

Abstract
Enveloped viruses, in general, have several transmembrane proteins and glycoproteins, which assist the virus in entry and attachment onto the host cells. These proteins also play a significant role in determining the shape and size of the newly formed virus particles. The lipid membrane and the embedded proteins affect each other in non-trivial ways during the course of the viral life cycle. Unraveling the nature of the protein–protein and protein–lipid interactions, under various environmental and physiological conditions, could therefore prove to be crucial in development of therapeutics. Here, we study the M protein of SARS-CoV-2 to understand the effect of temperature on the properties of the protein-membrane system. The membrane-embedded dimeric M proteins were studied using atomistic and coarse-grained molecular dynamics simulations at temperatures ranging between 10 and 50 °C. While temperature-induced fluctuations are expected to be monotonic, we observe a steady rise in the protein dynamics up to 40 °C, beyond which it surprisingly reverts back to the low-temperature behavior. Detailed investigation reveals disordering of the membrane lipids in the presence of the protein, which induces additional curvature around the transmembrane region. Coarse-grained simulations indicate temperature-dependent aggregation of M protein dimers. Our study clearly indicates that the dynamics of membrane lipids and integral M protein of SARS-CoV-2 enables it to better associate and aggregate only at a certain temperature range (i.e., ~ 30–40 °C). This can have important implications in the protein aggregation and subsequent viral budding/fission processes.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s00232-022-00244-y.
Keywords: Molecular dynamics, Aggregation, Pleomorphism, SARS-CoV-2, Membrane protein
Introduction
A deadlier form of severe acute respiratory syndrome (SARS) virus reappeared in 2019 and was named as SARS-CoV-2 (WHO 2021). The virus is highly contagious and detrimental to the respiratory tissues and ancillary organs of humans (Hu et al. 2021). The Covid19 pandemic caused by SARS-CoV-2 has so far resulted in a large number of fatalities and numerous casualties are still being reported around the world (WHO 2021; Hu et al. 2021). Similar to other coronaviruses, it encodes four major structural proteins; spike (S), membrane (M), envelope (E), and nucleocapsid (N). The S protein is the main interaction site for the human receptor, Angiotensin Converting Enzyme 2 (or ACE-2), the N protein associates with the RNA present in the viral genome, the M and E proteins are found on the lipid envelope and help in assembly and release (Hu et al. 2021; Mittal et al. 2020).
The viral M protein is one of the most abundant structural proteins distributed on the surface of the viral envelope. It associates with the S, E, and N proteins and directs the virus assembly and aids in the budding process (Schoeman and Fielding 2019). During synthesis, S, E, and N proteins are translated and inserted into the Endoplasmic Reticulum (ER) of the host cell, from where it is transferred to the Golgi complexes with the help of the M protein (Siu et al. 2008; Nguyen and Hogue 1997; Haan and Rottier 2005; Haan et al. 1999; Escors et al. 2001). The E and M proteins trigger the virion budding, where the nucleocapsid is already present in an enclosed form. The S proteins are subsequently incorporated on these virions. These large vesicles are then released from the host cells through exocytosis. Thus, the M proteins play a crucial role in the viral assembly and release (Schoeman and Fielding 2019; Wong and Saier 2021; Tseng et al. 2010). The co-expression of M and E proteins alone is sufficient to trigger the formation of virions as observed in insect cells (Vennema et al. 1996a; Baudoux et al. 1998; Bos et al. 1996). However, the majority of the research on SARS virus has so far focused exclusively on the S proteins (Padhi et al. 2021). While the extensive research has led to successful development of a number of effective vaccines, one of the fastest in medical history, our knowledge on the role and importance of other structural proteins and their interaction still remains inadequate. Only recently, there have been some noticeable attempts along this line: both toward modeling the other integral protein structures and assessing their validity in molecular simulation (Heo and Feig 2020; Monje-Galvan and Voth 2021; Mandala et al. 2020; Collins et al. 2021). In this work, we focus our attention on the integral M protein. We investigate the nature of protein–protein and protein–membrane interactions and investigate the effect of temperature on them.
An intriguing aspect of the viral particle formation process is the ability of the virus to exhibit pleomorphism. Pleomorphism is an example of extreme natural variations that allow changes in size and shape of enveloped Viruses such as influenza A virus or arenavirus (Stevenson and Biddle 1966; Harris et al. 2006; Hosaka et al. 1966; Murphy and Whitfield 1975). The coronaviruses are not an exception to this phenomenon (Murphy and Whitfield 1975; Neuman et al. 2006). However, the tolerance to pleomorphism depends on the structural proteins present on the viral envelope. Extreme conditions might result in failed assembly of virions or might lead to the formation of non-infectious viral particles. Although such variations (Schoeman and Fielding 2019) have been reported in the literature, very few studies have been carried out to understand how the overall virus structure is shaped (Stevenson and Biddle 1966; Harris et al. 2006; Hosaka et al. 1966; Murphy and Whitfield 1975; Neuman et al. 2006). Another important factor in the viral particle assembly is the number of M proteins in the membrane. It has been observed that a greater number of M proteins generate larger viral particles (Siu et al. 2008; Nguyen and Hogue 1997; Neuman et al. 2011a).
The coronavirus family M proteins share a high level of structural and functional similarity. The structure of M protein comprises a membrane-bound domain, an amino terminal, which is the ectodomain, and a carboxy terminal domain, which is toward the nucleocapsid or endodomain (Kuo et al. 2016). The M proteins largely accumulate in the Golgi apparatus or the ER (Wong and Saier 2021). Apart from associating with the N, S, and E proteins, the M protein is capable of associating with itself forming oligomers. For the development of a functional viral particle, the M protein should be able to successfully associate with the N and E proteins (Neuman et al. 2011a; Kuo et al. 2016). Hence, knowledge of the dynamics and behavior of the M protein is crucial in the understanding of the formation of the virus particle.
A distinct feature of the coronavirus envelope is its anionic nature (Basso et al. 2016; Wilsona et al. 2004; Verdiá-Báguena et al. 2012). The membrane in enveloped viruses enables them to sustain high environmental temperatures compared to that of non-enveloped viruses (Tuladhar et al. 2012). Reports by Polozov et al. show the existence of gel phase of lipids at temperatures below 22 °C, which is independent of the protein content in the membrane. This rubbery, gel-like state of the influenza lipid molecules on the envelope helps them withstand low temperatures and also helps in the airborne transmission of the virus (Polozov et. al. 2008). Although the basic characteristic of the phospholipid bilayer can be independent of the protein that is embedded in the viral lipid envelope, it is interesting to study how the lipid bilayer membrane and M protein together influence the size and shape of the virion particles at different temperatures.
The global pandemic caused by SARS-CoV-2 has been observed at varied temperature conditions, ranging from mere 8–10 °C in China to high ~ 40 °C in sub-tropical countries like India (Bukhari and Jameel 2020; Wu et al. 2020). Earlier studies have shown the influence of temperature on the structural properties of S proteins (Rath and Kumar 2020). Although extensive studies on drug development, mutation, and dynamics of SARS-CoV-2 protein are being carried out globally, it is important to understand how the architecture of the pleomorphic coronavirus, shaped by the M proteins, is affected by variations in temperature (Padhi et al. 2021; Cui et al. 2020). Toward this, we first investigate the properties of SARS-CoV-2 membrane protein embedded in a lipid bilayer using atomistic molecular dynamics (MD) simulations. Subsequently, we employ coarse-grained (CG) MD simulation to investigate the aggregation behavior of proteins on relatively larger membrane–protein systems. Our results suggest that the dynamics of the transmembrane domain of the M protein is mostly governed by the state of the lipid membrane, while the ectodomain exhibits distinct dynamics in the physiological temperature ranges (~ 30–40 °C). However, the presence of M protein does not seem to affect the nature of the model anionic membrane. Results from CG MD simulation suggest that the proteins cluster and form stable aggregates only beyond a certain temperature threshold, an observation that can be attributed to the gel phase of the membrane at lower temperatures. Furthermore, while a single M protein dimer can impart local deformations, clustering of M protein dimers does not seem to generate any large-scale deformation in the membrane, which indicates the possible role of other integral proteins along with M protein in the viral budding/fission process. The rest of the paper is organized as follows. We discuss the details of the atomistic and CG simulations in “Materials and Methods” section. In “Results and Discussion,” we discuss the important results from the study. We conclude in “Conclusion” section along with a brief discussion on the scope of the work and possible future directions.
Materials and Methods
Atomistic Simulations of an M Protein Dimer in a Model Anionic Membrane
The M protein model was obtained from the structures deposited by Feig’s lab (Heo and Feig 2020). In their work, the protein structure was predicted using inter-residue distance prediction-based modeling using trRosetta, where out of 10 predicted models, the lowest energy conformation was considered for subsequent studies. AlphaFold models were also used along with trRosetta models as initial structures for machine learning. This was followed by MD simulation-based refinement. In a recent study, Monje-Galvan and Voth (2021) have compared the two models of M protein from Feig’s lab based on their stability and the lipid sorting pattern around them. They found that while both the conformations are stable in water as well as in a complex bilayer, the lipid sorting pattern and the resulting membrane deformation profile around them are markedly different. In this study, we have used the open conformation model of the M protein (Heo and Feig 2020; Monje-Galvan and Voth 2021). In our study, a lipid bilayer membrane constituting DPPC and DPPG lipids in the ratio of 7:3, embedding a single M protein dimer, was built using CHARMM-GUI builder (Jo et al. 2008). The initial coordinates of the lipids were generated by the membrane builder tool of CHARMM-GUI. Water molecules and neutralizing ions (NaCl) were subsequently added to the system, such that the initial box was 10 × 10 × 13 nm in the X, Y, and Z directions, respectively. The total number of atoms in the system was 133,479, with 210 DPPC and 90 DPPG lipids, and 29,273 water molecules. The CHARMM36 force-field was used for protein and lipid molecules (Brooks et al. 2009) and TIP3P model for water (MacKerell et al. 1998). The assembled components were minimized and equilibrated to facilitate the individual components to relax and evolve during the production run. The input files for the initial equilibration were generated using CHARMM-GUI. Restraints are differentially added to the protein, membrane bilayer, water, and ions separately in each step of the equilibration, which included positional harmonic restraints to the protein backbone, sidechains, and ions; repulsive planar restraints to prevent water from entering the membrane and planar restraints to hold the lipid head groups in position along the Z axis with the buried lipid tails. The force on these restraints is gradually reduced in stepwise fashion (Sunhwan et al. 2008). Two initial NVT (constant volume, temperature) and four NPAT (constant pressure, area, temperature) equilibrations were performed before generating production trajectory. 0.5-µs-long production simulations were performed for subsequent analyses. The simulations were performed at five different temperatures, 10 °C, 20 °C, 30 °C, 40 °C, and 50 °C using GROMACS 5.0 molecular dynamics simulation software (Lee et al. 2016; Abraham et al. 2015).
Principal component analysis (PCA) transforms the inherent dynamics of protein from a high dimension to a low dimension by capturing the predominant modes of the protein dynamics. In PCA, a covariance matrix is built based on the protein atom coordinates and the matrix is subsequently diagonalized. Both the steps can be represented by using the following equations:
where xi and xj are the atomic coordinates, and the brackets indicate the ensemble average.
The symmetric covariance matrix C can be diagonalized by solving the eigenvalue problem using the following equation:
where A represents the eigenvectors, and λ represents the eigenvalues (David and Jacobs 2014; Rath and Senapati 2014). The eigenvectors indicate the directions of protein motion in configurational space and the corresponding eigen values indicate the mean square fluctuation in the motion along these vectors. The eigenvalues are ranked according to their contributions, where the first few eigenvectors are usually capable of capturing the majority of the protein dynamics (David and Jacobs 2014; Rath and Senapati 2014). In this work, PCA calculations were performed on the CA atoms of the full protein trajectories using Gromacs analysis tools, in order to capture the global free energy landscape of the protein. The distribution obtained from the first and second principal component was then used to calculate the free energy using the gsham module of GROMACS (Abraham et al. 2015). The energy is computed as − kBTln P(PC1, PC2), where P(PC1,PC2) is the distribution probability of the structures at different temperatures. When reporting errorbars, the average of two analysis windows and the related standard deviation were calculated.
Coarse-Grained Simulations of the M Proteins in Membrane
Coarse-grained (CG) simulations were carried out using Martini 2.2 (Qi et al. 2015) amino acid, Martini 2.0 (David and Jacobs 2014) lipids and non-polarizable water models (Hsu et al. 2017), and Gromacs 5.0 simulation package (Lee et al. 2016). We used the Martini Bilayer builder (Best et al. 2012) for generating two CG systems, embedding 8 and 64 dimeric M proteins, respectively, in a model bilayer. Subsequently, solvent (CG water) and neutralizing Na+ and Cl− ions were used to build bilayer-protein systems similar to the previous all-atom simulations. The box size for the 8-dimer system was 40 × 40 × 13.5 nm , and the total number of CG beads was 180,231, including 3423 DPPC and 1467 DPPG lipids, and 112,172 water beads. Similarly, for the 64-dimer system, the simulation box size was 85 × 85 × 13.5 nm and the total number of CG beads was 827,816, constituting 14,791 DPPC and 6339 DPPG lipids, and 493,141 water beads. The important system details are summarized in Table S1.
The all-atom simulations were carried out using CHARMM36 force-field (Jo et al. 2008), whereas that for CG simulations used Martini 2.2 (Qi et al. 2015). The parameters for proteins, lipids, and ions were prepared using the standard protocol (described earlier) of CHARMM-GUI for bilayer simulations (Klauda et al. 2010; Wu et al. 2014; Venable et al. 2013). After the initial minimization over 1 ns, equilibration was carried out in the NVT ensemble by gradually reducing the dihedral restraints. Subsequently, equilibration was carried out in the NPT ensemble. Production runs of 0.5 µs duration, in the NPT ensemble, were carried out in the temperature range between 10 and 50 °C (Table S1). All analyses were carried out using Gromacs analysis programs. Pymol (Lilkova, et al. 2015) and VMD (Humphrey et al. 1996) were used for visualization and generating structures. We use the open-source tool g_lomepro (Bhaskara et al. 2019) to compute the curvature profile of the lipid membrane, which is integrated as an analysis suit in Gromacs. This tool uses a grid-based algorithm [a modified version of GridMAT-MD (Allen et al. 2009)] to map the lipid atoms on a bilayer membrane and the embedded proteins to two 2D grids, corresponding to each leaflet in the bilayer. Spatial derivatives at each grid point of the surface are then calculated and used to compute the mean and Gaussian curvatures using geometric methods (Smith 2004; Lee 1997). Furthermore, spectral filtering is used to capture specific modes in membrane curvature. Here, we focus our attention on the mean curvature values. We also compute the area per lipid (APL) and thickness using g_lomepro.
Results and Discussion
The SARS-CoV-2 membrane (M) protein is 222-residue long and is embedded in the lipid bilayer. The N-terminal of the M protein is exposed to the external surface of the viral capsid and is in the form of a β sheet (Schoeman and Fielding 2019; Neuman et al. 2006). Previous studies have hinted toward a negatively charged viral lipid bilayer (Wilsona et al. 2004). Therefore, in this work, we have used a 7:3 ratio of DPPC: DPPG lipids which had also been used as pulmonary surfactant models and model membrane bilayers in earlier literatures (Morrow et al. 2007; Roy et al. 2014). While DPPC is a neutral lipid, DPPG carries a net negative charge. Similar composition of DPPC and DPPG has been earlier reported in pulmonary surfactants and hence used in this study as a model system (Allen et al. 2009). Although it is well known that the gel-to-crystalline transition temperature for both DPPC and DPPG lipids is around 41 °C (Lee 1997; Morrow et al. 2007; Riske et al. 2009) it is yet to be elucidated whether the presence of M protein in the anionic membrane modulates the relative temperature withstanding capabilities or brings about noticeable biophysical changes.
Individual SARS-CoV-2 Membrane proteins resemble a Greek amphora. Structurally, the M protein is organized into a small N-terminal ectodomain, three membrane spanning transmembrane regions and a large carboxy terminal endodomain (Fig. 1) (Murphy and Whitfield 1975). While the monomer–monomer interactions are reported to take place at the transmembrane region, M protein interactions with the S, E, and N proteins generally take place at the carboxy terminal domain. The M proteins are usually present as a dimer linked near the transmembrane region (Neuman et al. 2006, 2011a).
Fig. 1.

Structure of membrane-bound SARS-CoV-2 Membrane protein dimer. Monomers are shown in cyan and magenta in cartoon representation, lipid bilayers in CPK representation, and neutralizing sodium ions are shown as yellow vdW spheres
Two forms of the M protein have been reported (Neuman et al. 2006). The first form is in an elongated conformation, where the protein is capable of interacting with the nucleoprotein and imparting curvature to the membrane structure: reportedly 5–6 degrees per M protein dimer. The second compact form has very distinct boundaries and generally forms protein aggregates but does not help in the membrane curvature. It is possible that one form can be converted to another and vice versa under suitable environmental conditions (Neuman et al. 2006 Aug; Roy et al. 2014). The Feig lab (Heo and Feig 2020) has also reported two structures of the M protein: one based on the trRosetta method and a further refined structure using AlphaFold models. The two structures differ based on the orientation of the extra-membrane N-terminal region with respect to the transmembrane region. In this work, we use the former model of the M protein where the N-terminal domains remain in an open state.
To assess if the system has attained stability, we checked for the Root Mean Square Deviation (RMSD) of the full protein at the different temperatures over the simulation time (Fig. S1), which showed a slow drift over time during the simulation time. If only CA atoms are used and the terminal residues were excluded, the systems were observed to be largely stable after 150 ns (Fig. S2). The RMSDs at 30, 40, and 50 °C were found to be relatively higher than those at 10 and 20 °C. Next, we compared the Root Mean Square Fluctuation (RMSF) of the CA atoms of the protein at various temperatures (Fig. 2). The average RMSFs indicate that the protein dynamics remained overall consistent at different temperatures. Distinct peaks were found for residues 9–47 at temperatures 40 and 50 °C. The peak corresponds to the outermost part of the transmembrane helix toward the C-terminal. Comparatively lesser but noticeable changes were also observed for the remaining two helices lying close to the N-terminal. While the overall RMSFs were similar, solvent-exposed C-terminal loops around residues 180–190 and 203–220 displayed higher flexibility. Interestingly, the transmembrane helix flexibility is directly correlated to membrane properties such as diffusivity, fluidity, etc. (Kono 2002; Himeno et al. 2014).
Fig. 2.
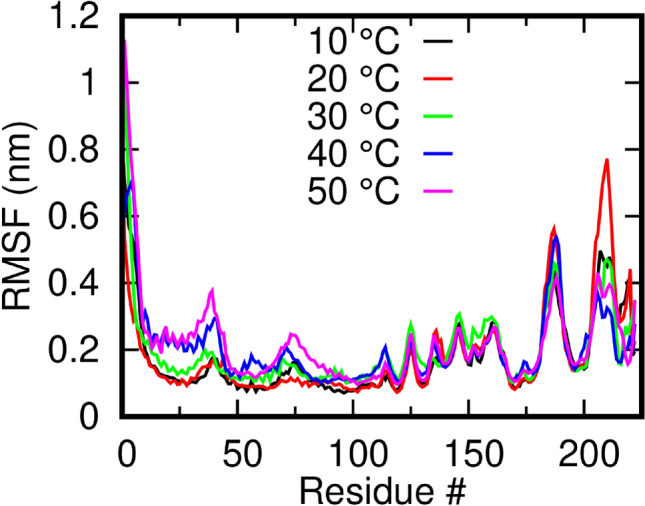
Effect of temperature on the Root Mean Squared Fluctuation (RMSF) of M Protein. RMSFs of the M protein monomers (mean of both dimers) at different temperatures indicating the overall stability of the systems
We access the overall temperature effects on the membrane–protein system in terms of (1) the structure and dynamics of a single M protein dimer, (2) the characteristics of the lipid membrane, (3) the ability of the M protein dimer in generating membrane curvature, and (4) the clustering behavior of multiple M protein dimers.
Effect of Temperature on Individual M Protein Dimers
To investigate the effect of temperature on the structure of the M protein dimer, we calculated the distribution of a pair of distances between the two monomeric units. The CA atoms of residue LYS15 and THR169 were considered as reference sites (see Fig. 3). LYS15 lies toward the end of the first helix and stays buried in the lipid membrane, while THR169 lies on the C-terminal β strands and is far from the influence of the membrane. The distance between the two reference sites on each monomeric unit was computed over the production trajectory at every temperature, and the 2-dimensional (2D) probability distribution of these two distances were calculated. In Fig. 3, we show these 2D distributions at various temperatures, where X-axis represent the distance between LYS15 sites (hereafter, referred as d15), Y-axis represent the distance between the THR169 sites (referred as d169) of the two monomers, and the colorbar indicate the probability. The ranges of both the axes and the colorbar are kept the same so as to better compare the distributions at various temperatures.
Fig. 3.

Effect of temperature on the inter-monomer distances in the extrinsic and intrinsic domains of the M protein. 2D distribution of inter-monomer distances between a set of reference sites in the two domains of the M protein at different temperatures, indicating the correlated dynamics of the two domains
The transmembrane part of the protein dimer is restricted in the gel-like lipid membrane at low temperature. With increasing temperature, the lipids in the membrane become more disordered (discussed below) and the flexibility of the transmembrane domain increases so as to allow it to sample different conformations. As evident, the 2D distribution at 10 °C is almost circular around a central high probability value of 3.2 nm and 3.05 nm along d15 and d169 axes, respectively. This indicates single narrow peak distributions along the individual axes, which might indicate a meta-stable conformation of the transmembrane domain, rather than a stable one, owing to the low temperature. We observe that d15 peaks at < 3.1 nm at 20 °C and around 2.9 nm at 30 and 40 °C. The distribution along d169, which changes very little between 10 and 30 °C (< 1 nm), becomes wide at 40 °C and beyond, indicating large thermal fluctuations of the extrinsic domains. At 50 °C, d15 peaks around < 3.1 nm (similar to that at 20 °C) with a shoulder at 2.9 nm (similar to the peak positions at 30 and 40 °C), and d169 peaks around > 2.9 nm. However, the distribution is quite broad owing to large thermal fluctuations and the peak is rather shallow as compared to those at other temperatures (see the color bar). The distribution at 50 °C is wide enough to cover all probable distances in d15 and d169 that are accessible at the lower temperatures. The motion of the protein subunits at high temperature, thus, seems to be governed by the thermal fluctuations rather than indicating any functional motion of the protein. On the other hand, the distribution at 30 °C seems to indicate a stable protein conformation with sharpest narrow peaks along both axes and a well-defined motion of the N-terminal domains at 40 °C. To further understand the implication of the increased thermal fluctuation in the functional motion of the various protein domains, we subsequently performed the PCA.
For getting more clarity of the dynamics, the part of the protein embedded in the membrane was not considered for the PC analysis (Fig. 4a). It was found that the first few eigenvectors were sufficient to describe the motions of the protein. Figure 4 shows the free energy landscape of the first two principal components using the CA atoms of the extrinsic domain of the M protein. The energy is computed as − kBTln P(PC1, PC2), where P(PC1,PC2) is the distribution probability of the structures at different temperatures. While the protein explored an extended phase space in (PC1, PC2) at 10, 20, and 30 °C, the minimum energy states were rather well defined at 40 and 50 °C. As observed from Fig. 4, the ∆G of the most populated conformation at 10, 20, 30, 40, and 50 °C was found to be 18.1, 16.6, 15.2, 20.2, and 19.1 kJ/mol, respectively. The time evolution of the first and second principal components [Fig. S4 (a, b)] showed frequent transitions in dynamics over the simulation time with increasing temperature. We compared the PC plots obtained from different time chunks during the last 50 ns of the simulation time, at 10 and 50 degrees, where the system is at equilibrium. Comparison of the ∆G values obtained from the PCA distribution along the first and second eigenvectors taken at different time intervals during the last 50 ns of the simulation shows an average ∆G value of 7.55 ± 0.86 kcal/mol and 6.69 ± 0.34 kcal/mol at 10 and 50 °C, respectively [Fig. S4 (c, d)], thus, implying consistent protein energetics. At 30 °C, along with a wide and shallow distribution of the conformations, the ∆G value was found to be the lowest. In contrast, a narrow distribution with a high ∆G value was seen for the protein at 40 °C, thus indicating a sharp transition from 30 to 40 °C. From the PCA plots, it is clear that at 30 °C, the extrinsic domain of protein is very dynamic. On the other hand, a stable conformation was observed at 40 °C that was different from the ones at 10, 20, and 50 °C, clearly indicating the difference in protein dynamics over the physiological temperature range. However, at higher temperatures, although the protein does not undergo any secondary structure change, the increased thermal motion might influence the dynamics of the extrinsic domain of the protein and correspondingly the free energy basins. We made a comparative study of the lowest energy conformation of the protein at the temperatures and observed that the flaps of the extrinsic domain were more open at 30 and 40 °C, confined at 10 and 20 °C and very close to each other at 50 °C (Fig. S3). In contrast to the extrinsic domain, the PCA calculation of the membrane-embedded region was very pronounced where one can observe the gradual increase in dynamics of the intrinsic region as the temperature increases. While at 10 °C the protein was confined, gradually its flexibility increases with temperature and it becomes extremely dynamic and flexible at 50 °C (Fig. S5). Such a gradual change can be attributed to the membrane interaction of the intrinsic region, which should also be affected by temperature. Therefore, it is worthwhile to investigate the effect of temperature on the lipid membrane that embeds the M protein.
Fig. 4.

Free Energy landscape of M protein from PCA. The free Energy landscape of a extrinsic domain of M protein (highlighted in red cartoon) at b 10 °C, c 20 °C, d 30 °C, e 40 °C, and f 50 °C. The red color in the colorbar depicts the most stable conformations of the protein. The free energy profile is the most extended at 30 °C and indicates a sharp transition from 30 to 40 °C
Effect of Temperature on the Embedding Lipid Membrane
Next, we investigated whether the lipid membrane, in the presence of the M protein, shows patterns similar to earlier observed anionic lipid membranes or does it have the inherent capacity to withstand high temperatures in the presence of protein as reported in the case of Influenza and other related viruses (Polozov et al. 2008). To monitor this, we computed for the lipid tail order parameter, also known as the deuterium order parameter, SCD, which measures the orientation and ordering of the membrane lipid tails with respect to the bilayer normal. The order parameters at different temperatures show the ordering of the lipid hydrocarbon tails. It is computed as the ensemble and time average of the second-order Legendre polynomial:
The order parameters at different temperatures show the ordering of the lipid hydrocarbon tails. SCD is measured in deuterium NMR experiments as
where θ represents the instantaneous angle between the CD bond and the bilayer normal. The angular brackets denote time and ensemble average. In simulation, θ is measured as the angle between the CH bond and the bilayer normal. The more the membrane is fluid-like, the lesser the tail ordering. The SCD values of the two lipid tails (averaged over all the lipids in the system and over the production trajectory) are shown in Fig. 5a as a function of backbone carbons. The average SCD values of the two hydrocarbon tails (averaged over all the tail atoms) are shown in Fig. 5b as a function of temperature. As expected, the SCD values decreased with temperature indicating increased disorderedness of the lipid tails. At 10 °C, the highest SCD value was found to be around 0.4 which decreased with increasing temperature becoming negligible at 50 °C. The transition temperature of DPPC and DPPG phospholipids from gel to a liquid crystalline state (Tc) has been found to be 41 °C (Himeno et al. 2014). The sharp decrease in the SCD values beyond 40 °C clearly captures this transition in our simulation. This also indicates that the presence of a single M protein dimer does not affect the intrinsic melting characteristics of the membrane lipids. However, to accurately assess the effect of multiple M protein dimers and precisely determine the transition temperature of these lipids in the presence of the M protein, one has to perform atomistic simulation of model membrane with multiple M protein dimers and sample the temperature space in closer intervals, which is beyond the scope of the current work.
Fig. 5.

Effect of temperature on lipid order. a Deuterium Order parameters (SCD) of the two lipid tails at various temperatures indicating the relative loss of ordering at high temperatures. b Average SCD values of the two tails as a function of temperature
While SCD values indicate the ordering of the individual lipids, the relative ordering of the lipids in the bilayer can be captured in terms of the APL and bilayer thickness. Figure 6 indicates the effect of temperature on these two quantities. It was found that the APL increases with temperature, while the thickness decreases. Similar to the trend observed in SCD, the change in APL and thickness is most prominent in the 40–50 °C window, again indicating the melting transition of the lipids. The experimentally reported APL value for pure DPPC bilayer at 50 °C is 0.64 nm2 (Nagle and Tristram-Nagle 1469; Wang et al. 1858; Neuman et al. 2011b), while that of DPPG at 50 °C is 0.67 nm2 (Pan et al. 2012; Andersen and Koeppe 2007). Thus, as expected, for a mixed DPPC/DPPG membrane with a transmembrane M protein dimer, the calculated APL values in our simulations are systematically larger than a pure DPPC bilayer (Leekumjorn and Sum 2006). Bilayer thickness of pure DPPG membrane at 50 °C is reported to be 3.55 nm (Andersen and Koeppe 2007). It is reported to be 3.43 nm for pure DPPC bilayer at 42 °C (Phillips et al. 2009) and 4 nm at 47 °C (Jesus and Allen 1828). Similar to APL, the observed thickness values in our simulation are systematically larger than the corresponding pure component lipid systems. The increase in thickness can be attributed to the presence of the transmembrane domain of the M protein dimers which, owing to the hydrophobic mismatch between the bilayer and the transmembrane region of the protein dimer, can locally increase the membrane thickness (Jesus and Allen 1828; Grau et al. 2017). Additionally, a larger bilayer thickness, in general, indicates more ordered lipids, and therefore, there is a possibility that the M protein dimers induce ordering in the neighborhood lipids. However, we have not explored this aspect of protein–membrane interaction in this study and plan to do that in a comparatively larger membrane.
Fig. 6.

Effect of temperature on the characteristics of lipid membrane. Change in a area per lipid and b local membrane thickness as a function of temperature
Next, we examine the ability of the M protein dimer in generating membrane deformation. The mean curvature profile of the atomistic membrane at 10, 30, and 50 °C is shown in Fig. 7 top panel. Here, we focus on the membrane leaflet that contains the N-terminal region of the M-dimer and the mean curvature is calculated w.r.t. the membrane mid-plane, i.e., a positive mean curvature indicates the membrane to bend outward, while a negative mean curvature indicates the membrane to bend inwards w.r.t. the mid-plane. The middle and the bottom panel show top and side view of representative membrane configuration along with the position of the protein dimer. The bilayer is represented as grid points based on which the curvature and thickness calculations are performed, where the grids correspond to the lipid head groups. The bilayer grids are colored based on the membrane thickness, where the color red represents lower membrane thickness and the color blue represents higher thickness values. The mean curvature and thickness profiles are calculated over the last 10 ns trajectory using g_lomepro. In the g_lomepro analysis, the curvature values are scaled by a factor of 100 for better representation, and a high q-filter value of 1 is used. We found the extrinsic N-terminal domain to impart a high negative mean curvature to the membrane, thus bending the membrane inwards. A similar observation of M protein-induced local membrane deformation has been made in another recent study (Collins et al. 2021).
Fig. 7.

Correlation between membrane curvature and thickness. The top panel indicates the curvature profile of the membrane at 10, 30, and 50 °C, with the colorbar representing the mean curvature values: yellow indicates negative and dark green indicates positive curvature. Middle panel: the corresponding thickness profile indicated in colorbar: red indicates thinner regions and blue indicates thicker region. Bottom panel: snapshots of the membrane–protein system with membrane color coded based on the bilayer thickness (same as middle panel). Disordering of lipids can be seen with increasing temperature
The magnitude of this bending is high at 10 and 30 °C, but almost half of that at 50 °C, which can be attributed to the increased disorderedness of the lipids and thermal fluctuations. Along with the high negative curvature at the dimer position, we also found other regions with negative and positive curvatures. The thick transmembrane region of the protein dimer comprises long helical domains that are bent w.r.t. the membrane normal (see Fig. 1). Such a structure, owing to the hydrophobic mismatch, can locally perturb the lipid ordering and generate curvature. A closer look at the membrane snapshot indicates locally disordered small lipid domains even at 10 and 30 °C, which in the absence of the protein is expected to be in a gel-like ordered state. Around these small regions, the membrane exhibits lower thickness and thus seems bent inwards (negative mean curvature). The regions with ordered lipids exhibit high thickness and thus a positive mean curvature. Due to large thermal fluctuations, the lipid membrane is fully disordered at 50 °C, and the variation in mean curvature and thickness profile is less drastic as compared to the low-temperature case. The extent of membrane deformation in our study is comparable to that reported in another recent study (Collins et al. 2021). However, we must add that our system size is rather small to rule out any finite size effects, and therefore, we analyzed the mean curvature in the presence of multiple M dimers in CG simulation (discussed later).
Effect of Temperature on Protein Aggregation
Considering the pleomorphic nature of the Coronaviruses, the conformation of the M protein and the dynamics of the membrane lipids at different temperatures might impact the shape of the virions formed. Previous studies have reported that under confinement, the M proteins are more prone to aggregation (Hemati et al. 2021). To understand if temperature influences M protein aggregation, CG MD simulations were performed using MARTINI 22 on a larger system consisting of 8 membrane protein dimers (16 monomers) embedded on a lipid membrane (see “Materials and Methods” section for technical details, see next section for model validation). The final conformation of the proteins at each temperature is shown in Fig. 8. At 10 and 20 °C, we did not observe any aggregates within the simulation time scale. This could be attributed to the high degree of ordering of the membrane lipids as shown in Fig. 8b. At 30 °C, formation of M-dimer clusters was observed within 100 ns time period, further aggregation took place within 300 ns beyond which no more protein clusters could be observed. At 40 °C, the proteins started forming clusters within the initial few nanoseconds after which steady aggregations took place at 200 ns and 250 ns, respectively. Barring two dimers, all the remaining protein dimers aggregated to form a single cluster comprising 12 proteins. At 50 °C, a very fast aggregation pattern was observed and two separate clusters could be noticed.
Fig. 8.

Effect of temperature of aggregation of M proteins. CG simulations of eight M protein dimers (magenta) embedded in a lipid membrane (green) at various temperatures, indicating the clustering pattern of the proteins at various temperatures a top view, b side view. Note the relative ordering/disordering in lipids in (b) as a function of temperature
The rate of formation of aggregates directly depends on the ordering of the membrane, which allows migration of protein molecules. At 50 °C, due to the large disordering of membrane lipids, the aggregates were able to form very quickly (Table S1) (Fig. 9). This also implies that irrespective of the noticeable conformational flexibility of the M protein at 10 and 20 °C (Fig. 3), the formation of protein clusters is largely governed by the ordering of the membrane. In conclusion, we observe that clustering of M protein dimers readily occurs only beyond 30 °C.
Fig. 9.
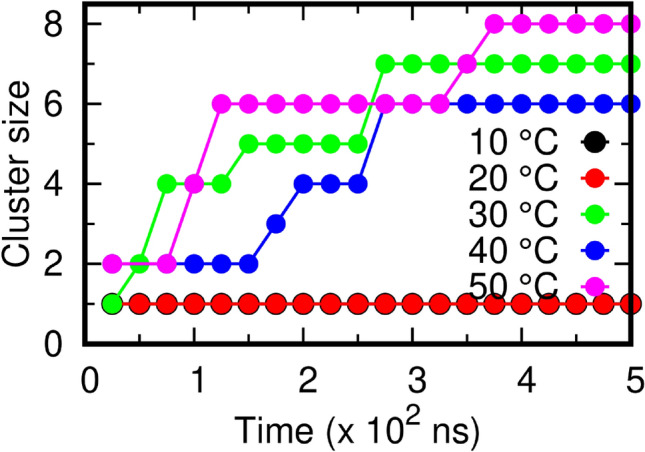
The effect of temperature on the dynamics of protein aggregation. The total number of protein clusters formed at different temperatures during the 500 ns CG simulations
To further clarify our observations on clustering of M protein dimers and the resulting membrane deformation, we carried out CG simulation involving 64 dimers on a lipid bilayer. We compared the RMSD and RMSF of M protein dimers in CG and atomistic simulations at 30 °C in Fig. S6 (a, b), which agree well with each other. The Radius of gyration (Rg) of the M proteins [Fig. S6 (c)] was found to be around 2.7 nm, in agreement with the observations by Collins et al. in atomistic simulations (Collins et al. 2021). In this part of the work, we specifically focused on the clustering behavior at 30 and 40 °C and the resulting membrane curvature. We show the final membrane conformation and the resulting mean curvature profile in Fig. 10.
Fig. 10.

The effect of M protein aggregation on membrane curvature. The top panel shows the top and side views of representative membrane snapshots with 128 M protein monomers at 30 °C and 40 °C. The bottom panel shows the corresponding mean curvature of the membrane indicating prominent local curvatures
While we could not observe all the dimers clustering to form a single large cluster during the time scales of the simulation, the effect of the partially formed clusters on the membrane curvature was evident. We did not observe any large-scale membrane deformation; however, the presence of such transient clusters could impart noticeable local curvature. As reported by Monje-Galvan and Voth (2021) and Collins et al. (2021), M protein alone can induce membrane deformation around it and we too observed the same in our atomistic simulation. The observations in our CG simulation, additionally, indicate that a large number of M proteins can cluster and collectively impart a local curvature to the membrane. Without such a collective behavior, there would be small deformation around each protein in which case, one would observe transverse fluctuation in the membrane, but no noticeable local deformation. However, as mentioned earlier, this deformation itself is rather small. This observation suggests that M protein alone may not be capable of generating large curvatures that can trigger budding/fission events and other integral proteins might play a crucial role, as shown in earlier studies (Vennema et al. 1996b; Mortola and Roy 2004). However, owing to their aggregation capability, the M protein might facilitate the curvature generating process. We will focus on investigating this collective mechanism in our future work.
Conclusion
Out of the four different structural proteins of the SARS-CoV-2 virus, the M protein is the most abundant and widely distributed. It associates with the S, E, and N protein and also with other M proteins and directs the virus assembly and aids in the viral release. The pleomorphic nature of the coronaviruses allows them to change the particle size and shape, which also depend on the structural proteins of the SARS-CoV-2, especially the M protein. Hence, understanding the dynamics and behavior of the M protein is very important to understand the formation of the virus particle. Although the variations have been reported in the literature, very few studies have been carried out to understand how the overall virus structure is shaped.
We performed atomistic and coarse-grained (CG) simulation on ionic DPPC/DPPG lipid membranes embedding M protein dimers to understand the effect of temperature on the behavior of both the protein and the membrane, and their mutual interactions. The atomistic simulation on single M protein dimers indicated that the conformation and dynamics of the M protein depend strongly on temperature, as expected. As both DPPC and DPPG lipids exhibit a transition temperature of 41 °C, the transmembrane domain was found to be flexible between 30 and 50 °C, where the transmembrane and extrinsic domain of the proteins could sample various conformations. PCA performed on the extrinsic N-terminal domain of the protein dimer further indicated that the free energy landscape of the dimer goes through significant change over this temperature range. The energy landscape at 40 °C was found to exhibit some well-defined basins indicating the possibility of functionally important dynamics that occurs over the physiological temperature range. The thermal motions of a protein are influenced by external conditions such as temperatures and vicinity with adjoining molecules, but the functional motion of M Protein can be described as the functionally active forms such as short-bent or long-extended conformations. The long-extended structures are more prone to bind to other membrane proteins but the short-bend forms do not associate with other protein molecules. While we were able to partly observe the long-extended conformation at 40 °C via the Thr169-169′ and Lys15-Lys15′ distribution, it was clear with the PCA calculations.
While the structure and ordered/disorderedness of lipids seem to play a crucial role in dictating the structure and dynamics of the embedded protein dimer, the overall nature of the lipid membrane was not much affected by the presence of the protein. The order parameter, APL, and bilayer thickness all showed signatures of the transition temperature beyond 40 °C. However, we could observe the overall bilayer thickness to be affected by the protein, which can be attributed to the hydrophobic mismatch between the lipid bilayer and the protein transmembrane region. We also observed some interesting interplay of protein–membrane interaction in the mean curvature profile of the membrane. While the protein locally induced negative mean curvature, it also induced appreciable positive and negative curvature around it. With increasing temperature, these fluctuations die down. Our observations, thus, suggest that the protein might lead to ordering/disordering of the neighborhood lipids, which further induces local curvature. However, to characterize such ordering, a systematic analysis of the lipid dynamics has to be performed in order to identify local ordered/disordered regions and their correlation with the protein. Moreover, our system might be small to clearly capture any large-scale undulations and finite size effects might also have some effects. Finally, while the PCA and statistical distributions indicate important qualitative trends, quantitative predictions would require much longer simulations over multiple replicas, which is beyond the scope of the present work. The qualitative trends, however, should remain comparable.
To better understand the effect of temperature on protein clustering and the resulting membrane curvature, we performed CG simulations. Simulations on 8 protein dimers indicated that protein clustering happens only at 30 °C and beyond, and the rate of clustering increases with increasing temperature. This again should be attributed to the gel-like ordered phase of the lipid membrane. Simulations on 64 protein dimers at 30 and 40 °C further indicated the effect of protein clustering on membrane curvature. While the dimers failed to form a single large cluster during the simulation time scales, the partially formed clusters could induce appreciable local membrane curvature. However, the scale of the deformation was far from that expected in a case where one can expect budding. As M proteins of SARS virus are implicated in virus budding, it was unclear if M protein alone is capable of leading to such large-scale deformation. In our study, we find this to be not feasible, which agrees with the earlier studies on viral particle formation (Vennema et al. 1996b; Mortola and Roy 2004). It will, therefore, be interesting to investigate the deformation profile of lipid membrane in the presence of both M and E proteins.
As we observe the most interesting changes in the protein–membrane system happening close to the physiological temperature range, we believe that the clustering of M protein dimers and the resulting change in membrane curvature might be an important factor in the viral activity. However, as the viral membrane consists of various other proteins as well, the behavior of M protein dimers in the presence of other proteins can provide crucial information on their mutual role in the viral life cycle. Recently, such a study has been performed (Collins et al. 2021), which aims at understanding the role of M and E proteins in generating membrane curvature. They found the M proteins to cooperatively generate membrane curvature, while E protein kept the membrane relatively flat. An important aspect in the membrane–protein study is the model membrane which should closely represent the viral membrane. In this line, the study by Monje-Galvan and Voth (2021) shows the difference in the lipid sorting pattern and the membrane deformation profile around different structures of the M protein. Along this line, it will be interesting to extend our study to understand the effect of temperature on cooperative protein–protein and protein–membrane interactions on complex model membranes in the presence of both M and E proteins. Although, the selection of the number of M proteins in this study was relatively subjective in this study, considering the exact M protein density in the viral envelope would provide more accurate insights into the protein dynamics. Additionally, CG MD simulation can enable the study of large-scale protein–membrane systems, which can provide crucial information on the aggregation behavior of the proteins and their effect on the lipid membrane.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
SLR thanks NIT Warangal for research seed grant (P1131) and National Energy Research Scientific Computing Center of the Ernest Orlando Lawrence Berkeley National Laboratory, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and the Extreme Science and Engineering Discovery Environment (XSEDE). We are grateful to the Covid19 HPC Consortium for providing resources and helping researchers work for a noble cause. We would like to thank Dr. Shikha Prakash and Dr. Durba Sengupta for their useful suggestions in g_lomepro analysis.
Author Contributions
SLR designed research; SLR and MT performed research; SLR, MT, and NM analyzed data; and SLR, MT, and NM wrote the paper.
Declarations
Conflict of interest
The authors declare no competing financial interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Soumya Lipsa Rath, Email: slrath@nitw.ac.in.
Madhusmita Tripathy, Email: tripathy@cpc.tu-darmstadt.de.
References
- Abraham M, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Allen WJ, Lemkul JA, Bevan DR. GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J Comput Chem. 2009;30(12):1952–1958. doi: 10.1002/jcc.21172. [DOI] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE., II Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Basso L, Vicente E, Crusca E, Jr., et al. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci Rep. 2016;6:37131. doi: 10.1038/srep37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoux P, Carrat C, Besnardeau L, Charley B, Laude A. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J Virol. 1998;72(11):8636–8643. doi: 10.1128/JVI.72.11.8636-8643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ (1) and χ (2) dihedral angles. J Chem Theory Comput. 2012;8(9):3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara RM, Grumati P, Garcia-Padro J, Kalayil S, Covarrubais-Pinto A, Chen W, Kudryashev M, Dikic I, Hummer G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective-ER phagy. Nat Commun. 2019;10(1):2370. doi: 10.1038/s41467-019-10345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos EC, Luytjes W, Van Der Meulen HV, Koerten HK, Spaan WJ. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology. 1996;218(1):52–60. doi: 10.1006/viro.1996.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, III, MacKerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari Q, Jameel Y. Will coronavirus pandemic diminish by summer? SSRN Electron J. 2020 doi: 10.2139/ssrn.3556998. [DOI] [Google Scholar]
- Collins LT, Elkholy T, Mubin S, Hill D, Williams R, Ezike K, Singhal A. Elucidation of SARS-CoV-2 budding mechanisms through molecular dynamics simulations of M and E protein complexes. BioRxiv. 2021 doi: 10.1101/2021.07.26.453874. [DOI] [PubMed] [Google Scholar]
- Cui W, Yang K, Yang H. Recent progress in the drug development targeting SARS-CoV-2 main protease as treatment for COVID-19. Front Mol Biosci. 2020 doi: 10.3389/fmolb.2020.616341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan CA, Rottier PJ. Molecular interactions in the assembly of coronaviruses. Adv Virus Res. 2005;64:165–230. doi: 10.1016/S0065-3527(05)64006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus AJ, Allen TW. The determinants of hydrophobic mismatch response for transmembrane helices. Biochim Biophys Acta BBA Biomembr. 2013;1828(2):851–863. doi: 10.1016/j.bbamem.2012.09.012. [DOI] [PubMed] [Google Scholar]
- de Haan CA, Smeets M, Vernooij F, Vennema H, Rottier PJ. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J Virol. 1999;73(9):7441–7452. doi: 10.1128/JVI.73.9.7441-7452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D, Ortego J, Laude H, Enjuanes L. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J Virol. 2001 doi: 10.1128/JVI.75.3.1312-1324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau B, Javanainen M, García-Murria MJ, et al. The role of hydrophobic matching on transmembrane helix packing in cells. Cell Stress. 2017;1(2):90–106. doi: 10.15698/cst2017.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci USA. 2006;103(50):19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemati M, Soosanabadi M, Ghorashi T, Ghaffari H, Vahedi A, Sabbaghian E, Rasouli Nejad Z, Salati A, Danaei N, Kokhaei P. Thermal inactivation of COVID-19 specimens improves RNA quality and quantity. J Cell Physiol. 2021;236(7):4966–4972. doi: 10.1002/jcp.30206. [DOI] [PubMed] [Google Scholar]
- Heo L, Feig M. Modeling of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) proteins by machine learning and physics-based refinement. BioRxiv. 2020 doi: 10.1101/2020.03.25.008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Shimokawa N, Komura S, et al. Charge-induced phase separation in lipid membranes. Soft Matter. 2014;10:7959–7967. doi: 10.1039/C4SM01089B. [DOI] [PubMed] [Google Scholar]
- Hosaka Y, Kitano H, Ikeguchi S. Studies on the pleomorphism of HVJ virions. Virology. 1966;29(2):205–221. doi: 10.1016/0042-6822(66)90027-4. [DOI] [PubMed] [Google Scholar]
- Hsu P-C, Bruininks BMH, Jefferies D, Souza PCT, Lee J, Patel DS, Marrink SJ, Qi Y, Khalid S, Im W. CHARMM-GUI martini maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J Comput Chem. 2017;38(27):2354–2363. doi: 10.1002/jcc.24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Guo H, Zhou P, et al. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD—visual molecular dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K. Thermosensitive polymer-modified liposomes. Adv Drug Deliv Rev. 2002;53(3):307–319. doi: 10.1016/S0169-409X(01)00204-6. [DOI] [PubMed] [Google Scholar]
- Kuo L, Hurst-Hess KR, Koetzner CA, Masters PS. Analyses of coronavirus assembly interactions with interspecies membrane and nucleocapsid protein chimeras. J Virol. 2016;90(9):4357–4368. doi: 10.1128/JVI.03212-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM. Riemannian manifolds: an introduction to curvature. New York: Springer; 1997. [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, III, MacKerell AD, Jr., Klauda JB, Im W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016;12(1):405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leekumjorn S, Sum AK. Molecular simulation study of structural and dynamic properties of mixed DPPC/DPPE bilayers. Biophys J. 2006;90(11):3951–3965. doi: 10.1529/biophysj.105.076596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilkova E, et al. The PyMOL molecular graphics system, version 2.0. New York: Schrödinger & LLC; 2015. [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mandala VS, McKay MJ, Shcherbakov AA, Dregni AJ, Kolocouris A, Hong M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat Struct Mol Biol. 2020;27(12):1202–1208. doi: 10.1038/s41594-020-00536-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Manjunath K, Ranjan RK, Kaushik S, Kumar S, Verma V. COVID-19 pandemic: insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020;16(8):e1008762. doi: 10.1371/journal.ppat.1008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje-Galvan V, Voth GA. Molecular interactions of the M and E integral membrane proteins of SARS-CoV-2. Faraday Discuss. 2021 doi: 10.1039/D1FD00031D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow MR, Temple S, Stewart J, Keough KMW. Comparison of DPPC and DPPG environments in pulmonary surfactant models. Biophys J. 2007;93(1):164–175. doi: 10.1529/biophysj.106.102681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortola E, Roy P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004 doi: 10.1016/j.febslet.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FA, Whitfield SG. Morphology and morphogenesis of arenaviruses. Bull World Health Organ. 1975;52(4–6):409–419. [PMC free article] [PubMed] [Google Scholar]
- Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. Biochim Biophys Acta (BBA) Rev Biomembr. 2000;1469(3):159–195. doi: 10.1016/S0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman BW, Adair BD, Yoshioka C, et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol. 2006;80(16):7918–7928. doi: 10.1128/JVI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman BW, Kiss G, Kunding AH, Bhella D, Fazil Baksh M, Connelly S, Droese B, Klaus JP, Makino S, Sawicki SG, Siddell SG, Stamou DG, Wilson IA, Kuhn P, Buchmeierg MJ. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. 2011;174(1):11–22. doi: 10.1016/j.jsb.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman BW, Kiss G, Kunding AH, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. 2011;174(1):11–22. doi: 10.1016/j.jsb.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VP, Hogue BG. Protein interactions during coronavirus assembly. J Virol. 1997;71(12):9278–9284. doi: 10.1128/JVI.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A, Rath S, Tripathi T. Accelerating COVID-19 research using molecular dynamics simulation. J Phys Chem B. 2021;125(32):9078–9091. doi: 10.1021/acs.jpcb.1c04556. [DOI] [PubMed] [Google Scholar]
- Pan J, Heberle FA, Tristram-Nagle S, Szymanski M, Koepfinger M, Katsaras J, Kučerka N. Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering. Biochim Biophys Acta BBA Biomembr. 2012;188(9):2135–2148. doi: 10.1016/j.bbamem.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Ursell T, Wiggins P, et al. Emerging roles for lipids in shaping membrane–protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polozov IV, Bezrukov L, Gawrisch K, Zimmerberg J. Progressive ordering with decreasing temperature of the phospholipids of influenza virus. Nat Chem Biol. 2008;4(4):248–255. doi: 10.1038/nchembio.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Ingólfsson HI, Cheng X, Lee J, Marrink SJ, Im W. CHARMM-GUI martini maker for coarse-grained simulations with the martini force field. J Chem Theory Comput. 2015;11:4486–4494. doi: 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
- Rath S, Kumar K. Investigation of the effect of temperature on the structure of SARS-CoV-2 spike protein by molecular dynamics simulations. Front Mol Biosci. 2020 doi: 10.3389/fmolb.2020.583523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath SL, Senapati S. Why are the truncated cyclin Es more effective CDK2 activators than the full-length isoforms? Biochemistry. 2014;53(28):4612–4624. doi: 10.1021/bi5004052. [DOI] [PubMed] [Google Scholar]
- Riske KA, Barroso RP, Vequi-Suplicy CC, Germano R, Henriques VB, Teresa Lamy M. Lipid bilayer pre-transition as the beginning of the melting process. Biochim Biophys Acta (BBA)—Biomembr. 2009;1788(5):954–963. doi: 10.1016/j.bbamem.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Roy B, Panda AK, Parimi S, Ametov I, Barnes T, Prestidge CA. Physico-chemical studies on the interaction of dendrimers with lipid bilayers. 1. Effect of dendrimer generation and liposome surface charge. J Oleo Sci. 2014;63(11):1185–1193. doi: 10.5650/jos.ess14081. [DOI] [PubMed] [Google Scholar]
- Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, Tsao SW, Nicholls JM, Altmeyer R, Peiris JSM, Bruzzone R, Nal B. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J Virol. 2008;82(22):11318–11330. doi: 10.1128/JVI.01052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JP (2004) Robust geometric methods for surface modeling and manufacturing. Ph.D. thesis, University of California, Berkeley
- Stevenson JP, Biddle F. Pleomorphism of influenza virus particles under the electron microscope. Nature. 1966;212(5062):619–621. doi: 10.1038/212619a0. [DOI] [PubMed] [Google Scholar]
- Sunhwan J, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Tseng Y-T, Wang S-M, Huang K-J, Lee AI-R, Chiang C-C, Wang C-T. Self-assembly of severe acute respiratory syndrome coronavirus membrane protein. J Biol Chem. 2010;285(17):12862–12872. doi: 10.1074/jbc.M109.030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuladhar E, Bouwknegt M, Zwietering MH, Koopmans M, Duizer E. Thermal stability of structurally different viruses with proven or potential relevance to food safety. J Appl Microbiol. 2012;112(5):1050–1057. doi: 10.1111/j.1365-2672.2012.05282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable RM, Luo Y, Gawrisch K, Roux B, Pastor RW. Simulations of anionic lipid membranes: development of interaction-specific ion parameters and validation using NMR data. J Phys Chem B. 2013;117(35):10183–10192. doi: 10.1021/jp401512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H, Godeke G-J, Rossen JWA, Voorhout WF, Horzinek MC, Opstelten D-JE, Rottier PJM. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996;15(8):2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H, Godeke GJ, Rossen JW, Voorhout WF, Horzinek MC, Opstelten DJ, Rottier PJ. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996 doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdiá-Báguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Torres J, Aguilella VM, Enjuanes L. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology. 2012;432(2):485–494. doi: 10.1016/j.virol.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gkeka P, Fuchs JE, Liedl KR, Cournia Z. DPPC-cholesterol phase diagram using coarse-grained molecular dynamics simulations. Biochim Biophys Acta BBA Biomembr. 2016;1858(11):2846–2857. doi: 10.1016/j.bbamem.2016.08.005. [DOI] [PubMed] [Google Scholar]
- WHO (2021)
- Wilson L, Mckinlay C, Gage P, Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330(1):322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong NA, Saier MH., Jr. The SARS-coronavirus infection cycle: a survey of viral membrane proteins, their functional interactions and pathogenesis. Int J Mol Sci. 2021;22(3):1308. doi: 10.3390/ijms22031308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J Comput Chem. 2014;35(27):1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Jing W, Liu J, Ma Q, Yuan J, Wang Y, et al. Effects of temperature and humidity on the daily new cases and new deaths of COVID-19 in 166 countries. Sci Total Environ. 2020;729:139051. doi: 10.1016/j.scitotenv.2020.13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.