

Abstract
A highly efficient and expeditious one-pot approach towards 2-(3-oxoindolin-2-yl)acetonitriles was designed, which involves a base-assisted aldol reaction of ortho-nitroacetophenones, followed by hydrocyanation, triggering an unusual reductive cyclization reaction.
Keywords: nitroalkanes, brønsted acid catalysis, indoles, rearrangements, cascade transformations
1. Introduction
The derivatives of 2-Alkylideneindolin-3-one play important role in modern medicinal chemistry. The bis-indole indirubin known as “Tyrian purple” dye has been used for many years as an active component of a traditional Chinese herbal medicine [1,2,3]. Lately, numerous synthetic analogs of this compound were reported to show potent and highly selective pharmacological inhibition of glycogen synthase kinases and cycline-dependent kinases [4,5,6]. Related compounds possessing a single 3-oxoindoline subunit (or two remotely positioned subunits) also were isolated from natural sources and are also known to exhibit a wide spectrum of important biological properties [7,8,9,10,11,12,13]. Normally, the preparation of these structures relies heavily on the chemistry of isatins, which greatly limits the diversity of accessible substitution patterns. Alternative methods have also been developed based on the aldol condensation of 3H-indol-3-ones with carbonyl compounds [14,15,16,17], the transition metal-catalyzed carbonylative cross-coupling of ortho-iodoanilines to alkynes [18,19,20,21], or the cascade cyclizations of anilines with -ketoesters [7,22]. We recently reported an original, facile, and highly efficient method for the preparation of 2-(3-oxoindolin-2-ylidene) acetonitriles 2 from ortho-nitrochalcones 1, operating as a triggered Michael addition of the cyanide anion to the chalcone followed by a cascade cyclization mechanistically related to the Baeyer–Drewson reaction (Scheme 1) [23].
Scheme 1.

Cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones.
Herein, we report on the development of a more streamlined synthetic approach which allows for the combining of this transformation with the preparation of the chalcone precursor 1 from aldehydes 4 and ortho-nitroacetophenones 3 in a single one-pot operation.
2. Results and Discussion
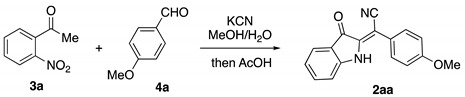
Since our newly developed method for preparation of 2-(3-oxoindolin-2-ylidene) acetonitriles 2 relies heavily on the availability of the corresponding chalchone precursors 1, we wondered about possibility of generating them in situ via an aldol condensation reaction. It was initially planned that this step could be carried out in the presence of catalytic amounts of alkali, which later would be neutralized with acetic acid to trigger the hydrocyanation step. However, much more attractive was the idea of employing potassium cyanide as a base to catalyze the aldol condensation step. This unusual approach to the aldol reaction was precedented in a single report by Migita et al. [24]. To test this idea, we subjected 2-nitroacetophenone (2a) to the reaction with p-anisaldehyde (4a) in the presence of KCN in methanol (2 mL). Water additive (130 μL) was used, as it was previously demonstrated it is crucial for successful cyclization [23]. The reaction mixture was refluxed for 1 h, then treated with acetic acid (150 μL) and refluxed for an additional 30 min to afford 3-oxoindoline 2aa in 64% yield (Table 1, entry 1). This reaction was accompanied by the formation of a large number of unidentified side products, as evidenced by TLC analysis. In attempt to improve this situation, we decided to carry out the reaction under conditions ensuring a faster kinetic rate of the desired transformation. First, cyanide loading was increased (with a proportional increase of water and acetic acid concentrations). The reaction proceeded faster, but the yield of the target product was not improved under these conditions (entry 2). Then, the concentration effect was evaluated, with the knowledge that this effect should be favorable for bimolecular reactions. Indeed, when the concentration of starting materials was increased twice, 2aa was formed cleanly in 95% yield (entry 3). Finally, it was found that decreasing the temperature had a detrimental effect on the reaction performance. The reaction proceeded very sluggishly, affording 2aa in 58% yield along with several unidentified side products (entry 4). Without water additive, this reaction affords lower yields (entry 5), and this is also the case when other acids are employed instead of acetic (entries 6, 7).
Table 1.
Optimization of the reaction conditions for one-pot conversion of 2-nitroacetophenone (3a) and p-anisaldehyde (4a) into (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitriles (2aa).
| KCN a | Temperature (°C)/Time (h) | Methanol/Water/AcOH b | Yield of 2aa, % c | |
|---|---|---|---|---|
| 1 | 130 | 65/1 | 2000/130/150 | 64 |
| 2 | 260 | 65/0.5 | 2000/260/300 | 65 |
| 3 | 130 | 65/0.5 | 1000/130/150 | 95(87) d |
| 4 | 130 | 20/12 | 1000/130/150 | 58 |
| 5 | 130 | 65/0.5 | 1000/0/150 | 79 |
| 6 | 130 | 65/0.5 | 1000/130/195 e | 38 |
| 7 | 130 | 65/0.5 | 1000/130/92 f | 49 |
a Amount of KCN in mg per 1 mmol of starting material 3a is listed. b Volumes of methanol, water, and acetic acid in μL per 1 mmol of starting material 3a are listed. c NMR yields are reported. The best result is shown in bold. d Isolated yield of purified product 2aa is provided in parentheses. e H3PO4 was used instead of AcOH. f HCOOH was used instead of AcOH.
With the optimized conditions in hand, we performed these transformations in a preparative scale (up to 2.00 mmol) and managed to obtain comparably high isolated yields of 2aa (85%) (Table 1, entry 3, Scheme 2). The reaction demonstrated good tolerance and compatibility with a variety of substituents, including alkoxyarenes, halogenated arenes, and cyclic acetals (Scheme 2). The formation of (E)-2-(3-oxoindolin-2-ylidene)acetonitrile moiety in this reaction was unambiguously confirmed by single crystal X-ray diffraction of compound 2al (CCDC #2157035, Figure 1).
Scheme 2.

Preparation of 2-(3-oxoindolin-2-yl)acetonitriles 2.
Figure 1.

ORTEP drawing of X-Ray structures of (E)-2-(5-fluoro-2-methylphenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2al, CCDC #2157035). The thermal ellipsoids are shown at 50% probability. Green:fluorine; blue:nitrogen; red:oxygen, gray:carbon.
It was possible to engage heterocyclic aldehydes into the featured transformation. Thus, the reaction of ortho-nitroacetophenone (3a) with thiophene-2-carbaldehyde (4o) afforded thienyl-substituted compound 2ao in good yield (Scheme 3). On the other hand, the reaction involving benzo[b]thiophene-3-carbaldehyde (4p) led to the formation of rearranged polycyclic product 2ap (Scheme 3). Further investigation of this unusual transformation is currently under way in our laboratories.
Scheme 3.

Reactions with thiophenecarbaldehydes 4o-p.
The mechanistic rationale for the featured transformation is shown in Scheme 4. It is believed that potassium cyanide can serve as a base to induce the enolization of ortho-nitroacetophenone 3 to provide enolate form 5, which can be engaged into an aldol reaction with aldehyde 4 affording chalchone 1 (Scheme 4). The subsequent conjugate addition of cyanide to chalchone would provide 4-oxo-4-arylbutanenitrile in enolate form 6. To demonstrate the possibility of carrying out this sequence of reactions, we tested the reactivity between acetophenone (11) and benzaldehyde (4b) in the presence of excess potassium cyanide under typical reaction conditions. Expectedly, 4-oxo-4-phenylbutanenitrile 12 was formed smoothly, albeit in marginal yield (Scheme 5). Once formed, enolate species 6 would experience intramolecular nucleophilic attack involving the ortho-nitro group to afford cyclic keto-azinate 7, which should exist in tautomeric equilibrium with azinic acid-enolate form 8 (Scheme 4). The elimination of the hydroxyl group from this species would lead to the formation of imine N-oxide intermediate 9, which should tautomerize into cyclic hydroxylamine 10. Subsequent reduction (most likely involving methanol as a reducing agent) [25] would afford final 2-(3-oxoindolin-2-yl)acetonitriles 2 (Scheme 4) [23]. It should be pointed out that carrying out a reaction between ortho-nitroacetophenone 3a and benzaldehyde 4b followed by quenching with phenacyl bromide (serving as a precursor of HBr, slowly released over an extended period) allowed for the isolation of compound 10ab in moderate yield (Scheme 6). The same compound was detected by mass spectroscopy in the reaction mixture carried out under standard conditions. It proved to be stable upon heating in DMSO at 100 °C for 2 hr. However, treatment of this sample with water or formic acid caused the slow consumption of 10ab (mass 285 [M + Na]) and accumulation of product 2ab (mass 269 [M + Na]). In the presence of methanol and acetic acid, this process proceeded notably faster. In preparative scale, this reaction afforded a nearly quantitative conversion into 2ab, identical to the sample obtained in one-pot fashion (Scheme 6).
Scheme 4.

Mechanistic rationale for cascade transformation providing 2-(3-oxoindolin-2-yl)acetonitriles 2.
Scheme 5.

Cyanide-induced Aldol condensation/Michael addition cascade.
Scheme 6.

Preparation intermediate cyclic hydroxylamine 10ab and its further conversion into 2-(3-oxoindolin-2-yl)acetonitrile 2ab.
3. Conclusions
An improved protocol for the preparation of 2-(3-oxoindolin-2-yl)acetonitriles 2 via a cyanide-mediated cascade reaction of ortho-nitroacetophenones (3) with aromatic aldehydes (4) was developed. This transformation involves an initial aldol condensation followed by a Michael-type conjugated addition of cyanide anion to the intermediate chalcone, which triggers an unusual cyclization mechanistically related to the Baeyer–Drewson reaction. This methodology was employed to synthesize a small, focused library of target molecules. It should be mentioned that aliphatic aldehydes do not participate in aldol condensation under these reaction conditions and cannot be used as precursors for preparation of alkyl-substituted analogs. An investigation into the biological activity of these new €-2-(3-oxoindolin-2-ylidene)-2-arylacetonitriles is currently under way in our laboratories.
4. Experimental Part
General
NMR spectra (1H and 13C) were measured in solutions of CDCl3 or DMSO-d6 on a Bruker AVANCE-III HD instrument (at 400.40 or 100.61 MHz, respectively). HRMS spectra were measured in MeCN solutions on Bruker maXis impact (electrospray ionization, employing HCO2Na–HCO2H for calibration). See Supplementary Materials for NMR and HRMS spectral charts and X-Ray crystallography data. IR spectra was measured on an FT-IR spectrometer Shimadzu IRAffinity-1S equipped with an ATR sampling module. Reaction progress, purity of isolated compounds, and Rf values were assessed by TLC on Silufol UV-254 plates. Column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). Melting points were measured on Stuart SMP30 apparatus. All reagents and solvents were purchased from commercial vendors and used as received. The reactions involving KCN are accompanied by the formation of highly toxic fumes of HCN. A well-ventilated fume hood must be used.
Preparation of (E)-2-aryl-2-(3-oxoindolin-2-ylidene)acetonitriles (2aa–2cb) by reaction of benzaldehydes (4a–n) and 2′-nitroacetophenones (3a–c) (General procedure):
A 5-mL round bottom flask equipped with magnetic stirring bar was charged with 2′-nitroacetophenone (1 mmol), benzaldehyde (1 mmol), KCN (2 mmol, 130 mg), H2O (130 mg), and MeOH (1 mL) and refluxed for 30 min (TLC control). After the consumption of the starting material, the reaction mixture was cooled to room temperature and AcOH (2.5 mmol, 150 mg, 143 µL) (exothermic reaction) was added and reflux was continued to another 30 min. Then, the reaction mixture was diluted with 100 mL of EtOAc and washed twice with 30 mL of concentrated NaHCO3 solution. The organic layer was concentrated and purified by column chromatography (gradient: EtOAc:Hex 1:4–1:1) or by recrystallization from EtOH.
(E)-2-(4-methoxyphenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2aa):
Red solid, mp (EtOH) 247.5–249.4 °C (Literature data: [23] mp 250.1–251.1 °C), Rf 0.23 (EtOAc/Hex, 1:2). Yield: 240 mg (0.87 mmol, 87%). 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.61 (dt, J = 16.6, 7.9 Hz, 4H), 7.11 (dd, J = 15.3, 8.2 Hz, 3H), 7.01 (t, J = 7.5 Hz, 1H), 3.84 (s, 3H).
(E)-2-(3-oxoindolin-2-ylidene)-2-phenylacetonitrile (2ab):
Orange solid, mp 235.0–236.3 °C (Literature data: [23] mp 233.1–235.9 °C), Rf 0.32 (EtOAc/Hex, 1:2). Yield: 162 mg (0.66 mmol, 66%). 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 7.72–7.52 (m, 6H), 7.48 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H).
(E)-2-(3-oxoindolin-2-ylidene)-2-(p-tolyl)acetonitrile (2ac):
Orange solid, mp (MeOH) 241.2–242.6 °C (literature data: [23] mp 236– 240 °C), Rf 0.46 (EtOAc/Hex, 1:2). Yield: 187 mg (0.72 mmol, 72%). 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.56 (dd, J = 16.0, 7.5 Hz, 3H), 7.38 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.1 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 2.38 (s, 3H).
(E)-2-(4-ethylphenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2ad):
Red solid, mp (EtOH) 214.4–215.1 °C (literature data: [23] mp 223.2–225.7 °C), Rf 0.56 (EtOAc/Hex, 1:2). Yield: 145 mg (0.53 mmol, 53%). 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 7.65 (d, J = 9.8 Hz, 1H), 7.57 (d, J = 5.1 Hz, 3H), 7.42 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.1 Hz, 1H), 7.03 (t, 1H), 2.67 (q, 2H), 1.22 (t, 3H).
(E)-2-(4-isopropylphenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2ae):
Red solid, mp (EtOH) 223.4–224.6 °C (literature data: [23] mp 223.0–226.6 °C), Rf 0.47 (EtOAc/Hex, 1:2). Yield: 127 mg (0.44 mmol, 44%). 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.62–7.53 (m, 3H), 7.45 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 8.1 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 2.97 (hept, J = 7.0 Hz, 1H), 1.24 (d, J = 7.0 Hz, 6H).
(E)-2-(4-(tert-butyl)phenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2af):
Red solid, mp 241–243 °C, Rf 0.57 (EtOAc/Hex, 1:1). Yield: 269 mg (0.89 mmol, 89%). 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.59 (s, 5H), 7.10 (d, J = 8.0 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 1.33 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.5, 151.9, 142.2, 137.5, 129.3, 128.6 (2C), 126.3 (2C), 124.9, 121.4, 119.5, 118.0, 112.8, 89.1, 34.7, 31.0 (3C). IR, vmax/cm−1: 3328, 2966, 2206, 1709, 1586, 1470, 1329, 1278, 1222, 1099, 969. HRMS (ES TOF) calculated for (M + Na)+ C20H18N2NaO 325.1311, found 325.1309 (0.6 ppm).
(E)-2-(3-oxoindolin-2-ylidene)-2-(o-tolyl)acetonitrile (2ag)
Red crystals, mp (MeOH) 205.3–206.6 °C (literature data: [23] mp 201.8–203.5 °C), Rf 0.29 (EtOAc/Hex, 1:2). Yield: 140 mg (0.54 mmol, 54%). 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.41 (dt, J = 7.6, 3.2 Hz, 3H), 7.35 (ddd, J = 7.9, 5.2, 2.7 Hz, 1H), 7.03–6.94 (m, 2H), 2.31 (s, 3H).
(E)-2-(4-fluorophenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2ah):
Orange solid, mp (EtOH) 279.3–281.4 °C (literature data: [23] mp 281.1–282.8 °C), Rf 0.54 (EtOAc/Hex, 1:2). Yield: 190 mg (0.72 mmol, 72%). 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 7.74–7.63 (m, 3H), 7.58 (t, J = 7.7 Hz, 1H), 7.42 (t, J = 8.7 Hz, 2H), 7.08 (d, J = 8.1 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ -111.4.
(E)-2-(4-chlorophenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2ai):
Red solid, mp 286–288 °C (literature data: [23] mp 285.9–287.8 °C), Rf 0.22 (EtOAc/Hex, 1:2). Yield: 182 mg (0.65 mmol, 65%). 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.68–7.62 (m, 5H), 7.61–7.56 (m, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H).
(E)-2-(4-bromophenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2aj):
Red solid, mp (EtOH) 283.0–284.8 °C (literature data: [23] mp 278.8–282.5 °C), Rf 0.66 (EtOAc/Hex, 1:2). Yield: 187 mg (0.58 mmol, 58%). 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 7.77 (d, J = 8.7 Hz, 2H), 7.65 (d, J = 7.2 Hz, 1H), 7.61–7.56 (m, 3H), 7.10–6.99 (m, 2H).
(E)-2-(3-chlorophenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2ak):
Orange solid, mp 270–271 °C (literature data: [23] mp 267–269 °C), Rf 0.57 (EtOAc/Hex, 1:2). Yield: 188 mg (0.67 mmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 7.66 (d, J = 7.4 Hz, 2H), 7.62–7.52 (m, 4H), 7.09 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H).
(E)-2-(5-fluoro-2-methylphenyl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2al)
Red crystals, mp (EtOH) 236.1–237.4 °C, Rf 0.22 (EtOAc/Hex, 1:2). Yield: 100 mg (0.36 mmol, 36%). 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 7.65 (dd, J = 7.6, 1.2 Hz, 1H), 7.56 (ddd, J = 8.4, 7.3, 1.3 Hz, 1H), 7.44 (dd, J = 8.5, 5.9 Hz, 1H), 7.33–7.23 (m, 2H), 7.05–6.94 (m, 2H), 2.28 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 184.1, 160.6 (d, J = 243.2 Hz), 152.5, 144.3, 137.8, 133.2 (d, J = 3.3 Hz), 132.7 (d, J = 8.1 Hz), 132.3 (d, J = 8.4 Hz), 125.1, 121.5, 119.6, 117.2, 116.9 (d, J = 22.2 Hz), 116.4 (d, J = 20.8 Hz), 112.4, 86.3, 18.5. 19F NMR (376 MHz, DMSO-d6) δ -116.2. IR, vmax/cm−1: 3372, 2206, 1713, 1608, 1482, 1463, 1333, 1224, 176, 1075, 1003, 858, 751. HRMS (ES TOF) calculated for (M + Na)+ C17H11FN2NaO 301.0745, found 301.0748 (0.8 ppm).
(E)-2-(3-oxoindolin-2-ylidene)-2-(3,4,5-trimethoxyphenyl)acetonitrile (2am):
Red solid, mp 216–218°C, Rf 0.29 (EtOAc/Hex, 1:1). Yield: 289 mg (0.86 mmol, 86%). 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.60–7.55 (m, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.89 (s, 2H), 3.86 (s, 6H), 3.73 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 184.1, 153.3 (2C), 152.5, 142.5, 138.1, 137.5, 127.4, 124.9, 121.4, 119.5, 117.9, 112.7, 106.4 (2C), 89.2, 60.1, 56.0 (2C). IR, vmax/cm−1: 3745, 3622, 3272, 2978, 2214, 1737, 1713, 1560, 1506, 1459, 1421, 1274, 1218, 1130, 993, 751. HRMS (ES TOF) calculated for (M + Na)+ C19H16N2NaO4 359.1002, found 359.1004 (−0.6 ppm).
(E)-2-(benzo[d][1,3]dioxol-5-yl)-2-(3-oxoindolin-2-ylidene)acetonitrile (2an):
Red solid, mp (EtOH) 247.3–248.6 °C (literature data: [23] mp 247.9–248.7 °C), Rf 0.62 (EtOAc/Hex, 1:2). Yield: 200 mg (0.69 mmol, 69%). 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.64 (d, J = 7.7 Hz, 1H), 7.60–7.51 (m, 1H), 7.18 (d, J = 1.7 Hz, 1H), 7.15–7.07 (m, 3H), 7.01 (t, J = 7.5 Hz, 1H), 6.14 (s, 2H).
(Z)-2-(3-oxoindolin-2-ylidene)-2-(thiophen-2-yl)acetonitrile (2ao):
Red solid, mp (EtOH) 229–230 °C, Rf 0.71 (EtOAc/Hex, 1:2). Yield: 159 mg (0.63 mmol, 63%). 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.86 (d, J = 5.4 Hz, 1H), 7.65–7.52 (m, 3H), 7.29 (t, J = 4.7 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 184.0, 152.1, 139.9, 137.3, 135.2, 129.43, 129.40, 129.1, 124.8, 121.9, 119.7, 117.1, 113.3, 84.6. IR, vmax/cm−1 3320, 2214, 1705, 1685, 1624, 1584, 1552, 1528, 1480, 1466, 1421, 1391, 1363, 1299, 1254, 1198. HRMS (ES TOF) calculated for (M + Na)+ C14H8N2NaOS 275.0250, found 275.0250 (−0.1 ppm).
5b-Hydroxy-5b,10-dihydrobenzo [4’,5’]thieno [2’,3’:3,4]cyclopenta [1,2-b]indole-11-carbonitrile (2ap):
Red solid, mp (EtOH) 218–219 °C, Rf 0.37 (EtOAc/Hex, 1:2). Yield: 139 mg (0.46 mmol, 46%). 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H), 11.39 (s, 1H), 9.00–8.90 (m, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.46–7.40 (m, 1H), 7.39–7.30 (m, 3H), 7.15 (t, J = 7.5 Hz, 1H).13C NMR (101 MHz, DMSO) δ 160.3, 159.5, 138.8, 137.4, 131.0, 130.7, 129.1, 128.4, 126.3, 126.2, 125.8, 125.4, 120.3, 117.7, 116.7, 116.0, 113.7, 113.4. IR, vmax/cm−1 3316, 3049, 2210, 1679, 1624, 1584, 1550, 1530, 1504, 1463, 1387, 1321. HRMS (ES TOF) calculated for (M + Na)+ C18H10N2NaO 325.0406, found 325.0404 (0.7 ppm).
(E)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)-2-(4-methoxyphenyl)acetonitrile (2ba):
Red solid, mp (EtOH) 270.6–272.1 °C, Rf 0.2 (EtOAc/Hex, 3:2). Yield: 140 mg (0.42 mmol, 42%). 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 7.57 (d, J = 8.9 Hz, 2H), 7.19–7.05 (m, 3H), 6.63 (s, 1H), 3.85 (d, J = 8.8 Hz, 6H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.9, 159.7, 157.7, 150.1, 144.8, 143.0, 130.3 (2C), 124.2, 118.1, 114.8 (2C), 110.5, 105.7, 95.8, 89.0, 56.1, 56.0, 55.5. IR, vmax /cm−1: 3332, 2203, 1684, 1596, 1493, 1354, 1246, 1191, 1135, 1033. HRMS (ES TOF) calculated for (M + Na)+ C19H16N2NaO4 359.1002, found 359.0993 (2.7 ppm).
(E)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)-2-phenylacetonitrile (2bb):
Purple solid, mp (EtOH) 204.9–206.6 °C (literature data: [23] mp 205.2–207.6 °C), Rf 0.47 (EtOAc/Hex, 1:1). Yield: 168 mg (0.55 mmol, 55%). 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 7.64–7.60 (m, 2H), 7.56 (t, J = 7.7 Hz, 2H), 7.49–7.45 (m, 1H), 7.09 (s, 1H), 6.63 (s, 1H), 3.86 (s, 3H), 3.76 (s, 3H).
(E)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)-2-(p-tolyl)acetonitrile (2bc):
Red solid, mp (EtOH) 281.6–282.7 °C, Rf 0.3 (EtOAc/Hex, 3:2). Yield: 148 mg (0.46 mmol, 46%). 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 7.07 (s, 1H), 6.62 (s, 1H), 3.85 (s, 3H), 3.75 (s, 3H), 2.38 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.9, 157.7, 150.2, 144.8, 143.5, 138.8, 129.9 (2C), 129.3, 128.7 (2C), 118.1, 110.4, 105.8, 95.8, 88.8, 56.1, 55.9, 20.9. IR, vmax /cm−1: 3273, 2207, 1688, 1598, 1485, 1360, 1202, 1169, 1129, 1081. HRMS (ES TOF) calculated for (M + Na)+ C19H16N2NaO3 343.1053, found 343.1046 (2.2 ppm).
(E)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)-2-(4-isopropylphenyl)acetonitrile (2be):
Red solid, mp (EtOH) 201.4–202.2 °C, Rf 0.4 (EtOAc/Hex, 3:2). Yield: 181 mg (0.52 mmol, 52%). 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.55 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.08 (s, 1H), 6.63 (s, 1H), 3.85 (s, 3H), 3.75 (s, 3H), 2.96 (hept, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.0, 157.7, 150.2, 149.6, 144.8, 143.6, 129.7, 128.8 (2C), 127.3 (2C), 118.1, 110.4, 105.8, 95.9, 88.7, 56.1, 56.0, 33.4, 23.7 (2C). IR, vmax /cm−1: 3293, 2211, 1682, 1598, 1491, 1358, 1324, 1202, 1167, 1135. HRMS (ES TOF) calculated for (M + Na)+ C21H20N2NaO3 371.1366, found 371.1360 (1.7 ppm).
(E)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)-2-(3,4,5-trimethoxyphenyl)acetonitrile (2bm):
Purple solid, mp (EtOH) 182.1–184.0 °C, Rf 0.17 (EtOAc/Hex, 3:2). Yield: 99 mg (0.25 mmol, 25%). 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.08 (s, 1H), 6.86 (s, 2H), 6.61 (s, 1H), 3.86 (s, 9H), 3.74 (d, J = 10.8 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 181.8, 157.8, 153.3 (2C), 150.2, 144.8, 143.9, 138.0, 127.5, 117.9, 110.4, 106.3 (2C), 105.8, 95.8, 88.8, 60.1, 56.1, 56.0 (3C). IR, vmax /cm−1: 3285, 2207, 1670, 1600, 1493, 1340, 1310, 1212, 1125, 996. HRMS (ES TOF) calculated for (M + Na)+ C21H20N2NaO6 419.1214, found 419.1208 (1.3 ppm).
(E)-2-(benzo[d][1,3]dioxol-5-yl)-2-(5,6-dimethoxy-3-oxoindolin-2-ylidene)acetonitrile (2bn):
Red solid, mp (EtOH) 240.7–242.7 °C, Rf 0.24 (EtOAc/Hex, 3:2). Yield: 140 mg (0.40 mmol, 40%). 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 7.16 (s, 1H), 7.15–7.08 (m, 2H), 7.06 (s, 1H), 6.63 (s, 1H), 6.14 (s, 2H), 3.86 (s, 3H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.9, 157.7, 150.2, 147.9 (2C), 144.8, 143.4, 125.8, 123.4, 118.1, 110.5, 109.1, 108.8, 105.8, 101.8, 95.8, 88.7, 56.1, 56.0. IR, vmax /cm−1: 3297, 2211, 1686, 1592, 1487, 1364, 1316, 1248, 1202, 1173, 1135. HRMS (ES TOF) calculated for (M + Na)+ C19H14N2NaO5 373.0795, found 373.0795 (2.8 ppm).
(E)-2-(4-methoxyphenyl)-2-(8-oxo-2,3,6,8-tetrahydro-7H-[1,4]dioxino [2,3-f]indol-7-ylidene)acetonitrile (2ca):
Red solid, mp (EtOH) 269.8–271.9 °C, Rf 0.32 (EtOAc/Hex, 1:1). Yield: 147 mg (0.44 mmol, 44%). 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 7.62–7.51 (m, 2H), 7.14–7.09 (m, 2H), 7.08 (s, 1H), 6.49 (s, 1H), 4.34 (dd, J = 5.6, 2.7 Hz, 2H), 4.24–4.18 (m, 2H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.3, 159.7, 152.0, 148.2, 142.8, 139.0, 130.2 (2C), 124.3, 118.1, 114.8 (2C), 112.6, 112.3, 100.4, 88.3, 65.2, 63.5, 55.5. IR, vmax/cm−1: 3344, 2914, 2211, 1685, 1642, 1586, 1470, 1310, 1242, 1170, 1138. HRMS (ES TOF) calculated for (M + Na)+ C19H14N2NaO4 357.0846, found 357.0837 (2.6 ppm).
(E)-2-(8-oxo-2H-[1,4]dioxino [2,3-f]indol-7(3H,6H,8H)-ylidene)-2-phenylacetonitrile (2cb)
Red crystals, mp (EtOH) 290.3–291.7 °C, Rf 0.23 (EtOAc/Hex, 1:2). Yield: 188 mg (0.62 mmol, 62%). 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 7.62–7.53 (m, 4H), 7.46 (t, J = 7.1 Hz, 1H), 7.08 (s, 1H), 6.48 (s, 1H), 4.34 (dd, J = 5.6, 2.7 Hz, 2H), 4.22 (dd, J = 5.4, 2.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.4, 152.1, 148.2, 143.7, 139.1, 132.3, 129.36 (2C), 128.96, 128.73 (2C), 118.1, 112.47, 112.43, 100.5, 87.8, 65.2, 63.5. IR, vmax/cm−1: 3300, 2975, 2218, 1699, 1608, 1490, 1333, 1208, 1164, 1067, 930, 896. HRMS (ES TOF) calculated for (M + Na)+ C18H12N2NaO3 327.0735, found 327.0740 (1.5 ppm).
(E)-2-(1-hydroxy-3-oxoindolin-2-ylidene)-2-phenylacetonitrile (10ab):
A 5-mL round bottom flask equipped with a magnetic stirring bar was charged with 2′-nitroacetophenone (1 mmol, 165 mg), benzaldehyde (1 mmol, 106 mg), KCN (2 mmol, 130 mg), H2O (130 mg), and MeOH (1 mL) and refluxed for 30 min (TLC control). The reaction mixture was cooled to room temperature, and after that, MeOH (2 mL) and phenacyl bromide (1 mmol, 199 mg) were added and the reaction mixture was allowed to stand overnight. The precipitate that had formed was filtered off and washed with methanol to obtain (E)-2-(1-hydroxy-3-oxoindolin-2-ylidene)-2-phenylacetonitrile as a yellowish solid, mp (MeOH) 197.3–198.6 °C (decomposition), Rf 0.23 (EtOAc/Hex, 1:2). Yield: 123 mg (0.47 mmol, 47%). 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 7.9 Hz, 2H), 7.27 (t, J = 7.7 Hz, 2H), 7.11 (t, J = 7.0 Hz, 2H), 6.99 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 8.1 Hz, 1H), 6.39 (t, J = 7.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 197.0, 170.0, 155.9, 137.7, 136.4, 127.7 (2C), 127.4 (2C), 123.9, 123.7, 122.7, 121.8, 116.6, 116.5, 75.1. IR, vmax/cm−1: 3308, 2219, 1708, 1600, 1465, 1445, 1389, 1328, 1208, 1141. HRMS (ES TOF) calculated for (M + Na)+ C16H10N2NaO2 285.0634, found 285.0629 (1.9 ppm).
4-Oxo-2,4-diphenylbutanenitrile (12):
A 5-mL round bottom flask equipped with a magnetic stirring bar was charged with acetophenone (1 mmol, 120 mg), benzaldehyde (1 mmol, 106 mg), KCN (2 mmol, 130 mg), H2O (130 mg), and MeOH (1 mL) and refluxed for 120 min (TLC control). The reaction mixture was cooled to room temperature, AcOH (1 mmol, 60 mg, 57 µL) was added, and reflux was continued to another 30 min. Then, the reaction mixture was diluted with 100 mL of EtOAc and washed twice with 30 mL of concentrated NaHCO3 solution. The organic layer was concentrated and purified by column chromatography (eluent EtOAc:Hex 1:4). Then, 4-Oxo-2,4-diphenylbutanenitrile was obtained as a white solid. NMR spectra were in agreement to previously published [26]. Yield: 35 mg (0.15 mmol, 15%). 1H NMR (400 MHz, Chloroform-d) δ 7.93 (dt, J = 7.1, 1.4 Hz, 2H), 7.64–7.55 (m, 1H), 7.52–7.30 (m, 7H), 4.56 (dd, J = 8.1, 6.0 Hz, 1H), 3.73 (dd, J = 18.0, 8.0 Hz, 1H), 3.51 (dd, J = 18.0, 5.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 194.7, 135.7, 135.3, 134.0, 129.4 (2C), 128.9 (2C), 128.5, 128.2 (2C), 127.6 (2C), 120.8, 44.6, 32.0. HRMS (ES TOF) calculated for (M + Na)+ C16H13NNaO 258.0889, found 258.0888 (0.6 ppm).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules27092808/s1, 1H and 13C NMR spectral charts (Figures S1–S52), HRMS spectral charts (Figures S53–S78), and X-Ray crystallography data (Figure S79, Tables S1–S7). References [27,28,29] are cited in the supplementary materials.
Author Contributions
N.A.A. (Nicolai A. Aksenov)—conceptualization, supervision, data analysis, funding acquisition; A.V.A.—supervision, investigation; I.A.K.—investigation; N.K.K.—investigation; D.A.A.—investigation, data analysis; N.A.A. (Nikolai A. Arutiunov)—investigation; D.S.A.—investigation; M.R.—conceptualization, supervision, data analysis, writing (original draft, review, and editing). All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Supporting Information data include NMR spectral charts.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
Funding Statement
The study was supported by the Russian Science Foundation (grant № 21-73-10029, https://rscf.ru/project/21-73-10029/ accessed on 27 July 2021).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hoessel R., Leclerc S., Endicott J.A., Nobel M.E.M., Lawrie A., Tunnah P., Leost M., Damiens E., Marie D., Marko D., et al. Indirubin, the active constituent of a Chinese antileukemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999;1:60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 2.Leclerc S., Garnier M., Hoessel R., Marko D., Bibb J.A., Snyder G.L., Greengard P., Biernat J., Wu Y.-Z., Mandelkow E.-M., et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 3.Meijer L., Skaltsounis A.-L., Magiatis P., Polychronopoulos P., Knockaert M., Leost M., Ryan X.P., Vonica C.A., Brivanlou A., Dajani R., et al. GSK-3-Selective Inhibitors Derived from Tyrian Purple Indirubins. Chem. Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Polychronopoulos P., Magiatis P., Skaltsounis A.-L., Myrianthopoulos V., Mikros E., Tarricone A., Musacchio A., Roe S.M., Pearl L., Leost M., et al. Structural Basis for the Synthesis of Indirubins as Potent and Selective Inhibitors of Glycogen Synthase Kinase-3 and Cyclin-Dependent Kinases. J. Med. Chem. 2004;47:935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 5.Seidler J., McGovern S.L., Doman T.N., Shoichet B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 6.Wang L., Zhou G.-B., Liu P., Song J.-H., Liang Y., Yan X.-J., Xu F., Wang B.-S., Mao J.-H., Shen Z.-X., et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 2008;105:4826–4831. doi: 10.1073/pnas.0712365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gein V.L., Tatarinov V.V., Rassudikhina N.A., Vakhrin M.I., Voronina E.V. Synthesis and antimicrobial activity of 2-aroylmethylene-6-hydroxy-2,3-dihydroindol-3-ones. Pharm. Chem. J. 2011;45:231–232. doi: 10.1007/s11094-011-0602-2. [DOI] [Google Scholar]
- 8.Lack N.A., Axerio-Cilies P., Tavassoli P., Han F.G., Chan K.H., Feau C., LeBlanc E., Guns E.T., Guy R.K., Rennie P.S., et al. Targeting the Binding Function 3 (BF3) Site of the Human Androgen Receptor through Virtual Screening. J. Med. Chem. 2011;54:8563–8573. doi: 10.1021/jm201098n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu D.-Q., Mao S.-C., Zhang H.-Y., Yu X.-Q., Feng M.-T., Wang B., Feng L.-H., Guo Y.-W. Racemosins A and B, two novel bisindole alkaloids from the green alga Caulerpa racemosa. Fitoterapia. 2013;91:15–20. doi: 10.1016/j.fitote.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Medvedev A.E., Ivanov A.S., Kamyshanskaya N.S., Kirkel A.Z., Moskvitina T.A., Gorkin V.Z., Li N.Y., Marshakov V.Y. Interaction of indole derivatives with monoamine oxidase A and B. Studies on the structure-inhibitory activity relationship. Biochem. Mol. Biol. Int. 1995;36:113–122. [PubMed] [Google Scholar]
- 11.Medvedev A.E., Ivanov A.S., Veselovsky A.V., Skvortsov V.S., Archakov A.I. QSAR Analysis of Indole Analogs as Monoamine Oxidase Inhibitors. J. Chem. Inf. Comput. Sci. 1996;36:664–671. doi: 10.1021/ci950126t. [DOI] [PubMed] [Google Scholar]
- 12.Ornano L., Donno Y., Sanna C., Ballero M., Serafini M., Bianco A. Phytochemical study of Caulerpa racemosa (Forsk.) J. Agarth, an invading alga in the habitat of La Maddalena Archipelago. Nat. Prod. Res. 2014;28:1795–1799. doi: 10.1080/14786419.2014.945928. [DOI] [PubMed] [Google Scholar]
- 13.Roaiah H.M., Ahmed K.M., Fawzy N.M., Wietrzyk J., Pawlik A., Ali M.M., Soliman A.M. Synthesis of novel acetamide derivatives and evaluation of their antiproliferative potency against different cancer cell lines. Int. J. Pharm. Sci. Rev. Res. 2016;36:129–136. [Google Scholar]
- 14.Guo C., Schedler M., Daniliuc C.G., Glorius F. N-Heterocyclic carbene-catalyzed formal [3+2] annulation reaction of enals. An efficient enantioselective access to spiro-heterocycles. Angew. Chem. Int. Ed. 2014;53:10232–10236. doi: 10.1002/anie.201405381. [DOI] [PubMed] [Google Scholar]
- 15.Merour J.-Y., Chichereau L., Desarbre E., Gadonneix P. Synthesis and reactivity of (3-oxo-2,3-dihydro-1H-indol-2-ylidene)acetic acid alkyl esters in Diels-Alder Reactions. Synthesis. 1996:519–524. doi: 10.1055/s-1996-4236. [DOI] [Google Scholar]
- 16.Sim H.M., Loh K.Y., Yeo W.K., Lee C.Y., Go M.L. Aurones as Modulators of ABCG2 and ABCB1: Synthesis and Structure-Activity Relationships. ChemMedChem. 2011;6:713–724. doi: 10.1002/cmdc.201000520. [DOI] [PubMed] [Google Scholar]
- 17.Souard F., Okombi S., Beney C., Chevalley S., Valentin A., Boumendjel A. 1-Azaaurones derived from the naturally occurring aurones as potential antimalarial drugs. Bioorg. Med. Chem. 2010;18:5724–5731. doi: 10.1016/j.bmc.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 18.An Z.W., Catellani M., Chiusoli G.P. A new palladium-catalyzed synthesis of indoxyl derivatives. J. Organomet. Chem. 1990;397:C31–C32. doi: 10.1016/0022-328X(90)80248-X. [DOI] [Google Scholar]
- 19.Genelot M., Bendjeriou A., Dufaud V., Djakovitch L. Optimized procedures for the one-pot selective syntheses of indoxyls and 4-quinolones by a carbonylative Sonogashira/cyclization sequence. Appl. Catal. A. 2009;369:125–132. doi: 10.1016/j.apcata.2009.09.016. [DOI] [Google Scholar]
- 20.Genelot M., Dufaud V., Djakovitch L. Heterogeneous metallo-organocatalysis for the selective one-pot synthesis of 2-benzylidene-indoxyl and 2-phenyl-4-quinolone. Tetrahedron. 2011;67:976–981. doi: 10.1016/j.tet.2010.11.112. [DOI] [Google Scholar]
- 21.Li R., Qi X., Wu X.-F. A general and convenient palladium-catalyzed synthesis of benzylideneindolin-3-ones with formic acid as the CO source. Org. Biomol. Chem. 2017;15:6905–6908. doi: 10.1039/C7OB01557G. [DOI] [PubMed] [Google Scholar]
- 22.Gein V.L., Demeneva A.V., Rassudikhina N.A., Vakhrin M.I. Synthesis of 2-aroylmethylidene-6-hydroxy-2,3-dihydroindol-3-ones. Russ. J. Org. Chem. 2006;42:617–618. doi: 10.1134/S1070428006040245. [DOI] [Google Scholar]
- 23.Aksenov N.A., Aksenov D.A., Arutiunov N.A., Aksenova D.S., Aksenov A.V., Rubin M. Unexpected cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones. RSC Adv. 2020;10:18440–18450. doi: 10.1039/D0RA03520C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Migita M., Okada T., Mataga N., Sakata Y., Misumi S., Nakashima N., Yoshihara K. Picosecond laser spectroscopy of intramolecular heteroexcimer systems. Time-resolved fluorescence studies of p-(CH3)2NC6H4-(CH2)n-(9-anthryl), p-(CH3)2NC6H4-(CH2)n-(1-pyrenyl) systems and 9,9’-bianthryl. Bull. Chem. Soc. Jpn. 1981;54:3304–3311. doi: 10.1246/bcsj.54.3304. [DOI] [Google Scholar]
- 25.Lin Z., Hu Z., Zhang X., Dong J., Liu J.-B., Chen D.-Z., Xu X. Tandem Synthesis of Pyrrolo[2,3-b]quinolones via Cadogen-Type Reaction. Org. Lett. 2017;19:5284–5287. doi: 10.1021/acs.orglett.7b02558. [DOI] [PubMed] [Google Scholar]
- 26.Yang J., Wang Y., Wu S., Chen F.-X. The highly efficient 1,4-addition of TMSCN to aromatic enones catalyzed by CsF with water as the additive. Synlett. 2009:3365–3367. doi: 10.1002/chin.201016064. [DOI] [Google Scholar]
- 27.Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009:42–339. [Google Scholar]
- 28.Sheldrick G.M. A short history of SHELX. Acta Cryst. A. 2008:A64–112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 29.Sheldrick G.M. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015:C71–3. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting Information data include NMR spectral charts.