of novel drugs targeting NASH and advancing novel drug therapies from preclinical to clinical development.
INTRODUCTION
Non‐alcoholic fatty liver disease (NAFLD) is the most common chronic liver condition worldwide. 1 The most severe form of NAFLD, non‐alcoholic steatohepatitis (NASH), is characterized by steatosis, lobular inflammation, and hepatocyte degeneration (ballooning). 2 There is a strong correlation between presence of the metabolic syndrome and NAFLD/NASH, 3 and the prevalence and incidence is therefore fueled by the obesity and diabetes epidemics. 4 Patients with NASH are at increased risk of developing liver fibrosis, which can progress to cirrhosis and hepatocellular carcinoma (HCC). 5 , 6 As a consequence, NASH represents the most rapid growing indication for liver transplantation. 7 Liver diagnostic biopsy is the gold standard and the definite criterion for confirming, grading, and staging of NASH, as no specific clinical phenotype permits accurate diagnosis of NASH. 8 Collectively, NASH has emerged as a major challenge for public health because of the increasing incidence in combination with difficulties in diagnosis and lack of effective therapies. In the absence of approved drug therapies, lifestyle intervention remains the first‐line of treatment for NASH, however, long‐term adherence is difficult to achieve, 9 emphasizing the high unmet need for efficacious therapies. Drug development for NASH is therefore a rapidly evolving field and several drug candidates are in various stages of clinical development. 10 , 11 , 12
Animal models with improved clinical translatability can optimally inform about potential clinical efficacy of novel drug candidates for treatment of NASH. 13 Accumulating patient‐based findings on molecular genetics and signaling pathways are critical to refine animal models of NASH. With the increasing appreciation that the pathogenesis involves multiple pathological “hits” in the onset and progression of NASH, 14 , 15 , 16 considerable progress has therefore been made in the development of preclinical models that better replicate the complex dynamics of the disease. 13 , 17 In particular, overnutrition is a central aspect in the natural history of the disease. Although conventional obesogenic high‐fat diets induce dyslipidemia and insulin resistance in rodent models of diet‐induced obesity (DIO) and prediabetes, these models only develop mild‐stage NAFLD without appreciable fibrosis. 13 Hence, additional dietary stimuli have been applied to enhance the pro‐fibrogenic properties of high‐fat diets used in preclinical NASH research. High intake of “Western diets”, defined as diets enriched in saturated fats, fructose, and cholesterol, is considered a driving factor for the onset and progression of NAFLD/NASH. 18 , 19 A number of Western diets have been reported to promote fibrotic NASH in mice. 13 Accordingly, the Gubra Amylin NASH (GAN) diet high in these nutrient components promotes obesity with consistent development of biopsy‐confirmed NASH and fibrosis in both C57BL/6 (GAN DIO‐NASH) and leptin deficient (GAN ob/ob) mice. 20 Importantly, the GAN DIO‐NASH mouse recapitulates both metabolic, biochemical, and histopathological hallmarks as well as transcriptome signatures in NASH patients. 21
Further translatability to the human condition is achieved when the preclinical model of NASH demonstrates progressive fibrosis and late‐stage complications as well as therapeutic efficacy of clinically relevant interventions. 22 However, most animal models of NASH lack sufficient validation with respect to disease progression and pharmacological intervention, which limits their use in preclinical drug discovery. Semaglutide, a long‐acting glucagon‐like receptor 1 (GLP‐1) agonist currently approved for treatment of type 2 diabetes and obesity, 23 , 24 has been reported to increase resolution of NASH without improving fibrosis stage in a 72‐week clinical phase II study. 25 In comparison, lanifibranor, a pan‐peroxisome proliferator‐activated receptor (PPAR‐α/δ/γ) agonist, achieved significant benefits on both NASH resolution and fibrosis regression in a recent 24‐week clinical phase II trial (NATIVE trial). 26 Whereas the hepatoprotective effects of semaglutide may predominantly relate to indirect beneficial effects on body weight and insulin sensitivity, 23 , 25 preclinical data suggests that lanifibranor have liver‐directed effects by targeting multiple hepatic cell populations involved in the pathogenesis of NASH. 27 , 28 Because semaglutide and lanifibranor represent promising investigational drugs for NASH with different clinical efficacy profile and molecular targets, this makes these two compounds well‐suited for evaluating pharmacological intervention in preclinical models of NASH. To further evaluate clinical translatabilty of the GAN DIO‐NASH mouse, the present study therefore characterized disease progression and profiled therapeutic efficacy of semaglutide and lanifibranor treatment compared to dietary intervention in the model.
METHODS
Ethics
The Danish Animal Experiments Inspectorate approved all experiments (license #2013‐15‐2934‐00784). All animal experiments were conducted in accordance with Gubra bioethical guidelines, which are fully compliant with internationally accepted principles for the care and use of laboratory animals.
Animals
C57BL/6J mice (5–6 weeks old) were from Janvier Labs (Le Genest Saint Isle, France) and housed in a controlled environment (12 h light/dark cycle, lights on at 3 a.m., 21 ± 2°C, humidity 50 ± 10%). Each animal was identified by an implantable subcutaneous microchip (PetID Microchip, E‐vet, Haderslev, Denmark). Mice had ad libitum access to tap water and chow (3.22 kcal/g, Altromin 1324; Brogaarden, Hoersholm, Denmark) or Gubra Amylin NASH diet ([GAN diet, 4.49 kcal/g, 40 kcal‐% fat] of these 46% saturated fatty acids by weight, 22% fructose, 10% sucrose, 2% cholesterol; D09100310, Research Diets). Mice were fed chow or the GAN diet for up to 88 weeks. Animals were terminated by cardiac puncture under isoflurane anesthesia.
Baseline liver biopsy
Animals underwent liver biopsy before treatment intervention, as described in detail previously. 29 Mice were anesthetized with isoflurane, a midline abdominal incision was made to expose the left lateral lobe, and a cone‐shaped biopsy of ~50 mg liver tissue was collected. Cut surfaces were electrocoagulated using an electrosurgicial unit. Thereafter, the liver was returned to the abdominal cavity, the abdominal wall was sutured, and the skin was stapled. Animals received 5 mg/kg carprofen prior to surgery and on postoperative days 1 and 2. Animals were single‐housed after the procedure and allowed to recover for 4 weeks prior to treatment start.
Treatment intervention
See Figure S1 for study outlines on dietary and pharmacological intervention. Animals were fed the GAN diet for 31–34 weeks (dietary intervention) or 34–38 weeks (pharmacological intervention). Only DIO‐NASH mice with severe steatosis (score 3) and fibrosis (≤ stage F1) were included, evaluated using standard clinical biopsy histopathological scoring criteria (see below). Liver biopsy histology inclusion criteria were met for greater than or equal to 93% of mice in the cohorts used. Mice were randomized and stratified to treatment based on baseline mean steatosis and fibrosis stage. DIO‐NASH mice were administered saline vehicle (s.c., q.d.) with or without dietary intervention by switching from the GAN diet to chow (Chow‐reversal) for 8 or 12 weeks (n = 12–14 per group). Vehicle‐injected DIO‐NASH controls were included in the chow‐reversal study to further enable comparison between the efficacy of chow reversal versus drug administration (see below). Chow‐fed mice receiving saline vehicle (s.c., q.d.) for 8 (n = 10) or 12 weeks (n = 15) served as normal controls. Other DIO‐NASH mice received vehicle, semaglutide (30 nmol/kg, s.c., q.d.) or lanifibranor (30 mg/kg, p.o., q.d.) for 8 or 12 weeks (n = 12–16 per group). A dose‐escalation scheme was implemented to reduce expected initial effects of semaglutide treatment, as transient GLP‐1 receptor‐induced discomfort in rodents, including taste aversion and pica behavior, is typically observed within the first 2–3 days of treatment. 30 The semaglutide dose was increased through daily increments (0.6–1.2–2.4–4.8–12–30 nmol/kg) for reaching the target dose on treatment day 6, thereafter being maintained for the remainder of the treatment period. Animals were kept on the GAN diet throughout the drug treatment period. In the drug treatment study, chow‐fed mice receiving the saline vehicle (s.c. or q.d.) for 12 weeks (n = 16) served as normal controls. Vehicles were phosphate‐buffered saline with 0.1% bovine serum albumin (for semaglutide) and 0.5% carboxymethyl cellulose with 0.01% Tween‐80 (for lanifibranor), respectively. Vehicles and compounds were administered in a dosing volume of 5 ml/kg. Body weight was measured daily, 24 h food intake was assessed once weekly during the intervention period.
Plasma and liver biochemistry
Four hour fasted terminal blood was sampled from the tail vein, kept on ice, and centrifuged (5 min, 4°C, 6000 g) to generate EDTA‐stabilized plasma. Plasma alanine aminotransferase (ALT), aspartate aminotransferase, triglycerides (TG), and total cholesterol (TC), as well as liver TG and TC was determined, as described previously. 29
Liver histology
Baseline liver biopsy and terminal samples (both from the left lateral lobe) were fixed overnight in 4% paraformaldehyde. Liver tissue was paraffin‐embedded and sectioned (3 µm thickness). Sections were stained with hematoxylin‐eosin (HE), picro‐Sirius Red (PSR; Sigma‐Aldrich, Brøndby, Denmark), anti‐galectin‐3 (cat. 125402; Biolegend, San Diego, CA), alpha‐smooth muscle action (α‐SMA, cat. ab124964; Abcam, Cambridge, UK), or anti‐type I collagen (Col1a1, cat. 1310‐01; Southern Biotech, Birmingham, AL) using standard procedures. 29 , 31 Deep learning‐based image analysis was applied for automated histopathological scoring using the NASH Clinical Research Network (CRN) scoring system (see below). In addition, deep learning‐based image analysis was applied to histopathological scoring variables for quantifying whole‐section number of lipid‐laden hepatocytes (percentage of hepatocytes), number of inflammatory foci (foci per mm2), and proportionate (%) area of perisinusoidal and periportal fibrosis. Additionally, quantitative histomorphometry was performed using a digital imaging software (Visiomorph; Visiopharm, Hørsholm, Denmark) for the determination of whole‐section liver fat (HE‐staining), fibrosis (PSR, Col1a1), inflammation (galectin‐3), and hepatic stellate cell (HSC) activation (α‐SMA), expressed relative (%) to total sectional area. To account for treatment‐induced changes in liver mass, whole‐liver marker content (mg) was estimated by multiplying % area of positive staining with corresponding total liver weight, being representative for whole‐liver histological changes in DIO‐NASH mice. 32 Classification of hepatic tumors was performed in DIO‐NASH mice fed the GAN diet for at least 58 weeks (n = 11 mice). A total of 17 tumors were evaluated on HE‐ and reticulin‐stained sections. For reticulin staining, slides were incubated with potassium permanganate solution, followed by sulfuric acid, oxalic acid, ferric ammonium sulfate solution, silver nitrate solution, formaldehyde solution, gold chloride solution, and sodium thiosulfate solution (staining kit #1.00251, Sigma‐Aldrich, St. Louis, MO). Sections were rinsed with distilled water before immersion in each solution and dehydrated in graded ethanol and xylene before cover slipping. Tumor classification was performed by an expert clinical histopathologist.
Automated deep learning‐based image analysis
An automated deep learning‐based digital imaging analysis pipeline (Gubra Histopathological Objective Scoring Technology [GHOST]) was developed to obtain more accurate and objective method for assessment of histopathological scores using the clinical NAFLD Activity Scoring (NAS) and fibrosis staging system according to the NASH CRN scoring system as outlined by Kleiner et al. 33 Three tissue sectional zones were defined, including sinusoidal and periportal zones as well as central veins. The area around the central veins were excluded from the analysis. Portioning was performed by applying a trained artificial intelligence (AI) model based on the Deeplabv3+ model architecture. Next, a multiclass AI model based on the U‐net model architecture was applied to HE sections to segment hepatocytes with/without lipid droplets, inflammatory foci, and ballooned hepatocytes. Steatosis score was obtained directly by computing the fraction of hepatocytes with lipid droplets relative to the total amount of hepatocytes, according to the Kleiner criteria (<5%, score 0; 5–33%, score 1; >33–66%, score 2; and >66%, score 3). Lobular inflammation score was computed by a four‐step process; initially, four or more inflammatory cells positioned closely together were assigned as a focus. The liver section was then divided into fields of 1024 × 1024 pixels, whereafter each field was assigned a score based on the number of foci in the field according to Kleiner et al. (0 foci, score 0; 1 foci, score 1; 2–4 foci, score 2; and >4 foci, score 3). The lobular inflammation score was computed as the unweighted average of the field values. Hepatocyte ballooning score was obtained by computing the number of ballooned cells per mm2 and applying thresholds to assign the corresponding score. Thresholds were chosen by comparing the automated analysis results to Kleiner scores assigned manually by an expert histopathologist. For the fibrosis stage, a Bayesian classifier was used to segment PSR‐positive staining inside sinusoidal and periportal zones. To further characterize fibrosis severity, a bridging segmentation was computed by thresholding on a feature image made by using a local polynomial filter on the red‐green contrast of the PSR image. The PSR area fractions, as well as features from the bridging segmentation was used as input to a XGBoost classifier to obtain the corresponding Kleiner fibrosis stage. The classifier was trained from 950 samples which had been assigned a manual score by an expert histopathologist.
RNA sequencing
Liver transcriptomics were performed using RNA sequencing on RNA extracts from terminal liver samples (15 mg fresh tissue), as described in detail elsewhere. 21 RNA sequence libraries were prepared with NeoPrep (Illumina, San Diego, CA) using Illumina TruSeq stranded mRNA Library kit for NeoPrep (Illumina) and sequenced on the NextSeq 500 (Illumina) with NSQ 500 hi‐Output KT version 2 (75 CYS; Illumina). Reads were aligned to the GRCm38 version 84 Ensembl Mus musculus genome using STAR version 2.5.2a with default parameters. A lower detection limit for gene expression was defined based on raw mapped read counts (RPKM of 0.1). The R‐package DESeq2 was used for differential gene expression analysis. Gene set analysis was conducted with the R package PIANO version 1.18.1 using the Stouffer method, and p values were corrected for multiple testing using the Benjamini‐Hochberg method (5% false discovery rate < 0.05). RNA sequencing data from the pharmacological intervention studies are accessible using a web‐based global gene expression data viewer (Gubra Gene Expression Experience [GGEX], https://rnaseq.gubra.dk/).
Statistics
Except from deep learning‐based image analysis and RNA sequencing, data were analyzed using GraphPad Prism version 9.0.2 software (GraphPad, La Jolla, CA). All results are shown as mean ± standard error of mean (SEM). A one‐sided Fisher’s exact test with Bonferroni correction was used for within‐subject comparison of histopathological scores before and after treatment intervention. A Dunnett’s test one‐ or two‐factor linear model with interaction was used for all other statistical analyses. A p value less than 0.05 was considered statistically significant.
RESULTS
Deep learning‐based assessment of liver histopathological scores
GHOST detected central veins and portal triads in the HE‐ and PSR‐stained sections (Figure 1a,b), enabling overall segmentation of zones relevant for standard histopathological scoring using the NAS and fibrosis staging system. 33 Relevant cell types were located in HE‐stained sections, including hepatocytes, inflammatory cells, and ballooned hepatocytes (Figure 1a). Clusters of ≥4 inflammatory cells were defined and segmented as foci (Figure 1a). NAS was computed and validated using a test set of 338 mouse liver biopsies with a high degree of agreement between automated and manual scoring of NAS (Figure 1c, Kappa value of 0.72). PSR‐stained collagen fibers were segmented in the sinusoidal and periportal space, also enabling segmentation of fibers forming bridges and branch points (Figure 1b). From these segmentations, fibrosis stage (0–3) was computed and validated using a test set of 537 mouse liver biopsies. There was a high concordance between automated and expert histopathologist manual scores (Figure 1d, Kappa value of 0.84). Quantitative morphometrics were derived from the scoring variables, expressed as density of hepatocytes with lipid droplets, number of inflammatory foci, as well as % area of sinusoidal and periportal fibrosis.
FIGURE 1.
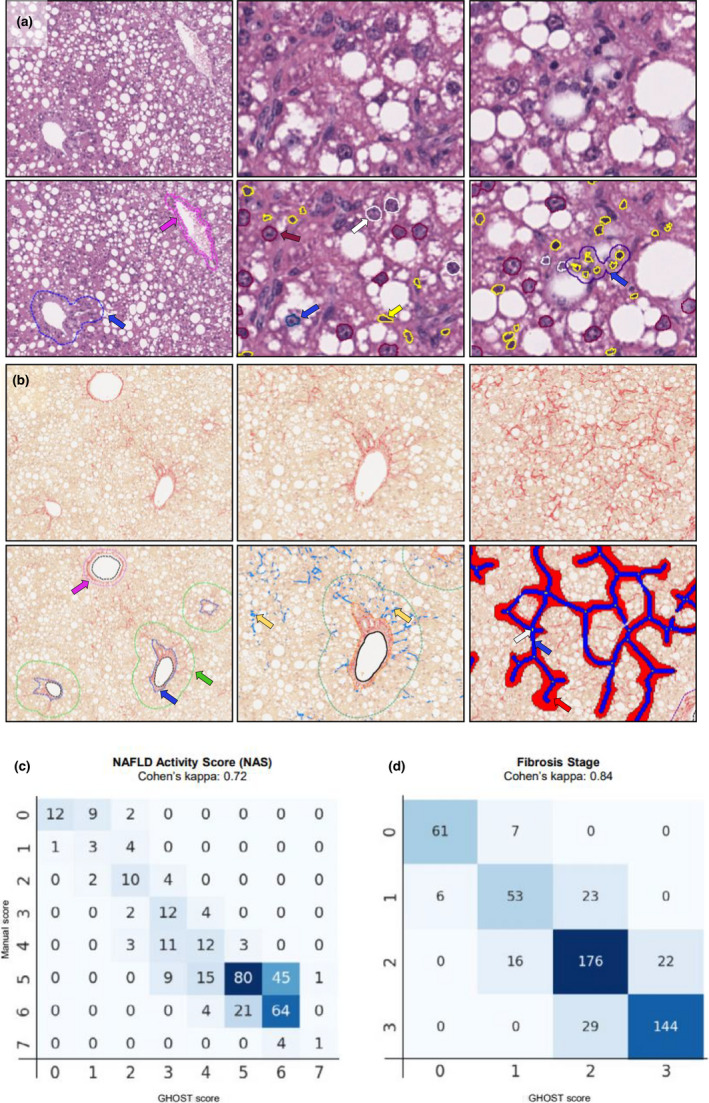
Deep learning‐based assessment of NAFLD Activity Score (NAS) and fibrosis stage in GAN DIO‐NASH mice. (a) Steatosis, inflammation, and hepatocyte ballooning degeneration scores were evaluated on scanned hematoxylin‐eosin (HE) stained slides for annotation, scoring and histomorphometric analysis of histopathological hallmarks of NASH. Left panel: Deep learning‐based detection (GHOST application) of portal tract (blue arrow) and central vein (pink arrow) at 10× magnification. Middle panel: Detection of hepatocytes with lipid droplets (purple), hepatocytes without lipids (white), ballooning hepatocytes (blue), and inflammatory cells (yellow) at 20x magnification. Right panel: By post‐processing, inflammatory foci were defined as a cluster of >3 inflammatory cells (blue). (b) Fibrosis stage was evaluated on scanned PSR‐stained slides. Left panel: Deep learning‐based detection of portal tract (blue arrow) and central veins (pink arrow) at 10× magnification. The boundary of the periportal zone was defined as 100 µm from the portal tract (green). Middle panel: Detection of collagen fibers (yellow arrows) in the periportal and sinusoidal zones. Right panel: Detection of bridging fibrosis (blue color), bridging zones (red color) and branch points (white arrow). (c, d) Correlation of manual versus GHOST‐based assessment of NAS and fibrosis stage. DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; GHOST, Gubra Histopathological Objective Scoring Technology; NAFLD, nonalcoholic fatty liver disease; PSR, picro‐Sirius Red.
Disease progression in GAN DIO‐NASH mice
Metabolic, histological, and transcriptome markers of NASH were profiled longitudinally in DIO‐NASH mice fed the GAN diet for 28–88 weeks. DIO‐NASH mice showed significant body weight gain compared to age‐matched chow‐fed mice. Weight gain in DIO‐NASH mice was relatively similar across the GAN diet feeding periods applied (GAN DIO‐NASH mice, 43–47 ± 0.7–1.7 g; chow‐fed controls, 33–34 ± 0.7–1.2 g, p < 0.001). Manifest NASH (NAS 4–5) was consistently observed at 28 weeks (Figure 2a). Severe steatosis (score 3) was evident from 28–48 weeks followed by a gradual decline over the remainder of the study (Figure 2a). Lobular inflammation increased in severity up to 58 weeks and remained maximal (score 3) thereafter (Figure 2a). Fibrosis progressed over the entire study period with consistent development of advanced fibrosis (stage F3) from 48 weeks and onwards (Figure 2a). Progressive decline in NAS from 58 weeks was explained by reduced steatosis (Figure 2a). When present, ballooning degeneration was mild (Figure 2a). Macroscopic hepatic neoplasms were first detected at 48 weeks with 100% incidence around 58–68 weeks, and increased progressively in number and size during the remainder of the study period (Figure 2b,c). The tumors showed pushing growth as indicated by a clear compression zone between the neoplastic and normal liver parenchyma (Figure 2d). The tumor cells were hepatocytic in nature and demonstrated cytologic atypia, including increased nuclear/cytoplasmic ratio with or without nuclear pleomorphism. All 17 tumors evaluated (1–13 mm in diameter) demonstrated extensive or complete loss of reticulin trabecular framework, a morphological characteristic of HCC (Figure 2d). Some tumors showed severe steatosis whereas others were essentially devoid of steatotic vesicles, both patterns markedly contrasting the surrounding liver tissue. Progressive increases in NAS‐associated quantitative steatosis and inflammation markers were observed (Figure 2e,f). Galectin‐3 also showed progressive increases over the feeding periods characterized (Figure 2f,g). Fibrosis scoring‐associated quantitative markers were significantly elevated from 28 weeks, reaching a plateau around 58–68 weeks (Figure 2e,f). The α‐SMA and Col1a1 expression was significantly increased over the entire study period (Figure 2f,g). Global gene expression signatures in GAN DIO‐NASH mice were markedly changed compared to chow controls (Figure S2A). GAN DIO‐NASH mice demonstrated widespread regulations in candidate genes linked to NASH, fibrosis, and tumorigenesis (Figure S2B, C). A 25‐gene set signature of more advanced fibrosing NASH (fibrosis stage ≥F2) has been reported in a large NAFLD patient cohort study. 34 A total of 18 genes in this clinical core set (Akr1B10, Amkrd29, Ccl20, Cfap221, Clic6, Col1a1, Col1a2, Dtna, Dusp8, Epb41l4a, Fermt1, Itgbl1, Ltbp2, Pdgfa, Sctr, Stmn2, Thy1, and Tnfrsf12a) were significantly upregulated in DIO‐NASH mice.
FIGURE 2.
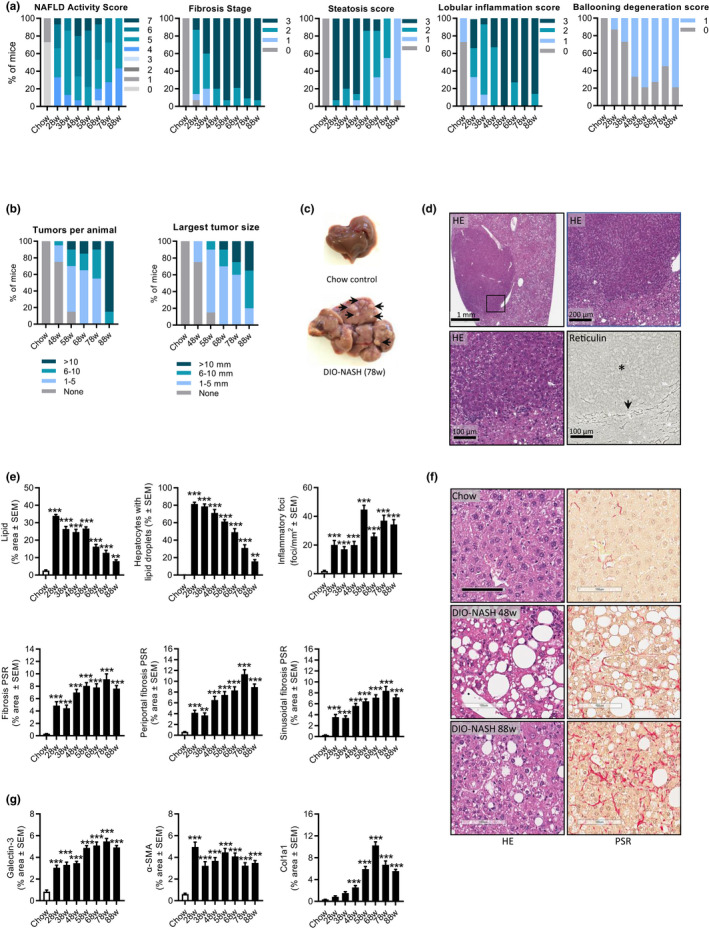
GAN DIO‐NASH mice show progressive development of severe fibrosing NASH and hepatocellular carcinoma. Mice were fed the GAN diet for 28–88 weeks (n = 11–15 per group). (a) NAFLD Activity Score (NAS), fibrosis stage, steatosis score, lobular inflammation score, and ballooning degeneration score. (b) Total number of hepatic tumors and largest liver tumor size (diameter, mm) per animal. (c) Representative whole‐liver of chow‐fed control and GAN DIO‐NASH mouse (78 weeks of feeding), respectively. The latter showing multiple tumors (arrows). (d) Cytologic and architectural characteristics of liver neoplastic lesions in DIO‐NASH mice (≥58 weeks of GAN diet feeding). The tumor demonstrates increased hepatocyte nuclear/cytoplasmic ratio (condensed cytoplasm with normal or enlarged nuclei) and absent reticulin trabecular framework (asterisk), a morphological characteristic of HCC. The compression zone between neoplastic and normal liver parenchyma is indicated by an arrow. Representative photomicrophraphs of HE‐ and reticulin‐stained tumor sections. Insert in upper left panel (HE staining) is further magnified in upper right and lower left panel. (e) GHOST‐based histomorphometrics. Proportionate (%) area of lipids; hepatocytes with lipid droplets relative to total hepatocyte counts; number of inflammatory foci per mm2; percentage total sectional area of fibrosis¸ percentage area of periportal fibrosis; and percentage area of perisinusoidal fibrosis (picro‐Sirius Red [PSR]). (f) Representative photomicrographs of HE and PSR stainings illustrating the development of steatosis and perisinusoidal fibrosis in DIO‐NASH mice (scale bar, 100 µm). (g) Conventional histomorphometrics. Percentage area of galectin‐3; α‐SMA, and Col1a1. **p < 0.01, ***p < 0.001 (Dunnett’s test one‐factor linear model). There was a minor loss of mice related to increased aging and the extended high‐fat diet feeding periods applied. DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; GHOST, Gubra Histopathological Objective Scoring Technology; HE, hematoxylin‐eosin; NAFLD, nonalcoholic fatty liver disease; PSR, picro‐Sirius Red.
Dietary intervention improves disease hallmarks in GAN DIO‐NASH mice
Chow‐reversal led to a robust weight loss (8 weeks, 14.3 ± 2.4%, p < 0.001; 12 weeks, 21.4 ± 1.5%, p < 0.001), accompanied by almost complete reversal of hepatomegaly, hypercholesterolemia, elevated ALT levels, and liver lipid accumulation (Figures 3a, S3, Table S1). Improvements in liver histological endpoints were relatively similar following 8 and 12 weeks of chow‐reversal (Figures 3, 4, and S4). Chow‐reversal improved NAS (≥2‐point, Figures 3b, S4) due to significant reductions in steatosis scores and, to a lesser extent, lobular inflammation scores (Figures 3c and S4). Of note, ballooning degeneration was absent in GAN DIO‐NASH mice receiving chow‐reversal (Figures 3c and S4). Chow‐reversal promoted complete resolution of hepatocyte lipid accumulation, paralleled by reductions in inflammatory foci (Figure 3d,e). The effect on lobular inflammation scores and foci appeared more consistent after 8 weeks of chow‐reversal. Additional histomorphometric markers of steatosis (% lipid) and inflammation (galectin‐3) were lowered by chow‐reversal (Figure 4a,b). Chow‐reversal had no significant effect on fibrosis scores (Figures 3b and S4). Chow‐reversal significantly reduced whole‐section α‐SMA (8 and 12 weeks), but showed no significant effect on quantitative markers of fibrosis (% area of PSR and Col1a1; Figure 3d,e and Figure 4a,b). Improvements in liver transcriptome signatures were relatively similar following 8 and 12 weeks of chow‐reversal, with notable changes in gene expression signatures pointing to improved intrahepatic lipid and carbohydrate handling as well as lowered immune and fibrogenesis activity (Figure S5).
FIGURE 3.
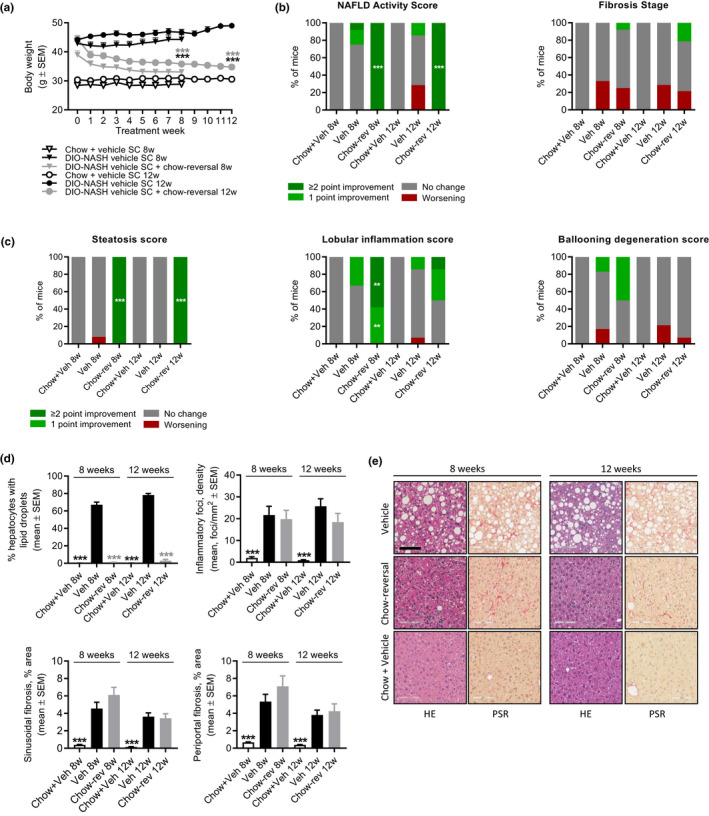
Dietary intervention improves liver histopathological hallmarks in GAN DIO‐NASH mice. DIO‐NASH mice were administered vehicle (s.c., q.d.) with (Chow‐reversal [Chow‐rev]) or without (Vehicle [Veh]) dietary intervention. Dietary intervention was performed by switching from the GAN diet to chow for 8 or 12 weeks (n = 12–14 per group). Age‐matched vehicle‐dosed (s.c.) chow‐fed mice (Chow + Veh) served as normal controls (n = 10–15 per group). Histopathological scores and histomorphometric assessment of corresponding histological parameters, as determined by the GHOST application. (a) Body weight. ***p < 0.001 (Dunnett’s test one‐factor linear model). (b) NAFLD Activity Score (NAS) and fibrosis stage. (c) Steatosis score, lobular inflammation score, and ballooning degeneration score. **p < 0.01, ***p < 0.001 (one‐sided Fisher’s exact test with Bonferroni correction). (d) Proportionate area (%) of hepatocytes with lipid droplets, number of inflammatory foci per mm2, sinusoidal fibrosis and periportal fibrosis. ***p < 0.001 (Dunnett’s test one‐factor linear model). (e) Representative photomicrographs showing disappearance of steatosis after chow‐reversal, whereas perisinuoidal fibrosis appears unchanged (scale bar, 100 µm). DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; GHOST, Gubra Histopathological Objective Scoring Technology; HE, hematoxylin‐eosin; NAFLD, nonalcoholic fatty liver disease; PSR, picro‐Sirius Red.
FIGURE 4.
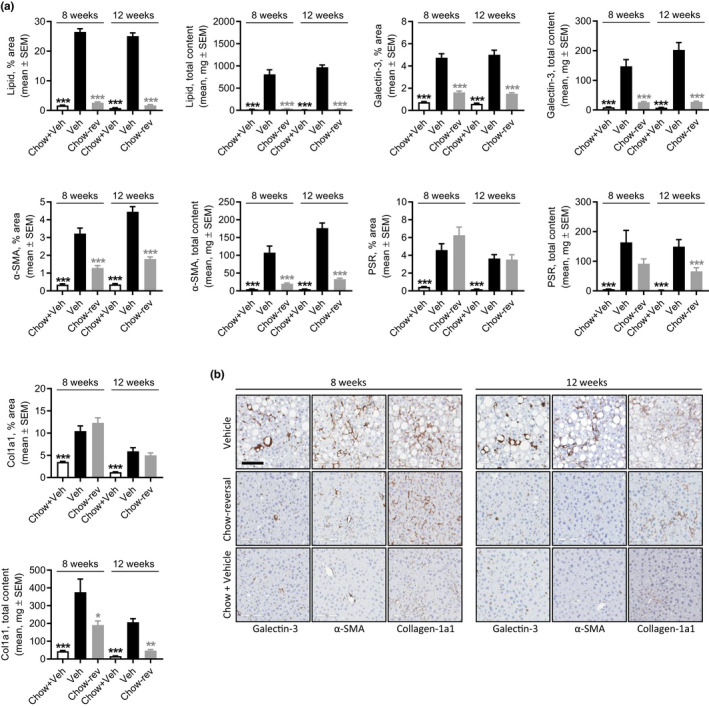
Dietary intervention improves quantitative histological markers of steatosis, inflammation and fibrosis in GAN DIO‐NASH mice. DIO‐NASH mice were administered vehicle (s.c. or q.d.) with (Chow‐reversal [Chow‐rev]) or without (Vehicle [Veh]) dietary intervention. Dietary intervention was performed by switching from the GAN diet to chow for 8 or 12 weeks (n = 12–14 per group). Age‐matched vehicle‐dosed (s.c.) chow‐fed mice (Chow + Veh) served as normal controls (n = 10–15 per group). (a) Histomorphometric assessment of the proportionate (%) area and estimated total liver content of lipids, galectin‐3, α‐SMA, PSR, and Col1a1 staining. (b) Representative photomicrographs showing decreased proportionate (%) area of galectin‐3, α‐SMA and Col1a1 after chow‐reversal (scale bar, 100 µm). DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; PSR, picro‐Sirius Red.
Differential efficacy profiles of semaglutide and lanifibranor in GAN DIO‐NASH mice
For both semaglutide and lanifibranor, therapeutic benefits on metabolic, histological, and transcriptional markers were generally achieved after 8 weeks of treatment. Robust weight loss was observed following treatment with semaglutide and lanifibranor (Figures 5a, S6, Tables S2, and S3). Maximal weight loss attained by semaglutide treatment occurred after ~ 2 weeks of dosing and was sustained after a total of 8 weeks (−26%) and 12 weeks (−24%) of treatment, respectively. In contrast, lanifibranor induced progressive weight loss over the entire treatment period (8 weeks, −23% and 12 weeks, −30%). Semaglutide, but not lanifibranor, transiently reduced food intake (Figure S6). Only semaglutide significantly reduced hepatomegaly, whereas both compounds improved transaminases, hypercholesterolemia, and liver lipid biochemistry (Tables S2 and S3). Whereas both compounds robustly reduced NAS after 8 and 12 weeks of treatment (≥2 points), only lanifibranor also significantly improved fibrosis stage after 12 weeks of treatment (≥1 point; Figures 5b, S7, and S8). For both compounds, improvements in NAS were predominantly driven by reductions in steatosis scores (Figure 5c, S7, and S8), although lanifibranor tended (p = 0.067) to improve lobular inflammation scores after 12 weeks of treatment (Figures 5c and S8). Both treatments significantly reduced hepatocyte lipid load, galectin‐3, and α‐SMA expression (Figure 5d,e, and Figure 6a,b). Although lanibranor significantly reduced inflammatory foci after both 8 and 12 weeks of treatment, semaglutide only reduced foci counts after 12 weeks of treatment (Figure 5d). Although the proportionate area of sinusoidal and periportal fibrosis was unaffected by both treatments (Figure 5d), whole‐section PSR‐staining was significantly reduced by lanifibranor after 12 weeks of treatment (Figure 6a,b). Both compounds had no effect on %‐area of Col1a1 (Figure 6a,b). Semaglutide significantly lowered total liver PSR and Col1a1 content as result of reduced hepatomegaly (Figure 6a). An extensive number of differentially expressed genes were determined in liver biopsies from GAN DIO‐NASH mice compared to chow‐fed controls. Relatively homogenous gene expression changes were observed as indicated by close within‐group clustering of global transcriptome signatures (Figure 7a). Liver global transcriptome responses were clearly different in the two drug treatment groups (Figure 7a). To obtain further resolution of hepatic transcriptome regulations, RNA sequencing data were probed for candidate genes linked to NASH pathology and fibrosis (Table S4). Consistent with more marked global gene expression changes, lanifibranor promoted widespread regulations within the seven defined gene categories, pointing to modulation of intrahepatic lipid, carbohydrate, and bile acid handling, as well as lowered pro‐inflammatory activity (Figure 7b). Both compounds significantly reduced the expression of several ECM‐associated genes. A web‐based gene expression data viewer (Gubra Gene Expression Experience [GGEX]) was developed to visualize global gene expression changes following administration of semaglutide and lanifibranor in GAN DIO‐NASH mice (Figure 7c). This open access resource can be used for further data mining on global gene expression changes (https://rnaseq.gubra.dk/).
FIGURE 5.
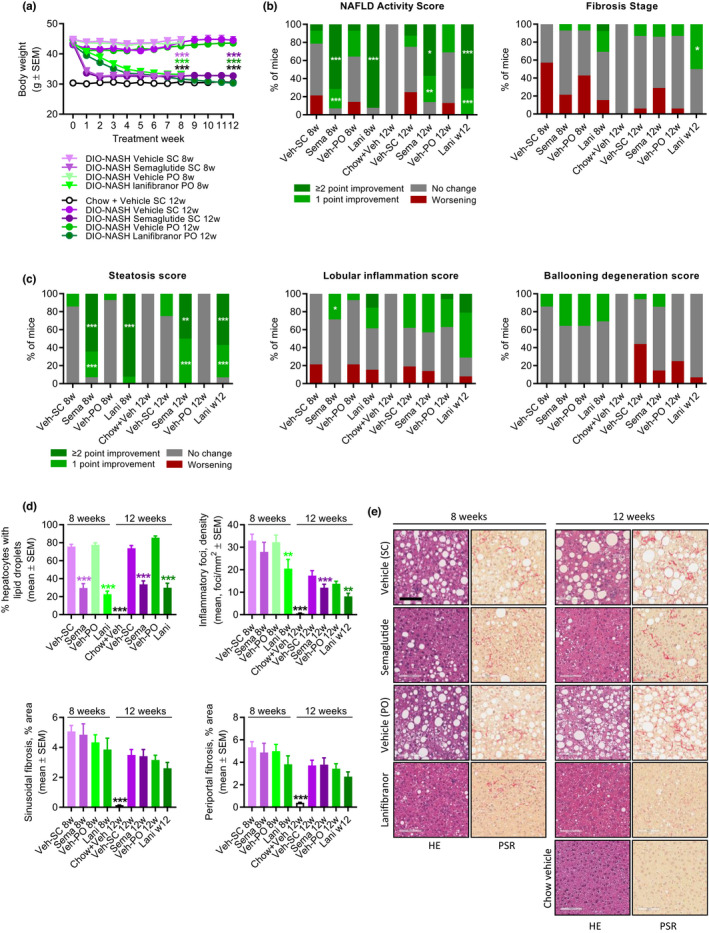
Semaglutide and lanifibranor differentially improves liver histopathological hallmarks in GAN DIO‐NASH mice. GAN DIO‐NASH mice were administered (q.d.) semaglutide (Sema, 30 nmol/kg, s.c.), lanifibranor (Lani, 30 mg/kg, p.o.), or corresponding vehicle (Veh, s.c., or p.o.) for 8 and 12 weeks, respectively (n = 13–16 per group). Chow‐fed mice receiving (q.d.) saline vehicle for 12 weeks (Chow + Veh) served as normal controls (n = 16). Histopathological scores and histomorphometric assessment of corresponding histological parameters as determined by the GHOST application. (a) Body weight. ***p < 0.001 (Dunnett’s test one‐factor linear model). (b) NAFLD Activity Score (NAS) and fibrosis stage. (c) Steatosis score, lobular inflammation score, and ballooning degeneration score. *p < 0.05, **p < 0.01, ***p < 0.001 (one‐sided Fisher’s exact test with Bonferroni correction). (d) Proportionate area (%) of hepatocytes with lipid droplets, number of inflammatory foci per mm2; sinusoidal fibrosis, and periportal fibrosis. **p < 0.01, ***p < 0.001 (Dunnett’s test one‐factor linear model). (e) Representative photomicrographs showing decreased proportionate (%) area of steatosis after semaglutide and lanifibranor treatment. Only lanifibranor improved fibrosis stage after 12 weeks of treatment (scale bar, 100 µm). DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; GHOST, Gubra Histopathological Objective Scoring Technology; HE, hematoxylin‐eosin; NAFLD, nonalcoholic fatty liver disease; PSR, picro‐Sirius Red.
FIGURE 6.
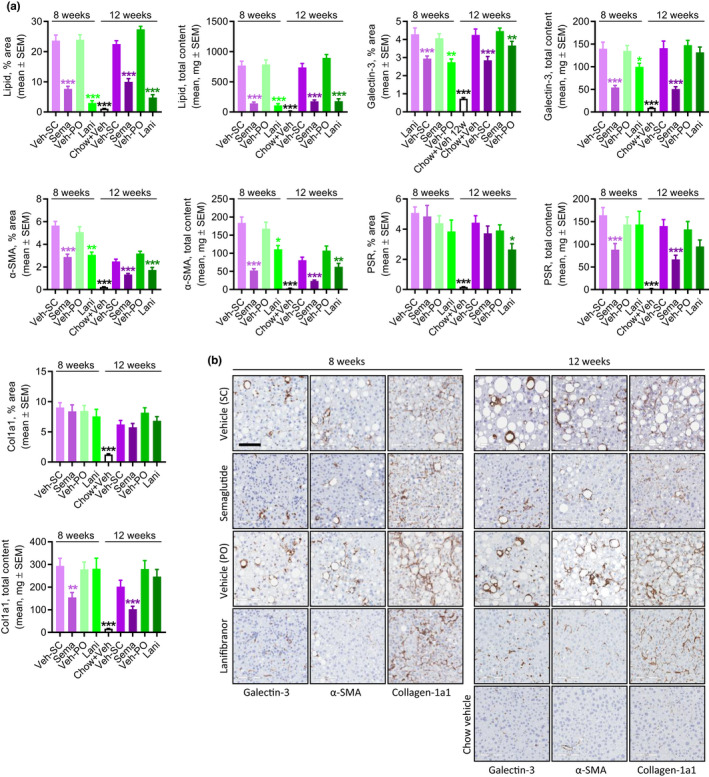
Semaglutide and lanifibranor differentially improves quantitative histological markers of steatosis, inflammation, and fibrosis in GAN DIO‐NASH mice. GAN DIO‐NASH mice were administered (q.d.) semaglutide (Sema, 30 nmol/kg, s.c.), lanifibranor (Lani, 30 mg/kg, p.o.), or corresponding vehicle (Veh, s.c., or p.o.) for 8 or 12 weeks (n = 13–16 per group). Chow‐fed mice receiving (q.d.) saline vehicle for 12 weeks (Chow + Veh) served as normal controls (n = 16). (a) Histomorphometric assessment of the proportionate (%) area and estimated total liver content of lipids, galectin‐3, α‐SMA, PSR, and Col1a1. *p < 0.05, **p < 0.01, ***p < 0.001 (Dunnett’s test one‐factor linear model). (b) Representative photomicrographs showing decreased proportionate (%) area of of galectin‐3, α‐SMA and Col1a1 after semaglutide and lanifibranor treatment. Only lanifibranor improved the percentage of the area of PSR staining after 12 weeks of treatment. Both treatments had no effect on the percentage of the area of Col1a1 staining (scale bar, 100 µm). DIO, diet‐induced obese; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet; PSR, picro‐Sirius Red
FIGURE 7.
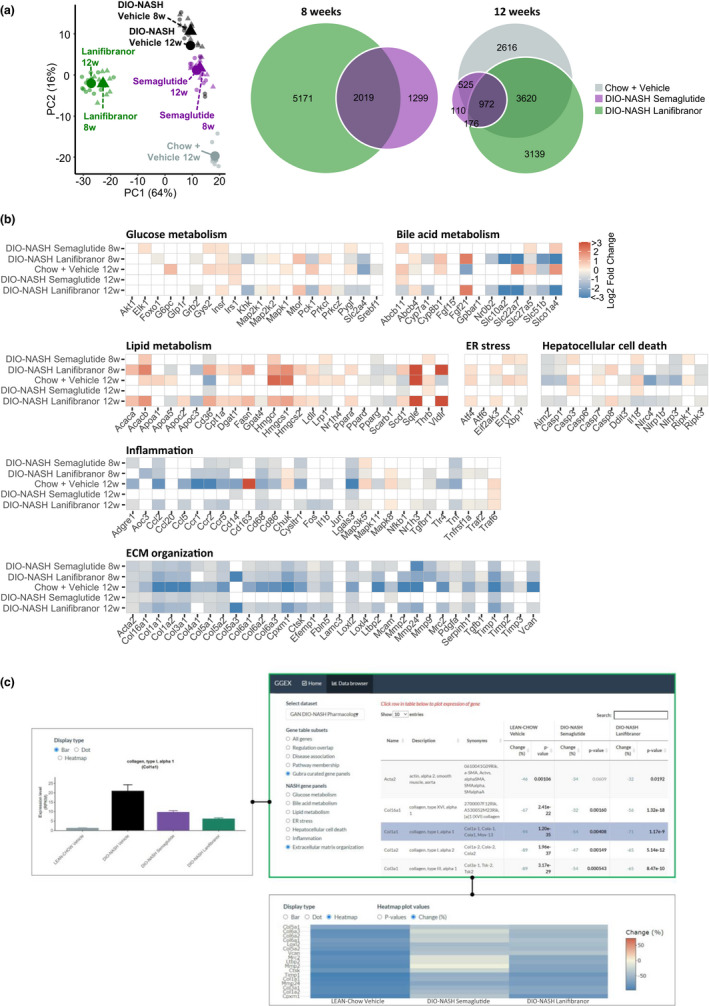
Changes in NASH‐linked hepatic gene expression signatures following drug treatment intervention in GAN DIO‐NASH mice. GAN DIO‐NASH mice were administered (q.d.) semaglutide (30 nmol/kg, s.c.), lanifibranor (30 mg/kg, p.o.), or corresponding vehicle (s.c. or p.o.) for 8 and 12 weeks, respectively. Chow‐fed mice receiving (q.d.) saline vehicle for 12 weeks (Chow + Vehicle) served as normal controls. (a) Left panel: Principal component analysis (PCA) of samples based on top 500 most variable gene expression levels. Right panel: Venn diagram of differentially expressed genes compared to corresponding vehicle‐dosed GAN DIO‐NASH mice. (b) Heatmaps illustrating changes in NASH and fibrosis‐associated candidate gene expression compared to corresponding vehicle‐dosed GAN DIO‐NASH mice. Color gradients in heatmaps indicate significantly upregulated (red color) or downregulated (blue color) gene expression (log2‐fold change, false discovery rate < 0.05). (c) Web‐based RNA sequencing data viewer. User interface of online gene expression data base system (Gubra Gene Expression Experience [GGEX], https://rnaseq.gubra.dk/) enabling browsing of complete RNA sequencing data from all mice included in pharmacological intervention studies in GAN DIO‐NASH mice. The RNA sequencing data are also accessible at the NCBI GEO database under accession no. GSE196908. Upper right panel: Gene panels filtered to show disease‐associated gene expression markers of extracellular matrix (ECM) organization (curated list). Table summarizing gene regulations (FDR adjusted p values, DEseq2 analysis). Relative (%) change in ECM gene expression in age‐matched chow‐fed controls (LEAN‐Chow Vehicle) and GAN DIO‐NASH mice administered semaglutide or lanifibranor for 12 weeks (values expressed as mean of n = 8–13 + S.E.M.). Statistical analysis as compared to vehicle‐dosed DIO‐NASH mice (DIO‐NASH Vehicle). The p values in bold indicates statistically significant regulation (p < 0.05). Red and blue numbers in the Change (%) column denote significant up‐ and downregulation of gene expression, respectively. Selected (highlighted in purple) of candidate gene of interest (collagen type 1 alpha‐1 [Col1a1]). Upper left panel: Graphical representation of Col1a1 gene expression levels. Col1a1 mRNA expression level as group average (mean RPKM value ± SEM). Lower panel: Heatmap plot depicting changes in ECM gene expression levels relative to vehicle control. DIO, diet‐induced obese; FDR, false discovery rate; GAN, Gubra‐Amylin non‐alcoholic steatohepatitis (NASH) diet.
DISCUSSION
GAN DIO‐NASH mice recapitulate the various histological stages of human disease, as determined by automated deep learning‐based image analysis. Late‐stage hepatic complications in GAN DIO‐NASH mice were characterized by advanced fibrosis and progressive tumor burden with a consistent HCC profile in the absence of cirrhosis. We observed a striking similarity between histological outcomes of semaglutide and lanifibranor treatment in GAN DIO‐NASH mice compared to corresponding clinical trials for NASH. Low‐caloric dietary intervention also promoted substantial benefits on metabolic, biochemical, and histological end points in GAN DIO‐NASH mice. Here, we also present an online interactive data browsing system for exploring global gene expression changes in response to pharmacological treatment in GAN DIO‐NASH mice. Collectively, our findings highlight the translatability and utility of GAN DIO‐NASH mice in preclinical drug development.
Currently accepted histological endpoints for conditional drug approval in precirrhotic NASH are based on manual semiquantitative histopathological scoring. 35 The NASH CRN scoring system is widely used in clinical trials and is largely reproducible in mouse models of NASH. 13 , 33 As for any manual scoring system, the NASH CRN scoring system has inherent limitations due to inter‐ and intra‐observer variability. 35 To overcome these challenges, we developed and implemented an automated deep learning‐based pipeline (GHOST) for objective and reproducible assessment of NASH CRN scores in GAN DIO‐NASH mice. Whole‐section NAS and fibrosis scores generated by the GHOST pipeline were in strong concordance with manual scoring by expert pathologists. Whereas single ordinal scoring provides discrete readouts on liver architectural changes, GHOST also enabled quantitative assessment of NASH histomorphological hallmarks in GAN DIO‐NASH mice.
In agreement with previous reports, 20 , 21 all morphological hallmarks of NASH and fibrosis were present in GAN DIO‐NASH mice at 28 weeks of dieting. Hepatocyte ballooning was consistently detected with extended GAN diet feeding periods, however, remained marginal, illustrating that hepatocyte lesion profiles in obese NASH mouse models do not meet human morphological criteria for prominent degeneration. 36 , 37 In GAN DIO‐NASH mice, NAS was therefore largely determined by changes in hepatosteatosis and inflammation. As observed in the clinic, DIO‐NASH mouse cohorts represented various stages of NAFLD and fibrosis for any dieting period characterized. Nevertheless, advanced fibrosis (stage F3) was observed ≥80% of the animals after 48 weeks of feeding. Histomorphometrics confirmed sequential development of severe steatosis, inflammation and fibrosis in GAN DIO‐NASH mice. To a large extent, GAN DIO‐NASH mice also reproduced regulations in core gene sets associated with progression to more advanced disease in patients with NASH. 21 , 34 , 38 The prevalence of severe steatosis decreased with advancing inflammation and fibrosis in GAN DIO‐NASH mice, resulting in low‐grade steatosis after 78 weeks of feeding. Prominent cytoarchitectural changes occurring during fibrosis progression in patients with NASH are often accompanied by gradual regression of initial steatosis, and can progress toward complete liver fat loss (burn‐out NASH) as a consequence of ensuing cirrhosis. 39 Although we did not observe transition from precirrhosis to manifest cirrhosis in GAN DIO‐NASH mice even after extremely long feeding periods, liver fat depletion may be an indirect marker of further advancing fibrotic injury in this model. HCC is a well‐known complication in both non‐cirrhotic and cirrhotic NASH patients. 5 , 6 DIO‐NASH models have generally low susceptibility for tumor development, which has prompted the addition of hepatic chemotoxins for reliable induction of HCC in these models. 40 One major drawback of such hybrid models is the gradual loss of metabolic features resulting in a hypercatabolic phenotype not mirroring the etiology and natural history of NASH. It is therefore noteworthy that GAN DIO‐NASH mice demonstrated a considerable tumor burden with reliable progression from advanced fibrosing NASH to tumor development as early as 58 weeks of GAN diet feeding. Also consistent with occurrence of liver neoplastic lesions, GAN‐DIO NASH mice acquired a pro‐oncogenic gene expression signature. 41 Tumors evaluated for malignancy demonstrated complete or marked loss of reticulin fiber architecture, a common characteristic of HCC. 39 In addition, the markedly deviating steatotic pattern in the tumor cells aided in identifying the tumors. Correspondingly, a compatible diet with similar nutrient composition (AMLN diet) has been reported to induce HCC after long‐term feeding. 42 In summary, the GAN DIO‐NASH mouse consistently presents with histological hallmarks of progressive NASH, severe fibrosis, and increasing tumor burden following long‐term GAN diet feeding. In further support of clinical translatability of the model, HCC develops in the context of natural disease progression and do not require induction by a chemical carcinogen. 13 It should be noted that the present longitudinal study was performed using male mice. Considering that sex differences exist in the prevalence, risk factors, clinical, and histological outcomes of NAFLD/NASH, 43 future studies should aim to address the metabolic and liver histological disease phenotype in GAN diet‐fed female mice.
Weight loss following intensive dietary intervention is directly correlated with improved liver histological outcomes in patients with NASH. 9 Therefore, we characterized therapeutic outcomes of long‐term low caloric diet feeding (“chow‐reversal”) in GAN DIO‐NASH mice. Chow‐reversal for 8–12 weeks induced a robust weight drop paralleled by nearly complete resolution of hypercholesterolemia, elevated plasma transaminase levels, hepatomegaly, and liver lipid accumulation. As expected, the improvements in metabolic and biochemical markers were paralleled by clear benefits on liver histological endpoints. Chow‐reversal effectively reduced NAS mostly due to reversal of steatosis. Highly stable collagen architecture was demonstrated by the observation that chow‐reversal for up to 12 weeks was not sufficient to improve fibrosis stage and quantitative collagen deposition. Because extracellular matrix (ECM) gene signatures indicated suppression of fibrogenesis activity at both 8 and 12 weeks in GAN DIO‐NASH mice, this invites the possibility that extended low‐caloric dieting could lead to improvements in fibrosis histology. It should be emphasized that low‐caloric dietary intervention was applied by switching from the GAN diet to to chow feeding over the entire intervention period. Chow‐reversal may therefore be perceived as a rigorous intervention paradigm facilitating substantial histological improvements in DIO‐NASH mice.
Semaglutide and lanifibranor represent different investigational drug concepts in advanced clinical development for NASH, 25 , 44 making it relevant to make a head‐to‐head comparison of their pharmacodynamics in GAN DIO‐NASH mice. Compound doses used in the present study were within ranges reported efficacious in DIO and DIO‐NASH mice. 27 , 28 , 45 , 46 Semaglutide, a long‐acting GLP‐1 receptor agonist currently approved for type 2 diabetes and obesity, 23 , 24 robustly reduced body weight, hepatomegaly, transaminases, and liver biochemical markers in GAN DIO‐NASH mice. It is noteworthy that similar beneficial metabolic effects of semaglutide have been reported in clinical trials for obesity 47 , 48 and NASH 25 as well as in various DIO mouse models. 45 , 46 In GAN DIO‐NASH mice, semaglutide‐induced therapeutic benefits on metabolic, histological, and transcriptional markers were established after 8 weeks of treatment. Improvements in NAS (≥2 points) and steatosis scores were substantiated by marked numerical reductions in lipid‐laden hepatocytes. Although lobular inflammation scores were unaffected, semaglutide reduced the number of inflammatory foci after 12 weeks of treatment, further arguing for applying AI‐assisted morphometrics on histopathological scoring variables. Semaglutide also reduced expression of galectin‐3, an inflammation marker particularly produced by activated macrophages. 49 Given the lack of hepatic GLP‐1 receptor expression in mice and humans, 50 , 51 beneficial effects of semaglutide on liver pathology are most likely indirect and may relate to beneficial effects on caloric intake, body weight, and insulin resistance. 45 , 52 Similar extrahepatic mechanisms have been proposed for other GLP‐1 receptor agonists tested in comparable DIO‐NASH mouse models. 31 It has been speculated that stimulated GLP‐1 receptor function may attenuate liver inflammation by limiting infiltration of GLP‐1 receptor‐expressing macrophages. 53 In addition, gut barrier‐protective mechanisms may potentially play a contributory role in mediating hepatoprotective effects of GLP‐1 receptor agonists. 54 Despite significant weight loss and robust reductions in NAS, semaglutide was unable to improve fibrosis histological end points in GAN DIO‐NASH mice. This observation is on par with a recent proof‐of‐concept clinical trial in patients with noncirrhotic NASH, demonstrating significant improvements in NAS with no worsening of fibrosis scores after 72 weeks of semaglutide treatment. 25 A closely related analogue, liraglutide, has demonstrated similar pharmacodynamics in patients with NASH 55 and various obese mouse models of NASH. 13 Although only indirectly supporting attenuation of profibrotic activity, semaglutide lowered α‐SMA levels, a reliable marker for activation of HSCs which are the principal collagen‐producing cells in the liver. 56 This effect was paralleled by suppression of several genes involved in ECM formation suggesting ongoing tissue remodeling towards lowered collagen formation. It may therefore be inferred that semaglutide reduced de novo collagen synthesis without stimulating clearance of pre‐existing fibers. Future studies must address whether antifibrotic efficacy of GLP‐1 receptor agonists can be achieved using extended treatment duration.
The pan‐PPAR (α/δ/γ) agonist lanifibranor has been reported to promote NASH resolution and fibrosis regression in a recent 24‐week clinical (NATIVE) trial in patients with noncirrhotic NASH. 26 Similar to semaglutide, therapeutic effects of lanifibranor in GAN DIO‐NASH mice were pronounced after 8 weeks of treatment. Histological outcomes of lanifibranor treatment in GAN DIO‐NASH mice were in close correspondence to clinical data. Compared to vehicle controls, lanifibranor robustly improved NAS (≥2 points). In contrast to semaglutide, lanifibranor promoted fibrosis regression (≥1 point) after 12 weeks of administration. Although significant improvements in NAS were predominantly attributed to reduced steatosis, lowered lobular inflammation score was observed in ≥70% of lanifibranor‐treated mice after 12 weeks of treatment. It is therefore possible that further histological improvements could be achieved by prolonged duration of lanifibranor treatment. Lanifibranor‐induced improvements in histological hallmarks were corroborated by reduced density of lipid‐laden hepatocytes, number of inflammatory foci, as well as galectin‐3, α‐SMA, and PSR‐staining. Liver‐directed mechanisms have been implicated in the hepatoprotective efficacy of lanifibranor, involving modulatory effects on hepatocyte lipid handling, macrophage polarization, and HSC function. 27 , 28 Although the primary indications for PPAR agonists are hyperlipidemia and type 2 diabetes, there is an increasing appreciation that this drug class may also be relevant in NASH management. Accordingly, PPAR isoforms have differential effects on metabolism, immune signaling, and collagen homeostasis, all of which have been implicated in the pathogenesis of NASH. 57 Various subtype‐selective PPAR agonists, including fibrates and thiazolidinediones, have demonstrated hepatoprotective effects in the context of NASH. 58 In particular, the dual PPAR‐α/δ agonist elafibranor has been extensively profiled in clinical trials and preclinical models of NASH. 31 , 59 In addition to stimulation of fatty acid mitochondrial β‐oxidation (PPAR‐α/β/δ), PPAR activation may attenuate NASH by direct suppression of macrophage (PPAR‐α/δ/γ) and HSC (PPAR‐α/γ) activity independent of improvements in lipid metabolism. 57 In agreement with a recent study in DIO‐NAFLD mice, 28 lanifibranor promoted weight loss, lowered plasma transaminases, and improved liver lipid biochemistry without attenuating hepatomegaly in GAN DIO‐NASH mice. Whereas plasma transaminase levels were also a consistent finding in the NATIVE trial, lanifibranor slightly increased body weight in patients with NASH compared to placebo. 44 The disparity in body weight effects in GAN DIO‐NASH mice and patients with NASH is likely explained by species differences in the expression, distribution, and function of mouse and human PPARs. Accordingly, PPAR agonist‐stimulated weight loss (PPAR‐α/δ) and peroxisomal proliferation‐associated liver hypertrophy (PPAR‐α) is observed in rodents, but not humans. 57 , 60 PPARα and PPAR‐δ agonist‐induced weight loss in DIO mice has been linked to suppression of central appetite function and induction of thermogenesis, which may indirectly contribute to their robust hepatoprotective effects in rodent studies. 61 , 62 , 63 , 64 Given that rodents and humans show differences in responsiveness to PPAR‐targeted drug therapies, studies in mouse models of NASH may be complemented using human‐based model systems. 65
CONCLUSION
The GAN DIO‐NASH mouse model demonstrates good clinical translatability with respect to disease etiology, histological hallmarks of NASH with advanced fibrosis, and progressive development of HCC. Importantly, the model also demonstrated clear therapeutic effects of compounds in late‐stage clinical development for NASH. In combination, these features highlight the utility of GAN DIO‐NASH mice for evaluation of novel drugs with potential efficacy in NASH and HCC.
CONFLICT OF INTEREST
M.B.M., K.T.T., D.O., A.O., C.G.S., M.R.M., M.F., and H.H.H. are employed by Gubra. S.S.V. was employed by Gubra and is presently employed by Novo Nordisk. L.P., E.S., and A.B. are employed by Boehringer Ingelheim Pharma GmbH & Co. KG. M.V. is employed by Aalborg University, Copenhagen, Denmark. N.V. and J.J. are owners of Gubra. This study was supported by Boehringer‐Ingelheim Pharma. No other potential conflicts of interest relevant to this article were reported.
AUTHORS CONTRIBUTIONS
S.S.V., A.B., E.S., N.V., J.J., M.F., and H.H.H. designed the research. M.B.M., K.T.T., D.O., A.O., C.G.S., M.R.M., L.P., and E.S. performed the research. M.B.M., S.S.V., K.T.T., D.O., A.O., C.G.S., M.R.M., L.P., M.V., E.S., A.B., N.V., J.J., M.F., and H.H.H. analyzed and interpreted the data. M.B.M., S.S.V., C.G.S., A.B., E.S., M.V., J.J., M.F., and H.H.H. wrote the manuscript.
Supporting information
Fig S1‐S8
Table S1‐S4
Møllerhøj MB, Veidal SS, Thrane KT, et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet‐induced obese and biopsy‐confirmed mouse model of NASH. Clin Transl Sci. 2022;15:1167‐1186. doi: 10.1111/cts.13235
Funding information
This work was supported by Boehringer‐Ingelheim Pharma GmbH. Research grants were provided to MBM from Innovation Fund Denmark (#9065‐00252) and the Danish Diabetes Academy which is funded by the Novo Nordisk Foundation (NNF17SA0031406).
REFERENCES
- 1. Younossi ZM, Henry L. Epidemiology of non‐alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021;3:100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown GT, Kleiner DE. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65:1080‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yki‐Järvinen H. Non‐alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2:901‐910. [DOI] [PubMed] [Google Scholar]
- 4. Younossi ZM. Non‐alcoholic fatty liver disease – a global public health perspective. J Hepatol. 2019;70:531‐544. [DOI] [PubMed] [Google Scholar]
- 5. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537‐2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ioannou GN. Epidemiology and risk‐stratification of NAFLD‐associated HCC. J Hepatol. 2021;75:1476‐1484. [DOI] [PubMed] [Google Scholar]
- 7. Younossi ZM, Stepanova M, Ong J, et al. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in the United States. Clin Gastroenterol Hepatol. 2020;19:580‐589.e5. [DOI] [PubMed] [Google Scholar]
- 8. Bedossa P. Diagnosis of non‐alcoholic fatty liver disease/non‐alcoholic steatohepatitis: Why liver biopsy is essential. Liver Int. 2018;38:64‐66. [DOI] [PubMed] [Google Scholar]
- 9. Hallsworth K, Adams LA. Lifestyle modification in NAFLD/NASH: facts and figures. JHEP Rep. 2019;1:468‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Albhaisi SAM, Sanyal AJ. New drugs for NASH. Liver Int. 2021;41:112‐118. doi: 10.1111/liv.14844 [DOI] [PubMed] [Google Scholar]
- 11. Neuschwander‐Tetri BA. Therapeutic landscape for NAFLD in 2020. Gastroenterology. 2020;158:1984‐1998.e3. [DOI] [PubMed] [Google Scholar]
- 12. Huisman TM, Dieterich DT, Friedman SL. Experimental and investigational targeted therapies for the management of fibrosis in NASH: an update. J Exp Pharmacol. 2021;13:329‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hansen HH, Feigh M, Veidal SS, Rigbolt KTG, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22:1707‐1718. [DOI] [PubMed] [Google Scholar]
- 14. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huby T, Gautier EL. Immune cell‐mediated features of non‐alcoholic steatohepatitis. Nat Rev Immunol. 2021. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411‐428. [DOI] [PubMed] [Google Scholar]
- 17. Farrell G, Schattenberg JM, Leclercq I, et al. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69:2241‐2257. [DOI] [PubMed] [Google Scholar]
- 18. Lim JS, Mietus‐Snyder M, Valente A, Schwarz J‐M, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251‐264. [DOI] [PubMed] [Google Scholar]
- 19. Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boland ML, Oró D, Tølbøl KS, et al. Towards a standard diet‐induced and biopsy‐confirmed mouse model of non‐alcoholic steatohepatitis: Impact of dietary fat source. World J Gastroenterol. 2019;25:4904‐4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hansen HH, Ægidius HM, Oró D, et al. Human translatability of the GAN diet‐induced obese mouse model of non‐alcoholic steatohepatitis. BMC Gastroenterol. 2020;20:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansen HH, Hansen G, Secher T, et al. Animal models of type 2 diabetes, obesity and nonalcoholic steatohepatitis – clinical translatability and applicability in preclinical drug development. In: Krentz A, Weyer C, Hompesch M, eds. Translational Research Methods in Diabetes, Obesity, and Nonalcoholic Fatty Liver Disease. 2nd ed. Springer; 2019:369‐403. [Google Scholar]
- 23. Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol. 2019;10:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. FDA approves new drug treatment for chronic weight management, first since 2014. FDA News Release [Internet]. 2021. https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐new‐drug‐treatment‐chronic‐weight‐management‐first‐2014
- 25. Newsome PN, Buchholtz K, Cusi K, et al. A placebo‐controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113‐1124. [DOI] [PubMed] [Google Scholar]
- 26. Francque SM, Bedossa P, Ratziu V, et al. A randomized, controlled trial of the Pan‐PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547‐1558. [DOI] [PubMed] [Google Scholar]
- 27. Boubia B, Poupardin O, Barth M, et al. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) α/γ/δ triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem. 2018;61:2246‐2265. [DOI] [PubMed] [Google Scholar]
- 28. Lefere S, Puengel T, Hundertmark J, et al. Differential effects of selective‐ and pan‐PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. J Hepatol. 2020;73:757‐770. [DOI] [PubMed] [Google Scholar]
- 29. Kristiansen MNB, Veidal SS, Rigbolt KTG, et al. Obese diet‐induced mouse models of nonalcoholic steatohepatitis‐tracking disease by liver biopsy. World J Hepatol. 2016;8:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR. The role of nausea in food intake and body weight suppression by peripheral GLP‐1 receptor agonists, exendin‐4 and liraglutide. Neuropharmacology. 2012;62:1916‐1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tølbøl KS, Kristiansen MN, Hansen HH, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J Gastroenterol. 2018;24:179‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baandrup Kristiansen MN, Veidal SS, Christoffersen C, et al. Validity of biopsy‐based drug effects in a diet‐induced obese mouse model of biopsy‐confirmed NASH. BMC Gastroenterol. 2019;19:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 34. Govaere O, Cockell S, Tiniakos D, et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med. 2020;12:eaba4448. [DOI] [PubMed] [Google Scholar]
- 35. Ratziu V. A critical review of endpoints for non‐cirrhotic NASH therapeutic trials. J Hepatol. 2018;68:353‐361. [DOI] [PubMed] [Google Scholar]
- 36. Liang W, Menke AL, Driessen A, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One. 2014;9:e115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denk H, Abuja P, Zatloukal K. Animal models of NAFLD from the pathologist’s point of view. Biochim Biophys Acta Mol Basis Dis. 2019;1865:929‐942. [DOI] [PubMed] [Google Scholar]
- 38. Suppli MP, Rigbolt KTG, Veidal SS, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal‐weight individuals. Am J Physiol Liver Physiol. 2019;316:G462‐G472. [DOI] [PubMed] [Google Scholar]
- 39. Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;128:837‐847. [DOI] [PubMed] [Google Scholar]
- 40. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical models for studying NASH‐driven HCC: how useful are they? Cell Metab. 2019;29:18‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang G, Wang Q, Liang N, et al. Oncogenic driver genes and tumor microenvironment determine the type of liver cancer. Cell Death Dis. 2020;11:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmoll D, Ziegler N, Viollet B, et al. Activation of adenosine monophosphate‐activated protein kinase reduces the onset of diet‐induced hepatocellular carcinoma in mice. Hepatol Commun. 2020;4:1056‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burra P, Bizzaro D, Gonta A, et al. Clinical impact of sexual dimorphism in non‐alcoholic fatty liver disease (NAFLD) and non‐alcoholic steatohepatitis (NASH). Liver Int. 2021;41:1713‐1733. [DOI] [PubMed] [Google Scholar]
- 44. Francque S, Bedossa P, Ratziu V, Anstee Q, Bugianesi E. The PanPPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non‐cirrhotic NASH: results of the NATIVE Phase 2b trial. Hepatology. 2020;72:9A‐11A. [Google Scholar]
- 45. Gabery S, Salinas CG, Paulsen SJ, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5:e133429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rakipovski G, Rolin B, Nøhr J, et al. The GLP‐1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE‐/‐ and LDLr‐/‐ mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci. 2018;3:844‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rubino D, Abrahamsson N, Davies M, et al. Effect of continued weekly subcutaneous semaglutide vs placebo on weight loss maintenance in adults with overweight or obesity: The STEP 4 randomized clinical trial. JAMA. 2021;325:1414‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davies M, Færch L, Jeppesen OK, et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double‐blind, double‐dummy, placebo‐controlled, phase 3 trial. Lancet. 2021;397:971‐984. [DOI] [PubMed] [Google Scholar]
- 49. Henderson NC, Sethi T. The regulation of inflammation by galectin‐3. Immunol Rev. 2009;230:160‐171. [DOI] [PubMed] [Google Scholar]
- 50. Panjwani N, Mulvihill EE, Longuet C, et al. GLP‐1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE −/− mice. Endocrinology. 2013;154:127‐139. [DOI] [PubMed] [Google Scholar]
- 51. Pyke C, Heller RS, Kirk RK, et al. GLP‐1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155:1280‐1290. [DOI] [PubMed] [Google Scholar]
- 52. Fonseca VA, Capehorn MS, Garg SK, et al. Reductions in insulin resistance are mediated primarily via weight loss in subjects with type 2 diabetes on semaglutide. J Clin Endocrinol Metab. 2019;104:4078‐4086. [DOI] [PubMed] [Google Scholar]
- 53. Wang Y, Parlevliet ET, Geerling JJ, et al. Exendin‐4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br J Pharmacol. 2014;171:723‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bang‐Berthelsen CH, Holm TL, Pyke C, et al. GLP‐1 induces barrier protective expression in brunner’s glands and regulates colonic inflammation. Inflamm Bowel Dis. 2016;22:2078‐2097. [DOI] [PubMed] [Google Scholar]
- 55. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non‐alcoholic steatohepatitis (LEAN): a multicentre, double‐blind, randomised, placebo‐controlled phase 2 study. Lancet. 2016;387:679‐690. [DOI] [PubMed] [Google Scholar]
- 56. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 57. Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity‐induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13:36‐49. [DOI] [PubMed] [Google Scholar]
- 58. Choudhary NS, Kumar N, Duseja A. Peroxisome proliferator‐activated receptors and their agonists in nonalcoholic fatty liver disease. J Clin Exp Hepatol. 2019;9:731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tacke F, Weiskirchen R. Non‐alcoholic fatty liver disease (NAFLD)/non‐alcoholic steatohepatitis (NASH)‐related liver fibrosis: mechanisms, treatment and prevention. Ann Transl Med. 2021;9:729‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bentley P, Calder I, Elcombe C, Grasso P, Stringer D, Wiegand HJ. Hepatic peroxisome proliferation in rodents and its significance for humans. Food Chem Toxicol. 1993;31:857‐907. [DOI] [PubMed] [Google Scholar]
- 61. Perreault M, Will S, Panza D, et al. Modulation of nutrient sensing nuclear hormone receptors promotes weight loss through appetite suppression in mice. Diabetes Obes Metab. 2010;12:234‐245. [DOI] [PubMed] [Google Scholar]
- 62. Larter CZ, Yeh MM, Van Rooyen DM, Brooling J, Ghatora K, Farrell GC. Peroxisome proliferator‐activated receptor‐α agonist, Wy 14 643, improves metabolic indices, steatosis and ballooning in diabetic mice with non‐alcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27:341‐350. [DOI] [PubMed] [Google Scholar]
- 63. Rachid TL, Penna‐de‐Carvalho A, Bringhenti I, Aguila MB, Mandarim‐de‐Lacerda CA, Souza‐Mello V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet‐induced male obese mice. Mol Cell Endocrinol. 2015;402:86‐94. [DOI] [PubMed] [Google Scholar]
- 64. Harrington WW, Britt CS, Wilson JG, et al. The effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan agonists on body weight, body mass, and serum lipid profiles in diet‐induced obese AKR/J mice. PPAR Res. 2007;97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boeckmans J, Natale A, Rombaut M, et al. Anti‐NASH drug development hitches a lift on PPAR agonism. Cells. 2019;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8
Table S1‐S4