

Abstract
Background:
MicroRNAs play an important role in health and disease. TGF-β signaling, upregulated by HIV Tat, and in chronic airway diseases and smokers upregulates miR-145-5p to suppress cystic fibrosis transmembrane conductance regulator (CFTR). CFTR suppression in chronic airway diseases like Cystic Fibrosis, COPD and smokers has been associated with suppressed MCC and recurrent lung infections and inflammation. This can explain the emergence of recurrent lung infections and inflammation in people living with HIV.
Methods:
Tat-induced aberrant microRNAome was identified by miRNA expression analysis. microRNA mimics and antagomirs were used to validate the identified miRNAs involved in Tat mediated CFTR mRNA suppression. CRISPR-based editing of the miRNA target sites in CFTR 3’UTR was used to determine rescue of CFTR mRNA and function in airway epithelial cell lines and in primary human bronchial epithelial cells exposed to TGF-β and Tat.
Findings:
HIV Tat upregulates miR-145-5p and miR-509-3p. The two miRNAs demonstrate cooperative effects in suppressing CFTR. CRISPR-based editing of the miRNA target site preserves CFTR mRNA and function in airway epithelial cells
Interpretation:
Given the important roles of TGF-β signaling and the multitude of genes regulated by miRNAs, we demonstrate that CRISPR-based gene-specific microRNA antagonism approach can preserve CFTR mRNA and function in the context of HIV Tat and TGF-β signaling without suppressing expression of other genes regulated by miR-145-5p.
Keywords: MicroRNA, HIV, gene therapy, CRISPR
Graphical abstract
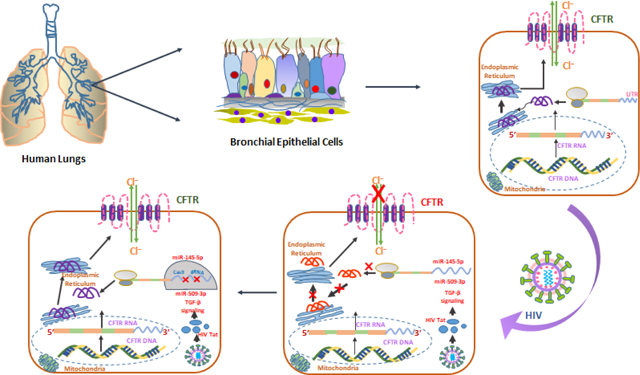
INTRODUCTION:
In aging HIV-infected populations, comorbid diseases are important determinants of morbidity and mortality. The advent of combination antiretroviral therapy (cART) has led to a dramatic decline in morbidity and mortality from HIV associated AIDS (1). However, despite making progress on effective cART, the life expectancy of HIV patients is lower than the non-HIV patients, and non-AIDS associated comorbidities have continued to remain highly prevalent among people living with HIV (2). The HIV care Continuum published by the CDC shows that even with improved access to cART, ∼50% of people living with HIV are not virally suppressed (3). Lungs can serve as anatomical reservoirs of HIV, and this has been demonstrated by the recovery of HIV from cell-free bronchoalveolar lavage fluid, alveolar macrophages, and intrapulmonary lymphocytes and suggesting that lungs constitute an anatomical HIV reservoir (4–6). Lung diseases such as chronic obstructive pulmonary disease (COPD), pulmonary hypertension, pneumonia, Asthma, and Asthma COPD overlap syndrome (ACOS) are emerging as significant comorbidities in the HIV-infected population (7, 8). COPD remains highly prevalent among these HIV-associated lung comorbidities in HIV patients when compared to non-HIV-infected adults (9, 10). HIV infection act as a driving and independent risk factor for the development and progression of COPD; even when compensated for smoking (11, 12). However, the molecular mechanism by which HIV infection promotes chronic inflammation predisposing to COPD is not clear (13). In our earlier work, we have shown that HIV infects the bronchial epithelium and suppresses components of the MCC apparatus (14).
Impaired mucociliary clearance (MCC) is a hallmark of chronic airway diseases like chronic obstructive pulmonary disease (COPD), cystic fibrosis, and chronic bronchitis associated with cigarette smoking (15, 16). MCC is a primary innate defense mechanism of the airways and protects the host from airborne pathogens, pollutants, and allergens (17). Optimal MCC requires mucus, cilia, and a thin layer of airway surface liquid (ASL) to facilitate ciliary beating (18). Abnormalities in any of these components lead to MCC dysfunction promoting microbial colonization and chronic inflammation (19, 20). The height of the ASL layer lining the airway surfaces is crucial for mediating MCC rates (21) and is tightly regulated by Cystic Fibrosis Transmembrane conductance regulator (CFTR) (22). CFTR dysfunction can compromise ASL depth and CBF decreasing MCC rates (e.g., 23, 24). HIV patients show abnormalities in their nasal MCC apparatus (25, 26). Nasal epithelium can serve as a surrogate for tracheobronchial epithelium and has been used as a barometer of overall MCC health (27–29).
We have shown that HIV and CS suppress CFTR via a common pathway involving TGF-β signaling (14, 30), and the suppression is additive (31). This is especially important, given that 60% of HIV patients are addicted to nicotine and smoke tobacco (32). While cART controls de novo infection and replication, Tat being an immediate-early gene of HIV continues to be expressed, and its expression is not suppressed by antiretroviral drugs (33, 34). HIV Tat contains a protein transduction domain that allows Tat secretion from infected cells and uptake by bystander cells (35, 36). Tat is released extracellularly and accumulates in tissues having pleiotropic effects (37–40). In our earlier report, we demonstrated that TGF-β signaling alters the bronchial epithelial microRNAome with consequent suppression of CFTR. Specifically, TGF-β upregulates miR-145-5p to suppress CFTR. In this study, we show that HIV Tat also alters the bronchial epithelial microRNAome to upregulate miR-145-5p that functions co-operatively with miR-509 to suppress CFTR. We have demonstrated that a neutralizing aptamer to TGFβR2 and miR-145-5p antagonism rescues TGF-β mediated CFTR suppression (41). Given that TGF- β signaling plays and important role in organ homeostasis. Likewise, miR-145-5p is a tumor suppressor, and its downregulation relates to tumor progression and metastasis (42–45). Hence, an indiscriminate suppression of TGF-β signaling and miR-145 mediated gene regulation can have deleterious effects. Given these important roles of TGF-β signaling and miR-145, it would be attractive if we can restrict microRNA antagonism to the gene of interest. We will explore a novel approach called gene-specific microRNA antagonism using CRISPR that edits the miR-145-5p target site within CFTR 3’ UTR region to preserve CFTR function in the context of HIV and cigarette smoke without blocking the entire TGF-β signaling pathway or interfering with the broader miRNA-mediated regulation of other genes.
METHODS:
Chemicals, Reagents, and Materials:
CFTR (inh)-172 (Sigma-Aldrich, St. Louis, MO, USA, Catalog# C2992) was dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA, Catalog# D4540) to make a stock concentration of 20 mM, GlyH-101 (Sigma-Aldrich, St. Louis, MO, USA, Catalog# 219671) was dissolved in DMSO to make a stock concentration of 50 mM, Amiloride hydrochloride hydrate (Sigma-Aldrich, St. Louis, MO, USA, Catalog# A7410) was dissolved in distilled H2O (Invitrogen™, Thermo Fisher Scientific, Inc., Waltham, MA, Catalog# 10977015) to make a stock concentration of 10 mM. Salbutamol hemisulfate salt (Sigma-Aldrich, St. Louis, MO, USA, Catalog# S5013) was dissolved in distilled H2O to make a stock concentration of 10 mM. HEPES (Sigma-Aldrich, St. Louis, MO, USA, Catalog# H4034) was dissolved in Hank’s Balanced Salt Solution (HBSS) (Gibco™, Thermo Fisher Scientific, Inc., Waltham, MA, Catalog# 14025092) to make a stock concentration of 10 mM. Recombinant TGF-β1 (R&D Systems, Minneapolis, MN, USA, Catalog# 240-B-002) was dissolved in sterile 4 mM HCl (Fisher Chemical, Waltham, MA, USA Catalog# SA49) containing 1 mg/mL bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO, USA, Catalog# A7638) to prepare a stock concentration of 10 μg/μL. HIV-1 IIIB Tat Recombinant Protein (Catalog # 2222) was received from NIH AIDS Reagent Program and reconstituted in PBS containing 1 mg/ml BSA and 0.1 mM DTT (Sigma-Aldrich, St. Louis, MO, USA, Catalog# D9779) to make the stock concentration to 10 μM. mirVana® miRNA mimics hsa-miR-145-5p (Thermo Fisher Scientific, Waltham, MA, USA, Assay ID: MC11480) and has-miR-509-3p (Thermo Fisher Scientific, Waltham, MA, USA, Assay ID: MC12984). mirVana® miRNA inhibitor: hsa-miR-145-5p (Thermo Fisher Scientific, Waltham, MA, USA, Assay ID: MH11480) and hsa-miR-509-3p (Thermo Fisher Scientific, Waltham, MA, USA, Assay ID: MC12984). Radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 89901), Protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA, Catalog # 78410), Bio-Rad protein assay dye reagent (Bio-Rad, Hercules, CA, USA, Catalog # 5000006), 4–20% Precast Bis-Tris acrylamide Gels (Bio-Rad, Hercules, CA, USA, Catalog # 4561094), Immuno-Blot PVDF Membrane (Bio-Rad, Hercules, CA, USA, Catalog #1620177), Blotting-Grade Blocker (Bio-Rad, Hercules, CA, USA, Catalog #1706404). Mouse primary monoclonal antibody against the R Domain of CFTR (Cystic Fibrosis Center, Chapel Hill, NC, Catalog # 570), Rabbit Anti-GAPDH primary antibody (Sigma Aldrich, St. Louis, MO, USA, Catalog # G9545); HRP conjugated anti-mouse IgG secondary antibody (Promega, Madison, WI, USA, Catalog# W4021) and anti-rabbit IgG secondary antibody (Promega, Madison, WI, USA, Catalog # W4011), SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA, USA, Catalog # 34094). TaqMan™ MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, Catalog# 4366596). High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, Catalog# 4368814). TaqMan™ Fast Advanced Master Mix (Applied Biosystems, Foster City, CA, Catalog# 4444964). Edit-R Cas9 expression plasmid (Dharmacon, Lafayette, CO, USA, Catalog# U-005600–120). DharmaFECT Duo Transfection Reagent (Dharmacon, Lafayette, CO, USA, Catalog# T-2010–02). PolyMag CRISPR Transfection Reagent (OZ Biosciences, San Diego, CA, USA, Catalog# PNC40200), Super Magnetic plate 8 × 12 cm (OZ Biosciences, San Diego, CA, USA, Catalog# MF10000), Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 13778075); Opti-MEM™ Reduced-Serum Medium (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 31985062). BEAS-2B cells were cultured in BioLite 75 cm2 flasks (Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 12–556-010); BioLite 12-well plate (Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 12–556-005). Cells were harvested using 0.25% Trypsin-EDTA (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA, Catalog# 25200114) and Trypsin neutralizer solution (Gibco™, Thermo Fisher Scientific, Waltham, MA, USA, Catalog# R002100). NHBE cells were cultured on Snapwell™ Culture Inserts (Corning™ Inc, NY, USA, Catalog# 07–200-708) and Transwell™ Membrane Inserts (Corning™ Inc, NY, USA, Catalog# 07–200-261). Inserts were coated with Type IV collagen from the human placenta (Sigma-Aldrich, St. Louis, MO, USA, Catalog# C5533). microRNA Ready-to-Use PCR, Human panel I+II, V4.M (Exiqon, Valencia, CA, USA, Catalog# 203615); Universal cDNA Synthesis Kit II (Exiqon, Valencia, CA, USA, Catalog# 203301); ExiLENT SYBR® Green master mix (Exiqon, Valencia, CA, USA, Catalog# 203421).
Cell culture and cell line:
Human small airway epithelial cells will be obtained from the University of Miami Life Alliance Organ Recovery Agency (LAORA). Lungs from organ donors that are compatible for transplant but released by the donor’s family for research was obtained with minor de-identified information like age, sex, smoking status, HIV/HCV/EBV/CMV infection, COPD status (if any), pack-years (in case of smoker’s lungs), etc. Primary bronchial epithelial cells were isolated as described by (46, 47) and adapted by us (14, 30, 31, 48). The cells were obtained under an MTA where the University of Miami Pulmonary division obtains the lungs and then dissects and isolates primary human bronchial epithelial cells and provides them to us for a nominal fee. The primary cultures undergo mucociliary differentiation at the ALI, reproducing both the in vivo morphology and key physiologic processes to regenerate the native bronchial epithelium ex vivo (46, 47). Since the cells were obtained from the deceased individual’s lung with minor, de-identified information, its use does not constitute human subjects research defined by CFR 46.102. Unless otherwise mentioned, we used lung cultures from the lungs of non-HIV non-smokers not to confound the findings in unknown ways. Normal human bronchial epithelial (NHBE) cells were expanded in T25 cell culture flasks coated with collagen I in bronchial epithelial growth medium (BEGM). 7–10 days later, the 350,000 NHBE cells per well were differentiated in collagen I-coated 12 mm Transwell or snap well clear permeable supports in an air-liquid interface (ALI) culture system. The ALI medium was supplemented with bovine pituitary extract (BPE, 0.1 mg/ml), recombinant epidermal growth factor (EGF, 0.5 ng/mL), insulin (5 μg/mL), hydrocortisone (0.5 μg/mL), epinephrine (0.6 μg/mL), penicillin (100 units/ml) and streptomycin (100 μg/ml), ethanolamine (500 nM), phosphoethanolamine (500 nM), 3,3,5-triiodothyronine (T3, 6.5 ng/ml), transferrin (10 μg/ml) and 0.1 ng/ml retinoic acid. The ALI medium in the apical side was removed once the cells form a monolayer, and the medium in the basolateral area was changed every 2–3 days. The NHBE cells in Air-liquid interface (ALI) cultures were allowed to differentiate fully for at least 3 weeks before experiments. Cell from each lung were re-differentiated individually such that each plate of the resulting ALI cultures have cells from only one lung. Bronchial epithelial cells from different lungs were not mixed to obtain ALI cultures.
The transformed bronchial epithelial cell line, BEAS-2B, expressing endogenous CFTR was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and maintained in BEGM basal medium. The following chemicals and reagents were dissolved in LHC media to prepare BEGM media; bovine pituitary extract (BPE, 0.1 mg/ml), recombinant epidermal growth factor (EGF, 25 ng/mL), insulin (5 μg/mL), hydrocortisone (0.5 μg/mL), epinephrine (0.6 μg/mL), penicillin (100 units/ml) and streptomycin (100 μg/ml), Gentamicin (50 μg/mL), Amphotericin B (2.5 μg/mL), ethanolamine (500 nM), phosphoethanolamine (500 nM), 3,3,5-triiodothyronine (T3, 6.5 ng/ml), transferrin (10 μg/ml) and 0.1 ng/ml retinoic acid. For all experiments, BEAS-2B cells were grown in monolayer culture for 48 hr before treatment, a time at which CFTR expression is close to highest. All cells were grown at 37°C in a humidified 5% CO2 atmosphere.
miRNA expression profile analysis:
The Tat MicroRNAome was determined by Exiqon miRCURY-Ready-to-Use PCR-human-panel-I+II-V1.M (Exiqon miRNA qPCR panel). According to the manufacturer’s protocol, a total of 20 to 25 ng RNA extracted from heat-inactivated HIV Tat (as control) and biologically active HIV Tat (as treatment) treated NHBE cells were subjected for reverse transcription and converted to cDNA by using the miRCURY Locked Nucleic Acid (LNA) Universal Reverse Transcription (RT) microRNA PCR, polyadenylation, and cDNA Synthesis Kit II. Then, the resulting cDNA used for real-time PCR was mixed with ExiLENT SYBR® Green master mix and loaded into Exiqon miRCURY-Ready-to-Use PCR-Human-panel-I + II-V1.M, which could identify total 742 miRNAs in HIV Tat treated NHBE cells which were differentially expressed compared to the heat-inactivated HIV Tat (as control). The thermal cycling protocol involved denaturation at 95 °C for 10 min, followed by 40 cycles at 95 °C for 10 seconds, and 60 °C for 60 seconds. Each 384-well PCR plates were scanned on the CFX384 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Only the miRNAs with cycle threshold (Ct) value < 37 and5 Ct’s less than the negative control (No Template Control, NTC) in the panel were processed using the Exiqon GenEx qPCR analysis software version 6. The Ct values obtained from the different panels were adjusted by an Inter Plate Calibrator (IPC). An RNA spike-in control (UniSp6) was used as technical controls to evaluate if all samples’ technical performance was similar.
Quantification of miRNA and mRNA expression by Quantitative Reverse Transcription-PCR (qRT-PCR):
According to the manufacturer’s protocol, total RNAs were isolated from NHBE cells and BAES-2B cells treated/transfected with different treatments using a RNeasy mini kit (Qiagen Inc. Valencia, CA). The extracted RNA’s purity and concentration were assessed (O.D. 260/280 nm absorbance ratio of at least 2.0) by a microspot RNA reader (Synergy HT Multi-Mode Microplate Reader from BioTek, Winooski, VT, USA). 0.5 μg of total RNA was reverse transcribed (RT) to synthesize cDNA using high-capacity cDNA reverse transcription kit as per the manufacturer’s protocol after eliminating genomic DNA. Then, the relative amount of mRNA was quantified using TaqMan Fast Advanced Master Mix as per the manufacturer’s protocol in 20 μL real-time PCR reactions with gene specific TaqMan primers a BioRad CFX96 real-time system. All data were normalized to GAPDH (in case of coding RNA; CFTR mRNA quantitation) or 18S RNA (in case of non-coding RNA; microRNA quantitation) and calculated as mean fold change in expression of the target gene using the comparative CT method.
The total RNA was subjected for reverse transcription by using the TaqMan MicroRNA Reverse Transcription Kit and the respective TaqMan primer according to the manufacturer’s instructions to analyze the expression of individual miRNA a two-step protocol to ensure high specificity and sensitivity. Each reverse transcription reaction contains 5–10 ng of total RNA, 10X reverse transcription buffer, 100 mM dNTPs, 20U/μL of RNase inhibitor, 50U/μL of MultiScribe Reverse Transcriptase, and 3 μL of 5X miRNA-specific stem-loop primers (has-miR-145-5p and has-miR-509-3p), in a final volume of 15 μL, adjusted with nuclease-free water. The reaction parameter for reverse transcription was: 30 min at 16°C, 30 min at 42°C, 5 min at 85°C, followed by a holding step at 4°C. After that, each RT product was amplified using the TaqMan Universal Master Mix and the specific 20x TaqMan MicroRNA Assays to validate miRNA expression on the 7500 Real-Time PCR System (Software 2.01, Applied Biosystems, Foster City, CA, USA). Each qPCR reaction contains 1.33 μL of the RT product, 10 μL of TaqMan Universal Master Mix, and 7.67 μL of Nuclease-free water up to a final volume of 20 xL. Reactions are run in a qPCR thermocycler set to perform the following protocol: 10 min at 95°C, 40 two-step cycles of 15 s at 95°C, and 1 min at 60°C.
Primers used for amplification were acquired from Applied Biosystems as follows GAPDH (Catalog# 4331182, Assay ID: Hs02786624_g1), CFTR (Catalog# 4331182, Assay ID: Hs00357011_m1), hsa-miR-509-3p (Catalog# 4427975, Assay ID: 002236), hsa-miR-145 (Catalog# 4427975, Assay ID: 002278).
Electrophysiological experiments by Ussing chamber:
Changes in short circuit currents (∆ISC) in NHBE cells were measured with Ussing chambers (Physiologic Instruments, San Diego, CA) as previously described (14, 41). Electrode offset and bath solution resistances were adjusted before starting the experiments. The apical and basolateral compartment was perfused in standard buffer solution (SBS) comprising Hank’s Balanced Salt Solution (HBSS, pH 7.4) supplemented with 10 mM 4-(2-hydroxyethyl)-1piperazineethanesulfonic acid (HEPES). Chambers and bathing solutions were kept at 37°C using heated water jackets and continuously gassed with a 95% O2 / 5% CO2 mixture. Inserts with NHBE cells grown in the growth or differentiation medium were washed twice with HBSS buffer.
Fully differentiated NHBE cells cultured on the filters of snapwells were mounted in Ussing chambers (EasyMount chamber) connected to a VCC MC6 voltage-clamp unit (Physiologic Instruments, San Diego, CA). To monitor the short-circuit current (ISC), the transepithelial membrane potential was clamped at 0 mV with a single-channel voltage-current clamp using Ag-AgCl electrodes in agar bridges. Following a short equilibration period, the baseline ISC was recorded, followed by the addition of 10 μM of Amiloride apically to block epithelial sodium ion (Na+) channel activity through ENaC and only keep the chloride ion (Cl−) channel active. Subsequently, Albuterol (10 μM) was added at the basolateral side to activate CFTR and change in short circuit current (ΔISC) was determined as an indicator of CFTR activity. CFTR specificity was confirmed by the sequential addition of CFTR inhibitor CFTRinh-172 (20 μM) and GlyH101 (50 μM) to the apical bath solution. Signals were acquired with DAQplot software (Acquire and Analyze v. 2.3.300, Physiologic Instruments). Data were expressed by subtracting the Isc after CFTRinh-172 addition from the peak of albuterol-stimulated Isc calculated as the CFTRinh-172 sensitive Isc.
Transfection of microRNA mimics and antagomirs in BEAS-2B cells:
BEAS-2B cells were plated on collagen-coated 12- well tissue culture plates at a 70% confluence and allowed to adhere overnight. 24 hr following plating, BEAS-2B cells were transfected with miRNA mimics or antagomir to miR-145-5p and miR-509-3p using lipofectamine RNAiMAX. Before transfection, 25 nM of each mimic or antagomir individually or as pools mixed with Lipofectamine RNAiMax in Opti-MEM medium. The mixtures were gently vortexed and kept at room temperature for 20 minutes before adding to the BEAS-2B cells to form a better complex. Separately, another set of BEAS-2B cells were treated with equivalent amounts of lipofectamine RNAiMAX in Opti-MEM used as transfection control. Each mimic or antagomir specific complex was added directly to the cells in the 12-well plates in triplicate and overlaid with OptiMEM media. Furthermore, 4–6 h post-transfection, the media were removed and replaced with BEGM media containing antibiotic supplements. Cells were incubated at 37°C, 5% CO2 for 24–48 h before harvesting.
Bioinformatics tools used for miRNA target sites in the CFTR gene:
The role of miRNAs in the regulation of CFTR expression was performed using a computational approach, which predicted the putative seed regions in the 3′UTR of CFTR mRNA that are identified by a variety of miRNAs. The putative target sites for the miRNAs were predicted for CFTR mRNA using the online target prediction algorithms miRANDA (http://www.microrna.org/microrna/), TargetScan (http://targetscan.org), miRTARbase, (https://mirtarbase.cuhk.edu.cn/∼miRTarBase/miRTarBase_2019/php/index.php) a database of experimentally validated miRNAs and PITA (https://urldefense.com/v3/__https://tools4mirs.org/software/target_prediction/pita/__;!!FjuHKAHQs5udqho!YCd9PuoiCBw-P9kIYktbs-vPc-vuNpR_svU7VRXgmMGmq540cxDPoiudoeg--Kg$). The NCBI database https://www.ncbi.nlm.nih.gov was used to get information on the human CFTR gene transcript (NCBI Reference Sequence: NM_000492.4). The miRBase database (http://www.mirbase.org) were used to get information on the microRNA sequences in supplementary table 1 and for location and sequence of hsa-miR-145-5p (MIMAT0000437) and has-miR-509-3p (MIMAT0002881).
CRISPR site selection for targeting miR-145-5p site:
The site was selected using the specificity check feature of Dharmacon (Horizon inspired cell solutions; CO) CRISPR design tool (https://dharmacon.horizondiscovery.com/gene-editing/crispr-cas9/crispr-design-tool/ ). The specificity check feature excludes any sequences with two or fewer mismatch alignments anywhere else in the genome thereby increasing gRNA specificity and preventing off-target effects. The DNA sequence from position 1– 500 of the untranslated region of CFTR mRNA was entered in the design tool to identify CRISPR sites targeting the miR-145-5p site at position 412 using default parameters. Fortuitously, the target sequence identified was at position 410 which overlaps with the 412 site of miR-145-5p. The sgRNA 410 was then custom synthesized and obtained from Dharmacon.
CRISPR/Cas9 transfection in NHBE cells:
In preparation for CRISPR/Cas9 transfection, NHBE cells were grown in a 6-well plate insert as a monolayer for three weeks for re-differentiation. The re-differentiated NHBE cells were transfected with Edit-R Cas9 expression plasmid with puromycin resistance using PolyMag CRISPR Transfection Reagent and DharmaFECT Duo Transfection Reagent recommended by the manufacturer protocols. Briefly, 200 nM of the synthetic guide RNAs (sgRNA410 resuspended in 10 mM Tris–HCl pH7.5) was mixed with Dharmafect Duo Transfection Reagent (2 mg/mL) and diluted in 200 μL of the serum-free medium in one tube. In another tube, Edit-R Cas9 expression plasmid (4 μg/well) was diluted in 200 μL of serum-free medium and mixed with PolyMag CRISPR transfection reagent (2 μL/mL). The tube contained Cas9 expression plasmid, and the PolyMag CRISPR transfection reagent was combined with the tube provided of sgRNAs and Dharmafect Duo Transfection Reagent. The transfection mixer was gently vortexed and incubated for 20 min at room temperature. The transfection reagent mixture was added dropwise onto the cells in a single well of a six-well cell culture plate containing 1.6 mL of ALI media. The plate was then set on the top of a plate magnet (Oz Biosciences) for 2 hours in the incubator. 48 hours following transfection, cells were treated with TGF- β1/HIV Tat. After an additional 16 hours, experiments were terminated, cells were trypsinized, and total RNA was analyzed for CFTR mRNA levels.
CRISPR/Cas9 transfection in BEAS-2B cells:
In preparation for CRISPR/Cas9 transfection, BEAS-2B cells (∼0.5×106/well) were seeded into a 12-well plate and incubated cells at 37°C with 5% CO2 overnight. 250 ng/ μL of working solution Cas9 plasmid and 25 μM synthetic sgRNA was prepared by adding Tris buffer. The plated BEAS-2B cells were transfected with synthetic guide RNA (sgRNA410) and Edit-R Cas9 expression plasmid with puromycin resistance using DharmaFECT Duo Transfection Reagent recommended by the manufacturer protocols. Briefly, 100 nM of sgRNA410 was mixed with Dharmafect Duo Transfection Reagent (2 mg/mL) and diluted in 100 μL of the serum-free medium in one tube. Edit-R Cas9 expression plasmid (2 μg/well) was diluted in 100 μL of a serum-free medium in another tube. The tube contained Cas9 expression plasmid was mixed with the tube provided of sgRNAs and Dharmafect Duo Transfection Reagent. The transfection mixer was gently vortexed and incubated for 20 min at room temperature. Then, the transfection reagent mixture was added dropwise onto the cells in a single well of a twelve-well cell culture plate containing 800 μL of BEGM media. 48 hours following transfection, cells were treated with TGF- β1/HIV Tat. After an additional 16 hours, experiments were terminated, cells were trypsinized, and total RNA was analyzed for CFTR mRNA levels.
Statistical analysis:
Unless otherwise mentioned, data were expressed as mean ± SEM from NHBE ALI cultures from at least three lungs. The data were subjected to statistical analysis using unpaired t-tests or ANOVA followed by Tukey Kramer honestly significant difference test for multiple comparisons as appropriate. The significance was considered at the level of p<0.05.
Ethics statement:
All experiments were done with appropriate approvals from the Institutional committees.
RESULTS
HIV Tat alters the bronchial epithelial microRNAome to suppress CFTR
In our earlier study, we have shown that HIV Tat suppresses CFTR biogenesis and function via TGF-β signaling (14). We have also demonstrated that TGF-β alters the bronchial epithelial microRNAome, specifically TGF-β upregulates miR-145-5p to suppress CFTR (41). We tried to determine the effects of Tat on the bronchial epithelial microRNAome. NHBE ALI cultures were treated with HIV Tat, as reported by us earlier (30). Change in miRNA expression profile was determined using the Exiqon microRNA Human panel I and II, V4, and data were analyzed using the GenEX software. This allows us to assay 742 mature human microRNAs. We found that HIV Tat substantially alters the bronchial epithelial microRNAome. HIV Tat treatment shows statistically significant Log2-fold change (p < 0.05) in the expression of over 83 different miRNAs (Figure 1A, and Supplementary Table 1 Supplementary Table 2). Given our earlier report that HIV Tat induces TGF-β signaling (14), we compared these changes to miRNA changes observed in TGF-β treated NHBE ALI cultures reported by us earlier (49). Our data show that Tat and TGF-β1 upregulated or downregulated at least 7 common miRNAs, albeit to different magnitudes (Figure 1B). An additional 7 microRNAs were either upregulated by Tat and suppressed by TGF-β or vice versa, thereby showing effects opposite TGF-β. Both Tat and TGF-β are potent signaling molecules that can drive transcriptional activation of multiple genes (50–52). It is important to remember that while HIV Tat can upregulate TGF-β signaling possibly by binding to the TGF-β1 promoter and increasing its transcription (53, 54), Tat is a transcriptional activator in its own right and can mediate independent effects on expression of genes (50). Tat also directly interacts with the RNA interference apparatus components, limiting mature miRNA processing (55). Moreover, Tat can also alter or initiate cellular signaling pathways, which may have secondary effects on miRNA expression and counter some of the activations or suppressions by TGF-β. Surprisingly, we found that while Tat, like TGF-β, upregulated miR145-5p, Tat also upregulated miR-509-3p known to modulate CFTR expression (56), which we had not observed with our TGF-β studies (49).
Figure 1: HIV Tat alters the microRNAome in bronchial epithelial cells.

(A) Volcano plot of the altered microRNAome in primary bronchial epithelial cultures treated with HIV Tat protein. Primary bronchial epithelial cultures from three different lungs were redifferentiated at the ALI and treated with HIV Tat protein (10 nM) or Heat inactivated Tat (as control) as described by us earlier (14). Total RNA was analyzed for miRNA expression using Exiqon microRNA Human panels I and II, V4 (Exiqon, Woburn, MA, US). This allows us to assay 742 mature human microRNAs. Data were analyzed using the GenEx software (Exiqon; Now Qiagen). (B) Comparison of miRNAs upregulated or downregulated by HIV Tat and TGFβ signaling. Text in Red denotes upregulation and Text in Green denotes downregulation. n = NHBE AI cultures from 3 different lungs.
Expression of miR-145-5p and miR-509-3p in HIV Tat treated Airway epithelial cells:
The transcriptional regulation of CFTR activity is tightly controlled by transcription factors and microRNAs (57), and reports including ours have shown the ability of both miR-145-5p and miR-509-3p to downregulate CFTR mRNA (41, 56, 58–60). Figure 2A shows the upregulation of miR-145-5p and miR-509-3p from our miRNA expression profile data in Supplementary Table 1. Next, we validated our miRNA expression profile results to determine the upregulation of miR-145-5p and miR-509-3p in NHBE ALI cultures treated with HIV Tat. NHBE ALI cultures were treated with HIV Tat (heat-inactivated Tat as control) described by us (14). 48hours following Tat treatment, total RNA was analyzed for miR-145-5p and miR-509-3p using specific Taqman probes. HIV Tat upregulates both CFTR targeting RNAs with ∼6-fold upregulation of miR-145-5p (Figure 2B) and a ∼2.5-fold upregulation of miR-509-3p (Figure 2C). Figure 2D shows the predicted target sites of miR-145-5p and miR-509-3p on the CFTR 3’UTR.
Figure 2: Identification of Potential CFTR-Targeting miRNAs Induced by HIV Tat protein.

(A) miRNAs targeting CFTR were identified by a combination of target site prediction algorithm miRanda (miRSVR scores <−0.5) and miRTARbase (database of experimentally validated miRNAs). miRNAs are listed in order of their scores, starting with the best. (B & C) miRNA expression profile validation for miR-145-5p and miR-509-3p. NHBE ALI cultures were treated with HIV Tat (10nM) and heat-inactivated Tat as controls. Total RNA was analyzed for miR145-5p and miR-509-3p expression using qRT-PCR and normalized to 18S RNA. n = 3 different experiments (3 different lungs for NHBE cells). *Significant (p < 0.05) vs control. (D) Schematic of the putative target sites of the identified miRNAs on the 3′ UTR of CFTR mRNA.
Mir-145-5p and miR-509-3p cooperatively suppress CFTR mRNA in HIV Tat treated cells:
Given that microRNAs regulate their cognate genes by suppressing their mRNA, we tried to determine if miR-145-5p and miR-509-3p mimics can decrease CFTR mRNA levels. We transfected BEAS-2B cells with miR-145-5p and miR-509-3p mimics using lipofectamine RNAiMAX. Transfection with lipofectamine RNAiMAX was used as control. 48 hours post-transfection, experiments were terminated, and total mRNA was isolated and analyzed for CFTR mRNA levels. We have already shown that miR-145-5p suppresses CFTR mRNA (41). Figure 3A reproduces these results. Besides, we also observed that miR-509-3p also suppresses CFTR mRNA compared to lipofectamine RNAiMAX control. Surprisingly, we noticed that when both mimics were transfected simultaneously such that the total mimic concentration remained the same as individually transfected mimics, we observed CFTR mRNA suppression comparable to that observed with individual mimics. These data suggest that both miRNAs may be acting cooperatively to suppress CFTR mRNA. We tested non-targeting miRNA mimic controls for their ability to suppress CFTR. Supplementary Figure 1 shows that none of the non-targeting mimics suppress CFTR validating the specificity of miR-145-5p and miR-509-3p mediated CFTR suppression. Several miRNAs have been shown to function independently and cooperatively to regulate target genes (61–63).
Figure 3: HIV Tat protein upregulates miR-145-5p and miR-509-3p to suppress CFTR biogenesis and function.

The ability of miR-145-5p and miR-509-3p identified in Figure 2B to suppress the CFTR was validated in transient transfection assays using miRNA mimics and antagomirs. (A) miR-145-5p and/or miR-509-3p mimic was transfected individually or in combination in BEAS2B airway epithelial cell lines. Lipofectamine RNAiMAX was used as the control, and lipofectamine RNAiMAX plus HIV Tat protein treatment was used for comparative analysis. Both miRNAs individually suppress CFTR mRNA expression, confirming that both miRNAs have target sites in 3′ UTR of CFTR mRNA. Co-operative effects in CFTR mRNA suppression were overserved when both microRNA mimics were transfected simultaneously, such that the total mimic concentration remained the same as individually transfected mimics. n = 5 experiments. (B) To determine if the suppression is co-operative and requires both miRNAs, BEAS-2B cells were transfected with antagomirs to miR-145-5p and miR-509-3p. An additional set was co-transfected with a combination of both antagomir-145-5p and antagomir-509-3p such that the total antagomir concentration remained the same as individually transfected antagomirs. Separately, another set of BEAS-2B cells was treated with HIV Tat and Lipofectamine RNAiMAX. Lipofectamine RNAiMAX alone was used for comparison. Individual antagomir’s targeting miR-145-5p or miR-509-3p completely rescued Tat-mediated CFTR mRNA suppression comparable to that seen in cells transfected with a combination of both antagomirs. n = 5 experiments. * = significant from control; S = significant from each other (p < 0.05).
We further tried to validate the role of miR-145-5p and mir-509-3p in HIV Tat-mediated CFTR mRNA suppression. We also tried to determine if the mRNA suppression is co-operative and requires both microRNAs. For these purposes, we transfected BEAS2B airway epithelial cells with respective antagomirs. An additional set was co-transfected with a combination of both antagomir-145-5p and antagomir-509-3p. Separately, another group of BEAS-2B cells was treated with HIV Tat and Lipofectamine RNAiMAX. Lipofectamine RNAiMAX alone was used for comparison. If suppression is co-operative and requires both miRNAs to be present, then either of the antagomir will completely rescue CFTR mRNA suppression, and the rescue will not be additive in cells co-transfected with a combination of both antagomirs. As seen in Figure 3B, antagomir targeting miR-145-5p and miR-509-3p when transfected individually, completely rescued Tat-mediated CFTR mRNA suppression comparable to that seen in cells transfected with a combination of both antagomirs. Together these data demonstrate that both miR-145-5p and miR-509-3p are required to suppress CFTR mRNA. Indeed Ramachandran et al. have shown that miR-509-3p acts cooperatively with another CFTR suppressing miRNA (in their case miR-494), to suppress CFTR mRNA (56).
CRISPR-mediated CFTR specific microRNA antagonism restores TGF-β1-mediated CFTR suppression:
CRISPR targeting of 412 target site in the CFTR 3’UTR was designed using the specificity check feature of Dharmacon (Horizon inspired cell solutions; CO) CRISPR design tool (https://dharmacon.horizondiscovery.com/gene-editing/crispr-cas9/crispr-design-tool/ ). The specificity check feature excludes any sequences with two or fewer mismatch alignments anywhere else in the genome thereby increasing gRNA specificity and preventing off-target effects. SgRNA 410 was custom synthesized and obtained from Dharmacon. The DNA target of sgRNA410 and the miR-145-5p target sequence are shown in Figure 4A. Since the role of miR145-5p in TGF- β -mediated CFTR suppression has been validated by others and by us (49, 64), SgRNA410 was tested for its ability to block TGF-β1-mediated CFTR suppression in BEAS-2B cells. Briefly, BEAS2B cells were co-transfected with sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo reagent according to the manufacturer’s instructions. BEAS-2B cells were treated with Dharmafect Duo alone (as control). 48 hours posttransfection, 10ng TGF-β1 was added. TGF-β1 treatment plus Dharmafect Duo alone treatment was used for comparison. After an additional 16 hours, experiments were terminated, and total RNA was analyzed for CFTR mRNA levels. Figure 4B shows that TGF-β suppresses CFTR mRNA, and sgRNA410 prevents TGF-β-mediated CFTR mRNA suppression in BEAS2B cells. To demonstrate the specificity of our CFTR-specific microRNA antagonism approach, BEAS2B cells were transfected with miR-145-5p mimic (lipofectamine RNAimax reagent) or sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid (Dharmafect duo reagent). BEAS-2B cells treated with Dharmafect Duo alone was used as control. 48 hours post-transfection, 10ng TGF-β1 was added to sgRNA410 along and Cas9 plasmid transfected cells. TGF-β1 treatment plus Dharmafect Duo alone treatment was used for comparison. After an additional 16 hours, experiments were terminated, and total RNA was analyzed for BNIP3 mRNA levels. BNIP3 is another target of miR-145-5p and shares only the 6-nt seed sequence with the CFTR miR-145-5p target site. Hence, a sgRNA designed to hybridize to a 20nt region of the CFTR target site should not edit the BNIP3 3’ UTR target site. Figure 4C shows that both TGF-β1 and miR-145-5p mimic suppresses BNIP3. SgRNA410 designed to edit the CFTR miR-145-5p target site does not rescue BNIP3 suppression, demonstrating our approach’s specificity.
Figure 4: Gene-specific microRNA antagonism prevents TGF-β mediated suppression of CFTR mRNA.

(A) sgRNA410 was designed and custom synthesized against the miR-145-5p at position 412 on the 3’UTR of CFTR. The sequence of the target DNA, position in the 3’ UTR is shown. (B) BEAS2B cells were co-transfected with sgRNA410 along with the Cas9 plasmid followed by TGF-β1 treatment. miR-145-5p mimic transfection was used for comparison. Total RNA was analyzed for BNIP3, which is another target of miR-145-5p. TGF-β1 treatment and miR-145-5p suppress BNIP3 demonstrating that BNIP3 is a target of miR-145-5p. n = 3 experiments. (C) sgRNA410 and Cas9 transfection does not prevent TGF-Β1 mediated BNIP3 suppression, confirming that our microRNA antagonism approach is specific for CFTR and does not prevent TGF-β mediated regulation of other genes. n = 3 experiments. (D) DNA sequence analysis from NHBE ALI cultures magnetofected with cas9 alone shows a clean chromatogram with a single sequence [panel D(i)]. Magnetofection based delivery of sgRNA410 and Cas9 shows editing of the miR-145-5p target site [panel D(ii)]. The Redline shows the miR-145-5p target site. The blue line shows the sgRNA410 target site. (E) NHBE ALI cultures were magnetofected with either sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo (Dharmafect) and Polymag CRISPR transfection reagent (OZ biosciences). NHBE ALI cultures were then treated with TGF-β1 or vehicle (as control). CRISPR-based editing of the miR-145-5p target site by sgRNA410 and Cas9 preserves CFTR mRNA levels in NHBE ALI cultures treated with TGF-β1. n = 8 experiments. * = significant from control; S = significant from each other (p < 0.05).
Next, we tried to determine if sgRNA410 can preserve CFTR mRNA levels in TGF-β treated NHBE ALI cultures. We first decided to deliver and successfully achieve editing of the miR-145-5p target site in the CFTR 3’UTR in NHBE ALI cultures. Successful transient transfection of NHBE cells is very difficult and challenging (65, 66). We developed an in-house magnetofection protocol to get a robust and better transfection efficiency in NHBE cells to address this challenge. In this protocol, the NHBE cells redifferentiated at the ALI on snap wells were co-transfected with sgRNA410 and the Edit-R Cas9 Nuclease expression plasmid cocomplexing with the Dharmafect Duo and Polymag CRISPR transfection reagent (OZ Biosciences) according to the respective manufacturer’s instructions. One set of NHBE cells was treated with Cas9 plasmid alone co-complexed with Dharmafect Duo, and Polymag CRISPR transfection reagent as control was used as control. Total DNA was isolated from these cells and PCR amplified using primer pairs flanking the 1500 bp CFTR UTR with the high-fidelity Vent polymerase (NEB #M0254S). The PCR product was then sequenced using a 5’ primer designed to hybridize just upstream of the CFTR termination codon. Figure 4D shows that control magnetofection with Cas9 plasmid alone shows a clean chromatogram with only one sequence. Sequencing chromatogram from NHBE ALI cultures from paired lungs magnetofected with sgRNA410 and Cas9 plasmid show editing of the miR-145-5p site and multiple sequences demonstrating successful delivery of our CRISPR cassette and successful editing of the miR145-5p target site in fully differentiated NHBE ALI cultures.
Since TGF-β signaling is upregulated in chronic airway diseases like Asthma, COPD, and ACOS, and HIV Tat also mediates its effects on CFTR via TGF-β signaling (14, 31), we tried to determine if our CFTR-specific microRNA antagonism approach can preserve CFTR mRNA in NHBE ALI cultures treated with TGF-β1. NHBE ALI cultures redifferentiated on snap-wells were co-transfected with sgRNA410 and the Edit-R Cas9 Nuclease expression plasmid by co-complexing with Dharmafect Duo (Dharmafect) and PolyMag CRISPR transfection reagent (OZ biosciences) using our in-house protocol. One set of NHBE cells was treated with Cas9 plasmid with Dharmafect Duo and PolyMag CRISPR transfection reagent alone as a control. 48 hours following magnetofections, TGF-β1 (10ng/ml) was added to the co-transfected NHBE cells. TGF-β treatment (with Cas9 plasmid and reagents) was used for comparison. Total RNA was isolated from these cells and analyzed for CFTR mRNA. TGF-β1 suppresses CFTR mRNA in NHBE ALI cultures, and magnetofection with our sgRNA410 prevents TGF-β-mediated suppression of CFTR mRNA (Figure. 4E).
CRISPR-mediated CFTR specific microRNA antagonism preserves CFTR mRNA and function in HIV Tat-treated NHBE ALI cultures:
We have shown that HIV Tat protein induces TGF β1 signaling to suppress CFTR in bronchial epithelial cells (14). Hence, we tested the ability of our CRISPR-based gene-specific miRNA antagonism approach to preserve CFTR mRNA in HIV Tat treated airway epithelial cell lines and BEAS2B cells were transfected with the sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo transfection reagent according to the manufacturer’s instructions. 48 hours the following co-transfection, BEAS-2B cells were treated with recombinant HIV Tat as described by us (14). 48 hours of post-Tat treatment, total RNA was analyzed for CFTR mRNA levels by qRT-PCR. Figure 5A shows that sgRNA preserves CFTR mRNA expression in Tat treated BEAS2B cells. Next, we tested our gene-specific microRNA antagonism approach in simulated physiological conditions in primary bronchial epithelial cells redifferentiated at the ALI.
Figure 5: Gene-specific microRNA antagonism prevents HIV tat mediated suppression of CFTR biogenesis and function.

(A) BEAS2B cells were transfected with the sgRNA410 and the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo transfection reagent. 48 hours following co-transfection, BEAS-2B cells were treated with recombinant HIV Tat. 16 hours of post-Tat treatment, total RNA was analyzed for CFTR expression by qRT-PCR. CRISPR-based editing of miR-145-5p target site with sgRNA410 and Cas9 preserves CFTR mRNA expression in Tat treated BEAS2B cells. n = 11 experiments. (B) NHBE ALI cultures were magnetofected with sgRNA410 and the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo transfection reagent. 48 hours the following co-transfection, NHBE ALI cultures were treated with recombinant HIV Tat. CRISPR-based editing of miR-145-5p target site using sgRNA410 and Cas9 preserves CFTR mRNA levels in NHBE ALI cultures in HIV Tat. n = 8 experiments from 4 different lungs. (C) NHBE ALI cultures redifferentiated on snap wells were magnetofected with sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo transfection reagent. 48 hours the following co-transfection, NHBE ALI cultures were treated with recombinant HIV Tat. Filters were mounted in Ussing chambers, and CFTR activity was determined as described by us (14, 41). (D) An Ussing chamber trace shows that CRISPR-based editing of the miR-145-5p target site in NHBE ALI cultures preserves apical localization and CFTR function. n = 8 experiments from 3 different lungs. * = significant from control; S = significant from each other (p < 0.05).
NHBE cultures redifferentiated at the ALI were magnetofected with sgRNA410 along with the Edit-R Cas9 Nuclease expression plasmid using the Dharmafect Duo and Polymag CRISPR transfection reagent according to our protocol described in methods. One set of NHBE cells was magnetofected with Cas9 plasmid using Dharmafect Duo and PolyMag CRISPR transfection reagent alone as a control. 48 hours post magnetofection, NHBE ALI cultures were treated with HIV Tat as described by us (14). Tat treatment (with Cas9 plasmid and reagents) was used for comparison. After an additional 48 hours, Total RNA from these cells was analyzed for CFTR mRNA levels by qRT-PCR. As seen in Figure 5B, CRISPR-based editing of miR-145-5p target site preserves CFTR mRNA levels in HIV Tat treated NHBE ALI cultures. We tried to determine if CRISPR-based editing of the miR-145-5p site within the CFTR 3’UTR has any effects on the apical localization of CFTR or its function. NHBE ALI cultures redifferentiated on snap wells and treated identically as in Figure 5B. 48 hours post Tat treatment, the snap wells were mounted in Ussing chambers, and CFTR function was determined by albuterol mediated activation as described by us (14, 30, 41, 48). Figure 5C shows that our CRISPR-based gene-specific microRNA antagonism approach preserves CFTR localization and function in HIV Tat treated NHBE ALI cultures demonstrating that CRISPR-based editing of the microRNA target sites in the CFTR 3’UTR does not affect CFTR trafficking and activity. Figure 5D shows an Ussing chamber trace showing that sgRNA410-based editing of miR-145-5p target site preserves CFTR function.
Discussion:
HIV infection was found to be an independent risk factor for COPD in the cART era, even when accounting for smoking status (11). HIV-infected cells in the airway (including bronchial epithelial cells (14) can serve as a source of HIV proteins like Tat. HIV Tat is an immediate-early gene of HIV, and its expression is not suppressed by antiretrovirals (67–70). The protein transduction domain of Tat allows its secretion by infected cells and uptake by bystander cells where it mediates pleiotropic effects (37–39, 71). Hence HIV Tat can have deleterious effects not only on infected cells but also on uninfected cells. We have shown that HIV Tat induces TGF-β signaling (14), and in our recent report, we have demonstrated that TGF-β signaling alters the bronchial epithelial microRNAome (49). Since the first RNA interference report, multiple reports have established that miRNAs play an important role in temporal control of gene regulation (72). Multiple miRNAs control a single gene, and a single miRNA can regulate multiple genes (73). In this study, we first demonstrated that HIV Tat alters the bronchial epithelial microRNAome. While some miRNAs common to both TGF-β and HIV Tat, HIV Tat also altered miRNAs distinct from those altered by TGF-β. This could be because HIV Tat can activate the expression of genes that can have further downstream effects on miRNA expression. We have shown that TGF-β also upregulates miR-145-5p to suppress CFTR (49). Not surprisingly, we found that like TGF-β, HIV Tat also upregulates miR-145-5p. In addition, our miRNA expression profile data showed that HIV Tat also upregulates miR-509-3p, another CFTR targeting miRNA (56) which had not been detected in our TGF-β altered microRNAome from our earlier report (49). Using miRNA mimics and antagomirs for both miRNAs, we validate previous reports of miR-145-5p and miR-509-3p based CFTR mRNA suppression (41, 56, 58–60). In addition, we demonstrate that the suppression is co-operative and requires the presence of both miRNAs, as antagomirs to either one microRNA completely rescues Tat-mediated CFTR mRNA suppression. It is possible that even though TGF-β did not alter the expression of miR-509-3p in our earlier report (49), the upregulated miR-145-5p works in cooperation with baseline levels of miR-590–3p to suppress CFTR mRNA.
Ramachandran et al. have also reported that miR-509-3p works co-operatively with other miRNAs to regulate CFTR expression (56). Together these experiments suggest that both miR-145-5p and miR-509-3p are required to suppress CFTR. Such cooperativity in miRNA-based regulation may be more common than reported, given that a single miRNA can regulate multiple genes (73). For instance, miR-145-5p has been experimentally validated to regulate over 50 genes. There would be other targets that have not been reported. Indeed, we recently reported the first miRNA-based regulation of SLC26A9 and demonstrated the ability of miR-145-5p to suppress this important CFTR modifier (41). In these circumstances, counter-regulation or cooperative regulation by multiple miRNAs may play an important role in maintaining the mRNA homeostasis of specific targets when only one miRNA expression is altered.
TGF-β isoforms are expressed and secreted by several cell types in the airway and regulate a wide range of biological processes, including cell proliferation, differentiation, extracellular matrix (ECM) synthesis, and apoptosis (74–76). Likewise, miR-145-5p is considered a major tumor suppressor tumor progression and metastasis (42–45). We and others have previously reported efficient rescue of CFTR by modulating TGF-β signaling or by miR-145 antagonism (41, 64). However, indiscriminate suppression of TGF-β signaling, or interfering with miR-145 regulation can have non-specific effects. Under those circumstances, modulating the miRNA target site on the CFTR 3’UTR can preserve CFTR mRNA and function without suppressing the broader TGF-β signaling or miR-145-5p mediated regulation of other genes. While this manuscript was in preparation, a recent study has shown that masking the 412 sites with peptide nucleic acids (PNA) augments CFTR availability (77). This report confirmed previous predictions and identification of the miR-412 target site for miR-145 on the CFTR 3’ UTR. However, the study had certain limitations in that the authors have used Calu-3 cell lines and not primary cells and have not shown apical localization nor CFTR function. This is important since masking by PNAs may protect the RNAs but may interfere with their translation. The differences would be more pronounced in primary cells with much lower levels of cellular transcription and translation than cell lines. Also, PNAs are primarily used in an anti-sense role for gene inhibition instead of gene augmentation (78–82), and they have to be explicitly designed for the target sequence. Hence, their effects on mRNA stability or any off-target effects due to sequence complementarity with other genes can limit their application in a clinical setting.
However, this report does confirm the position 412 in the CFTR 3’ UTR as a miR-145-5p target site. We developed a novel gene-specific microRNA antagonism approach using CRISPR to preserve CFTR function in the context of increased TGF-β signaling. CRISPR-based gene editing has found significant applications in several pre-clinical studies (83–85). CRISPR introduces targeted double-stranded breaks in the DNA, which are repaired primarily by the NHEJ (non-homologous end joining) DNA repair. CRISPR-based gene-specific microRNA antagonism is attractive in that, even though one miRNA can regulate multiple genes, its target sequences vary significantly between the genes, with only a 6–8 nucleotide seed sequence required for gene silencing. For instance, the reported target site of miR-145-5p on CFTR gene is 5’-TGATTAAGTAATGATAACTGGAA-3’ while that on BNIP3 is 5’-GTGTCTACTTTAAAAAACTGGAA-3’. The two sequences share homology only in the underlined seed sequence. Hence a gRNA designed to edit the miR-145-5p site on CFTR will not edit the miR-14–5p5 site on BNIP3 restricting miR145-5p antagonism to a specific gene of interest (in our case, CFTR). We exploited these differences in target sites to design gRNAs to target the miR-145-5p target site (position 412 in the CFTR 3’UTR).
CRISPR-based editing of miRNA target sites has two distinct advantages in that it works at the DNA levels requiring fewer administrations, and the ability of NHEJ recombination to create repair tracts almost 200 bp away from the target sites (86), allows flexibility in the selection of targets with more specificity to the 3’ UTR of interest. We used the specificity check feature of Dharmacon to select an sgRNA targeting within a 100 nucleotides of position 412 (miR-145-5p target site). Fortuitously, the algorithm designed a gRNA targeting position 410 which overlaps with the miR-145-5p target site at 412. sgRNA410 was designed to edit the 412 sites of miR-145-5p. Our lab showed that magnetofection using PolyMag could efficiently deliver plasmid DNA to cells (data not shown). We developed an in-house protocol using reagents from Dharmacon and OZ Biosciences to give our sgRNA and Cas9 plasmid to redifferentiated NHBE ALI cultures. Our data demonstrate that CRISPR-based CFTR-specific microRNA antagonism preserves CFTR mRNA in BEAS2B airway epithelial cells as well as NHBE ALI cultures. We demonstrate that our CRISPR-based gene-specific microRNA antagonism approach preserves the apical CFTR localization and function, as seen in our Ussing chamber experiments in NHBE ALI cultures. Our gene-specific microRNA antagonism approach can be applicable to many diseases that are characterized by dysregulation due to an aberrant microRNAome.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENT
We would like to thanks Dr. Barbara Gould (Qiagen) for her help in analyzing miRNA expression profile data.
FUNDING
This research was sponsored by NIH R21 HL128141–02 and R01 HL147715 to HU.
Footnotes
CRediT authorship contribution statement
RD: Performed experiments and wrote the Manuscript. SC: performed experiments. S-MJ: performed experiments. IR: planning and analysis. HU: Performed experiments, planning, analysis and writing manuscript.
Declaration of Interests: The authors declare no conflicts of interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Palella FJ Jr., Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853–60. [DOI] [PubMed] [Google Scholar]
- 2.Marcus JL, Chao CR, Leyden WA, Xu L, Quesenberry CP Jr., Klein DB, et al. Narrowing the Gap in Life Expectancy Between HIV-Infected and HIV-Uninfected Individuals With Access to Care. J Acquir Immune Defic Syndr. 2016;73(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CDC. Undertanding the HIV Care Continuum. [Google Scholar]
- 4.Twigg HL, Soliman DM, Day RB, Knox KS, Anderson RJ, Wilkes DS, et al. Lymphocytic alveolitis, bronchoalveolar lavage viral load, and outcome in human immunodeficiency virus infection. American journal of respiratory and critical care medicine. 1999;159(5 Pt 1):1439–44. [DOI] [PubMed] [Google Scholar]
- 5.Nakata K, Weiden M, Harkin T, Ho D, and Rom WN. Low copy number and limited variability of proviral DNA in alveolar macrophages from HIV-1-infected patients: evidence for genetic differences in HIV-1 between lung and blood macrophage populations. Mol Med. 1995;1(7):744–57. [PMC free article] [PubMed] [Google Scholar]
- 6.Rose RM, Krivine A, Pinkston P, Gillis JM, Huang A, and Hammer SM. Frequent identification of HIV-1 DNA in bronchoalveolar lavage cells obtained from individuals with the acquired immunodeficiency syndrome. Am Rev Respir Dis. 1991;143(4 Pt 1):850–4. [DOI] [PubMed] [Google Scholar]
- 7.Morris A, George MP, Crothers K, Huang L, Lucht L, Kessinger C, et al. HIV and chronic obstructive pulmonary disease: is it worse and why? Proceedings of the American Thoracic Society. 2011;8(3):320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gingo MR, Wenzel SE, Steele C, Kessinger CJ, Lucht L, Lawther T, et al. Asthma diagnosis and airway bronchodilator response in HIV-infected patients. J Allergy Clin Immunol. 2012;129(3):708–14 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Sadr WM, Lundgren J, Neaton JD, Gordin F, Abrams D, Arduino RC, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–96. [DOI] [PubMed] [Google Scholar]
- 10.Heffernan RT, Barrett NL, Gallagher KM, Hadler JL, Harrison LH, Reingold AL, et al. Declining incidence of invasive Streptococcus pneumoniae infections among persons with AIDS in an era of highly active antiretroviral therapy, 1995–2000. J Infect Dis. 2005;191(12):2038–45. [DOI] [PubMed] [Google Scholar]
- 11.Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, and Justice AC. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest. 2006;130(5):1326–33. [DOI] [PubMed] [Google Scholar]
- 12.Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. American journal of respiratory and critical care medicine. 2011;183(3):388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigna JJ, Kenne AM, Asangbeh SL, and Sibetcheu AT. Prevalence of chronic obstructive pulmonary disease in the global population with HIV: a systematic review and meta-analysis. The Lancet Global Health. 2018;6(2):e193–e202. [DOI] [PubMed] [Google Scholar]
- 14.Chinnapaiyan S, Parira T, Dutta R, Agudelo M, Morris A, Nair M, et al. HIV Infects Bronchial Epithelium and Suppresses Components of the Mucociliary Clearance Apparatus. PloS one. 2017;12(1):e0169161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi J, Li H, Yuan C, Luo M, Wei J, and Liu X. Cigarette Smoke-Induced Acquired Dysfunction of Cystic Fibrosis Transmembrane Conductance Regulator in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Oxid Med Cell Longev. 2018;2018:6567578-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clunes LA, Davies CM, Coakley RD, Aleksandrov AA, Henderson AG, Zeman KL, et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012;26(2):533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustamante-Marin XM, and Ostrowski LE. Cilia and Mucociliary Clearance. Cold Spring Harb Perspect Biol. 2017;9(4):a028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Londino JD, Collawn JF, and Matalon S. In: Parent RA ed. Comparative Biology of the Normal Lung (Second Edition). San Diego: Academic Press; 2015:467–77. [Google Scholar]
- 19.Mall MA. Role of cilia, mucus, and airway surface liquid in mucociliary dysfunction: lessons from mouse models. J Aerosol Med Pulm Drug Deliv. 2008;21(1):13–24. [DOI] [PubMed] [Google Scholar]
- 20.Bhowmik A, Chahal K, Austin G, and Chakravorty I. Improving mucociliary clearance in chronic obstructive pulmonary disease. Respir Med. 2009;103(4):496–502. [DOI] [PubMed] [Google Scholar]
- 21.Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev. 2002;54(11):1359–71. [DOI] [PubMed] [Google Scholar]
- 22.Boucher RC. Regulation of airway surface liquid volume by human airway epithelia. Pflugers Arch. 2003;445(4):495–8. [DOI] [PubMed] [Google Scholar]
- 23.Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, Lazarowski ER, et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J Biol Chem. 2005;280(42):35751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarran R, Grubb BR, Gatzy JT, Davis CW, and Boucher RC. The relative roles of passive surface forces and active ion transport in the modulation of airway surface liquid volume and composition. J Gen Physiol. 2001;118(2):223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milgrim LM, Rubin JS, and Small CB. Mucociliary clearance abnormalities in the HIV-infected patient: a precursor to acute sinusitis. Laryngoscope. 1995;105(11):1202–8. [DOI] [PubMed] [Google Scholar]
- 26.Rosen EJ, and Calhoun KH. Alterations of nasal mucociliary clearance in association with HIV infection and the effect of guaifenesin therapy. Laryngoscope. 2005;115(1):27–30. [DOI] [PubMed] [Google Scholar]
- 27.Rutland J, Griffin WM, and Cole PJ. Human ciliary beat frequency in epithelium from intrathoracic and extrathoracic airways. Am Rev Respir Dis. 1982;125(1):100–5. [DOI] [PubMed] [Google Scholar]
- 28.Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, et al. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med. 2006;173(10):1139–44. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Skinner D, Hicks SB, Bevensee MO, Sorscher EJ, Lazrak A, et al. Sinupret Activates CFTR and TMEM16A-Dependent Transepithelial Chloride Transport and Improves Indicators of Mucociliary Clearance. PloS one. 2014;9(8):e104090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unwalla HJ, Ivonnet P, Dennis JS, Conner GE, and Salathe M. Transforming growth factor-beta1 and cigarette smoke inhibit the ability of beta2-agonists to enhance epithelial permeability. Am J Respir Cell Mol Biol. 2015;52(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chinnapaiyan S, Dutta R, Bala J, Parira T, Agudelo M, Nair M, et al. Cigarette smoke promotes HIV infection of primary bronchial epithelium and additively suppresses CFTR function. Sci Rep. 2018;8(1):7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chinnapaiyan S, and Unwalla HJ. Mucociliary dysfunction in HIV and smoked substance abuse. Front Microbiol. 2015;6:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, and Marsh JW. Selective Transcription and Modulation of Resting T Cell Activity by Preintegrated HIV DNA. Science. 2001;293(5534):1503–6. [DOI] [PubMed] [Google Scholar]
- 34.Mediouni S, Darque A, Baillat G, Ravaux I, Dhiver C, Tissot-Dupont H, et al. Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect Disord Drug Targets. 2012;12(1):81–6. [DOI] [PubMed] [Google Scholar]
- 35.Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J, et al. HIV-1 Tat Promotes Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) vIL-6-Induced Angiogenesis and Tumorigenesis by Regulating PI3K/PTEN/AKT/GSK-3β Signaling Pathway. PloS one. 2013;8(1):e53145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frankel AD, and Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–93. [DOI] [PubMed] [Google Scholar]
- 37.Frankel AD, and Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–93. [DOI] [PubMed] [Google Scholar]
- 38.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, et al. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67(1):277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ensoli B, Barillari G, Salahuddin SZ, Gallo RC, and Wong-Staal F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature. 1990;345(6270):84–6. [DOI] [PubMed] [Google Scholar]
- 40.Ensoli B, Gendelman R, Markham P, Fiorelli V, Colombini S, Raffeld M, et al. Synergy between basic fibroblast growth factor and HIV-1 Tat protein in induction of Kaposi’s sarcoma. Nature. 1994;371(6499):674–80. [DOI] [PubMed] [Google Scholar]
- 41.Dutta RK, Chinnapaiyan S, Rasmussen L, Raju SV, and Unwalla HJ. A Neutralizing Aptamer to TGFBR2 and miR-145 Antagonism Rescue Cigarette Smoke- and TGF-β-Mediated CFTR Expression. Molecular therapy : the journal of the American Society of Gene Therapy. 2019;27(2):442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu WC, Li WM, Lee YC, Huang AM, Chang LL, Lin HH, et al. MicroRNA-145 suppresses cell migration and invasion in upper tract urothelial carcinoma by targeting ARF6. FASEB J. 2020;34(4):5975–92. [DOI] [PubMed] [Google Scholar]
- 43.Tang C, He JY, Yu C, Wang PJ, Huang SH, Zheng HJ, et al. MicroRNA-145 performs as a tumor suppressor in human esophageal squamous cell carcinoma by targeting phospholipase C epsilon 1. J Cell Biochem. 2019;120(6):10678–87. [DOI] [PubMed] [Google Scholar]
- 44.Xu L, Zhang Y, Tang J, Wang P, Li L, Yan X, et al. The Prognostic Value and Regulatory Mechanisms of microRNA-145 in Various Tumors: A Systematic Review and Meta-analysis of 50 Studies. Cancer Epidemiol Biomarkers Prev. 2019;28(5):867–81. [DOI] [PubMed] [Google Scholar]
- 45.Cui SY, Wang R, and Chen LB. MicroRNA-145: a potent tumour suppressor that regulates multiple cellular pathways. J Cell Mol Med. 2014;18(10):1913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, and Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. [DOI] [PubMed] [Google Scholar]
- 47.Fulcher ML, and Randell SH. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol. 2013;945:109–21. [DOI] [PubMed] [Google Scholar]
- 48.Unwalla HJ, Horvath G, Roth FD, Conner GE, and Salathe M. Albuterol modulates its own transepithelial flux via changes in paracellular permeability. Am J Respir Cell Mol Biol. 2012;46(4):551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dutta RK, Chinnapaiyan S, Rasmussen L, Raju SV, and Unwalla HJ. A Neutralizing Aptamer to TGFBR2 and miR-145 Antagonism Rescue Cigarette Smoke- and TGF-beta-Mediated CFTR Expression. Mol Ther. 2019;27(2):442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark E, Nava B, and Caputi M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget. 2017;8(16):27569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill CS. Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol. 2016;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Derynck R, Zhang Y, and Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95(6):737–40. [DOI] [PubMed] [Google Scholar]
- 53.Sputum Reid L. and mucociliary clearance mechanisms. Postgrad Med J. 1976;52(606):183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bogerd HP, Fridell RA, Madore S, and Cullen BR. Identification of a novel cellular cofactor for the Rev/Rex class of retroviral regulatory proteins. Cell. 1995;82(3):485–94. [DOI] [PubMed] [Google Scholar]
- 55.Bennasser Y, and Jeang KT. HIV-1 Tat interaction with Dicer: requirement for RNA. Retrovirology. 2006;3:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramachandran S, Karp PH, Osterhaus SR, Jiang P, Wohlford-Lenane C, Lennox KA, et al. PostTranscriptional Regulation of Cystic Fibrosis Transmembrane Conductance Regulator Expression and Function by MicroRNAs. American Journal of Respiratory Cell and Molecular Biology. 2013;49(4):544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swahn H, and Harris A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes (Basel). 2019;10(3):235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amato F, Seia M, Giordano S, Elce A, Zarrilli F, Castaldo G, et al. Gene Mutation in MicroRNA Target Sites of CFTR Gene: A Novel Pathogenetic Mechanism in Cystic Fibrosis? PloS one. 2013;8(3):e60448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oglesby IK, Chotirmall SH, McElvaney NG, and Greene CM. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, −223, and −494 is altered in DeltaF508 cystic fibrosis airway epithelium. J Immunol. 2013;190(7):3354–62. [DOI] [PubMed] [Google Scholar]
- 60.Gillen AE, Gosalia N, Leir SH, and Harris A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem J. 2011;438(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lai X, Schmitz U, Gupta SK, Bhattacharya A, Kunz M, Wolkenhauer O, et al. Computational analysis of target hub gene repression regulated by multiple and cooperative miRNAs. Nucleic Acids Res. 2012;40(18):8818–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antonov AV, Dietmann S, Wong P, Lutter D, and Mewes HW. GeneSet2miRNA: finding the signature of cooperative miRNA activities in the gene lists. Nucleic Acids Res. 2009;37(Web Server issue):W323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shao T, Wang G, Chen H, Xie Y, Jin X, Bai J, et al. Survey of miRNA-miRNA cooperative regulation principles across cancer types. Brief Bioinform. 2019;20(5):1621–38. [DOI] [PubMed] [Google Scholar]
- 64.Lutful Kabir F, Ambalavanan N, Liu G, Li P, Solomon GM, Lal CV, et al. MicroRNA-145 Antagonism Reverses TGF-beta Inhibition of F508del CFTR Correction in Airway Epithelia. American journal of respiratory and critical care medicine. 2018;197(5):632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsui H, Johnson LG, Randell SH, and Boucher RC. Loss of Binding and Entry of Liposome-DNA Complexes Decreases Transfection Efficiency in Differentiated Airway Epithelial Cells. Journal of Biological Chemistry. 1997;272(2):1117–26. [DOI] [PubMed] [Google Scholar]
- 66.Matsui H, Randell SH, Peretti SW, Davis CW, and Boucher RC. Coordinated clearance of periciliary liquid and mucus from airway surfaces. The Journal of clinical investigation. 1998;102(6):1125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly J, Beddall MH, Yu D, Iyer SR, Marsh JW, and Wu Y. Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology. 2008;372(2):300–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Y, and Marsh JW. Early transcription from nonintegrated DNA in human immunodeficiency virus infection. J Virol. 2003;77(19):10376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu Y, and Marsh JW. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science. 2001;293(5534):1503–6. [DOI] [PubMed] [Google Scholar]
- 70.Ensoli B, Bellino S, Tripiciano A, Longo O, Francavilla V, Marcotullio S, et al. Therapeutic immunization with HIV-1 Tat reduces immune activation and loss of regulatory T-cells and improves immune function in subjects on HAART. PloS one. 2010;5(11):e13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang HC, Samaniego F, Nair BC, Buonaguro L, and Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11(12):1421–31. [DOI] [PubMed] [Google Scholar]
- 72.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shivdasani RA. MicroRNAs: regulators of gene expression and cell differentiation. Blood. 2006;108(12):3646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sacco O, Romberger D, Rizzino A, Beckmann JD, Rennard SI, and Spurzem JR. Spontaneous production of transforming growth factor-beta 2 by primary cultures of bronchial epithelial cells. Effects on cell behavior in vitro. The Journal of Clinical Investigation. 1992;90(4):1379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tirado-Rodriguez B, Ortega E, Segura-Medina P, and Huerta-Yepez S. TGF- beta: an important mediator of allergic disease and a molecule with dual activity in cancer development. J Immunol Res. 2014;2014:318481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheppard D Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3(5):413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sultan S, Rozzi A, Gasparello J, Manicardi A, Corradini R, Papi C, et al. A Peptide Nucleic Acid (PNA) Masking the miR-145-5p Binding Site of the 3’UTR of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) mRNA Enhances CFTR Expression in Calu-3 Cells. Molecules. 2020;25(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cogoi S, Rapozzi V, and Xodo LE. Inhibition of gene expression by peptide nucleic acids in cultured cells. Nucleosides Nucleotides Nucleic Acids. 2003;22(5–8):1615–8. [DOI] [PubMed] [Google Scholar]
- 79.Fraser GL, Holmgren J, Clarke PB, and Wahlestedt C. Antisense inhibition of delta-opioid receptor gene function in vivo by peptide nucleic acids. Mol Pharmacol. 2000;57(4):725–31. [DOI] [PubMed] [Google Scholar]
- 80.Liu Y, Braasch DA, Nulf CJ, and Corey DR. Efficient and isoform-selective inhibition of cellular gene expression by peptide nucleic acids. Biochemistry. 2004;43(7):1921–7. [DOI] [PubMed] [Google Scholar]
- 81.Scarfi S, Giovine M, Pintus R, Millo E, Clavarino E, Pozzolini M, et al. Selective inhibition of inducible cyclo-oxygenase-2 expression by antisense peptide nucleic acids in intact murine macrophages. Biotechnol Appl Biochem. 2003;38(Pt 1):61–9. [DOI] [PubMed] [Google Scholar]
- 82.Turner JJ, Ivanova GD, Verbeure B, Williams D, Arzumanov AA, Abes S, et al. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent transactivation in cells. Nucleic Acids Res. 2005;33(21):6837–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guan L, Han Y, Zhu S, and Lin J. Application of CRISPR-Cas system in gene therapy: Pre-clinical progress in animal model. DNA Repair (Amst). 2016;46:1–8. [DOI] [PubMed] [Google Scholar]
- 84.Xue HY, Zhang X, Wang Y, Xiaojie L, Dai WJ, and Xu Y. In vivo gene therapy potentials of CRISPR-Cas9. Gene Ther. 2016;23(7):557–9. [DOI] [PubMed] [Google Scholar]
- 85.Zlotorynski E Genome engineering: NHEJ and CRISPR-Cas9 improve gene therapy. Nat Rev Mol Cell Biol. 2016;18(1):4. [DOI] [PubMed] [Google Scholar]
- 86.Hollywood JA, Lee CM, Scallan MF, and Harrison PT. Analysis of gene repair tracts from Cas9/gRNA double-stranded breaks in the human CFTR gene. Sci Rep. 2016;6:32230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.