

Abstract
Amyloid-β (Aβ) accumulation and tauopathy are considered the pathological hallmarks of Alzheimer’s disease (AD), but attenuation in choline signaling, including decreased nicotinic acetylcholine receptors (nAChRs), is evident in the early phase of AD. Currently, there are no drugs that can suppress the progression of AD due to a limited understanding of AD pathophysiology. For this, diagnostic methods that can assess disease progression non-invasively before the onset of AD symptoms are essential, and it would be valuable to incorporate the concept of neurotheranostics, which simultaneously enables diagnosis and treatment. The neuroprotective pathways activated by nAChRs are attractive targets as these receptors may regulate microglial-mediated neuroinflammation. Microglia exhibit both pro- and anti-inflammatory functions that could be modulated to mitigate AD pathogenesis. Currently, single-cell analysis is identifying microglial subpopulations that may have specific functions in different stages of AD pathologies. Thus, the ability to image nAChRs and microglia in AD according to the stage of the disease in the living brain may lead to the development of new diagnostic and therapeutic methods. In this review, we summarize and discuss the recent findings on the nAChRs and microglia, as well as their methods for live imaging in the context of diagnosis, prophylaxis, and therapy for AD.
Keywords: neurodegenerative disease, nicotinic acetylcholine receptors, glial cells, neuroprotection, neuroinflammation, subtype, subpopulation, imaging
1. Introduction
In 2015, Alzheimer’s Disease (AD) International estimated that approximately 46.8 million people worldwide suffer from dementia and that this number would double in two decades. Further, dementia incidence is doubling every 5.0–5.5 years in the over-65 population [1,2]. AD is the most common disorder causing dementia, accounting for 60%–80% of all cases [3]. Aging is the predominant risk factor for AD [1,2], and the world’s population is aging rapidly [3]. Thus, there is an urgent need for truly effective treatments for AD.
The pathological hallmarks of AD include focal loss of neurons, senile plaques formed by extracellular accumulation of amyloid-β (Aβ) peptides, neurofibrillary tangles consisting of intraneuronal accumulations of hyperphosphorylated tau proteins, and synaptic loss [4]. While there is still no complete picture of AD pathogenesis, it is speculated that an imbalance in Aβ production and elimination can induce a cascade of pathological processes, including neuroinflammation, oxidative stress, and excitotoxicity termed the “amyloid-β cascade hypothesis” [4]. Thus, in this hypothesis, the accumulation of Aβ and its oligomers [5] in brains is believed to be the primordial event provoking or accelerating subsequent neurodegenerative events, including the formation of neurofibrillary tangles and synaptic loss, and this hypothesis has provided the rationale for much current research on AD therapeutics [4]. The technological progress for detecting AD biomarkers, especially in positron emission tomography (PET) amyloid imaging [6], has revealed that Aβ begins to accumulate decades before clinical symptoms emerge [7,8], consistent with this amyloid-β cascade. However, multiple lines of evidence suggest important contributions by other pathogenic mechanisms to disease progression, symptom expression, and, potentially, also AD onset.
Prior to the first report on the purification of an amyloidogenic protein (later named Aβ) from the cerebral vasculature of AD patients in 1984 [9], there was already compelling evidence for a “cholinergic hypothesis” for AD progression. This theory is based on evidence showing decreased activity of choline acetyltransferase (ChAT), the rate-limiting enzyme in acetylcholine synthesis [10], and specific loss of cholinergic neurons in the nucleus basalis of Meynert [11] in autopsied brains of AD patients. Various molecular biological and in situ labeling studies also reported substantial reductions in the expression levels of nicotinic acetylcholine receptors (nAChRs) in the AD brain [12,13,14], while expression levels of the other class of cholinergic receptors, muscarinic acetylcholine receptors, were relatively preserved [15,16]. Further supporting this cholinergic hypothesis, inhibitors of the ACh catabolic enzyme acetylcholinesterase, such as donepezil, galantamine, and rivastigmine, are currently the only broadly effective treatments for AD [17]. However, these agents provide only symptomatic treatment and cannot stop AD progression. Further investigations on the causes of cholinergic signaling deficits in AD may lead to the identification of more appropriate therapeutic targets, the development of improved diagnostic and monitoring methods to capture the progression of AD-associated neuropathology, and ultimately better treatments. In this review, we highlight the diagnostic potential of in vivo nAChR detection methods and the therapeutic potential of nAChR modulators.
Nicotinic AChRs are homo- or hetero-pentamers of α1–7, α9–10, β1–4, γ, δ, and ε subunits [18]. Although some combinations do not form receptor/channels, there is still considerable heterogeneity in receptor/channel structure, pharmacology, and electrophysiological behavior [18]. Neuroprotective effects of nicotine were first reported in rat primary cultured neurons using the glutamate-induced cell death (excitotoxicity) model [19]. Subsequently, nAChR stimulation was reported to prevent Aβ-induced neuronal cell death [20] by increasing expression of the anti-apoptotic protein Bcl-2 via activation of the phosphatidylinositol 3 kinase (PI3K)-Akt signaling axis, an effect that was especially robust upon stimulation of homomeric α7 subtype nAChRs [21]. It is still uncertain, however, if direct stimulation of cholinergic synapses containing α7 nAChRs is feasible for AD treatment as receptor expression is markedly reduced in AD and many neurons may be lost or irreparably damaged by the time of diagnosis [12,13,14]. On the other hand, nAChRs are also expressed by glial cells, such as microglia and astrocytes, which provide trophic support to neurons and mediate many of the pro-inflammatory processes implicated in AD-associated neurodegeneration [22]. Modulation of microglia immunoactivity by nAChRs, especially α7 nAChRs, was first reported in 2004 [23] and has been confirmed numerous times since (Section 2 and Section 3). In this review, we summarize evidence that the regulation of nAChRs on glial cells and neurons in the early phase of AD is a potentially effective strategy for delaying the onset and progression of AD. In addition, we speculate that the imaging of nAChRs in living brains with precise spatiotemporal resolution will aid AD diagnosis and contribute to studies on basic pathogenesis and treatment responses.
Microglia are brain tissue macrophages that function as the primary initiators of the brain immune response, and there is voluminous evidence that the inflammatory activities of microglia can either protect vulnerable neurons or exacerbate neurodegeneration [24,25]. For instance, microglia are activated and accumulate around Aβ plaques, where they may facilitate clearance through phagocytosis or neuronal death by amplification of local inflammation [22]. In addition, microglia have functions beyond immunity with potential relevance to AD progression or treatment, such as regulation of neuronal progenitor cells [26], synapses [27,28,29,30,31], and networks [32,33]. Furthermore, a unique subpopulation of disease-associated microglia (DAM) may be a major contributor to pathogenesis as such cells are found primarily or exclusively in the brains of AD model mice and human AD patients [22,34,35,36]. In this review, we summarize emerging evidence that microglial activities can be imaged in living brains to provide clues to pathogenesis and potentially modulated as an AD treatment strategy.
2. nAChRs and AD Pathophysiology
2.1. Neuroprotection and nAChRs
The long history of research on the cholinergic system strongly implicates nAChR signaling in the facilitation of cognitive functions and neuroprotection [37,38]. In recent years, these beneficial effects of nAChR signaling have been demonstrated unequivocally using highly specific agonists, antagonists, and positive allosteric modulators (PAMs), as well as genetic manipulation of receptor subunits in experimental animals. Some of these neuroprotective effects of nAChR modulation are summarized in Table 1. Orthosteric cholinergic agonists, such as ACh and nicotine, directly stimulate nAChRs to open the ligand-gated cation channel, which drives membrane depolarization via sodium influx and activates multiple intracellular signaling pathways mainly via calcium influx [18]. However, nAChRs, especially α7 nAChRs, show rapid desensitization during orthosteric agonist stimulation [39]. On the other hand, PAMs modulate the AChR function by binding to distinct allosteric sites [39]. PAMs are classified into type I and type II. Type I PAMs increase the agonist-induced current peak amplitude by promoting channel ion permeability, while type II PAMs both enhance ion permeability and extend channel open time [39]. Thus, PAMs, especially type II, may have a more substantial impact on the functions of nAChRs showing rapid desensitization during agonist stimulation, especially α7 nAChRs as described above [39]. These α7 nAChRs are a major subtype in the central nervous system (CNS) with a high permeability to calcium, resulting in activation of multiple intracellular signaling pathways in addition to membrane depolarization [39].
Table 1.
Neuroprotective effects of nAChR-related agents.
| Description | Agent | Action | Model | Ref. |
|---|---|---|---|---|
| Non-selective nAChR agonist | ACh | Activates PI3K/Akt, Nfr2/keap1 | Primary cultured mouse hippocampal neurons treated with Aβ25-35 | [40] |
| Activates Erk1/2 | AD model mice (3xTgAD) | [41] | ||
| Inhibits MAPKs (p38 MAPK, JNK) | Mice intrahippocampally-injected with Aβ1–42 | [42] | ||
| Inhibits phosphorylation of p44/42 and p38 MAPKs → Suppresses TNF-α |
Primary cultured mouse microglia treated with LPS | [23] | ||
| Suppress iNOS, TNF-α, IL-1β → Restores IGF |
Primary cultured mouse microglia treated with LPS | [43] | ||
| Selective α7 nAChR agonist | AR-R17779 | Reverses the pro-inflammatory phenotype | Primary cultured fetal sheep astrocytes | [44] |
| PNU-282987 | Inhibits Erk → Restores 5-HT1A, 2C → Improves anxiety and depressive-like behaviors |
Aβ-injected mice | [45] | |
| Activates CaM-CaMKII-CREB → Improves learning and memory |
AD model mice (APPswe/PSldE9) | [46] | ||
| Selective α7 nAChR partial agonist | A582941 | Increases Erk1/2, MAPKs, Arc → Behavior: pro-cognitive activity |
Rats | [47] |
| DMXBA (GST-21) |
Promotes microglial Aβ phagocytosis → Improves brain Aβ burden and memory Dysfunction → Suppresses γ-secretase activity |
Primary cultured rat microglia Human neuroblastoma SH-SY5Y cells AD model mice (APdE9) |
[48] | |
| Inhibits NF-κB → Suppresses IL-6 and TNF-α Activates Nfr2 → Increases HO-1, TXNRD1, NQO1 |
Primary cultured mouse astrocytes treated with LPS | [49] | ||
| Selective α7 nAChR antagonist | α-bungarotoxin | Enhances the inflammatory phenotype | Primary cultured fetal sheep astrocytes | [44] |
| Type I PAM for α7 nAChR | CCMI | Increases Erk1/2, MAPKs, Arc → Behavior: pro-cognitive activity |
Rats | [47] |
| JWX-A0108 | Inhibits NF-κB → Suppresses TNF-α, IL-1β, IL-6 |
AD model mice (APP/PS1) | [50] | |
| Type II PAM for α7 nAChR | PNU-120596 | Increases: BDNF → Behavior pro-cognitive activity |
Rats | [47] |
| Ago-PAM for α7 nAChR | GAT107 | Suppresses peripheral immune reactions, neuroinflammation | EAE mice | [51] |
| AChE inhibitor | Galantamine | Activates JNK → Increases α7 nAChRs Inhibits Akt → Induces autophagy → Promotes Aβ sequestration |
Human neuroblastoma SH-SY5Y cells | [52] |
| Enhances nAChR sensitivity to choline → Activates CaM-CaMKII and CaM- Rac1-WAVE signaling → Promotes microglial Aβ phagocytosis |
Primary cultured rat microglia AD model mice (APdE9) |
[53] | ||
| Simultaneous stimulation Selective α7 nAChR agonist Selective σ1 receptor agonist |
PHA-543613 PRE-084 |
Modulates glial cells → Increases ACh by σ1-R stimulation |
6-OHDA rat model of PD | [54,55] |
| Simultaneous stimulation Selective α7 nAChR agonist Selective α4β2 nAChR agonist |
PNU-282987 RJR-2403 oxalate |
Inhibits dephosphorylation of AMPAR GluA1 subunit → Reduces AMPARs |
Primary cultured mouse hippocampal neurons treated with Aβ1–42 oligomers | [56] |
| NAD-dependent deacetylase | SIRT1 | Activates Erk1/2 → Increases α7 nAChRs |
AD model mice (APdE9) Human neuroblastoma SH-SY5Y cells |
[57] |
5-HT, hydroxytryptamine; Aβ, amyloid β; ACh, acetylcholine; AChE, acetylcholinesterase; AD, Alzheimer’s disease; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; Arc, activity-regulated cytoskeleton associated protein; BDNF, brain-derived neurotrophic factor; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element binding protein; DMXBA, 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride; EAE, experimental autoimmune encephalomyelitis; Erk, extracellular signal-regulated kinase; HO-1, heme oxygenase 1; IGF, insulin-like growth factor; IL, interleukin; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; keap1, kelch-like ECH-associated protein 1, LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; nAChR, nicotinic acetylcholine receptor; NAD, nicotinamide adenine dinucleotide; NF-κB, nuclear factor-κB; Nfr2, nuclear factor erythroid 2-related factor 2; NQO1, NAD(P)H:quinone oxidoreductase 1; PAM, positive allosteric modulator; PD, Parkinson’s disease; PI3K, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; SIRT1, sirtuin 1; TNF-α, tumor necrosis factor-α; TXNRD1, thioredoxin reductase 1; WAVE, Wiskott–Aldrich syndrome protein family verprolin-homologous protein.
Both single and repeated intraperitoneal administration of the selective α7 nAChR partial agonist A582941, the type I PAM CCMI, and the type II PAM PNU120596 enhanced rat performance in the novel object recognition test [47] (Table 1), which is strongly dependent on memorial activity within medial temporal lobe structures such as the hippocampus and entorhinal cortex [58]. In addition, separate stimulation by A582941 or CCMI, but not PNU120596, increased expression levels of the extracellular signal-regulated kinase (Erk)1/2, members of the mitogen-activated protein kinase (MAPK) family, and activity-regulated cytoskeleton-associated protein (Arc) mRNA in the subregions of the frontal cortex and hippocampus [47] (Table 1). Improved cognitive function by AChR stimulation is believed to depend in part on the stimulation of synaptic plasticity by ERK1/2 and Arc [59,60]. In addition, the type II PAM PNU-120596 may enhance cognitive function by promoting the synthesis and release of brain-derived neurotrophic factor (BDNF) [47].
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptors (AMPARs) are the primary mediators of postsynaptic membrane depolarization at glutamatergic synapses, as well as the expression of synaptic plasticity at these synapses [61]. Aβ reduces the expression of AMPARs on the cell surface by promoting endocytosis, thereby reducing AMPAR function and weakening synaptic transmission. The major nAChR subtypes in the hippocampus are α7 nAChRs, α4β2 nAChRs, and α3β4 nAChRs.
A recent study indicated that the specific and simultaneous stimulation of α7 nAChRs with PNU-282987 and α4β2 nAChRs with RJR-2403 oxalate can restore the reduced cell surface expression of AMPARs in primary cultured mouse hippocampal neurons treated with Aβ1–42 oligomers [56]. This is thought to inhibit the calcium influx induced by Aβ1–42 oligomers and thereby prevent calcium-dependent dephosphorylation of the AMPAR GluA1 subunit. However, separate stimulation of either receptor subtype alone does not appear to restore the expression [56]. Surprisingly, stimulation of α3β4 nAChRs with α7 nAChRs, α4β2 nAChRs, or both failed to restore AMPAR surface expression following Aβ1–42 treatment, suggesting that these receptor subtypes may activate distinct and interactive downstream pathways and that highly selective stimulation is required for therapeutic effects on AD, such as cognitive enhancement [56] (Table 1). A more recent publication also reported that the concomitant activation of α7 nAChRs by N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-furo[2,3-c]pyridine-5-carboxamide (PHA)-543613 and σ1 receptors (σ1-Rs) by 2-(4-morpholinethyl) 1-phenylcyclohexanecarboxylate (PRE)-084 protected nigrostriatal dopaminergic neurons, the primary target of Parkinson’s disease (PD) pathology, through suppression of microglia and astrocytic inflammatory activity [54] (Table 1). In addition, the promotion of ACh release by σ1-Rs may also contribute to neuroprotection [55] (Table 1).
As mentioned, α7 nAChRs activate multiple signaling pathways implicated in neuroprotection. Recent studies have suggested the involvement of PI3K/Akt and Nrf2/keap1 signaling [40], as well as Erk1/2 signaling [41], through the promotion of oxidative stress resistance (Table 1). Inconsistent with these findings, however, it was also reported that the restoration of dysregulated 5-HT1A and 5-HT2C receptor expression and mitigation of anxiety- and depressive-like behaviors in Aβ1–42-injected mice by PNU282987 was associated with Erk suppression in the basolateral amygdala [45] (Table 1). These seemingly contradictory results suggest that α7 nAChR activity protects and restores neuronal function by rebalancing Erk pathway activity rather than inducing consistent up or downregulation. It has also been reported that the natural polyphenol antioxidant resveratrol upregulates α7 nAChR expression in a mouse model of AD (APdE9 mice) and human neuroblastoma SH-SY5Y cells by activating the protein deacetylase sirtuin 1 (SIRT1) and Erk1/2 signaling [57] (Table 1 and Table 2). Taken together, these recent findings suggest crosstalk between α7 nAChR and the Erk signaling pathway. Conversely, downregulation of the other MAPKs, p38 MAPK and JNK, was associated with α7 nAChR-induced neuroprotection in mice intrahippocampally-injected with Aβ1–42 [42] (Table 1). Inhibition of p38 and JNK also increased α7 nAChR expression [42] (Table 1 and Table 2). Furthermore, selective stimulation of α7 nAChR by PNU-282987 was reported to decrease Aβ deposition, increase the expression of synaptic-associated proteins, and improve impaired learning and memory in the APdE9 mouse model of AD [46] (Table 1).
Neuroprotection by α7 nAChR stimulation may stem from the restoration of Ca2+ homeostasis via activation of the CaM (calmodulin)-CaMKII (CaM-binding protein kinase II)-CREB (cAMP-responsive element-binding protein) signaling pathway [46]. Moreover, a unique neuroprotective mechanism activated by α7 nAChRs was recently described in human neuroblastoma SH-SY5Y cells, in which α7 nAChRs on the cell surface bind to Aβ with high affinity and act as cargo carriers for internalization and subsequent sequestration in autophagosomes via complex formation with the autophagosome-associated protein LC3 [52] (Table 1). The same study further demonstrated that galantamine, an AChE inhibitor used clinically for AD treatment, protects neurons through activation of JNK signaling to enhance α7 nAChR expression and through inhibition of the Akt pathway to induce autophagy, thereby also promoting intracellular Aβ sequestration [52] (Table 1). Thus, α7 nAChR activation may protect neurons through multiple distinct but complementary mechanisms.
2.2. Modulation of Neurotransmission through nAChRs by Aβ
At presynaptic boutons, activation of α7 nAChRs promotes the release of neurotransmitters such as glutamate [62,63]. It has been reported that α7 nAChR-mediated responses in hippocampal neurons are inhibited by Aβ at nanomolar concentrations [64], but, at more physiological picomolar concentrations, Aβ surprisingly enhanced glutamatergic synaptic transmission and long-term potentiation (LTP) via α7 nAChRs in mouse hippocampal slices [65]. Consistent with these later findings, a recent study demonstrated that both hippocampal slices from α7 nAChRs KO mice and Aβ antibody-treated slices from wild-type (WT) mice showed impaired LTP [66]. These α7 nAChR-KO mice also demonstrated greater age-dependent memory impairment, expression of amyloid precursor protein (APP) and Aβ, hyperphosphorylation of tau, and neuronal loss than WTs. The authors speculated that the increases in APP and Aβ may be, at least in part, a compensatory response for the lack of α7 nAChRs signaling mediated by physiological Aβ concentrations [66]. This further suggests the presence of a feedback mechanism regulating APP and Aβ expression via Aβ-α7 nAChR signaling. Such a mechanism may partially explain Aβ accumulation in sporadic AD, which accounts for more than 95% of AD cases [67] and has no genetic mutations directly related to Aβ regulation [68].
2.3. Neuroinflammation and nAChRs
There is mounting evidence that cholinergic signaling from the CNS can modulate inflammation in both peripheral organs and locally within the CNS. In lipopolysaccharide (LPS)-injected rats, stimulation of the vagus nerve induced ACh release from ChAT-expressing T cells in the spleen [69], activated the peripheral cholinergic system, and reduced the production of tumor necrosis factor (TNF) in liver and blood [70]. Treatment of human macrophages with ACh also decreased the production of pro-inflammatory cytokines such as TNF, interleukin (IL)-1β, IL-6, and IL-18 [70]. These findings indicate the existence of a systemic “cholinergic anti-inflammatory pathway” involving various peripheral immune cells such as T cells and macrophages. A recent study also demonstrated that GAT107, an allosteric agonist and positive allosteric modulator (ago-PAM) of α7 nAChRs, significantly decreased the proliferation of encephalitogenic T cells and the number of macrophages, dendritic cells, and B cells, and further suppressed the production of pro-inflammatory cytokines but increased anti-inflammatory cytokines in experimental autoimmune encephalomyelitis (EAE) in a mouse model of multiple sclerosis [51] (Table 1). The suppression of peripheral immune reactions was further correlated with the attenuation of neuroinflammation in the spinal cord and the severity of EAE [64]. Therefore, this study not only demonstrates a cholinergic anti-inflammatory pathway but also anti-inflammatory crosstalk between immunity in the periphery and CNS via α7 nAChRs with the therapeutic potential for brain diseases [71].
Neuroinflammation is a central pathogenic process in a variety of neurodegenerative diseases [72] and is one of the most important factors in the etiology of AD [73]. Within the CNS, inflammatory responses are initiated and supported primarily by microglia and astrocytes [74] and, like neurons, these cells express nAChRs, suggesting that this “cholinergic anti-inflammatory pathway” can also suppress inflammation in the CNS via actions on microglia and astrocytes. The stimulation of α7 nAChRs on primary cultured mouse microglia by ACh or nicotine suppressed LPS-induced TNF-α production by inhibiting phosphorylation (activation) of p44/42 and p38 MAPKs [23]. In addition, a recent study reported that ACh suppressed LPS-induced expression or release of pro-inflammatory factors by primary cultured rat microglia, including inducible nitric oxide synthase (iNOS), TNF-α, and IL-1β, via α7 nAChR signaling [43] (Table 1). Furthermore, ACh restored the LPS-induced decrease in microglial expression of the neurotrophic factor insulin-like growth factor-1 (IGF-1) and attenuated neuronal apoptosis in microglial co-cultures [43] (Table 1). More recently, JWX-A0108, a novel type I PAM of α7 nAChRs [75], was found to improve cognitive function in the APP/PS1 mouse model of AD and concomitantly inhibit NF-κB signaling, decrease the production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6 by brain tissue, and suppress the expression of the microglial marker ionized calcium-binding adaptor molecule-1 (Iba-1), strongly suggesting that α7 nAChR stimulation suppresses pro-inflammatory activation of microglial and that this anti-inflammatory effect can preserve cognitive function [50] (Table 1).
In addition to cytokine production, microglia have a phagocytic function, including the phagocytosis of extracellular Aβ fibrils in AD. We previously reported that positive modulation of nAChRs on primary cultured rat microglia by galantamine and direct stimulation by nicotine promoted Aβ phagocytosis [53] (Table 1), possibly by increasing cytosolic Ca2+ influx and activating calcium-dependent actin cytoskeleton reorganization through CaM-CaMKII and CaM-Rac1 (Ras-related C3 botulinum toxin substrate 1)-WAVE (Wiskott–Aldrich syndrome protein family verprolin-homologous protein) pathways. The same study also indicated that galantamine treatment reduced brain Aβ burden and attenuated learning and memory impairments in the APdE9 mouse model of AD [53] (Table 1). In a subsequent study, we further confirmed the promotion of microglial Aβ phagocytosis in primary cultured rat microglia and attenuation of brain Aβ burden and memory dysfunction in the same mouse model by treatment with the α7 nAChR-selective partial agonist 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride (DMXBA; also known as GST-21) [48] (Table 1). In addition to the promotion of microglial Aβ phagocytosis, DMXBA treatment suppressed the activity of γ-secretase, a proteolytic enzyme that cleaves the membrane-bound APP to generate pathogenic Aβ fragments, both in human neuroblastoma SH-SY5Y cells and the brain of AD model mice [48] (Table 1). Thus, α7 nAChR signaling in microglia and neurons may activate two complementary neuroprotective mechanisms, reduced generation and enhanced removal of Aβ, leading to a reduction in brain Aβ burden and an attenuation of associated sequela such as neurodegeneration and memory deficits.
Astrocytes may also contribute to the cholinergic anti-inflammatory pathway. Under physiological conditions (i.e., in the absence of experimental inflammation), stimulation of astrocyte nAChRs induced glutamate release and, in turn, excited GABAergic interneurons and consequently suppressed the excitability of hippocampal pyramidal neurons, resulting in impaired synaptic plasticity and memory formation [76]. On the other hand, under experimental neuroinflammation, DMXBA significantly reduced LPS-induced secretion of inflammatory cytokines such as IL-6 and TNF-α from primary cultured mouse astrocytes through the inhibition of the NF-κB pathway [49] (Table 1). Furthermore, DMXBA treatment increased the expression of the Nrf2-regulated stress response factors heme oxygenase-1 (HO-1), thioredoxin reductase (TXNRD1), and NAD(P)H:quinone oxidoreductase-1 (NQO1) [49] (Table 1). Thus, stimulation of astrocytic α7 nAChRs during neuroinflammation may protect neurons by suppressing inflammatory signaling and activating endogenous cytoprotective signaling pathways. More recently, it was reported that primary cultured fetal sheep astrocytes exposed to LPS in utero and then again in vitro exhibited signs of inflammatory memory as evidenced by comprehensive RNA-sequencing (RNA-seq) analysis, indicating excessive pro-inflammatory activity induced by the second LPS treatment compared to naïve astrocytes [44] (Table 1). Further, this pro-inflammatory phenotype was reversed by pretreatment with the α7 nAChR-selective agonist AR-R17779 through suppression of NF-κB and STAT3 signaling, whereas the α7 nAChR-selective antagonist α-bungarotoxin enhanced the inflammatory phenotype [44] (Table 1). Thus, α7 nAChR signaling on astrocytes may both suppress acute inflammatory activities and reduce the sensitivity to subsequent pro-inflammatory events. This latter mechanism may be critical for reducing neuroinflammation in response to periodic activation of pathogenic processes such as Aβ deposition.
2.4. Regulation of nAChR Expression
The reduced expression of nAChRs in the AD brain [12,13,14,77,78,79] suggests that endogenous factors regulating receptor expression or downstream signal transduction are involved in disease pathogenesis or may serve as therapeutic targets. Factors known to regulate nAChR expression are listed in Table 2. A recent study suggested the involvement of post-transcriptional regulation by microRNAs (miRNAs), endogenous non-coding small RNAs that directly regulate specific mRNAs through complementary binding and induction of degradation [80]. The miRNA miR-98-5p was shown to be significantly upregulated in the brain of both AD patients and a mouse model of AD and to negatively regulate α7 nAChR protein expression without affecting receptor mRNA levels in a mouse model of AD [80] (Table 2). Experimental knockdown of miR-98-5p by injection of an adeno-associated virus-anti-miR-98-5p into the hippocampi of AD model mice restored α7 nAChR protein levels and attenuated Aβ pathology, synaptic dysfunction, cognitive decline, and neuroinflammation [80] (Table 2). Possible therapeutic mechanisms activated by restoration of α7 nAChR expression by miR-98-5p suppression include recovery of Ca2+ homeostasis and CAMKII signaling, suppression of the NF-κB pathway, and upregulation of Nrf2-responsive genes, such as HO-1 and NQO1 [80]; responses that could, in turn, improve synaptic transmission and plasticity, suppress neuroinflammation, and mitigate the effects of oxidative stress, respectively. Human serum contains stably expressed miRNAs that may serve as noninvasive biomarkers for AD [81]. Genome-wide serum microRNA expression profiling revealed that miR-98-5p expression was significantly downregulated among individuals at high risk of AD [82], and miR-98-5p is included in the top five key downregulated miRNA-mRNA networks in AD [83]. Thus, the measurement of miR-98-5p in serum may provide additional information for AD diagnosis and staging, as well as clues to the underlying pathogenesis.
Table 2.
Regulators of α7 nAChR expression.
| Effect | Agent | Action | Ref. |
|---|---|---|---|
| Downregulation of α7 nAChR | miR-98-5p | Negatively regulates the expression of α7 nAChRs | [80] |
| Upregulation of α7 nAChR | SIRT1 | Activates the Erk1/2 signaling pathway | [57] |
| Galantamine | Activates JNK signaling | [52] | |
| SP600125 | Inhibits JNK signaling | [42] | |
| SB202190 | Inhibits p38 MAPK signaling | [42] | |
| Morin | Restores decreased α7 nAChR mRNA expression | [84] | |
| RIC-3 | Promotes functional assembly of α7 nAChRs | [85,86] | |
| NACHO | Promotes functional assembly of α7 nAChRs | [87,88] | |
| Ly6h | Promotes functional assembly of α7 nAChRs | [89] |
Erk, extracellular signal-regulated kinase, JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; nAChR, nicotinic acetylcholine receptor; RIC-3, resistance to inhibitors of cholinesterase-3; SIRT1, Sirtuin 1.
It is reported that the activation of Erk1/2 signaling by SIRT1 [51] and activation of JNK signaling by galantamine [54] can increase the expression of α7 nAChRs (Table 2). However, it was also reported that pharmacological suppression of JNK and p38 MAPK signaling by inhibitors SP600125 and SB202190, respectively, increased the expression of α7 nAChRs [52] (Table 2). In addition, the natural flavonoid morin restored α7 nAChR mRNA expression in Aβ-injected rats, possibly by upregulating BDNF [84] (Table 2). Another potential strategy for upregulating α7 nAChRs is modulation of the chaperone proteins necessary for receptor subunit assembly, such as resistance to inhibitors of cholinesterase-3 (RIC-3) [85,86], the ER-resident protein NACHO [87,88], and Ly6h [89] (Table 2). Further studies on the mechanisms regulating α7 nAChR subunit expression and assembly are needed to identify the most promising targets for therapeutic regulation of α7 nAChR surface activity.
3. Live Imaging of nAChRs
Changes in nAChR densities have been reported in the brains of patients with AD [90], PD [90], attention-deficit/hyperactivity disorder [91], Tourette’s syndrome [92], and depression [93]. Therefore, a method is needed to perform noninvasive measurements of nAChR distribution and density in the human brain as these parameters may be useful for diagnosis, reveal novel aspects of disease pathogenesis and potential therapeutic targets, and facilitate the development of new therapeutic drugs. Among nAChR subtypes, the heteromeric α4β2 nAChR has been strongly implicated in the regulation of attention, cognition, and emotion [90], while the homomeric α7 nAChR is implicated in neuroprotection as well as cognitive functions [90]. Thus, there is a strong impetus for the development of therapeutic drugs targeting α7 nAChRs.
Molecular imaging technology enables the visualization of biochemical and molecular biological processes at the tissue/cellular level for research on basic pathomechanisms and drug discovery (Section 3.1 and Section 3.2). Well-known biomolecular imaging methods include various modalities in which a target, such as an exogenous probe or endogenous compound, is detected by visible, fluorescent, or near-infrared light, nuclear magnetic resonance, emission of radiation, or emission of photons. PET and single-photon emission computerized tomography (SPECT) use radioactive probes to non-invasively capture disease characteristics in three dimensions and thus can reveal diagnostic markers throughout the entire body. In recent years, several nAChR imaging probes that use 123I and 99mTc for SPECT detection or 18F and 11C for PET detection have been reported, and their practicality for use in clinical studies of nAChRs has been demonstrated (Section 3.1 and Section 3.2).
3.1. In Vivo PET and SPECT Imaging Probes for α4β2 nAChR
Imaging probes targeting α4β2 nAChRs have been investigated for human applications over many years. The labeled nicotine probe [11C]nicotine (Ki = 2.71 nM) [94] was one of the earliest developed for PET imaging, but both basic and clinical studies have demonstrated that it is not ideal owing to nonspecific binding, rapid metabolism, and rapid washout from the brain. Subsequently, a number of PET imaging probes labeled with 11C or 18F targeting α4β2 nAChR have been developed, including 5-[11C]methyl-3-(2-(S)-azetidinylmethoxy)pyridine ([11C]5MA) [95] (Ki = 0.27 nM), 2-[18F]fluoro-3-(2(S)-azetidinylmethoxy)-pyridine (2-[18F]FA) [96,97,98] (Ki = 1.33 nM), 6-[18F]fluoro-3-(2(S)-azetidinylmethoxy)pyridine (6-[18F]FA) [99] (Ki = 0.26 nM), (−)-2-(6-18F-fluoro-2,3′-bipyridin-5-yl)-7-methyl-7-azabicyclo[2.2.1]heptane ([18F]AZAN) [100,101] (Ki = 0.26 nM), (−)-2-(2′-18F-fluoro-3,3′-bipyridin-5-yl)-7-methyl-7-azabicyclo[2.2.1]heptane ([18F]XTRA) [102], and (−)-6-(6-[18F]fluoropyridine-3-yl)-8-azabicyclo[3.2.1]octane ([18F]flubatine) [103,104,105] (Ki = 0.11 nM) (Figure 1). The development of 18F PET imaging probes has been a priority in recent years because of their positron-emitting property and favorable half-life of 109.8 min. As the parent compound of several imaging probes targeting α4β2 nAChR, it is classified into A85380 and epibatidine structures. Initially, 2-[18F]FA was expected to be a promising compound, and various investigations, including clinical studies, were conducted [106]. However, 2-[18F]FA requires considerable time to reach equilibrium in the brain and prolonged PET scanning (lasting approximately 4 h after administration). A potentially better alternative is [18F]AZAN, which has a high binding affinity for α4β2 nAChR and rapid brain kinetics in baboons [101]. Further, [18F]AZAN was reported to detect α4β2 nAChRs in the human brain within 90 min by PET scanning [100]. Two additional PET imaging probes for α4β2 nAChR are [18F]XTRA and [18F]flubatine. However, human clinical studies are currently underway, and there have been no definitive reports demonstrating clinical utility.
Figure 1.

Representative α4β2 nAChRs radioligands for PET.
In contrast to this limited number of PET probes, several SPECT imaging probes have been developed, and one, 5-[123I]Iodo-3-(2-(S)-azetidinylmethoxy)pyridine ([123I]5IA, Ki = 0.37 nM), has been the subject of clinical study [107,108] as well as labeling distribution studies in the brains of rats and common marmosets. The highest radioactivity was observed in the thalamus and the lowest radioactivity in the cerebellum, regions that have the highest and lowest densities of α4β2 nAChRs, respectively [107]. Furthermore, the brain distribution of [123/125I]5IA was strongly correlated with the density of α4β2 nAChRs as reported previously (correlation coefficient: 0.97) [107]. In clinical studies as well, the accumulation of [123I]5IA in the brain was strongly correlated (coefficient: 0.95) with the density of α4β2 nAChRs [108]. In addition to iodine-based radioprobes, 99mTc-labeled A-85380 derivatives have also been developed, of which [99mTc]CPTT-A-E [109] (Ki = 0.55 nM) and [99mTc]-A-YN-IDA-C4 [110] (Ki = 0.4 nM) demonstrate high affinity for α4β2 nAChR, but brain uptake needs to be improved [109,110] (Figure 2).
Figure 2.

Representative α4β2 nAChRs radioligands for SPECT.
3.2. In Vivo PET and SPECT Imaging Probes for α7 nAChR
Imaging probes targeting α7 nAChRs must be highly selective because brain expression of these receptors is substantially lower than that of α4β2 nAChRs [111], and α7 nAChRs are in the same superfamily of ligand-activated ion channels as serotonin 3 receptors [112]. Both [125I]α-bungarotoxin and [3H]methyllycaconitine bind to α7 nAChRs with high affinity and can reveal α7 nAChR distributions [113] but are only suitable for in vitro experiments owing to their large molecular sizes and toxicity. Therefore, there have been intensive efforts to develop PET and SPECT imaging probes targeting α7 nAChR, but no clinically applicable probes are yet available. In contrast, a α4β2 nAChR probe derived from 1,4-diazabicyclo[3.2.2]nonane, [11C]CHIBA-1001 [114] (Ki = 46–193 nM) has been available since 2008, and subsequent clinical studies conducted. While [11C]CHIBA-1001 also binds to α7 nAChRs with moderate affinity [114], the signal distribution does not reflect the distribution of α7 nAChRs in the human brain [114]. Several new probes with greater affinity have been developed, including [11C]NS-14492 [115] (Ki = 2.2 nM), [18F]NS-10743 (Ki = 11.6 nM) [116], [18F]NS-14490 [117] (Ki = 2.5 nM), [18F]AZ11637326 [118] (Ki = 0.2 nM, spirofuropridine derivatives), [11C]MeQAA [119] (Ki = 40.6 nM), and the dubenzo[b,d]thiophendioxide derivatives [18F]DBT10 (Ki = 0.6 nM), and [18F]ASEM (Ki = 0.4 nM; α7/α4β2 = 3414) [120,121] (Figure 3). Among them, [11C]MeQAA has been examined in clinical studies and found to reach equilibrium in the brain relatively rapidly, with early images showing a distribution according to blood flow and late images showing distribution in brain regions known to express α7 nAChRs (thalamus and partial corpus callosum) [119]. The probes [18F]DBT10 and [18F]ASEM have also garnered attention in recent years as in vivo biodistribution studies in CD-1 mice found that both readily entered the brain and accumulated as predicted by α7 nAChR expression density [121]. Furthermore, in vivo blocking studies using the α7 nAChR-selective ligand SSR18071 reported dose-dependent probe displacement [121]. Both probes show similar kinetics in rhesus monkey brains [122]. However, [18F]ASEM may be a more promising compound for use in clinical studies.
Figure 3.

Representative α7 nAChR radioligands for PET.
The SPECT imaging probe [125I]Iodo-ASEM has been reported to have high-binding affinity and selectivity for α7 nAChR (Ki = 0.5 nM, α7/α4β2 = 3414) (Figure 4) [123]. In a recent report, [125I]Iodo-ASEM exhibited specific binding to α7 nAChR in mouse, rat, and pig brains, and binding overlapped with that of [125I]α-bungarotoxin, while no specific binding was observed in α7 nAChR-KO mice [124]. Conversely, binding to heteromeric α7β2 nAChRs has been suggested, so further investigation is required [124].
Figure 4.
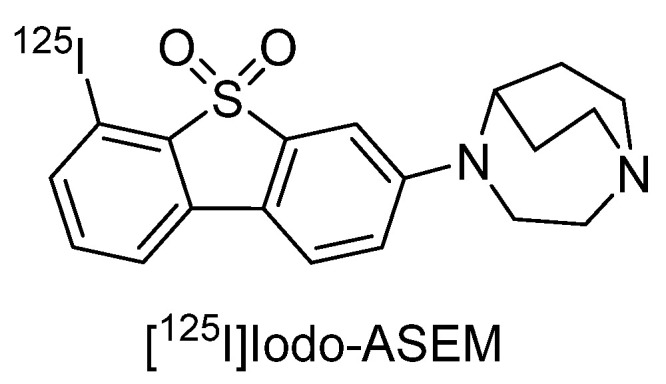
Representative α7 nAChR radioligands for SPECT.
4. Microglia and AD Pathophysiology
Microglia are the resident macrophages within the CNS, and so play key roles in the brain immune response, as well as in brain development and homeostasis [22]. Microglia express pattern recognition receptors (PRRs) that detect damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and neurodegeneration-associated molecular patterns (NAMPs), indicating tissue damage, entry of pathogenic microorganisms, and various neurodegenerative events in the CNS, respectively [36,125,126]. Upon recognition of these signals, microglia are transformed into a reactive phenotype that initiates and modulates the neuroinflammatory response. This neuroinflammatory response involves the release of pro-inflammatory cytokines, chemokines, and reactive oxygen species, and it is now widely believed that a prolonged neuroinflammatory response by microglia is a major driver of AD onset and progression [127,128]. In fact, it has been suggested that microglial-mediated neuroinflammation precedes Aβ plaque formation and thus could be an early trigger for AD development [129]. In addition to microglia, however, astrocytes and even neurons can modulate neuroinflammatory events [130]. Moreover, while activated microglia can trigger pathological inflammation resulting in neurodegeneration [24], these cells can also restore brain homeostasis by remodeling tissue and terminating neuroinflammation through phagocytic activity and the release of anti-inflammatory cytokines [25].
Major pro-inflammatory cytokines produced by microglia include TNF-α, IL-1β, IL-6, and interferon-γ (IFN-γ) [131]. Elevated IL-1β has been found in the brains of aged mice, some AD mouse models, and human AD patients [132,133,134,135,136,137,138]. Active IL-1β is produced from pro-IL-1β by caspase-1, which, in turn, is activated by inflammasomes, multiprotein complexes including pro-caspase-1 assembled in response to various pathology-associated stimuli [126,130]. Neuroinflammation and related signaling pathways and molecules are summarized in Figure 5. Stimulation of PRRs on the cell surface by DAMPs and PAMPs activates the intracellular NF-κB signaling pathway, which triggers the expression of pro-IL-1β, pro-IL-18, NOD-, LRR-, and pyrin domain-containing 3 (NLRP3) in microglia [126,130,139,140]. In addition, internalized DAMPs promote NLRP3 oligomerization and recruit apoptosis-associated speck-like proteins containing a caspase recruitment domain (ASC) and pro-caspase-1 to form the NLRP3 inflammasome complex and produce mature caspase-1 [126,140].
Figure 5.

Neuroinflammation and related signaling pathways and molecules. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; Aβ, amyloid β; DAM, disease-associated microglia; DAMPs, damage-associated molecular patterns; GSDMD, Gasdermin D; IL, interleukin; MAPKs, mitogen-activated protein kinases; nAChRs, nicotinic acetylcholine receptors; NAMPs, neurodegeneration-associated molecular patterns; NLRP, NOD-, LRR-, and pyrin domain-containing; PRRs, pattern recognition receptors.
Caspase-1 processes pro-IL-1β and pro-IL-18 and produces mature IL-1β and IL-18 [139] and also cleaves and actives Gasdermin D (GSDMD), which is the effector of the inflammation-associated cell death mechanism termed pyroptosis [141]. Activation of IL-1βR and IL-18R expressed on microglia, astrocytes, and neurons by released IL-1β and IL-18 further activates NF-κB signaling [142], thereby amplifying inflammatory cytokine production and release. In neurons, IL-1β may also activate the p38 MAPK pathway and suppress the expression of the synaptic protein synaptophysin and upregulate tau phosphorylation [134,143], promoting synaptic loss and formation of neurofibrillary tangles, respectively, two pathological hallmarks of AD [4]. Cleaved GSDMD forms pores in the plasma membrane and promotes further cytokine release [130,144], ultimately triggering the lysogenic pyroptosis death pathway [145,146]. These GSDMD pores have also been demonstrated to promote ASC transformation from macrophages to neighboring cells [126,147,148]. Thus, both extracellular vesicles secreted by microglia [149] and GSDMD pores are involved in the glia-to-neuron transfer of bioactive components and may exacerbate AD pathology such as tau hyperphosphorylation, as reported in a model mouse of tauopathy (Tau22 mice) [150]. Furthermore, neurons express other components of the NLRP3 inflammasome [125]. Thus, although the sequence of events has not yet been proven, neuronal damage and pyroptotic neurodegeneration due to the transmissible activation of NLRP3 inflammasome in neurons from microglia have recently been proposed [130]. As described in subsequent sections, propagating tau [151,152] may be involved in the activation of NLRP3 inflammasomes in neurons [153]. Taken together, the activation of the NLRP3 inflammasome and ensuing production of excessive IL-1β and cleaved GSDMD in microglia and neurons may link the AD-associated pathologies of Aβ and tau aggregation, synaptic loss, and neuronal death. In turn, early neuronal damage and neurodegeneration may promote the release of DAMPs (and possibly NAMPs) and further drive a feed-forward loop that amplifies and accelerates neurodegenerative neuroinflammation.
In addition to inflammatory cytokines, Aβ also activates microglial NLRP3 inflammasomes and may disrupt normal phagocytic activity by accumulating within and damaging lysosomes [154]. Consistent with this notion, NLRP3 gene deletion suppressed Aβ deposition in the brains of APP/PS1 AD model mice, possibly by eliminating the inhibitory effect on phagocytosis [138]. Upon lysosomal damage by Aβ internalization, the lysosomal protease cathepsin B is aberrantly released into the cytosol of microglia [154,155]. Cathepsin B, in turn, can degrade NLRP10, which normally binds NLRP3 and inhibits assembly with ACS and pro-caspase-1, so the loss of NLRP10 can result in enhanced activation of NLRP3 inflammasomes [156]. A recent study further demonstrated that tau seeds, like Aβ, are phagocytosed by microglia, damage lysosomes, induce cathepsin B release, and activate NLRP3 inflammasomes in both primary cultured mouse microglia and the P301L mouse model of tauopathy [153]. Thus, rather than clear pathogenic proteins, the phagocytosis of Aβ and/or tau by microglia may further promote inflammasome activation and initiate a positive feedback loop of pro-inflammatory activation.
Recently, single-cell RNA-seq (scRNA-seq) and single nucleus RNA-seq (snRNA-seq) studies have revealed high microglial heterogeneity in mice and humans at embryonic and early postnatal stages, which gradually declines with maturation but increases again during senescence [157,158,159,160,161,162,163]. Single-cell analysis also revealed a minor subpopulation of damage-associated microglia (DAM) in the brains of 5XFAD AD model mice [34] and human AD patients [35,164,165,166]. Similar DAM-like subpopulations have been detected in the brains of patients with other neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), tauopathy, and epilepsy, as well as in normal aging [34,160,166,167,168,169,170]. Therefore, it is speculated that this DAM subpopulation emerges when microglia recognize NAMPs released by neuronal damage [36]. Although the role of this DAM subpopulation is still unclear, gene expression profiling suggests high phagocytosis capacity and lipid metabolism, consistent with a neuroprotective function [36]. However, cathepsin B is elevated in both human and mouse DAM populations [22], while there is little overlap in gene expression profiles among DAM subpopulations in AD mouse models and human AD [171]. Given the aforementioned actions of cathepsin B in NLPL3 inflammasome hyperactivation, an alternative possibility is that these DAM subpopulations induce excessive neuroinflammation. Live imaging of microglia, a technology described in the next section, could help identify the pathological functions of these microglial subpopulations.
5. Live Imaging of Microglia in AD
5.1. PK11195 and Its Targeting Protein TSPO (PBR)
[11C]PK11195 has been used for more than three decades as a radiotracer for activated microglia and a marker for neuroinflammation in PET studies (for review, see [172]). PK11195 binds to the 18-kDa translocator protein (TSPO) that predominantly localizes to the outer mitochondrial membrane. TSPO was previously known by several other names, such as peripheral-type benzodiazepine receptor or recognition site (PBR) (for review, see [173]). The name PBR was broadly accepted initially as TSPO was first identified as a peripheral binding site for the benzodiazepine derivative [3H]diazepam in the kidneys [174]. Studies using the diazepam derivative [3H]Ro5-4864, which specifically binds to PBR, then revealed that the PBR (i.e., TSPO) binding capacity in the brain is approximately one-fourth that of central-type benzodiazepine binding sites (i.e., GABAA receptors) [175,176]. Subsequent studies revealed that TSPO participates in a variety of important mitochondria-dependent and independent functions, including the transport of cholesterol for steroidogenesis [177] and porphyrin for heme biosynthesis [178], as well as mitochondrial protein import [179] and the opening of the mitochondrial permeability transition pore (MPTP) [180]. Based on these discoveries, this multifunctional protein was renamed TSPO [173]. Although early KO studies showed that PBR is indispensable for normal development and viability in mice [181], recent studies have reported that conditional and even global TSPO-knockdown (KD)/KO mice are viable [182,183,184,185,186,187,188], and TSPO function is reinterpreted [187].
5.2. Allelic Variances of TSPO
A PET study using [11C]PBR28 found little signal from either the brain or the peripheral organs of certain individuals [189]. Subsequent investigation, however, revealed that the non-responders to [11C]PBR28 have no changes in the binding to [11C]PK11195 [190,191]. Based on the tracer signal variation, individuals can be divided into high-affinity binders (HABs), low-affinity binders (LABs), and mixed-affinity binders (MABs) [191,192], and genetic analysis revealed that these classes are related to TSPO genotype, specifically to the expression of a single nucleotide polymorphism (SNP), rs6971, which gives rise to an Ala to Thr mutation at amino acid position 147 [193]. The high specific second-generation PET ligands for TSPO, such as [18F]PBR111, [18F]PBR06, [11C]DPA713, and [11C]DAA1106, are all sensitive to the SNP [192]. Thus, when PET imaging uses TPSO radiotracers except for [11C]PK11195, it needs to consider the SNP of TPSO.
5.3. Distribution and Cell Origins of TSPO in the Brain
Under normal physiological conditions, overall brain tissue expression of TSPO is maintained at extremely low levels compared to peripheral organs [182,183,184]. However, a recent immunohistochemical analysis detected more robust TSPO immunoreactivity in specific brain regions, such as the olfactory bulb, choroid plexus, subventricular zone, ependyma of the ventricles, hippocampal dentate gyrus, and cerebellum in normal mouse brains [194]. This immunoreactivity was localized primarily to astrocytes, vascular endothelial cells, pericytes, smooth muscle cells, some neurons (especially cerebellar Purkinje cells), and neural stem/progenitor cells, but not microglia [194]. However, TSPO expression has been reported in microglia as well as astrocytes and vascular endothelial cells in the brains of normal rats [195]. In the human brain, Gui et al. also found TSPO immunoreactivity in microglia, as well as in astrocytes, vascular endothelial cells, and smooth muscles cells [196]. In the PET study with [11C]PBR28 in Macaca mulatta, Hillmer et al. took advantage of microglial depletion to examine microglial contribution in the baseline signal under normal physiological conditions [197]. A selective inhibitor of colony-stimulating factor 1 receptor (CSF1R), PLX3397, exclusively depletes microglia in the brain [198], and it reduced the [11C]PBR28 signal by 46% from baseline in Macaca mulatta [197]. Thus, about half of all brain TSPO seems to be expressed by microglia even under normal physiological conditions in Macaca mulatta.
5.4. Recent Findings in Biological Function of TSPO
Several recent studies have reported that conditional or global TSPO-KO mice are viable and exhibit no abnormalities in growth, fertility, lifespan, heme biosynthesis, and MPTP [182,183,184,185,186,187,188]. While TSPO deficiency also has no effects on steroid biosynthesis in these KO mice, regulation of steroid biosynthesis by TSPO is still under debate [199,200,201] as it was reported that an allelic variant influences steroid biosynthesis in humans [202]. TSPO also appears to promote microglial metabolic activity, such as oxygen consumption rate, ATP production, mitochondrial membrane potential, and cytosolic Ca2+ [182,203]. It is also reported that TSPO deficiency suppresses mitochondrial oxidative phosphorylation and glycolysis [204]. Of note, this metabolic deficit in microglia suppressed both pro- and anti-inflammatory activation [204]. Furthermore, it was reported that TSPO binds to NADPH oxidase 2 (NOX2) subunits gp91phox and p22phox in primary cultured mouse microglia [205], suggesting that TSPO modulates reactive oxygen species (ROS) production [205]. Similarly, TSPO was reported to regulate NOX1 activity and subsequent ROS production in retinal microglia [206]. These findings suggest vital functions of TSPO in the regulation of the microglial redox balance through ROS production.
5.5. TSPO Expression in Pro-Inflammatory Activated Microglia as Detected by PET Imaging
In contrast to normal physiological conditions, neural TSPO expression is substantially upregulated during neuroinflammatory conditions, such as trauma, stroke, infection, cancer, and various neurodegenerative disorders [207,208]. In the BV-2 mouse microglial cell line, LPS treatment strongly induced TSPO expression at least in part via activation of the transcriptional factor AP-1 and the ensuing release of histone deacetylase 1 (HDAC1) from the enhancer region of the TSPO gene [209]. Systemic administration of LPS in baboons [210] and humans [211,212] increased serum levels of pro-inflammatory cytokines, including IL-6 and TNF-α, accompanied by robust enhancement of [11C]PBR28 signal emission from the brain as detected by PET [210,212]. Further immunohistochemical analysis of baboon brains revealed that TSPO was upregulated exclusively in microglia rather than astrocytes and neurons [210]. This result is supported by a study showing that the increased TSPO signal after LPS injection in rats is mediated by microglial proliferation [213]. However, in the human study cited above [212], no correlations were found between serum cytokine elevations and [11C]PBR28 binding in the brain. Nonetheless, this result highlights the potential importance of [11C]PBR28 imaging for the assessment of microglial activation in the brain. Furthermore, a human [11C]PBR28-PET study revealed a significant correlation between increased [11C]PBR28 signal in the brain and impairment of hippocampus-dependent memory following LPS injection [211].
5.6. TSPO Targeting Radioactive Imaging in AD Model Animals and AD Patients
AD is one of the most frequently analyzed brain disorders using TSPO radiotracers (for review, see [214]). A SPECT study using a mouse model of AD (3xTg-AD mice) even suggested that neuroinflammation, as detected by increased TSPO radiotracer signals, may precede Aβ accumulation and tauopathy [215]. Moreover, a PET study using [18F]DPA-714 conducted by Hamelin et al. reported that an initially higher TSPO binding signal is associated with better clinical prognosis, whereas the subsequent increase is linked to the worsening of the clinical condition independent of the initial amyloid load [216]. Thus, capturing the onset of neuroinflammation by live imaging with TSPO radiotracers may make it possible to diagnose AD earlier than by amyloid-PET, thereby expanding the therapeutic window for earlier intervention and providing additional information on the neuropathological mechanisms leading to clinical symptom progression. In accord with Hamelin et al. [216], most clinical studies thus far have reported that TSPO expression is elevated in the brain of AD patients compared to normal age-matched controls, underscoring the potential utility of radioactive imaging targeting TSPO [217,218,219]. However, several other studies have found no significant difference in TSPO expression between the brains of AD patients and controls using radiotracers [196,220,221,222], so additional studies are required to determine if and when such changes in TSPO occur.
Moreover, the diagnostic utility and mechanistic insights provided by TSPO imaging are strongly dependent on the cellular origins of the signal. Gui et al. reported that TSPO immunoreactivity is not limited to microglia and astrocytes but can also be found in vascular endothelial cells and smooth muscles cells in postmortem AD brains [196]. Similarly, a study of a rat model of AD (TgF344-AD rats) reported robust immunoreactivity of TSPO in vascular endothelial cells, as well as microglia and astrocytes [195]. In this study, the authors further performed the fluorescence-activated cell sorting (FACS) analysis using the radioligand-treated brain tissue of rats and AD subjects and showed that the binding signal of [125I]CLINDE, a TSPO radiotracer for SPECT, was significantly increased in astrocytes and microglia but not in vascular endothelial cells along with the progression of the pathological condition of AD. Interestingly, results further indicated that the increase in the [125I]CLINDE binding signal was visible at 12 and 24 months in astrocytes and only at 24 months in microglia in the hippocampus of TgF344-AD rats. The authors, therefore, indicated a positive involvement of astrocytes activation in the relatively early phase of AD pathology and suggested that TSPO radioactive imaging in AD is an indicator of glial cells activity but not specific for microglial activation in AD brains. Thus, it is suggested that the timing of activations of astrocytes, microglia, or either of them detected by radioactive imaging will vary depending on the stage of AD pathology. Considering it together with a clinical study by Hamelin et al. [216] that the higher the TSPO binding signal in the early stage of AD, the better the clinical prognosis, it is possible that the initial increase in TSPO expression through astrocyte activation is important for improving the clinical prognosis of AD. Further basic and clinical studies are required to better understand the stage dependence of microglial and astroglial reactivity and the therapeutic utility targeting TSPO expression.
5.7. Beyond Microglia Imaging by PET and SPECT
Certain MRI modalities have demonstrated promise for the imaging of microglial activity in living brains. An ex vivo study using ultra-high-resolution MRI detected a signal in brain tissue from AD patients (but not matched controls) that appeared to originate from microscopic iron-bearing microglia, which are known to be abundant in the AD brain [223]. Our research group also succeeded in detecting transplanted Ferucarbotran (Resovist)-labeled microglia in the neonatal rat brain following hippocampal injection of Aβ1–42 using MRI [224] and found that these cells migrated to the injection site for Aβ clearance by phagocytosis. Thus, MRI could be a powerful tool for detecting microglial dynamics and activity in AD patients.
Expression of the ATP-gated P2X7 receptor (P2X7R) is upregulated in the AD brain and associated with pathological progression [225,226,227]. Recent studies have further found that P2X7R is upregulated relatively specifically in pro-inflammatory (activated) microglia, where activation and downstream signaling regulate Aβ-induced NLRP3 inflammasome activity and IL-1β secretion [228,229]. Several PET tracers targeting P2X7R such as [11C]GSK1482160, [11C]SMW139, [11C]JNJ-54173717, and [11C]JNJ-64413739 have been developed and evaluated in mice and humans (for review, see [230]). Although these tracers have detected differences in expression between neuroinflammatory models/patients and controls, further studies are required to assess if detection includes polymorphic variants and identify the most appropriate reference area for the evaluation of changes in regional expression. PET tracers are also under development for the G-protein-coupled P2Y12 receptor (P2Y12R), which is specifically expressed in microglia [231] and found to be upregulated in rat microglia (CD206+), activated by injection of the pro-inflammatory cytokine IL-4 [232]. PET tracers for P2Y12R are also being developed [232]. Thus, PET probes targeting P2X7R and P2Y12R have the potential to discriminate inflammatory and anti-inflammatory microglial states, respectively [230,233]. However, P2Y12R expression was also reported to decrease in a DAM subpopulation arising by NAMP stimulation [34,36]. Thus, tracers for P2Y12R, as well as tracers for CX3CR1 and TMEM119, which are also repressed in microglia upon stimulation with NAMPs and recognized as markers of homeostatic microglia [34,36], may reflect microglial homeostasis in live imaging. The development of imaging tracers to detect microglial activation states and subpopulations for next-generation live imaging of microglia may help facilitate microglia-targeted AD therapies.
6. Conclusions
The cholinergic hypothesis of AD has proven to be a successful concept for the development of symptomatic drugs, and more detailed studies are now being conducted to identify better cholinergic targets for neurotheranostics. The cholinergic anti-inflammatory pathway allows for complex interactions between cholinergic signaling and brain immunity, as well as crosstalk with peripheral immunity. In brain immunity, neuroinflammation induced primarily by microglia through NLRP3 inflammasome activation is strongly related to AD progression and may even trigger AD development. The cholinergic anti-inflammatory pathway appears to suppress NLRP3 inflammasome activation via microglial nAChR stimulation. Further, nAChRs promote microglial Aβ phagocytosis and reset the inflammatory memory of astrocytes. Thus, the functional regulation of nAChRs in glial cells, especially microglia, is a promising strategy to reduce neuroinflammation, protect neurons, and mitigate AD symptoms. For this regulation, we now know that methods to control the functional assembly of nAChRs by chaperon proteins are required. As experimental techniques advance, such as single-cell RNA-seq and highly specific imaging probes, further high-resolution studies, including subtype-, cell type-, and cell subpopulation-specific analysis of nAChRs and microglial activities, are expected to contribute greatly to AD diagnosis, monitoring, and treatment.
Abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s Disease |
| ago-PAM | Allosteric Agonist And Positive Allosteric Modulator |
| ALS | Amyotrophic Lateral Sclerosis |
| AMPARs | α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid-Type Glutamate Receptors |
| Arc | Activity-Regulated Cytoskeleton-Associated Protein |
| ASC | Apoptosis-Associated Speck-Like Protein Containing A Caspase Recruitment Domain |
| Aβ | Amyloid-β |
| BAMs | Border-Associated Macrophages |
| BDNF | Brain-Derived Neurotrophic Factor |
| CaM | Calmodulin |
| CaMKII | CaM-Binding Protein Kinase II |
| ChAT | Choline Acetyltransferase |
| CNS | Central Nervous System |
| CREB | cAMP Responsive Element-Binding Protein |
| CSF1R | Colony-Stimulating Factor 1 Receptor |
| Cytob558 | Cytochrome B558 |
| DAM | Disease-Associated Microglia |
| DAMPs | Damage-Associated Molecular Patterns |
| EAE | Experimental Autoimmune Encephalomyelitis |
| ER | Endoplasmic Reticulum |
| Erk1/2 | Extracellular Signal-Regulated Kinase 1/2 |
| FACS | Fluorescence-Activated Cell Sorting |
| GSDMD | Gasdermin D |
| HABs | High-Affinity Binders |
| HDAC1 | Histone Deacetylase 1 |
| HO-1 | Heme Oxygenase-1 |
| Iba-1 | Ionized Calcium-Binding Adaptor Molecule-1 |
| IFN-γ | Interferon-γ |
| IGF-1 | Insulin-Like Growth Factor-1 |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| KD | Knockdown |
| KO | Knockout |
| LABs | Low-Affinity Binders |
| LPS | Lipopolysaccharide |
| LSP | Lipopolysaccharide |
| LTP | Long-Term Potentiation |
| MABs | Mixed-Affinity Binders |
| MAPKs | Mitogen-Activated Protein Kinases |
| miRNA | microRNA |
| MPTP | Mitochondrial Permeability Transition Pore |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple Sclerosis |
| nAChRs | Nicotinic Acetylcholine Receptors |
| NAMPs | Neurodegeneration-Associated Molecular Patterns |
| NLRP3 | NOD-, LRR-, And Pyrin Domain Containing 3 |
| NOX2 | NADPH Oxidase 2 |
| NQO1 | NAD(P)H: Quinone Oxidoreductase-1 |
| P2Y12R | P2Y12 Receptor |
| PAMs | Positive Allosteric Modulators |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PBR | Peripheral-Type Benzodiazepine Receptor |
| PD | Parkinson’s Disease |
| PET | Positron Emission Tomography |
| PI3K | Phosphatidylinositol 3 Kinase |
| PRRs | Pattern Recognition Receptors |
| Rac1 | Ras-Related C3 Botulinum Toxin Substrate 1 |
| RIC-3 | Resistance To Inhibitors Of Cholinesterase-3 |
| ROS | Reactive Oxygen Species |
| scRNA-seq | Single Cell RNA-Sequencing |
| SIRT1 | Sirtuin 1 |
| SNP | Single Nucleotide Polymorphism |
| snRNA-seq | Single Nucleus RNA- Sequencing |
| SPECT | Single Photon Emission Computed Tomography |
| TNF | Tumor Necrosis Factor |
| TSPO | Translocator Protein |
| TXNRD1 | Thioredoxin Reductase |
| WAVE | Wiskott–Aldrich Syndrome Protein Family Verprolin-Homologous Protein |
| σ1-R | σ1 Receptor |
Author Contributions
Conceptualization, K.T.; writing—original draft preparation, K.T., H.K., D.Y. and K.H.; writing—review and editing, K.T., H.K., D.Y., K.H., K.N., Y.K., S.S. and I.T. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Funding Statement
This study was supported by grants-in-aid from the Private University Research Branding Project for the Ministry of Education, Culture, Sports, Science, and Technology, the Japan Society for the Promotion of Science (JSPS) KAKEN (20H03569 to K.T.; 19K07854 to K.N.; 21K06586 to Y.K.; 17K09783; 20K0789 to SS, 20K20588 to I.T.), the Hoansha Foundation (K.T.), Kobayashi Foundation (K.T.), and the Smoking Research Foundation (K.T., Y.K. and S.S.).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jorm A.F., Jolley D. The incidence of dementia: A meta-analysis. Neurology. 1998;51:728–733. doi: 10.1212/WNL.51.3.728. [DOI] [PubMed] [Google Scholar]
- 2.Corrada M.M., Brookmeyer R., Paganini-Hill A., Berlau D., Kawas C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010;67:114–121. doi: 10.1002/ana.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022;18:700–789. doi: 10.1002/alz.12638. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klunk W.E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D.P., Bergstrom M., Savitcheva I., Huang G.F., Estrada S., et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 7.Jack C.R., Jr., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jack C.R., Jr., Knopman D.S., Jagust W.J., Petersen R.C., Weiner M.W., Aisen P.S., Shaw L.M., Vemuri P., Wiste H.J., Weigand S.D., et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glenner G.G., Wong C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 10.Davies P., Maloney A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/S0140-6736(76)91936-X. [DOI] [PubMed] [Google Scholar]
- 11.Whitehouse P.J., Price D.L., Clark A.W., Coyle J.T., DeLong M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 12.Shimohama S., Taniguchi T., Fujiwara M., Kameyama M. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem. 1986;46:288–293. doi: 10.1111/j.1471-4159.1986.tb12960.x. [DOI] [PubMed] [Google Scholar]
- 13.Flynn D.D., Mash D.C. Characterization of L-[3H]nicotine binding in human cerebral cortex: Comparison between Alzheimer’s disease and the normal. J. Neurochem. 1986;47:1948–1954. doi: 10.1111/j.1471-4159.1986.tb13113.x. [DOI] [PubMed] [Google Scholar]
- 14.Nordberg A., Winblad B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci. Lett. 1986;72:115–119. doi: 10.1016/0304-3940(86)90629-4. [DOI] [PubMed] [Google Scholar]
- 15.Mulugeta E., Karlsson E., Islam A., Kalaria R., Mangat H., Winblad B., Adem A. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res. 2003;960:259–262. doi: 10.1016/S0006-8993(02)03542-4. [DOI] [PubMed] [Google Scholar]
- 16.Svensson A.L., Alafuzoff I., Nordberg A. Characterization of muscarinic receptor subtypes in Alzheimer and control brain cortices by selective muscarinic antagonists. Brain Res. 1992;596:142–148. doi: 10.1016/0006-8993(92)91541-L. [DOI] [PubMed] [Google Scholar]
- 17.Li D.D., Zhang Y.H., Zhang W., Zhao P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front. Neurosci. 2019;13:472. doi: 10.3389/fnins.2019.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckingham S.D., Jones A.K., Brown L.A., Sattelle D.B. Nicotinic Acetylcholine Receptor Signalling: Roles in Alzheimer’s Disease and Amyloid Neuroprotection. Pharmacol. Rev. 2009;61:39–61. doi: 10.1124/pr.108.000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akaike A., Tamura Y., Yokota T., Shimohama S., Kimura J. Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;644:181–187. doi: 10.1016/0006-8993(94)91678-0. [DOI] [PubMed] [Google Scholar]
- 20.Kihara T., Shimohama S., Sawada H., Kimura J., Kume T., Kochiyama H., Maeda T., Akaike A. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann. Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- 21.Kihara T., Shimohama S., Sawada H., Honda K., Nakamizo T., Shibasaki H., Kume T., Akaike A. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 22.Takata K., Ginhoux F., Shimohama S. Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 2021;192:114754. doi: 10.1016/j.bcp.2021.114754. [DOI] [PubMed] [Google Scholar]
- 23.Shytle R.D., Mori T., Townsend K., Vendrame M., Sun N., Zeng J., Ehrhart J., Silver A.A., Sanberg P.R., Tan J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 24.Streit W.J., Mrak R.E., Griffin W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Streit W.J. Microglia and neuroprotection: Implications for Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Sierra A., Encinas J.M., Deudero J.J., Chancey J.H., Enikolopov G., Overstreet-Wadiche L.S., Tsirka S.E., Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wake H., Moorhouse A.J., Jinno S., Kohsaka S., Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parkhurst C.N., Yang G., Ninan I., Savas J.N., Yates J.R., 3rd, Lafaille J.J., Hempstead B.L., Littman D.R., Gan W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 30.Hong S., Beja-Glasser V.F., Nfonoyim B.M., Frouin A., Li S., Ramakrishnan S., Merry K.M., Shi Q., Rosenthal A., Barres B.A., et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B., et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 32.Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Squarzoni P., Oller G., Hoeffel G., Pont-Lezica L., Rostaing P., Low D., Bessis A., Ginhoux F., Garel S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- 34.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B., et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–1290 e1217. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y., Song W.M., Andhey P.S., Swain A., Levy T., Miller K.R., Poliani P.L., Cominelli M., Grover S., Gilfillan S., et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020;26:131–142. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell. 2018;173:1073–1081. doi: 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Shimohama S., Kawamata J. Roles of Nicotinic Acetylcholine Receptors in the Pathology and Treatment of Alzheimer’s and Parkinson’s Diseases. Springer; Singapore: 2018. pp. 137–158. [PubMed] [Google Scholar]
- 38.Kume T., Takada-Takatori Y. Nicotinic Acetylcholine Receptor Signaling: Roles in Neuroprotection. Springer; Singapore: 2018. pp. 59–71. [PubMed] [Google Scholar]
- 39.Papke R.L., Horenstein N.A. Therapeutic Targeting of alpha7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2021;73:1118–1149. doi: 10.1124/pharmrev.120.000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo J., Yang G., He Y., Xu H., Fan H., An J., Zhang L., Zhang R., Cao G., Hao D., et al. Involvement of α7nAChR in the Protective Effects of Genistein Against β-Amyloid-Induced Oxidative Stress in Neurons via a PI3K/Akt/Nrf2 Pathway-Related Mechanism. Cell. Mol. Neurobiol. 2021;41:377–393. doi: 10.1007/s10571-020-01009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fonar G., Polis B., Sams D.S., Levi A., Malka A., Bal N., Maltsev A., Elliott E., Samson A.O. Modified Snake alpha-Neurotoxin Averts beta-Amyloid Binding to alpha7 Nicotinic Acetylcholine Receptor and Reverses Cognitive Deficits in Alzheimer’s Disease Mice. Mol. Neurobiol. 2021;58:2322–2341. doi: 10.1007/s12035-020-02270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang K.-W., Zong H.-F., Ma K.-G., Zhai W.-Y., Yang W.-N., Hu X.-D., Xu J.-H., Chen X.-L., Ji S.-F., Qian Y.-H. Activation of α7 nicotinic acetylcholine receptor alleviates Aβ1–42-induced neurotoxicity via downregulation of p38 and JNK MAPK signaling pathways. Neurochem. Int. 2018;120:238–250. doi: 10.1016/j.neuint.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Li L., Liu Z., Jiang Y.Y., Shen W.X., Peng Y.P., Qiu Y.H. Acetylcholine suppresses microglial inflammatory response via alpha7nAChR to protect hippocampal neurons. J. Integr. Neurosci. 2019;18:51–56. doi: 10.31083/j.jin.2019.01.114. [DOI] [PubMed] [Google Scholar]
- 44.Cao M., MacDonald J.W., Liu H.L., Weaver M., Cortes M., Durosier L.D., Burns P., Fecteau G., Desrochers A., Schulkin J., et al. alpha7 Nicotinic Acetylcholine Receptor Signaling Modulates Ovine Fetal Brain Astrocytes Transcriptome in Response to Endotoxin. Front. Immunol. 2019;10:1063. doi: 10.3389/fimmu.2019.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang K.-W., Zong H.-F., Wang M., Rizvi M.Y., Neha S.I., Yang W.-N., Ji S.-F., Ma Y.-B., Qian Y.-H. PNU282987 alleviates Aβ-induced anxiety and depressive-like behaviors through upregulation of α7nAChR by ERK-serotonin receptors pathway. Neurosci. Lett. 2020;731:135118. doi: 10.1016/j.neulet.2020.135118. [DOI] [PubMed] [Google Scholar]
- 46.Wang X.-L., Deng Y.-X., Gao Y.-M., Dong Y.-T., Wang F., Guan Z.-Z., Hong W., Qi X.-L. Activation of α7 nAChR by PNU-282987 improves synaptic and cognitive functions through restoring the expression of synaptic-associated proteins and the CaM-CaMKII-CREB signaling pathway. Aging. 2020;12:543–570. doi: 10.18632/aging.102640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potasiewicz A., Faron-Gorecka A., Popik P., Nikiforuk A. Repeated treatment with alpha 7 nicotinic acetylcholine receptor ligands enhances cognitive processes and stimulates Erk1/2 and Arc genes in rats. Behav. Brain Res. 2021;409:113338. doi: 10.1016/j.bbr.2021.113338. [DOI] [PubMed] [Google Scholar]
- 48.Takata K., Amamiya T., Mizoguchi H., Kawanishi S., Kuroda E., Kitamura R., Ito A., Saito Y., Tawa M., Nagasawa T., et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Abeta accumulation through suppression of neuronal gamma-secretase activity and promotion of microglial amyloid-beta phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging. 2018;62:197–209. doi: 10.1016/j.neurobiolaging.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Patel H., McIntire J., Ryan S., Dunah A., Loring R. Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J. Neuroinflamm. 2017;14:192. doi: 10.1186/s12974-017-0967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H., Gao J., Chang Y., Li K., Wang L., Ju C., Zhang F. JWX-A0108, a positive allosteric modulator of alpha7 nAChR, attenuates cognitive deficits in APP/PS1 mice by suppressing NF-kappaB-mediated inflammation. Int. Immunopharmacol. 2021;96:107726. doi: 10.1016/j.intimp.2021.107726. [DOI] [PubMed] [Google Scholar]
- 51.Mizrachi T., Marsha O., Brusin K., Ben-David Y., Thakur G.A., Vaknin-Dembinsky A., Treinin M., Brenner T. Suppression of neuroinflammation by an allosteric agonist and positive allosteric modulator of the α7 nicotinic acetylcholine receptor GAT107. J. Neuroinflamm. 2021;18:99. doi: 10.1186/s12974-021-02149-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin M.-W., Chen Y.-H., Yang H.-B., Lin C.C., Hung S.-Y. Galantamine Inhibits Aβ1–42-Induced Neurotoxicity by Enhancing α7nAChR Expression as a Cargo Carrier for LC3 Binding and Aβ1–42 Engulfment During Autophagic Degradation. Neurotherapeutics. 2020;17:676–689. doi: 10.1007/s13311-019-00803-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takata K., Kitamura Y., Saeki M., Terada M., Kagitani S., Kitamura R., Fujikawa Y., Maelicke A., Tomimoto H., Taniguchi T., et al. Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 2010;285:40180–40191. doi: 10.1074/jbc.M110.142356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chalon S., Vetel S., Foucault-Fruchard L., Tronel C., Buron F., Vergote J., Bodard S., Routier S., Sérrière S. Neuroprotective and anti-inflammatory effects of a therapy combining agonists of nicotinic α7 and σ1 receptors in a rat model of Parkinson’s disease. Neural Regen. Res. 2021;16:1099. doi: 10.4103/1673-5374.300451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maurice T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016;296:270–278. doi: 10.1016/j.bbr.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 56.Roberts J.P., Stokoe S.A., Sathler M.F., Nichols R.A., Kim S. Selective coactivation of α7- and α4β2-nicotinic acetylcholine receptors reverses beta-amyloid–induced synaptic dysfunction. J. Biol. Chem. 2021;296:100402. doi: 10.1016/j.jbc.2021.100402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao K., Dong Y.-T., Xiang J., Xu Y., Li Y., Song H., Yu W.-F., Qi X.-L., Guan Z.-Z. The neuroprotective effects of SIRT1 in mice carrying the APP/PS1 double-transgenic mutation and in SH-SY5Y cells over-expressing human APP670/671 may involve elevated levels of α7 nicotinic acetylcholine receptors. Aging. 2020;12:1792–1807. doi: 10.18632/aging.102713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antunes M., Biala G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams J.P., Sweatt J.D. Molecular psychology: Roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 60.Tzingounis A.V., Nicoll R.A. Arc/Arg3.1: Linking Gene Expression to Synaptic Plasticity and Memory. Neuron. 2006;52:403–407. doi: 10.1016/j.neuron.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 61.Malinow R., Malenka R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 62.Giocomo L.M., Hasselmo M.E. Nicotinic modulation of glutamatergic synaptic transmission in region CA3 of the hippocampus. Eur. J. Neurosci. 2005;22:1349–1356. doi: 10.1111/j.1460-9568.2005.04316.x. [DOI] [PubMed] [Google Scholar]
- 63.Cheng Q., Yakel J.L. The effect of alpha7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem. Pharmacol. 2015;97:439–444. doi: 10.1016/j.bcp.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Q., Kawai H., Berg D.K. Beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gulisano W., Melone M., Ripoli C., Tropea M.R., Li Puma D.D., Giunta S., Cocco S., Marcotulli D., Origlia N., Palmeri A., et al. Neuromodulatory Action of Picomolar Extracellular Abeta42 Oligomers on Presynaptic and Postsynaptic Mechanisms Underlying Synaptic Function and Memory. J. Neurosci. 2019;39:5986–6000. doi: 10.1523/JNEUROSCI.0163-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tropea M.R., Li Puma D.D., Melone M., Gulisano W., Arancio O., Grassi C., Conti F., Puzzo D. Genetic deletion of alpha7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Prog. Neurobiol. 2021;206:102154. doi: 10.1016/j.pneurobio.2021.102154. [DOI] [PubMed] [Google Scholar]
- 67.Rahman M.A., Rahman M.S., Uddin M.J., Mamum-Or-Rashid A.N.M., Pang M.-G., Rhim H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020;27:44659–44672. doi: 10.1007/s11356-020-08243-z. [DOI] [PubMed] [Google Scholar]
- 68.Mawuenyega K.G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J.C., Yarasheski K.E., Bateman R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosas-Ballina M., Olofsson P.S., Ochani M., Valdes-Ferrer S.I., Levine Y.A., Reardon C., Tusche M.W., Pavlov V.A., Andersson U., Chavan S., et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Borovikova L.V., Ivanova S., Zhang M., Yang H., Botchkina G.I., Watkins L.R., Wang H., Abumrad N., Eaton J.W., Tracey K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 71.Norris G.T., Kipnis J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019;216:60–70. doi: 10.1084/jem.20180199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ransohoff R.M. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 73.Hampel H., Caraci F., Cuello A.C., Caruso G., Nistico R., Corbo M., Baldacci F., Toschi N., Garaci F., Chiesa P.A., et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020;11:456. doi: 10.3389/fimmu.2020.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Habbas S., Santello M., Becker D., Stubbe H., Zappia G., Liaudet N., Federica, Kollias G., Fontana A., Christopher, et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell. 2015;163:1730–1741. doi: 10.1016/j.cell.2015.11.023. [DOI] [PubMed] [Google Scholar]
- 75.Sun L.-L., Yang T.-Y., Wei N.-N., Lu W., Jiao W.-X., Zhou Q.-Q., Miao Y.-Z., Gao Q., Wang X.-T., Sun Q., et al. Pharmacological characterization of JWX-A0108 as a novel type I positive allosteric modulator of α7 nAChR that can reverse acoustic gating deficits in a mouse prepulse inhibition model. Acta Pharmacol. Sin. 2019;40:737–745. doi: 10.1038/s41401-018-0163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pabst M., Braganza O., Dannenberg H., Hu W., Pothmann L., Rosen J., Mody I., Karen, Deisseroth K., Albert, et al. Astrocyte Intermediaries of Septal Cholinergic Modulation in the Hippocampus. Neuron. 2016;90:853–865. doi: 10.1016/j.neuron.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 77.Ren J.M., Zhang S.L., Wang X.L., Guan Z.Z., Qi X.L. Expression levels of the alpha7 nicotinic acetylcholine receptor in the brains of patients with Alzheimer’s disease and their effect on synaptic proteins in SH-SY5Y cells. Mol. Med. Rep. 2020;22:2063–2075. doi: 10.3892/mmr.2020.11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guan Z.Z., Zhang X., Ravid R., Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J. Neurochem. 2000;74:237–243. doi: 10.1046/j.1471-4159.2000.0740237.x. [DOI] [PubMed] [Google Scholar]
- 79.Nakaizumi K., Ouchi Y., Terada T., Yoshikawa E., Kakimoto A., Isobe T., Bunai T., Yokokura M., Suzuki K., Magata Y. In vivo Depiction of alpha7 Nicotinic Receptor Loss for Cognitive Decline in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018;61:1355–1365. doi: 10.3233/JAD-170591. [DOI] [PubMed] [Google Scholar]
- 80.Song C., Shi J., Xu J., Zhao L., Zhang Y., Huang W., Qiu Y., Zhang R., Chen H., Wang H. Post-transcriptional regulation of alpha7 nAChR expression by miR-98-5p modulates cognition and neuroinflammation in an animal model of Alzheimer’s disease. FASEB J. 2021;35:e21658. doi: 10.1096/fj.202100257R. [DOI] [PubMed] [Google Scholar]
- 81.Chen X., Ba Y., Ma L., Cai X., Yin Y., Wang K., Guo J., Zhang Y., Chen J., Guo X., et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 82.Tan L., Yu J.T., Tan M.S., Liu Q.Y., Wang H.F., Zhang W., Jiang T., Tan L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014;40:1017–1027. doi: 10.3233/JAD-132144. [DOI] [PubMed] [Google Scholar]
- 83.Wang Z., Shen L., Wang Y., Huang S. Integrated analysis of miRNA and mRNA expression in the blood of patients with Alzheimer’s disease. Mol. Med. Rep. 2020;22:1053–1062. doi: 10.3892/mmr.2020.11162. [DOI] [PubMed] [Google Scholar]
- 84.Mohammadi N., Asle-Rousta M., Rahnema M., Amini R. Morin attenuates memory deficits in a rat model of Alzheimer’s disease by ameliorating oxidative stress and neuroinflammation. Eur. J. Pharmacol. 2021;910:174506. doi: 10.1016/j.ejphar.2021.174506. [DOI] [PubMed] [Google Scholar]
- 85.Halevi S. The C.elegansric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 2002;21:1012–1020. doi: 10.1093/emboj/21.5.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mizrachi T., Vaknin-Dembinsky A., Brenner T., Treinin M. Neuroinflammation Modulation via alpha7 Nicotinic Acetylcholine Receptor and Its Chaperone, RIC-3. Molecules. 2021;26:6139. doi: 10.3390/molecules26206139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gu S., Jose, Lord B., Anthony S., Weston D. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron. 2016;89:948–955. doi: 10.1016/j.neuron.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 88.Matta J.A., Gu S., Davini W.B., Lord B., Siuda E.R., Harrington A.W., Bredt D.S. NACHO Mediates Nicotinic Acetylcholine Receptor Function throughout the Brain. Cell Rep. 2017;19:688–696. doi: 10.1016/j.celrep.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 89.Wu M., Liu C.Z., Barrall E.A., Rissman R.A., Joiner W.J. Unbalanced Regulation of α7 nAChRs by Ly6h and NACHO Contributes to Neurotoxicity in Alzheimer’s Disease. J. Neurosci. 2021;41:8461–8474. doi: 10.1523/JNEUROSCI.0494-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Posadas I., Lopez-Hernandez B., Cena V. Nicotinic Receptors in Neurodegeneration. Curr. Neuropharmacol. 2013;11:298–314. doi: 10.2174/1570159X11311030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilens T.E., Decker M.W. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: Focus on cognition. Biochem. Pharmacol. 2007;74:1212–1223. doi: 10.1016/j.bcp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanberg P.R., Silver A.A., Shytle R.D., Philipp M.K., Cahill D.W., Fogelson H.M., McConville B.J. Nicotine for the treatment of Tourette’s syndrome. Pharmacol. Ther. 1997;74:21–25. doi: 10.1016/S0163-7258(96)00199-4. [DOI] [PubMed] [Google Scholar]
- 93.Philip N.S., Carpenter L.L., Tyrka A.R., Price L.H. Nicotinic acetylcholine receptors and depression: A review of the preclinical and clinical literature. Psychopharmacology. 2010;212:1–12. doi: 10.1007/s00213-010-1932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saji H., Magata Y., Yamada Y., Tajima K., Yonekura Y., Konishi J., Ohmomo Y., Yokoyama A. Synthesis of (S)-N-[methyl-11C]nicotine and its regional distribution in the mouse brain: A potential tracer for visualization of brain nicotinic receptors by positron emission tomography. Chem. Pharm. Bull. 1992;40:734–736. doi: 10.1248/cpb.40.734. [DOI] [PubMed] [Google Scholar]
- 95.Iida Y., Ogawa M., Ueda M., Tominaga A., Kawashima H., Magata Y., Nishiyama S., Tsukada H., Mukai T., Saji H. Evaluation of 5-(11)C-methyl-A-85380 as an imaging agent for PET investigations of brain nicotinic acetylcholine receptors. J. Nucl. Med. 2004;45:878–884. [PubMed] [Google Scholar]
- 96.Horti A.G., Scheffel U., Koren A.O., Ravert H.T., Mathews W.B., Musachio J.L., Finley P.A., London E.D., Dannals R.F. 2-[18F]Fluoro-A-85380, an in vivo tracer for the nicotinic acetylcholine receptors. Nucl. Med. Biol. 1998;25:599–603. doi: 10.1016/S0969-8051(98)00031-6. [DOI] [PubMed] [Google Scholar]
- 97.Valette H., Bottlaender M., Dolle F., Guenther I., Fuseau C., Coulon C., Ottaviani M., Crouzel C. Imaging central nicotinic acetylcholine receptors in baboons with [18F]fluoro-A-85380. J. Nucl. Med. 1999;40:1374–1380. [PubMed] [Google Scholar]
- 98.Doll F., Dolci L., Valette H., Hinnen F., Vaufrey F., Guenther I., Fuseau C., Coulon C., Bottlaender M., Crouzel C. Synthesis and nicotinic acetylcholine receptor in vivo binding properties of 2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine: A new positron emission tomography ligand for nicotinic receptors. J. Med. Chem. 1999;42:2251–2259. doi: 10.1021/jm9910223. [DOI] [PubMed] [Google Scholar]
- 99.Koren A.O., Horti A.G., Mukhin A.G., Gundisch D., Kimes A.S., Dannals R.F., London E.D. 2-, 5-, and 6-Halo-3-(2(S)-azetidinylmethoxy)pyridines: Synthesis, affinity for nicotinic acetylcholine receptors, and molecular modeling. J. Med. Chem. 1998;41:3690–3698. doi: 10.1021/jm980170a. [DOI] [PubMed] [Google Scholar]
- 100.Wong D.F., Kuwabara H., Kim J., Brasic J.R., Chamroonrat W., Gao Y., Valentine H., Willis W., Mathur A., McCaul M.E., et al. PET imaging of high-affinity alpha4beta2 nicotinic acetylcholine receptors in humans with 18F-AZAN, a radioligand with optimal brain kinetics. J. Nucl. Med. 2013;54:1308–1314. doi: 10.2967/jnumed.112.108001. [DOI] [PubMed] [Google Scholar]
- 101.Kuwabara H., Wong D.F., Gao Y., Valentine H., Holt D.P., Ravert H.T., Dannals R.F., Horti A.G. PET Imaging of nicotinic acetylcholine receptors in baboons with 18F-AZAN, a radioligand with improved brain kinetics. J. Nucl. Med. 2012;53:121–129. doi: 10.2967/jnumed.111.092338. [DOI] [PubMed] [Google Scholar]
- 102.Gao Y., Kuwabara H., Spivak C.E., Xiao Y., Kellar K., Ravert H.T., Kumar A., Alexander M., Hilton J., Wong D.F., et al. Discovery of (-)-7-methyl-2-exo-[3′-(6-[18F]fluoropyridin-2-yl)-5′-pyridinyl]-7-azabicyclo[2.2.1]heptane, a radiolabeled antagonist for cerebral nicotinic acetylcholine receptor (alpha4beta2-nAChR) with optimal positron emission tomography imaging properties. J. Med. Chem. 2008;51:4751–4764. doi: 10.1021/jm800323d. [DOI] [PubMed] [Google Scholar]
- 103.Sattler B., Kranz M., Starke A., Wilke S., Donat C.K., Deuther-Conrad W., Patt M., Schildan A., Patt J., Smits R., et al. Internal dose assessment of (-)-18F-flubatine, comparing animal model datasets of mice and piglets with first-in-human results. J. Nucl. Med. 2014;55:1885–1892. doi: 10.2967/jnumed.114.137059. [DOI] [PubMed] [Google Scholar]
- 104.Hillmer A.T., Esterlis I., Gallezot J.D., Bois F., Zheng M.Q., Nabulsi N., Lin S.F., Papke R.L., Huang Y., Sabri O., et al. Imaging of cerebral alpha4beta2* nicotinic acetylcholine receptors with (-)-[(18)F]Flubatine PET: Implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. Neuroimage. 2016;141:71–80. doi: 10.1016/j.neuroimage.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deuther-Conrad W., Patt J.T., Lockman P.R., Allen D.D., Patt M., Schildan A., Ganapathy V., Steinbach J., Sabri O., Brust P. Norchloro-fluoro-homoepibatidine (NCFHEB)—A promising radioligand for neuroimaging nicotinic acetylcholine receptors with PET. Eur. Neuropsychopharmacol. 2008;18:222–229. doi: 10.1016/j.euroneuro.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 106.Brašić J.R., Cascella N., Kumar A., Zhou Y., Hilton J., Raymont V., Crabb A., Guevara M.R., Horti A.G., Wong D.F. Positron emission tomography experience with 2-[18F]fluoro-3-(2(s)-azetidinylmethoxy)pyridine (2-[18F]fa) in the living human brain of smokers with paranoid schizophrenia. Synapse. 2012;66:352–368. doi: 10.1002/syn.21520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saji H., Ogawa M., Ueda M., Iida Y., Magata Y., Tominaga A., Kawashima H., Kitamura Y., Nakagawa M., Kiyono Y., et al. Evaluation of radioiodinated 5-iodo-3-(2(S)-azetidinylmethoxy)pyridine as a ligand for SPECT investigations of brain nicotinic acetylcholine receptors. Ann. Nucl. Med. 2002;16:189–200. doi: 10.1007/BF02996300. [DOI] [PubMed] [Google Scholar]
- 108.Fujita M., Ichise M., van Dyck C.H., Zoghbi S.S., Tamagnan G., Mukhin A.G., Bozkurt A., Seneca N., Tipre D., DeNucci C.C., et al. Quantification of nicotinic acetylcholine receptors in human brain using [123I]5-I-A-85380 SPET. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:1620–1629. doi: 10.1007/s00259-003-1320-0. [DOI] [PubMed] [Google Scholar]
- 109.Kimura H., Ueda M., Kawashima H., Arimitsu K., Yagi Y., Saji H. Synthesis and biological evaluation of Tc-99m-cyclopentadienyltricarbonyl-technetium-labeled A-85380: An imaging probe for single-photon emission computed tomography investigation of nicotinic acetylcholine receptors in the brain. Bioorg. Med. Chem. 2019;27:2245–2252. doi: 10.1016/j.bmc.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 110.Mori D., Kimura H., Kawashima H., Yagi Y., Arimitsu K., Ono M., Saji H. Development of (99m)Tc radiolabeled A85380 derivatives targeting cerebral nicotinic acetylcholine receptor: Novel radiopharmaceutical ligand (99m)Tc-A-YN-IDA-C4. Bioorg. Med. Chem. 2019;27:4200–4210. doi: 10.1016/j.bmc.2019.07.053. [DOI] [PubMed] [Google Scholar]
- 111.Mathew S.V., Law A.J., Lipska B.K., Dávila-García M.I., Zamora E.D., Mitkus S.N., Vakkalanka R., Straub R.E., Weinberger D.R., Kleinman J.E., et al. α7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in neuregulin 1. Hum. Mol. Genet. 2007;16:2921–2932. doi: 10.1093/hmg/ddm253. [DOI] [PubMed] [Google Scholar]
- 112.Dani J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Elsevier; Amsterdam, The Netherlands: 2015. pp. 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davies A.R., Hardick D.J., Blagbrough I.S., Potter B.V., Wolstenholme A.J., Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: A new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38:679–690. doi: 10.1016/S0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 114.Toyohara J., Sakata M., Wu J., Ishikawa M., Oda K., Ishii K., Iyo M., Hashimoto K., Ishiwata K. Preclinical and the first clinical studies on [11C]CHIBA-1001 for mapping alpha7 nicotinic receptors by positron emission tomography. Ann. Nucl. Med. 2009;23:301–309. doi: 10.1007/s12149-009-0240-x. [DOI] [PubMed] [Google Scholar]
- 115.Ettrup A., Mikkelsen J.D., Lehel S., Madsen J., Nielsen E.O., Palner M., Timmermann D.B., Peters D., Knudsen G.M. 11C-NS14492 as a novel PET radioligand for imaging cerebral alpha7 nicotinic acetylcholine receptors: In vivo evaluation and drug occupancy measurements. J. Nucl. Med. 2011;52:1449–1456. doi: 10.2967/jnumed.111.088815. [DOI] [PubMed] [Google Scholar]
- 116.Deuther-Conrad W., Fischer S., Hiller A., Nielsen E.O., Timmermann D.B., Steinbach J., Sabri O., Peters D., Brust P. Molecular imaging of alpha7 nicotinic acetylcholine receptors: Design and evaluation of the potent radioligand [18F]NS10743. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:791–800. doi: 10.1007/s00259-008-1031-7. [DOI] [PubMed] [Google Scholar]
- 117.Rotering S., Scheunemann M., Fischer S., Hiller A., Peters D., Deuther-Conrad W., Brust P. Radiosynthesis and first evaluation in mice of [(18)F]NS14490 for molecular imaging of alpha7 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013;21:2635–2642. doi: 10.1016/j.bmc.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 118.Ravert H.T., Dorff P., Foss C.A., Mease R.C., Fan H., Holmquist C.R., Phillips E., McCarthy D.J., Heys J.R., Holt D.P., et al. Radiochemical synthesis and in vivo evaluation of [18F]AZ11637326: An agonist probe for the alpha7 nicotinic acetylcholine receptor. Nucl. Med. Biol. 2013;40:731–739. doi: 10.1016/j.nucmedbio.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 119.Ogawa M., Nishiyama S., Tsukada H., Hatano K., Fuchigami T., Yamaguchi H., Matsushima Y., Ito K., Magata Y. Synthesis and evaluation of new imaging agent for central nicotinic acetylcholine receptor alpha7 subtype. Nucl. Med. Biol. 2010;37:347–355. doi: 10.1016/j.nucmedbio.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 120.Wong D.F., Kuwabara H., Pomper M., Holt D.P., Brasic J.R., George N., Frolov B., Willis W., Gao Y., Valentine H., et al. Human Brain Imaging of α7 nAChR with [18F]ASEM: A New PET Radiotracer for Neuropsychiatry and Determination of Drug Occupancy. Mol. Imaging Biol. 2014;16:730–738. doi: 10.1007/s11307-014-0779-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gao Y., Kellar K.J., Yasuda R.P., Tran T., Xiao Y., Dannals R.F., Horti A.G. Derivatives of dibenzothiophene for positron emission tomography imaging of alpha7-nicotinic acetylcholine receptors. J. Med. Chem. 2013;56:7574–7589. doi: 10.1021/jm401184f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hillmer A.T., Li S., Zheng M.Q., Scheunemann M., Lin S.F., Nabulsi N., Holden D., Pracitto R., Labaree D., Ropchan J., et al. PET imaging of alpha7 nicotinic acetylcholine receptors: A comparative study of [(18)F]ASEM and [(18)F]DBT-10 in nonhuman primates, and further evaluation of [(18)F]ASEM in humans. Eur. J. Nucl. Med. Mol. Imaging. 2017;44:1042–1050. doi: 10.1007/s00259-017-3621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gao Y., Mease R.C., Olson T.T., Kellar K.J., Dannals R.F., Pomper M.G., Horti A.G. [(125)I]Iodo-ASEM, a specific in vivo radioligand for alpha7-nAChR. Nucl. Med. Biol. 2015;42:488–493. doi: 10.1016/j.nucmedbio.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Donat C.K., Hansen H.H., Hansen H.D., Mease R.C., Horti A.G., Pomper M.G., L’Estrade E.T., Herth M.M., Peters D., Knudsen G.M., et al. In Vitro and In Vivo Characterization of Dibenzothiophene Derivatives [(125)I]Iodo-ASEM and [(18)F]ASEM as Radiotracers of Homo- and Heteromeric alpha7 Nicotinic Acetylcholine Receptors. Molecules. 2020;25:1425. doi: 10.3390/molecules25061425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Voet S., Srinivasan S., Lamkanfi M., Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019;11:e10248. doi: 10.15252/emmm.201810248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hanslik K.L., Ulland T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020;11:570711. doi: 10.3389/fneur.2020.570711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tangestani Fard M., Stough C. A Review and Hypothesized Model of the Mechanisms That Underpin the Relationship Between Inflammation and Cognition in the Elderly. Front. Aging Neurosci. 2019;11:56. doi: 10.3389/fnagi.2019.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ahmad M.A., Kareem O., Khushtar M., Akbar M., Haque M.R., Iqubal A., Haider M.F., Pottoo F.H., Abdulla F.S., Al-Haidar M.B., et al. Neuroinflammation: A Potential Risk for Dementia. Int. J. Mol. Sci. 2022;23:616. doi: 10.3390/ijms23020616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wright A.L., Zinn R., Hohensinn B., Konen L.M., Beynon S.B., Tan R.P., Clark I.A., Abdipranoto A., Vissel B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE. 2013;8:e59586. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Van Zeller M., Dias D., Sebastião A.M., Valente C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021;83:939–961. doi: 10.3233/JAD-210268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaur D., Sharma V., Deshmukh R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology. 2019;27:663–677. doi: 10.1007/s10787-019-00580-x. [DOI] [PubMed] [Google Scholar]
- 132.Mejias N.H., Martinez C.C., Stephens M.E., de Rivero Vaccari J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018;15:23. doi: 10.1186/s12950-018-0198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Smith J.A., Das A., Ray S.K., Banik N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Griffin W.S., Liu L., Li Y., Mrak R.E., Barger S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Heneka M.T., O’Banion M.K., Terwel D., Kummer M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010;117:919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- 136.Italiani P., Puxeddu I., Napoletano S., Scala E., Melillo D., Manocchio S., Angiolillo A., Migliorini P., Boraschi D., Vitale E., et al. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J. Neuroinflamm. 2018;15:1–12. doi: 10.1186/s12974-018-1376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Simard A.R., Soulet D., Gowing G., Julien J.-P., Rivest S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 138.Heneka M.T., Kummer M.P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.-C., et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lamkanfi M. Vishva Mechanisms and Functions of Inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 140.Schroder K., Tschopp J. The Inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 141.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 142.Walsh J.G., Muruve D.A., Power C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014;15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 143.Li Y., Liu L., Barger S.W., Griffin W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chan A.H., Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020;217:jem.20190314. doi: 10.1084/jem.20190314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.He W.-T., Wan H., Hu L., Chen P., Wang X., Huang Z., Yang Z.-H., Zhong C.-Q., Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Franklin B.S., Bossaller L., De Nardo D., Ratter J.M., Stutz A., Engels G., Brenker C., Nordhoff M., Mirandola S.R., Al-Amoudi A., et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Baroja-Mazo A., Martin-Sanchez F., Gomez A.I., Martinez C.M., Amores-Iniesta J., Compan V., Barbera-Cremades M., Yague J., Ruiz-Ortiz E., Anton J., et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 149.Prada I., Gabrielli M., Turola E., Iorio A., D’Arrigo G., Parolisi R., De Luca M., Pacifici M., Bastoni M., Lombardi M., et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018;135:529–550. doi: 10.1007/s00401-017-1803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S.V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R.M., Tejera D., et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vergara C., Houben S., Suain V., Yilmaz Z., De Decker R., Vanden Dries V., Boom A., Mansour S., Leroy K., Ando K., et al. Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019;137:397–412. doi: 10.1007/s00401-018-1953-5. [DOI] [PubMed] [Google Scholar]
- 152.He Z., Guo J.L., McBride J.D., Narasimhan S., Kim H., Changolkar L., Zhang B., Gathagan R.J., Yue C., Dengler C., et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018;24:29–38. doi: 10.1038/nm.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stancu I.-C., Cremers N., Vanrusselt H., Couturier J., Vanoosthuyse A., Kessels S., Lodder C., Brône B., Huaux F., Octave J.-N., et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599–617. doi: 10.1007/s00401-018-01957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Halle A., Hornung V., Petzold G.C., Stewart C.R., Monks B.G., Reinheckel T., Fitzgerald K.A., Latz E., Moore K.J., Golenbock D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wu Z., Sun L., Hashioka S., Yu S., Schwab C., Okada R., Hayashi Y., McGeer P.L., Nakanishi H. Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiol. Aging. 2013;34:2715–2725. doi: 10.1016/j.neurobiolaging.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 156.Murphy N., Grehan B., Lynch M.A. Glial Uptake of Amyloid Beta Induces NLRP3 Inflammasome Formation via Cathepsin-Dependent Degradation of NLRP10. Neuromol. Med. 2014;16:205–215. doi: 10.1007/s12017-013-8274-6. [DOI] [PubMed] [Google Scholar]
- 157.Matcovitch-Natan O., Winter D.R., Giladi A., Vargas Aguilar S., Spinrad A., Sarrazin S., Ben-Yehuda H., David E., Zelada Gonzalez F., Perrin P., et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- 158.Li Q., Cheng Z., Zhou L., Darmanis S., Neff N.F., Okamoto J., Gulati G., Bennett M.L., Sun L.O., Clarke L.E., et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 2019;101:207–223.e210. doi: 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hammond T.R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A.J., Gergits F., Segel M., Nemesh J., et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 2019;50:253–271.e256. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Masuda T., Sankowski R., Staszewski O., Bottcher C., Amann L., Sagar, Scheiwe C., Nessler S., Kunz P., van Loo G., et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 161.Thion M.S., Ginhoux F., Garel S. Microglia and early brain development: An intimate journey. Science. 2018;362:185–189. doi: 10.1126/science.aat0474. [DOI] [PubMed] [Google Scholar]
- 162.Bian Z., Gong Y., Huang T., Lee C.Z.W., Bian L., Bai Z., Shi H., Zeng Y., Liu C., He J., et al. Deciphering human macrophage development at single-cell resolution. Nature. 2020;582:571–576. doi: 10.1038/s41586-020-2316-7. [DOI] [PubMed] [Google Scholar]
- 163.Masuda T., Sankowski R., Staszewski O., Prinz M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020;30:1271–1281. doi: 10.1016/j.celrep.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 164.Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X., et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Grubman A., Chew G., Ouyang J.F., Sun G., Choo X.Y., McLean C., Simmons R.K., Buckberry S., Vargas-Landin D.B., Poppe D., et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 166.Olah M., Menon V., Habib N., Taga M.F., Ma Y., Yung C.J., Cimpean M., Khairallah A., Coronas-Samano G., Sankowski R., et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020;11:6129. doi: 10.1038/s41467-020-19737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Friedman B.A., Srinivasan K., Ayalon G., Meilandt W.J., Lin H., Huntley M.A., Cao Y., Lee S.H., Haddick P.C.G., Ngu H., et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018;22:832–847. doi: 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- 168.Zheng Z., Liang P., Hou B., Lu X., Ma Q., Yu X., Han S., Peng B., Chen T., Liu W., et al. The effect of dipeptidyl peptidase IV on disease-associated microglia phenotypic transformation in epilepsy. J. Neuroinflamm. 2021;18:112. doi: 10.1186/s12974-021-02133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Holtman I.R., Raj D.D., Miller J.A., Schaafsma W., Yin Z., Brouwer N., Wes P.D., Moller T., Orre M., Kamphuis W., et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mrdjen D., Pavlovic A., Hartmann F.J., Schreiner B., Utz S.G., Leung B.P., Lelios I., Heppner F.L., Kipnis J., Merkler D., et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity. 2018;48:380–395.e386. doi: 10.1016/j.immuni.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 171.McQuade A., Kang Y.J., Hasselmann J., Jairaman A., Sotelo A., Coburn M., Shabestari S.K., Chadarevian J.P., Fote G., Tu C.H., et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020;11:5370. doi: 10.1038/s41467-020-19227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cagnin A., Kassiou M., Meikle S.R., Banati R.B. Positron emission tomography imaging of neuroinflammation. Neurotherapeutics. 2007;4:443–452. doi: 10.1016/j.nurt.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Papadopoulos V., Baraldi M., Guilarte T.R., Knudsen T.B., Lacapere J.J., Lindemann P., Norenberg M.D., Nutt D., Weizman A., Zhang M.R., et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 174.Braestrup C., Squires R.F. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc. Natl. Acad. Sci. USA. 1977;74:3805–3809. doi: 10.1073/pnas.74.9.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schoemaker H., Bliss M., Yamamura H.I. Specific high-affinity saturable binding of [3H] R05-4864 to benzodiazepine binding sites in the rat cerebral cortex. Eur. J. Pharmacol. 1981;71:173–175. doi: 10.1016/0014-2999(81)90405-2. [DOI] [PubMed] [Google Scholar]
- 176.Marangos P.J., Patel J., Boulenger J.P., Clark-Rosenberg R. Characterization of peripheral-type benzodiazepine binding sites in brain using [3H]Ro 5-4864. Mol. Pharmacol. 1982;22:26–32. [PubMed] [Google Scholar]
- 177.Lacapere J.J., Papadopoulos V. Peripheral-type benzodiazepine receptor: Structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68:569–585. doi: 10.1016/S0039-128X(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 178.Taketani S., Kohno H., Okuda M., Furukawa T., Tokunaga R. Induction of peripheral-type benzodiazepine receptors during differentiation of mouse erythroleukemia cells. A possible involvement of these receptors in heme biosynthesis. J. Biol. Chem. 1994;269:7527–7531. doi: 10.1016/S0021-9258(17)37318-0. [DOI] [PubMed] [Google Scholar]
- 179.Wright G., Reichenbecher V. The effects of superoxide and the peripheral benzodiazepine receptor ligands on the mitochondrial processing of manganese-dependent superoxide dismutase. Exp. Cell Res. 1999;246:443–450. doi: 10.1006/excr.1998.4331. [DOI] [PubMed] [Google Scholar]
- 180.McEnery M.W., Snowman A.M., Trifiletti R.R., Snyder S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Papadopoulos V., Amri H., Boujrad N., Cascio C., Culty M., Garnier M., Hardwick M., Li H., Vidic B., Brown A.S., et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids. 1997;62:21–28. doi: 10.1016/S0039-128X(96)00154-7. [DOI] [PubMed] [Google Scholar]
- 182.Banati R.B., Middleton R.J., Chan R., Hatty C.R., Kam W.W., Quin C., Graeber M.B., Parmar A., Zahra D., Callaghan P., et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014;5:5452. doi: 10.1038/ncomms6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tu L.N., Morohaku K., Manna P.R., Pelton S.H., Butler W.R., Stocco D.M., Selvaraj V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 2014;289:27444–27454. doi: 10.1074/jbc.M114.578286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang H., Zhai K., Xue Y., Yang J., Yang Q., Fu Y., Hu Y., Liu F., Wang W., Cui L., et al. Global Deletion of TSPO Does Not Affect the Viability and Gene Expression Profile. PLoS ONE. 2016;11:e0167307. doi: 10.1371/journal.pone.0167307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Morohaku K., Pelton S.H., Daugherty D.J., Butler W.R., Deng W., Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology. 2014;155:89–97. doi: 10.1210/en.2013-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sileikyte J., Blachly-Dyson E., Sewell R., Carpi A., Menabo R., Di Lisa F., Ricchelli F., Bernardi P., Forte M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)) J. Biol. Chem. 2014;289:13769–13781. doi: 10.1074/jbc.M114.549634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Selvaraj V., Stocco D.M. The changing landscape in translocator protein (TSPO) function. Trends Endocrinol. Metab. 2015;26:341–348. doi: 10.1016/j.tem.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhao A.H., Tu L.N., Mukai C., Sirivelu M.P., Pillai V.V., Morohaku K., Cohen R., Selvaraj V. Mitochondrial Translocator Protein (TSPO) Function Is Not Essential for Heme Biosynthesis. J. Biol. Chem. 2016;291:1591–1603. doi: 10.1074/jbc.M115.686360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fujita M., Imaizumi M., Zoghbi S.S., Fujimura Y., Farris A.G., Suhara T., Hong J., Pike V.W., Innis R.B. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage. 2008;40:43–52. doi: 10.1016/j.neuroimage.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kreisl W.C., Fujita M., Fujimura Y., Kimura N., Jenko K.J., Kannan P., Hong J., Morse C.L., Zoghbi S.S., Gladding R.L., et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49:2924–2932. doi: 10.1016/j.neuroimage.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Owen D.R., Howell O.W., Tang S.P., Wells L.A., Bennacef I., Bergstrom M., Gunn R.N., Rabiner E.A., Wilkins M.R., Reynolds R., et al. Two binding sites for [3H]PBR28 in human brain: Implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2010;30:1608–1618. doi: 10.1038/jcbfm.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Owen D.R., Gunn R.N., Rabiner E.A., Bennacef I., Fujita M., Kreisl W.C., Innis R.B., Pike V.W., Reynolds R., Matthews P.M., et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011;52:24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Owen D.R., Yeo A.J., Gunn R.N., Song K., Wadsworth G., Lewis A., Rhodes C., Pulford D.J., Bennacef I., Parker C.A., et al. An 18-kDa Translocator Protein (TSPO) Polymorphism Explains Differences in Binding Affinity of the PET Radioligand PBR28. J. Cereb. Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Betlazar C., Harrison-Brown M., Middleton R.J., Banati R., Liu G.J. Cellular Sources and Regional Variations in the Expression of the Neuroinflammatory Marker Translocator Protein (TSPO) in the Normal Brain. Int. J. Mol. Sci. 2018;19:2707. doi: 10.3390/ijms19092707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tournier B.B., Tsartsalis S., Ceyzeriat K., Fraser B.H., Gregoire M.C., Kovari E., Millet P. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020;77:1043–1056. doi: 10.3233/JAD-200136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gui Y., Marks J.D., Das S., Hyman B.T., Serrano-Pozo A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. 2020;30:151–164. doi: 10.1111/bpa.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hillmer A.T., Holden D., Fowles K., Nabulsi N., West B.L., Carson R.E., Cosgrove K.P. Microglial depletion and activation: A [(11)C]PBR28 PET study in nonhuman primates. EJNMMI Res. 2017;7:59. doi: 10.1186/s13550-017-0305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Elmore M.R., Najafi A.R., Koike M.A., Dagher N.N., Spangenberg E.E., Rice R.A., Kitazawa M., Matusow B., Nguyen H., West B.L., et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bader S., Wolf L., Milenkovic V.M., Gruber M., Nothdurfter C., Rupprecht R., Wetzel C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology. 2019;106:65–76. doi: 10.1016/j.psyneuen.2019.03.029. [DOI] [PubMed] [Google Scholar]
- 200.Papadopoulos V., Fan J., Zirkin B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018;30:e12500. doi: 10.1111/jne.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lejri I., Grimm A., Halle F., Abarghaz M., Klein C., Maitre M., Schmitt M., Bourguignon J.J., Mensah-Nyagan A.G., Bihel F., et al. TSPO Ligands Boost Mitochondrial Function and Pregnenolone Synthesis. J. Alzheimer’s Dis. 2019;72:1045–1058. doi: 10.3233/JAD-190127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Owen D.R., Narayan N., Wells L., Healy L., Smyth E., Rabiner E.A., Galloway D., Williams J.B., Lehr J., Mandhair H., et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017;37:2679–2690. doi: 10.1177/0271678X17710182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Milenkovic V.M., Slim D., Bader S., Koch V., Heinl E.S., Alvarez-Carbonell D., Nothdurfter C., Rupprecht R., Wetzel C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019;20:3359. doi: 10.3390/ijms20133359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yao R., Pan R., Shang C., Li X., Cheng J., Xu J., Li Y. Translocator Protein 18 kDa (TSPO) Deficiency Inhibits Microglial Activation and Impairs Mitochondrial Function. Front. Pharmacol. 2020;11:986. doi: 10.3389/fphar.2020.00986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Loth M.K., Guariglia S.R., Re D.B., Perez J., de Paiva V.N., Dziedzic J.L., Chambers J.W., Azzam D.J., Guilarte T.R. A Novel Interaction of Translocator Protein 18 kDa (TSPO) with NADPH Oxidase in Microglia. Mol. Neurobiol. 2020;57:4467–4487. doi: 10.1007/s12035-020-02042-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wolf A., Herb M., Schramm M., Langmann T. The TSPO-NOX1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020;11:2709. doi: 10.1038/s41467-020-16400-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Batarseh A., Papadopoulos V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 2010;327:1–12. doi: 10.1016/j.mce.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cosenza-Nashat M., Zhao M.L., Suh H.S., Morgan J., Natividad R., Morgello S., Lee S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009;35:306–328. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shimoyama S., Furukawa T., Ogata Y., Nikaido Y., Koga K., Sakamoto Y., Ueno S., Nakamura K. Lipopolysaccharide induces mouse translocator protein (18 kDa) expression via the AP-1 complex in the microglial cell line, BV-2. PLoS ONE. 2019;14:e0222861. doi: 10.1371/journal.pone.0222861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hannestad J., Gallezot J.D., Schafbauer T., Lim K., Kloczynski T., Morris E.D., Carson R.E., Ding Y.S., Cosgrove K.P. Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. Neuroimage. 2012;63:232–239. doi: 10.1016/j.neuroimage.2012.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Woodcock E.A., Hillmer A.T., Sandiego C.M., Maruff P., Carson R.E., Cosgrove K.P., Pietrzak R.H. Acute neuroimmune stimulation impairs verbal memory in adults: A PET brain imaging study. Brain. Behav. Immun. 2020;91:784–787. doi: 10.1016/j.bbi.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sandiego C.M., Gallezot J.-D., Pittman B., Nabulsi N., Lim K., Lin S.-F., Matuskey D., Lee J.-Y., O’Connor K.C., Huang Y., et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA. 2015;112:12468–12473. doi: 10.1073/pnas.1511003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tournier B.B., Tsartsalis S., Ceyzeriat K., Medina Z., Fraser B.H., Gregoire M.C., Kovari E., Millet P. Fluorescence-activated cell sorting to reveal the cell origin of radioligand binding. J. Cereb. Blood Flow Metab. 2020;40:1242–1255. doi: 10.1177/0271678X19860408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tournier B.B., Tsartsalis S., Ceyzériat K., Garibotto V., Millet P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells. 2020;9:1941. doi: 10.3390/cells9091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Tournier B.B., Tsartsalis S., Rigaud D., Fossey C., Cailly T., Fabis F., Pham T., Gregoire M.C., Kovari E., Moulin-Sallanon M., et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019;121:95–105. doi: 10.1016/j.nbd.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 216.Hamelin L., Lagarde J., Dorothee G., Potier M.C., Corlier F., Kuhnast B., Caille F., Dubois B., Fillon L., Chupin M., et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer’s disease. Brain. 2018;141:1855–1870. doi: 10.1093/brain/awy079. [DOI] [PubMed] [Google Scholar]
- 217.Fan Z., Okello A.A., Brooks D.J., Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain. 2015;138:3685–3698. doi: 10.1093/brain/awv288. [DOI] [PubMed] [Google Scholar]
- 218.Tuisku J., Plaven-Sigray P., Gaiser E.C., Airas L., Al-Abdulrasul H., Bruck A., Carson R.E., Chen M.K., Cosgrove K.P., Ekblad L., et al. Effects of age, BMI and sex on the glial cell marker TSPO—A multicentre [(11)C]PBR28 HRRT PET study. Eur. J. Nucl. Med. Mol. Imaging. 2019;46:2329–2338. doi: 10.1007/s00259-019-04403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lyoo C.H., Ikawa M., Liow J.S., Zoghbi S.S., Morse C.L., Pike V.W., Fujita M., Innis R.B., Kreisl W.C. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J. Nucl. Med. 2015;56:701–706. doi: 10.2967/jnumed.114.146027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Golla S.S., Boellaard R., Oikonen V., Hoffmann A., van Berckel B.N., Windhorst A.D., Virta J., Haaparanta-Solin M., Luoto P., Savisto N., et al. Quantification of [18F]DPA-714 binding in the human brain: Initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015;35:766–772. doi: 10.1038/jcbfm.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gulyas B., Vas A., Toth M., Takano A., Varrone A., Cselenyi Z., Schain M., Mattsson P., Halldin C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with [11C]vinpocetine. Neuroimage. 2011;56:1111–1121. doi: 10.1016/j.neuroimage.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 222.Schuitemaker A., Kropholler M.A., Boellaard R., van der Flier W.M., Kloet R.W., van der Doef T.F., Knol D.L., Windhorst A.D., Luurtsema G., Barkhof F., et al. Microglial activation in Alzheimer’s disease: An (R)-[(1)(1)C]PK11195 positron emission tomography study. Neurobiol. Aging. 2013;34:128–136. doi: 10.1016/j.neurobiolaging.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 223.Zeineh M.M., Chen Y., Kitzler H.H., Hammond R., Vogel H., Rutt B.K. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging. 2015;36:2483–2500. doi: 10.1016/j.neurobiolaging.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Takata K., Kitamura Y., Yanagisawa D., Morikawa S., Morita M., Inubushi T., Tsuchiya D., Chishiro S., Saeki M., Taniguchi T., et al. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett. 2007;581:475–478. doi: 10.1016/j.febslet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 225.McLarnon J.G., Ryu J.K., Walker D.G., Choi H.B. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 2006;65:1090–1097. doi: 10.1097/01.jnen.0000240470.97295.d3. [DOI] [PubMed] [Google Scholar]
- 226.Francistiova L., Bianchi C., Di Lauro C., Sebastian-Serrano A., de Diego-Garcia L., Kobolak J., Dinnyes A., Diaz-Hernandez M. The Role of P2X7 Receptor in Alzheimer’s Disease. Front. Mol. Neurosci. 2020;13:94. doi: 10.3389/fnmol.2020.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lee H.G., Won S.M., Gwag B.J., Lee Y.B. Microglial P2X(7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011;43:7–14. doi: 10.3858/emm.2011.43.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kahlenberg J.M., Dubyak G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004;286:C1100–C1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 229.Chiozzi P., Sarti A.C., Sanz J.M., Giuliani A.L., Adinolfi E., Vultaggio-Poma V., Falzoni S., Di Virgilio F. Amyloid beta-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019;9:6475. doi: 10.1038/s41598-019-42931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Beaino W., Janssen B., Vugts D.J., de Vries H.E., Windhorst A.D. Towards PET imaging of the dynamic phenotypes of microglia. Clin. Exp. Immunol. 2021;206:282–300. doi: 10.1111/cei.13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mildner A., Huang H., Radke J., Stenzel W., Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. 2017;65:375–387. doi: 10.1002/glia.23097. [DOI] [PubMed] [Google Scholar]
- 232.Villa A., Klein B., Janssen B., Pedragosa J., Pepe G., Zinnhardt B., Vugts D.J., Gelosa P., Sironi L., Beaino W., et al. Identification of new molecular targets for PET imaging of the microglial anti-inflammatory activation state. Theranostics. 2018;8:5400–5418. doi: 10.7150/thno.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shen Z., Bao X., Wang R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018;10:314. doi: 10.3389/fnagi.2018.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.