

Abstract
Epitopes are portions of a protein that are recognized by antibodies. These small amino acid sequences represent a significant breakthrough in a branch of bioinformatics called immunoinformatics. Various software are available for linear B-cell epitope (BCE) prediction such as ABCPred, SVMTrip, EpiDope, and EpitopeVec; a well-known BCE predictor is BepiPred-2.0. However, despite the prediction, there are several essential steps, such as epitope assembly, evaluation, and searching for epitopes in other proteomes. Here, we present EpiBuilder (https://epibuilder.sourceforge.io), a user friendly software that assists in epitope assembly, classifying and searching using input results of BepiPred-2.0. EpiBuilder generates several output results from these data and supports a proteome-wide processing approach. In addition, this software provides the following features: Chou & Fasman beta-turn prediction, Emini surface accessibility prediction, Karplus and Schulz flexibility prediction, Kolaskar and Tongaonkar antigenicity, Parker hydrophilicity prediction, N-glycosylation domains, and hydropathy. These information generate a unique topology for each epitope, visually demonstrating its characteristics. The software can search the entire epitope sequence in various FASTA files, and it allows to use BLASTP to identify epitopes that eventually have sequence variations. As an EpiBuilder application, we developed a epitope dataset from the protozoan Trypanosoma brucei gambiense, the gram-positive bacterium Clostridioides difficile, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Keywords: Immunoinformatics, BepiPred-2.0, Galaxy
Introduction
Immune responses are divided into innate and adaptive responses. The adaptive immune response appears as a response to a large number of substances and adapts to them. It is characterized by specificity (ability to distinguish different substances) and memory (ability to respond more vigorously to repeated infection). The components of adaptive immunity are lymphocytes and their secretions, including antibodies or immunoglobulins (secreted by B lymphocytes or B cells). 1
Secreted antibodies recognize a specific part of the antigen, called an epitope, 2 bind together, neutralize it (preventing it from infecting cells), or mark it for elimination through, for example, phagocytosis, activation of the classical complement pathway, and recognition by eosinophils of immunoglobulin E bound to the pathogen followed by the release of granules harmful to the invading microorganism.
Antibodies are the only components of adaptive immunity that prevent infections. Therefore, the production of potent antibodies is the objective of vaccination. 1 The identification of B-cell epitopes (BCEs) is of great importance for the development of vaccines, immunodiagnostics, and therapeutic antibodies. 2
Experimental methods to identify BCEs in vivo and in vitro are expensive and time-consuming. Therefore, new computational methods must be developed to rapidly identify potential epitopes of B cells. 3
Currently, immunoinformatics is a promising tool in the study of several organisms, including viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV2)4,5; protozoa such as Toxoplasma gondii, 6 Trypanosoma brucei brucei, 7 and Leishmania infantum 8 ; bacteria such as Leptospira interrogans 9 ; and even cancer cells. 10
Several immunoinformatics tools have been developed for the identification of BCEs, such as ABCPred, 11 SVMTrip, 12 EpiDope, 13 and EpitopeVec, 14 one of the most popular being BepiPred-2.0. In addition, the Epitope Prediction and Analysis Tools (http://tools.iedb.org/bcell/) provide several methods for analyzing individual characteristics, such as Chou & Fasman beta-turn, 15 Emini surface accessibility, 16 Karplus and Schulz flexibility, 17 Kolaskar and Tongaonkar antigenicity, 18 and Parker hydrophilicity. 19 Despite this, such tools do not have integration, generating independent results that need to be generated individually, which makes the task expensive for analysis with large volumes of proteins.
In this regard, we present EpiBuilder, a tool capable of assembling BCEs from data generated by BepiPred-2.0, and from these data, the tool assists the researcher in several types of experiments, including those on a proteome-wide scale.
Materials and Methods
EpiBuilder was developed using the Java programming language (supported by version 1.8) with a graphical user interface (GUI) and command-line options, without any additional prerequisite to perform all functions except BLASTP if this is set. EpiBuilder is prepared to run on any operating system that supports Java technology, such as Windows, MacOS, and Unix-based systems.
In addition to the aforementioned versions, we developed an EpiBuilder version with a GUI for the Galaxy platform. 20 The interfaces were developed and validated using the Planemo (https://planemo.readthedocs.io) program, following the recommendations of good galaxy tool development practices.
Galaxy is an open-source framework that aims to facilitate the reproducibility of bioinformatic analyses and access tools for various types of analysis. 20 The platform allows the user to adapt any program that runs along the command line to a graphical interface. Open servers (https://usegalaxy.org) make analytics more accessible when dealing with large volumes of data, and tools and interfaces are shared with the entire community through the Galaxy ToolShed repository. 21 The repository extends the Galaxy service for developers to create and publish new tools or adaptations of the existing programs.
The GUI and terminal mode executable files, in addition to the source code, are available at https://epibuilder.sourceforge.io. The EpiBuilder galaxy version is available in ToolShed under the name EpiBuilder.
Input files
There are 3 primary forms of system inputs: the first is the CSV file (comma separated values) generated by BepiPred-2.0 22 and the second is the result generated by BepiPred-2.0 through the output of the B-Cell Standalone IEDB program (http://tools.iedb.org/bcell/). 23 The third option allows the user to input a job id (the number used to run BepiPred-2.0 online), and after the process is finished, the file can be downloaded from the BepiPred-2.0 server.
Processing
The file generated by BepiPred-2.0 22 is uploaded to EpiBuilder, the proteins are reassembled from these data, and the scores are calculated for the following features: Emini surface accessibility, 16 Parker hydrophilicity, 19 Chou & Fasman beta-turn, 15 Karplus & Schulz flexibility, 17 and Kolaskar and Tongaonkar. 18 These scores are calculated internally by EpiBuilder, and such calculations have been rewritten in Java based on the IEDB B-Cell Standalone Python script. In addition, the molecular mass (MW), isoelectric point (pI), and hydropathy index of each amino acid are calculated using the BioJava library. 24
The epitopes are mounted from the threshold of BepiPred-2.0 (default value of 0.6). That is, if the amino acid score is greater than or equal to the cutoff point, it will be concatenated with the following amino acid with a score greater than or equal to the cutoff point. When an amino acid with a score less than the cutoff point is identified, the process reserves the generated epitope. Then, these epitopes are filtered so that only those between the minimum (default value 10) and maximum (default value 30) remain. Next, the presence of the N-glycosylation site is verified by searching for the Asn-Xaa-(Ser/Thr) domain, where Xaa is any amino acid except Proline. 25
Topology
For each identified epitope, an individual analysis of amino acids is performed for the features chosen by the user. If the amino acid has a score equal to or above the threshold chosen for the feature, and the threshold can be calculated (default) or informed by the user, the current amino acid in question receives a representative character “E” when the assigned value is lower.
Finally, a structure called All Matches is generated, which includes the amino acids with a score above or equal to the cutoff point for all selected functionalities; thus, it is possible to evaluate the specific stretches in which there is consensus in the analyzed structures. All Matches also receive a coverage value relative to the epitope.
Along with these excerpts of the topology, the N-glycosylation sites are also informed through the character “E”. If the amino acid has a hydropathy index less than 0, it is assigned the character “–”, and if it is greater than or equal to 0, it is assigned the character. The case of epitope topology PSSHPAPQQQQAYYQ extracted from the tbg972.9.8930 protein of Trypanosoma brucei gambiense reference proteome (strain TREU927, TriTrypdb version 49) is shown in Figure 1.
Figure 1.

Topology generated by EpiBuilder, the epitope is individually analyzed with visual presentation of its features, besides presenting the element All Matches, where the amino acid is above the cutoff point in all selected methods. The topology also presents the sites of N-glycosylation and also hydropathy.
For each feature, the number of amino acids above the cutoff point is determined, thus generating the percentage coverage of each feature in relation to the complete epitope. The average of all coverage values is calculated by generating the Avg cover attribute and these values appear in the descending order during the generation of data.
Search for epitopes
The epitopes generated are identified in the proteomes informed (FASTA file) by the user. The proteomes are loaded into the RAM, and the search is performed by searching for the complete epitope sequence in the proteins. Therefore, we recommend adjusting the execution memory of the java parameters settings, -Xms (start memory) and -Xmx (max memory), according to the file size of the proteomes.
The search can also be performed by BLASTP 26 (requires prior installation on the computer), which is necessary to inform the location of the installation in the execution command or in the graphical interface (if the command is already in the system $PATH, one can inform only the blastp and makeblastdb commands, without full path). The identity (default value 90), cover (default value 90), word size (default value 4), and task (blastp-short default value) parameters can also be adjusted. The Blastp results are converted internally and integrated with the EpiBuilder result, and next to the positive hit, the protein accession number is added to the identity and coverage if the user wants to know the hits. For extensive searches, it is suggested that this configuration be removed. As these are small sequences for searching, it is important to evaluate blastp values, as they can eliminate results by exchanging only 1 amino acid.
For EpiBuilder installed via Galaxy ToolShed, if a BLASTP search is chosen, the blastp and makeblastdb commands are required to be on the system $PATH. For the desktop version, there is a test option to verify that the blastp and makeblastdb commands setup is correct.
Output files
The output files are generated with the name <basename>-epibuilder-<file>.<archive>, so the user can perform multiple experiments in the same directory by swapping the <basename> of the experiment in question. The files generated are listed in Table 1.
Table 1.
EpiBuilder provides several files generated at the final of the process.
| File | Type | Description |
|---|---|---|
| epibuilder-detail | CSV | Individual epitope detail |
| epibuilder-epitopes | FASTA | epitope FASTA file |
| epibuilder-parameters | TXT | Parameters of execution |
| epibuilder-protein-summary | TSV | Summary of the run |
| epibuilder-scores | TSV | All generated scores |
| epibuilder-topology | TSV | Epitopes topology |
| epibuilder | TXT | Full report with all information except the score files |
| epibuilder | XLSX | XLSX file with the following tabs: Tab 1 = epibuilder-detail Tab 2 = epibuilder-protein-summary Tab 3 = epibuilder-topology |
| epibuilder | LOG | The run log |
| epibuilder-blast* | – | The files generated in the BLASTP process |
These files are provided in a different type. The main file is epibuilder.xlsx.
If the user chooses to run the query via BLASTP, all output files generated by BLASTP follow the <basename>-epibuilder-blast-<proteome alias>.<extension>, and the result of BLASTP is generated in the CSV standard. The full EpiBuilder Stream is shown in Figure 2.
Figure 2.

The complete EpiBuilder pipeline, indicating input files (BepiPred-2.0 files and proteomes for query) and the internal process such as input loading and processing of software scores, searching for N-glycosylation, assembling of the topology, and the searching in proteomes. After all these steps, the reports are generated in several files. IEDB indicates Immune Epitope Database; MW, molecular mass; IP, Isoleletric Point.
Experiments
For the experiments, we selected 3 human pathogens: the protozoan T. b. gambiense, causative agent of human African trypanosomiasis (HAT—sleeping sickness), a fatal neglected tropical disease27,28; the gram-positive bacterium Clostridioides difficile, formerly called Clostridium difficile, which causes severe diarrhea, colitis, toxic megacolon, and potentially death29,30; and SARS-CoV-2, also fatal 31 which caused a pandemic in 2020. 32
The proteomes of SARS-CoV-2 (UP000464024, Uniprot), Clostridioides difficile (NCBI: txid1496), and T. b. gambiense (strain DAL972, TriTrypdb version 49) were downloaded and manually preprocessed to eliminate characters other than valid amino acids such as “*” and “X”, which is a condition for running BepiPred-2.0. The proteomes were submitted to BepiPred-2.0 analysis using the B-Cell Standalone IEDB software, and this result was used as an input for EpiBuilder with the following configurations: BepiPred-2.0 threshold (0.5, 0.55, 0.6, 0.65, 0.7, 0.75, 0.8, 0.85, and 0.9), minimum epitope size (10), and maximum epitope size (30), all parameters were selected with default cutoff points. The searches were performed with the direct search of the sequence, as well as with the BLASTP option with identity parameters = 90, coverage = 90, word size = 4, and search method = blastp-short. In all experiments, the proteome was included in the search for epitopes that were repeated in other proteins. In addition, for each experiment, epitopes were searched in the human proteome (UP000005640, Uniprot).
To search for T. b. gambiense, we used the proteomes of Trypanosoma brucei brucei (strain TREU927, TriTrypdb version 49), Trypanosoma evansi (strain STIB902, TriTrypdb version 49), Trypanosoma congolense (strain IL3000, TriTrypdb version 49), and Trypanosoma vivax (strain Y486, TriTrypdb version 49). For C. difficile, we used the proteomes of Clostridium botulinum (strain Hall/ATCC 3502/NCTC 13319/Type A) (UP00001986, Uniprot), Clostridium argentinense (strain CDC 2741, UP000031366), and Clostridium perfringens (strain 13/Type A, UP000000818).
For runs with SARS-CoV-2 in addition to the proteome and human proteome, we also added a database of spike proteins (4 854 709 proteins) and all deposited proteins (133 081 009 proteins) in GISAID to the day (November 9, 2021). Blastp evaluation in this round was suppressed because of the number of sequences.
The last experiment with SARS-CoV-2 used the assembly of epitopes from the proteins S (spike), M (membrane), N (nucleocapsid), and E (envelope) of the Reference of SARS-CoV-2 (UP000464024, Uniprot) and were submitted to EpiBuilder to search for the respective proteins of the VoC (variant of concern) lines obtained from GISAID: alpha (EPI_ISL_6949888), beta (EPI_ISL_5905874), gamma (EPI_ISL_6943989), delta (EPI_ISL_6950127), and omicron (EPI_ISL_6914032). The genomes obtained from GISAID were submitted to NextStrain 33 for assembly, protein prediction, and lineage confirmation (Supplementary Figure 1). For this experiment, the BepiPred-2.0 threshold = 0.6 was explicitly chosen.
Finally, the SARS-CoV-2 proteins sequences of the reference, alpha, beta, gamma, delta, and omicron were aligned with Clustal Omega 34 (https://www.ebi.ac.uk/Tools/msa/clustalo/) with the default parameters values to verify mutations in the epitopes, and the results were organized by Jalview 35 omitting the regions that were not predicted as epitopes.
Results
GUI
Figure 3 shows the graphical interface of the EpiBuilder main screen, where the user can perform all the necessary settings, and the results are displayed in the results tab in text format.
Figure 3.

EpiBuilder desktop main screen. From this screen, the user can configure the input file, threshold, the minimum and maximum size of epitopes, the additional features it intends to analyze, the proteomes for searching the epitopes, and the option to search proteomes by BLASTP.
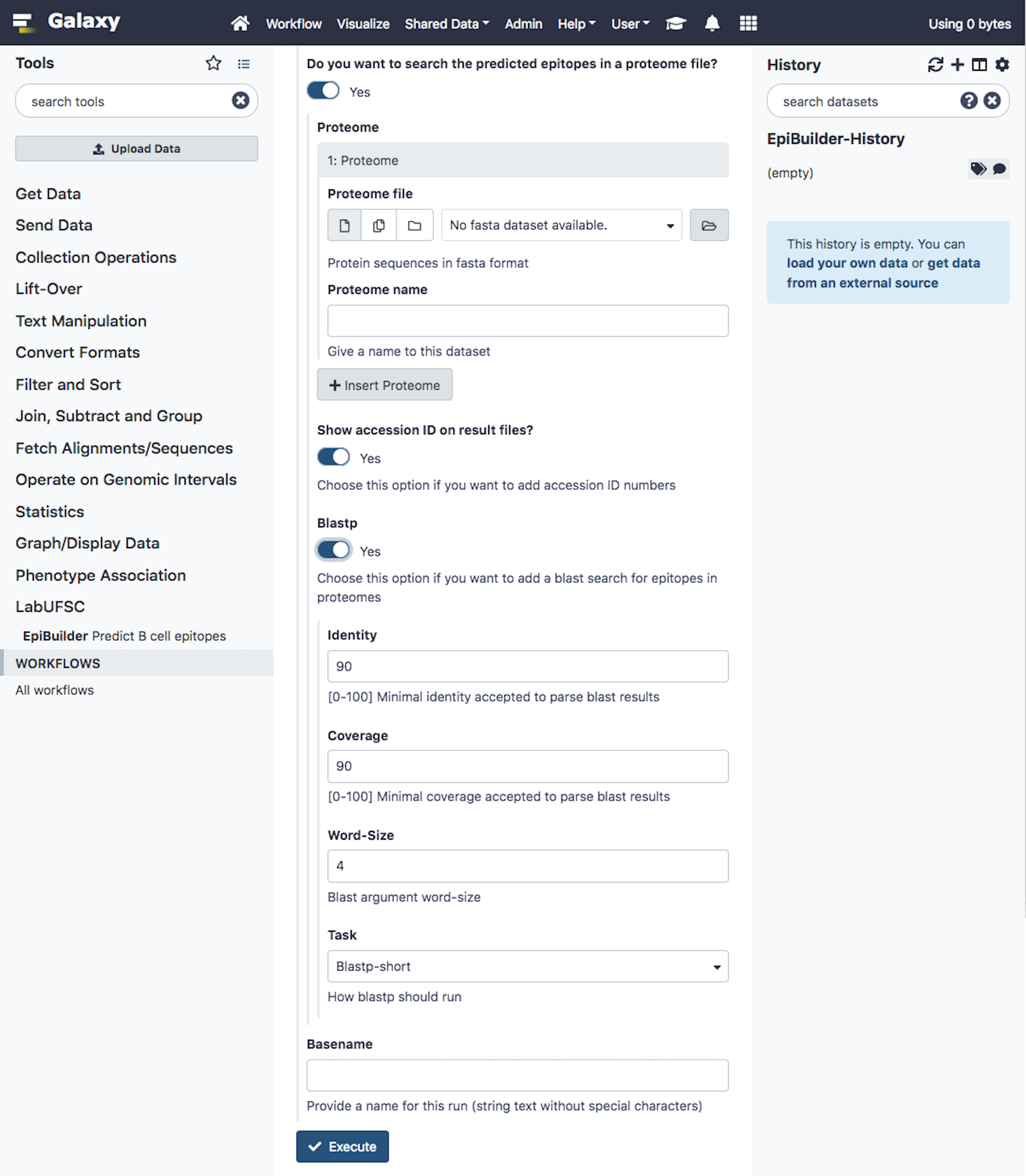
Similar to the desktop graphical interface, it is possible to observe the graphical interface developed for the Galaxy platform (Figure 4). The search settings are presented in Supplementary Figure 2.
Figure 4.

EpiBuilder main screen on galaxy platform. From this screen, the user can configure the input file, threshold, the minimum and maximum size of epitopes, the additional features it intends to analyze, the proteomes for searching the epitopes, and the option to search proteomes by BLASTP.
Experiments
In all experiments, epitopes were found up to a threshold of 0.75; from threshold = 0.8, no epitopes were identified in any of the experiments (Table 2).
Table 2.
The total generated epitopes considering the various thresholds for the organisms C. difficile, T. b. gambiense, and SARS-CoV-2.
| BepiPred-2.0 threshold | Epitopes | ||
|---|---|---|---|
| C. difficile | T. b. gambiense | SARS-CoV-2 | |
| 0.50 | 13 395 | 45 547 | 182 |
| 0.55 | 5 664 | 37 946 | 96 |
| 0.60 | 1 431 | 19 907 | 29 |
| 0.65 | 401 | 5 462 | 3 |
| 0.70 | 47 | 329 | 2 |
| 0.75 | 7 | 8 | 0 |
Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Epitopes of SARS-CoV-2
Table 3 shows that the epitope with the most occurrences of spike protein was CTEVPVAIHADQ, which was present in ~99% of the proteins of the protein spikes bank and ~4% of the complete protein bank (133 081 009). The other epitopes MSLGAENSVAY and YRLFRKSNLKP were found in ~95% and NNLDSKVGGN and DEVRQIAPGQTGK in ~95%, which shows their exemplary conservation. No epitope contained N-glycosylation sites. Full results can be seen in Supplementary Data 1.
Table 3.
SARS-CoV-2 epitopes generated using the threshold of 0.6.
| Accession ID | Epitope | N-Glyc | Length | Avg cover | Direct proteome search | ||
|---|---|---|---|---|---|---|---|
| SCoV | All spikes | All proteins | |||||
| R1AB_SARS2 | DDDSQQTVGQQDG | N | 13 | 0.78 | 2 | 0 | 4 876 200 |
| AP3A_SARS2 | GTTSPISEHD | N | 10 | 0.76 | 1 | 0 | 4802 655 |
| VME1_SARS2 | KLNTDHSSSSDNI | N | 13 | 0.75 | 1 | 0 | 4862 069 |
| R1A_SARS2 | EQEEDWLDDDSQQTVGQQD | N | 19 | 0.75 | 2 | 0 | 4 875 778 |
| SPIKE_SARS2 | QTQTNSPRRARSVA | N | 14 | 0.74 | 1 | 1 127 228 | 1 130 945 |
| R1A_SARS2 | DLGDELGTDPYEDFQENWN | N | 19 | 0.72 | 2 | 0 | 4 774 399 |
| R1AB_SARS2 | LHPTQAPTHLS | N | 11 | 0.71 | 1 | 0 | 4 861 900 |
| R1AB_SARS2 | LGDELGTDPYED | N | 12 | 0.7 | 2 | 0 | 4 785 788 |
| SPIKE_SARS2 | NNLDSKVGGN | N | 10 | 0.7 | 1 | 4 607 437 | 4 743 622 |
| NCAP_SARS2 | MSGKGQQQQGQTVTK | N | 15 | 0.69 | 1 | 0 | 3 518 539 |
| R1AB_SARS2 | EEVKPFITESKPSVEQRKQDDKK | N | 23 | 0.69 | 2 | 0 | 4 744 396 |
| R1A_SARS2 | CEEEEFEPSTQYEYGTEDDYQGKPLEF | N | 27 | 0.68 | 2 | 0 | 4 798 844 |
| NCAP_SARS2 | SDSTGSNQNGERSGARSKQRRPQGLPN | N | 27 | 0.68 | 1 | 0 | 4 812 247 |
| R1A_SARS2 | EVKPFITESKPSVEQRKQDDKKIKA | N | 25 | 0.67 | 2 | 0 | 4 743 252 |
| R1AB_SARS2 | EPSTQYEYGTEDDYQGKPLEFG | N | 22 | 0.66 | 2 | 0 | 4 799 515 |
| NCAP_SARS2 | KDKKKKADETQALPQRQKKQQTV | N | 23 | 0.58 | 1 | 0 | 2 253 691 |
| R1A_SARS2 | NKGAGGHSYGADL | N | 13 | 0.57 | 2 | 0 | 4781487 |
| SPIKE_SARS2 | DEVRQIAPGQTGK | N | 13 | 0.57 | 1 | 4 595 459 | 4733939 |
| NCAP_SARS2 | RRIRGGDGKM | N | 10 | 0.52 | 1 | 0 | 4866340 |
| SPIKE_SARS2 | YRLFRKSNLKP | N | 11 | 0.51 | 1 | 4 661 055 | 4797597 |
| R1AB_SARS2 | VNNLDKSAGF | N | 10 | 0.5 | 1 | 0 | 4854988 |
| R1AB_SARS2 | LKKDAPYIVG | N | 10 | 0.48 | 2 | 0 | 4888434 |
| R1AB_SARS2 | IDKCSRIIPA | N | 10 | 0.46 | 1 | 0 | 4769110 |
| R1A_SARS2 | LEETKFLTENLL | N | 12 | 0.45 | 2 | 0 | 4835320 |
| R1AB_SARS2 | PQEHYVRITG | N | 10 | 0.42 | 1 | 0 | 4888859 |
| SPIKE_SARS2 | MSLGAENSVAY | N | 11 | 0.42 | 1 | 4 681 033 | 4820746 |
| SPIKE_SARS2 | CTEVPVAIHADQ | N | 12 | 0.42 | 1 | 4 789 431 | 4929716 |
| R1A_SARS2 | TIQTIVEVQPQLEMELT | N | 17 | 0.34 | 2 | 0 | 3725516 |
| R1AB_SARS2 | VEVQPQLEMELT | N | 12 | 0.33 | 2 | 0 | 4875451 |
Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SCoV indicates SARS-CoV-2. It was identified a total of 29 epitopes. The search was performed using the GISAID database protein for spike and all proteins.
Considering the S, N, M, and E proteins of SARS-CoV-2, only protein E had no epitopes predicted for the adjusted values. Protein S presented 6 epitopes, some of which were completely identified in VoC sequences only by BLASTP, indicating amino acid exchanges in the epitopes of these variants, which is the case for the epitope QTQTNSPRRARSVA, whose sequence with the search for the complete structure only had an occurrence in the beta and gamma variants. Even with the blastp search, there was no occurrence of these epitopes in the omicron variant, including the epitope NNLDSKVGGN. The complete list of epitopes with their hits is shown in Table 4, and the alignment with the epitopes of the S protein can be seen in Figure 5, which precisely shows the exchange of a single amino acid in the alpha, delta, and omicron variants in the epitope QTQTNSPRRARSVA and the exchange of 2 amino acids in the epitope NNLDSKVGGN only in the omicron variant. Other data are presented in Supplementary Data 2.
Table 4.
SARS-CoV-2 epitopes of the proteins spike (S), membrane (M), and nucleocapsid (N).
| Protein | Epitope | Length | Avg cover | Direct search | BLASTP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCoV | α | β | γ | δ | Ο | SCoV | α | β | γ | δ | Ο | ||||
| S | QTQTNSPRRARSVA | 14 | 0.74 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 |
| NNLDSKVGGN | 10 | 0.70 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | |
| DEVRQIAPGQTGK | 13 | 0.57 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | |
| YRLFRKSNLKP | 11 | 0.51 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| MSLGAENSVAY | 11 | 0.42 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| CTEVPVAIHADQ | 12 | 0.42 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| N | MSGKGQQQQGQTVTK | 15 | 0.69 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| SDSTGSNQNGERSGARSKQRRPQGLPN | 27 | 0.68 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | |
| KDKKKKADETQALPQRQKKQQTV | 23 | 0.58 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | |
| RRIRGGDGKM | 10 | 0.52 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| M | KLNTDHSSSSDNI | 13 | 0.75 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
Abbreviations: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; VoC, variant of concern.
SCoVSARS-CoV-2Reference; SARS-CoV-2strain Alpha; SARS-CoV-2strain Beta; SARS-CoV-2strain Gamma; SARS-CoV-2strain Delta; ΟSARS-CoV-2strain Omicron. The search of these epitopes compares the VoC proteins of SARS-CoV-2.
Figure 5.

Alignment of the spike proteins (S) of SARS-CoV-2, compared with the VoC variants where it is possible to analyze that there are mutations that impact amino acids present in identified areas epitopes, especially in the omicron variant. The alignment was performed using Clustal Omega with default parameter values. SARS-CoV-2 indicates severe acute respiratory syndrome coronavirus 2; VoC, variant of concern.
There were no occurrences of epitopes of SARS-CoV-2 in the human proteome according to the established criteria. However, by individually analyzing the files generated by BLASTP, it can be seen that if the values of identity and coverage were 50%, there would be several valid hits, such as the epitope QTQTNSPRRARSVA with 100% coverage hit and 50% identity in the genes |Q92902| HPS1_HUMAN (SPRRARS), |J3KPV6| J3KPV6_HUMAN (RRARSVA), sp|P37088| SCNNA_HUMAN (RRARSVA), and |C5HTZ1| C5HTZ1_HUMAN (RRARSVA). The epitope DEVRQIAPGQTGK had 17 hits, the epitope MSLGAENSVAY had 2 hits, and the epitope CTEVPVAIHADQ had only 1 hit.
Epitopes of C. difficile
Considering the threshold point of 0.75, 7 epitopes of C. difficile were identified (Table 5), 5 of which presented N-glycosylation sites. Epitopes were not found in the other Clostridioides species either directly or by BLASTP. However, similar to SARS-CoV-2, if the identity and coverage values were modified to 60% coverage and identity, it was possible to find several hits. The exciting thing is that despite this, 54 hits were found with BLASTP, including 2 directly from the epitope ASSSSSSSSN in humans. In addition, the epitopes identified showed almost 100% coverage in the selected features, except for the Kolaskar 18 method, increasing the value of EpiBuilder’s Avg cover. Additional information is available in Supplementary Data 3.
Table 5.
List of the epitopes from C. difficile, with the threshold of 0.75.
| ID | Epitope | N-Glyc | Length | Avg Cover | Proteome search | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Direct search | BLASTP | |||||||||||||
| Cdif | Cper | Cbot | Carg | Hsap | Cdif | Cper | Cbot | Carg | Hsap | |||||
| WP_003427432.1 | QTNENNTTNNSE | Y | 12 | 0.8 | 1 | 0 | 0 | 0 | 0 | 1 | X | X | 0 | X |
| WP_003434503.1 | TNNNQQNNANTNQQNNNS | Y | 18 | 0.8 | 1 | 0 | 0 | 0 | 0 | 1 | X | 0 | 0 | 0 |
| WP_021368964.1 | NNSSNSNSTNN | Y | 11 | 0.8 | 1 | 0 | 0 | 0 | 0 | X | X | X | 0 | X |
| WP_021368964.1 | KPNSSSNQNSQPNSNSK | Y | 17 | 0.8 | 1 | 0 | 0 | 0 | 0 | 1 | X | 0 | 0 | 0 |
| WP_021390451.1 | NSNSNSNSNSNS | N | 12 | 0.8 | 3 | 0 | 0 | 0 | 0 | 13 | 0 | X | 0 | 0 |
| WP_003438294.1 | SNSNSDNSSNSNGNNSSSS | Y | 19 | 0.79 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | X |
| WP_021364658.1 | ASSSSSSSSSN | N | 11 | 0.73 | 1 | 0 | 0 | 0 | 2 | 2 | X | X | X | 54 |
Abbreviations: Carg, C. argentinense; Cbot, C. botulinum; Cdif, C. difficile; Cper, C. perfringens; Hsap, H. sapiens.
Xno hit identified for the epitope, when the value is 0, that is, there was hit but did not reach the cutoff point of coverage and identity.
Epitopes of T. b. gambiense
Considering the threshold of 0.75, we found 8 epitopes, 3 of them had N-glycosylated sites (Table 6). The presence of epitopes with a small variety of amino acids was noted, with the most frequent epitopes being asparagine (N) and glutamine (Q). The higher frequency of occurrence of epitopes in T. evansi than that in T. b. brucei was also verified. In T. congolense and T. vivax, epitopes were not identified via direct search, but for T. vivax, the occurrence of the epitope QQQQQQQQQQQQQQLl was observed 27 times via BLASTP, and this same epitope had the occurrence of 416 in humans. Additional information is available in Supplementary Data 4.
Table 6.
List of the epitopes from T. b. gambiense, with the threshold of 0.75.
| Accession ID | Epitope | N-Glyc | Length | Avg cover | Proteome search | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Direct search | BlastP | |||||||||||||||
| Tbg | Tbb | Tco | Tev | Tvi | Hsap | Tbg | Tbb | Tco | Tev | Tvi | Hsap | |||||
| Tbg972.11.480 | QPPQQQQQQA | N | 10 | 0.94 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tbg972.11.11840 | QQQPQQQPQQQQQQG | N | 15 | 0.84 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Tbg972.9.8810 | NNNNNNNNNNNNS | Y | 13 | 0.8 | 4 | 1 | 0 | 2 | 0 | 0 | 3 | 3 | 0 | 7 | 0 | – |
| Tbg972.1.2160 | NNNKNNNNNN | N | 10 | 0.8 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | – |
| Tbg972.3.1330 | NNNSNNNNNNNNNN | Y | 14 | 0.8 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | – |
| Tbg972.1.1570 | NNNNNNNNNNNSGNG | Y | 15 | 0.75 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | – |
| Tbg972.3.3000 | QQQQQQQQQQQQQL | N | 14 | 0.7 | 1 | 1 | 0 | 1 | 0 | 10 | 4 | 0 | 0 | 0 | 27 | 416 |
| Tbg972.9.4350 | QQQHHNTHQTTAQQ | N | 14 | 0.69 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
Abbreviations: Hsap, H. sapiens; Tbb, T. b. brucei; Tbg, T. b. gambiense; Tev, T. evansi; Tvi, T. vivax.
Discussion
EpiBuilder is designed to be an intuitive and easy-to-use tool, with no installation required, and it can be rotated via the terminal, on servers for heavier processing, or on a personal computer with a GUI. This tool is also available for the Galaxy platform through ToolShed and can be installed on any server. Because it is an assembly tool based on the results of BepiPred-2.0, we understand that there is no comparative software because this tool is a pipeline tool to generate several results that would be done manually.
After execution, we verified the possibility of running EpiBuilder as a proteome-wide tool for epitope generation, searching, and classification. From the executions, we noticed that finding epitopes with a threshold of BepiPred-2.0 greater than or equal to 0.8 was challenging; there were no results for all 3 evaluated organisms.
An experiment with SARS-CoV-2 did not generate any identifiable epitopes in humans. However, this result can change according to the settings. We also noticed that for T. b. gambiense and C. difficile, some of the epitopes generated did not occur in humans in general, even with BLASTP consultation, which may indicate potential targets with less possibility of cross-reaction.
We found that the higher the threshold of the established BepiPred-2.0, the greater the coverage of the selected EpiBuilder features, which shows a direct relationship between these features and the threshold definition.
As these are genome-wide experiments, these results need to be validated more cautiously because they may contain data from the assembly and prediction of proteins generated with some type of error.
The next steps of EpiBuilder are will be to allow the use of results from other BCE predictors and integrate them using docker to turn the application more accessible for the non-specialist.
Supplemental Material
{kind=link}
Supplemental material, sj-png-5-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
{kind=link}
Supplemental material, sj-png-6-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-4-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Footnotes
Author Contributions: RSM: Investigation, Writing - original draft, Formal analysis. Data curation. NAC: Investigation. VBF: Formal analysis. Data curation GW: Data curation, Validation. LCM: Conceptualization, Validation, Writing - review & editing, Project administration, Funding acquisition.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Vilmar Benetti Filho  https://orcid.org/0000-0003-0309-4877
https://orcid.org/0000-0003-0309-4877
Nathália Anderson Calomeno
https://orcid.org/0000-0002-8785-2390
Glauber Wagner
https://orcid.org/0000-0001-5003-6595
Luiz Claudio Miletti
https://orcid.org/0000-0001-5926-0286
Data Availability and Requirements: All the experimental data, source code, and binaries are available in https://epibuilder.sourceforge.io.
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Abbas A, Lichtman A, Pillai S. Cellular and Molecular Immunology. 9th ed. Amsterdam, The Netherlands: Elsevier; 2017. [Google Scholar]
- 2. Shirai H, Prades C, Vita R, et al. Antibody informatics for drug discovery. Biochim Biophys Acta. 2014;1844:2002-2015. doi: 10.1016/j.bbapap.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 3. Manavalan B, Govindaraj RG, Shin TH, Kim MO, Lee G. iBCE-EL: a new ensemble learning framework for improved linear B-cell epitope prediction. Front Immunol. 2018;9:1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahammad I, Lira SS. Designing a novel mRNA vaccine against SARS-CoV-2: an immunoinformatics approach. Int J Biol Macromol. 2020;162:820-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sadat SM, Aghadadeghi MR, Yousefi M, Khodaei A, Sadat Larijani M, Bahramali G. Bioinformatics analysis of SARS-CoV-2 to approach an effective vaccine candidate against COVID-19. Mol Biotechnol. 2021;63:389-409. doi: 10.1007/s12033-021-00303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foroutan M, Ghaffarifar F, Sharifi Z, Dalimi A, Pirestani M. Bioinformatics analysis of ROP8 protein to improve vaccine design against Toxoplasma gondii. Infect Genet Evol. 2018;62:193-204. doi: 10.1016/j.meegid.2018.04.033. [DOI] [PubMed] [Google Scholar]
- 7. Manivel G, Meyyazhagan A, Durairaj DR, Piramanayagam S. Genome-wide analysis of excretory/secretory proteins in Trypanosoma brucei brucei: insights into functional characteristics and identification of potential targets by immunoinformatics approach. Genomics. 2019;111:1124-1133. doi: 10.1016/j.ygeno.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 8. Ostolin TLVDP, Gusmão MR, Mathias FAS, et al. A chimeric vaccine combined with adjuvant system induces immunogenicity and protection against visceral leishmaniasis in BALB/c mice. Vaccine. 2021;39:2755-2763. doi: 10.1016/j.vaccine.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 9. Lata KS, Kumar S, Vaghasia V, et al. Exploring Leptospiral proteomes to identify potential candidates for vaccine design against Leptospirosis using an immunoinformatics approach. Sci Rep. 2018;8:6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaliamurthi S, Selvaraj G, Junaid M, Khan A, Gu K, Wei D-Q. Cancer immunoinformatics: a promising era in the development of peptide vaccines for human papillomavirus-induced cervical cancer. Curr Pharm Des. 2019;24:3791-3817. [DOI] [PubMed] [Google Scholar]
- 11. Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65:40-48. http://www.ncbi.nlm.nih.gov/pubmed/16894596 [DOI] [PubMed] [Google Scholar]
- 12. Yao B, Zheng D, Liang S, Zhang C. SVMTriP: a method to predict B-cell linear antigenic epitopes. Methods Mol Biol. 2020;2131:299-307. http://www.ncbi.nlm.nih.gov/pubmed/32162263 [DOI] [PubMed] [Google Scholar]
- 13. Collatz M, Mock F, Barth E, Hölzer M, Sachse K, Marz M. EpiDope: a deep neural network for linear B-cell epitope prediction. Bioinformatics. http://www.ncbi.nlm.nih.gov/pubmed/34109374. Published 2021. [DOI] [PubMed]
- 14. Bahai A, Asgari E, Mofrad MRK, Kloetgen A, McHardy AC. EpitopeVec: linear epitope prediction using deep protein sequence embeddings. Bioinformatics. 2021;37:4517-4525. http://www.ncbi.nlm.nih.gov/pubmed/34180989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 2006;47:45-148. [DOI] [PubMed] [Google Scholar]
- 16. Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55:836-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karplus PA, Schulz GE. Prediction of chain flexibility in proteins — a tool for the selection of peptide antigens. Naturwissenschaften. 1985;72:212-213. [Google Scholar]
- 18. Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276:172-174. [DOI] [PubMed] [Google Scholar]
- 19. Parker JMR, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425-5432. [DOI] [PubMed] [Google Scholar]
- 20. Afgan E, Baker D, Batut B, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537-W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blankenberg D, Von Kuster G, Bouvier E, et al. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014;15:2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:W24-W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vita R, Mahajan S, Overton JA, et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339-D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lafita A, Bliven S, Prlić A, et al. BioJava 5: a community driven open-source bioinformatics library. PLoS Comput Biol. 2019;15:e1006791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pitti T, Chen CT, Lin HN, Choong WK, Hsu WL, Sung TY. N-GlyDE: a two-stage N-linked glycosylation site prediction incorporating gapped dipeptides and pattern-based encoding. Sci Rep. 2019;9:15975. doi: 10.1038/s41598-019-52341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roesch LFW, Fulthorpe RR, Jaccques RJS, Bento FM, de Oliveira Camargo FA. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. World J Microbiol Biotechnol. 2006;22:3389-3402. [Google Scholar]
- 27. Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet. 2017;390:2397-2409. https://linkinghub.elsevier.com/retrieve/pii/S0140673617315106 [DOI] [PubMed] [Google Scholar]
- 28. Kennedy PGE. Update on human African trypanosomiasis (sleeping sickness). J Neurol. 2019;266:2334-2337. http://www.ncbi.nlm.nih.gov/pubmed/31209574 [DOI] [PubMed] [Google Scholar]
- 29. Czepiel J, Drdóżdż M, Pituch H, et al. Clostridium difficile infection: review. Eur J Clin Microbiol Infect Dis. 2019;38:1211-1221. http://link.springer.com/10.1007/s10096-019-03539-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mullish BH, Williams HR. Clostridium difficile infection and antibiotic-associated diarrhoea. Clin Med (Northfield Il). 2018;18:237-241. https://www.rcpjournals.org/lookup/doi/10.7861/clinmedicine.18-3-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang M-Y, Zhao R, Gao L-J, Gao XF, Wang DP, Cao JM. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol. 2020;10:587269. https://www.frontiersin.org/articles/10.3389/fcimb.2020.587269/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Awadasseid A, Wu Y, Tanaka Y, Zhang W. SARS-CoV-2 variants evolved during the early stage of the pandemic and effects of mutations on adaptation in Wuhan populations. Int J Biol Sci. 2021;17:97-106. https://www.ijbs.com/v17p0097.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121-4123. https://academic.oup.com/bioinformatics/article/34/23/4121/5001388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. https://onlinelibrary.wiley.com/doi/10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Procter JB, Carstairs GM, Soares B, et al. Alignment of biological sequences with Jalview. In: Katoh K, ed. Multiple Sequence Alignment. Methods in Molecular Biology. New York, NY: Humana; 2021:203-224. http://link.springer.com/10.1007/978-1-0716-1036-7_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-png-5-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-png-6-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-4-bbi-10.1177_11779322221095221 for EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes by Renato Simões Moreira, Vilmar Benetti Filho, Nathália Anderson Calomeno, Glauber Wagner and Luiz Claudio Miletti in Bioinformatics and Biology Insights