

Abstract
The ubiquitous ribonucleoprotein (RNP) form of RNase P catalyzes the Mg2+-dependent cleavage of the 5′ leader of precursor-transfer RNAs. The rate and fidelity of the single catalytic RNA subunit in the RNase P RNP is significantly enhanced by association with protein cofactors. While the bacterial RNP exhibits robust activity at near-physiological Mg2+ concentrations with a single essential protein cofactor, archaeal and eukaryotic RNase P are dependent on up to five and ten protein subunits, respectively. Archaeal RNase P–whose proteins share eukaryotic homologs–is an experimentally tractable model for dissecting in a large RNP the roles of multiple proteins that aid an RNA catalyst. We describe protocols to assemble RNase P from Methanococcus maripaludis, a methanogenic archaeon. We present strategies for tag-less purification of four of the five proteins (the tag from the fifth is removed post-purification), an approach that helps reconstitute the RNase P RNP with near-native constituents. We demonstrate the value of native mass spectrometry (MS) in establishing the accurate masses (including native oligomers and modifications) of all six subunits in M. maripaludis RNase P, and the merits of mass photometry (MP) as a complement to native MS for characterizing the oligomeric state of protein sub-complexes. We showcase the advantage of native MS and MP over SDS-PAGE in revealing time-dependent modifications (e.g., oxidation) and aggregation of protein subunits, thereby providing insights into the decreased function of RNase P assembled with aged preparations of recombinant subunits. Our protocols and cautionary findings are applicable to studies of other cellular RNPs.
Keywords: ribonucleoprotein, protein oligomerization, protein modification, mass spectrometry, mass photometry
1. Introduction
RNase P is an essential and ubiquitous ribonucleoprotein (RNP) complex that catalyzes Mg2+-dependent cleavage of the 5′ leader of precursor-transfer RNAs (pre-tRNAs) (Altman, 2007; Esakova & Krasilnikov, 2010; Evans, Marquez, & Pace, 2006; Lai, Vioque, Kirsebom, & Gopalan, 2010). The RNase P RNP holoenzyme is comprised of a single catalytic RNase P RNA (RPR) that associates with a variable number of RNase P protein (RPP) subunits depending on the domain: one in bacteria, up to five in archaea, and up to 10 in eukarya (Esakova & Krasilnikov, 2010; Evans et al., 2006; Gopalan, Jarrous, & Krasilnikov, 2018; Lai et al., 2010). While the bacterial RPR in the absence of its single essential protein subunit is catalytically active, albeit at high ionic strength and Mg2+ concentrations, the cleavage activities of the archaeal and eukaryotic RPRs alone are considerably lower or, in some cases, undetectable (Guerrier-Takada, Gardiner, Marsh, Pace, & Altman, 1983; Kikovska, Svard, & Kirsebom, 2007; Pannucci, Haas, Hall, Harris, & Brown, 1999; Pulukkunat & Gopalan, 2008). However, in the presence of protein subunits, RNase P exhibits robust cleavage activity at lower Mg2+ concentrations (Guerrier-Takada et al., 1983; Pulukkunat & Gopalan, 2008; Sun & Harris, 2007; Tsai, Pulukkunat, Woznick, & Gopalan, 2006). With the multi-protein archaeal and eukaryotic RNase P, addition of one or more protein subunits to the RNA results in fractional pre-tRNA cleavage activity compared to the complete holoenzyme, whereas the addition of all protein subunits restores full catalytic activity at near-physiological Mg2+ (Chen, Pulukkunat, Cho, Tsai, & Gopalan, 2010; Chen et al., 2012; Lai et al., 2017; Perederina, Berezin, & Krasilnikov, 2018). Thus, archaeal and eukaryotic RNase P are appealing models for investigating the individual and collective roles of multiple protein co-factors in aiding RNA catalysis.
To dissect the contributions of each of the RPPs to RNP holoenzyme assembly and to pre-tRNA substrate recognition and cleavage, it is essential to obtain homogeneous preparations of the RNA and protein subunits for subsequent reconstitution. Archaeal RNase P, which has fewer (albeit all homologous) protein subunits compared to the eukaryotic relative, is biochemically tractable in vitro. In fact, the RNase P holoenzymes from several archaea have been successfully reconstituted (Cho, Lai, Susanti, Mukhopadhyay, & Gopalan, 2010; Fukuhara et al., 2006; Kouzuma et al., 2003; Lai et al., 2017). Pre-tRNA cleavage assays of step-wise assemblies of the RPR with partial and complete suites of all the recombinant protein subunits (POP5, RPP30, RPP21, RPP29, and L7Ae) helped establish the functional contributions of each RPP. Four of the five proteins operate as binary complexes: POP5·RPP30 and RPP21·RPP29 (Tsai et al., 2006). While POP5·RPP30 enhances the pre-tRNA cleavage rate, RPP21·RPP29 increases the binding affinity for substrate (Chen et al., 2010; Pulukkunat & Gopalan, 2008; Tsai et al., 2006). The fifth archaeal RPP, L7Ae, has been shown to further increase catalytic efficiency while reducing the Mg2+ ion requirement to near-physiological concentrations (Cho et al., 2010).
Here, we provide the protocols for in vitro reconstitution of RNase P from Methanococcus maripaludis (M. maripaludis), a methanogenic archaeon (Cho et al., 2010). We describe preparation of the RPR by T7 RNA polymerase (T7 RNAP)-catalyzed in vitro transcription (IVT) and present affinity tag-less strategies for purification of four of the five RPPs to minimize the potential interference of tags during reconstitution of the RNase P holoenzyme. We also describe a protocol for a pre-tRNA cleavage assay to measure the activity of the reconstituted enzyme. Although we detail assembly of a complete holoenzyme, partial assemblies (i.e., lacking some subunits) can be reconstituted similarly to determine the individual functional contributions of the proteins.
In addition, we highlight two methods for measuring the mass of macromolecules: native mass spectrometry (MS) and mass photometry (MP), which offer a complementary, two-pronged approach to assess the quality of RNA/protein preparations and gain insights into the mechanism of time-dependent protein deactivation. The high mass accuracy of native MS enables detailed assessment of the quality of purified proteins by providing information on the accurate mass, oligomeric species present, and metal ion-binding states in a single experiment. Such details are not obtainable by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the preferred method for validating protein purification despite its lower mass accuracy and inability to shed light on native assemblies or stoichiometries. Interestingly, re-analysis by native MS of aged RPP stocks (~1 year post-purification) revealed that the individual RPPs can become heavily modified (e.g., oxidation), which in turn may account for the observed activity loss upon long-term storage at −20°C. The recently published cryo-electron microscopy structure of Methanocaldococcus jannaschii RNase P (Wan et al., 2019) provides hints as to possible protein folding or substrate-binding defects upon oxidation of select Cys, Met, and Trp residues in different RPPs.
On the other hand, MP, whose measurements are of lower mass accuracy than native MS, can be used to readily gain information on the oligomeric state of proteins either alone or in complexes (Marciano et al., 2021; Soltermann et al., 2020; Sonn-Segev et al., 2020). Unlike native MS, MP is compatible with non-volatile salts, such as those that are typically included in storage or assay buffers (e.g., Mg2+ and Tris). Thus, MP can be used to assess the oligomeric distribution of proteins under buffer conditions and notably at RPP concentrations identical to those used in activity assays. We find that the oligomeric states detected in two different preparations of POP5·RPP30 correlate with their activity differences. SDS-PAGE was predictably unable to discern the full range of non-native oligomeric states in these protein preparations, although faint bands corresponding to putative heterodimers could be observed even under denaturing conditions (see 3.6 and Conclusions).
Overall, although we have provided protocols for assembly and functional testing of M. maripaludis RNase P, our approaches can be easily adapted and the insights applied to other RNPs.
2. Materials
All protein overexpression (2.2), purification (2.3), native MS (2.4), and mass photometry (2.6) solutions should be prepared using reverse osmosis-filtered or double distilled/deionized water (ddH2O). In vitro transcription (2.1) and cleavage assay (2.5) solutions should be prepared using autoclaved ddH2O.
2.1. In vitro transcription (IVT) of M. maripaludis RNase P RNA (RPR)
10X T7 RNA polymerase (RNAP) IVT buffer: 400 mM Tris·HCl, pH 7.6 at room temperature (RT; typically 20–23°C); 240 mM MgCl2; 20 mM spermidine; 0.1% (v/v) Triton X-100
100 mM dithiothreitol (DTT), dissolved in ddH2O and stored at −20°C (see Note 1)
Nucleoside triphosphate (NTP) mix (25 mM each), diluted in ddH2O (see Notes 2 and 3)
T7 RNAP IVT template (see Note 4). The template used to transcribe M. maripaludis RPR was prepared as described previously (Cho et al., 2010)
Thermostable inorganic pyrophosphatase (TIPP; New England Biolabs), diluted to 0.2 U/μL in 1X T7 RNAP IVT buffer and 10% (v/v) glycerol
T7 RNAP (see Note 5)
1X Tris-boric acid-ethylenediaminetetraacetic acid (TBE; 89 mM Tris base, 89 mM boric acid, 2 mM EDTA)
2% (w/v) agarose gel (0.6 g agarose per 30 mL 1X TBE)
DNase I (10 U/μL; Roche)
Buffer-saturated phenol, pH 7.9 (IBI Scientific)
Chloroform, ≥ 99.9% (MilliporeSigma)
3,500 Da molecular weight cut-off (MWCO) dialysis tubing (BioDesign Inc., New York)
3 M sodium acetate, pH 5.2
100% (v/v) ethanol
75% (v/v) ethanol
2.2. Overexpression of M. maripaludis RNase P proteins (RPPs)
- Appropriate overexpression strains: in this protocol, the following strains of Escherichia coli were used:
- BL21(DE3) (Studier & Moffatt, 1986)
- BL21(DE3) Rosetta (Novagen)
- Recombinant plasmids containing genes encoding M. maripaludis RPPs under the control of a T7 promoter (see Note 6): the recombinant plasmids used in this protocol were described previously (Cho et al., 2010):
- pLANT-2b–POP5·RPP30
- pLANT-2b–RPP21
- pET-15b–(His)6-RPP29 (a thrombin cleavage site is included to allow removal of the (His)6 tag)
- pET-15b–L7Ae
Miller’s LB: Add 2.5 g tryptone, 2.5 g NaCl, 1.25 g yeast extract per 250 mL ddH2O. Sterilize by autoclaving.
LB agar: Add 1.5 g agar per 100 mL Miller’s LB. Sterilize by autoclaving.
- Antibiotics, prepared as described and stored at −20°C
- 35 mg/mL chloramphenicol, dissolved in 100% methanol
- 35 mg/mL kanamycin, dissolved in ddH2O
- 100 mg/mL carbenicillin, dissolved in 100% methanol
40% (w/v) glucose, dissolved in ddH2O and filter sterilized
1 M Isopropyl β-D-1-thiogalactopyranoside (IPTG; GoldBio), dissolved in ddH2O and stored at −20°C
100 mM ZnCl2
2.3. Purification of M. maripaludis RPPs
2.3.1. Purification of POP5·RPP30
POP5·RPP30/RPP21 lysis buffer: 25 mM Tris·HCl, pH 8 at RT; 5 mM DTT; 1 mM EDTA; 0.1 mM phenylmethylsulfonyl fluoride (PMSF); 1 M NaCl
Sonicator [e.g., Q125 Sonicator (Qsonica)]
10% (v/v) polyethyleneimine (PEI), diluted in ddH2O and pH adjusted to 8 (see Note 7)
Finely crushed (NH4)2SO4
Ion-exchange (IEX) buffer A: 25 mM Tris·HCl, pH 8 at RT; 5 mM DTT; 1 mM EDTA; 0.1 mM PMSF
0.4 μm syringe filters (VWR International)
1 mL HiTrap SP (Sulfopropyl)-Sepharose FF column (Cytiva; see Note 8)
IEX buffer B: 25 mM Tris·HCl, pH 8 at RT; 5 mM DTT; 1 mM EDTA; 0.1 mM PMSF; 2 M NaCl
15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel
1 mL HiTrap Phenyl HP column (Cytiva; see Note 8)
3,000 Da MWCO centrifugal concentrators [e.g., Amicon Ultra-15 (MilliporeSigma)]
3,500 Da MWCO dialysis tubing (BioDesign Inc., New York)
RPP storage buffer: 50 mM HEPES-KOH, pH 8 at RT; 500 mM ammonium acetate; 7.5 mM MgCl2
100% (v/v) glycerol
2.3.2. Purification of RPP21
2.3.3. Purification of RPP29
RPP29 buffer A: 25 mM Tris·HCl, pH 8 at RT; 1 mM EDTA; 500 mM NaCl; 10 mM imidazole
1 mL HisTrap HP column (Cytiva; see Note 8)
RPP29 buffer B: 25 mM Tris·HCl, pH 8 at RT; 1 mM EDTA; 500 mM NaCl; 1 M imidazole
1X phosphate buffered saline (PBS): 137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; 1.7 mM KH2PO4; pH 7.4
Thrombin (1 U/μL; Cytiva)
See 2.3.1 for a list of other common purification materials.
2.3.4. Purification of L7Ae
L7Ae lysis buffer: 100 mM Tris·HCl, pH 9 at RT; 5 mM DTT; 0.1 mM PMSF
L7Ae IEX buffer A: 25 mM Tris·HCl, pH 8 at RT; 1 mM DTT; 1 mM EDTA; 0.1 mM PMSF
1 mL HiTrap Q (quaternary ammonium) FF column (Cytiva; see Note 8)
L7Ae IEX buffer B: 25 mM Tris·HCl, pH 8 at RT; 1 mM DTT; 1 mM EDTA; 0.1 mM PMSF; 2 M NaCl
18% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) SDS gel
See 2.3.1 for a list of other common purification materials.
2.4. Validation of RPR and RPPs by native MS
Pierce 96-well microdialysis plate, 3,500 Da MWCO (Thermo Fisher Scientific)
Alternative: Micro Bio-Spin P6 column (Bio-Rad Laboratories)
500 mM ammonium acetate, 99.99% (MilliporeSigma)
0.4 mM magnesium acetate
-
0.5–10 μL μltra Micro Gel Tip (Genesee Scientific)
Alternative: Hamilton Syringe (10 μL, cemented needle, 26 gauge, 2 inch needle)
Borosilicate filament capillaries, OD 1.0 mm, ID 0.78 mm (Sutter Instrument)
P-97 Flaming/Brown Micropipette Puller (Sutter Instrument)
Nanospray Flex ion source fitted with a platinum wire electrode (Thermo Fisher Scientific)
-
Exactive Extended Mass Range (EMR) mass spectrometer (Thermo Fisher Scientific) modified with quadrupole.
Alternative: Q Exactive Ultra-High Mass Range (UHMR) mass spectrometer (Thermo Fisher Scientific) or other mass spectrometers, with their compatible sources optimized for native MS.
2.5. Reconstitution of M. maripaludis RNase P
2X assay buffer: 100 mM Tris·HCl, pH 7.5 at 37°C; 1 M NH4OAc; 15 mM MgCl2
RPP dilution buffer: 50 mM Tris·HCl, pH 7.5 at 37°C; 500 mM NH4OAc; 7.5 mM MgCl2; 5% (v/v) glycerol
50% (v/v) glycerol
2 mg/mL bovine serum albumin (BSA; Pierce)
Precursor-tRNA (pre-tRNA) substrate (see Note 9), both unlabeled and radiolabeled (see Notes 10 and 11)
Quench dye: 7 M urea, 10% (v/v) phenol, 0.04% (w/v) bromophenol blue, 0.04% (w/v) xylene cyanol
8% (w/v) polyacrylamide (19:1 acrylamide:bisacrylamide), 7 M urea gel
Storage phosphor screen (Cytiva)
Laser scanner with phosphor imaging mode [e.g., Typhoon RGB (Cytiva)]
2.6. Assessing RPP oligomerization by mass photometry
Refeyn One™ mass photometry system (Refeyn)
Precision Cover Glasses, #1.5H Thickness, 24 × 50 mm (Thorlabs)
Water bath sonicator
50% (v/v) methanol
100% (v/v) methanol
Nitrogen gas, industrial grade
CultureWell™ reusable gaskets, 50–3 mm diameter, 1 mm depth (3–10 μL) (Grace Bio-Labs)
100% (v/v) isopropyl alcohol
IMMOIL-F30CC low autofluorescence immersion oil (Olympus)
3. Methods
3.1. In vitro transcription (IVT) of M. maripaludis RNase P RNA (RPR)
- Prepare the IVT reaction mix. An example IVT reaction mix is tabulated below:
Incubate the reaction for 3–4 h at 37°C.
Check for successful IVT of the RPR by electrophoresing 1 μL of the transcription reaction on a 2% (w/v) agarose gel (see Note 13), using 1X TBE as running buffer.
After verifying synthesis of the expected RPR product, add DNase I (10 U per 100 μL IVT reaction) to the transcription reaction and incubate for 1 h at 37°C.
Add 1/2 volume each of phenol and chloroform to the transcription reaction, vortex vigorously, and centrifuge for 5 min at 20,000 × g.
Remove the top aqueous phase, and transfer it to a clean tube.
Add 1 volume of chloroform to the isolated aqueous phase, vortex vigorously, and centrifuge for 5 min at 20,000 × g.
Remove the top aqueous phase, and transfer it to a clean tube.
Adjust the volume of the isolated aqueous phase to 300 μL with ddH2O.
Transfer the sample to dialysis tubing (3,500 Da MWCO), and dialyze extensively against ddH2O (at least 3 × 4 L; the final round of dialysis should continue for at least 16 h).
Recover the sample from dialysis, and transfer it to a clean tube.
Add 1/10 volume 3 M sodium acetate (pH 5.2) and 2.5 volumes 100% (v/v) EtOH. Mix well, and incubate the mixture at −80°C for at least 1 h.
Thaw the sample, and centrifuge for 5 min at 20,000 × g.
Carefully withdraw the solution without disturbing the pellet. Wash the pellet with 70% (v/v) EtOH. Vortex vigorously, and centrifuge for 5 min at 20,000 × g. Then carefully withdraw the solution. Perform at least two EtOH washes in this manner (see Note 14).
Allow the pellet to dry, and then resuspend it in ddH2O. Vortex vigorously to ensure complete resuspension.
Measure the concentration of the resuspended RPR by UV-Vis (calculated ε = 2,562,400 M−1 cm−1).
Store at −20°C.
3.2. Overexpression of M. maripaludis RNase P proteins (RPPs)
- Transform the appropriate strain of E. coli competent cells with a single plasmid containing a gene encoding one (two in the case of POP5·RPP30) of the M. maripaludis RPPs.
- BL21(DE3) cells are used for overexpression of POP5·RPP30 and RPP21.
- BL21(DE3) Rosetta cells are used for overexpression of RPP29 and L7Ae.
Plate the transformation reaction on LB agar plates supplemented with either 35 μg/mL kanamycin (POP5·RPP30 and RPP21) or 35 μg/mL chloramphenicol and 100 μg/mL carbenicillin (RPP29 and L7Ae). Incubate the plate overnight at 37°C.
Start a seed culture by inoculating 2 mL LB media [supplemented with appropriate antibiotics and 1% (w/v) glucose (see Note 15)] with a single transformant.
Shake the seed culture for 15–16 h at 37°C.
Start a large overexpression culture by inoculating 250 mL LB media (supplemented with appropriate antibiotics) with 250 μL of seed culture.
Shake the overexpression culture at 37°C until the OD600 reaches ~0.5 (about 2–2.5 h).
- Induce overexpression with the addition of the appropriate amount of IPTG inducer.
- Overexpression of POP5·RPP30 or L7Ae is initiated by addition of 2 mM IPTG.
- Overexpression of RPP21 or RPP29 is initiated by addition of 0.5 mM IPTG. For RPP21, 1 mM ZnCl2 (final concentration) is also added to the overexpression culture at the time of IPTG addition.
- Continue shaking the overexpression culture under previously optimized conditions.
- Overexpression cultures for POP5·RPP30 and L7Ae are shaken for 4 h at 37°C.
- Overexpression cultures for RPP21 and RPP29 are shaken for 15 h at 21°C
Following overexpression, harvest the cells by centrifugation for 10 min at 5,000 × g.
Divide the resulting pellet into 2 × 125-mL pellets, and freeze them at −−80°C until ready to use.
3.3. Purification of M. maripaludis RPPs
3.3.1. Purification of POP5·RPP30
Remove a single 125-mL pellet from −80°C, and thaw on ice.
Once thawed, resuspend the pellet in 25 mL POP5·RPP30/RPP21 lysis buffer.
Lyse the resuspended cells by sonicating for a total on-time of 6 min at 50% amplitude with pulse cycles of 5 s on and 2 s off. Keep the cells on ice during sonication.
Centrifuge the lysate for 15 min at 12,000 × g and 4°C.
Transfer the cleared lysate to a clean 50 mL Erlenmeyer flask, and incubate for 15 min at 65°C.
Centrifuge the sample for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean tube, and add 0.05% (v/v) PEI (final concentration). Incubate on ice for 20 min (see Note 7).
Centrifuge the suspension for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean beaker, and place on ice.
Slowly add finely crushed (NH4)2SO4 with stirring until the solution reaches 80% saturation. After the addition of (NH4)2SO4, continue stirring on ice for an additional 15 min.
Centrifuge the suspension for 15 min at 12,000 × g and 4°C (see Note 16).
Remove the supernatant and resuspend the pellet in 20 mL IEX buffer A.
Filter the solution using a 0.4 μm syringe filter to remove any undissolved particulate.
Load the sample onto a 1 mL HiTrap SP FF column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 column volumes (CVs) IEX buffer A, and collect the flowthrough.
- Attach the column to an FPLC, and wash it as follows:
- 5 mL IEX buffer A
- 5 CV gradient to 25% IEX buffer B
- 20 CV gradient to 60% IEX buffer B
Use the UV absorbance profile to identify the peak fractions, and test aliquots from these fractions on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the fractions with the highest concentration of POP5·RPP30 and the fewest impurities. POP5·RPP30 typically elutes between 600–750 mM NaCl.
Adjust the NaCl concentration to 2 M by adding solid NaCl and nutating gently until all the solid NaCl dissolves.
Load the sample onto a 1 mL HiTrap Phenyl HP column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 CVs of IEX buffer B, and collect the flowthrough.
Attach the column to an FPLC, and wash it with a 20 CV gradient to 0% IEX buffer B.
Use the UV absorbance profile to identify the peak fractions, and test aliquots from these fractions on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the peak fractions as above. POP5·RPP30 typically elutes as a single peak between 1,300–600 mM NaCl.
Concentrate the protein as desired using a centrifugal concentrator (see Note 17).
Transfer the protein to dialysis tubing (3,500 Da MWCO), and dialyze against RPP storage buffer (500 mL) at 4°C for 2 h. Replace the buffer, and allow the second round of dialysis to continue overnight.
Recover the sample, and measure the protein concentration by UV-Vis [calculated ε for (POP5 + RPP30) = 41,830 M−1 cm−1].
Add glycerol to a final concentration of 25% (v/v).
Aliquot and store at −20°C.
3.3.2. Purification of RPP21
Remove a single 125-mL pellet from −80°C and thaw on ice.
Once thawed, resuspend the pellet in 25 mL POP5·RPP30/RPP21 lysis buffer.
Lyse the resuspended cells by sonicating for a total on-time of 6 min at 50% amplitude with pulse cycles of 5 s on and 2 s off. Keep the cells on ice during sonication.
Centrifuge the lysate for 15 min at 12,000 × g and 4°C.
Transfer the sample to a clean tube, and add 0.05% (v/v) PEI (final concentration). Incubate on ice for 20 min (see Note 7).
Centrifuge the suspension for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean beaker, and place on ice.
Slowly add finely crushed (NH4)2SO4 with stirring until the solution reaches 60% saturation. After the addition of (NH4)2SO4, continue stirring on ice for an additional 15 min.
Centrifuge the suspension for 15 min at 12,000 × g and 4°C (see Note 16).
Remove the supernatant, and resuspend the pellet in 20 mL IEX buffer A.
Filter the solution using a 0.4-μm syringe filter to remove any undissolved particulate.
Load the sample onto a 1 mL HiTrap Heparin HP column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 CV of IEX buffer A and collect the flowthrough.
- Attach the column to an FPLC and wash it as follows:
- 20 CV gradient to 50% IEX buffer B
- 10 CV gradient to 100% IEX buffer B
Use the UV absorbance profile to identify the peak fractions, and test aliquots from these fractions on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the fractions with the highest concentration of RPP21 and the fewest impurities. RPP21 typically elutes between 750–1,000 mM NaCl.
Adjust the NaCl concentration to 2 M by adding solid NaCl and nutating gently until all the solid NaCl dissolves.
Load the sample onto a 1 mL HiTrap Phenyl HP column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 CV of IEX buffer B, and collect the flowthrough.
- Attach the column to an FPLC and wash it as follows:
- 5 CV gradient to 50% IEX buffer B
- 10 CV gradient to 0% IEX buffer B
Use the UV absorbance profile to identify the peak fractions, and test aliquots from these fractions on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the fractions with the highest concentration of RPP21 and the fewest impurities. RPP21 typically elutes at <500 mM NaCl.
Concentrate the protein as desired using a centrifugal concentrator (see Note 17).
Transfer the protein to dialysis tubing (3,500 Da MWCO), and dialyze against RPP storage buffer (500 mL) at 4°C for 2 h. Replace the buffer, and allow the second round of dialysis to continue overnight.
Recover the sample and measure the protein concentration by UV-Vis (calculated ε = 11,460 M−1cm−1).
Add glycerol to a final concentration of 25% (v/v).
Aliquot and store at −20°C.
3.3.3. Purification of RPP29
Remove a single 125-mL pellet from −80°C and thaw on ice.
Once thawed, resuspend the pellet in 25 mL RPP29 buffer A.
Lyse the resuspended cells by sonicating for a total on-time of 6 min at 50% amplitude with pulse cycles of 5 s on and 2 s off. Keep the cells on ice during sonication.
Centrifuge the lysate for 15 min at 12,000 × g and 4°C.
Transfer the sample to a clean tube, and add 0.05% (v/v) PEI (final concentration). Incubate on ice for 20 min (see Note 7).
Centrifuge the suspension for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean tube, and filter it using a 0.4 μm syringe filter to remove any particulate.
Load the sample onto a 1 mL HisTrap HP column using a syringe or a peristaltic pump, and collect the flowthrough.
- Attach the column to an FPLC and wash it as follows:
- 5 CV RPP29 buffer A
- 10 CV gradient to 50% RPP29 buffer B
- 6 CV 50% RPP29 buffer B
Use the UV absorbance profile to identify the peak fractions, and test aliquots from these fractions on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the fractions with the highest concentration of RPP29 and the fewest impurities. RPP29 typically elutes at >200 mM imidazole.
Transfer the protein to dialysis tubing (3,500 Da MWCO), and dialyze against 500 mL 1X PBS for 1.5 h at RT (see Note 18).
Open the dialysis bag, and add thrombin according to manufacturer’s instructions.
Resume dialysis against 500 mL fresh 1X PBS for 2.5 h at RT, and then continue dialyzing overnight at 4°C.
Check for the extent of cleavage by thrombin on a 15% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Concentrate the protein as desired using a centrifugal concentrator (see Note 17).
Transfer the protein to dialysis tubing (3,500 Da MWCO), and dialyze against RPP storage buffer (500 mL) at 4°C for 2 h. Replace the buffer, and allow the second round of dialysis to continue overnight.
Recover the sample and measure the protein concentration by UV-Vis (calculated ε = 2,980 M−1cm−1; see Note 19).
Add glycerol to a final concentration of 25% (v/v).
Aliquot and store at −20°C.
3.3.4. Purification of L7Ae
Remove a single 125-mL pellet from −80°C and thaw on ice.
Once thawed, resuspend the pellet in 25 mL L7Ae lysis buffer.
Lyse the resuspended cells by sonicating for a total on-time of 6 min at 50% amplitude with pulse cycles of 5 s on and 2 s off. Keep the cells on ice during sonication.
Centrifuge the lysate for 15 min at 12,000 × g and 4°C.
Transfer the cleared lysate to a clean 50 mL Erlenmeyer flask, and incubate for 15 min at 65°C.
Centrifuge the sample for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean tube, and add 0.05% (v/v) PEI (final concentration). Incubate on ice for 20 min (see Note 7).
Centrifuge the suspension for 15 min at 12,000 × g and 4°C.
Transfer the supernatant to a clean tube, and filter it using a 0.4 μm syringe filter to remove any particulate.
Load the sample onto a 1 mL HiTrap Q FF column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 CV of L7Ae IEX buffer A, and collect the flowthrough.
Attach the column to an FPLC and wash it with a 20 CV gradient to 25% L7Ae IEX buffer B
Check the UV peak fractions on an 18% (w/v) polyacrylamide (37.5:1 acrylamide:bisacrylamide), 0.4% (w/v) sodium dodecyl sulfate (SDS) gel.
Pool the fractions with the highest concentration of L7Ae and the fewest impurities. L7Ae typically elutes at 200–360 mM NaCl.
Adjust the NaCl concentration to 2 M by adding solid NaCl and nutating gently until all the solid NaCl dissolves.
Load the sample onto a 1 mL HiTrap Phenyl HP column using a syringe or a peristaltic pump, and collect the flowthrough.
Wash the column with 10 CV of IEX buffer B, and collect the flowthrough.
Verify the presence of L7Ae in the load and wash flowthrough fractions.
-
21.
Pool the load and wash flowthrough fractions, and concentrate the protein as desired using a centrifugal concentrator (see Note 17).
-
22.
Transfer the protein to dialysis tubing (3,500 MWCO) and dialyze against RPP storage buffer (500 mL) at 4°C for 2 h. Replace the buffer, and allow the second round of dialysis to continue overnight.
-
23.
Recover the sample and measure the protein concentration by UV-Vis (calculated ε = 4,470 M−1cm−1; see Note 19).
-
24.
Add glycerol to a final concentration of 25% (v/v).
-
25.
Aliquot and store at −20°C.
3.4. Validation of RPR and RPPs by native MS
Remove the RPR and RPP stocks from −20°C, and thaw. The RPR can be thawed at RT, while the RPPs should remain on ice.
Dilute the RPR to 1 μM using 500 mM ammonium acetate, 0.4 mM magnesium acetate.
Transfer an aliquot of each RPP stock into individual microdialysis wells (3,500 Da MWCO), and dialyze them against 500 mM ammonium acetate. Change the well solution once every hour for up to 8 changes. If needed, dialysis can continue overnight (see Note 20).
Recover the dialyzed protein solutions and measure their concentrations by UV-Vis (see 3.3.1, 3.3.2, 3.3.3, and 3.3.4).
Dilute each of the RPPs to 1–10 μM using native MS solution.
- Pull multiple glass emitters with the micropipette puller (Figure 1). Emitter pulling parameters for a P-97 Flaming/Brown Micropipette Puller can vary (see user manual). An example of parameter ranges is given below:
Parameter Setting Heat *Ramp heat ± 10 Pull 15–25 Velocity 15–25 Delay (ms) 150–250 *Determined by conducting a pilot ramp test Pipette (Ultra Micro Gel Tip) 2–5 μL of an RPR or RPP dilution into a glass emitter (see Note 21).
Load the emitter onto the platinum wire that is fitted to the Nanospray Flex ion source.
Place the tip of the emitter centered and less than 1 cm from the front of the inlet of the mass spectrometer.
Adjust the spray voltage between 0.6–1.3 kV until stable signal is observed. The EMR mass spectrometer tune settings for positive ion mode electrospray ionization are detailed in Table 1.
Record the full mass spectrum for each sample in this fashion (Figure 2). Collisional activation may be used as needed (see Note 22).
For protein complexes such as POP5·RPP30, adjust higher-energy collisional dissociation (HCD) voltage to cause complexes to dissociate into constituent proteins so that their masses may be measured accurately (see Note 22). Record the resulting dissociation spectrum (Figure 2E)
Process the recorded mass spectra by manually calculating charge state or by using deconvolution software like UniDec (Marty et al., 2015). The UniDec deconvolution parameters used to generate the deconvolved mass spectra in Figures 2 and 4 are listed in Table 2. The observed masses are reported in Table 3.
Figure 1. Four-fold magnification of the tips of emitters.
The general tip shape of nano electrospray ionization emitters that work well at the spray voltages specified in Table
Table 1.
EMR mass spectrometer tune settings used in this method
| Setting | Value | Notes |
|---|---|---|
| Spray voltage (kV) | 0.7–0.8 | First increase spray voltage past the recommended range, then gradually decrease it until a stable signal is observed. |
| Capillary temperature (°C) | 250 | |
| S-Lens RF level (V) | 200 | |
| Injection flatapole DC (V) | 12 | |
| Bent flatapole DC (V) | 10 | |
| Inter flatapole lens (V) | 8 | |
| HCD direct eV (V) | 1 | Higher HCD voltages can be used for fragmentation and for de-adducting. |
| Inject time (ms) | 200 | |
| Microscans | 2 | |
| Scan range (m/z) | 1,000–10,000 | |
| Resolution (at 200 m/z) | 8,750 | Adjust to higher resolution to resolve isotopic peaks. |
| Trapping gas pressure | 4–6 | Use larger values to improve higher m/z transmission. |
Figure 2. Deconvolved zero-charge mass spectra of the purified M. maripaludis RNase P subunits.
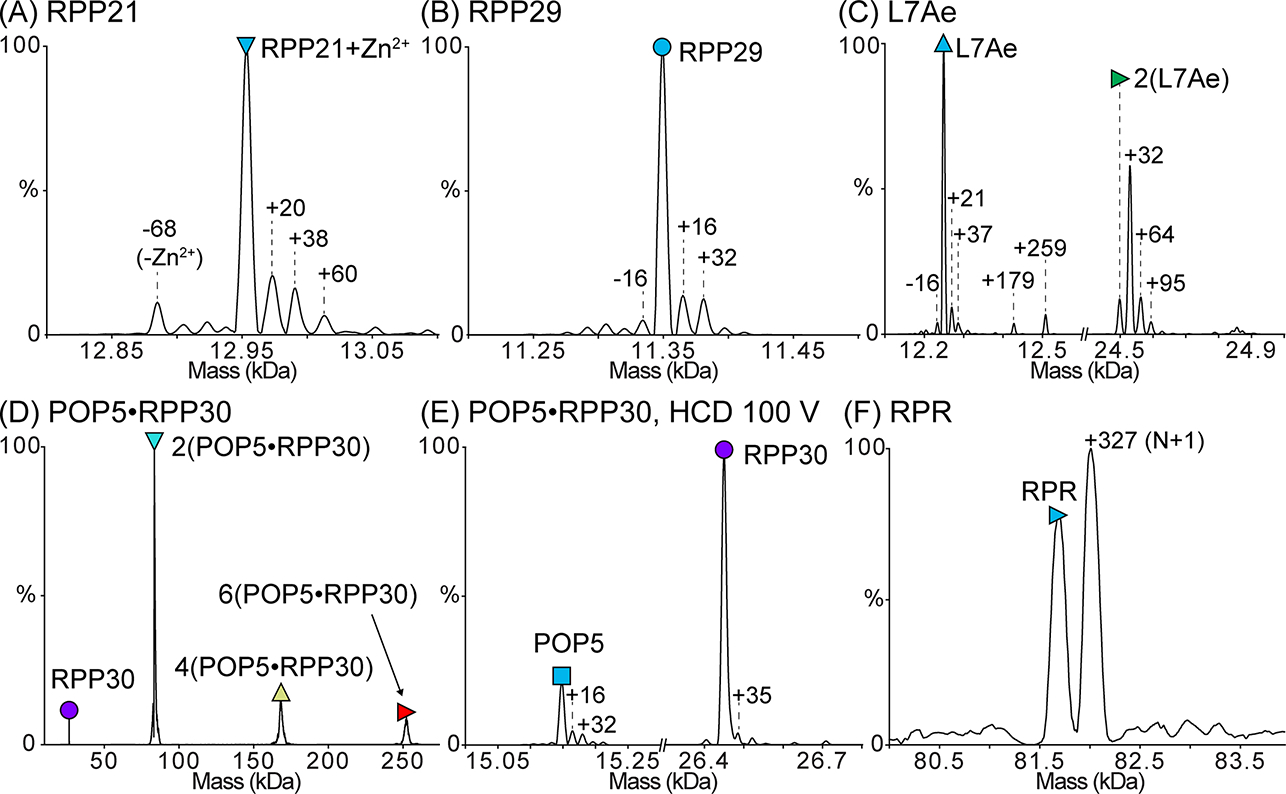
Proteins were dialyzed into 500 mM ammonium acetate prior to analysis. In each spectrum, the peak corresponding to the mass of the full-length subunit is indicated with a symbol. The expected and observed masses of each subunit are reported in Table 3. (A) Mass spectrum of RPP21 bound to Zn2+. Only a small population of RPP21 is Zn2+-free, indicating that Zn2+ remains tightly bound to RPP21 during all purification steps. (B) Mass spectrum of RPP29. (C) Mass spectrum of L7Ae. L7Ae readily forms a disulfide-mediated homodimer, which is lost upon treatment with DTT. The single-Cys-mediated dimer is also supported by the inability to dissociate it using HCD. (D) Mass spectrum of POP5·RPP30. This binary complex forms the expected heterotetramer [2(POP5·RPP30)] in addition to some higher-order oligomers. (E) Dissociation of the heterotetramer in (D) yields species with the expected masses for POP5 and RPP30. (F) Mass spectrum of RPR in 500 mM ammonium acetate and 0.4 mM Mg(OAc)2. Both the target RNA species (N) and a species extended by a single nucleotide (N+1), which is typical of T7 RNAP IVT, are observed. The mass spectrum confirms that the RPR is a mix of N and N+1 species with few contaminants (if any), suggesting that the doublet observed in a 2% (w/v) agarose gel likely results from alternative RPR conformations (see Note 13). In (A-E), mass differences (in Da) from the expected peak are indicated, and these differences may result from salt adducts or modifications, such as oxidation. Some of the adducted and/or modified peaks appear to have peak overlap as indicated by the peak asymmetry.
Figure 4. Deconvolved zero-charge mass spectra after quadrupole isolation and HCD dissociation of a 2(POP5·RPP30) charge state.
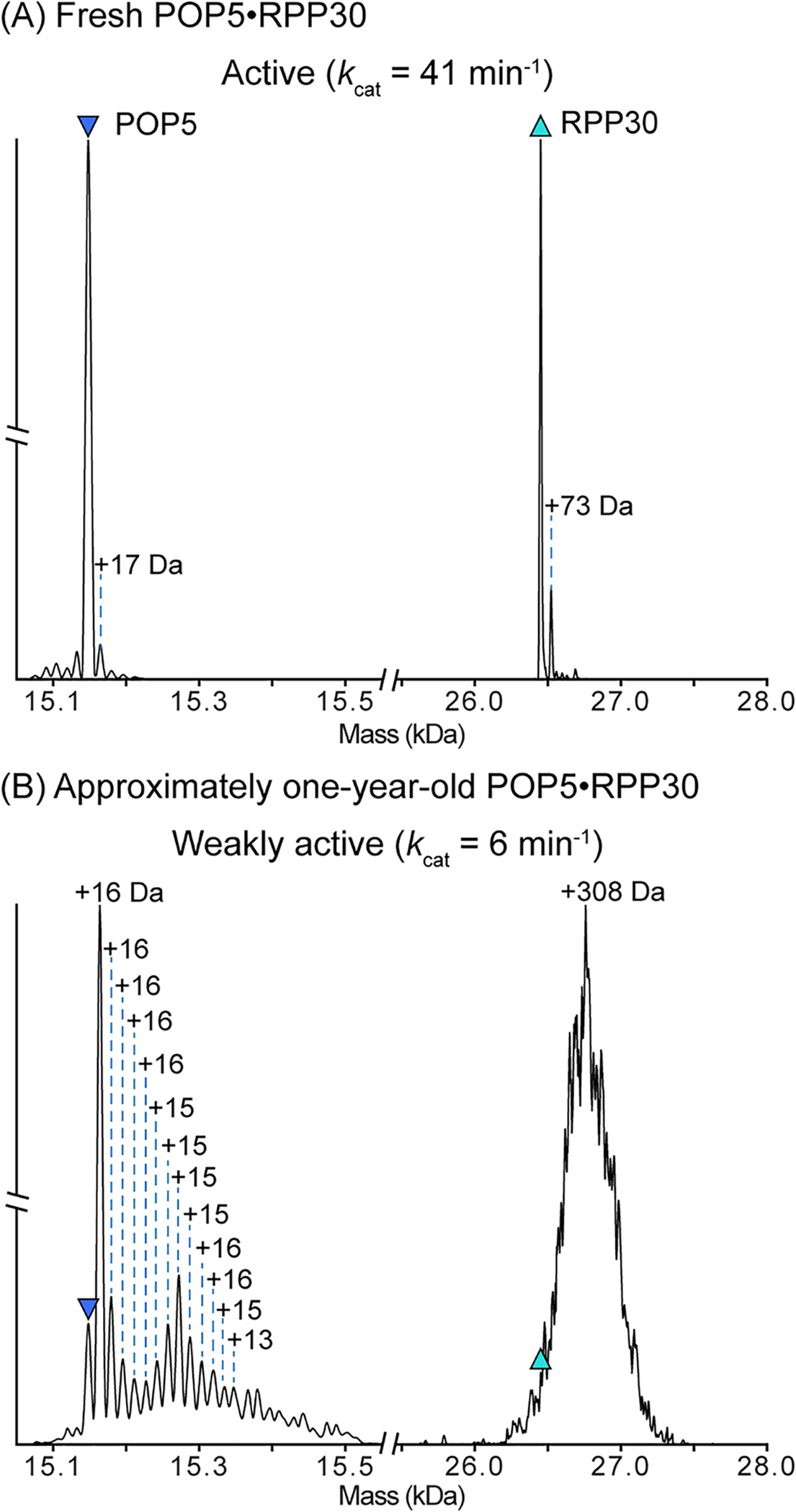
Dissociation of the same preparation of POP5·RPP30 within (A) one month and (B) approximately one year after overexpression and purification. Purified POP5·RPP30 is stored at −20°C as described in 3.3.1. Prior to analysis, POP5·RPP30 was dialyzed into 500 mM ammonium acetate. After one month (A), nearly all of the POP5 and RPP30 have the expected mass, indicating the majority of the protein in solution is unmodified. However, after one year (B), both proteins in the complex are heavily modified. The major POP5 peak now shows a single oxidation (+16 Da), and the presence of multiple additional masses correspond to further oxidations. Observed peak overlap and the peak distribution suggest that POP5 likely has other adducts and/or modifications that cannot be assigned. Similarly, the molecular weight of RPP30 has shifted by an average of +308 Da. The unresolved peaks suggest heterogeneous mass additions, which may include modifications, such as oxidation, and other tightly-bound adducts. M. maripaludis POP5 contains five Cys, Met, and Trp residues, which are all readily oxidized (Raftery, 2014). RPP30 has eight such residues. Thus, oxidation is a likely route for modification of these two protein subunits (RPP21 and RPP29 are similarly modified; data not shown). As indicated by the kcat values above each spectrum, the activity of M. maripaludis significantly decreases over time, perhaps due to modification of the protein subunits during storage.
Table 2.
UniDec deconvolution parameters used in this method
| Setting | Value | Notes |
|---|---|---|
| Data Processing | ||
| m/z range | 1,000–8,000 | Adjusted to the peak range in each spectrum. |
| Normalize Data | On | |
| Data Bin | Nonlinear, 0 | |
| UniDec Parameters | ||
| Charge Range | 1–60 | These ranges can be narrowed to be targeted for a known species mass range in a low-complexity spectrum. |
| Mass Range | 5,000–300,000 | |
| Sample Mass Every (Da) | 1 | Decrease based on resolution and desired mass accuracy. |
| Peak FWHM (Th) | 0.85 | |
| Peak Shape Function | Gaussian | |
| Beta | 0 | Adjusted to 50 for spectra with broad peaks. |
| Charge Smooth Width | 1 | |
| Point Smooth Width | 1 | Adjusted to 100 for spectra with broad peaks. |
| Mass Smooth Width | 0 | |
| Maximum # of Iterations | 100 | |
| m/z to Mass Transform | Smart | |
| Adduct Mass (Da) | 1.007276467 | The charge carrier (proton) in positive mode ESI. |
| Manual Mode | Off | Manual mode turned on to assign the +29 and +36 peaks in the POP5·RPP30 sample. |
| Native Charge Offset Range | +/− 1000 | |
| Peak Selection and Plotting | Adjusted to label peaks in the mass spectra. | |
Table 3.
Expected and observed masses of M. maripaludis RNase P subunits
| Subunit | Determined (Da) | Expected (Da) | Difference (Da) |
|---|---|---|---|
| RPP21+Zn | 12953 ± 0 | 12952 | 1 |
| RPP29 | 11349 ± 0 | 11350 | −1 |
| L7Ae | 12249 ± 0 | 12250 | −1 |
| 2 L7Ae | 24498 ± 4 | 24498 | 0 |
| 2(POP5·RPP30) | 83522 ± 49 | 83201 | 321 |
| 4(POP5·RPP30) | 168346 ± 474 | 166404 | 1942 |
| 6(POP5·RPP30) | 252289 ± 406 | 249606 | 2683 |
| POP5 | 15148 ± 0 | 15149 | −1 |
| RPP30 | 26450 ± 1 | 26452 | −2 |
| RPR | 81683 ± 3 | 81582 | 101 |
3.5. Reconstitution of M. maripaludis RNase P
An example cleavage assay mix is tabulated below. Step-by-step preparation of assay components, execution of the assay, and analysis of the data are detailed in the text that follows.
| Component | Initial concentration | Volume (μL) | Final concentration |
|---|---|---|---|
| Reaction buffer | - | 5 | - |
| Folded RPR | 6.25 nM | 2 | 0.625 nM |
| POP5·RPP30 | 312.5 nM | 2 | 31.25 nM |
| RPP21 | 312.5 nM | 2 | 31.25 nM |
| RPP29 | 312.5 nM | 2 | 31.25 nM |
| L7Ae | 312.5 nM | 2 | 31.25 nM |
| Pre-tRNA mix | 2 μM; 1,600 dpm/μL | 5 | 500 nM; 400 dpm/μL* |
Quenching 2.5–5 μL will allow loading of 1,000–2,000 disintegrations per minute (dpm) (see Note 23)
To fold the RPR, transfer a small aliquot of RPR to a thin-walled 0.5-mL PCR tube.
Transfer the PCR tube to a thermocycler, and incubate at 50°C for 50 min.
Drop the temperature to 37°C, and incubate for 10 min.
Add one volume of 2X assay buffer to the RPR, and continue incubating at 37°C for an additional 30 min.
Remove the PCR tube from the thermocycler, and keep the folded RPR at RT.
Dilute the folded RPR to 6.25 nM using 1X assay buffer.
Remove purified RPP stocks from −20°C, and keep on ice.
Dilute small aliquots of each of the RPP stocks to 312.5 nM using RPP dilution buffer. Add BSA to a final concentration of 0.1 mg/mL to each of the RPP dilutions. Keep the dilutions on ice.
Prepare reaction buffer consisting of 1X assay buffer, glycerol, and BSA. The amount of glycerol and BSA added to the reaction buffer should be adjusted so that the final glycerol and BSA concentrations are 2.5% (v/v) and 0.1 mg/mL, respectively, in the final assay mix.
In a thin-walled 0.5 mL PCR tube, prepare pre-tRNA mix consisting of 1X assay buffer, unlabeled pre-tRNA substrate, and trace amounts of 5′-[γ-32P]-radiolabeled or internally-[α-32P]-radiolabeled pre-tRNA substrate (see Notes 10 and 23).
Mix the folded RPR (final concentration 0.625 nM), RPPs (final concentration 31.25 nM each), and reaction buffer in a thin-walled 0.5 mL PCR tube, and incubate in a thermocycler at 37°C for 5 min.
During the last minute of the incubation in step 11, transfer the PCR tube containing the pre-tRNA mix to the thermocycler, and incubate at 37°C for 1 min.
At the end of the 5 min incubation in step 11, transfer an aliquot of the pre-tRNA mix to the tube containing the RPR and RPPs, and mix by pipetting to initiate the pre-tRNA cleavage reaction.
At pre-determined time points (see Note 24), remove an aliquot of the assay mix, and transfer it to tubes containing quench dye.
Load the quenched reaction samples onto an 8% (w/v) polyacrylamide (19:1 acrylamide:bisacrylamide), 7 M urea gel (16.5 cm × 16.5 cm), and electrophorese using 1X TBE as running buffer.
Once the bromophenol blue dye reaches 0.5–1 inch from the bottom of the 16.5 cm × 16.5 cm gel, terminate electrophoresis.
Carefully wrap the gel in plastic wrap.
Transfer the wrapped gel to a storage phopshor cassette, and expose it to a storage phosphor screen for the desired amount of time (see Note 25).
Scan the storage phosphor screen using a phosphorimager instrument, such as a Typhoon RGB (Cytiva) to obtain an image displaying the migration of the radiolabeled pre-tRNA substrate and cleavage products (Figure 3A).
Using software such as ImageQuant (Molecular Dynamics), determine the volumes of the substrate and product bands. The measured volumes can be used to calculate the concentration of cleaved pre-tRNA substrate as a function of time.
Using data analysis software such as Kaleidagraph (Synergy Software), plot cleavage product concentration versus time, and fit a straight line to the data to obtain initial velocity (Figure 3B).
Figure 3. Measuring the pre-tRNA cleavage activity of M. maripaludis RNase P.
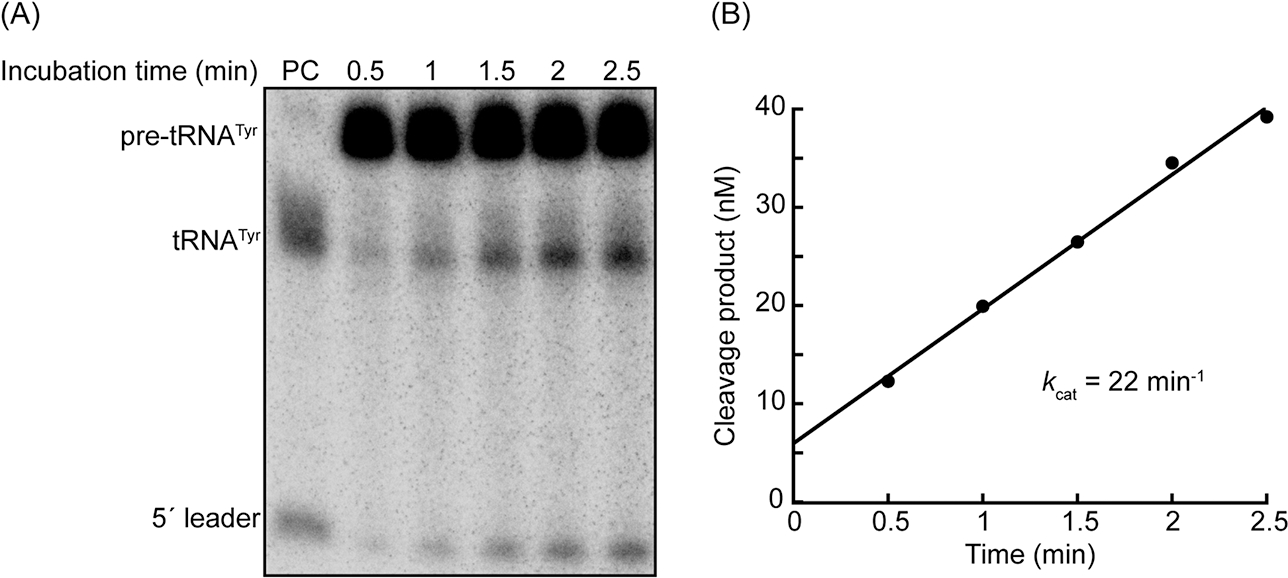
(A) Representative 8% (w/v) polyacrylamide, 7 M urea gel image illustrating cleavage of 250 nM E. coli pre-tRNATyr by 0.625 nM M. maripaludis RNase P (RPR + 5 RPPs). A trace amount of internally-[α-32P]-radiolabeled pre-tRNATyr was added to the assay mix to track the progress of the cleavage reaction. The PC (positive control) lane contains substrate that was cleaved for 15 min at 37°C by E. coli RNase P. The cleavage products in this lane serve as migration markers to confirm that M. maripaludis RNase P cleaves the pre-tRNA substrate at the expected site. (B) Representative plot of cleavage product concentration versus time. The volumes of each of the bands in (A) were calculated using ImageQuant software (Molecular Dynamics). The percentage of cleavage product at each time point was calculated using the band volumes as follows: %cleavage = [(tRNAtyr) + (5′ leader)]/[(Pre-tRNATyr) + (tRNATyr) + (5′ leader)]. The resulting values were multiplied by the total substrate concentration at the beginning of the reaction (250 nM). The resulting concentrations are plotted against time. A straight line was fit to the data, and the resulting slope was divided by enzyme concentration to yield kcat.
3.6. Assessing RPP oligomerization by mass photometry
Clean glass slides by sonicating them in 50% (v/v) methanol twice for 30 min each using a water bath sonicator.
Sonicate the glass slides in 100% (v/v) methanol twice for 30 min each.
Rinse the glass slides with ddH2O, and then dry with nitrogen gas.
Prepare sample wells by slicing CultureWell™ reusable gaskets into groups of four wells each.
Clean the gaskets by submerging them in 100% (v/v) isopropyl alcohol for 3 min and then rinsing with ddH2O. Repeat this washing step four times. Store the gaskets in ddH2O until ready to use.
Dry a single gasket with compressed air.
Place the gasket onto the glass slide with the wells facing up.
Place a single drop of immersion oil on the sample stage.
Place the gasket-containing slide on top of the oil drop. Move the slide to position a well over the center of the sample stage.
Aliquot 20 μL of sample (see Note 26) into the well and focus the optics on the droplet.
Collect mass photometry data in the ratiometric mode for 1–2 min using the Refeyn AcquireMP software.
Analyze, calibrate, and plot the data in the Refeyn DiscoverMP software.
Fit the plotted mass distribution to a Gaussian curve (or multiple Gaussian curves for a multi-modal mass distribution) to calculate the average measured mass and the standard deviation (Figure 5).
Figure 5. Oligomerization of M. maripaludis POP5·RPP30 analyzed by SDS-PAGE and mass photometry.
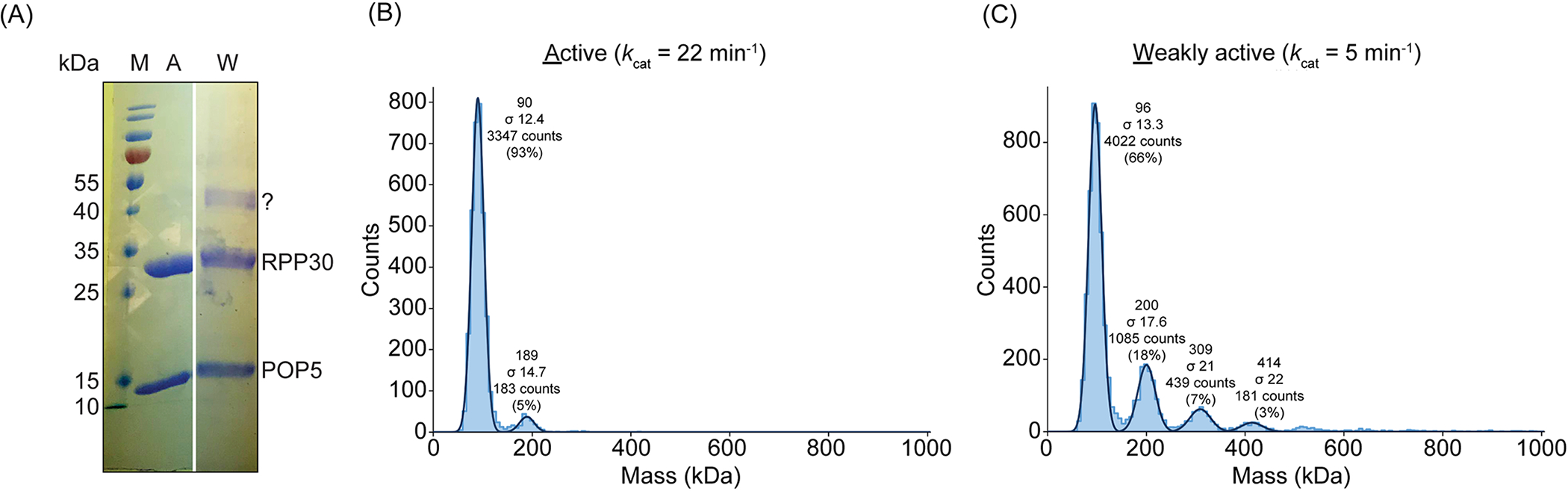
(A) SDS-PAGE (15% [w/v] polyacrylamide) analysis to compare highly active (H) and weakly active (W) preparations of POP5·RPP30. The gel image was spliced to position lanes of interest adjacent to each other. Although there is essentially no difference between the two preparations in the mobilities and relative intensities of the POP5 and RPP30 bands, the weakly active preparation has bands that migrate between the 40 and 55 kDa markers (M), likely corresponding to a heterodimer given the expected molecular weight of POP5·RPP30. The same two preparations of POP5·RPP30 (150 nM each) were diluted in 1X assay buffer (see 2.5) and analyzed by mass photometry (B and C). Histogram plotting and fitting was performed in DiscoverMP. The average mass of each population (in kDa), determined by the Gaussian fits, are indicated above each peak. For mass calculation, the following calibrants were used: lens epithelium-derived growth factor, LEDGF (60.4 kDa); bovine serum albumin, BSA (68.5 kDa); HIV integrase (80.4 kDa, monomer; 160.8 kDa, dimer); dihydroorotate dehydrogenase B (237 kDa); apoferritin (480 kDa); thyroglobulin (670 kDa). (B) Mass photometry analysis of a highly active preparation of POP5·RPP30. The major species (93%) corresponds to the POP5·RPP30 heterotetramer, which is the expected oligomeric state. A minor population of octamer (5%) is also observed. (C) Mass photometry analysis of a weakly active preparation of POP5·RPP30. While the major species is the heterotetramer (66%), the remaining species (29%) are octamer or larger.
4. Conclusions
We have presented protocols for purification, reconstitution, and mass analysis of all six subunits (one RNA, five proteins) of RNase P from an archaeon. We emphasize a tag-less approach to protein purification to minimize the potential interference by tags or their remnants during reconstitution of any RNP from constituent subunits prepared by affinity chromatography. We demonstrate the use of MS and MP as complementary approaches for obtaining mass information on the subunits of archaeal RNase P, including accurate molecular weight, metal ion-binding, and oligomeric state. These analyses provided some unexpected insights to better understand the time-dependent decrease in the activity of reconstituted M. maripaludis RNase P. Comparison of fresh or aged protein preparations by MS revealed that the proteins are modified significantly even when stored long-term at −20°C. For example, in a single preparation of POP5·RPP30, both subunits had several modifications after a year of storage (Figure 4); in the case of POP5, oxidation seems likely given the step-wise addition of 16 Da to the expected mass. Second, we examined two preparations of POP5·RPP30 that exhibited a 7-fold difference in activities in the reconstituted holoenzymes. MP clearly reveals that, under conditions comparable to those used in activity assays, there are differences in the distributions of the oligomeric states present in these two preparations (Figure 5). While the nexus (if any) between oxidation and oligomerization (or vice-versa) remains to be determined, these two methods provide insights into sample heterogeneity that is not easily obtained by other means. For example, biochemists rely heavily on SDS-PAGE, a convenient method for establishing molecular weights and protein degradation, but oxidations and non-native oligomerizations are likely to go undetected with this method. With insights from MS and MP, customized preventive measures could be implemented to extend protein shelf life (e.g., flushing stock tubes of protein with argon or adding a small amount of non-ionic detergent).
The growing appreciation of the roles of long non-coding (lnc) RNAs and lncRNA–protein complexes in diverse cellular processes heightens the need for information and insights that will facilitate biochemical and biophysical studies of RNPs. Our findings on M. maripaludis RNase P are likely to be instructive in this regard.
5. Notes
Fresh DTT is required for the full activity of T7 RNAP. DTT solution stocks should be frozen at −20°C for long-term storage.
When preparing fresh NTP stock solutions from solid stocks, it is important to adjust the pH to ~7 using NaOH. If the pH is not adjusted, the final pH of the IVT reaction will be acidic (due to the high final concentration of NTPs), thus impairing the activity of T7 RNAP and leading to an overall low RNA yield.
While a standard NTP mix contains each nucleotide at a concentration of 25 mM, the RNA yield can often be improved by adjusting the concentrations of individual NTPs to match the GC content of the target RNA transcript.
IVT templates can be cloned into a plasmid under the control of the T7 promoter sequence and upstream from a (preferably type IIS) restriction enzyme recognition sequence (Vioque, Arnez, & Altman, 1988). Accordingly, restriction digestion of the plasmid will generate a linear DNA template for run-off transcription in which T7 RNAP-mediated transcription is terminated when the polymerase reaches the end of the linear template. Alternatively, an IVT template can be prepared by performing a PCR reaction in which the template sequence (encoded in a plasmid or in a linear double-stranded DNA) is amplified using a forward primer that contains the T7 promoter sequence followed by the 5′ end of the gene, and a reverse primer that is complementary to the 3′ end of the gene.
Recombinant T7 RNAP can be purchased or overexpressed and purified in-house. In the latter case, the optimal amount of T7 RNAP to add to a transcription reaction to maximize RNA yield must be empirically determined by performing test IVT reactions containing varying amounts of T7 RNAP (Lyon & Gopalan, 2018).
While POP5·RPP30 from all archaea tested thus far readily co-purify when co-overexpressed (Chen et al., 2010), RPP21 and RPP29 co-purify in only a few systems. In the case of M. maripaludis, the two proteins become separated during column chromatography, and therefore cannot be co-purified (Cho et al., 2010). Regardless, both proteins are soluble on their own.
At pH ~8 and high ionic strength (e.g., >1 M NaCl), PEI will precipitate contaminating RNAs, which can then be removed by centrifugation. Importantly, minimization of nucleic acid contaminants is critical for active RNase P assembly.
Similar columns that are compatible with the FPLC system of choice may be substituted.
E. coli pre-tRNATyr is effectively cleaved by M. maripaludis RNase P and other in vitro reconstituted archaeal RNase P holoenzymes (Chen et al., 2010; Tsai et al., 2006).
Pre-tRNA substrates can be radiolabeled using one of two strategies. First, substrates can be radiolabeled internally by supplementing a trace amount of a single α-32P-radiolabeled NTP into the IVT reaction used to synthesize pre-tRNA. Second, substrates can be 5′-radiolabeled by dephosphorylating the IVT-synthesized pre-tRNA with calf intestinal alkaline phosphatase. The dephosphorylated pre-tRNA can then be re-phosphorylated by T4 polynucleotide kinase in the presence of trace amounts of γ-32P-radiolabeled ATP. Regardless of radiolabeling strategy, the radiolabeled pre-tRNAs should be gel-purified to remove excess radiolabeled NTPs and any abortive transcription products.
As an alternative to radiolabeling, pre-tRNAs can be detected by using a fluorescent dye to directly label [e.g., fluorescein (Howard et al., 2016)] or stain [e.g., SYBR Gold (ThermoFisher)] the pre-tRNA.
The optimal amount of IVT template to maximize RNA yield must be empirically determined by performing test IVT reactions containing varying amounts of template. In general, we have been successful using 4 μg of 3,000 bp DNA template in a 100 μL IVT reaction.
The full-length M. maripaludis RPR migrates slower than a 200 bp double-stranded DNA marker on a 2% (w/v) agarose gel. Occasionally, the RPR may migrate as a doublet, which indicates conformational heterogeneity. If such a migration pattern is observed, heat a small aliquot of the RPR for 30 min at 65°C and then 10 min at 37°C before gel electrophoresis. In cases where such a treatment does not collapse the doublet into a single band on an agarose gel, denaturing PAGE or native MS can be used to confirm that a single major RNA species is present (Figure 2F).
Although two EtOH washes suffice for many uses, we strongly recommend a third wash for applications that are particularly sensitive to salts (including native MS).
Omission of 1% (w/v) glucose from the seed culture results in reproducibly longer growth times to reach the appropriate OD600 for induction (~5–6 h versus 2–3 h).
After resuspending the pellet, some material may not dissolve. This insoluble matter can be removed by an additional round of centrifugation.
M. maripaludis RPP stocks (>50 μM) were reported to retain full activity for several months when stored at −20°C (Cho et al., 2010).
Thrombin cleavage activity is optimal in 1X PBS, so RPP29 is first dialyzed into 1X PBS before thrombin is added to catalyze removal of the N-terminal (His)6 tag.
RPP29 and L7Ae have very low extinction coefficients, making concentration measurement very difficult for dilute protein stocks. Use centrifugal concentrators to concentrate stocks in order to obtain reliable absorbance measurements.
It is critical that all non-volatile buffer components are removed and replaced with a volatile salt (e.g., ammonium acetate). The presence of non-volatile salts will cause ion suppression and adduct formation thereby complicating native MS spectra and data analysis.
To ensure the solution is completely at the tip (sharp end) of the emitter, tape the emitter (tip down) to a mini centrifuge rotor and spin briefly at a low RPM.
HCD or other types of collisional activation (collision-induced dissociation, CID; in-source trapping; surface-induced dissociation, SID) can be used to dissociate a complex. Alternatively, collisional activation at lower voltages that do not cause complex dissociation or covalent fragmentation can be used to de-adduct and sharpen peaks. This de-adducting, sometimes referred to as “collisional cleaning”, is especially useful for accurate mass measurements, particularly in the case of RNAs which retain salts introduced during synthesis and purification steps. The collision voltages needed for dissociation and collisional cleaning are highly sample-dependent; scanning across a range of voltages is recommended.
For sensitive gel-based analysis of pre-tRNA substrate and cleavage products, the amount of radiolabeled pre-tRNA substrate added to the reaction should be adjusted so that quenched reaction aliquots loaded onto the urea–PAGE gel contain at least 1,000–2,000 dpm per lane.
To obtain an accurate measurement of the reaction velocity, it is ideal to measure reaction progress when less than 25% of the substrate has been converted to product.
To obtain the highest quality gel image, exposure time should be adjusted based on the dpm of the radiolabeled tRNA substrate loaded onto the gel. However, if the dpm loaded is low (<1000 dpm), longer exposures will also significantly increase background.
Sample concentration for mass photometry analysis depends on the sample identity. Data quality will be low at high sample concentrations (>1 μM). We recommend starting at 500 nM and then gradually diluting the sample to optimize data quality.
6. Acknowledgements
We are grateful for research funding from the National Institutes of Health [GM120582 to V.G and V.H.W.; P41GM128577 to V.H.W.] and from the OSU Comprehensive Cancer Center for a Pelotonia Post-doctoral Fellowship [W.J.Z.]. We thank Richard Fishel and Ross Larue for training and use of the Refeyn One mass photometry instrument, funded by NIH grant CA067007 and the James Comprehensive Cancer Center.
References
- Altman S (2007). A view of RNase P. Mol Biosyst, 3(9), 604–607. doi: 10.1039/b707850c [DOI] [PubMed] [Google Scholar]
- Chen WY, Pulukkunat DK, Cho IM, Tsai HY, & Gopalan V (2010). Dissecting functional cooperation among protein subunits in archaeal RNase P, a catalytic ribonucleoprotein complex. Nucleic Acids Res, 38(22), 8316–8327. doi: 10.1093/nar/gkq668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Singh D, Lai LB, Stiffler MA, Lai HD, Foster MP, & Gopalan V (2012). Fidelity of tRNA 5’-maturation: a possible basis for the functional dependence of archaeal and eukaryal RNase P on multiple protein cofactors. Nucleic Acids Res, 40(10), 4666–4680. doi: 10.1093/nar/gks013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho IM, Lai LB, Susanti D, Mukhopadhyay B, & Gopalan V (2010). Ribosomal protein L7Ae is a subunit of archaeal RNase P. Proc Natl Acad Sci U S A, 107(33), 14573–14578. doi: 10.1073/pnas.1005556107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, & Krasilnikov AS (2010). Of proteins and RNA: the RNase P/MRP family. RNA, 16(9), 1725–1747. doi: 10.1261/rna.2214510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D, Marquez SM, & Pace NR (2006). RNase P: interface of the RNA and protein worlds. Trends Biochem Sci, 31(6), 333–341. doi: 10.1016/j.tibs.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Fukuhara H, Kifusa M, Watanabe M, Terada A, Honda T, Numata T, . . . Kimura M (2006). A fifth protein subunit Ph1496p elevates the optimum temperature for the ribonuclease P activity from Pyrococcus horikoshii OT3. Biochem Biophys Res Commun, 343(3), 956–964. doi: 10.1016/j.bbrc.2006.02.192 [DOI] [PubMed] [Google Scholar]
- Gopalan V, Jarrous N, & Krasilnikov AS (2018). Chance and necessity in the evolution of RNase P. RNA, 24(1), 1–5. doi: 10.1261/rna.063107.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, & Altman S (1983). The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell, 35(3 Pt 2), 849–857. doi: 10.1016/0092-8674(83)90117-4 [DOI] [PubMed] [Google Scholar]
- Howard MJ, Karasik A, Klemm BP, Mei C, Shanmuganathan A, Fierke CA, & Koutmos M (2016). Differential substrate recognition by isozymes of plant protein-only Ribonuclease P. RNA, 22(5), 782–792. doi: 10.1261/rna.055541.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikovska E, Svard SG, & Kirsebom LA (2007). Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc Natl Acad Sci U S A, 104(7), 2062–2067. doi: 10.1073/pnas.0607326104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzuma Y, Mizoguchi M, Takagi H, Fukuhara H, Tsukamoto M, Numata T, & Kimura M (2003). Reconstitution of archaeal ribonuclease P from RNA and four protein components. Biochem Biophys Res Commun, 306(3), 666–673. doi: 10.1016/s0006-291x(03)01034-9 [DOI] [PubMed] [Google Scholar]
- Lai LB, Tanimoto A, Lai SM, Chen WY, Marathe IA, Westhof E, . . . Gopalan V (2017). A novel double kink-turn module in euryarchaeal RNase P RNAs. Nucleic Acids Res, 45(12), 7432–7440. doi: 10.1093/nar/gkx388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai LB, Vioque A, Kirsebom LA, & Gopalan V (2010). Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett, 584(2), 287–296. doi: 10.1016/j.febslet.2009.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon S, & Gopalan V (2018). A T7 RNA Polymerase Mutant Enhances the Yield of 5’-Thienoguanosine-Initiated RNAs. Chembiochem, 19(2), 142–146. doi: 10.1002/cbic.201700538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano S, Debabrata D, Sonn-Segev A, Busch F, Kim Y, Harvey S, . . . Schreiber. (2021). The complexity of homomeric protein quaternary assemblies. J Am Chem Soc. [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, & Robinson CV (2015). Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem, 87(8), 4370–4376. doi: 10.1021/acs.analchem.5b00140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannucci JA, Haas ES, Hall TA, Harris JK, & Brown JW (1999). RNase P RNAs from some Archaea are catalytically active. Proc Natl Acad Sci U S A, 96(14), 7803–7808. doi: 10.1073/pnas.96.14.7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Berezin I, & Krasilnikov AS (2018). In vitro reconstitution and analysis of eukaryotic RNase P RNPs. Nucleic Acids Res, 46(13), 6857–6868. doi: 10.1093/nar/gky333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulukkunat DK, & Gopalan V (2008). Studies on Methanocaldococcus jannaschii RNase P reveal insights into the roles of RNA and protein cofactors in RNase P catalysis. Nucleic Acids Res, 36(12), 4172–4180. doi: 10.1093/nar/gkn360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery MJ (2014). Determination of oxidative protein modifications using mass spectrometry. Redox Rep, 19(4), 140–147. doi: 10.1179/1351000214Y.0000000089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltermann F, Foley EDB, Pagnoni V, Galpin M, Benesch JLP, Kukura P, & Struwe WB (2020). Quantifying protein-protein interactions by molecular counting with mass photometry. Angew Chem Int Ed Engl, 59(27), 10774–10779. doi: 10.1002/anie.202001578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonn-Segev A, Belacic K, Bodrug T, Young G, VanderLinden RT, Schulman BA, . . . Kukura P (2020). Quantifying the heterogeneity of macromolecular machines by mass photometry. Nat Commun, 11(1), 1772. doi: 10.1038/s41467-020-15642-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, & Moffatt BA (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol, 189(1), 113–130. doi: 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- Sun L, & Harris ME (2007). Evidence that binding of C5 protein to P RNA enhances ribozyme catalysis by influencing active site metal ion affinity. RNA, 13(9), 1505–1515. doi: 10.1261/rna.571007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HY, Pulukkunat DK, Woznick WK, & Gopalan V (2006). Functional reconstitution and characterization of Pyrococcus furiosus RNase P. Proc Natl Acad Sci U S A, 103(44), 16147–16152. doi: 10.1073/pnas.0608000103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vioque A, Arnez J, & Altman S (1988). Protein-RNA interactions in the RNase P holoenzyme from Escherichia coli. J Mol Biol, 202(4), 835–848. doi: 10.1016/0022-2836(88)90562-1 [DOI] [PubMed] [Google Scholar]
- Wan F, Wang Q, Tan J, Tan M, Chen J, Shi S, . . . Lei M (2019). Cryo-electron microscopy structure of an archaeal ribonuclease P holoenzyme. Nat Commun, 10(1), 2617. doi: 10.1038/s41467-019-10496-3 [DOI] [PMC free article] [PubMed] [Google Scholar]