

Abstract
The extracellular polyhydroxybutyrate (PHB) depolymerase gene (phaZPst) of Pseudomonas stutzeri was cloned and sequenced. phaZPst was composed of 1,728 bp encoding a protein of 576 amino acids. Analyses of the N-terminal amino acid sequence and the matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) mass spectrum of the purified enzyme showed that the mature enzyme consisted of 538 amino acids with a deduced molecular mass of 57,506 Da. Analysis of the deduced amino acid sequence of the protein revealed a domain structure containing a catalytic domain, putative linker region, and two putative substrate-binding domains (SBDI and SBDII). The putative linker region was similar to the repeating units of the cadherin-like domain of chitinase A from Vibrio harveyi and chitinase B from Clostridium paraputrificum. The binding characteristics of SBDs to poly([R]-3-hydroxybutyrate) [P(3HB)] and chitin granules were characterized by using fusion proteins of SBDs with glutathione S-transferase (GST). These GST fusion proteins with SBDII and SBDI showed binding activity toward P(3HB) granules but did not bind on chitin granules. It has been suggested that the SBDs of the depolymerase interact specifically with the surface of P(3HB). In addition, a kinetic analysis for the enzymatic hydrolysis of 3-hydroxybutyrate oligomers of various sizes has suggested that the catalytic domain of the enzyme recognizes at least two monomeric units as substrates.
Poly([R]-3-hydroxybutyrate) [P(3HB)] and its copolymers are synthesized and accumulated intracellularly as a material for storage of carbon and energy by a wide variety of bacteria (2, 9, 43). The purified polyesters, which are water-insoluble and partially crystalline polymers, are hydrolyzed by microbial extracellular polyhydroxybutyrate (PHB) depolymerases (18). Extracellular PHB depolymerases are produced by various microorganisms in natural environments such as soil (8, 16, 19, 31), activated sludge (44), freshwater (34), and seawater (23, 33, 46). A number of PHB depolymerases have been purified and characterized (16, 23, 33, 42, 44, 46, 50), and some PHB depolymerase genes have been cloned and analyzed (7, 14, 15, 17, 20, 24, 25, 26, 37, 41). All the PHB depolymerases have a bifunctional organization composed of a catalytic domain at the N terminus, substrate-binding domain (SBD) at the C terminus, and linker region connecting the two domains.
Recently, the structure-function relation of the catalytic or substrate-binding domains in PHB depolymerases has been investigated (4, 10, 20, 35). The catalytic domain contains catalytic machinery composed of a catalytic triad (Ser-His-Asp) (14, 15). The serine is a part of a lipase box pentapeptide Gly-X-Ser-X-Gly (35), which has been found in all known serine hydrolases, such as lipases, esterases, and serine proteases (39). Deletion of SBD from Alcaligenes faecalis PHB depolymerase caused the ablation of hydrolytic activity for insoluble P(3HB), although the activity toward soluble 3HB oligomers was retained (35). In addition, our previous study involving fusion proteins of the SBD of A. faecalis PHB depolymerase with glutathione S-transferase (GST) demonstrated that the SBD moiety is essential for the adsorption of PHB depolymerase to the surface of P(3HB) granules (40). These results suggest that the catalytic domains and SBDs work independently. In contrast, the function of the fibronectin type III (Fn III) module or threonine (Thr)-rich region, which have been found in all known PHB depolymerases as linker regions connecting the catalytic domains and SBDs, remains to be clarified (7, 15, 18, 35).
In a previous study (46), we isolated a P(3HB)-degrading bacterium, Pseudomonas stutzeri, from seawater and investigated the biochemical properties of its PHB depolymerase. That study showed that the depolymerase has a few unique characteristics: (i) the N-terminal amino acid sequence of the enzyme was different from those of other PHB depolymerases; (ii) the enzymatic degradation product of P(3HB) was a mixture of 3HB monomer (major product) and 3HB dimer (minor product); (iii) the depolymerase showed a lower adsorption affinity for P(3HB) than did the depolymerases from Comamonas acidovorans (20) and Alcaligenes faecalis (22). Furthermore, the enzyme was purified to electrophoretic homogeneity in a single step and in good yield (approximately 5 mg/liter of culture). This will be favorable for the elucidation of the enzyme conformation, which will involve crystallization of the enzyme.
This paper reports the cloning of PHB depolymerase gene from P. stutzeri and the functional analyses of the SBD.
MATERIALS AND METHODS
Chemicals.
P(3HB) was purchased from Polyscience Inc. Chitin was purchased from Sigma Chemical Co. Other chemicals were purchased from Kanto Chemicals (Tokyo, Japan) or Wako Chemicals (Osaka, Japan).
Bacterial strains, plasmids, media, and growth conditions.
All bacterial strains and plasmids used in this study are listed in Table 1. P. stutzeri was grown in a mineral medium as described previously (46). Escherichia coli was grown at 37°C in Luria-Bertani broth in the presence of ampicillin (50 μg/ml). P. stutzeri was deposited in the Japan Culture Collection of Microorganisms (JCM), Saitama, Japan.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference(s) |
|---|---|---|
| Strains | ||
| E. coli DH5α | supE44 ΔlacU169(φ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Toyobo |
| P. stutzeri JCM 10168 = YM1006 | Growth on P(3HB) | 13, 46 |
| Plasmids | ||
| pUC118 | Ampr, lacZ | 47 |
| pGEX 4T-1 | Ampr, tac promoter, GST fusion vector | Pharmacia |
| pGSDI | The putative SBDI gene in pGEX 4T-1 | This study |
| pGSDII | The putative SBDII gene in pGEX 4T-1 | This study |
| pGSDII-I | The putative SBDI and SBDII genes in pGEX 4T-1 | This study |
| pGCL | The putative catalytic domain and linker region genes in pGEX 4T-1 | This study |
| pCS401 | 4.0-kbp SalI fragment carrying phaZpst in pUC118 | This study |
Analytical procedures of the enzyme.
PHB depolymerase was purified as described previously (13, 46). The molecular mass was determined by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF MS) with sinapinic acid as a matrix. The PHB depolymerase activity was measured at 650 nm by using P(3HB) granules (33). Polyacrylamide gel electrophoresis of the enzyme in the presence of sodium dodecyl sulfate (SDS) was carried out by the method of Laemmli (27) with a molecular weight calibration kit (Pharmacia). After electrophoresis, the proteins were stained with Coomassie brilliant blue R-250 (Kanto Chemical, Tokyo, Japan). Protein concentrations were determined by the method of Bradford (6) with the protein assay kit II (Bio-Rad Laboratories, Tokyo, Japan) with bovine serum albumin as the standard. For immunodetection, Western blotting was used (45). The proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred electrophoretically onto an Immobilon-P transfer membrane (Millipore Corp., Bedford, Mass.). PHB depolymerases were detected with antiserum raised against P. stutzeri PHB depolymerase (13), which was recognized by anti-rabbit immunoglobulin G alkaline phosphatase conjugate (Sigma). The staining was done with nitroblue tetrazolium (Wako Chemicals, Osaka, Japan) and bromochloroindolyl phosphate (Wako Chemicals).
N-terminal amino acid sequencing.
The purified enzyme (250 μg) was incubated at 50°C for 15 min in 50 μl of 0.4 M ammonium bicarbonate (pH 8.0) containing 8 M urea and 5 μl of 45 mM dithiothreitol. After 5 μl of 100 mM iodoacetamide was added to the reaction mixture, the enzyme was treated with 0.2 μg of lysyl endopeptidase (Wako Chemicals) per ml in 100 mM Tris-HCl (pH 9.0) for 24 h at 37°C. The resulting peptides were separated in an SDS-polyacrylamide gel and transferred onto an Immobilon-P transfer membrane by electroblotting. The blotted peptides were cut out from the membrane and subjected to N-terminal amino acid sequence analysis on an Applied Biosystems 473A protein sequencer.
DNA preparation and manipulation.
E. coli was grown aerobically in Luria-Bertani medium, and the cells were transformed by the calcium chloride procedure (3). Recombinant plasmid DNA was isolated by the method of Birnboim and Doly (5) or with a Flexi-prep kit (Pharmacia) for sequencing. Restriction enzymes were purchased from Takara Shuzo (Kyoto, Japan) or Toyobo (Osaka, Japan), and calf intestinal alkaline phosphatase was purchased from Boehringer GmbH (Mannheim, Germany). The enzymes were used as specified by the manufacturers.
Preparation of DNA probe.
The N-terminal amino acid sequences of the mature enzyme and one of the proteolysis polypeptide were determined to be GQTFSYTSPQQAYSGSRERSYKVYV and AAADRYGFILVAPFI, respectively. To prepare DNA probes for screening a PHB depolymerase gene from the P. stutzeri genomic DNA, 5′ and 3′ primers were designed on the basis of the N terminus of the purified enzyme and the proteolytic polypeptide amino acid sequence, respectively. The sequences of the primers used were as follows: N-terminal, 5′-GG(A/G/C/T)CA(A/G)AC(A/G/C/T)TT(C/T)(A/T)(G/C)(A/G/C/T)TA(C/T)AC-3′; and proteolysis polypeptide, 5′-AT(A/G)AA(A/G/C/T)GG(A/G/C/T)GC(A/G/C/T)AC(A/G/C/T)A(A/G)(A/T/G)AT(A/G)AA-3′. The PCR mixture contained PCR buffer, deoxynucleoside triphosphate, 100 pmol of each primer, 1 μg of genomic DNA as template, and 1 U of exTaq as the DNA polymerase. A DNA thermal cycler (Perkin-Elmer Applied Biosystems, Foster City, Calif.) was used for amplification of the gene under the following conditions: 5 cycles of denaturation at 94°C for 30 s, annealing at 45°C for 30 s, and extension at 72°C for 90 s, and subsequently 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 90 s. The amplified DNA was purified by chloroform extraction and ethanol precipitation and labeled with a digoxigenin (DIG) oligonucleotide tailing kit (Boehringer Mannheim).
Identification and cloning of the PHB depolymerase gene.
Genomic DNA of P. stutzeri was digested completely with the restriction endonucleases ApaI, EcoRI, EcoRV, HincII, HindIII, KpnI, PstI, and SalI and separated by agarose gel electrophoresis. The DNA was blotted onto a positively charged nylon membrane and hybridized with a DIG-labeled DNA probe. Hybridization was performed at 42°C for 12 h in 50% formamide–5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–50 mM sodium phosphate buffer (pH 7.0)–0.1% N-lauroylsarcosine–7% SDS–2% blocking reagent (Boehringer). The membrane was washed in 1× SSC–0.1% SDS at 60°C. Positive DNA fragments were subcloned into the pUC 118 vector (47) and then transformed into E. coli DH5α. Colony hybridization of genomic sublibraries with a DIG-labeled DNA probe was performed.
DNA sequencing and sequence analysis.
A series of deletion fragments were generated and subcloned into pUC118. Nucleotide sequence was determined on an Applied Biosystems 310 DNA sequencer with the Taq dye terminator cycle sequencing kit (Perkin-Elmer Applied Biosystems). The DNA and deduced amino acid sequences were analyzed by using the sequence analysis program GENETYX (Software Development Co., Tokyo, Japan). Database searches were performed with the program BLAST via GenomeNet www server.
Construction, production, and purification of GST fusion proteins.
As shown in Fig. 1A, DNA fragments encoding the SBDI (I), SBDII (II), SBDI plus SBDII, and catalytic domain (C) plus linker region (L) were obtained by PCR amplification from plasmid pCS401 carrying the phaZPst gene. A BamHI restriction site (boldface) was introduced at the 5′ end of oligonucleotide primer 1 (5′-CGGGATCCGGCAGCGGCCAGGCCTTCACCTG-3′), primer 3 (5′-ATGTTGGATCCGTCTACGATCCCAACGCCCCGGTGGAAACCT-3′), and primer 5 (5′-CGGGATCCGGGCAAACCTTCTCCTACACCT-3′), and a SalI restriction site (boldface) and stop codon (underlined) were introduced at the 5′ end of oligonucleotide primer 2 (5′-GCGTCGACTCAGTTGCTGCAGCGTCCGGCCTG-3′), primer 4 (5′-CGGTCGACTTAGCCGCAAGCGGCGGGCCGCTGCGCG-3′), and primer 6 (5′-CGGTCGACTTAGGTTTCCACCGGGGCGTTGGGA-3′). The PCR products including BamHI and SalI restriction sites prepared from the primer combinations 1 plus 2, 3 plus 4, 3 plus 2, and 5 plus 2 were ligated into the BamHI- and SalI-digested pGEX-4T-1 (Pharmacia), yielding pGSDI, pGSDII, pGSDII-I, and pGCL, respectively. Plasmids pGSDI, pGSDII, pGSDII-I, and pGCL encode the SBDI, SBDII, SBDI plus SBDII, and catalytic domain plus linker region with GST (Fig. 1B), respectively. Correct insertions of these genes were verified by DNA sequencing for both strands. To express the GST fusion protein, these chimeric plasmids were transformed into E. coli DH5α. The production and purification of GST fusion proteins were performed as described previously (20, 41), and the resulting proteins are shown in Fig. 1C.
FIG. 1.

(A) Restriction map of pCS401 containing the phaZPst gene. The coding region is represented by solid arrows. Thin lines and black segments correspond to cloned DNA fragment and plasmid DNA, respectively, and amplified DNA fragments encoding the respective domains. (B) Domain structures of GST and GST fusion proteins of its derivatives. (C) SDS-PAGE of purified samples (1.0 μg each) from GST and GST fusion proteins. Lanes: 1, GST; 2, GST-SBDI; 3, GST-SBDII; 4, GST-SBDII-I; 5, GST-C-L. Molecular mass standards were run in lane M; sizes are shown to the left. S, signal peptide; C, catalytic domain; L, linker region.
Binding assay.
P(3HB) and chitin granules were used as the adsorbents for the binding assay. P(3HB) granules were purified by washing with distilled water, acetone, and hexane. The granules were dried in vacuo at room temperature. Chitin granules were purified by washing with distilled water. P(3HB) and chitin granules (400 μg) were suspended in 1.0 ml of 0.05 M Tris-HCl buffer (pH 7.5) and preincubated at 37°C for 5 min. After preincubation, a solution of a given amount (1 to 50 μg) of fusion protein was added. The reaction mixture was incubated at 37°C for 10 min and then centrifuged in a Eppendorf F 45-30-11 rotor at 10,000 × g for 1 min. The amount of protein in the supernatant was determined by the Bradford method (6) with bovine serum albumin as the standard. The amount of enzyme bound to polymer granules was calculated from the values of soluble and added enzyme.
Enzymatic hydrolyses of (R)-3HB oligomers and their derivatives.
Methyl (R)-3-hydroxybutanoate [H(3HB)M] was supplied by Kaneka Chemical Industries. (3R)-3-{[(3′R)-3′-hydroxybutanoyl]oxy}butanoic acid [H(3HB)2H] was supplied by Takasago International Corp. Syntheses of methyl (3R)-3-{[(3′R)-3′-hydroxybutanoyl]oxy}butanoate [H(3HB)2M], methyl (3R)-3-{[(3′R)-3′-(methoxy)butanoyl]oxy}butanoate [M(3HB)2M], methyl (3R)-3-{[(3′R)-3′-{[(3"R)-3"-{[(3"′R)-3"′-hydroxybutanoyl]oxy}butanoyl]oxy}butanoate [H(3HB)3M] and methyl (3R)-3-{[(3′R)-3′-{[(3"R)-3"-{[(3"′R)-3"′-hydroxybutanoyl]oxy}bu-tanoyl]oxy}butanoyl]oxy}butanoate [H(3HB)4M] were performed as described elsewhere (28). Enzymatic hydrolysis of the 3HB oligomers (5 mM) by the purified PHB depolymerase (4 μg) from P. stutzeri was carried out at 37°C in 1 ml of 100 mM potassium phosphate buffer (pH 7.4). The hydrolytic products after enzymatic hydrolysis were analyzed with a Shimadzu LC-9A high-performance liquid chromatography (HPLC) system with a gradient controller and an SPD-10A UV spectrophotometric detector (1). The stainless steel column (250 by 4 mm) containing LiChrospher RP-8 (5 mm) was used at 40°C. Sample solutions after the enzymatic hydrolysis were acidified to pH 2.5 with HCl solution, and 50-μl samples were injected into the column. The gradient of distilled water (pH 2.5, adjusted by the addition of HCl solution) to acetonitrile was carried out for 40 min with a pump speed of 1.0 ml/min. In this method, (R)-3-hydroxybutanoic acid [H(3HB)H], H(3HB)M, (R)-3-(methoxy)butanoic acid [M(3HB)H], H(3HB)2H, H(3HB)2M, (3R)-3-{[(3′R)-3′-(methoxy)butanoyl]oxy}butanoic acid [M(3HB)2H], M(3HB)2M, the trimeric ester of (R)-3-hydroxybutanoic acid [H(3HB)3H], H(3HB)3M, the tetrameric ester of (R)-3-hydroxybutanoic acid [H(3HB)4H], and H(3HB)4M were detected at 210 nm and eluted at 5.2, 10.4, 10.6, 12.3, 15.5, 15.7, 19.7, 16.3, 19.5, 19.2, and 22.3 min, respectively.
Nucleotide sequence accession number.
The DDBJ accession no. for the P. stutzeri PHB depolymerase gene is AB012225.
RESULTS
Amino acid sequences of PHB depolymerase and preparation of DNA probe.
PHB depolymerase purified from P. stutzeri was digested with lysyl endopeptidase, resulting in the formation of two major peptides (PSA and PSB). The N-terminal amino acid sequences of PSA and PSB were determined as ASDTGCSPYHQNDYGCRHIA and AAADRYGFILVAPFI, respectively. In addition, the N-terminal amino acid sequence of mature enzyme had been determined as GQTFSYTSPQQAYSGSRERSYKVYV (13). To amplify a PHB depolymerase gene by PCR, the primers were designed on the basis of the N-terminal amino acid sequence of mature enzyme and PSB. PCR with the primers resulted in a PCR product of approximately 200 bp length, and the nucleotide sequence of this product was determined. We concluded that the 200-bp fragment was a part of the PHB depolymerase gene, since the N-terminal amino acid sequences of mature enzyme and PSB were found in the amino acid sequence deduced from the nucleotide sequence of this PCR product. Therefore, the PCR product was labeled and used as a probe.
Cloning of the PHB depolymerase gene (phaZPst) of P. stutzeri.
Southern blots of genomic DNA of P. stutzeri digested with various restriction enzymes were hybridized with the DIG-labeled PCR product. A 4.0-kbp SalI fragment was found to hybridize to the probe. Then, we constructed a genomic sublibrary with the 4.0-kbp SalI fragment in the pUC118 vector in E. coli DH5α. The positive recombinant clones were screened from the genomic sublibrary by colony hybridization with the same probe. Of 2,000 colonies, 2 hybridized to the probe. Restriction analysis revealed that two positive clones had the same 4.0-kb SalI fragment. The inserted fragments in the two clones were oriented in the opposite direction, and the clones were referred to as pCS401 and pCS402, respectively. The culture supernatants from E. coli DH5α carrying pCS401 or pCS402 exhibited the ability to hydrolyze P(3HB) granules. In addition, the gene products were confirmed by using Western blotting and had the same molecular mass as the native enzyme (data not shown). These results suggest that a phaZPst promoter was located within the insert and that the phaZPst gene product performed functional expression and secretion of PHB depolymerase in E. coli.
Nucleotide sequence and deduced amino acid sequence.
The DNA sequence of the 2.4-kbp SphI-BalI region of the 4.0-kbp SalI fragment was analyzed. As a result, an open reading frame of 1,728 bp was assigned to phaZPst (Fig. 1A). The putative initiation codon ATG at nucleotide 1 was preceded at a spacing of 7 bp by a potential ribosome-binding sequence at nucleotides −13 to −8 (5′-ACGGAG-3′), and the sequences at nucleotides −139 to −134 (5′-TTGAAA-3′) and at nucleotides −125 to −120 (5′-TGGCAT-3′) with 18-bp spacing showed some homology to the −35 and −10 E. coli ς70-like promoter sequences (Fig. 2).
FIG. 2.

Nucleotide sequences of the phaZPst gene and deduced amino acid sequence of the gene product. A putative ribosome-binding (Shine-Dalgarno [S/D]) site is boxed, and the −35 and −10 regions of a possible promoter sequence are indicated by double underlines. Amino acids confirmed by Edman degradation are underlined, and the processing site of the depolymerase precursor is marked by a vertical arrow. The orientation of transcription and a putative termination signal of transcription are indicated by arrows. A putative linker region is boxed, and a putative SBD is shaded.
The encoded polypeptide is a preprotein of 576 amino acids with a predicted molecular mass of 61,746 Da. The N-terminal amino acid sequences determined for the mature PHB depolymerase (PhaZPst) secreted by P. stutzeri and its proteolytic fragments (PSA and PSB) derived from lysyl endopeptidase digestion existed in the gene product (Fig. 2). In addition, amino acid residues 39 to 63 of the deduced gene product were identical to the N-terminal amino acid sequence determined for the mature PHB depolymerase secreted by P. stutzeri, suggesting that 38-amino-acid polypeptide is signal peptide. The molecular mass deduced for the mature PHB depolymerase of P. stutzeri was 57,506 Da, which was in good agreement with the value determined by MALDI-TOF MS (57,456 Da).
The deduced amino acid sequence of the depolymerase exhibited a low level of homology to those of other PHB depolymerases (below 10%), while relatively high degrees of similarities were found in the regions surrounding putative active sites and the SBD at the C terminus. The N-terminal catalytic domain contained conserved amino acids of the catalytic triad (Ser163, Asp240, and His289) and oxyanion hole (His78), which functions as an active center in many known serine hydrolases (39), and the lipase box, which functions as an active center (Ser163) at the center of the domain. The amino acid sequence of the catalytic domain showed sequence homology to those of A. faecalis T1 PhaZAfa (level of identity, 21.3%) (37), A. faecalis AE122 PhaZAfaAE (14.3%) (24), R. pickettii PhaZRpi (20.9%) (26), P. lemoignei PhaZ1Ple (24.4%) (17), P. lemoignei PhaZ2Ple (25.1%) (7), P. lemoignei PhaZ3Ple (28.4%) (7), P. lemoignei PhaZ4Ple (21.6%) (15), and P. lemoignei PhaZ5Ple (22.2%) (15).
A fibronectin type III module or a threonine-rich region identified as putative linker region of other PHB depolymerases was not found in the PHB depolymerase from P. stutzeri. As shown in Fig. 3A, the sequence of amino acids 332 to 454 in the PHB depolymerase from P. stutzeri had significant homology to reiterated sequences from Vibrio harveyi chitinase A (level of identity, 38.1 and 36.1%) (accession no. U81496) and Clostridium paraputrificum chitinase B (35.9 and 32.4%) (32), which has been referred to as cadherin-like domain (32).
FIG. 3.

(A) Alignment of the putative linker region of PHB depolymerase from P. stutzeri (PhaZPst) with reiterated cadherin-like domains of V. harveyi chitinase A (Vh ChiA) and Clostridium paraputrificum chitinase B (Cp ChiB). Identical amino acids are shown on a black background, and amino acids which are conserved in at least three of the five sequences are shaded in grey. (B) Alignment of the putative SBD of the PHB depolymerase with those of other proteins. The sequences of the PHB depolymerases from PhaZPst, A. faecalis T1 (PhaZAfaT1), A. faecalis AE122 (PhaZAfaAE122), R. pickettii (PhaZRpi), Comamonas sp. (PhaZCsp), C. testosteroni (PhaZCte), C. acidovorans (PhaZCac), S. exfoliatus (PhaZSex), and P. lemoignei (PhaZ1Ple, PhaZ2Ple, PhaZ3Ple, PhaZ4Ple, PhaZ5Ple) are shown. Identical amino acids are shown against a black background, and amino acids which are conserved in at least 7 of the 12 sequences are shaded in grey; all sequences are numbered from Met-1 of the peptide.
In addition, two amino acid sequences at the C-terminal region of the enzyme showed sequence homology to the SBDs identified in some PHB depolymerases, as shown in Fig. 3B. These regions, which are separated by 13 amino acids, were designated SBDI (amino acids 527 to 576) and SBDII (amino acids 455 to 514), respectively. As listed in Table 2, the amino acid sequences of SBDI and SBDII showed 12.2 to 40.4% and 11.9 to 20.3% identity to SBDs of other PHB depolymerases, respectively. The amino acid sequence of SBDI showed 14.3% identity to that of SBDII.
TABLE 2.
The amino acid sequence homologies among SBDs of PHB depolymerases
| Enzyme | Level of identity (%) to:
|
|
|---|---|---|
| P. stutzeri PhaZPst SBDI | P. stutzeri PhaZPst SBDII | |
| P. stutzeri PhaZPst SBDI | 100 | 14.3 |
| P. stutzeri PhaZPst SBDII | 14.3 | 100 |
| A. faecalis PhaZAfaAE122 PHV | 12.2 | 13.6 |
| A. faecalis PhaZAfaAE122 PHB | 18.4 | 23.7 |
| A. faecalis PhaZAfa | 40.4 | 15.3 |
| R. pickettii PhaZRpi | 35.3 | 15.3 |
| P. lemoignei PhaZ1Ple | 22.4 | 6.8 |
| P. lemoignei PhaZ2Ple | 35.3 | 11.9 |
| P. lemoignei PhaZ3Ple | 37.3 | 27.1 |
| P. lemoignei PhaZ4Ple | 24.5 | 20.3 |
| P. lemoignei PhaZ5Ple | 35.3 | 15.3 |
| Comamonas sp. strain PhaZCsp | 31.4 | 11.9 |
| C. testosteroni PhaZCte | 33.3 | 11.9 |
| C. acidovorans PhaZCac | 35.5 | 15.3 |
| S. exfoliatus PhaZSex | 39.2 | 11.9 |
Function of SBDs.
To investigate the binding characteristics of the two SBDs, we constructed fusion proteins of GST with several polypeptides of the PHB depolymerase of P. stutzeri (Fig. 1B) and performed the adsorption test on polymer granules of P(3HB) and chitin. As shown in Fig. 1C, four fusion proteins, GST-SBDI, GST-SBDII, GST-SBDII-I, and GST-C-L were purified to electrophoretic homogeneity, and the molecular masses of the fusion proteins were consistent with the values calculated from their sequences; 27,897 for GST, 33,007 for GST-SBDI, 34,130 for GST-SBDII, 40,809 for GST-SBDII-I, and 70,422 for GST-C-L (Fig. 1C). Table 3 shows the binding specificities of GST and four GST fusion proteins (10 μg of protein per ml) for P(3HB) and chitin granules. Three GST fusion proteins with SBDs (i.e., GST-SBDI, GST-SBDII, and GST-SBDII-I) adsorbed to P(3HB). On the other hand, GST itself and the GST fusion protein lacking SBDs (i.e., GST-C-L) were not able to adsorb to P(3HB) granules. None of the GST fusion proteins used in this study adsorbed to chitin granules. These results clearly indicate that SBDI and SBDII are the domains responsible for binding of enzyme on the surface of P(3HB) granules.
TABLE 3.
Binding specificities of GST fusion proteins for P(3HB) and chitin granules at 37°C
| Substrate | Amt (μg) of bound proteina
|
||||
|---|---|---|---|---|---|
| GST | GST-SBDI | GST-SBDII | GST-SBDII-I | GST-C-L | |
| P(3HB) | 0 | 3.8 | 1.3 | 8.1 | 0 |
| Chitin | 0 | 0 | 0 | 0 | 0 |
10 μg of a protein and 400 μg of polymer granules were used in each assay.
The kinetics of the adsorption of three fusion proteins, GST-SBDI, GST-SBDII, and GST-SBDII-I, to P(3HB) granules were investigated at 37°C. Figure 4 shows the relationship between the amount of adsorbed protein (Ead) and the equilibrium concentration of protein ([E]e) in the presence of three fusion proteins at 37°C. The adsorption of fusion proteins on the surface of P(3HB) granules could be expressed by the Langmuir adsorption equation:
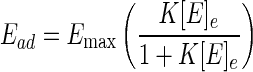 |
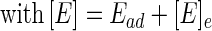 |
where Emax and K are the maximum amount of protein bound on P(3HB) granules and the adsorption equilibrium constant of the protein, respectively, and [E] is the concentration of protein added. The values of K and Emax for adsorption of GST fusion proteins to P(3HB) granules were determined from the data in Fig. 4 and are listed in Table 4. Both values of K and Emax decreased in the following order: GST-SBDII-I > GST-SBDI > GST-SBDII.
FIG. 4.

Adsorption isotherms for the GST-SBD fusion proteins on P(3HB) granules. Symbols: ○, GST-SBDI; •, GST-SBDII; ▴, GST-SBDI-II.
TABLE 4.
Adsorption equilibrium constant, K, of GST fusion proteins and maximum amount, Emax, of the proteins bound on the surfaces of P(3HB) granules at 37°C
| Protein | K (ml/μg) | Emax (μg/mg of granule) |
|---|---|---|
| GST-SBDI | 0.26 ± 0.02 | 15.3 ± 1.0 |
| GST-SBDII | 0.07 ± 0.01 | 11.4 ± 1.0 |
| GST-SBDII-I | 0.4 ± 0.04 | 26.8 ± 2.0 |
Hydrolysis of 3HB oligomers by PHB depolymerase from P. stutzeri.
Six types of 3HB oligomer derivatives from monomer to tetramer were prepared, and the enzymatic hydrolyses were studied in the presence of PHB depolymerase from P. stutzeri. The composition of the hydrolytic product was measured by HPLC analysis during the enzymatic hydrolysis of 3HB oligomers at 37°C in 0.1 M potassium phosphate buffer (pH 7.4). Figure 5 shows the results of the time course experiment for enzymatic hydrolysis of 3HB oligomers (5 mM) by PHB depolymerase from P. stutzeri (4 μg/ml). When the methyl ester of 3HB monomer [H(3HB)M] was used as a substrate for hydrolysis by the enzyme, the hydrolytic product was not detected during the 5-h reaction (Fig. 5A). Three types of 3HB dimer derivatives were tested. When H(3HB)2H was used as substrate, the 3HB monomer [H(3HB)H] was generated as a hydrolytic product (Fig. 5B). On the other hand, enzymatic hydrolysis of H(3HB)2M yielded a mixture of H(3HB)H, H(3HB)M, and H(3HB)2H at almost identical rates in the initial stage of the reaction, followed by a slow hydrolysis of the H(3HB)2H (Fig. 5C). In addition, enzymatic hydrolysis of M(3HB)2M yielded a mixture of M(3HB)H, H(3HB)M, and M(3HB)2H as hydrolysates in the initial stage of the reaction, followed by the formation of H(3HB)H from M(3HB)2H (Fig. 5D). The 3HB dimer derivatives of H(3HB)2M and M(3HB)2M were completely hydrolyzed within 2 h. In contrast, the rate of enzymatic hydrolysis for the 3HB trimer derivative of H(3HB)3M was greater by 2 orders of magnitude than the rates for the 3HB dimer derivatives, and the H(3HB)3M trimer was completely hydrolyzed within 1 min to yield the mixture of H(3HB)H, H(3HB)M, H(3HB)2H, H(3HB)2M, and H(3HB)3H (Fig. 5E). As shown in Fig. 5, the 3HB tetramer derivative [H(3HB)4M] was also hydrolyzed rapidly within 1 min to yield the mixture of H(3HB)H, H(3HB)M, H(3HB)2H, H(3HB)2M, H(3HB)3H, and H(3HB)3M as hydrolysates (Fig. 5F).
FIG. 5.

Relative amounts of the products generated at 37°C during the hydrolysis of 5 mM H(3HB)M (A), H(3HB)2H (B), H(3HB)2M (C), M(3HB)2M (D), H(3HB)3M (E), and H(3HB)4M (F) by P. stutzeri PHB depolymerase (4 μg/ml) and frequency of bond cleavage and velocity of oligomer degradation during the initial action of the depolymerase. H(3HB)H (○), H(3HB)M (•), M(3HB)H ( ), M(3HB)2H (▵), H(3HB)2M (▴), M(3HB)2M (▿), M(3HB)2H (▾), H(3HB)3M (□), H(3HB)3M (■), and H(3HB)4M (⊠) were analyzed by HPLC. The broken lines indicate ester bonds hydrolyzed by the enzyme, and the numbers represent frequencies of enzyme attack.
), M(3HB)2H (▵), H(3HB)2M (▴), M(3HB)2M (▿), M(3HB)2H (▾), H(3HB)3M (□), H(3HB)3M (■), and H(3HB)4M (⊠) were analyzed by HPLC. The broken lines indicate ester bonds hydrolyzed by the enzyme, and the numbers represent frequencies of enzyme attack.
The frequencies of bond cleavage and the rates of enzymatic hydrolysis of these oligomers were determined at the initial stage of reaction (Fig. 5). The bond cleavage of H(3HB)2M took place at same rate for the first and second ester bonds from the hydroxy terminus, while the hydrolysis of M(3HB)2H occurred at the first ester bond from the methoxy terminus at a high frequency. The rates of enzymatic hydrolysis of H(3HB)2H, H(3HB)2M, and M(3HB)2M were 0.7 ± 0.1, 4.6 ± 0.4, and 3.8 ± 0.6 mol h−1, respectively. Thus, the rates of H(3HB)2M and M(3HB)2M hydrolysis were about six times higher than that of H(3HB)2H. These results suggest that the rate of enzymatic hydrolysis is influenced by the presence of methyl ester and that the enzyme recognizes at least two monomeric units of the substrate. On the other hand, the bond cleavage of H(3HB)3M also occurred at all ester bonds. The initial bond cleavage of H(3HB)4M could not be evaluated due to further hydrolysis of the hydrolysates. The rates of hydrolysis of H(3HB)3M and H(3HB)4M were 598 ± 17 and 1,180 ± 115 mM h−1, respectively.
DISCUSSION
This paper has reported the cloning and sequence analysis of the gene (phaZPst) for an extracellular PHB depolymerase of P. stutzeri YM1006 and the relationship between the primary structure and the function of the enzyme. phaZPst encodes a polypeptide composed of 576 amino acids, and the amino acid sequence shows a low homology (below 10%) to those of other known PHB depolymerases. However, several consensus regions that occur in amino acid sequences of other known depolymerases were found in the sequence of the depolymerase. On the basis of the consensus regions, we have concluded that the PHB depolymerase of P. stutzeri consists of an N-terminal signal peptide, a catalytic domain, a cadherin-like domain as linker region, and two SBDs at the C terminal (Fig. 3). The N-terminal 38 amino acid residues showed characteristics typical of signal peptides, which are composed of a positively charged region (amino acids 1 to 5), a hydrophobic region (amino acids 6 to 33), and a signal peptidase recognition site (Val35-X-Ala37). The N-terminal amino acid sequence of cloned enzyme was identical to that of the mature enzyme produced by P. stutzeri. In addition, the molecular mass deduced for the mature PHB depolymerase of P. stutzeri was almost identical to that determined by MALDI-TOF MS. These results indicate that the signal peptide of P. stutzeri PHB depolymerase functions correctly in E. coli.
The sequence consisting of the next 320 amino acid residues showed a sequence similarity to the catalytic domain of PHB depolymerases which have a lipase box in the center of the domain (level of identity, 28.4 to 14.3%). In a previous report (46), we demonstrated that the P. stutzeri PHB depolymerase yielded 3-hydroxybutyric acid as a major product (approximately 97%) with a small portion (approximately 3%) of 3HB dimer during the enzymatic hydrolysis of P(3HB) film. In contrast, the PHB depolymerases from C. acidovorans (20), C. testosteroni (19), and A. faecalis (22) hydrolyzed the P(3HB) film to yield 3HB dimer as a major product. This suggests that the substrate recognition by the active site of P. stutzeri PHB depolymerase may be different from that of other depolymerases. Therefore, to investigate the function of the catalytic domain of P. stutzeri PHB depolymerase, we performed kinetic analysis of enzymatic hydrolysis by using several different types of water-soluble oligomers of 3HB as substrates. The methyl ester of 3HB monomer [H(3HB)M] was not hydrolyzed by the enzyme, while the methyl ester of 3HB dimer [H(3HB)2M] was hydrolyzed to yield a mixture of H(3HB)H, H(3HB)M, and H(3HB)2H as the hydrolysates, suggesting that the active site of the catalytic domain recognizes at least two monomeric units as substrates for the hydrolysis of ester bonds in a 3HB sequence. For the 3HB dimer derivatives used, the rate of enzymatic hydrolysis decreased in the following order: M(3HB)2M = H(3HB)2M > H(3HB)2H (Fig. 5), indicating that the presence of a hydroxy terminus and a carboxy terminus is not essential for the enzymatic hydrolysis of 3HB dimers. The hydrolysis rate of the 3HB trimer H(3HB)3M was 2 orders of magnitude higher than the hydrolysis rates of the 3HB dimer derivatives and was almost the same as the rate for 3HB tetramer H(3HB)4M, suggesting that the active site of the catalytic domain prefers to bind three 3HB units for the hydrolysis of a 3HB sequence.
A homology search of the deduced amino acid sequence of P. stutzeri PHB depolymerase with respect to those of other PHB depolymerases has suggested that the depolymerase has two SBDs. Several His, Ala, Arg, and Cys residues, which are conserved in the SBDs of other PHB depolymerases, were also present in two SBDs of P. stutzeri PHB depolymerase (Fig. 3B). To clarify the function of two SBDs, we have constructed four types of GST fusion proteins (Fig. 1). GST itself and the GST fusion protein without SBDs (GST-C-L) did not adsorb to the surface of P(3HB) or chitin granules. The three other fusion proteins, containing SBDs (GST-SBDI, GST-SBDII, and GST-SBDII-I), did not adsorb to chitin granules but bound to the surface of P(3HB) granules. This result indicates that each SBD moiety confers a binding activity to the surface of P(3HB) on P. stutzeri PHB depolymerase. Besides the SBDs of P. stutzeri PHB depolymerase, it has been suggested that xylanase A of Clostridium stercoraarium (38) and PHB depolymerase of A. faecalis AE122 (24) have two SBDs. For those enzymes, however, the logical reason to have two SBDs remains to be clarified. The values of the adsorption equilibrium constant K at 37°C for adsorption of both GST-SBDI (K = 0.26 ± 0.02 ml/μg) and GST-SBDII (K = 0.07 ± 0.01 ml/μg) to P(3HB) granules were lower than those for GST fusion proteins with other SBDs of PHB depolymerases from C. acidovorans YM1609 (K = 1.0 ± 0.1 ml/μg), C. testosteroni YM1004 (K = 1.1 ± 0.1 ml/μg), and A. faecalis T1 (K = 0.8 ± 0.1 ml/μg) (21). On the other hand, the affinities of SBDs for P(3HB) granules increased in the following order: GST-SBDII < GST-SBDI < GST-SBDII-I, suggesting that the binding of two domains SBDI and SBDII on the surface of P(3HB) takes place cooperatively. Here, we speculate that P. stutzeri PHB depolymerase has two SBDs due to a weak substrate-binding affinity of each SBD.
The sequence consisting of 97 amino acid residues present between the catalytic domain and SBDs showed a comparatively high similarity to the cadherin-like domain found in chitinases from V. harveyi (level of identity, 36.1 to 38.1%), and Clostridium paraputrificum (32.4 to 35.9%) (32). Cadherins are known to be one type of membrane protein of animal cells responsible for the cell adhesion (30, 36), as well as fibronectins. It is of interest that the cell membrane proteins such as cadherins and fibronectins have been found in a linker region of insoluble bacterial polymer hydrolases such as PHB depolymerases (14, 15, 20, 25, 37, 41), cellulases (11), and chitinases (48, 49). However, the function of the chitinase cadherin-like domain remains unclear. In this study, the fusion protein GST-C-L containing catalytic and cadherin-like domains did not bind to P(3HB), indicating that the cadherin-like domain of the enzyme is not involved in substrate binding of the enzyme. Similarly, it has been found that the cadherin-like domain of Clostridium paraputrificum chitinase also does not have affinity for chitin (32). These results suggest that this domain may play a structural role in maintaining an optimal distance between the catalytic domain and SBDs. The cadherin-like domains seem to be spread by horizontal transfer among insoluble bacterial polymer hydrolases as linking domain, as well as a fibronectin type III domain (12, 29).
ACKNOWLEDGMENT
This work was supported by CREST (Core Research for Evolutional Science and Technology) of Japan Science and Technology Corporation (JST).
REFERENCES
- 1.Abe H, Doi Y, Aoki H, Akehata T, Hori Y, Yamaguchi A. Physical properties and enzymatic degradability of copolymers of (R)-3-hydroxybutyric and 6-hydroxyhexanoic acids. Macromolecules. 1995;28:7630–7637. [Google Scholar]
- 2.Anderson A J, Dawes E A. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev. 1990;54:450–477. doi: 10.1128/mr.54.4.450-472.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausubel F M, Burent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1987. [Google Scholar]
- 4.Behrends A, Klingbeil B, Jendrossek D. Poly(3-hydroxybutyrate) depolymerases bind to their substrate by a C-terminal located substrate binding site. FEMS Microbiol Lett. 1996;143:191–194. doi: 10.1111/j.1574-6968.1996.tb08479.x. [DOI] [PubMed] [Google Scholar]
- 5.Birnboim H C, Doly J A. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Briese B H, Schmidt B, Jendrossek D. Pseudomonas lemoignei has five poly(hydroxyalkanoic acid) (PHA) depolymerase genes: a comparative study of bacterial and eukaryotic PHA depolymerases. J Environ Polym Degrad. 1994;2:75–87. [Google Scholar]
- 8.Delafield F P, Doudroff M M, Palleroni N J, Lusty C J, Contopoulos R. Decomposition of poly-β-hydroxybutyrate by pseudomonas. J Bacteriol. 1965;90:1455–1466. doi: 10.1128/jb.90.5.1455-1466.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi Y. Microbial polyesters. New York, N.Y: VCH Publishers; 1990. Microorganisms and poly(3-hydroxyalkanoates) pp. 33–62. [Google Scholar]
- 10.Fukui T, Narikawa T, Miwa K, Shirakura Y, Saito T, Tomita K. Effect of limited tryptic modification of a bacterial poly(3-hydroxybutyrate) depolymerase on its catalytic activity. Biochim Biophys Acta. 1988;952:164–171. doi: 10.1016/0167-4838(88)90112-4. [DOI] [PubMed] [Google Scholar]
- 11.Gilkes N R, Henrissat B, Kilburn D G, Miller R C, Jr, Warren R A J. Domains in microbial β-1,4-glycanases: sequence conservation, function, and enzyme families. Microbiol Rev. 1991;55:303–315. doi: 10.1128/mr.55.2.303-315.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen C K. Fibronectin type III-like sequences and new domain type in prokaryotic depolymerases with insoluble substrates. FEBS Lett. 1992;305:91–96. doi: 10.1016/0014-5793(92)80871-d. [DOI] [PubMed] [Google Scholar]
- 13.Iwata T, Doi Y, Kasuya K, Inoue Y. Visualization of enzymatic degradation of poly[(R)-3-hydroxybutyrate] single crystals by an extracellular PHB depolymerase. Macromolecules. 1997;30:833–839. [Google Scholar]
- 14.Jendrossek D, Backhaus M, Andermann M. Characterization of the extracellular poly(3-hydroxybutyrate) depolymerase of Comamonas sp. and its structural gene. Can J Microbiol. 1995;41:160–169. doi: 10.1139/m95-183. [DOI] [PubMed] [Google Scholar]
- 15.Jendrossek D, Frisse A, Behrends A, Andermann M, Kratzin H, Stanislawski T, Schlegel H G. Biochemical and molecular characterization of the Pseudomonas lemoignei polyhydroxyalkanoate depolymerase system. J Bacteriol. 1995;177:596–607. doi: 10.1128/jb.177.3.596-607.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jendrossek D, Knoke I, Habibian R B, Steinbüchel A, Schlegel H G. Degradation of poly(3-hydroxybutyrate), PHB, by bacteria and purification of a novel PHB depolymerase from Comamonas sp. J Environ Polym Degrad. 1993;1:53–63. [Google Scholar]
- 17.Jendrossek D, Müller B, Schlegel H G. Cloning and characterization of the poly(hydroxyalkanoic acid)-depolymerase gene locus, phaZ1, of Pseudomonas lemoignei and its gene product. Eur J Biochem. 1993;218:701–710. doi: 10.1111/j.1432-1033.1993.tb18424.x. [DOI] [PubMed] [Google Scholar]
- 18.Jendrossek D, Schirmer A, Schlegel H G. Biodegradation of polyhydroxyalkanoic acids. Appl Microbiol Biotechnol. 1996;46:451–463. doi: 10.1007/s002530050844. [DOI] [PubMed] [Google Scholar]
- 19.Kasuya K, Doi Y, Yao T. Enzymatic degradation of poly[(R)-3-hydroxybutyrate] by Comamonas testosteroni ATSU of soil bacterium. Polym Degrad Stab. 1994;45:379–386. [Google Scholar]
- 20.Kasuya K, Inoue Y, Tanaka T, Akehata T, Iwata T, Fukui T, Doi Y. Biochemical and molecular characterization of the polyhydroxybutyrate depolymerase of Comamonas acidovorans YM1609, isolated from fresh water. Appl Environ Microbiol. 1997;63:4844–4852. doi: 10.1128/aem.63.12.4844-4852.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasuya, K., T. Ohura, K. Masuda, and Y. Doi. Substrate and binding specificities of bacterial polyhydroxybutyrate depolymerase. Submitted for publication. [DOI] [PubMed]
- 22.Kasuya K, Inoue Y, Yamada Y, Doi Y. Kinetics of surface hydrolysis of poly(R)-3-hydroxybutyrate) film by PHB depolymerase from Alcaligenes faecalis T1. Polym Degrad Stab. 1995;48:167–174. [Google Scholar]
- 23.Kita K, Ishimaru K, Teraoka M, Yanase H, Kato N. Properties of poly(3-hydroxybutyrate) depolymerase from a marine bacterium, Alcaligenes faecalis AE122. Appl Environ Microbiol. 1995;61:1727–1730. doi: 10.1128/aem.61.5.1727-1730.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kita K, Mashiba S, Nagita M, Ishimaru K, Okamoto K, Yanase H, Kato N. Cloning of poly(3-hydroxybutyrate) depolymerase from a marine bacterium, Alcaligenes faecalis AE122, and characterization of its gene product. Biochim Biophys Acta. 1997;1352:113–122. doi: 10.1016/s0167-4781(97)00011-0. [DOI] [PubMed] [Google Scholar]
- 25.Klingbeil B, Kroppenstedt R M, Jendrossek D. Taxonomic identification of Streptomyces exfoliatus K10 and characterization of its poly(3-hydroxybutyrate) depolymerase gene. FEMS Microbiol Lett. 1996;142:103–109. doi: 10.1111/j.1574-6968.1996.tb08433.x. [DOI] [PubMed] [Google Scholar]
- 26.Kurusu Y, Kohama K, Uchida Y, Saito T, Yukawa H. Cloning and nucleotide sequencing of the poly(3-hydroxybutyrate) depolymerase gene from Pseudomonas pickettii. In: Doi Y, Fukuda K, editors. Biodegradable plastics and polymers. Amsterdam, The Netherlands: Elsevier Science Publishing BV; 1994. pp. 357–361. [Google Scholar]
- 27.Laemmli U K. Cleavage of structural proteins during the assembly of head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Li, J., J. Uzawa, and Y. Doi. Conformational analysis of oligomers of (R)-3-hydroxybutanoic acid in solutions by 1H NMR spectroscopy. Bull. Chem. Soc. Jpn., in press.
- 29.Little E, Bork P, Doolittle R F. Tracing the spread of fibronectin type III domains in bacterial glycohydrolases. J Mol Evol. 1994;39:631–643. doi: 10.1007/BF00160409. [DOI] [PubMed] [Google Scholar]
- 30.Mahoney P A, Weber U, Onofrechuk P, Biessmann H, Bryant P J, Goodman C S. The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell. 1991;67:853–868. doi: 10.1016/0092-8674(91)90359-7. [DOI] [PubMed] [Google Scholar]
- 31.Mergaert J, Weeb A, Anderson C, Wouters A, Swings J. Microbial degradation of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvarelate) in soils. Appl Environ Microbiol. 1993;59:3233–3238. doi: 10.1128/aem.59.10.3233-3238.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morimoto K, Karita S, Kimura T, Sakka K, Ohmiya K. Cloning, sequencing, and expression of the gene encoding Clostridium paraputrificum chitinase ChiB and analysis of the functions of novel cadherin-like domains and a chitin-binding domain. J Bacteriol. 1997;179:7306–7314. doi: 10.1128/jb.179.23.7306-7314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukai K, Yamada K, Doi Y. Enzymatic degradation of poly(hydroxyalkanoates) by a marine bacterium. Polym Degrad Stab. 1993;41:85–91. [Google Scholar]
- 34.Mukai K, Yamada K, Doi Y. Efficient hydrolysis of polyhydroxyalkanoates by Pseudomonas stutzeri YM1414 isolated from lake water. Polym Degrad Stab. 1994;43:319–327. [Google Scholar]
- 35.Nojiri M, Saito T. Structure and function of poly(3-hydroxybutyrate) depolymerase from Alcaligenes faecalis T1. J Bacteriol. 1997;179:6965–6970. doi: 10.1128/jb.179.22.6965-6970.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozawa M, Engel J, Kemler R. Single amino acid substitutions in one Ca2+ binding site of uvomorulin abolish the adhesive function. Cell. 1990;63:1033–1038. doi: 10.1016/0092-8674(90)90506-a. [DOI] [PubMed] [Google Scholar]
- 37.Saito T, Suzuki K, Yamamoto J, Fukui T, Miwa K, Tomita K, Nakanishi S, Odani S, Suzuki J, Ishikawa K. Cloning, nucleotide and expression in Escherichia coli of the gene for poly (3-hydroxybutyrate) depolymerase from Alcaligenes faecalis. J Bacteriol. 1989;171:184–189. doi: 10.1128/jb.171.1.184-189.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakka K, Kojima Y, Kondo T, Karita S, Ohmiya K, Shimada K. Nucleotide sequence of the Clostridium stercorarium xynA gene encoding xylanase A: identification of catalytic and cellulose binding domains. Biosci Biotechnol Biochem. 1993;57:273–277. doi: 10.1271/bbb.57.273. [DOI] [PubMed] [Google Scholar]
- 39.Schrag J D L, Cygler M. Ser-His-Glu triad forms the catalytic site of the lipase from Geotrichum candidum. Nature. 1991;351:761–764. doi: 10.1038/351761a0. [DOI] [PubMed] [Google Scholar]
- 40.Shinomiya M, Iwata T, Doi Y. The adsorption of substrate-binding domain of PHB depolymerases to the surface of poly(3-hydroxybutyric acid) Int J Biol Macromol. 1998;22:129–135. doi: 10.1016/s0141-8130(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 41.Shinomiya M, Iwata T, Kasuya K, Doi Y. Cloning of the gene for poly(3-hydroxybutyric acid) depolymerase of Comamonas testosteroni and functional analysis of its substrate-binding domain. FEMS Microbiol Lett. 1997;154:89–94. doi: 10.1111/j.1574-6968.1997.tb12628.x. [DOI] [PubMed] [Google Scholar]
- 42.Shirakura Y, Fukui T, Saito T, Okamoto Y, Narikawa T, Koide K, Tomita K, Takemasa T, Masamune S. Degradation of poly(3-hydroxybutyrate) by poly(3-hydroxybutyrate) depolymerase from Alcaligenes faecalis T1. Biochim Biophys Acta. 1986;880:46–53. doi: 10.1016/0304-4165(86)90118-2. [DOI] [PubMed] [Google Scholar]
- 43.Steinbüchel A. Polyhydroxyalkanoic acids. In: Byrom D, editor. Biomaterials. London, England: Macmillan; 1991. pp. 123–213. [Google Scholar]
- 44.Tanio T, Fukui T, Shirakura Y, Saito T, Tomita K, Kaiho T, Masamune S. An extracellular poly(3-hydroxybutyrate) depolymerase from Alcaligenes faecalis. Eur J Biochem. 1982;124:71–77. doi: 10.1111/j.1432-1033.1982.tb05907.x. [DOI] [PubMed] [Google Scholar]
- 45.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uefuji M, Kasuya K, Doi Y. Enzymatic degradation of poly[(R)-3-hydroxybutyrate]: secretion and properties of PHB depolymerase from Pseudomonas stutzeri. Polym Degrad Stab. 1997;58:275–281. [Google Scholar]
- 47.Vieira J, Messing J. Production of single-stranded plasmid DNA. Methods Enzymol. 1987;153:3–11. doi: 10.1016/0076-6879(87)53044-0. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe T, Oyanagi W, Suzuki K, Ohnishi K, Tanaka H. Structure of the gene encoding chitinase D of Bacillus circulans WL-12 and possible homology of the enzyme to other prokaryotic chitinases and class III plant chitinase. J Bacteriol. 1992;174:408–414. doi: 10.1128/jb.174.2.408-414.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe T, Suzuki K, Oyanagi W, Ohnishi K, Tanaka H. Gene cloning of chitinase A1 from Bacillus circulans WL-12 revealed its evolutionary relationship to Serratia chitinase and to the type III homology units of fibronectin. J Biol Chem. 1990;265:15659–15665. [PubMed] [Google Scholar]
- 50.Yamada K, Mukai K, Doi Y. Enzymatic degradation of poly(hydroxyalkanoates) by Pseudomonas pickettii. Int J Biol Macromol. 1993;15:215–220. doi: 10.1016/0141-8130(93)90040-s. [DOI] [PubMed] [Google Scholar]