

Abstract
Intracellular accumulation of tau aggregates is a hallmark of several neurodegenerative diseases. However, global genetic reduction of tau is beneficial also in models of other brain disorders that lack such tau pathology, suggesting a pathogenic role of nonaggregated tau. Here, conditional ablation of tau in excitatory, but not inhibitory, neurons reduced epilepsy, sudden unexpected death in epilepsy (SUDEP), overactivation of the phosphoinositide 3-kinase–AKT-mammalian target of rapamycin pathway, brain overgrowth (megalencephaly), and autism-like behaviors in a mouse model of Dravet syndrome, a severe epileptic encephalopathy of early childhood. Furthermore, treatment with a tau-lowering antisense oligonucleotide, initiated on postnatal day 10, had similar therapeutic effects in this mouse model. Our findings suggest that excitatory neurons are the critical cell type in which tau has to be reduced to counteract brain dysfunctions associated with Dravet syndrome and that overall cerebral tau reduction could have similar benefits, even when initiated postnatally.
One Sentence Summary:
Tau ablation in excitatory neurons and postnatal knockdown of brain tau reduce epilepsy, early death, and autism behaviors in a Dravet syndrome model.
INTRODUCTION
Autism spectrum disorders (ASDs) are a group of neurodevelopmental diseases, possibly affecting ~1% of people worldwide (1, 2). Although etiologically diverse, these conditions share several core symptoms, including repetitive and restricted (stereotypic) behaviors, impaired social interactions, and communication deficits (1, 3–6). ASDs are often accompanied by additional health problems, including epilepsy, delayed psychomotor development, intellectual disability, hyperactivity, attention deficits, anxiety, gastrointestinal disturbances, and sleep abnormalities (1, 7, 8). Conversely, brain disorders in which such other dysfunctions predominate can be accompanied by autism, for example, Dravet syndrome (DS), a severe epileptic encephalopathy that can result in autism.
Beginning in early childhood, DS causes a variety of seizures that can lead to sudden unexpected death in epilepsy (SUDEP) (9, 10). In more than 80% of cases, DS is caused by de novo variants of the SCN1A gene, 50–60% of which result in haploinsufficiency of the voltage-gated sodium channel α subunit NaV1.1 (11–16). NaV1.1 is particularly abundant in parvalbumin-expressing inhibitory interneurons and critically supports the function of these cells (17–19). Scn1aRX/+ knockin mice carry a truncation mutation (R1407X) in one Scn1a allele that results in a premature stop codon and Nav1.1 haploinsufficiency in heterozygous mice (17). The corresponding SCN1A variant causes DS in humans (20–22), although its prevalence among patients with DS seems to be unknown. Scn1aRX/+ mice begin to show generalized seizures and SUDEP in the third postnatal week (17), and those who survive beyond early postnatal maturation often go on to develop other features of DS (9), including cognitive decline and ASD-related behaviors such as excessive stereotypic behaviors, hyperactivity, and social interaction deficits (23–25). To simplify the description of genotypes, Scn1aRX/+ mice will be referred to as “DS mice.”
Seizures associated with DS in humans are often refractory to therapeutic interventions (26). In general, roughly 30% of people with diverse types of epilepsy do not benefit from currently available antiepileptic treatments (27). In a similar vein, the various drugs used for the management of autism have limited efficacy and many have substantial side effects (28, 29). Better strategies are needed to treat epileptic encephalopathies, autism, and disabling comorbidities (27, 29–33).
Several ASDs that are associated with epilepsy also feature overactivation of the phosphoinositide 3-kinase (PI3K)–AKT–mammalian target of rapamycin (mTOR) pathway and brain overgrowth (megalencephaly) (33–40). We previously showed that genetically reducing overall expression of the microtubule-associated protein tau can prevent or diminish multiple disease manifestations in mouse models of such ASDs, including DS mice (24, 25).
Tau has been studied most extensively in relation to tauopathies, aging-related neurodegenerative disorders that are associated with microscopically visible accumulations of abnormal tau aggregates in neurons or other cell types (41–44). However, studies carried out in models of ASD, depression, epilepsy, and stroke have revealed additional disease-enabling roles of nonaggregated tau in brain disorders that are not associated with such tau pathology [see ref. (45) for review]. For example, global genetic ablation of tau prevented or markedly reduced epileptic activity, SUDEP, cognitive deficits and autism-like behaviors in DS mice, which show no evidence for abnormal tau accumulation (24, 25). Reducing tau expression by 50% through inactivation of one Mapt allele was also efficacious (24, 25), indicating that tau-lowering interventions may not have to fully eliminate tau to achieve therapeutic benefits. Tau ablation did not normalize the reduced expression of NaV1.1 in mutant brain tissues (24), suggesting a downstream block of epileptogenesis and related symptoms. Because tau is expressed in inhibitory and excitatory neurons (46, 47), a critical first step toward unraveling the mechanisms by which tau reduction counteracts the development of neural network and behavioral dysfunctions is to determine from which cell type(s) tau has to be removed for tau reduction to yield therapeutic benefits. The comparative analysis of conditional knockout mice with selective ablation of tau in excitatory or inhibitory neurons presented in this study suggests that excitatory forebrain neurons are the key cell type.
Although the development of increasingly effective viral vectors and gene editing strategies (48, 49) could ultimately allow for tau reduction in specific neuronal populations of people at risk for developing epilepsy, ASD, or both, overall knockdown of tau with tau-targeting antisense oligonucleotide (ASOs) (50–52) seems more readily translatable at this point in time. ASOs are short single-stranded sequences of DNA that selectively bind to target RNA by Watson-Crick base pairing (53). Intrathecal or intracerebroventricular (ICV) injection of ASOs into the cerebrospinal fluid causes widespread distribution of the molecules across the central nervous system (CNS) and prolonged knockdown or modulation of targeted gene products (53). A tau-targeting ASO is being tested in a clinical trial for Alzheimer’s disease (AD) (NCT03186989) (53).
Treatment with tau-targeting ASOs may also be helpful in ASDs, epilepsies and other tau-dependent disorders of early onset. However, the efficacy and feasibility of this approach will depend, at least in part, on when tau reduction has to be initiated to prevent disease manifestations. Does it have to be initiated in utero, as it is in tau knockout mice, or can it be delayed until later postnatal stages of development? Here, we address this question by treating young DS mice with ICV injection of a tau-targeting ASO. Early postnatal cerebral knockdown of tau reduced SUDEP, epilepsy, and several autism-like behaviors.
RESULTS
Conditional ablation of tau in excitatory but not inhibitory neurons counteracts the development of epileptic activity
To compare the effects of tau ablation in excitatory versus inhibitory neurons, we used conditional knockout mice with floxed Mapt alleles (Maptflox/flox mice) that were generated by homologous recombination in the Roberson laboratory. Heterozygous Emx1IRES-cre/+ knockin mice expressing Cre recombinase in excitatory neurons of multiple forebrain regions, including the hippocampus and neocortex (54), on a homozygous Maptflox/flox background (ΔTau-EX mice), and heterozygous VgatIRES-cre/+ mice expressing Cre in inhibitory neurons (55) on a homozygous Maptflox/flox background (ΔTau-IN mice) were generated through equivalent breeding schemes, as described in Materials and Methods. During embryonic development, the Emx1 and Vgat promoters initiate gene expression by E12.5 (embryonic day 12.5) (54, 56, 57). Unless indicated otherwise, experimental and control groups in this study included comparable proportions of male and female mice.
After validating the specificity of the Tau5 antibody by comparative analysis of brain sections from Mapt+/+, Mapt+/–, and Mapt–/– mice (fig. S1), we used this reagent in combination with antibodies to cell type–specific markers to confirm the selective ablation of tau in excitatory neurons of ΔTau-EX mice and in inhibitory neurons of ΔTau-IN mice, respectively (Fig. 1A). Because increased resistance to epileptogenesis is among the most robust effects of global genetic tau reduction (24, 25, 45, 58), we challenged ΔTau-EX mice, ΔTau-IN mice, Mapt–/– (ΔTau), and Maptflox/flox mice (Control) with an intraperitoneal (IP) injection of the proconvulsant drug pentylenetetrazole (PTZ). Similar to global genetic ablation of tau, selective ablation of tau in excitatory neurons reduced both electroencephalographic (Fig. 1, B and C) and behavioral (Fig. 1D) evidence of PTZ-induced epileptic activity, whereas selective ablation of tau in inhibitory neurons did not (Fig. 1, B to D), suggesting that excitatory neurons are the key cell type in which tau enables and tau reduction prevents this type of network dysfunction. For additional statistical comparisons between groups, see table S1.
Fig. 1. Selective tau ablation in excitatory neurons reduces PTZ-induced epileptic activity in conditional knockout mice.
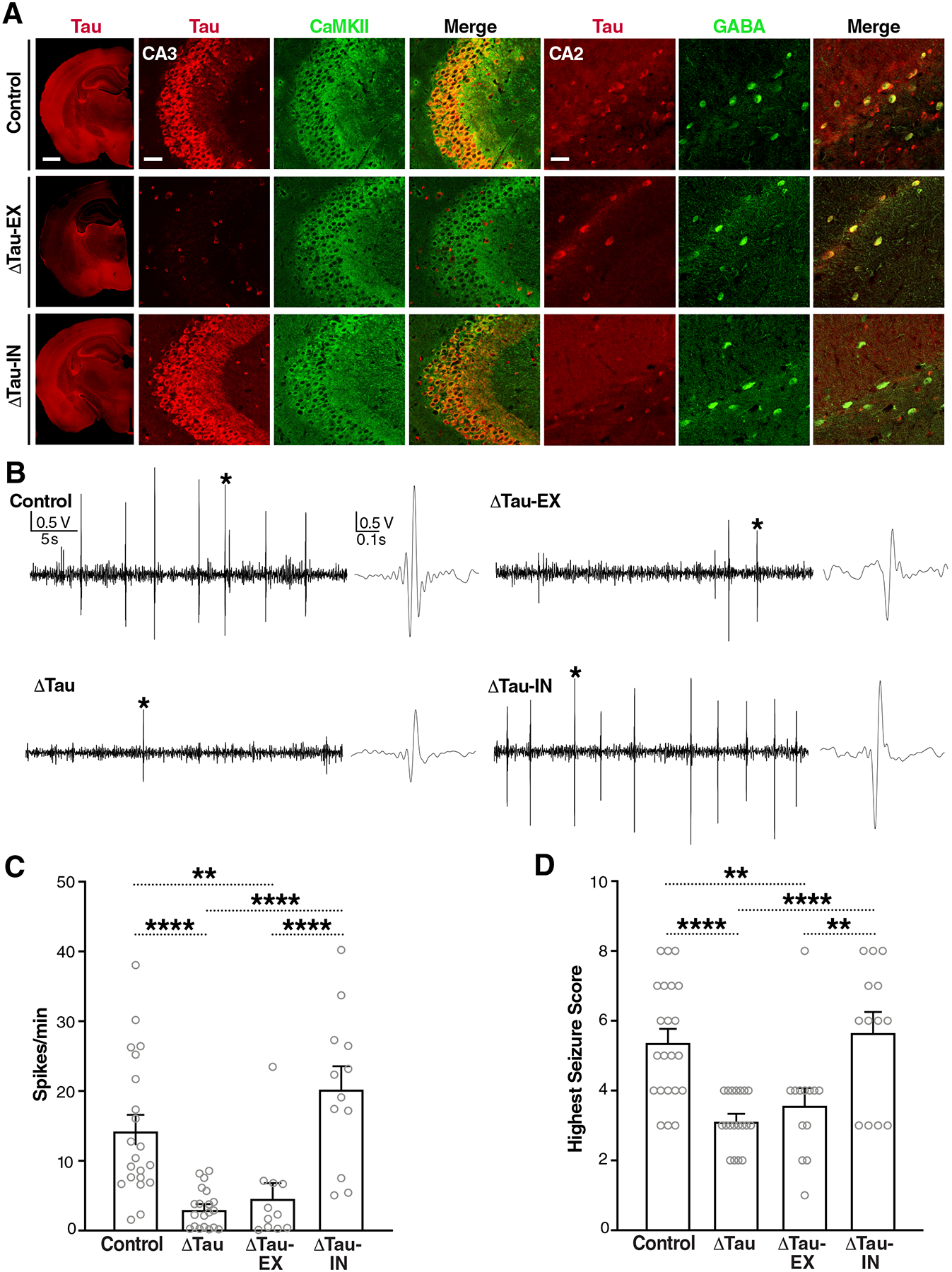
(A) Coronal brain sections from Maptflox/flox (Control), Emx1IRES-cre/+/Maptflox/flox (ΔTau-EX), and VgatIRES-cre/+/Maptflox/flox (ΔTau-IN) mice coimmunostained for tau (red) and markers (green) of excitatory (CaMKII) or inhibitory (GABA) neurons. The leftmost column shows hemibrains (scale bar, 900 μm) and the images on the right show details of the hippocampal CA3 and CA2 subfields (scale bars, 60 μm). CA3 illustrates tau ablation in excitatory neurons (middle row). Because CA2 contains fewer pyramidal cells, it is more suitable for illustrating tau ablation in inhibitory neurons (bottom row). Genotypes are indicated on the very left. (B to D) Video-EEG recordings were used to assess epileptiform spike activity (B and C) and behavioral seizure activity (D) in 3.5-month-old male Control, Mapt–/– (ΔTau), ΔTau-EX, and ΔTau-IN mice after intraperitoneal injection of PTZ (50 mg/kg). (B) Representative EEG traces recorded ~5 min after PTZ injection. Sections of traces marked with an asterisk are shown on the right at higher time resolution. (C) Epileptiform spikes per minute averaged from a 20-min EEG recording after the PTZ injection, or from shorter periods for mice that died during the recording. n = 11–21 mice per group. (D) Highest seizure score reached within 20 min after the PTZ injection. n = 13–22 mice per group. **P < 0.01 and ****P < 0.0001 by one-way ANOVA and Holm-Sidak test. Gray circles represent data from individual mice. Values in (C) and (D) are means ± SEM.
Tau ablation in excitatory neurons reduces SUDEP, epileptiform activity, and autism-like behaviors in DS mice
We next investigated whether selective ablation of tau in excitatory or inhibitory neurons modulates the phenotype of DS mice. Crosses between genetically modified lines of mice were set up as detailed in Materials and Methods to generate offspring comprising Maptflox/flox mice (Controls), ΔTau-EX mice, and ΔTau-IN mice, as well as DS mice on a Maptflox/flox (DS*), Emx1IRES-cre/+/Maptflox/flox (DS*ΔTau-EX), or VgatIRES-cre/+/Maptflox/flox (DS*ΔTau-IN) background.
Ablation of tau in excitatory, but not inhibitory, neurons reduced SUDEP (Fig. 2A) and epileptiform activity (Fig. 2, B and C) in DS* mice. Analysis of the survival curves (Fig. 2A) by log-rank (Mantel-Cox) test with Holm-Sidak correction revealed Control mice to differ from DS* (P < 0.001) and DS*ΔTau-IN (P < 0.001) mice, but not from the other groups (P > 0.35); DS*ΔTau-EX mice differed from both DS* (P < 0.01) and DS*ΔTau-IN (P < 0.05) mice. DS*ΔTau-EX mice also showed fewer stereotypic (digging-like) behaviors than DS* mice, whereas DS*ΔTau-IN mice did not (Fig. 2D). Compared to Controls, both DS* and DS*ΔTau-IN mice showed strong reductions in their responses to a social odor, suggesting impairments in social interaction or communication, whereas the responsivity of DS*ΔTau-EX mice was in between those of DS* mice and Controls (Fig. 2, E and F). Habituation to each odor was measured as the slopes of linear regression lines through the three trials, as determined by Generalized Estimating Equation (GEE) framework analysis (25). DS* mice did not significantly differ from Controls in their habituation to water (P = 0.1741) and vanilla (P = 0.0855; Fig. 2E), indicating a comparable sense of smell.
Fig. 2. Selective tau ablation in excitatory neurons reduces premature mortality, epileptiform activity, and autism-like behaviors in DS mice.
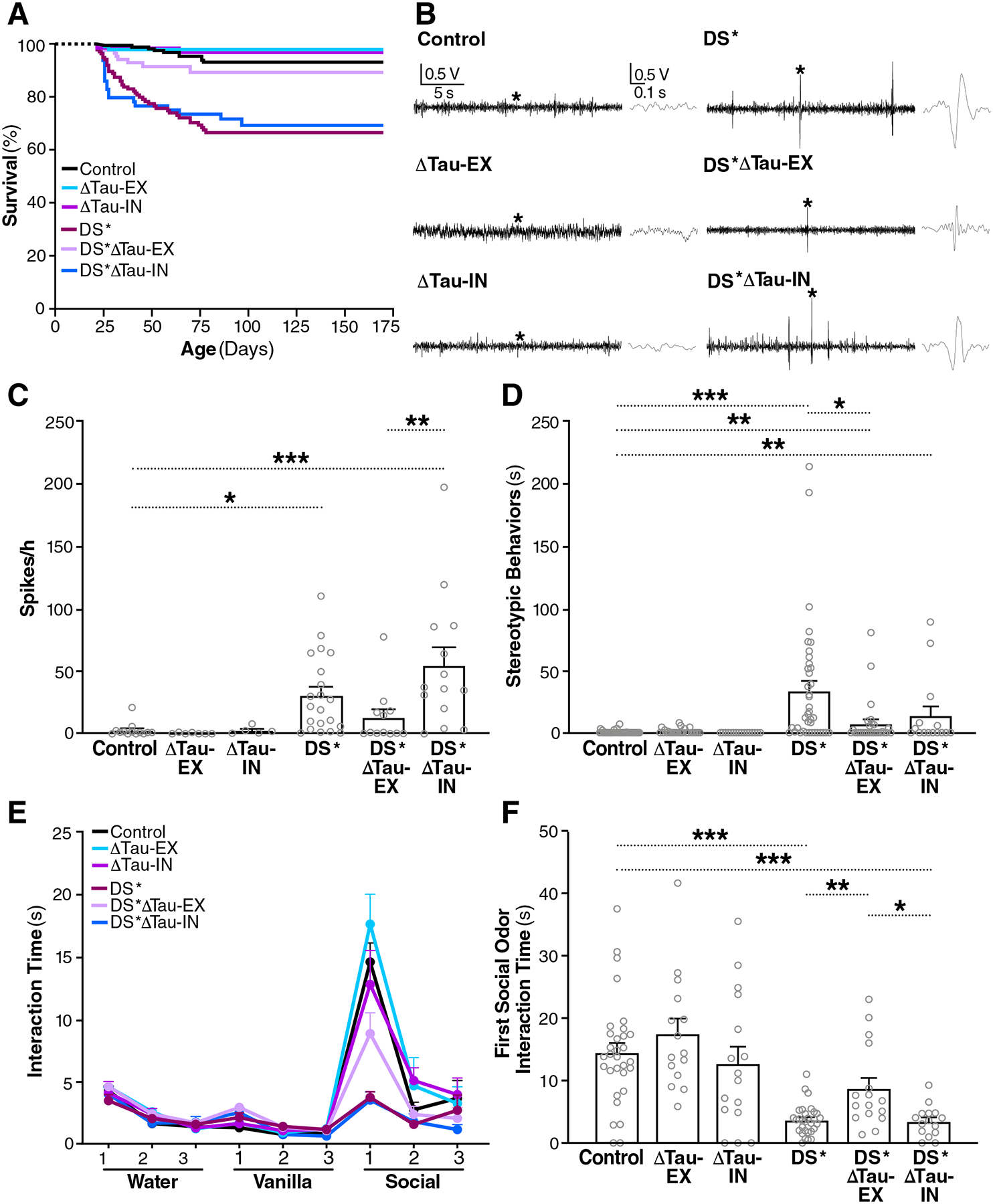
(A) Survival curves of Control, ΔTau-EX, ΔTau-IN, Scn1aRX/+/Maptflox/flox (DS*), Scn1aRX/+/Emx1IRES-cre/+/Maptflox/flox (DS*ΔTau-EX) and Scn1aRX/+/VgatIRES-cre/+/Maptflox/flox (DS*ΔTau-IN) mice. n = 61–168 mice per group at P20, when monitoring began. (B) Representative EEG traces recorded from 2.5-month-old mice of the indicated genotypes. Sections of traces marked with an asterisk are shown on the right at higher time resolution. (C) Frequency of epileptiform spikes quantified at 2.5 months of age. n = 5–21 mice per group. (D) Time mice were engaged in stereotypic (digging-like) behaviors at 6 months of age. n = 15–43 mice per group. (E and F) Olfactory habituation/dishabituation in 8-month-old mice. n = 15–31 mice. (E) Three different olfactory stimuli were presented for 6 min each in the indicated order and the amount of time mice interacted with each stimulus was recorded in three 2-min bins. Mouse bedding was used as the social odor. (F) Stimulus interaction time during the first 2 min of social odor presentation. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA and Holm-Sidak test (C) or permutation test with Holm-Sidak correction (D and F). Gray circles represent data from individual mice. Values in (C) to (F) are means ± SEM.
Dishabituation was measured as the difference of sniffing time between water 3 and vanilla 1 and between vanilla 3 and social 1, reflecting responses to a new nonsocial and a social odor, respectively. All groups of mice displayed similar dishabituation from water to vanilla (Fig. 2E), as determined by one-way analysis of variance (ANOVA) and Holm-Sidak test (P > 0.2 for all comparisons). For dishabituation from vanilla to social odor, a permutation test with Holm-Sidak correction revealed that DS* mice differed from Control (P = 0.001), ΔTau-EX (P = 0.001), ΔTau-IN (P = 0.001), and DS*ΔTau-EX (P = 0.0018) mice, but not from DS*ΔTau-IN mice (P = 0.2053). In addition, DS*ΔTau-EX mice differed from DS*ΔTau-IN mice (P = 0.049) and Controls (P = 0.0495). Focusing on the stimulus interaction time during the first two minutes of the social odor presentation (Fig. 2F), DS*ΔTau-EX mice engaged more extensively with the social odor than DS* (P = 0.0018) and DS*ΔTau-IN mice (P = 0.0218), whereas the sniffing time of DS*ΔTau-IN mice did not significantly differ from that of DS* mice (P = 0.7897).
Tau ablation in excitatory neurons reduces overactivation of the PI3K-AKT-mTOR pathway and megalencephaly in DS mice
Excessive activity of the PI3K-AKT-mTOR pathway has been implicated in the pathogenesis of epilepsies and ASDs (30, 31, 33, 39, 59) and can be prevented by genetic tau reduction in DS mice (25). We therefore measured the phosphorylation (activation) states of AKT (protein kinase B) and ribosomal protein S6, which have been widely used to assess PI3K-AKT-mTOR activity (25, 31, 39, 60–63). In contrast to phosphorylation of S6 at Ser235/236, phosphorylation of S6 at Ser 240/244 provides a reliable measure of mTOR activity (62, 64–66). Ablation of tau in excitatory neurons prevented PI3K-AKT-mTOR overactivation in DS* mice, as determined by Western blot analysis of phosphorylated AKT (p-AKT) and Ser240/244-phosphorylated S6 (p-S6; Fig. 3, A to C) in the hippocampus. DS* and DS*ΔTau-IN mice had more p-AKT than Controls, whereas DS*ΔTau-EX mice did not (Fig. 3, A and B). DS*ΔTau-EX mice had less p-AKT than DS* mice (Fig. 3, A and B). Among the groups analyzed, only DS* mice had more p-S6 than Controls (Fig. 3, A and C). For additional statistical comparisons between groups and the identification of trends, see table S1.
Fig. 3. Selective tau ablation in excitatory neurons prevents overactivation of the PI3K-AKT-mTOR pathway and megalencephaly in DS mice.
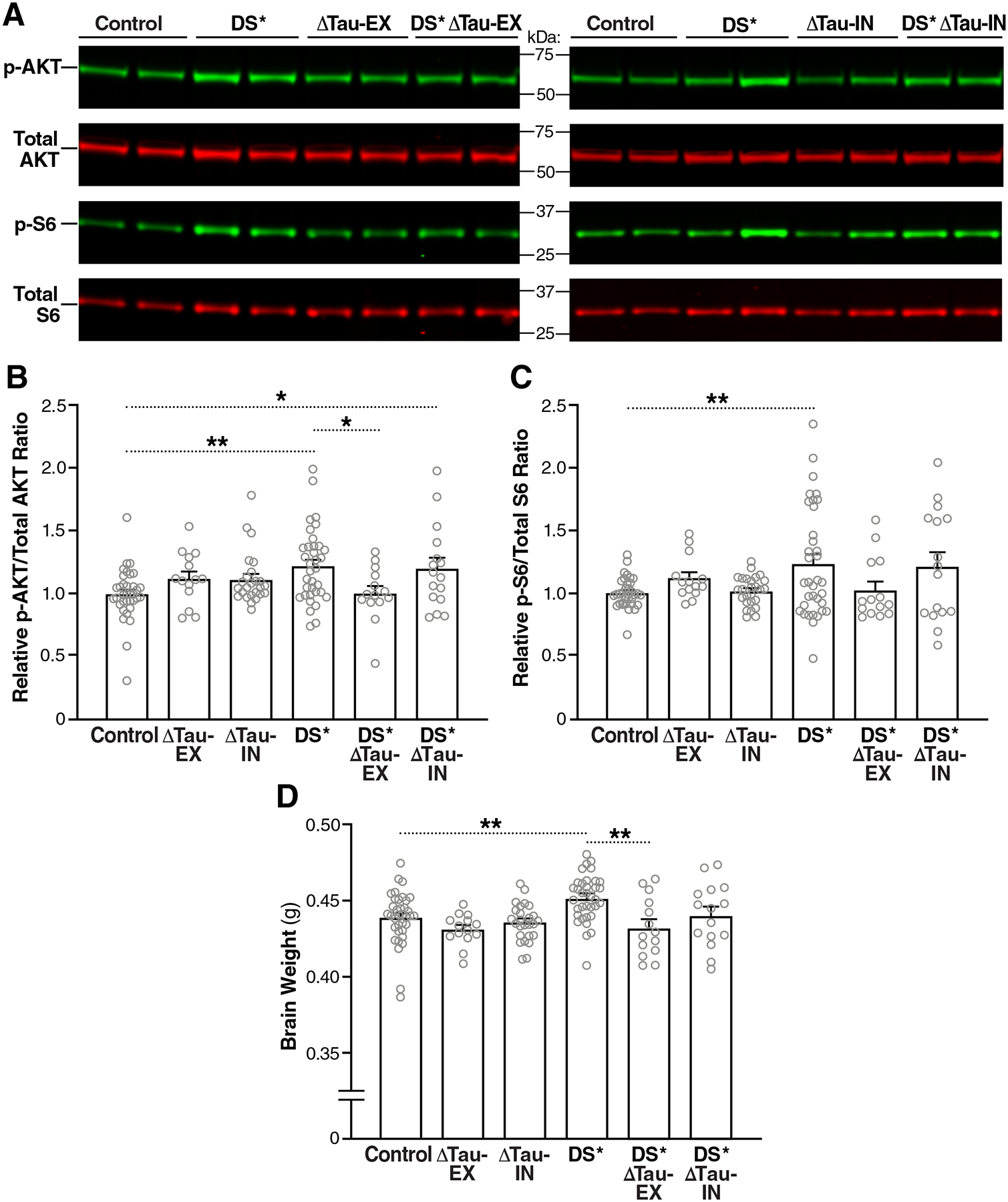
(A) Representative Western blot showing amounts of p-AKT, total AKT, p-S6 (Ser 240/244), and total S6 in the hippocampus of 2-month-old mice. MW markers are indicated in the middle. (B and C) p-AKT/total AKT (B) and p-S6/total S6 (C) ratios in the hippocampus of 2-month-old mice determined by Western blot analysis. Mean ratios in Control mice were defined as 1.0. (D) Brain weights of mice at 2 months of age. n = 14–36 mice per group. *P < 0.05 and **P < 0.01 by one-way ANOVA and Holm-Sidak test. Gray circles represent data from individual mice. Bars are means ± SEM.
As expected based on previous findings (25), DS* mice developed megalencephaly as they matured (Fig. 3D). At two months of age, only the brain weights of DS* mice differed from those of Controls, and DS*ΔTau-EX mice had lower brain weights than DS* mice (Fig. 3D), whereas DS*ΔTau-IN mice did not (P = 0.1264).
Together, these findings suggest that reducing tau in excitatory neurons counteracts the development of autism-like behavioral abnormalities as well as biochemical and anatomical alterations that may contribute to such brain dysfunctions.
ASO-mediated reduction of brain tau in wild-type and DS mice
To determine whether tau reduction can diminish neural network and behavioral dysfunctions in DS mice also when initiated postnatally, we injected DS mice (Scn1aRX/+ mice on a Mapt+/+ background) and wild-type (WT) controls ICV with saline, a nontargeting control ASO (NT-ASO), or a Mapt mRNA-targeting ASO (Tau-ASO) on postnatal day 10 (P10) and P56 (Fig. 4A). Roughly four months after the first injection, both ASOs were still widely distributed throughout the brain, as demonstrated by immunostaining in Fig. 4B and fig. S2A for WT mice. Staining of brain sections with the Tau5 antibody revealed that tau immunoreactivity was still reduced ~4 months after the first Tau-ASO injection (Fig. 4C). As documented by colabeling of brain sections with cell type-specific markers, the Tau-ASO reduced tau expression across different brain regions and cell types, including in excitatory and inhibitory neurons in the prefrontal cortex, hippocampal CA1 and CA3 region, dentate gyrus, and cerebellum, as well as in oligodendrocytes in the prefrontal cortex, dentate gyrus, fimbria, and cerebellum (Fig. 4, D to F and fig. S2, B to E).
Fig. 4. Widespread distribution of ASOs and tau knockdown in mouse brains.
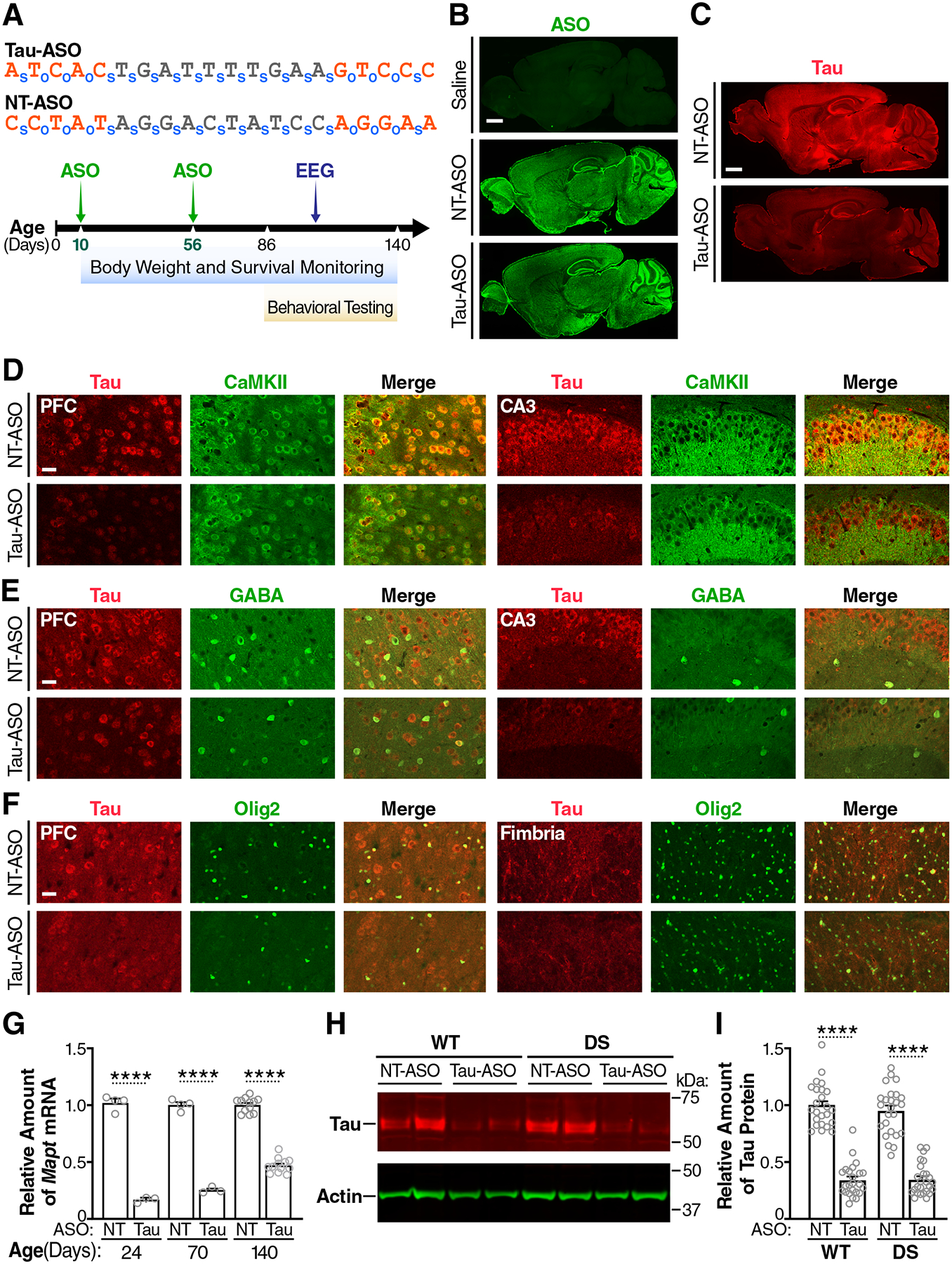
(A) Nucleotide sequences of the Tau-ASO and NT-ASO (top) and experimental design (bottom). Nucleotides containing 2′-O-methoxyethyl modifications are shown in orange. o, phosphodiester bond; s, phosphorothioate bond. WT and Scn1aRX/+ (DS) mice were injected ICV with the Tau-ASO or NT-ASO at P10 (200 μg) and P56 (300 μg). Replicate groups of mice were analyzed at P24, P70, or P140 for hippocampal tau expression and for other outcome measures as indicated. (B and C) Sagittal brain sections from 140-day-old WT mice showing representative distributions of ASO (B) and tau (C) immunostaining after the indicated ICV injections. Scale bars, 600 μm. (D to F) Sagittal brain sections from NT-ASO or Tau-ASO–treated 140-day-old WT mice coimmunolabeled for tau (red) and markers (green) of excitatory neurons (CaMKII) (D), inhibitory neurons (GABA) (E), or oligodendrocytes (Olig2) (F). PFC, prefrontal cortex; CA3, hippocampal subregion. Scale bars, 30 μm. (G) Hippocampal amounts of Mapt mRNA in replicate groups of WT mice treated as indicated and analyzed by RT-qPCR at P24 (n = 3–4 mice per group), P70 (n = 3–4 mice per group), or P140 (n = 12–13 mice per group). Mapt/Gapdh mRNA ratios in age-matched NT-ASO–treated mice were defined as 1.0. (H) Representative Western blot showing hippocampal amounts of tau and β-actin (loading control) in the indicated groups of mice at P140. Each lane contains a sample from a different mouse. (I) Hippocampal amounts of tau protein in the indicated mice (n = 24–26 per group) were quantified by Western blotting at P140. Mean tau/β-actin ratios in NT-ASO–treated WT mice were defined as 1.0. Gray circles represent data from individual mice. ****P < 0.0001 by two-way ANOVA and Holm-Sidak test. Values in (G) and (I) are means ± SEM.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis of hippocampal RNA from replicate groups of mice revealed that treatment with the Tau-ASO reduced the abundance of Mapt mRNA to 16–25% of control values at two weeks after the first and second ICV injections and that the amount of Mapt mRNA had returned to 47% of control values by ~4 months after the first injection (Fig. 4G). However, Western blot analysis at the latter time point showed that hippocampal tau protein was still reduced to 35% of control values in both DS and WT mice (Fig. 4, H and I).
Reduced SUDEP, epileptiform activity, and autism-like behaviors in Tau-ASO–treated DS mice
To determine whether postnatal knockdown of tau reduces SUDEP in DS mice, we monitored the survival of mice treated with Tau-ASO or NT-ASO (Figs. 4A and 5A) from P25, when brain tau expression was at its lowest in Tau-ASO–treated mice (Fig. 4G), until P140, when tau protein in such mice was still reduced (Fig. 4, H and I) but the amount of Mapt mRNA was beginning to rise toward 50% of normal (Fig. 4G). Tau-ASO treatment reduced SUDEP (Fig. 5A) and epileptiform activity (Fig. 5B) in DS mice, as compared to NT-ASO–treated DS mice. Analysis of the survival curves (Fig. 5A) by log-rank (Mantel-Cox) test with Holm-Sidak correction revealed a significant difference between NT-ASO treated WT and DS mice (P = 0.0015) and between DS mice treated with NT-ASO versus Tau-ASO (P = 0.0277), but not between Tau-ASO–treated DS mice and WT mice treated with NT-ASO (P = 0.3977) or Tau-ASO (P = 0.0666). These survival curves provide an indirect measure of early-onset seizures that are most likely to cause SUDEP, particularly generalized tonic-clonic seizures (67, 68). To assess the effect of tau reduction on such seizures more directly, we treated an independent cohort of DS mice with the Tau-ASO or the NT-ASO at P10, implanted intracranial EEG electrodes at P25, and continuously monitored mice by video-EEG from P27 to P30 (fig. S3A). Tau-ASO treatment reduced the proportion of DS mice that exhibited generalized tonic-clonic movements in association with EEG seizure activity during this recording period (fig. S3, B and C): ~18% of Tau-ASO–treated mice compared to ~58% of NT-ASO–treated mice (P < 0.05). To broadly monitor for potential off-target effects and assess the tolerability of cerebral tau knockdown during early postnatal maturation, we measured the body weights of ASO-treated mice. No differences were found in weights and weight gains among the treatment groups (Fig. 5C).
Fig. 5. Postnatal Tau-ASO treatment reduces SUDEP, epileptiform spike frequency, and autism-like behaviors in DS mice.
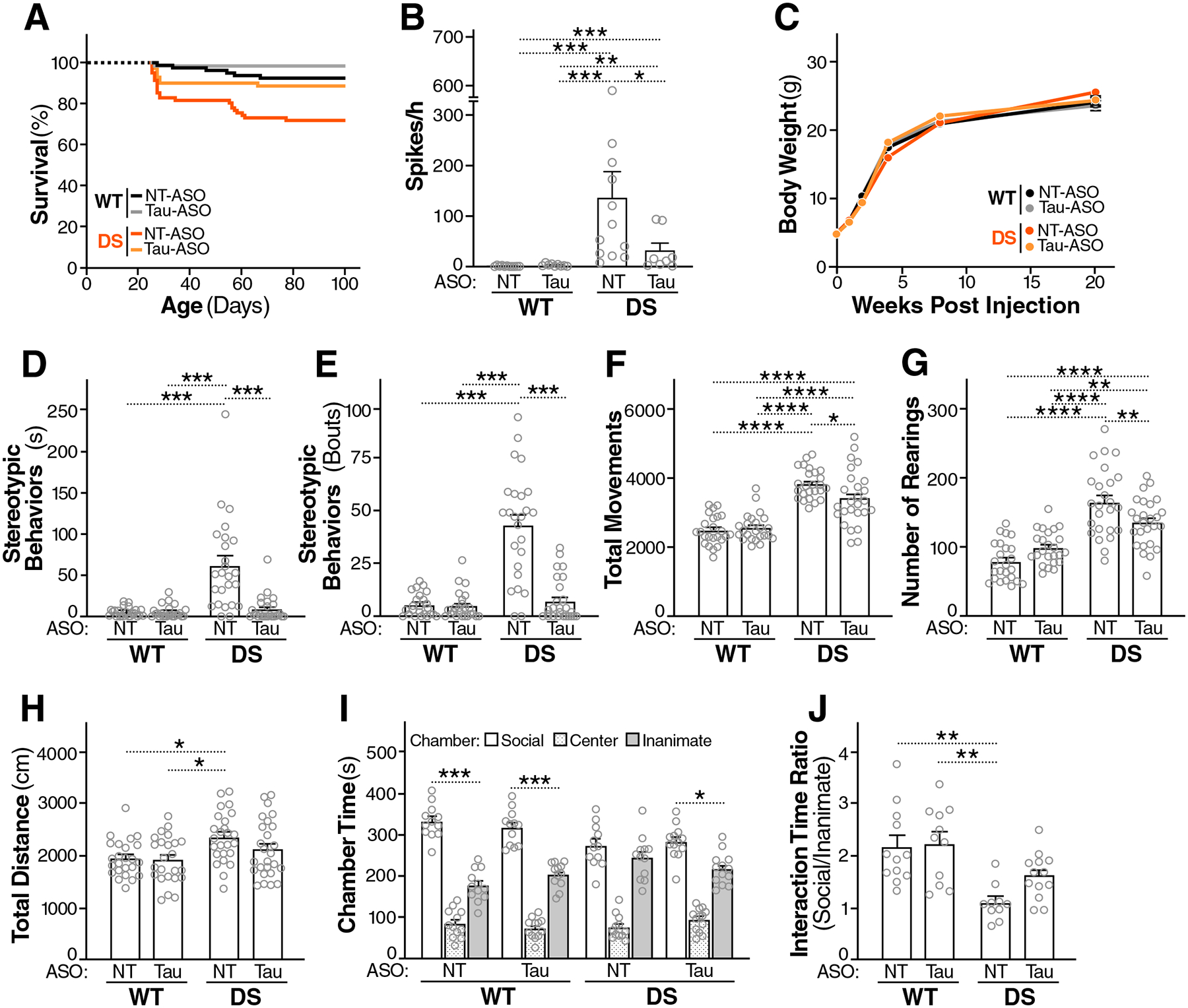
(A) Survival curves of WT and DS mice treated with NT-ASO or Tau-ASO as in Fig. 4. n = 68–82 mice per group at P25, when monitoring began. (B) Frequency of epileptiform spikes in WT and DS mice recorded between P90 and P110 (Fig. 4A). (C) Maturation related increases in body weights across groups. (D to J) Behavioral effects of ASO treatments were assessed in WT and DS mice between P86 and P140 (Fig. 4A). The following autism-related behaviors were assessed: time engaged in stereotypic (digging-like) behaviors (D) and number of bouts of such behaviors (E), total movements (F) and number of rearings (G) in the open field, distance traveled in the elevated plus maze (H), and social preference in the three-chamber test as evidenced by time spent in the social or inanimate chamber (I) and ratio of times mice spent interacting with social or inanimate cups within these chambers (J). n = 8–12 male and female mice (B), 24–26 male and female mice (C to H), 12–13 female mice (I), or 10–13 female mice (J) per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Mann-Whitney U test with Holm-Sidak correction (B), permutation test with Holm-Sidak correction (D and E), two-way ANOVA and Holm-Sidak test (F to H and J), or Student’s t test with Holm-Sidak correction (I). Gray circles represent data from individual mice. Values in (B) to (J) are means ± SEM.
Next, we compared the behavior of DS and WT mice after ICV injections of Tau-ASO or NT-ASO (Fig. 4A) in ASD-relevant tests. Tau-ASO treatment reduced excessive stereotypic (digging-like) behaviors (Fig. 5, D and E) and hyperactivity (Fig. 5, F to H) in DS mice. In the three-chamber social preference test, Tau-ASO treatment yielded trends toward improvements in female DS mice (Fig. 5, I and J), but not male DS mice (fig. S4). In the female cohort, WT mice treated with NT-ASO or Tau-ASO, as well as DS mice treated with Tau-ASO, spent more time in the chamber that contained an inverted wire cup with a live mouse (social cup) than in the chamber that contained a cup with an inanimate object, whereas NT-ASO treated DS mice did not (Fig. 5I). In addition, the ratio of time periods mice spent interacting with the social or inanimate cup were reduced in NT-ASO treated, but not Tau-ASO treated, DS mice, as compared to WT controls treated with NT-ASO or Tau-ASO (Fig. 5J).
Similar to genetic ablation of tau (24), postnatal knockdown of tau did not prevent the depletion of NaV1.1 in brain tissues of DS mice (fig. S5). It is therefore likely that these interventions reduce SUDEP and behavioral abnormalities by modulating pathogenic mechanisms downstream of the NaV1.1 haploinsufficiency.
Tau-ASO treatment blocks overactivation of the PI3K-AKT-mTOR pathway and megalencephaly in DS mice
Compared with WT controls, hippocampal amounts of p-AKT (Fig. 6, A and B) and p-S6 (Fig. 6, A and C) were increased in NT-ASO–treated, but not Tau-ASO–treated, DS mice. Immunohistochemical analysis of brain sections from these mice revealed increased abundance of p-S6 in various brain regions of NT-ASO–treated, but not Tau-ASO–treated, DS mice, as compared to WT mice treated with either ASO (Fig. 6D).
Fig. 6. Tau-ASO treatment reduces the overactivation of the PI3K-AKT-mTOR pathway and megalencephaly in DS mice.
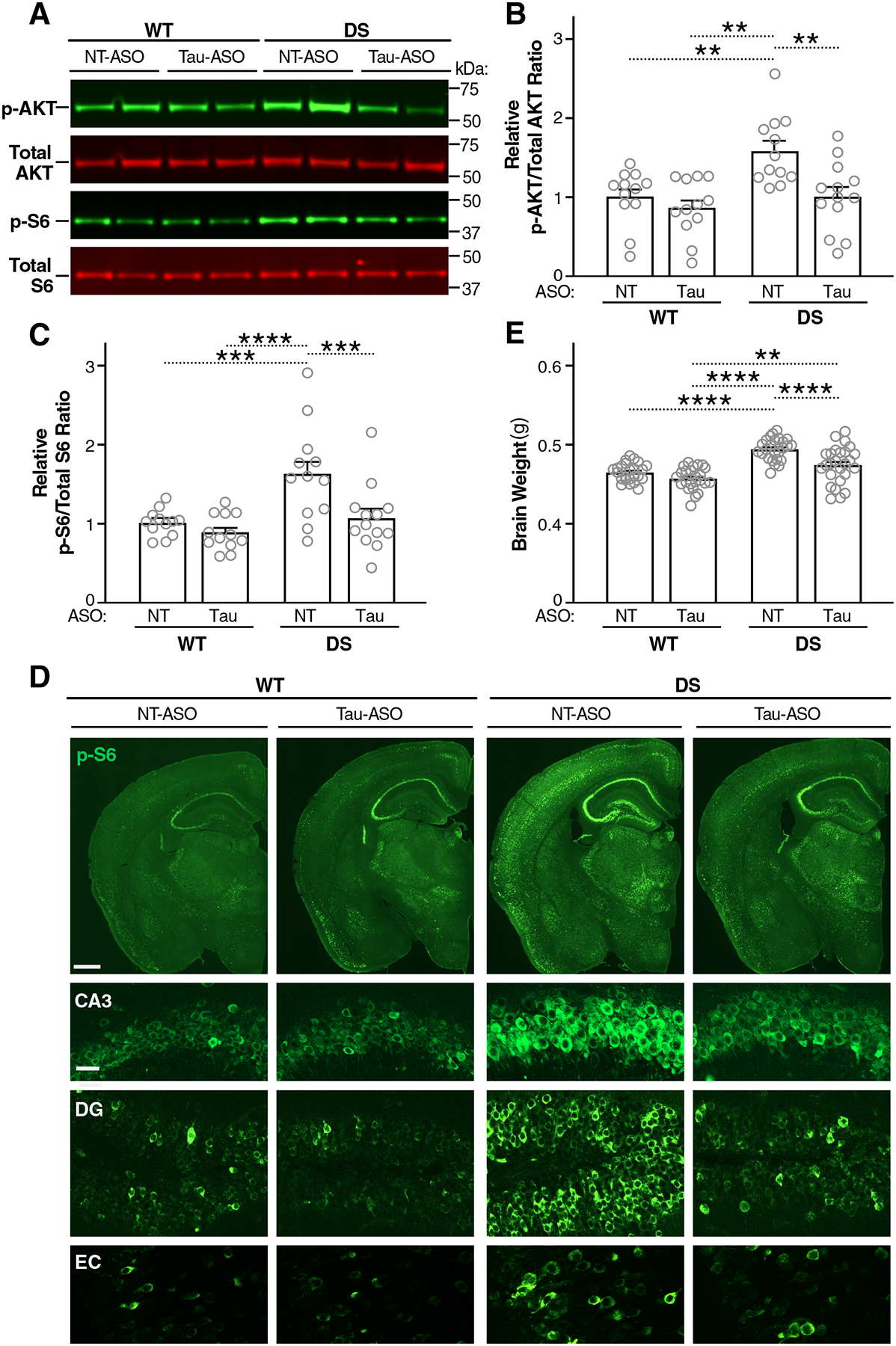
(A to E) After treatment with ASOs as in Fig. 4, WT and DS mice were assessed at P140 for hippocampal activation of the PI3K-AKT-mTOR pathway and brain weight. (A) Representative Western blot showing hippocampal amounts of p-AKT, total AKT, p-S6 (Ser 240/244), and total S6. MW markers are indicated on the right. (B and C) p-AKT/total AKT (B) and p-S6/total S6 (C) ratios in the hippocampus determined by Western blot analysis. Mean ratios in NT-ASO–treated WT mice were defined as 1.0. n = 12–13 animals. (D) Representative images of coronal brain sections immunostained for p-S6 (Ser 240/244). Hemibrains (scale bar, 600 μm) are shown in the top row and details (scale bar, 30 μm) of the hippocampal subfield CA3, the dentate gyrus (DG), and the entorhinal cortex (EC) are shown below. (E) Brain weights. n = 24–26 mice per group. **P < 0.01, ***P < 0.001, and ****P < 0.0001 by two-way ANOVA and Holm-Sidak test. Gray circles represent data from individual mice. Bars are means ± SEM.
Because both autism and overactivation of the PI3K-AKT-mTOR pathway can be associated with megalencephaly and this anatomical abnormality can be prevented by genetic reduction of tau (25), we compared the brain weights of ~4-month-old DS and WT mice after they were treated with Tau-ASO or NT-ASO (Fig. 4A). Tau-ASO treatment prevented megalencephaly in DS mice (Fig. 6E).
DISCUSSION
These findings suggest that tau expression in excitatory neurons contributes more to the development of epilepsy, SUDEP, overactivation of the PI3K-AKT-mTOR pathway, megalencephaly, and autism-like behaviors in NaV1.1-haploinsufficient DS mice than tau expression in inhibitory interneurons. They further demonstrate that reducing tau in excitatory neurons is sufficient to prevent or markedly reduce the development of these abnormalities, indicating that these cells are a critical, if not the most important, cell population to target with tau-lowering therapeutic interventions, at least in the context of NaV1.1 haploinsufficency. The results of our knockdown experiments further highlight that tau reduction does not need to be initiated in utero to have therapeutic benefits and that tau-targeting ASO treatment initiated after birth can prevent or ameliorate multiple disabling and even life-threatening consequences of NaV1.1 haploinsufficency in a mouse model of DS. Thus, our study provides important insights into the cellular mechanisms of tau-dependent neural dysfunctions as well as potentially useful guidance for the development of tau-targeting therapeutics, particularly in regard to autism and epilepsy of early onset.
In general, epileptic activity can emerge from alterations in excitatory or inhibitory neurons, and it can be prevented or suppressed by modulating activities of either cell type (69–71). Although the likeliest cause of epilepsy after PTZ injection, and in DS mice, is deficient inhibitory regulation of excitatory neurons (17–19, 72), in both models tau ablation counteracted the development of epilepsy only when implemented in excitatory, but not inhibitory, neurons. These findings raise the possibility that tau ablation reduces the propensity of excitatory neurons to become engaged in hypersynchronous network activity. Consistent with this hypothesis, we recently showed that tau ablation reduces the activity and excitation/inhibition (E/I) ratio of excitatory neurons at baseline and after excessive depolarization, which would be expected to counteract conditions that cause excessive E/I ratios such as DS, other types of epilepsy, and some ASDs, independent of their diverse pathogenic triggers (73). The extent to which these effects depend on modulation of the PI3K-AKT-mTOR pathway merits further investigation.
Genetic alterations that cause early-onset epilepsies are often associated with ASDs, raising the possibility that aberrant increases in the E/I ratio and network hyperexcitability promote the development of autistic behaviors, at least under certain pathophysiological conditions (7, 37, 74–77). Similar questions have been raised about a potential cause-effect relationship between megalencephaly and autism (34, 36, 38, 78). Aberrant activity of the PI3K-AKT-mTOR pathway may link all three of these disease manifestations (33, 36, 39). Overactivation of this pathway can both result from and cause epileptic activity, and can also cause megalencephaly and autism-like behaviors (25, 31, 33, 36, 38, 39, 78). In our study, tau ablation in excitatory, but not inhibitory, neurons prevented or reduced not only epileptic activity and SUDEP, but also overactivation of the PI3K-AKT-mTOR pathway, megalencephaly, and several autism-like behaviors, highlighting the importance of tau expression in excitatory neurons in the pathogenesis of these abnormalities. The phosphatase and tensin homolog deleted on chromosome 10 (PTEN) counteracts the activity of PI3K and is negatively regulated by tau (25, 79). We previously identified increased PTEN activity as a likely molecular mechanism through which tau reduction prevents the ASD-promoting overactivation of the PI3K-AKT-mTOR pathway (25).
Because the overall magnitude of p-AKT and p-S6 changes we identified may seem subtle, it should be noted that we reduced tau in select cell populations but analyzed bulk tissues in which the PI3K-AKT-mTOR pathway was active in diverse types of brain cells (60, 63), including many cells in which we did not reduce tau or in which tau would not be expected to modulate this pathway because it is expressed in very low amounts at baseline (46). Consequently, signal changes in neurons affected by DS and tau reduction were likely diminished by the lack of changes in other cell types that were unaffected by these variables.
Pharmacological inhibition of the PI3K-AKT-mTOR pathway reduces epilepsy (30, 31) and autism-like behaviors (33, 39) in mouse models, but has shown variable efficacy and can have multiple side effects, including hyperglycemia, hyperlipidemia, pneumonitis, hepatotoxicity and bone marrow suppression. In our study, tau reduction suppressed overactivation of the PI3K-AKT-mTOR pathway in brain tissues and was well tolerated, independent of whether it was achieved through ASO treatment or conditional ablation in specific types of neurons. The lack of obvious adverse effects of tau reduction in our study is consistent with previous findings obtained after global genetic tau reduction or ASO-mediated cerebral tau knockdown at other ages in mice and nonhuman primates (45, 51, 58). Reports of results from a clinical trial of a tau-targeting ASO in patients with AD (NCT03186989) are pending at this time.
A major question in developing and deploying therapeutics for epilepsy, autism, and many other brain disorders is when interventions would have to be initiated to prevent or reduce disabling symptoms. Our study demonstrates that early postnatal knockdown of tau reduces epilepsy, SUDEP, overactivation of the PI3K-AKT-mTOR pathway, megalencephaly, and several autism-like behaviors in DS mice. Thus, tau reduction could help prevent the development of such abnormalities even when initiated after birth. Additional therapeutic targets for ASO-mediated modulation in DS include Scn1a itself. Increasing the expression of functional NaV1.1 by modulating the splicing of WT Scn1a with ASOs in heterozygous Scn1a knockout mice ameliorated multiple DS-like disease manifestations (80). Studies in AD-relevant models (81, 82) suggest that—similar to tau reduction [ref. (45) and this study]—enhancing the function of NaV1.1 may be of benefit not only in DS, but also in other conditions associated with interneuron impairments and neural network dysfunctions.
In general, ASOs are considered viable therapeutics and have become the focus of an increasing number of preclinical and clinical trials for CNS diseases (50–53, 83), including for Angelman syndrome, AD, amyotrophic lateral sclerosis, and—most successfully so far—spinal muscular atrophy, a neurodegenerative disorder that, similar to ASDs, predominantly affects children (53, 84, 85). An ASO targeting survival of motor neuron 2 pre-mRNA has already been approved for treatment of spinal muscular atrophy by the U.S. Food and Drug Administration and the European Medicines Agency (53, 85).
In our experimental model, postnatal knockdown of tau reduced some behavioral abnormalities more effectively than others. For example, Tau-ASO treatment reduced excessive stereotypic behaviors more markedly than hyperactivity, although it diminished both behavioral alterations. We observed little to no amelioration of social deficits after the Tau-ASO treatment, whereas such deficits were more robustly reduced by the conditional ablation of tau in excitatory neurons (this study) and by global genetic reduction of tau (25). Distinct autism-like behaviors may differ in regard to the time period during which they can be modulated by tau reduction and with respect to the extent of tau reduction required to achieve this outcome, depending on the neuronal populations involved, the underlying pathogenic mechanisms, and the developmental stages during which they emerge. In a mouse model of Rett syndrome, a low dose of an MeCP2-targeting ASO was sufficient to reduce excessive stereotypic behaviors, whereas the reduction of learning deficits and excessive anxiety required higher doses or more prolonged treatment (83). At least some stereotypic behaviors seem to be regulated by a circuit involving the prefrontal cortex, basolateral amygdala, and hippocampus (86, 87). Perhaps, this circuit is, in general, more responsive to therapeutic interventions than circuits and networks subserving more complex brain functions such as learning and social interactions. In addition, it is possible that the pathogenesis of some autism-related behavioral abnormalities is more dependent on tau than that of others. This and other differences in underlying pathomechanisms may shift the developmental and disease stages during which different types of brain dysfunctions are amenable to therapeutic modulation.
Because ASOs need to be delivered directly into the cerebrospinal fluid by intrathecal or ICV injection, the development of orally bioavailable small-molecule drugs that can effectively and safely lower all forms of tau in excitatory neurons is an important objective. In addition, studies are needed to further assess the usefulness of tau reduction in models of other potentially tau-dependent brain disorders, including different ASDs, and to define the best treatment regimens, including optimal periods for treatment initiation as well as the extent and duration of tau reduction that is required to effectively and persistently reduce disease manifestations.
Our study has several limitations. Because it focused on a specific line of knockin mice, additional studies are needed to determine whether the selective ablation of tau in excitatory neurons and the postnatal knockdown of brain tau expression also have beneficial effects in models of other epilepsies, ASDs, and related conditions causing aberrantly increased E/I ratios.
Although our study was not designed to identify the molecular mechanisms by which tau reduction counteracts the development of epilepsy, SUDEP, and autism-like behaviors in DS mice, it clearly identified excitatory neurons as a key cell type on which this quest should focus in the future. In light of our previous work (25) and the current study, it would be particularly interesting to further dissect tau’s ability to regulate the PI3K-AKT-mTOR pathway in this cell type. It could also be worthwhile to assess whether other autism-relevant pathways such as Wnt signaling (88, 89) are altered in DS mice and normalized by tau reduction. It may be informative to more directly correlate tau-dependent modulations of signaling pathways with network and behavioral dysfunctions in the same cohort of mice. However, if the signaling changes were transient and had persistent effects, they might precede the development of structural and functional alterations but not correlate with the extent of these alterations after they have become manifest. Because Emx1 targets Cre expression not only to neurons, but also to some glial populations (54), it could also be of interest to further assess tau’s roles in glial cells with robust amounts of tau expression such as oligodendrocytes (46). In this study, we evaluated the effects of postnatal tau reduction only at P10. From a therapeutic perspective, it would be useful to assess additional intervention times to determine the latest stages of brain development and maturation at which tau reduction still shows benefits. Most experimental models simulate some, but not all, aspects of the conditions they were designed to investigate, and species differences can complicate extrapolations from animal models to humans. Therefore, caution is always in order when advancing discoveries from preclinical trials into therapeutic strategies for assessments in clinical trials. Although the ASO-mediated knockdown of tau in the CNS is well tolerated in mice and nonhuman primates [refs. (50, 51) and this study], the results of a clinical trial of an intrathecally delivered tau-targeting ASO in humans (NCT03186989) have not yet been published. The need to repeatedly administer ASOs by intrathecal or ICV injection is a limitation that the development of orally bioavailable small-molecule tau-reducing drugs should be able to overcome.
MATERIALS AND METHODS
Study design
The research objectives, subjects of investigation, and overall experimental design are all described in Results. Briefly, we used behavioral, electrophysiological, anatomical and biochemical methods to test the hypotheses that (i) selective genetic ablation of tau in excitatory or inhibitory neurons and (ii) postnatal knockdown of brain tau have similar effects on the phenotype of Scn1aRX/+ mice as global genetic reduction of tau. Outcome measures and sample sizes were determined a priori based on previous studies of Scn1aRX/+ mice on Mapt+/+, Mapt+/–, and Mapt–/– backgrounds. For all behavioral testing, EEG recordings, histological analyses, and biochemical assays, experimenters were blinded to the genotype and treatment of mice. For behavioral testing and EEG recordings, the testing order of mice was randomized. With the following exception, no data were excluded. Mice with obvious health problems such as skin lesions, eye injuries, tumors, or slowed movements were excluded from behavioral testing and analysis. Behavioral experiments were replicated in at least two independent cohorts of mice. In all experiments, data obtained in individual mice were considered one biological replicate. Data collection was not stopped until the experiments were completed. Most experiments were replicated in at least two independent sex-balanced cohorts of mice.
Mice
Scn1aRx/+ mice generated by Dr. K. Yamakawa (Laboratory for Neurogenetics, RIKEN Brain Science Institute) were obtained from Dr. M. H. Meisler (University of Michigan) and crossed onto the C57BL/6-J background for >10 generations. Tau knockout (Mapt–/–) mice on the C57BL/6-J background (no. 007251) were obtained from the Jackson Laboratory. Emx1IRES-cre (no. 005628) and VgatIRES-cre (no. 028862) knockin mice on the C57BL/6J background were also obtained from the Jackson Laboratory. Maptflox/flox mice on the C57BL/6-J background were generated by homologous recombination in C57BL/6-J embryonic stem cells in the Roberson Laboratory. All mice were maintained on a C57BL/6-J background by breeding genetically modified mice with WT C57BL/6-J mice from the Jackson Laboratory (000664). Scn1aRx/+ male mice were bred with WT female mice to generate Scn1aRx/+ and WT mice for ASO treatments. Hemizygous Scn1aRx/+ mice, Emx1IRES-cre/+ mice, and VgatIRES-cre/+ knockin mice were separately crossed onto the Maptflox/flox background. Scn1aRx/+/Maptflox/flox males were then bred with either Emx1IRES-cre/+/Maptflox/flox or VgatIRES-cre/+/Maptflox/flox females to generate Scn1a+/+, Scn1aRX/+, Emx1IRES-cre/+, VgatIRES-cre/+, Scn1aRX/+/Emx1IRES-cre/+, and Scn1aRX/+/VgatIRES-cre/+ mice on the Maptflox/flox background. Unless indicated otherwise, experimental and control groups were sex-balanced, age-matched, and generated by the same breeding schemes. Mice were housed in groups of 2–5 per cage on a 12 hour light/dark cycle with ad libitum access to food (PicoLab Rodent Diet 20, 5053; LabDiet) and water. For euthanasia, anesthetized mice were perfused transcardially with ice-cold 0.9% NaCl solution. Brain tissues were immediately dissected on ice and processed and analyzed with the reagents summarized in table S2 as described further below. All experiments involving mice were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
PTZ-induced seizures
Mice were intraperitoneally injected with saline or PTZ (50 mg/kg body weight). Immediately after the injection, mice were observed for the development of movements or behaviors indicative of seizure activity. Seizure severity was quantified blinded to the genotype and treatment of mice using the following scores: 0, no abnormal movements or behaviors; 1, immobility; 2, tail twitch; 3, whole body twitch; 4, a single body contraction; 5, serial body contractions; 6, series of abnormal movements (tail twitch, body shaking or contraction; sometimes associated with loss of balance) lasting 5–10 seconds; 7, tonic-clonic convulsions; and 8, death.
ASO treatments
The NT-ASO and Tau-ASO [Fig. 4A and ref. (90)] were obtained from Ionis Pharmaceuticals. Both ASOs have five nucleotides on the 5′- and 3′-termini containing 2′-O-methoxyethyl modifications and ten unmodified central oligodeoxynucleotides to support ribonuclease H activity, as well as phosphorothioate backbone linkages at select locations to improve nuclease resistance and promote cellular uptake. Each ASO was solubilized in sterile 1x phosphate-buffered saline (PBS) without Ca2+/Mg2+ (Gibco, 14190). ASO concentrations were determined with a NanoDrop system (Thermo Fisher Scientific) and adjusted to 100 mg/ml. For manual ICV injections of ASOs at P10, mouse pups were anesthetized with isoflurane. The head was stabilized by hand, and a 27-gauge needle connected to a 25 μl Hamilton syringe was used to puncture the skin and skull and to inject 200 μg of ASO in 2 μl of vehicle into the right lateral ventricle (~5 mm behind the front of the eyes and ~1.5 mm from the midline) over a 5-s period. To reduce leakage along the needle track, the needle was held in place for 15 s. After the injection, mice were kept in a warm container for 5–10 min until spontaneous movements and proper responsiveness to stimuli were evident. For ICV injections at later ages, mice were anesthetized with isoflurane and placed in a stereotaxic instrument (Stoelting). After applying local anesthetic and sterilizing the surgical site, a midline incision was made over the skull and a small hole was drilled through the skull. A 10 μl Hamilton syringe connected to a motorized nanoinjector was then used to inject 300 μg of ASO in 3 μl of vehicle into the right lateral ventricle (coordinates relative to the bregma: anteroposterior, −0.22 mm; mediolateral, +1.0 mm; dorsoventral, −2.5 mm) over a 6-min period. After the injection, the needle was left in place for another 5 min. The skin incision was closed with a suture, followed by subcutaneous injection of buprenorphine (0.05 mg/kg) and carprofen (5 mg/kg) to alleviate any postsurgical pain or discomfort. Mice were monitored for at least 3 days after the surgery.
Behavioral testing
Data acquisition and analysis were carried out by investigators blinded to the genotype and treatment of mice. All behavioral tests were performed during the light period. Before testing, mice were transferred to the testing room and allowed to acclimate for at least 1 hour. All work surfaces were cleaned with Vimoba (100 parts per million of chlorine dioxide solution made from MB-10 Tablets, Quip Laboratories) at the beginning and end of the day and with 70% alcohol between test sessions. The number and sequence of behavioral paradigms in which mice were tested was the same for experimental and control mice within any given cohort, but differed across cohorts because of practical variables such as the testing schedule of the Behavioral Core, which often supports multiple projects in parallel.
Open-field
Spontaneous activity in the open field was measured with an automated Flex Field Open Field Photobeam Activity System (San Diego Instruments). Mice were tested for 15 min in a clear plastic chamber (41 cm by 41 cm by 30 cm) equipped with two 16 by 16 photobeam arrays for the detection of horizontal and vertical movements. Total movements, number of rearings, entries into the center of the arena, and the ratio of movements made in the center versus periphery of the arena were recorded automatically for subsequent analysis.
Elevated plus maze
The maze consisted of two open arms (without side walls) and two enclosed arms (with side walls) elevated 63 cm above the ground (Kinder Scientific). Mice were placed at the junction among the arms facing one of the open arms and allowed to explore the maze for 10 min. Total distance traveled and time spent in the open versus closed arms were monitored with infrared photobeams.
Stereotypic behaviors
Mice were placed individually into clean cages (25 cm by 18 cm by 16 cm) without bedding for 20 min and videotaped. To avoid habituation-related confounds, only the last 10 min of each video was scored for the total amount of time mice were engaged in stereotypic behaviors and the number of bouts during which such behaviors dominated their spontaneous activity. Because the most prevalent stereotypic behavior of Scn1aRx/+ mice in this study consisted of repetitive forearm movements in a downward direction, these digging-like behaviors were used for quantifications.
Three-chamber social preference
The three-chamber apparatus consisted of a polycarbonate box (60 cm by 40 cm by 22 cm) with removable partitions that separate the box into three chambers with a 15 cm by 40 cm middle chamber and two 22.5 cm by 40 cm side chambers. A wire cup (7.6 cm in diameter and 10 cm in height) was placed upside down in each of the side chambers. Experimental mice were initially placed individually into the middle of the apparatus and allowed to explore all chambers for 10 min in the absence of a stimulus mouse. For the social preference trial, an age- and sex-matched stimulus mouse that had been habituated to one of the wire cups for two 15-min sessions the day before was placed under the wire cup in the “social” side chamber, whereas an inanimate object was placed under the wire cup in the “nonsocial” side chamber. Which of the side chambers contained the stimulus mouse was varied between trials to control for any chamber-specific confounds. During the social preference trial, experimental mice were allowed to freely explore all chambers for 10 min. The test was done under dim light. Movements were videotaped, and interactions with the wire cups were analyzed offline with the TopScan tracking system (CleverSys). The amount of time mice spent in each chamber and within 1.5 cm of the cups (interaction zone), as well as the number of times their noses entered the interaction zone, were quantified.
Olfactory habituation/dishabituation
Testing was carried out inside a biosafety cabinet or laminar flow hood. Mice were placed individually into clean cages (25 cm by 18 cm by 16 cm) without bedding for a 20–25-min habituation. They were then presented with cotton swab applicators dipped into water or vanilla extract (Spice Islands, 403911) or swiped in soiled mouse bedding (social odor). Vanilla extract was diluted 1:100 in water and stored in a plastic vial. Social odors were prepared by swiping cotton tip applicators across the bottom of a cage of an unfamiliar mouse of the same strain and sex in a zigzag fashion several times; bedding was removed from the cotton tip. Each odor was presented for 6 min, and the response of mice to the odor was scored throughout this period in 2-min bins. Odor presentations were separated by a 1-min interval. Applicators were inserted individually through the middle of the flat wire cage top and secured with a binder clip so that the cotton tip was roughly 5.8 cm above the cage floor. The number of interactions and the time mice spent interacting with the applicator were recorded. Interaction was defined as the mouse facing the cotton tip and its nose being within 2 cm of it. Climbing on the applicator was not counted as interaction time. The whole procedure was videotaped for confirmatory analysis.
Electroencephalography (EEG)
Lightweight EEG plugs were made in-house by soldering four Teflon-coated silver wire electrodes (0.125 mm in diameter) to a multichannel electrical connector. After mice were anesthetized with isoflurane, EEG electrodes were surgically implanted under the skull and over the left frontal cortex (reference electrode) and the left and right parietal cortices as described (91). Mice were allowed to recover from surgery for 2 weeks before EEG recordings began. Digital EEG activity and videos of their locomotor activity were recorded with a PowerLab data acquisition system and analyzed with Labchart 7 Pro software (AD Instruments). EEG traces and videos were evaluated by an investigator blinded to the genotype of mice. Epileptiform spikes were detected automatically with a macro written in Labchart 7. Deflections were identified as epileptiform spikes if their amplitude was ≥4-fold the average baseline of the trace and the absolute value of the second derivative of the slope (the rate of change of voltage over a period of 5 ms) was ≥104. An investigator then manually verified each spike. Potentially spurious spikes associated with movements of the recording wire were excluded from the analysis. Spike frequency at rest was measured during the 12 hour light cycle and expressed as number of spikes per hour.
Reverse transcription quantitative polymerase chain reaction
Hippocampi were quickly isolated from saline-perfused hemibrains, frozen on dry ice, and stored at −80°C. Total RNA was extracted from individual mouse hippocampi with the RNeasy Kit (QIAGEN, 74106), and 300 ng of RNA per sample was used to synthesize cDNA with TaqMan Reverse Transcription Reagents (Thermo Fisher Scientific, N8080234). Quantitative real-time PCR was performed on an ABI Prism 7900HT Sequence Detection System (Thermo Fisher Scientific) using TaqMan Gene Expression Master Mix (Thermo Fisher Scientific, 4368706), a Mapt TaqMan probe (Life Technologies, #4351370), and a Gapdh TaqMan probe (Thermo Fisher Scientific, #4448484). Normalized relative amounts of Mapt mRNA were calculated by the 2–ΔΔCT method (92) using Gapdh mRNA as the internal reference. Technical triplicates were averaged for each sample and Mapt/Gapdh mRNA ratios in experimental groups graphed relative to the mean of ratios in the main control group.
Western blot analysis
Hippocampi were quickly isolated from saline-perfused hemibrains and frozen on dry ice. Within 1–2 hours, individual hippocampi were homogenized with a cordless pestle motor (VWR, 47747–370) at 4°C in 300 μl of ice-cold radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, 89901) containing protease inhibitors (Complete Ultra Mini, Roche, 05892791001; 2 tablets per 10 ml) and phosphatase inhibitors: PhosSTOP (Roche, 04906837001; 2 tablets per 10 ml) and phosphatase inhibitor cocktails 2 and 3 (1:100; Sigma-Aldrich, P5726 and P0044). The protein lysate was then sonicated on ice for 2 min at amplitude 40 using an EpiSonic multifunctional bioprocessor (EpiGentek) and centrifuged at 10,000 rpm for 10 min at 4°C. Supernatant was collected and protein concentration measured by Bradford protein assay (Bio-Rad, 5000006). Lysate containing 20 μg of protein was mixed with NuPAGE LDS Sample Buffer (Thermo Fisher Scientific, NP0007) and NuPAGE Sample Reducing Agent (Thermo Fisher Scientific, NP009), incubated at 70°C for 10 min, cooled down, electrophoresed on NuPAGE Novex 4–12% bis-tris Midi protein gels (Thermo Fisher Scientific, WG1402BOX) in cold fresh NuPAGE Mops SDS running buffer (Thermo Fisher Scientific, NP0001–02) at 180 V for 1 hour and 20 min at 4°C, and transferred at 25 V for 10 min onto nitrocellulose membranes (Thermo Fisher Scientific, IB23001) with an iBlot 2 gel transfer device (Thermo Fisher Scientific, IB21001). Membranes were blocked with 50% Odyssey blocking buffer (LI-COR, 927–50000) diluted in tris-buffered saline (TBS; Thermo Fisher Scientific, BP24711) for 1 hour at room temperature and incubated for 2 days at 4°C in 50% Odyssey blocking buffer diluted in TBS containing 0.1% Tween 20 (TBST) and the following primary antibodies: mouse anti–pan-AKT (1:1000; Cell Signaling Technology, 2920), rabbit anti–p-AKT (1:1000; Cell Signaling Technology, 9271), mouse anti–p-S6 ribosomal protein (1:500; Cell Signaling Technology, 2317), rabbit anti–phospho-S6 ribosomal protein (1:1000; Cell Signaling Technology, 4858), mouse anti-tau (1:1000; Millipore, MAB361), or rabbit anti–β-actin (1:3000; LI-COR, 926–42210). Membranes were then washed four times in TBST alternating with TBS for 10 min each at room temperature, incubated with IRDye 800CW goat anti-rabbit (1:10,000; LI-COR, 925–32211) and IRDye 680LT goat anti-mouse immunoglobulin G (IgG; 1:10,000; LI-COR, 925–68020) for 1 hour at room temperature in the dark, and washed four times in TBST alternating with TBS for 10 min each at room temperature. Protein bands were visualized with an Odyssey CLx Infrared Imaging System (LI-COR) and signal intensities were quantified with Image Studio Lite software (LI-COR, version 5.2). The abundance of NaV1.1 protein was determined as described (93).
Immunohistochemistry
To prepare brain sections, saline-perfused hemibrains were drop-fixed in 4% paraformaldehyde for 48 hours, washed with PBS, and stored at 4°C in PBS with 0.01% sodium azide. Before sectioning, hemibrains were equilibrated in 30% sucrose for ≥48 hours at 4°C. Sagittal or coronal brain sections (30 μm) were cut with a sliding microtome (Leica, SM2010R) and stored at −20°C in cryoprotectant medium (30% glycerol and 30% ethylene-glycol in PBS).
For tau immunostaining and colabeling for cell type-specific markers, free-floating sections were placed into 24-well plates, rinsed in PBS three times for 10 min each, and permeabilized with 0.25% Triton X-100 in PBS (PBS-Tx) for 30 min. Sections were pretreated with 3% H2O2 and 10% methanol in PBS for 15 min to block endogenous peroxidase activity and washed four times with PBS-Tx for 10 min each. For antigen retrieval, sections were incubated with 500 μl of boiling antigen retrieval buffer (10 mM citric acid and 40 mM sodium citrate at pH 6.0) at 105°C for 10 min, allowed to cool for 10 min at room temperature, and washed four times with PBS-Tx alternating with PBS for 10 min each. Endogenous mouse IgG was blocked with Mouse on Mouse (M.O.M) blocking reagent following the M.O.M. basic kit instruction (Vector Laboratories, BMK-2202) for 1 hour at room temperature. Primary antibodies comprising mouse anti-tau (Tau5, 1:1000; BioLegend, 806401), rabbit anti-calcium/calmodulin-dependent protein kinase II (CaMKII) (1:500; Abcam, Ab52476), rabbit anti–γ-aminobutyric acid (GABA) (1:250; Millipore Sigma, A2052), rabbit anti-Olig2 (1:500; Novus Biologicals, NBP1–28667SS), and guinea pig anti-NeuN (1:1000; Synaptic Systems, 266 004) were diluted as indicated in M.O.M diluent, which was prepared by mixing one part of protein concentrate stock solution (Vector Laboratories, PK-2200) with 13 parts of PBS. Sections were incubated in primary antibody solutions overnight at room temperature, washed four times in PBS alternating with PBS-Tx for 10 min each, and incubated with anti-rabbit Alexa Fluor 488 (1:500; Thermo Fisher Scientific, A-11034) or anti-guinea pig Alexa Fluor 488 (1;500; Thermo Fisher Scientific, A-11073) conjugated secondary antibodies in PBS containing 3% normal goat serum (Jackson ImmunoResearch, 005-000-121) for 90 min at room temperature. Sections were then washed four times in PBS alternating with PBS-Tx for 10 min each and incubated with horseradish peroxidase conjugated anti-mouse antibody (1:500; Calbiochem, 401253–2ML) for 1 hour at room temperature. Tau immunoreactivity was amplified with the Tyramide Signal Amplification (TSA) Kit (TSA Plus Cyanine 5, Akoya Biosciences, NEL745001KT). Briefly, the TSA Plus Amplification Reagent was solubilized in 150 μl of dimethyl sulfoxide (DMSO; Sigma-Aldrich, D2650-5X5ML) and diluted 1:50 in 1X Amplification Diluent. Sections were incubated with the resulting solution for 2 min at room temperature, washed three times with PBS-Tx, stained with Hoechst 33342 (1:10,000; Life Technologies, H3570) in PBS for 15 min at room temperature to visualize cell nuclei, and mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, P36970).
For ASO and p-S6 immunostaining, free-floating sections were rinsed in PBS three times for 10 min each and permeabilized with PBS-Tx for 30 min, followed by 1 hour of blocking with PBS-Tx containing 10% normal goat serum. Sections were then incubated in PBS-Tx containing 3% normal goat serum and rabbit anti-ASO antibody (1:1000; gift from Ionis Pharmaceuticals) or rabbit anti-p-S6 ribosomal protein antibody (1:1000; Cell Signaling Technology, 4858) overnight at room temperature. Sections were washed four times in PBS alternating with PBS-Tx for 10 min each and incubated with anti-rabbit Alexa Fluor 488 conjugated secondary antibody (1:500; Thermo Fisher Scientific, A-11034) and Hoechst 33342 (1:10,000; Life Technologies, H3570) in PBS containing 3% normal goat serum (Jackson ImmunoResearch, 005-000-121) for 1 hour at room temperature. Sections were then washed three times with PBS-Tx and mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, P36970).
Images of whole hemibrain sections in Figs. 1A, 4 (B and C), and 6D, and fig. S1 were obtained with a 10x objective on a BZ-9000 inverted epifluorescence automated microscope system (Keyence) equipped with a 12-bit monochrome camera with red, green, and blue capability. Higher magnification images in Fig. 1A were obtained with a 20x objective on a Zeiss LSM880 confocal microscope, and higher magnification images in Figs. 4, D to F, and 6D, and fig. S2 were obtained with a 20x objective at 2x zoom on an Olympus FV3000RS confocal microscope.
Statistical analysis
Statistical tests used, biological n, what n represents, and definitions of significance are described in the figure legends and in table S1. All statistical analyses were performed with GraphPad Prism software version 8.00 (GraphPad Software, La Jolla, CA) or R studio.
No repeated measures in the same mice/samples
If group sizes were small (n < 10), parametric tests were used because distribution assumptions could not be reliably assessed (94, 95). For datasets with larger group sizes, we first used the D’Agostino-Pearson test to ascertain whether all groups were distributed normally. For normally distributed data, we used the F-test (for datasets with two groups) or Brown-Forsythe test (for datasets with more than two groups) to determine whether the variances among groups were comparable. If so, unpaired, two-tailed Student’s t test (for datasets with two groups and one independent variable), one-way ANOVA followed by Holm-Sidak post hoc test for pairwise comparisons (for datasets with more than two groups and one independent variable), or two-way ANOVA followed by Holm-Sidak post hoc test for pairwise comparisons (for datasets with more than two groups and two independent variables) was used. For data that were not normally distributed, we determined whether the variances among groups were comparable after log transformation of data. If so, we used the Kruskal-Wallis test, followed by Dunn’s test for datasets with more than two groups. If not, the Mann-Whitney U test was used to compare two groups, and a permutation test with Holm-Sidak correction was used for multiple comparisons. Chi-square test was used to analyze the proportion of mice with or without seizures of early onset.
Repeated measures in the same mice
Olfactory habituation was analyzed by calculating the slopes of linear regression lines through the three trials for each stimulus. D’Agostino-Pearson test revealed that these data were not normally distributed. Therefore, habituation data were modeled in the GEE framework (96) as a function of genotype, odorant stimulus, and their interaction. DS* mice were chosen as the reference genotype, and water as the reference stimulus. Olfactory dishabituation was analyzed by calculating the difference between the sniffing time of the first trial of “vanilla” (vanilla 1) versus the last trial of “water” (water 3), and the difference between the sniffing time of the first trial of “social” (social1) versus the last trial of “vanilla” (vanilla 3). Because the vanilla1-water 3 dataset was not normally distributed but had similar variances among groups, Kruskal-Wallis test and Dunn’s test were used to assess differences across genotypes and treatment groups. Because the social1-vanilla 3 dataset was not normally distributed and had unequal variances among groups, it was analyzed by permutation test followed by Holm-Sidak test.
Supplementary Material
Acknowledgements:
We thank M.H. Meisler and K. Yamakawa for Scn1aRX/+ mice; H. Kordasiewicz for ASOs, information about their chemical structure and advice on dosing; R. Thomas for advice on statistical analyses; J. Carroll for preparation of graphics; and R. Mott and E. Kimball for administrative assistance.
Funding:
This work was supported by National Institutes of Health grants R01 MH115679 (to L.M.), P01 AG073082 (to L.M.), and R01 NS075487 (to E.D.R.), the BrightFocus Foundation (to E.D.R.), and a research fellowship from the Alzheimer’s Association (to C.-W.C.).
Competing interests:
L.M. has received research funding from Cure Network Dolby Acceleration Partners (CNDAP), holds a small equity interest in CNDAP, has served on CNDAP’s board of managers and on the scientific advisory boards of Acumen, Arvinas, and Biogen, and has consulted for Eisai, PassageBio, Pivotal bioVenture Partners, reMYND, Sangamo Therapeutics, and Takeda. E.D.R. has served on Biogen’s scientific advisory board. E.D.R. and L.M. are coinventors on patents held by the Gladstone Institutes that focus on tau reduction as a strategy to diminish neuronal overexcitation (Titled: “Agents that reduce neuronal overexcitation;” U.S. patent number 9,084,813, issued July 21, 2015 and U.S. patent number 9,198,982, issued December 1, 2015).
Footnotes
Data and materials availability:
The Tau-ASO and NT-ASO were obtained from Ionis Pharmaceuticals through a material transfer agreement. Requests for raw data, analyzed data and materials will be reviewed by the Gladstone Institutes to determine whether the request is subject to intellectual property or confidentiality obligations. Data and materials that can be shared will be released using a material transfer agreement.
References and Notes
- 1.Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, Jones EJH, Jones RM, Pickles A, State MW, Taylor JL, Veenstra-VanderWeele J, Autism spectrum disorder. Nat. Rev. Dis. Primers 6, 1–23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fombonne E, MacFarlane H, Salem AC, Epidemiological surveys of ASD: advances and remaining challenges. J. Autism Dev. Disord, Online ahead of print (2021). [DOI] [PubMed] [Google Scholar]
- 3.Won H, Mah W, Kim E, Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front. Mol. Neurosci 6, 1–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, Stevens C, Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, Brand H, Schwartz G, Nguyen R, Guerrero EE, Dias C, C. Autism Sequencing, P.-B. C. i, Betancur C, Cook EH, Gallagher L, Gill M, Sutcliffe JS, Thurm A, Zwick ME, Borglum AD, State MW, Cicek AE, Talkowski ME, Cutler DJ, Devlin B, Sanders SJ, Roeder K, Daly MJ, Buxbaum JD, Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manoli DS, State MW, Autism spectrum disorder genetics and the search for pathological mechanisms. Am. J. Psychiatry 178, 30–38 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen NE, Lord C, Volkmar FR, The diagnosis of Autism: from Kanner to DSM-III to DSM-5 and beyond. J. Autism Dev. Disord 51, 4253–4270 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukmanji S, Manji SA, Kadhim S, Sauro KM, Wirrell EC, Kwon CS, Jette N, The co-occurrence of epilepsy and autism: a systematic review. Epilepsy Behav 98, 238–248 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Hossain MM, Khan N, Sultana A, Ma P, McKyer ELJ, Ahmed HU, Purohit N, Prevalence of comorbid psychiatric disorders among people with autism spectrum disorder: an umbrella review of systematic reviews and meta-analyses. Psychiatry Res 287, 1–14 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Dravet C, Bureau M, Dalla Bernardina B, Guerrini R, Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 52 (Suppl 2), 1–2 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Cooper MS, McIntosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, Ganesan V, Gill D, Kivity S, Lerman-Sagie T, McLellan A, Pelekanos J, Ramesh V, Sadleir L, Wirrell E, Scheffer IE, Mortality in Dravet syndrome. Epilepsy Res 128, 43–47 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Wang JW, Kurahashi H, Ishii A, Kojima T, Ohfu M, Inoue T, Ogawa A, Yasumoto S, Oguni H, Kure S, Fujii T, Ito M, Okuno T, Shirasaka Y, Natsume J, Hasegawa A, Konagaya A, Kaneko S, Hirose S, Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 49, 1528–1534 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Claes LR, Deprez L, Suls A, Baets J, Smets K, Van Dyck T, Deconinck T, Jordanova A, De Jonghe P, The SCN1A variant database: a novel research and diagnostic tool. Hum. Mutat 30, E904–E920 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Escayg A, Goldin AL, Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51, 1650–1658 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH, Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 76, 594–600 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Meng H, Xu HQ, Yu L, Lin GW, He N, Su T, Shi YW, Li B, Wang J, Liu XR, Tang B, Long YS, Yi YH, Liao WP, The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat 36, 573–580 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Rosander C, Hallbook T, Dravet syndrome in Sweden: a population-based study. Dev. Med. Child Neurol 57, 628–633 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K, Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci 27, 5903–5914 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, Yamakawa K, Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum. Mol. Genet 22, 4784–4804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catterall WA, Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem. Res 42, 2495–2504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K, Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58, 1122–1124 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y, Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126, 531–546 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, Ohfu M, Inoue T, Watanachai A, Kira R, Matsuo M, Muranaka H, Sofue F, Zhang B, Kaneko S, Mitsudome A, Hirose S, Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia 45, 140–148 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Ito S, Ogiwara I, Yamada K, Miyamoto H, Hensch TK, Osawa M, Yamakawa K, Mouse with Nav1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol. Dis 49, 29–40 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L, Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol 76, 443–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tai C, Chang CW, Yu GQ, Lopez I, Yu X, Wang X, Guo W, Mucke L, Tau reduction prevents key features of autism in mouse models. Neuron 106, 421–437.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirrell EC, Nabbout R, Recent advances in the drug treatment of Dravet syndrome. CNS Drugs 33, 867–881 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Löscher W, Potschka H, Sisodiya SM, Vezzani A, Drug resistance in epilepsy: Clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol. Rev 72, 606–638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeFilippis M, Wagner KD, Treatment of autism spectrum disorder in children and adolescents. Psychopharmacol. Bull 46, 18–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persico AM, Ricciardello A, Lamberti M, Turriziani L, Cucinotta F, Brogna C, Vitiello B, Arango C, The pediatric psychopharmacology of autism spectrum disorder: a systematic review - Part I: the past and the present. Prog. Neuropsychopharmacol. Biol. Psychiatry 110, 1–27 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Brandt C, Hillmann P, Noack A, Romermann K, Ohler LA, Rageot D, Beaufils F, Melone A, Sele AM, Wymann MP, Fabbro D, Loscher W, The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 140, 107–120 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Hodges SL, Lugo JN, Therapeutic role of targeting mTOR signaling and neuroinflammation in epilepsy. Epilepsy Res 161, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diaz-Caneja CM, State MW, Hagerman RJ, Jacquemont S, Marin O, Bagni C, Umbricht D, Simonoff E, de Andres-Trelles F, Kaale A, Pandina G, Gomez-Mancilla B, Wang PP, Cusak J, Siafis S, Leucht S, Parellada M, Loth E, Charman T, Buitelaar JK, Murphy D, Arango C, A white paper on a neurodevelopmental framework for drug discovery in autism and other neurodevelopmental disorders. Eur. Neuropsychopharmacol 48, 49–88 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Sharma A, Mehan S, Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int 147, 1–15 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Huang WC, Chen Y, Page DT, Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun 7, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Libero LE, Nordahl CW, Li DD, Ferrer E, Rogers SJ, Amaral DG, Persistence of megalencephaly in a subgroup of young boys with autism spectrum disorder. Autism Res 9, 1169–1182 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeung KS, Tso WWY, Ip JJK, Mak CCY, Leung GKC, Tsang MHY, Ying D, Pei SLC, Lee SL, Yang W, Chung BH, Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol. Autism 8, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strasser L, Downes M, Kung J, Cross JH, De Haan M, Prevalence and risk factors for autism spectrum disorder in epilepsy: a systematic review and meta-analysis. Dev. Med. Child Neurol 60, 19–29 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Wu H, Li H, Bai T, Han L, Ou J, Xun G, Zhang Y, Wang Y, Duan G, Zhao N, Chen B, Du X, Yao M, Zou X, Zhao J, Hu Z, Eichler EE, Guo H, Xia K, Phenotype-to-genotype approach reveals head-circumference-associated genes in an autism spectrum disorder cohort. Clin. Genet 97, 338–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karalis V, Bateup HS, Current approaches and future directions for the treatment of mTORopathies. Dev. Neurosci 43, 143–158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JK, Andrews DS, Ozonoff S, Solomon M, Rogers S, Amaral DG, Nordahl CW, Longitudinal evaluation of cerebral growth across childhood in boys and girls with autism spectrum disorder. Biol. Psychiatry 90, 286–294 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Mandelkow E, Tau in physiology and pathology. Nat. Rev. Neurosci 17, 5–21 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Goedert M, Spillantini MG, in Tau Biology. Advances in Experimental Medicine and Biology Takashima A, Wolozin B, Buee L, Eds. (Springer Nature Singapore, 2019), vol. 1184, chap. 1, pp. 3–21. [DOI] [PubMed] [Google Scholar]
- 43.Götz J, Halliday G, Nisbet RM, Molecular pathogenesis of the tauopathies. Annu. Rev. Pathol 14, 239–261 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, De Los Santos MB, Klickstein N, Corjuc DL, Corjuc BT, Dooley PM, Viode A, Oakley DH, Moore BD, Mullin K, Jean-Gilles D, Clark R, Atchison K, Moore R, Chibnik LB, Tanzi RE, Frosch MP, Serrano-Pozo A, Elwood F, Steen JA, Kennedy ME, Hyman BT, Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med 26, 1256–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang CW, Shao E, Mucke L, Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 371, eabb8255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA, Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kubo A, Misonou H, Matsuyama M, Nomori A, Wada-Kakuda S, Takashima A, Kawata M, Murayama S, Ihara Y, Miyasaka T, Distribution of endogenous normal tau in the mouse brain. J. Comp. Neurol 527, 985–998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saha K, Sontheimer EJ, Brooks PJ, Dwinell MR, Gersbach CA, Liu DR, Murray SA, Tsai SQ, Wilson RC, Anderson DG, Asokan A, Banfield JF, Bankiewicz KS, Bao G, Bulte JWM, Bursac N, Campbell JM, Carlson DF, Chaikof EL, Chen ZY, Cheng RH, Clark KJ, Curiel DT, Dahlman JE, Deverman BE, Dickinson ME, Doudna JA, Ekker SC, Emborg ME, Feng G, Freedman BS, Gamm DM, Gao G, Ghiran IC, Glazer PM, Gong S, Heaney JD, Hennebold JD, Hinson JT, Khvorova A, Kiani S, Lagor WR, Lam KS, Leong KW, Levine JE, Lewis JA, Lutz CM, Ly DH, Maragh S, McCray PB Jr., McDevitt TC, Mirochnitchenko O, Morizane R, Murthy N, Prather RS, Ronald JA, Roy S, Roy S, Sabbisetti V, Saltzman WM, Santangelo PJ, Segal DJ, Shimoyama M, Skala MC, Tarantal AF, Tilton JC, Truskey GA, Vandsburger M, Watts JK, Wells KD, Wolfe SA, Xu Q, Xue W, Yi G, Zhou J, S–CGE. Consortium, The NIH Somatic Cell Genome Editing program. Nature 592, 195–204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wegmann S, DeVos SL, Zeitler B, Marlen K, Bennett RE, Perez-Rando M, MacKenzie D, Yu Q, Commins C, Bannon RN, Corjuc BT, Chase A, Diez L, Nguyen HB, Hinkley S, Zhang L, Goodwin A, Ledeboer A, Lam S, Ankoudinova I, Tran H, Scarlott N, Amora R, Surosky R, Miller JC, Robbins AB, Rebar EJ, Urnov FD, Holmes MC, Pooler AM, Riley B, Zhang HS, Hyman BT, Persistent repression of tau in the brain using engineered zinc finger protein transcription factors. Sci. Adv 7, 1–19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM, Antisense reduction of tau in adult mice protects against seizures. J. Neurosci 33, 12887–12897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM, Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med 9, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoch KM, Miller TM, Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett CF, Kordasiewicz HB, Cleveland DW, Antisense drugs make sense for neurological diseases. Annu. Rev. Pharmacol. Toxicol 61, 831–852 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR, Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci 22, 6309–6314 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vong L, Ye C, Yang Z, Choi B, Chua S Jr., Lowell BB, Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simeone A, Acampora D, Gulisano M, Stornaiuolo A, Boncinelli E, Nested expression domains of four homeobox genes in developing rostral brain. Nature 358, 687–690 (1992). [DOI] [PubMed] [Google Scholar]
- 57.Cheng AH, Fung SW, Hegazi S, Abdalla O, Cheng HM, SOX2 regulates neuronal differentiation of the suprachiasmatic nucleus. Int. J. Mol. Sci 23, 1–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L, Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Lipton JO, Sahin M, The neurology of mTOR. Neuron 84, 275–291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Way SW, McKenna J 3rd, Mietzsch U, Reith RM, Wu HC, Gambello MJ, Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum. Mol. Genet 18, 1252–1265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E, Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, Brown RD, Godfrey JT, Winokur D, Walsh J, Mino-Kenudson M, Maheswaran S, Settleman J, Wargo JA, Flaherty KT, Haber DA, Engelman JA, TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci. Transl. Med 5, 1–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao XF, Liao Y, Alam MM, Mathur R, Feustel P, Mazurkiewicz JE, Adamo MA, Zhu XC, Huang Y, Microglial mTOR is neuronal protective and antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. J. Neurosci 40, 7593–7608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G, S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol 24, 3112–3124 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J, RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem 282, 14056–14064 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, Cagnard N, Carpentier W, Kiss T, Meyuhas O, Pende M, Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 33, 474–483 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA, Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Invest 123, 1798–1808 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G, Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15, 1075–1088 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Goldberg EM, Coulter DA, Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci 14, 337–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Staley K, Molecular mechanisms of epilepsy. Nat, Neurosci 18, 367–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Löscher W, Klein P, The pharmacology and clinical efficacy of antiseizure medications: from bromide salts to cenobamate and beyond. CNS Drugs, Online ahead of print (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, Dillon GH, Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J. Pharmacol. Exp. Ther 298, 986–995 (2001). [PubMed] [Google Scholar]
- 73.Chang CW, Evans MD, Yu X, Yu GQ, Mucke L, Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony. Cell Rep 37, 109855 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berg AT, Plioplys S, Epilepsy and autism: is there a special relationship? Epilepsy Behav 23, 193–198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mullins C, Fishell G, Tsien RW, Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89, 1131–1156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ewen JB, Marvin AR, Law K, Lipkin PH, Epilepsy and autism severity: a study of 6,975 children. Autism Res 12, 1251–1259 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Besag FMC, Vasey MJ, Seizures and epilepsy in autism spectrum disorder. Child Adolesc. Psychiatr. Clin. N. Am 29, 483–500 (2020). [DOI] [PubMed] [Google Scholar]
- 78.Dobyns WB, Mirzaa GM, Megalencephaly syndromes associated with mutations of core components of the PI3K-AKT-MTOR pathway: PIK3CA, PIK3R2, AKT3, and MTOR. Am. J. Med. Genet. C. Semin. Med. Genet 181, 582–590 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont J, Issad T, Gerhardt E, Pagesy P, Vileno M, Bournonville C, Hamdane M, Bantubungi K, Lancel S, Demeyer D, Eddarkaoui S, Vallez E, Vieau D, Humez S, Faivre E, Grenier-Boley B, Outeiro TF, Staels B, Amouyel P, Balschun D, Buee L, Blum D, Tau deletion promotes brain insulin resistance. J. Exp. Med 214, 2257–2269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, Liu C, Leiser SC, Meena, Aznarez I, Liau G, Isom LL, Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med 12, 1–14 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ, Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Losa M, Tracy TE, Ma K, Verret L, Clemente-Perez A, Khan AS, Cobos I, Ho K, Gan L, Mucke L, Alvarez-Dolado M, Palop JJ, Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98, 75–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shao Y, Sztainberg Y, Wang Q, Bajikar SS, Trostle AJ, Wan YW, Jafar-Nejad P, Rigo F, Liu Z, Tang J, Zoghbi HY, Antisense oligonucleotide therapy in a humanized mouse model of MECP2 duplication syndrome. Sci.Transl. Med 13, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC, Group ES, Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Coratti G, Pane M, Lucibello S, Pera MC, Pasternak A, Montes J, Sansone VA, Duong T, Dunaway Young S, Messina S, D’Amico A, Civitello M, Glanzman AM, Bruno C, Salmin F, Tacchetti P, Carnicella S, Sframeli M, Antonaci L, Frongia AL, De Vivo DC, Darras BT, Day J, Bertini E, Muntoni F, Finkel R, Mercuri E, S. g. i, Age related treatment effect in type II Spinal Muscular Atrophy pediatric patients treated with nusinersen. Neuromuscul. Disord 31, 596–602 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Felix-Ortiz AC, Tye KM, Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J. Neurosci 34, 586–595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gandhi T, Lee CC, Neural mechanisms underlying repetitive behaviors in rodent models of autism spectrum disorders. Front. Cell. Neurosci 14, 1–44 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH, Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 3453–3461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eyring KW, Geschwind DH, Three decades of ASD genetics: building a foundation for neurobiological understanding and treatment. Hum. Mol. Genet 30, R236–R244 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kordasiewicz H, Swayze EE, Freier SM, Bui H, in The Patent Cooperation Treaty, W. I. P. Organization, Ed. (2015), chap. WO 2015/010135 A3.
- 91.Das M, Maeda S, Bozhong H, Yu G, Guo W, Lopez I, Yu X, Tai C, Wang X, Mucke L, Neuronal levels and sequence of tau modulate the power of brain rhythms. Neurobiol. Dis 117, 181–188 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 93.Johnson ECB, Ho K, Yu GQ, Das M, Sanchez PE, Biljana D, Lopez I, Yu X, Gill M, Zhang W, Paz JT, Palop JJ, Mucke L, Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer’s disease mutations but not by inhibition of BACE1. Mol. Neurodegener 15, 1–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brown M, Forsythe A, Robust tests for the equality of variances. J. Am. Stat. Assoc 69, 364–367 (1974). [Google Scholar]
- 95.Razali N, Wah Y, Power comparisons of Shapiro-Wilk, Kolmogorov-Smirnov, Lilliefors and Anderson-Darling tests. J. Stat. Modeling Analytics 2, 21–33 (2011). [Google Scholar]
- 96.Zeger SL, Liang KY, Longitudinal data analysis for discrete and continuous outcomes. Biometrics 42, 121–130 (1986). [PubMed] [Google Scholar]
- 97.Butler M, Stecker K & Bennett CF Cellular distribution of phosphorothioate oligodeoxynucleotides in normal rodent tissues. Lab. Invest 77, 379–388 (1997). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Tau-ASO and NT-ASO were obtained from Ionis Pharmaceuticals through a material transfer agreement. Requests for raw data, analyzed data and materials will be reviewed by the Gladstone Institutes to determine whether the request is subject to intellectual property or confidentiality obligations. Data and materials that can be shared will be released using a material transfer agreement.