

Supplemental Digital Content is Available in the Text.
A transient receptor potential ankyrin 1 antagonist (ISC 17536) was evaluated for efficacy and safety in patients with chronic, painful diabetic peripheral neuropathy. ISC 17536 was not found to be effective in the overall patient population. However, statistically significant and clinically meaningful improvements in pain were seen with ISC 17536 in an exploratory hypothesis-generating subpopulation with preserved small nerve fiber function.
Keywords: TRPA1 antagonist, Painful diabetic peripheral neuropathy, Quantitative sensory testing, Preserved small nerve fiber function, Proof-of-concept, Pain phenotype
Abstract
Patients with chronic pain syndromes, such as those with painful peripheral neuropathy due to diabetes mellitus, have limited treatment options and suffer ongoing attrition of their quality of life. Safer and more effective treatment options are needed. One therapeutic approach encompasses phenotypic characterization of the neuropathic pain subtype, combined with the selection of agents that act on relevant mechanisms. ISC 17536 is a novel, orally available inhibitor of the widely expressed pain receptor, transient receptor potential ankyrin 1, which mediates nociceptive signaling in peripheral small nerve fibers. In this randomized, placebo-controlled, proof-of-concept trial, we assessed the safety and efficacy of 28-day administration of ISC 17536 in 138 patients with chronic, painful diabetic peripheral neuropathy and used quantitative sensory testing to characterize the baseline phenotype of patients. The primary end point was the change from baseline to end of treatment in the mean 24-hour average pain intensity score based on an 11-point pain intensity numeric rating scale. The study did not meet the primary end point in the overall patient population. However, statistically significant and clinically meaningful improvement in pain were seen with ISC 17536 in an exploratory hypothesis-generating subpopulation of patients with preserved small nerve fiber function defined by quantitative sensory testing. These results may provide a mechanistic basis for targeted therapy in specific pain phenotypes in line with current approaches of “precision medicine” or personalized pain therapeutics. The hypothesis is planned to be tested in a larger phase 2 study.
1. Introduction
Diabetes mellitus affects more than 400 million people worldwide.23 One of the most common complications of diabetes mellitus is diabetic peripheral neuropathy (DPN), which affects approximately half of all diabetic patients in their lifetime.12 DPN is associated with significant complications, including chronic pain and a diminished quality of life.12
Clinical guidelines recommend treatment of painful DPN with anticonvulsants, tricyclic antidepressants, serotonin and norepinephrine reuptake inhibitors, opioid analgesics, and topical capsaicin.13 Three nonopioid oral drugs (duloxetine, pregabalin, and gabapentin) are approved in the United States and European Union for the treatment of painful DPN.11,17 The available treatment options are not effective in all patients, which do not always provide complete relief (only about one-third achieve > 50% pain relief)14 and may have significant toxicities. Commonly used agents are often centrally acting, rather than interrupting pain signaling at the site of initiation in the periphery, and are associated with central nervous system (CNS) side effects.
Transient receptor potential ankyrin 1 is an evolutionarily conserved ion channel1 that contributes to perception of noxious stimuli.2 Activation of TRPA1 produces mechanical, thermal, and cold hyperalgesia in rodent models, and pharmacological inhibition or genetic ablation of TRPA1 alleviates these.2,5,18 A topical TRPA1 antagonist reduces pain in a rat model,7 and naturally occurring, rare TRPA1 gain-of-function mutations in humans lead to familial episodic pain syndrome, providing proof of mechanism for therapeutic intervention in this pathway.21
In the current study, the primary question was whether the TRPA1 antagonist, ISC 17536, might provide sufficient pain relief for use as monotherapy in patients with DPN. Of note, the TRPA1 ion channels are expressed on C-unmyelinated fibers, thinly myelinated Aδ fibers, and in the dorsal root ganglion. Therefore, it was expected that patients with DPN with a substantial loss of peripheral sensory nerves might not respond to this mechanism. Conversely, patients with early damage to peripheral nerves were considered more likely to benefit from the drug because most peripheral sensory nerves would still be intact and possibly in a hyperexcitable state, with upregulated TRPA1 expression. Therefore, an exploratory analysis was planned in this population of patients, identified using quantitative sensory testing (QST).
The novel, orally available small molecule, ISC 17536, is a potent and selective, peripherally acting TRPA1 antagonist with analgesic efficacy demonstrated in multiple rodent models of pain. To date, ISC 17536 has also been evaluated in 5 phase 1 trials in >250 healthy volunteers and 1 proof-of-concept, randomized phase 2 trial to assess the efficacy and safety of ISC 17536 in the treatment of painful DPN. In this article, we describe the results of this phase 2 study as well as the effectiveness of ISC 17536 in the exploratory hypothesis subgroup of patients with preserved SNF function.
2. Materials and methods
2.1. Study design
This phase 2, randomized, placebo-controlled, double-blind, parallel-group, proof-of-concept study (EudraCT: 2012-002320-33; ClinicalTrials.gov Identifier: NCT01726413 and CTRI/2013/02/003347) was conducted in the Czech Republic, India, and Germany between December 2012 and July 2014. The protocol was approved by independent ethics committees, and the study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. All patients provided written informed consent before participation. The study consisted of a screening visit, washout period of 7 days, 1-week placebo run-in period, 4-week double-blind dosing period, and 2-week follow-up period. Eligible patients were randomized on day 1 of the study (Fig. 1).
Figure 1.
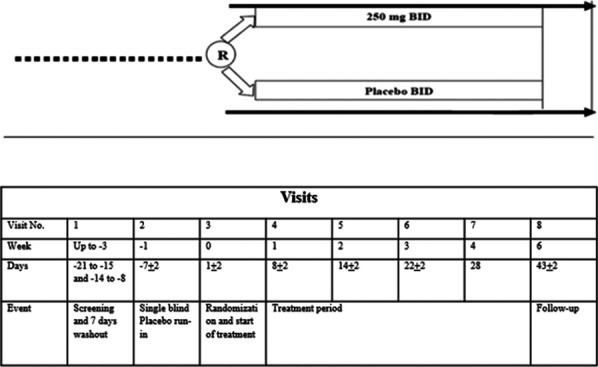
Study design schematic. BID, twice daily; R, randomization.
2.2. Patient selection
Inclusion criteria were male or female (postmenopausal or surgically sterile) patients aged 18 to 75 years (18-65 years in India) diagnosed with DM (type 1 or 2) and distal, symmetric, chronic, sensorimotor, painful DPN for at least 6 months but no more than 5 years. Other inclusion criteria were a Douleur Neuropathique en (DN4) score ≥ 4 and a baseline 24-hour average pain intensity (API) score ≥ 4 and < 9, as measured on an 11-point pain intensity numerical rating scale (NRS). The DN4 score is a patient-based questionnaire used to screen for the presence of neuropathic pain, and the cut-off value for neuropathic pain is a total score of 4 of 10 items.6 Patients eligible for study participation were required to withdraw from all the neuropathic pain medications before the placebo run-in period (Fig. 1).
Exclusion criteria were a 24-hour daily API score of ≥ 9 on the 11-point NRS at screening or day 1 of the study; chronic pain conditions not associated with DPN that could confound the assessment of neuropathic pain; and other causes of neuropathy or lower extremity pain including, but not limited to, lower extremity pain caused by osteoarthritis of the ankle or foot, gout, bursitis, or fasciitis. Details for other inclusion and exclusion criteria are captured in Supplementary Material 1, available at http://links.lww.com/PAIN/B484.
2.3. Identification of the exploratory hypothesis subgroup
As TRP ion channels are expressed on peripheral SNFs (C and Aδ fibers), diminished warm detection threshold (WDT) and cold detection threshold (CDT) represent functional loss or damage to these nerve fibers. Patients with a diminished ability to detect warm temperatures (WDT > 49°C) or cold temperatures (CDT < 18°C) are thus considered to be significantly denervated. As TRPA1 antagonists act by inhibition of hypersensitive and hyperexcitable nociceptive neurons (peripherally sensitized patients) in neuropathic pain, ISC 17536 was hypothesized to be efficacious for patients with moderate to severe pain (API > 5), in the context of functionally intact peripheral sensory nerves. Baseline somatosensory function and signs of pain were characterized using the QST protocol established by the German Research Network on Neuropathic Pain (DFNS), which included 13 parameters.19 The standardized assessment comprises different thermal and mechanical tests which assess small and large fiber function as well as central pathways. The protocol included the assessment of thermal perception thresholds for cold (ie, CDT) and warm (ie, WDT) as well as during alternating warm and cold stimuli (ie, thermal sensory limen and paradoxical heat sensations). Furthermore, thermal pain thresholds for cold (ie, cold pain threshold) and heat (ie, heat pain threshold) pain were assessed. The thermal stimuli were applied with a medical thermode (TSA 2001-II; Medoc Ltd, Ramat Yishai, Israel [probe size: 3 × 3 cm] with a ramp of 1°/second (max 50°C, min 0°C)). The mechanical testing comprised mechanical detection thresholds for touch (ie, mechanical detection threshold) by applying different forces of modified von Frey hairs (Optihair2-Set; Marstock Nervtest, Germany) as well as vibration (ie, vibration detection threshold) by applying a standard neurological tuning fork (64 Hz). The pain sensitivity (ie, mechanical pain sensitivity) and thresholds (ie, mechanical pain threshold) were assessed by different forces of pinpricks (MRC Systems GmbH, Heidelberg, Germany) as well as cotton wool, Q-Tip, and brush stimuli for mechanical pain sensitivity. Further on, central sensitization parameters (ie, dynamic mechanical allodynia and wind-up ratio) were assessed by the application of brush stimuli (for dynamic mechanical allodynia) and a series of pinprick stimuli (for wind-up ratio). In addition, a pressure algometer (FDN200; Wagner Instruments, Greenwich, CT) assessed deep somatosensory function (ie, pressure pain threshold).
All sites were trained for QST procedures by a trained QST expert. The site personnel familiarized the patients with QST procedures during the screening visit and retrained the patients as required. All QST procedures were performed by trained personnel using the same equipment and standardized instructions to the patients. At each visit, the instructions were explained to the patients before performing the test in a language he or she understands.
All QST assessments were performed at each visit from screening through follow-up, at the most painful site within the affected body area, as well as at the contralateral area. Patients with evidence of preserved small nerve fiber function were identified using the protocol-specified QST thermal thresholds, using the last QST assessment before randomization. These predetermined thresholds were based on the reference range for WDT and CDT published by the German Research Network.19
2.4. Study treatments
The investigational study drug ISC 17536 was manufactured and provided by Glenmark Pharmaceuticals, Limited, and supplied as granules; each dose contained 250-mg ISC 17536 potassium, to be reconstituted in 90 mL of water. Matching placebo was also provided as granules. Patients were assigned to either ISC 17536 or placebo in a blinded fashion, and background analgesics were prohibited except as rescue therapy. Permitted rescue medications were ibuprofen (400-600 mg administered orally 4 times daily (QID), to a maximum dose of 2.4 g/day, for sites in India and the Czech Republic) and paracetamol (500-1000 mg administered orally twice or 3 times daily, to a maximum dose of 3 g/day, for sites in Germany).
2.5. Randomization
Patients were randomized if they met the following criteria on day 1 of the study: compliance of > 80% with placebo administration during the run-in period, compliance with concomitant medications, and mean 24-hour API score of ≥ 4 at baseline. The compliance to treatment was assessed weekly by reconciliation of dispensed or remaining medication. The baseline mean 24-hour API score was calculated as the mean of the 24-hour daily API scores during the 7 days before randomization (placebo run-in period), and patients were required to have documented API scores for at least 4 of the 7 days.
Eligible patients were randomized in a 1:1 ratio to either ISC 17536 (250 mg po twice daily [BID]) or matching placebo for 28 days. The study drugs were to be administered with food. Patients, investigators, and all other personnel directly involved in the conduct of the trial were blinded to treatment allocation during the study. Randomization codes were generated by an IWRS vendor, and an electronic version was stored in a secured area. Only authorized site personnel had access to these secure systems for unblinding.
2.6. Study procedures
During the double-blind dosing period, patients returned to the study site weekly (weeks 1, 2, 3, and 4). At each visit, patients underwent safety and efficacy assessments and compliance with study drugs, concomitant medications, use of the electronic patient diary, and use of rescue medications since the last visit were recorded. At weeks 1, 2, and 3, double-blind study medication was dispensed for the subsequent 7-day period. Patients returned to the site on week 6 to undergo final assessments. Patients who withdrew prematurely from the study had the week 6 study procedures within 2 weeks after their last dose of study drugs.
Experimental population pharmacokinetic (PK) sampling was performed to characterize PK of ISC 17536 in patients with DPN. Most patients were assigned to the sparse PK group while extensive PK sampling was performed in the rich PK group, planned for 33 patients with a blinded ISC 17536-to-placebo ratio of 2:1, guided by IWRS. Predose and 2-hour postmorning dose samples were collected from patients in both groups on all days (ie, days 1, 8, 14, 22, and 28), except on days 1 and 28 from the rich PK group. Predose, 1-, 2-, 4-, 8-, 12- (predose to second dose), 13-, 14-, 16-, 20-, and 24-hour samples were collected on days 1 and 28 from the rich PK group.
2.7. Assessments
Patients were instructed on the use of electronic diaries to record their daily 24-hour API scores. The intensity of pain that occurred during the study was quantified during the initial assessment and on an ongoing basis. A validated 11-point NRS where 0 = “no pain” and 10 = “pain as bad as you can imagine” was used to bracket the 24-hour API score.
The primary efficacy end point was the change from baseline in the mean 24-hour API scores at the end of dosing (week 4).
Secondary end points included in the study were as follows:
The change from baseline to the end of weeks 1, 2, 3, 4, and 6 in
(1) mean night-time API and worst pain intensity scores (patient diary),
(2) mean sleep interference scores on an 11-point NRS (patient diary),
(3) mean daily dose of rescue medication (patient diary),
(4) number of patients who were responders on the Patient Global Impression of Change and Clinician Global Impression of Change questionnaires,
(5) number of patients achieving various levels of percent reduction from baseline in the mean 24-hour API score,
(6) Neuropathic Pain Symptom Inventory,
(7) QST assessments,
(8) time to onset of sustained improvement in the 24-hour daily API score, and
(9) change from baseline in the mean 24-hour API on NRS at the end of weeks 1, 2, 3, and 6.
Safety assessments included physical examination findings, vital signs and electrocardiograms (ECGs), clinical laboratory test results, and recording of adverse events (AEs).
2.7.1 Pharmacokinetics: ISC 17536
plasma concentrations were estimated using a validated LC-MS-MS method. Pharmacokinetic evaluation was performed using Phoenix WinNonlin version 6.3 (Pharsight Corp, St. Louis, MO). Noncompartmental analysis was performed on ISC 17536 concentration–time data from visits 3 (day 1) and 7 (day 28) from patients in the rich PK group. Patients in the sparse PK group were not included in the listing or summary of PK parameters.
2.8. Statistics
2.8.1. Determination of sample size
A sample size of 138 patients was planned to yield 80% power to detect a difference of 1.2 units between ISC 17536 and placebo in the primary efficacy end point, defined as the mean change from baseline in the API score from baseline to week 4. The 1.2 unit difference was selected based on previous efficacy studies of approved drugs for DPN. This assumed a standard deviation of 2.3, a drop-out rate of 15%, and 2-sided testing at the 0.05 significance level.
2.8.2. Populations for analyses
The intent-to-treat population was defined as all randomized patients with a nonmissing mean 24-hour API score at baseline, who received at least 1 dose of study drug and had at least 1 postbaseline visit API score (requiring at least 4 days of pain data during the preceding week). The intent-to-treat (ITT) population was used for all efficacy analyses.
The safety population was defined as all patients who received at least 1 dose of study drug and were used for all safety assessments.
2.8.3. Analysis of the mean 24-hour average pain intensity scores
The API score at baseline was defined as the mean of the 24-hour API scores during the 7 days before randomization.9 The API score at the end of dosing was defined as the mean of the 24-hour API scores obtained between Day 23 and Day 29 (the fourth week of dosing). The change from baseline to Week 4 in the mean 24-hour API score was evaluated with an analysis of covariance (ANCOVA) model fitting terms for study treatment, center, stratification factor, and baseline API as a continuous covariate. The least-square mean difference (95% confidence interval [CI]) between ISC 17536 and placebo was obtained from the model. If the upper bound of the 95% CI was less than zero, then it was concluded that ISC 17536 significantly reduces the mean 24-hour API scores at week 4 relative to placebo. The change from baseline in the mean 24-hour API score was also analyzed with mixed-model repeated-measures (MMRM), with fixed-effect terms for treatment, week, and treatment-by-week interaction: baseline 24-hour API score as a covariate and patient as a random effect. These analyses were performed in both ITT and PP populations. These methods were also applied to the change from baseline in the mean 24-hour API score at weeks 1, 2, 3, and 6 for the ITT and PP populations as well as at weeks 1, 2, 3, 4, and 6 for the hypothesis-generating nondenervated subgroup of the ITT population.
2.8.4. Secondary end point analysis
The analyses of all secondary end points were performed using predefined statistical methods. For the responder analyses of the active and placebo groups, the percentages of patients achieving ≥ 30% reduction and ≥ 50% reduction in API scores from baseline based on NRS were determined using the ITT population. These API thresholds were selected based on the previously published literature.9 All analyses were performed using SAS software version 9.4.
2.9. Analysis of safety
AEs were collected from the time of signing of the Informed consent form (ICF) ICF through the end of the protocol-specified follow-up period. All AEs were followed until the event resolved, stabilized, or returned to baseline if a baseline value was available.
Exacerbation of a chronic or intermittent pre-existing condition, including an increase in frequency or intensity of the condition, and laboratory changes that were reported as clinically significant were also considered to be AEs.
Clinical laboratory assessments included measures of hemoglobin, hematocrit, erythrocytes, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, white blood count, and platelets. A differential blood count was performed for lymphocytes, monocytes, neutrophils, basophils, and eosinophils. Clinical chemistry parameters included aspartate aminotransferase; alanine aminotransferase; gamma-glutamyl transpeptidase; alkaline phosphatase; creatine kinase; direct bilirubin, indirect bilirubin, and total bilirubin; creatinine; potassium; sodium; and plasma glucose (fasting). Additional parameters were assessed on specified visits as follows:
(1) Visit 1 (screening) and visit 8 (follow-up): serum levels of cholesterol, triglycerides, total protein, albumin, C-reactive protein, HbA1c, and a urinalysis that included pH, glucose, protein, ketones, bilirubin, and urine microscopy.
(2) Visit 1 (screening): vitamin B12, folate, TSH, and FSH (in female patients as applicable).
(3) Visits 3 and 7: international normalized ratio.
(4) Visit 8 (follow-up): partial thromboplastin time and prothrombin time.
Vital signs, including systolic and diastolic BP, pulse rate, respiratory rate, and oral body temperature, were also recorded at scheduled visits.
A single 12-lead ECG was recorded at scheduled visits. Triplicate ECGs were recorded at the discretion of the investigator. Clinically significant abnormalities were reported as AEs.
Physical examinations were performed at scheduled visits and included general appearance; weight; examination of the skin, neck (including thyroid gland), eyes, ears, nose, throat, mouth, lungs, heart, abdomen, back, lymph nodes, and extremities; and a brief examination of the neurological system. Overall interpretation (normal, abnormal not clinically significant, and abnormal clinically significant) of each body system was recorded.
Demographic data, medical and surgical history, and a detailed history of the patients with DPN were obtained at Visit 1 (screening).
3. Results
3.1. Patient demographics and baseline characteristics
Most of the patients were male (69.1%), and the mean age of patients was 55 and 57 years in the ISC 17536 and placebo groups, respectively. Baseline demographics of the ITT population are presented in Table 1. A total of 82.6%, 13.0%, and 4.3% patients were enrolled at sites in India, the Czech Republic, and Germany, respectively.
Table 1.
Demographics and baseline characteristics, intent-to-treat population.
| Parameters | ISC 17536, 250 mg BID (N = 70) | Placebo (N = 66) | Overall (N = 136) |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 51 (72.9) | 43 (65.2) | 94 (69.1) |
| Female | 19 (27.1) | 23 (34.8) | 42 (30.9) |
| Age, y, mean (SD) | 55.03 (9.4) | 57.29 (7.5) | 56.13 (8) |
| DN4 score, mean (SD) | 5.8 (1.4 | 5.8 (1.4) | 5.8 (1.4) |
| Weight kg, mean (SD) | 75.8 (19.5) | 73.1 (15.90) | 74.5 (17.86) |
| BMI (kg/m2), mean (SD) | 27.23 (5.4) | 27.28 (4.7) | 27.2 (5.1) |
| Days since DPN diagnosis, mean (SD) | 815.7 (534.0) | 774.3 (587.0) | 795.6 (558.6) |
| Race, n (%) | |||
| White | 15 (21.4) | 9 (13.6) | 24 (17.6) |
| Asian | 55 (78.6) | 57 (86.4) | 112 (82.4) |
BID, twice daily; DN4, Douleur Neuropathique en 4 questions; DPN, diabetic peripheral neuropathy.
3.2. Patient disposition
A total of 138 patients were randomized to receive ISC 17536 (N = 72) or placebo (N = 66). The ITT population included 136 patients with the mean 24-hour API scores at both baseline and ≥1 postbaseline week and who received at least 1 dose of randomized study drug. One hundred twenty-five patients (90.6%) completed the study: 64 patients (88.8%) in the ISC 17536 group and 61 patients (92.4%) in the placebo group. In the ISC 17536 group, 8 patients (11.1%) discontinued from the study, whereas in the placebo group, 5 patients (7.6%) withdrew consent (Fig. 2).
Figure 2.
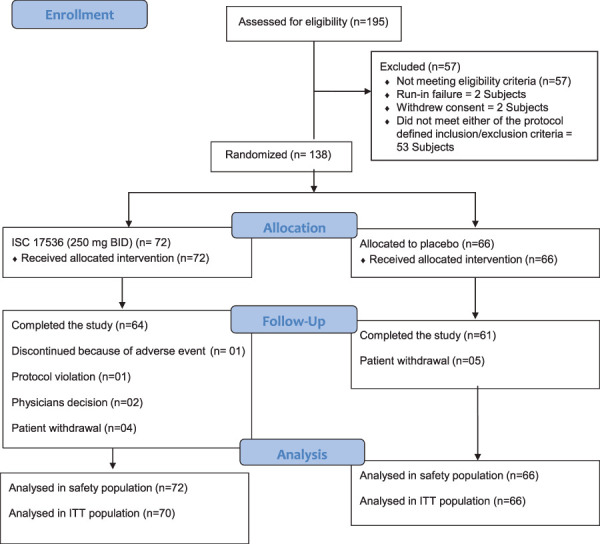
Patient disposition.
3.3. Primary end point, intent-to-treat population
The mean (SD) baseline 24-hour average pain intensity scores (11-point NRS) in the ISC 17536 group and placebo group were 6.0 (1.04) and 6.1 (1.07), respectively. The mean (SD) changes in the 24-hour API score from the start of double-blind treatment to Week 4 for the ISC 17536 and placebo groups were −1.9 (1.66) and −1.7 (1.58), respectively (Fig. 3 and Table 2). The LS mean difference (active minus placebo) was −0.26 (95% CI: −0.81 to 0.28). The difference between these 2 groups was not statistically significant. The LS mean difference obtained using MMRM was −0.27 (95% CI: −0.76 to 0.22) (Table 3). The results of sensitivity analyses for the PP population using ANCOVA and MMRM were consistent with the primary end point results for the ITT population.
Figure 3.
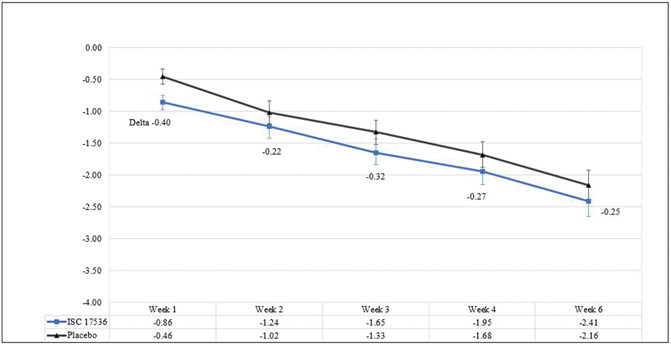
Mean 24-hour API score change from baseline, ITT population. API, average pain intensity; ITT, intent-to-treat.
Table 2.
Mean 24-hour average pain intensity score change from baseline to week 4 (ANCOVA), intent-to-treat population.
| Statistic | ISC 17536, treatment A | Placebo, treatment B | |
|---|---|---|---|
| Absolute value and change from baseline to end of treatment (week 4) | |||
| Baseline | N | 70 | 66 |
| Mean (SD) | 6.0 (1.04) | 6.1 (1.07) | |
| Min†, max | 2.4, 8.3 | 3.7, 8.9 | |
| Change from baseline (Week 4) | N | 70 | 66 |
| Mean (SD) | −1.9 (1.66) | −1.7 (1.58) | |
| Min, max | −6.1, 0.9 | −7.0, 1.7 |
| Visit | No. of patients | LS means | Treatment A-B | |||
|---|---|---|---|---|---|---|
| A | B | A | B | LSM difference | 95% CI | |
| Statistical assessment of change from baseline in the API score* | ||||||
| Week 4 | 70 | 66 | −1.94 | −1.68 | −0.26 | (−0.81, 0.28) |
The baseline mean 24-hour API score was the mean of 24-hour API scores during the 7 days before randomization. The week 4 mean 24-hour API score was the mean of the 24-hour API scores obtained between day 23 and day 29 during the fourth week of treatment.
The change from baseline to week 4 in the mean 24-hour API score was analyzed with an ANCOVA model with terms for treatment and baseline 24-hour API score as covariates.
The minimum baseline mean 24-hour API scores are < 4 (lower than the limit set by an inclusion criterion) because this table shows that in the ITT population (rather than the PP [per-protocol] population), 1 patient in the ISC 17536 group and 1 patient in the placebo group had baseline scores of 2.4 and 3.67, respectively.
ANCOVA, analysis of covariance; API, average pain intensity; BID, twice daily; CI, confidence interval; ITT, intent-to-treat; LS mean or LSM, least-square mean; Max, maximum; Min, minimum; N, number of patient.
Table 3.
Change from baseline in the average pain intensity score using mixed-model repeated-measures, intent-to-treat population.
| Visit | No. of patients | Least-square estimates (mean) | Treatment A-B | |||
|---|---|---|---|---|---|---|
| ISC 17536 (treatment A) | Placebo (treatment B) | ISC 17536 | Placebo | LS mean difference | 95% confidence interval | |
| Week 1 | 70 | 66 | −0.86 | −0.46 | −0.40 | (−0.89, 0.09) |
| Week 2 | 70 | 65 | −1.24 | −1.02 | −0.22 | (−0.71, 0.28) |
| Week 3 | 65 | 62 | −1.65 | −1.33 | −0.32 | (−0.82, 0.18) |
| Week 4 | 70 | 66 | −1.95 | −1.68 | −0.27 | (−0.76, 0.22) |
| Week 6 | 63 | 62 | −2.41 | −2.16 | −0.25 | (−0.75, 0.26) |
Model: Change from baseline in the API score = treatment + week + treatment*week + baseline API + patient as a random effect.
ITT, intent-to-treat.
Additional secondary end points including responder analyses (Table 4), night-time API, worst pain intensity, sleep interference Patient Global Impression of Change, Clinician Global Impression of Change, and Neuropathic Pain Symptom Inventory did not show statistically significant differences for ISC 17536 vs placebo, confirming that in the broad population the drug was not effective. The results of the secondary end points are presented in Appendix 1 (available as supplemental digital content at http://links.lww.com/PAIN/B484).
Table 4.
Mean 24-hour average pain intensity score change from baseline—responder analysis, intent-to-treat population.
| Visit | API score reduction | No. (%) of patients | Fisher exact test (P) | |
|---|---|---|---|---|
| ISC 17536 (N = 70) | Placebo (N = 66) | |||
| Week 1 | ≥30% | 11 (15.71) | 2 (3.03) | 0.0173 |
| ≥50% | 0 | 0 | — | |
| Week 2 | ≥30% | 19 (27.14) | 17 (25.76) | 0.8466 |
| ≥50% | 5 (7.14) | 4 (6.06) | 1.0000 | |
| Week 3 | ≥30% | 29 (41.43) | 21 (31.82) | 0.2069 |
| ≥50% | 12 (17.14) | 7 (10.61) | 0.3209 | |
| Week 4 | ≥30% | 38 (54.29) | 30 (45.45) | 0.3911 |
| ≥50% | 21 (30.00) | 11 (16.67) | 0.0728 | |
| Week 6 | ≥30% | 38 (54.29) | 34 (51.52) | 0.8508 |
| ≥50% | 23 (32.86) | 19 (28.79) | 0.7016 | |
API, average pain intensity; BID, twice daily; ITT, intent-to-treat; N, number of patients.
3.4. Exploratory hypothesis subpopulation
The exploratory subgroup of patients with preserved SNF function with moderate to severe pain (API > 5) at baseline comprised 65 patients, of whom 30 received ISC 17536 and 35 received placebo. There were no baseline differences between the pain scores of the ISC 17356 and placebo groups. There was a statistically significant difference in the pain score at week 4 in favor of ISC 17536 over placebo, demonstrating efficacy of the drug in patients with high baseline pain. The LS mean difference (ISC 17536 minus placebo) was −0.96 (95% CI: −1.68 to −0.24). A statistically significant difference with ISC 17536 was also seen at week 1 (−0.82 [95% CI: −1.52 to −0.12]), week 2 (−0.74 [95% CI: −1.45 to −0.03]), week 3 (−0.83 [95% CI: −1.54 to −0.12]), and week 6 (−1.37 [95% CI: −2.10 to −0.64]). The results for the exploratory hypothesis subgroup are shown in Figure 4.
Figure 4.
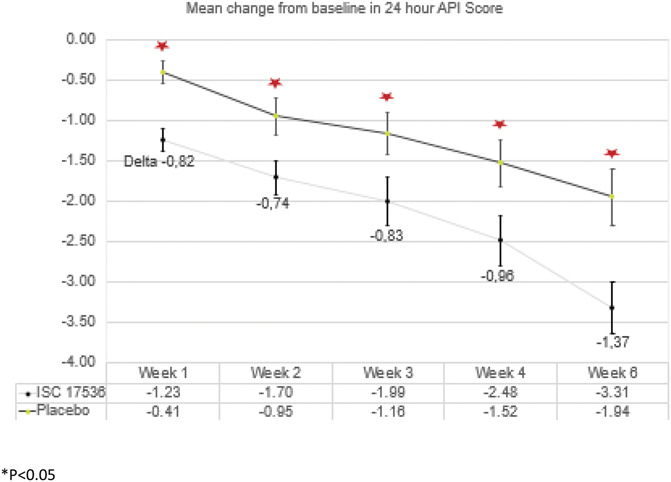
Mean change from baseline in the 24-hour API score: exploratory hypothesis subgroup analysis; P < 0.05, descriptive.
In this subgroup, the percentage of patients who achieved a ≥ 50% reduction in pain score was statistically significantly greater for ISC 17536 compared with placebo (P = 0.025). The results for the exploratory hypothesis subgroup are shown in Table 5.
Table 5.
Mean 24-hour average pain intensity score change from baseline (responder analysis), exploratory hypothesis subgroup.
| Visit | Percent reduction | No. (%) of patients | Fisher exact test | |
|---|---|---|---|---|
| ISC 17536 (250 mg BID) | Placebo | P | ||
| Exploratory hypothesis subgroup* also excluding patients with baseline API <5 | ||||
| No. of patients | N=30 | N=35 | — | |
| Week 4 | ≥30% | 17 (56.7) | 14 (40.0) | 0.218 |
| ≥50% | 12 (40.0) | 5 (14.3) | 0.025 | |
Exploratory subgroup = ITT population excluding patients with CDT <18°C and WDT >49°C; also excludes patients with missing CDT and WDT at baseline.
API, average pain intensity; BID, twice daily; CDT, cold detection threshold; ITT, intent-to-treat; N, number of patients; WDT, warm detection threshold.
3.5. Safety
The incidence of AEs was similar in the ISC 17536 and placebo groups (31.9% and 37.9% of patients, respectively). Most of the AEs were considered mild or moderate in intensity. There were no deaths in this study. One patient in the ISC 17536 group had a severe treatment-emergent SAE of pyrexia that led to discontinuation of the drug and was considered to be unrelated to the study drug by the investigator. The most common AEs affected the gastrointestinal system and included diarrhea, dyspepsia, and abdominal distension. There were no CNS-related AEs. A summary of AEs that occurred in > 2% of patients in either treatment group can be found in Table 6.
Table 6.
Adverse events occurring in more than 2% of patients in either treatment group.
| Preferred term | ISC 17536, n = 72, n (%) | Placebo, n = 66, n (%) | Overall, N = 138, n (%) |
|---|---|---|---|
| Abdominal distension | 1 (1.4) | 2 (3.0) | 3 (2.2) |
| Diarrhea | 2 (2.8) | 0 | 2 (1.4) |
| Dyspepsia | 2 (2.8) | 3 (4.5) | 5 (3.6) |
| Throat irritation | 1 (1.4) | 3 (4.5) | 4 (2.9) |
| Dysgeusia | 2 (2.8) | 1 (1.5) | 3 (2.2) |
| Pain | 2 (2.8) | 0 | 2 (1.4) |
| Pyrexia | 1 (1.4) | 2 (3.0) | 3 (2.2) |
| Blood creatine phosphokinase (CPK) increased | 1 (1.4) | 3 (4.5) | 4 (2.9) |
| Blood potassium increased | 2 (2.8) | 2 (3.0) | 4 (2.9) |
| Hyperglycemia | 2 (2.8) | 2 (3.0) | 4 (2.9) |
| Hypoglycemia | 0 | 3 (4.5) | 3 (2.2) |
| Proteinuria | 0 | 2 (3.0) | 2 (1.4) |
A total of 6 patients in the study had Treatment emergent adverse events (TEAEs) TEAEs leading to discontinuation of study drug: 5 [6.9%] in the ISC 17536 group, including 1 with an SAE, and 1 [1.5%] in the placebo group. Of these, 3 patients had AEs reported as related to the study drug. Patients who discontinued the study drug because of TEAEs are listed in Table 7.
Table 7.
Patients who discontinued the study drug because of adverse events (safety population).
| Patient identifier | TEAEs (preferred term) | Serious | Relationship/Severity | Outcome |
|---|---|---|---|---|
| ISC 17536 (250 mg BID group) | ||||
| A | Aspartate aminotransferase increased | No | Related/Moderate | Resolved |
| B | Dyspepsia | No | Related/Moderate | Resolved |
| C* | Vomiting | No | Related/Moderate | Resolved |
| Abdominal distension | No | Related/Moderate | Resolved | |
| D | Hypertension | No | Not related/Moderate | Resolved |
| Hyperchlorhydria | No | Related/Moderate | Resolved | |
| Pyrexia | Yes | Not related/Severe | Resolved | |
| E | Blood creatine phosphokinase abnormal | No | Not related/Severe | Resolved |
| Placebo group | ||||
| F | Skin hypopigmentation | No | Not related/Moderate | Resolved |
| Hemorrhage | No | Not related/Moderate | Resolved |
MedDRA (version 14.1) preferred term.
This patient discontinued the study drug on day 1 because of events that started during the placebo run-in period (ie, not treatment emergent).
BID, twice daily; TEAE, treatment-emergent adverse event.
3.6. Pharmacokinetics
The rich PK population consisted of 19 patients on ISC 17536 (250 mg BID) who had concentration–time PK data for both Day 1 and Day 28. On Day 1, Cmax was generally observed after the evening dose, as expected with the BID dosing regimen. On Day 28, Cmax was generally observed at the morning predose time point; however, for some patients, Cmax was observed at approximately 24 hours. At steady state (day 28), there were shoulders and multiple peaks in the ISC 17536 concentration profiles of some patients, whereas others had a near steady ISC 17536 concentration profile. The mean PK profile from the rich PK population on day 1 and day 28 is shown in Figure 5. Based on AUC0-tau, there was an approximately 6-fold accumulation of ISC 17536 after 28 days of dosing (250 mg BID). Day 28 geometric mean AUC0-24 and Cmax were 28,400 ng.h/mL and 2670 ng/mL, respectively. ISC 17536 is effective in inhibiting rat as well as human TRPA1 receptor signaling within a narrow IC50 range and shows high binding to plasma proteins with free fraction of 0.007 and 0.005 in rat and human plasma, respectively. With the 250 mg BID dose, on Day 28, the mean free ISC 17536 plasma concentrations were maintained at least 4-fold higher than the in vitro IC50 for human TRPA1 inhibition throughout the entire 24 hours, indicating that exposures adequate to engage the target were achieved in the study. In addition, systemic exposures in patients with DPN exceeded those required for maximal reversal of hyperalgesia in several rodent models of pain (where ISC 17536 exposures up to 13,000 ng.h/mL were observed at the EDmax doses (10-30 mg/kg)).
Figure 5.
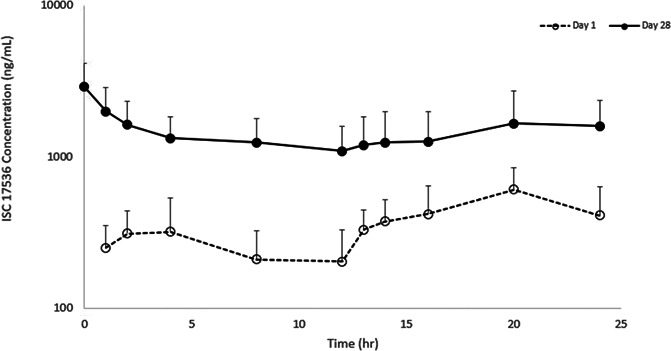
Mean PK Profile on days 1 and 28 with ISC 17536 250 mg BID; mean + SD plotted on a semilogarithmic scale is shown.
4. Discussion
Despite advances in the understanding of the complex neurobiology of pain, only limited improvement in neuropathic pain treatments have been attained and a large proportion of patients are left with insufficient pain relief.10 Many recent clinical trials showed negative results in phase 3, although the compounds seemed promising in earlier phases of development. In this current study, ISC 17536 was not superior to placebo in the overall patient population, with only small numeric trends in favor of the active drug.
The question arises for drugs which fail in clinical trials as to whether a beneficial treatment effect might be present in subgroups of patients with specific underlying mechanisms of pain generation and whether this beneficial effect might be diluted when assessed in the entire study population.4 Therefore, in this study we augmented the conventional development approach of “1 drug to treat all patients with DPN” with a prospectively defined exploratory strategy. The patients were classified on the basis of preservation of SNF function based on a mechanistic understanding of the role of TRPA1 in the generation of pain. This approach is consistent with a concept of pain therapy that calls for precise clinical phenotypic characterization of the patients to be combined with a selection of agents that act on the underlying mechanisms.3
Based on the pathophysiology of DPN and the expression of TRPA1 on SNFs, it was prospectively hypothesized that patients with a substantial loss of peripheral nerves would be unlikely to respond to ISC 17356. Hence, this study aimed to identify a patient subgroup with a sensory profile corresponding to preserved SNF function to enable an assessment of efficacy and safety of ISC 17536 based on a mechanistic approach. This study used the QST protocol, with highly standardized stimuli, where the responses to these are precisely assessed, facilitating the characterization of individual somatosensory phenotypes.
In the overall ITT population, twice-daily dosing of ISC 17536 for 28 days showed a small but consistent trend toward reduction in the mean 24-hour API pain scores with an LS mean difference for the change from baseline to end of treatment of −0.26 (95% CI: −0.81, 0.28). However, these results and those for the secondary efficacy end points with ISC 17536 were not statistically significant as compared with placebo, and the reduction in pain at the end of treatment did not indicate an analgesic effect of the magnitude seen for approved drugs for painful DPN.
A significant weakness in the current study was the failure to stratify the patients prospectively, based on the results of QST. Nevertheless, in the exploratory, hypothesis-generating, nondenervation subgroup, statistically significant and clinically meaningful benefits were observed. ISC 17536 showed efficacy over placebo in reducing pain consistently in this subgroup with preserved SNF function. Pain relief due to ISC 17536 was detected as early as Week 1 in this group and was sustained over placebo until the end of treatment (week 4). Of note, pain reduction continued through Week 6 of the study, even after dosing of study drugs had been discontinued. Although the cause of sustained pain relief is not known, it is possible that the effect was maintained after drug discontinuation because of long-lasting pharmacodynamic effects of TRPA1 inhibition, such as changes in receptor expression or function or neural plasticity (synaptic sprouting or pruning). Of note, preclinical studies indicate that sustained activation of the TRPA1 ion channel is involved in the pathogenesis of DPN,15 and thus, blockade of signaling through this receptor could ultimately slow the progression of DPN; this potential for a disease-modifying effect is an interesting hypothesis that remains to be tested.
In our study, the prevalence of patients with preserved SNF function was approximately 50% of the ITT population, indicating that a large segment of patients with DPN could potentially benefit from treatment with ISC 17536. The magnitude of the reduction of pain seen with ISC 17536 in the exploratory hypothesis subgroup is in line with the pain reduction seen with approved drugs such as pregabalin and duloxetine.11,22 The degree of skin denervation has been associated with the duration of a patient's diabetes20; thus, earlier initiation of TRPA1 inhibition may be beneficial for both the analgesic effect and for potential disease modification.
Using QST parameters to phenotype patients has been previously described. A similar mechanism-based approach for the sensory profiling of patients with peripheral neuropathic pain was evaluated in a previous, investigator-initiated, randomized, double-blind study in which the patients were prospectively stratified using QST, based on their sensory phenotype.8 The study concluded that a better pain-relieving effect was observed in patients with a sensory profile corresponding to the irritable nociceptor phenotype, as compared with patients lacking this phenotype, and preservation of thermal sensation predicted a response to the pharmacological agent (oxcarbazepine).8 These findings, together with the findings from our study, support the notion that a mechanism-based approach, in conjunction with patient sensory profiling, is a step forward toward developing personalized treatments for peripheral neuropathic pain.8
Another published investigation identified patient subgroups with distinct sensory profiles in a large sample of patients with neuropathic pain from a wide range of etiologies and concluded that sensory profiling is an adequate stratification tool for determining specific sensory phenotypes of patients in exploratory clinical trials for neuropathic pain.4
Overall, ISC 17536 was found to be safe and well tolerated in this clinical study and was not associated with CNS-related AEs, supporting the nonclinical findings showing that ISC 17536 is a peripherally acting, noncannabinoid, nonopioid, nonsteroidal analgesic agent and could be a potential option for the treatment of painful DPN in a subset of patients.
The current study is uniquely positioned in generating a hypothesis to support the mechanism-based approach for the treatment of painful DPN in patients according to underlying sensory profiles. The study also provides a rationale for future hypothesis-driven mechanistic studies targeting specific pain phenotypes.
Limitations of the current study include the short (4 week) treatment duration and testing of only a single-dose level and regimen to assess efficacy of ISC 17536. As the use of concomitant background neuropathic pain medications was restricted in this placebo-controlled trial, it was difficult to recruit patients at European sites, and thus, most of the patient data are from the population in India. This geographical limitation also likely accounts for the larger number of male patients enrolled vs female patients, given cultural norms in India. The study was conducted specifically in patients with painful DPN and excluded patients with other painful conditions. Although TRPA1 may be involved in other pain conditions, the study results cannot be generalized to indications other than painful DPN. In addition, as the ISC 17536 preclinical toxicology study was not completed before the initiation of this study, only postmenopausal or surgically sterilized women were permitted to be enrolled. The preserved small nerve fiber subpopulation was planned as an exploratory analysis; thus, definitive conclusions regarding the effect of ISC 17536 in this subgroup cannot be made based on the current data, and the results need to be tested in a subsequent phase 2 study. In addition, a simple bedside stratification tool for QST is planned to be used in the subsequent study to determine specific sensory phenotypes in a way that can be easily applied in routine clinical practice.16
In conclusion, ISC 17536 did not show significant efficacy in reducing neuropathic pain in the overall patient population with painful DPN. Patients with DPN with a substantial loss of peripheral sensory nerves are not likely to respond to a drug with peripheral action, which may be the major reason for the negative results in the ITT population. Thus, there is a high likelihood that ISC 17536 is not effective in patients with DPN who have extensively denervated or damaged peripheral nerves. However, in the exploratory hypothesis-generating subgroup, data suggest that in patients with a sensory profile corresponding with preserved SNF function, ISC 17536 could provide an effective pharmacotherapeutic option in this difficult-to-treat disease. Further larger confirmatory studies are planned to expand on the safety and efficacy of this novel molecule.
Conflict of interest statement
S.M. Jain and R. Balamurugan were principal investigators in the study and received an investigator grant. M. Tandon is a full-time employee of Glenmark Pharmaceuticals Limited. N. Mozaffarian is a full-time employee of Janssen Pharmaceuticals, Inc, and G. Gudi and Y. Salhi are full-time employees of Ichnos Sciences, Inc. R. Holland, R. Freeman, and R. Baron are not employees of Glenmark Pharmaceuticals or Ichnos Sciences and were not compensated for this manuscript.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B484.
Supplementary Material
Acknowledgments
The authors thank the patients, principal investigators, and all site staff members who participated in the study. The authors thank the clinical operations team for their implementation of the trial and acknowledge Yukti Singh from IQVIA, India, and Sagar Borle from Glenmark Pharmaceuticals Ltd, India, for their medical writing and editorial support.
This study was funded by Glenmark Pharmaceuticals SA, Chernin de la Combeta, 5, 2300 La Chaux-de-Fonds, Switzerland.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
Contributor Information
Sunil M. Jain, Email: sunilmjain@gmail.com.
Ramanathan Balamurugan, Email: rbmkdsc@gmail.com.
Neelufar Mozaffarian, Email: neelmoza@yahoo.com.
Girish Gudi, Email: Girish.gudi@ichnossciences.com.
Yacine Salhi, Email: Yacine.salhi@ichnossciences.com.
Robert Holland, Email: bob.holland@doctors.org.uk.
Roy Freeman, Email: rfreeman@bidmc.harvard.edu.
Ralf Baron, Email: r.baron@neurologie.uni-kiel.de.
References
- [1].Andrade E, Meotti F, Calixto J. TRPA1 antagonists as potential analgesic drugs. Pharmacol Ther 2012;133:189–204. [DOI] [PubMed] [Google Scholar]
- [2].Baraldi PG, Preti D, Materazzi S, Geppetti P. Transient receptor potential ankyrin 1 (TRPA1) channel as emerging target for novel analgesics and anti-inflammatory agents. J Med Chem 2010;53:5085–107. [DOI] [PubMed] [Google Scholar]
- [3].Baron R. Mechanisms of disease: neuropathic pain--a clinical perspective. Nat Clin Pract Neurol 2006;2:95–106. [DOI] [PubMed] [Google Scholar]
- [4].Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, Finnerup NB, Haanpää M, Hansson P, Hüllemann P, Jensen TS, Freynhagen R, Kennedy JD, Magerl W, Mainka T, Reimer M, Rice ASC, Segerdahl M, Serra J, Sindrup S, Sommer C, Tölle T, Vollert J, Treede R-D. German Neuropathic Pain Research N, the E, consortia N. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. PAIN 2017;158:261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bautista D, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006;124:1269–82. [DOI] [PubMed] [Google Scholar]
- [6].Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, Cunin G, Fermanian J, Ginies P, Grun-Overdyking A. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). PAIN 2005;114:29–36. [DOI] [PubMed] [Google Scholar]
- [7].de David Antoniazzi CT, De Prá SDT, Ferro PR, Silva MA, Adamante G, de Almeida AS, Camponogara C, da Silva CR, de Bem Silveira G, Silveira PCL. Topical treatment with a transient receptor potential ankyrin 1 (TRPA1) antagonist reduced nociception and inflammation in a thermal lesion model in rats. Eur J Pharm Sci 2018;125:28–38. [DOI] [PubMed] [Google Scholar]
- [8].Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, Jensen TS, Sindrup SH. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. PAIN 2014;155:2263–73. [DOI] [PubMed] [Google Scholar]
- [9].Farrar JT, Young JP, Jr, LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. PAIN 2001;94:149–58. [DOI] [PubMed] [Google Scholar]
- [10].Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. PAIN 2010;150:573–81. [DOI] [PubMed] [Google Scholar]
- [11].Freeman R, Durso-DeCruz E, Emir B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care 2008;31:1448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hicks CW, Selvin E. Epidemiology of peripheral neuropathy and lower extremity disease in diabetes. Curr Diabetes Rep 2019;19:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Javed S, Alam U, Malik RA. Treating diabetic neuropathy: present strategies and emerging solutions. The Rev diabetic Stud RDS 2015;12:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jensen TS, Backonja M-M, Jiménez SH, Tesfaye S, Valensi P, Ziegler D. New perspectives on the management of diabetic peripheral neuropathic pain. Diabetes Vasc Dis Res 2006;3:108–19. [DOI] [PubMed] [Google Scholar]
- [15].Koivisto A, Hukkanen M, Saarnilehto M, Chapman H, Kuokkanen K, Wei H, Viisanen H, Åkerman KE, Lindstedt K, Pertovaara A. Inhibiting TRPA1 ion channel reduces loss of cutaneous nerve fiber function in diabetic animals: sustained activation of the TRPA1 channel contributes to the pathogenesis of peripheral diabetic neuropathy. Pharmacol Res 2012;65:149–58. [DOI] [PubMed] [Google Scholar]
- [16].Koulouris AE, Edwards RR, Dorado K, Schreiber KL, Lazaridou A, Rajan S, White J, Garcia J, Gibbons C, Freeman R. Reliability and validity of the boston bedside quantitative sensory testing battery for neuropathic pain. Pain Med 2020;21:2336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Quilici S, Chancellor J, Löthgren M, Simon D, Said G, Le TK, Garcia-Cebrian A, Monz B. Meta-analysis of duloxetine vs. pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurol 2009;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rech JC, Eckert WA, Maher MP, Banke T, Bhattacharya A, Wickenden AD. Recent advances in the biology and medicinal chemistry of TRPA1. Future Med Chem 2010;2:843–58. [DOI] [PubMed] [Google Scholar]
- [19].RolkeMagerl RW, Campbell KA, Schalber C, Caspari S, Birklein F, Treede RD. Quantitative sensory testing: a comprehensive protocol for clinical trials. Eur J Pain 2006;10:77–88. [DOI] [PubMed] [Google Scholar]
- [20].Shun CT, Chang YC, Wu HP, Hsieh SC, Lin WM, Lin YH, Tai TY, Hsieh ST. Skin denervation in type 2 diabetes: correlations with diabetic duration and functional impairments. Brain 2004;127:1593–605. [DOI] [PubMed] [Google Scholar]
- [21].Skerratt S. Recent progress in the discovery and development of TRPA1 modulators. Prog Med Chem 2017;56:81–115. [DOI] [PubMed] [Google Scholar]
- [22].Smith T, Nicholson RA. Review of duloxetine in the management of diabetic peripheral neuropathic pain. Vasc Health Risk Manag 2007;3:833. [PMC free article] [PubMed] [Google Scholar]
- [23].WHO. Diabetes Fact sheet 2020; Vol 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B484.