

Abstract
Fibrotic disease is caused by the continuous deposition of extracellular matrix by persistently activated fibroblasts (also known as myofibroblasts), even after the resolution of the injury. Using fibroblasts from porcine aortic valves cultured on hydrogels that can be softened via exposure to ultraviolet light, here we show that increased extracellular stiffness activates the fibroblasts, and that cumulative tension on the nuclear membrane and increases in the activity of histone deacetylases transform transiently activated fibroblasts into myofibroblasts displaying condensed chromatin with genome-wide alterations. The condensed structure of the myofibroblasts is associated with cytoskeletal stability, as indicated by the inhibition of chromatin condensation and myofibroblast persistence after detachment of the nucleus from the cytoskeleton via the displacement of endogenous nesprins from the nuclear envelope. We also show that the chromatin structure of myofibroblasts from patients with aortic valve stenosis is more condensed than that of myofibroblasts from healthy donors. Our findings suggest that nuclear mechanosensing drives distinct chromatin signatures in persistently activated fibroblasts.
Fibrosis represents a major cause of morbidity and mortality worldwide, affecting most tissues, but has no effective therapies1. Key mediators of fibrosis are chronically activated fibroblasts, known as myofibroblasts, which produce excessive amounts of extracellular matrix (ECM), which accumulates and impairs organ function. Transient myofibroblast activation is important for normal tissue homeostasis, wound-healing and local inflammation, and cells deactivate or undergo apoptosis following injury resolution2,3. However, the survival and persistence of myofibroblasts leads to pathological ECM deposition and long-term fibrotic disease in organs and tissues4,5. Although substantial progress has been made to understand the molecular mechanisms that promote myofibroblast activation from quiescent fibroblasts6,7, less is known about the mechanisms that lead to their sustained persistence. Nevertheless, increased microenvironment stiffness has been associated with persistent activation of myofibroblasts, and evidence suggests that these cells have lost the sensitivity to respond to changes in matrix mechanical cues, so-called mechanosensing8,9. Major questions remain as to whether myofibroblast persistence is caused by altered mechanosensing after long-term exposure to stiff fibrotic microenvironments and the establishment of a new cellular steady-state.
Epigenetic mechanisms are crucial regulators of cell-state transitions or ‘cell identity’10,11 and have been associated with the transformation of fibroblasts to activated myofibroblasts in nearly every major organ12,13. In mouse embryonic fibroblasts, microenvironmental mechanical cues have been shown to promote epigenetic remodelling and influence the localization of key chromatin remodelling proteins during myofibroblast activation14. However, considerably less is known about the role of epigenetic remodelling in myofibroblast persistence. Epigenetic mechanisms include stable chromatin modifications, changes in DNA accessibility or compaction and nuclear organization, ultimately resulting in the regulation of gene expression and subsequent cell-fate decisions11. Studies suggest that epigenetic mechanisms may also play a role in myofibroblast persistence in lung and kidney fibrosis15,16, although the complexity of the environment limits the ability to dissect specific mechanisms responsible for myofibroblast persistence.
Biomaterials engineered for cell culture applications have been designed to mimic aspects of the ECM and provide researchers with methods with which to study mechanical signalling from the matrix microenvironment in both a spatially and temporally controlled manner17. Poly(ethylene glycol) (PEG) hydrogels are one of the most widely studied and applied synthetic hydrogels, and their mechanical properties are readily controlled by changing the crosslinking density of the hydrogel network18. Of direct relevance to this work, PEG hydrogel chemistries have been developed with properties that can be changed on-demand and in the presence of cells19. These materials are especially useful for studying mechanosensing, and in one example, primary fibroblasts isolated from aortic valve leaflets were cultured on photo-softening hydrogels to assess how exposure to stiff microenvironments influences the evolution of the myofibroblast phenotype20. Cell-laden stiff hydrogels were softened with light to demonstrate that the time of exposure to stiff microenvironments influences the ability of the myofibroblast to deactivate to the quiescent fibroblast. Here we utilize similar photo-softening PEG hydrogels to investigate the mechanisms that lead to persistent myofibroblast activation following extended exposure to stiff microenvironments (that is, mechanical dosing).
Primary fibroblasts isolated from pig aortic heart valves were cultured on PEG hydrogels with photo-softening mechanical properties that span a range of physiologically relevant tissue moduli. With this hydrogel culture system, experimental conditions were identified where myofibroblasts would deactivate after their microenvironment was softened, so-called transient myofibroblasts. In addition, experimental conditions were determined whereby myofibroblasts failed to deactivate after matrix softening, thereby leading to persistent myofibroblasts. We observed that persistent myofibroblasts had increased global chromatin condensation compared with transiently activated myofibroblasts through genome-wide chromatin accessibility analysis. We also discovered that the chromatin remodelling is directly linked to mechanical transmission from the actin cytoskeleton to the nucleus, rather than by signalling pathways or transcriptional regulation. Immunostaining for myofibroblasts in valve tissue from patients with aortic valve stenosis revealed similar trends in chromatin signatures observed in our in vitro myofibroblast model, thereby reinforcing the clinical implications of our studies and potential relevance of altered mechanosensing in disease progression.
Results
Chromatin accessibility changes with myofibroblast persistence.
We utilized PEG acrylate hydrogels with a photo-cleavable crosslinker (PEGdiPDA) as a cell culture system for healthy porcine aortic valve fibroblasts to study the role of ECM stiffness in myofibroblast persistence (Supplementary Fig. 1)19. A fibronectin-derived peptide sequence, arginine-glycine-aspartic acid-serine, was incorporated in the hydrogel to facilitate cell-matrix interactions and adhesion. Hydrogels were generated to promote myofibroblast activation (storage modulus (G′) = ~4.5 kPa)21. Following exposure to a cytocompatible light dose22,23 (10 mW cm−2; 365 nm; 6 min), the hydrogels soften to an elastic modulus similar to that of healthy aortic valve (G′ = ~1.7 kPa (ref. 24); Fig. 1a,b and Supplementary Fig. 1).
Fig. 1 |. Extended culture on stiff microenvironments induces persistence of myofibroblast activation.
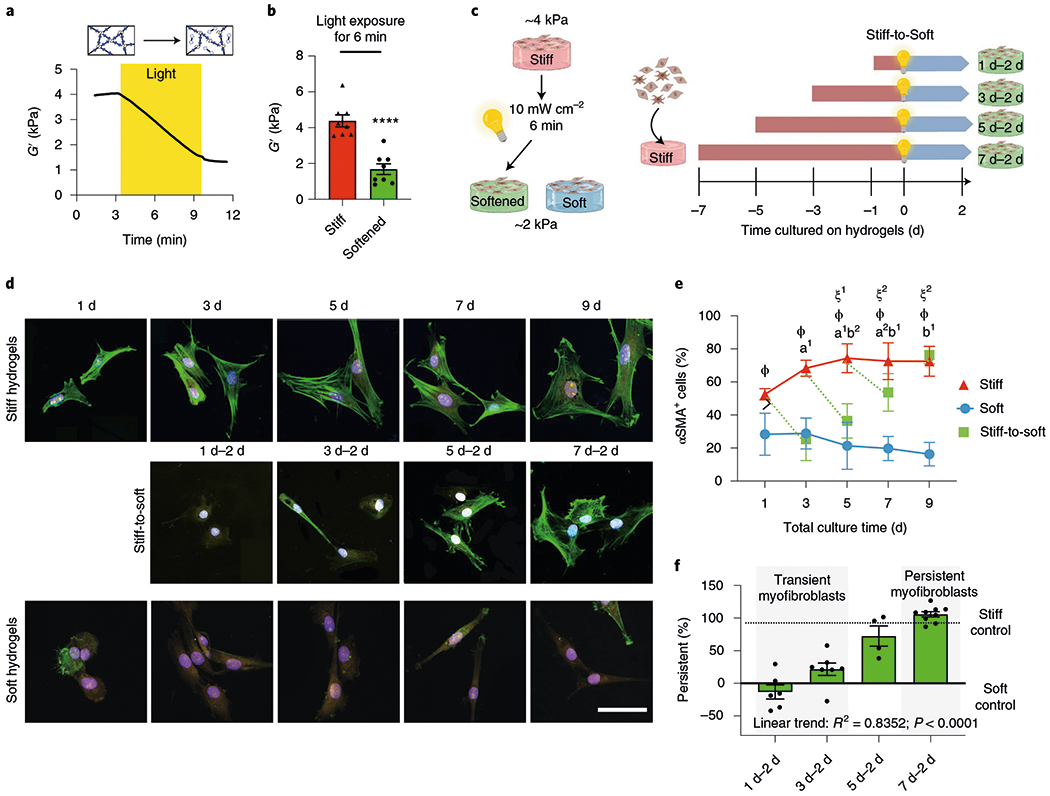
a, PEG hydrogels soften with exposure to light due to a reduction in the crosslinking density (mean rheological traces, n = 5 traces). b, Hydrogels that match fibrotic- or healthy-tissue stiffness can be generated. Two-tailed paired Student’s t-test, ****P < 0.0001; n = 8. c, Schematic of the cell culture experiment to test for myofibroblast persistence. d, Representative images of primary aortic valve fibroblasts cultured on hydrogels for different time periods (n = 12 images per hydrogel). CellMask, yellow; αSMA, green; 4′,6-diamidino-2-phenylindole (DAPI), blue. Scale bar, 50 μm. e, Percentage of αSMA+ cells in the different groups following culture on hydrogels for different time periods. Significance of stiff-to-soft versus stiff: a1, P < 0.001 and a2, P = 0.003; significance of stiff-to-soft versus soft: b1, P < 0.001 and b2, P = 0.0379; significance of stiff versus soft: ϕ, P < 0.001; significance of stiff time point versus stiff day 1: ξ1, P = 0.004 and ξ2, P = 0.008; two-way analysis of variance (ANOVA) with Bonferroni’s multiple-comparison test; n = 5, 6, 8, 9 and 9 hydrogels for total culture time of 1, 3, 5, 7 and 9 d, respectively. f, Per cent myofibroblast persistence for cells cultured on hydrogels for different time periods. One-way ANOVA with test for the applied linear trend; R2, Pearson’s correlation coefficient; n =6 (1 d–2 d), 7 (3 d–2 d), 4 (5 d–2 d) and 9 (7 d–2 d) hydrogels. Data from five biologically independent replicates. Data reported as the mean ± s.e.m.
To identify conditions where transiently activated myofibroblasts might evolve into persistently activated myofibroblasts, we tested the ability of myofibroblasts to deactivate following in situ softening of the underlying PEG matrix as a function of time (Fig. 1c). Specifically, valve fibroblasts were cultured on stiff hydrogels (G′ = ~4.5 kPa) for 1, 3, 5 or 7 d, and after light-induced in situ matrix softening (G′ = ~1.7 kPa), the cells were cultured for two more days (that is, stiff-to-soft 1 d–2 d, 3 d–2 d, 5 d–2 d and 7 d–2 d, respectively). As controls, fibroblasts were cultured on stiff or soft PEG hydrogels without changing the modulus, so that the cells were continuously exposed to the high or low modulus materials. Throughout this work, we assessed the expression of α-smooth muscle actin (αSMA) stress fibres by immunofluorescence to identify activated myofibroblasts, as it is a commonly employed marker for fibrotic valvular myofibroblasts25,26.
At all time points, approximately 60% of the cells cultured on the stiff hydrogels showed expression of αSMA stress fibres, indicating myofibroblast activation (Fig. 1d,e). Conversely, only about 20% of the cells cultured on soft hydrogels showed αSMA stress fibres, indicating that approximately 80% of the cells were quiescent fibroblasts. Myofibroblasts cultured for 1 or 3 d on stiff hydrogels, followed by 2 d of culture after in situ softening (1 d–2 d and 3 d–2 d, respectively) deactivated, as shown by significantly reduced numbers of αSMA stress fibres. Only partial reversibility was observed in cells cultured for 5 d on stiff hydrogels before in situ softening (5 d–2 d). Conversely, the αSMA stress fibres were not reduced in myofibroblasts that were cultured for 7 d on stiff hydrogels, followed by either 2 (7 d–2 d) or 6 d (7 d–6 d) of culture after softening, and remained as activated myofibroblasts (Fig. 1d,e and Supplementary Fig. 2). Transient activation of 1 d–2 d myofibroblasts was confirmed by the downregulation of αSMA and fibronectin protein levels after in situ softening (both indicators of myofibroblast activation27; Supplementary Fig. 2). Both fibronectin and αSMA expression remained elevated in the 7 d–2 d cells, similar to the stiff hydrogel controls, indicating persistent activation. We conclude that through the use of photo-softening PEG hydrogels, we can generate two distinct myofibroblast populations: ‘transient’ myofibroblasts, cultured for 1 or 3 d on stiff hydrogels, that are able to deactivate when the pathological mechanical stimulus is removed and ‘persistent’ myofibroblasts, cultured for ≥7 d on stiff hydrogels, that are unable to deactivate following the removal of the pathological stimulus (Fig. 1f).
We next investigated how epigenetic mechanisms may be involved with persistent myofibroblast activation, given that the evolution of the myofibroblast phenotype towards persistent activation parallels the commitment of stem cells to a particular lineage and relies on epigenetic remodelling10. We evaluated the epigenetic landscape as a function of time using an automated analysis of the global chromatin structure of cells exposed to these same conditions (Fig. 1c). Using confocal imaging, we calculated the chromatin condensation parameter (CCP)28 for the myofibroblast populations (Fig. 2a,b). Fibroblasts on soft hydrogels did not change their chromatin condensation over time, whereas myofibroblasts on stiff hydrogels first showed decreased chromatin condensation, corresponding to transient myofibroblasts, and then a progressive increase of the CCP with time, reaching a plateau in persistent myofibroblasts. This result suggests that the chromatin structure condenses during the evolution of the myofibroblast phenotype from transient to persistent. After in situ softening, transient myofibroblasts significantly increased their CCP, reaching soft controls, whereas the CCP of persistent myofibroblasts remained constant. Correlation analysis showed that persistence correlated with nuclear roundness, cell area and the CCP, with a Pearson’s correlation coefficient of −0.9950, 0.9244 and 0.9636, respectively (Fig. 2c and Supplementary Fig. 3).
Fig. 2 |. Extended culture on stiff microenvironments reduces chromatin accessibility.
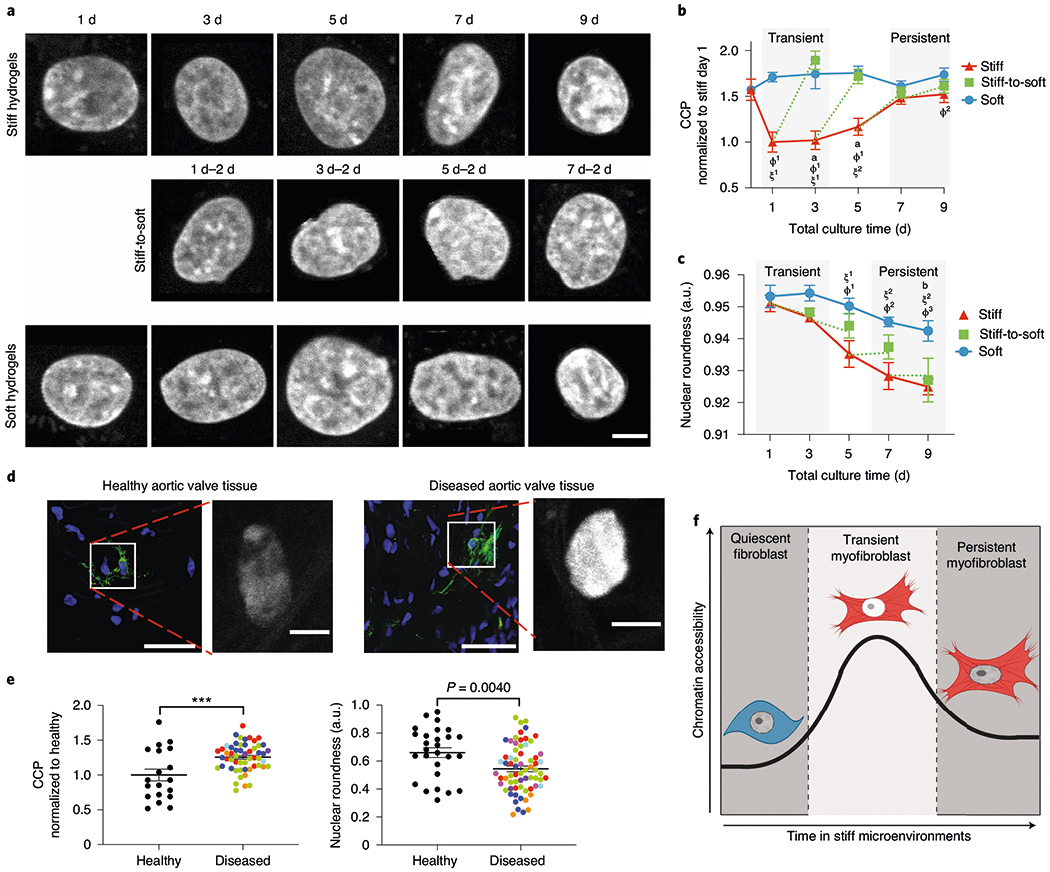
a, Representative images of DAPI-stained nuclei subjected to CCP analysis (n = 400 images). Scale bar, 5 μm. b, CCP for cells cultured on hydrogels for different time periods. Significance of stiff-to-soft versus stiff: a, P < 0.0001; significance of stiff versus soft: ϕ1, P < 0.001 and ϕ2, P = 0.0071; significance of stiff time point versus stiff day 0: ξ1, P < 0.001 and ξ2, P = 0.0013; two-way ANOVA with Bonferroni’s post-hoc test; 0 d, n = 16 cells; stiff, n = 72 (1 d), 89 (3 d), 102 (5 d), 169 (7 d) and 92 (9 d) cells; soft, n = 162 (1 d), 51 (3 d), 33 (5 d), 63 (7 d) and 47 (9 d) cells; stiff-to-soft, n = 53 (1 d–2 d), 61 (3 d–2 d), 67 (5 d–2 d) and 58 (7 d–2 d) cells. c, Nuclear roundness of cells cultured on hydrogels for different time periods. Significance of stiff-to-soft versus soft: b, P = 0.0074; significance of stiff time point versus stiff day 1: ξ1, P = 0.0046 and ξ2, P < 0.001; significance of stiff versus soft: ϕ1, P = 0.0093; ϕ2, P = 0.0027 and ϕ3, P = 0.0019; two-way ANOVA with Bonferroni’s post-hoc test. Soft, n =5 (1 d), 6 (3 d) and 8 (5, 7 and 9 d) hydrogels; stiff, n = 5 (1 d), 6 (3 d) and 8 (5, 7 and 9 d); stiff-to-soft, n = 5 (1 d–2 d, 3 d–2 d and 5 d–2 d) and 8 (7 d–2 d). d, Representative images of myofibroblasts and corresponding DAPI-stained nuclei from healthy (left) or diseased (right) human aortic valve tissue (n = 53 images from eight biological samples). Nuclei, blue; αSMA, green. Scale bars, 50 μm (main images) and 5 μm (insets). e, CCP (normalized to healthy tissue; left) and nuclear roundness (right) of DAPI-stained nuclei from healthy and diseased human aortic valve tissue. Colours indicate individual patients. Two-tailed Student’s t-test; CCP analysis, n = 19 (healthy) and 54 (diseased) cells; nuclear roundness, n = 27 (healthy) and 67 (diseased) cells. f, Illustration for the hypothesis that chromatin plays a role in myofibroblast persistence. ***P < 0.001. Data from three biologically independent replicates. Data reported as the mean ± s.e.m.; a.u., arbitrary units.
Notably, myofibroblasts from human aortic valve tissue showed similar trends in epigenetic signatures (Fig. 2d,e and Supplementary Fig. 4). Myofibroblasts from human fibrotic valves had a significantly increased CCP compared with myofibroblasts from healthy valves. Moreover, myofibroblasts from fibrotic valves had significantly reduced nuclear roundness compared with myofibroblasts from healthy valves (Fig. 2e and Supplementary Fig. 4). These results support the idea that myofibroblast persistence is associated with increased chromatin condensation (Fig. 2f), which was observed in our in vitro culture model as well as in human myofibroblasts from fibrotic valves.
Myofibroblast persistence is associated with a global reduction in chromatin accessibility.
To further understand the chromatin accessibility changes between the transient and persistent myofibroblasts, we performed an assay for transposase-accessible chromatin using sequencing (ATAC-seq)29. To avoid disrupting the native chromatin landscape specific for both the transient and persistent myofibroblasts cultured on the PEG hydrogels, we developed an in situ omni-ATAC-seq protocol, permeabilizing the cell membranes and carrying out the transposition reactions while the cells were attached to their hydrogel substrates (Supplementary Methods). We envisioned two possible scenarios of the chromatin response: (1) the chromatin becomes less accessible genome-wide in persistent myofibroblasts and (2) chromatin accessibility is not globally altered but discrete chromatin loci are uniquely open/closed in either transient or persistent myofibroblasts.
When cells from different experimental conditions do not yield identical amounts of DNA (in this case open chromatin), spike-in controls are necessary30. We relied on fixed amounts of Drosophila melanogaster S2 nuclei as spike-ins for internal controls during the transposition reactions to accurately identify genome-wide accessibility changes30,31. We expected to obtain a correspondingly higher yield of Drosophila transposition events if the chromatin of the persistently activated myofibroblasts became more closed genome-wide. The persistent myofibroblast datasets indeed contained a higher fraction of mapped Drosophila fragments compared with the transient myofibroblast datasets. This result indicates that the chromatin of the persistent myofibroblasts is more closed genome-wide than transient myofibroblasts, as the fixed number of transposases have more limited access to the chromatin of the myofibroblasts and their activity shifts to the chromatin of the Drosophila nuclei. Given that the same amount of Drosophila nuclei were used in all samples, we normalized the ATAC-seq datasets to the fraction of mapped Drosophila fragments. After this spike-in normalization, we observed that the persistent myofibroblasts showed decreased ATAC-seq signal at both the transcription start sites and gene bodies for both myofibroblast-specific genes (αSMA (ACTA2)) and non-myofibroblast-specific housekeeping genes (Fig. 3a). Furthermore, this effect held true for both high- and low-signal peaks, with both classes of peaks showing lower signal in the persistent myofibroblasts compared with the transient myofibroblasts (Fig. 3b,c and Supplementary Fig. 5).
Fig. 3 |. ATAC sequencing reveals a global reduction in chromatin accessibility in persistent myofibroblasts relative to transiently activated myofibroblasts.
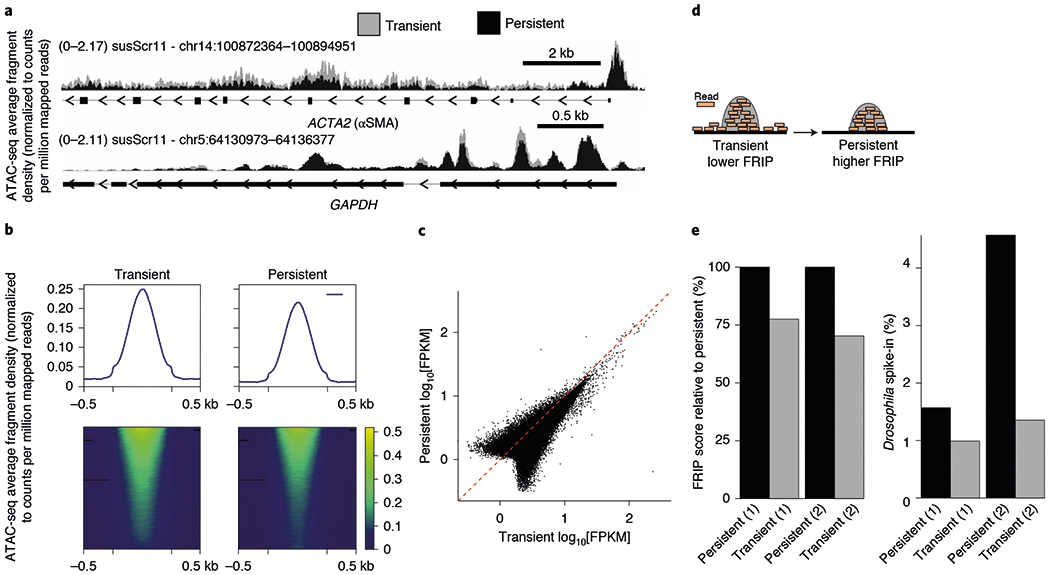
a, ATAC-seq performed on transient (3 d stiff) and persistent (9 d stiff) myofibroblasts. ATAC-seq density signal for two genomic loci: αSMA (ACTA2; top), a marker of myofibroblasts, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; bottom), a basal metabolic housekeeping gene whose activity is not specific to myofibroblasts. b, Average ATAC-seq peak profile (top) and signal heatmap (bottom) for both transient (left) and persistent (right) myofibroblasts. The ATAC-seq data were normalized to the counts per million mapped reads, which accounts for differences in sequencing depth and to the D. melanogaster S2 nuclei spike-ins, which accounts for genome-wide differences in accessibility. c, Scatter plot of the log10-transformed fragments per kilobase of transcript per million (FPKM) values for all ATAC-seq peaks for persistent versus transient myofibroblasts. The red dashed line is the theoretical 1:1 equivalent signal across both conditions. The majority of the peaks have higher FPKM values for transient myofibroblasts, which is observed as a global rightward skew of the data point mass. d, Diagram representing the difference in the FRIP score metric between the transient and the persistent myofibroblast datasets. e, FRIP score of the persistent myofibroblasts (left). Due to a genome-wide reduction in accessibility, reads could only be obtained from transposition events on highly open chromatin loci, such as those at gene regulatory regions. D. melanogaster S2 nuclei were added to the ATAC-seq reactions and the proportion of reads mapped to the Drosophila genome relative to the total mapped reads are shown (right).
Interestingly, we noted that persistently activated myofibroblasts exhibited a higher fraction of transposition events, occurring primarily at well-defined open chromatin loci (for example, transcription start sites and other transcriptional regulatory elements). The fraction-of-reads-in-peaks (FRIP) score, as defined by the Encyclopedia of DNA Elements (ENCODE) project, quantifies this shift in transposition events from out-of-peaks to in-peaks32 (Fig. 3d,e). Across the biological replicates, we observed that persistent myofibroblasts had a higher FRIP score than transient myofibroblasts, suggesting a shift in transposition events to in-peaks in persistent myofibroblasts and similar to the fraction of mapped Drosophila fragments.
Overall, the ATAC-seq results suggest that the chromatin landscape in persistent myofibroblasts is less accessible compared with transient myofibroblasts, which is in agreement with our previous observations. Notably, because these chromatin accessibility changes seem to occur genome-wide, we were unable to identify bona fide condition-specific peaks that could be used to pinpoint specific chromatin remodellers or transcription factors responsible for changing the chromatin landscape (Supplementary Fig. 6)33. In addition, although persistent myofibroblasts had a similar CCP to fibroblasts on soft hydrogels, there were notable differences in the chromatin accessibility (Supplementary Fig. 7). This difference suggests that persistent myofibroblasts and fibroblasts have differential chromatin landscapes governing their respective phenotypes, which is not readily observed in a CCP analysis of the global chromatin accessibility.
Myofibroblast persistence is linked to chromatin accessibility.
To test whether histone deacetylases (HDACs) were responsible for the increased genome-wide chromatin condensation in persistent myofibroblasts, we measured the gene expression and total activity of HDACs (Supplementary Fig. 8). Although the differences in the levels of HDAC gene expression between persistent and transient myofibroblasts were not significant, the HDAC activity of the persistent myofibroblasts was higher compared with transient myofibroblasts. In contrast, the HDAC activity of fibroblasts cultured on soft hydrogels remained constant over 7 d, demonstrating that stiffness is linked to the time-dependent increase in HDAC activity. Next, we assessed whether closed chromatin structure was responsible for myofibroblast persistence using trichostatin A (TSA), a well-known inhibitor of Class I and Class II HDACs34 (Fig. 4a). After defining an appropriate dose to increase chromatin accessibility (Supplementary Fig. 9), we treated persistent myofibroblasts with TSA (150 nmol) for 2d (Fig. 4b). After treatment, the CCP of the persistent myofibroblasts decreased by approximately 50%, reaching levels comparable to transient myofibroblasts (Fig. 4c) and the HDAC activity reduced about fivefold compared with vehicle-treated persistent myofibroblasts (Supplementary Fig. 9). Treatment with TSA did not deactivate persistent myofibroblasts (Fig. 4d), suggesting that opening chromatin itself is not enough to revert persistent myofibroblasts to quiescent fibroblasts.
Fig. 4 |. Chromatin remodelling is necessary for myofibroblast persistence.
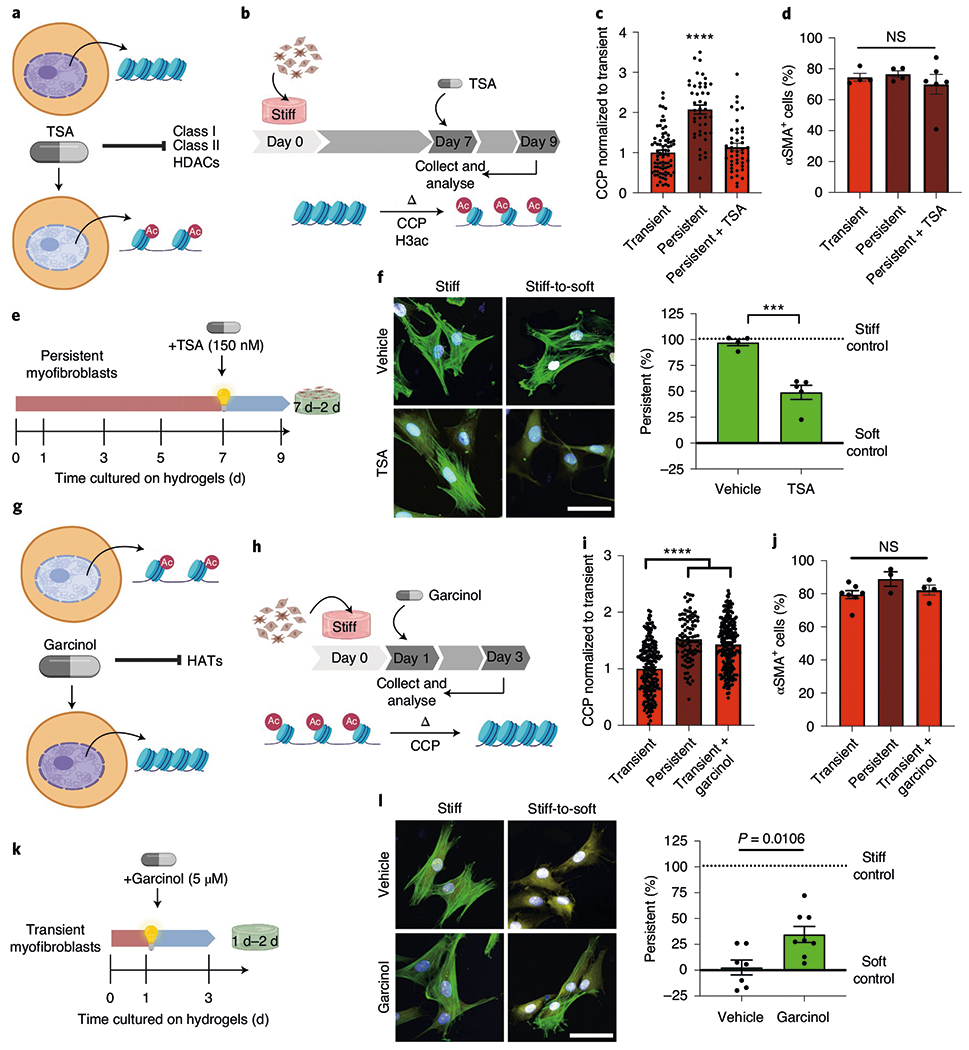
a, TSA as a tool to increase chromatin accessibility. b, Schematic of the cell culture experiment to test whether TSA can revert the chromatin structure of persistent cells. c, CCP of transient and persistent myofibroblasts as well as persistent myofibroblasts treated with TSA. One-way ANOVA with Bonferroni’s post-hoc test; n = 78 (transient) and 44 (persistent and persistent + TSA) cells. d, Per cent αSMA+ cells in transient myofibroblasts (1 d stiff, 2 d stiff + dimethyl sulfoxide (DMSO)), persistent myofibroblasts (7 d stiff, 2 d stiff + DMSO) and TSA-treated persistent myofibroblasts (7 d stiff, 2 d stiff + TSA). One-way ANOVA with Bonferroni’s post-hoc test; n = 4 (transient and persistent) and 6 (persistent + TSA) hydrogels. e, Schematic of the cell culture experiment to test whether TSA can reverse myofibroblast persistence. f, Left: representative images of persistent myofibroblasts treated with vehicle (DMSO) or TSA for 2 d (n = 12 images per hydrogel). CellMask, yellow; αSMA, green; DAPI, blue. Scale bar, 50 μm. Right: myofibroblast persistence after vehicle or TSA treatment for 2 d (7 d stiff, 2 d soft + DMSO or TSA). Two-tailed Student’s t-test; n = 4 (vehicle) and 5 (TSA) hydrogels. g, Garcinol as a tool to decrease chromatin accessibility. HAT, histone acetyltransferase. h, Schematic of the cell culture experiment to test whether garcinol can encode chromatin condensation in transient cells. i, Quantification of chromatin compaction by CCP for transient (1 d stiff, 2 d stiff + DMSO), persistent (7 d stiff, 2 d stiff + DMSO) and garcinol-treated transient myofibroblasts (1 d stiff, 2 d stiff + garcinol). One-way ANOVA with Bonferroni’s post-hoc test; n = 209 (transient), 95 (persistent) and 235 (transient + garcinol) cells. j, Per cent αSMA+ cells in transient myofibroblasts (1 d stiff, 2 d stiff + DMSO), persistent myofibroblasts (7 d stiff, 2 d stiff + DMSO) and garcinol-treated transient myofibroblasts (1 d stiff, 2 d stiff + garcinol). One-way ANOVA with Bonferroni’s post-hoc test; n = 7 (transient), 3 (persistent) and 4 (transient + garcinol) hydrogels. k, Schematic of the cell culture experiment to test whether garcinol can encode myofibroblast persistence. l, Left: representative images of transient myofibroblasts treated with vehicle or garcinol for 2 d (n = 12 images per hydrogel). CellMask, yellow; αSMA, green; DAPI, blue. Scale bar, 50 μm. Right: myofibroblast persistence with vehicle or garcinol treatment (1 d stiff, 2 d soft + DMSO or garcinol). Two-tailed Student’s t-test; n = 7 (vehicle) and 8 (garcinol) hydrogels. Data from three biologically independent replicates. ***P < 0.001; ****P < 0.0001; NS, not significant. Data reported as the mean ± s.e.m.
As the CCP of TSA-treated persistent myofibroblasts was comparable to transient myofibroblasts, we next queried whether these cells would deactivate in response to in situ softening (Fig. 4e). Control persistent myofibroblasts with condensed chromatin structures failed to deactivate, whereas TSA-treated persistent myofibroblasts with open chromatin structures deactivated approximately 50% (Fig. 4f), indicating that the chromatin structure determines the ability of myofibroblasts to respond to environmental mechanical cues. We confirmed this mechanism by closing the chromatin in transient myofibroblasts using a histone acetyltransferase inhibitor (garcinol; Fig. 4g–j). Garcinol-treated transient myofibroblasts acquired partial persistence (Fig. 4l,m). Based on these results, we concluded that the myofibroblast phenotype can be reprogrammed by manipulation of chromatin accessibility but deactivation depends on mechanical cues. Notably, other phenotypic markers of TSA-treated persistent myofibroblasts (that is, cell area and nuclear roundness) did not return to the transient levels (Supplementary Fig. 9), suggesting that other mechanisms are contributing to the evolution of the persistent phenotype.
The actin network regulates time-dependent chromatin remodelling.
Mechanical forces, such as matrix stiffness, can regulate chromatin accessibility35,36. The nucleus itself senses mechanical forces through connections from focal adhesions, the cytoskeleton and the linker of nucleoskeleton and cytoskeleton (LINC) complex to chromatin37 (Fig. 5a). This ‘nuclear mechanosensing’ transduces forces directly and is sufficient to trigger changes in gene expression38. With this in mind, we explored whether the stabilization of the cytoskeleton was observed in persistent myofibroblasts and whether it was responsible for the reduced chromatin accessibility. Compared with transient myofibroblasts, focal adhesions and the cytoskeleton were stable in persistent myofibroblasts. Persistent myofibroblasts had higher expression of vinculin and phosphorylated Cofilin (p-cofilin), a cytoskeleton remodeller inhibited by phosphorylation, which suggests stabilization of focal adhesions and lower cytoskeletal actin turnover, respectively (Supplementary Fig. 10).
Fig. 5 |. A stabilized actin cytoskeleton is required for myofibroblast persistence.
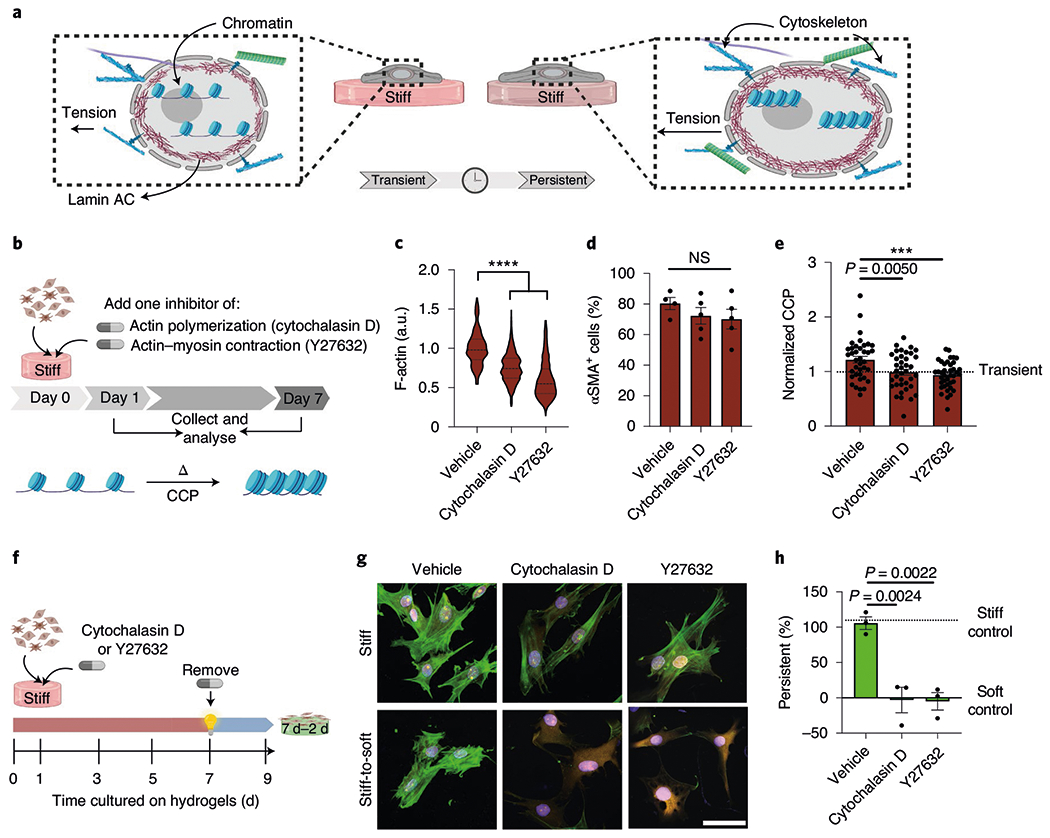
a, Illustration of the hypothesis that mechanics directly regulate chromatin structure in persistent myofibroblasts. b, Schematic of the cell culture experiment to test the effect of actin cytoskeleton on the chromatin structure. c, Actin levels determined from immunofluorescence in cells treated with the actin inhibitors cytochalasin D and Y27632 for 7 d on stiff hydrogels. The values were normalized to the vehicle. One-way ANOVA with Bonferroni’s post-hoc test; n = 192 (vehicle), 1,465 (cytochalasin D) and 1,300 (Y27632) cells; a.u., arbitrary units. d, Per cent αSMA+ cells after treatment with cytochalasin D and Y27632 for 7 d on stiff hydrogels. One-way ANOVA with Bonferroni’s post-hoc test; n = 4 (vehicle) and 5 (cytochalasin D and Y27632) hydrogels. e, CCP for persistent myofibroblasts (7 d stiff) treated with DMSO, cytochalasin D or Y27632 and normalized to CCP from transient myofibroblasts (1d stiff). One-way ANOVA with Bonferroni’s post-hoc test; n = 38 (vehicle) and 39 (cytochalasin D and Y27632) cells. f, Schematic of the cell culture experiment to test whether actin inhibition can prevent myofibroblast persistence. g, Representative images of persistent myofibroblasts treated with vehicle, cytochalasin D and Y27632 on stiff (9 d) or stiff-to-soft (7 d–2 d) hydrogels (n = 12 images per hydrogel). CellMask, yellow; αSMA, green; DAPI, blue. Scale bar, 50 μm. h, Per cent persistent myofibroblasts after treatment with vehicle, cytochalasin D and Y27632 (7 d stiff + drug, 2 d soft). One-way ANOVA with Bonferroni’s post-hoc test; n = 3 hydrogels. Data from three biologically independent replicates. ***P < 0.001; ****P < 0.0001; NS, not significant. Data reported as the mean ± s.e.m.
Next, we investigated whether disruption of the cytoskeletal stability would impact chromatin accessibility and myofibroblast persistence. Actin, intermediate filaments and microtubule cytoskeletons are the main components of a cell cytoskeleton and can modulate nuclear dynamics39,40. Persistent myofibroblasts on stiff hydrogels were treated with actin inhibitors (cytochalasin D and Y27632), an intermediate filament inhibitor (withaferin A) or a microtubule inhibitor (nocodazole). The concentrations were chosen based on their efficacy after 7 d in culture without a reduction in the percentage of αSMA+ cells (Supplementary Fig. 11). At the selected concentrations, the actin inhibitors reduced the total cellular actin but did not alter the expression of αSMA stress fibres in myofibroblasts cultured 7d on stiff hydrogels (Fig. 5b–d). Actin inhibitors also prevented increases in the CCP and the HDAC activity in persistent myofibroblasts compared with the vehicle-treated controls (Fig. 5e and Supplementary Fig. 12). Treatment with witheferin A and nocodazole did not result in changes in chromatin condensation compared with the controls (Supplementary Fig. 13). These results indicate that stabilization of actin cytoskeletal networks, but not vimentin or microtubule networks, causes chromatin condensation in persistent myofibroblasts.
We then tested whether inhibition of the actin cytoskeleton could also prevent the establishment of myofibroblast persistence (Fig. 5f–h). Myofibroblasts cultured for 7 d on stiff hydrogels during treatment with actin inhibitors were able to deactivate after in situ softening, demonstrating that inhibitor-treated myofibroblasts were transiently activated. These results indicate that actin-cytoskeleton stabilization is not required for myofibroblast activation after 7 d of culture on stiff hydrogels but may be responsible for myofibroblast persistence.
The actin cytoskeleton increases nuclear tension to promote myofibroblast persistence.
As the actin cytoskeleton transmits forces to the nuclear membrane, we evaluated whether persistent myofibroblasts had increased nuclear tension by measuring the nuclear localization of lamin AC and tension across the LINC complex. Lamin AC connects to the cytoskeleton network via the LINC complex and its expression scales with increased tissue stiffness41. In agreement with previous findings, valve fibroblasts cultured on stiff hydrogels expressed more lamin AC at day 9 than those on soft hydrogels (Supplementary Fig. 14). In addition, the nuclear lamina anchors chromatin and can regulate its spatial orientation42. The spatial distribution of nuclear lamin AC was significantly different between transient and persistent myofibroblasts, with a progressive increase in the lamin AC intensity at the centre of the nucleus in persistent myofibroblasts (Fig. 6a). Furthermore, myofibroblasts from fibrotic human valve tissue displayed a similar trend, where there was an increased spatial distribution of lamin AC in the nuclear interior compared with myofibroblasts from healthy valves (Fig. 6b and Supplementary Fig. 4). Although the expression of total lamin AC did not differ significantly between transient and persistent myofibroblasts, the expression of lamin A was increased in persistent myofibroblasts compared with lamin C (Fig. 6c). A higher lamin A:C ratio has been associated with differentiation to osteoblasts, whereas decreased lamin A:C has been linked to neurons and other cell types found in soft tissues43.
Fig. 6 |. The actin cytoskeleton increases nuclear tension to promote myofibroblast persistence.
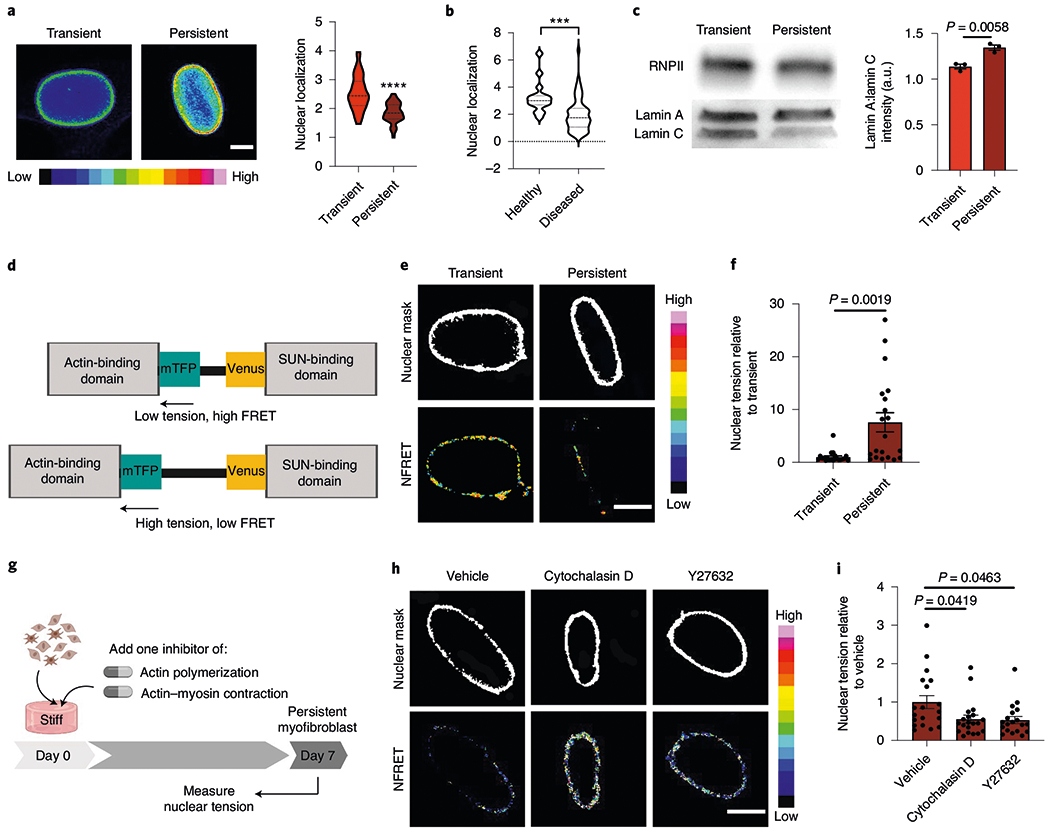
a, Images (left) and quantification of the spatial localization of lamin AC (right) in the nuclei of transient (1 d stiff) and persistent myofibroblasts (7 d stiff; n = 200 images per hydrogel). Scale bar, 5 μm. Two-tailed Student’s t-test; n = 40 (transient) and 63 (persistent) cells. b, Spatial localization of lamin AC in human fibrotic and healthy aortic valve tissue. Two-tailed Student’s t-test; n = 15 (healthy) and 59 (diseased) cells. In a,b, nuclear localization was determined as the ratio of the average intensity of the nuclear periphery to the average intensity of the nuclear centre. c, Western blot (left) and quantification of the lamin AC protein expression for transient (1 d stiff) and persistent (7 d stiff) myofibroblasts (right). Uncropped blots can be found in Supplementary Fig. 16. Two-tailed Student’s t-test; n = 3 hydrogels. d, Schematic for the nesprin-tension sensor. e, NFRET images from transient (1 d stiff; left) and persistent myofibroblasts (7 d stiff, right; n = 40 images across four hydrogels per condition). Scale bar, 2.5 μm. f, Nuclear tension measured by inverse NFRET measurements for transient (1 d stiff) and persistent (7 d stiff) myofibroblasts. Two-tailed Student’s t-test; n = 21 (transient) and 20 (persistent) cells. g, Schematic of the cell culture experiment to test whether actin inhibition changes the nuclear forces. h, NFRET images of persistent myofibroblasts treated with vehicle, cytochalasin D and Y27632 (7 d stiff + drug; n = 75 images across four hydrogels per condition). Scale bar, 5 μm. i, Nuclear tension measured by inverse NFRET measurements for persistent myofibroblasts treated with vehicle, cytochalasin D and Y27632 (7 d stiff + drug). One-way ANOVA with Bonferroni’s post-hoc test; n = 19 (vehicle and Y27632) and 20 (cytochalasin D) cells. Data from three biologically independent replicates. ***P < 0.001; ****P < 0.0001. Data reported as the mean ± s.e.m.
The LINC complex spans the nuclear membrane, attaching the cytoskeletal network to the nuclear lamina. This complex plays a key role in nuclear mechanosensing and can signal directly to chromatin via changes in tension44. To measure this mechanical tension across the nuclear membrane, we employed a fluorescence resonance energy transfer (FRET)-based tension biosensor for Nesprin-2G, a component of the LINC complex45. The FRET signal of the sensor is inversely proportional to the tension, thus high nuclear tension results in a lower FRET signal (Fig. 6d). We confirmed that the sensor could detect differences in nuclear tension in cells transfected with the FRET construct and treated with or without a ROCK inhibitor and a low-tension sensor control (headless nesprin; Supplementary Fig. 14). Persistent myofibroblasts had an increased inversed normalized FRET (1/NFRET) score compared with transient myofibroblasts, indicating that persistence is associated with higher nuclear tension (Fig. 6e,f). When persistent myofibroblasts were treated with actin cytoskeleton inhibitors (cytochalasin D and Y27632), the 1/NFRET score decreased by approximately 85%, indicating that actin is the source of nuclear tension in persistent myofibroblasts (Fig. 6g–i).
Finally, we uncoupled the cytoskeleton from the nucleus using a dominant negative nesprin construct containing the carboxy-terminal Klarsicht/ANC-1/Syne-1 homology (KASH) domain fused to an amino-terminal mCherry46. Expression of the dominant negative KASH domain (DNKASH) saturates the available binding sites at the nuclear envelope by promiscuously binding to endogenous SUN proteins, resulting in the displacement of endogenous nesprins from the nuclear envelope46 (Fig. 7a). This effectively ‘detaches’ the cytoskeleton from the nucleus. The DNKASH protein was expressed in primary valve fibroblasts via viral infection and verified with mCherry expression across the nuclear membrane (Supplementary Fig. 15). After 7 d of culture on stiff hydrogels, the increase in CCP (7 d versus 1 d) was significantly reduced in DNKASH-expressing myofibroblasts compared with control-infected, persistent myofibroblasts (Fig. 7b). These levels were similar to those seen in transient myofibroblasts. Both control-infected and DNKASH persistent myofibroblasts were significantly larger than their corresponding transient myofibroblasts; however, there was no difference in nuclear roundness between DNKASH-expressing persistent and transient myofibroblasts (Supplementary Fig. 15). These results indicate that DNKASH expression influences the cytoskeletal-to-nuclear forces but does not significantly affect the overall cytoskeleton. In addition, the increase in HDAC activity seen in control-infected, persistent myofibroblasts was reduced in DNKASH-expressing myofibroblasts (Fig. 7c). These ‘uncoupled’ myofibroblasts acquired transient-like phenotypes even after prolonged culture on stiff hydrogels and deactivated in response to in situ softening (Fig. 7d–f). These results demonstrate that myofibroblast persistence is epigenetically determined by forces transduced from the extracellular microenvironment to the nucleus via the actin cytoskeleton (Fig. 8).
Fig. 7 |. The LINC complex promotes myofibroblast persistence.
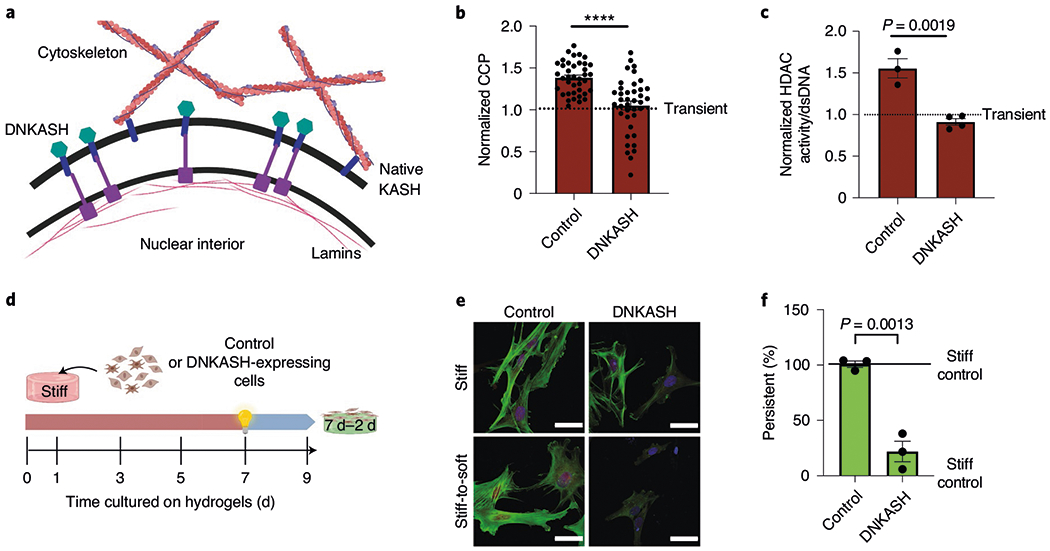
a, Schematic of the effect of DNKASH expression on the connection between the cytoskeleton and nuclear membrane. b, CCP of control-infected (mCherry) and DNKASH persistent myofibroblasts (7 d stiff) normalized to transient (1 d stiff) myofibroblasts. Two-tailed Student’s t-test; n = 39 cells. c, HDAC activity of control-infected (mCherry) and DNKASH persistent myofibroblasts (7 d stiff) normalized to transient (1 d stiff) myofibroblasts. Two-tailed Student’s t-test; n = 3 (control) and 4 (DNKASH) hydrogels. d, Schematic of the cell culture experiment to test for myofibroblast persistence in cells overexpressing DNKASH. e, Representative images of persistent myofibroblasts with expression of mCherry or DNKASH (9 d stiff (top) and 7 d–2 d stiff-to-soft (bottom); n = 12 images per hydrogel). CellMask, red; αSMA, green; DAPI, blue. Scale bars, 50 μm. f, Per cent of persistent myofibroblasts with expression of control and DNKASH (7 d–2 d). Two-tailed Student’s t-test; n = 3 hydrogels. Data from three biologically independent replicates. ****P < 0.0001. Data reported as the mean ± s.e.m.
Fig. 8 |. Mechanism for stiffness-induced myofibroblast persistence.
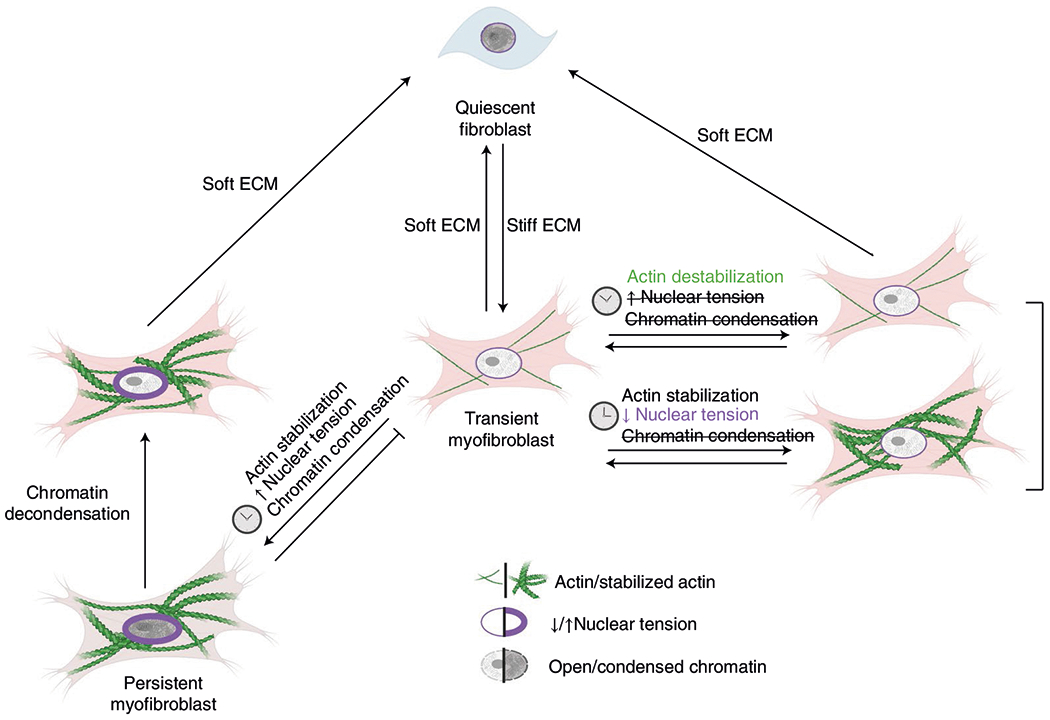
Summary of results. Quiescent fibroblasts activate to transient myofibroblasts on stiff matrices. If the matrix is softened, transient myofibroblasts revert to quiescent fibroblasts. Transient myofibroblasts transform into persistent myofibroblasts when their actin cytoskeleton stabilizes, which causes high nuclear tension and chromatin condensation. Persistence can be avoided if either actin stabilization or increased nuclear tension are prevented. Persistent myofibroblasts can be reverted to transient myofibroblasts if the chromatin is decondensed. Transient myofibroblasts will return to quiescent fibroblasts when they return to soft matrices.
Discussion
Tissue fibrosis is a major unresolved medical problem associated with increased morbidity and mortality1, and myofibroblasts are key cellular mediators of fibrotic diseases. Here we demonstrate that myofibroblast persistence is determined by mechanically driven chromatin remodelling involving the actin cytoskeleton and nuclear mechanosensing (Fig. 8). We show that increased extracellular stiffness activates fibroblasts to transient myofibroblasts and that cumulative tension on the nuclear membrane can transform transient myofibroblasts to persistent myofibroblasts by increasing HDAC activity. Increased tension on the nucleus inscribed fibrotic programming in the chromatin, conferring myofibroblast persistence. This global reduction in chromatin accessibility is ultimately linked to the persistent myofibroblast. Both epigenetic reset and removal of the pathological mechanical stimulus are needed to deactivate persistent myofibroblasts.
Biophysical cues—including stiffness, stretch and topography—can modulate chromatin organization and accessibility, and have been implicated in cell-fate commitment and cellular plasticity36,47. Importantly, evaluation of genome-wide chromatin accessibility using ATAC-seq of cancer cells cultured in stiff or soft microenvironments revealed that matrix mechanics had a dramatic effect on the global chromatin structure48. Moreover, the integrated time on stiff substrates can also influence epigenetic remodelling. For example, extended exposure to increased stiffness can instil a ‘mechanical memory’ in mesenchymal stem cells (MSCs)27,49 via changes in chromatin architecture50,51. When these cells are removed from the activating mechanical environment, their chromatin structure persists and correlates with fate determination. Similarly, persistent myofibroblasts derived from the aortic valve and studied here exhibited mechanical memory, as they remained activated even when the underlying substrate was softened. However, the chromatin structure of MSCs and fibroblasts does not always follow similar trends in chromatin accessibility in response to microenvironmental stiffness cues. For instance, a recent study found that MSCs maintain an open, accessible chromatin structure on soft substrates compared with stiff substrates52. In contrast, our data show that aortic valve fibroblasts maintain a closed, inaccessible chromatin structure on soft hydrogels compared with stiff substrates. Moreover, unlike myofibroblasts that display a closed chromatin architecture when persistently activated by stiff microenvironments, MSCs are irreversibly activated by stiff microenvironments with an open, accessible chromatin structure50. Interestingly, a study investigating the effect of time-dependent dynamic loading (another mechanical input) in MSCs showed that dynamic-loading-induced memory was encoded by chromatin condensation that could be reversed with TSA treatment51. These findings suggest that distinct mechanisms regulate chromatin accessibly in response to mechanical inputs in MSCs and fibroblasts, which is surprising as they are both mesenchymal cells and have potential for multi-potency (aortic fibroblasts can transdifferentiate to myofibroblasts or osteoblast-like cells)53. Global non-specific chromatin decondensation can partially deactivate persistent myofibroblasts, possibly allowing for the transcription of signalling networks responsible for myofibroblast reversal. Interestingly, a recent study investigated whether mechanical ‘unloading’ of fibrotic tissue might attenuate disease but did not find a reduction in the development of fibrosis54. Although the hydrogel experiments presented here do not capture the full complexity of fibrosis, the results suggest that fibrotic tissues may require a combination of mechanical unloading and a chromatin reset to reverse fibrosis. Providing support for this hypothesis, we further show that persistent myofibroblasts deactivate when chromatin decondensation is accompanied by substrate softening.
Myofibroblast persistence is a well-recognized problem in vivo55, and epigenetic changes have been implicated. Multiple studies have shown that fibroblasts isolated from fibrotic tissues retain an ‘activated’ myofibroblast-like phenotype56,57, and altered chromatin accessibility is associated with myofibroblast persistence in vivo15. Fibrosis is also characterized by the accumulation of senescent apoptotic-resistant myofibroblasts58. In lung fibrosis, resistance to apoptosis is caused by a loss of cellular redox homeostasis59. Notably, these senescent apoptotic-resistant myofibroblasts have decreased histone acetylation and increased histone methylation, suggesting decreased chromatin accessibility60, mirroring our findings here. In addition, we observe an enlarged cell area on stiff hydrogels over time, a biomarker of senescence61; it would be interesting to investigate whether myofibroblasts cultured on stiff hydrogels for extended time periods display other characteristics of senescence or resistance to apoptosis. We hypothesize that persistent myofibroblasts have reduced global chromatin accessibility because their phenotype is stabilized and there is little need to access genes unassociated with myofibroblast programming. We posit that persistent myofibroblasts have reached a ‘steady-state’, similar to fibroblasts on soft hydrogels, which display a closed chromatin landscape. This hypothesis would explain why fibroblasts and persistent myofibroblasts share a similar level of CCP but display differential chromatin accessibility landscapes that probably govern their respective cellular phenotypes. In contrast, a transient myofibroblast is in a dynamic state and can either transition to a persistent myofibroblast or a quiescent fibroblast; it is this flexibility that might necessitate a more open chromatin structure. Our findings align well with previous observations and corroborate the powerful role of epigenetic remodelling in determining the cell phenotype.
Mechanical cues affect nuclear dynamics, including chromatin, through both traditional biochemical signalling and enzyme action (for example, HDACs) and direct the transmission of force through the cytoskeleton—nuclear connectivity35,62. Our results show that stiffness-induced myofibroblast persistence is caused by a decrease in genome-wide chromatin accessibility but cannot be linked to a specific transcription factor. Direct nuclear mechanotransduction of forces, originating from ECM-focal adhesions to the actin cytoskeleton and then to the nucleus, seem to cause epigenetic remodelling in myofibroblast persistence. Stiff hydrogels promote actin assembly, and this has been associated with chromatin decondensation in fibroblasts14. We also observed increased chromatin accessibility with myofibroblast activation on our stiff hydrogels (day 1–3); however, this accessibly gradually decreased with time (day 7–9) and stabilization of the actin network. Increased actin stabilization—and thus tension—is known to alter the chromatin architecture and has been shown to decrease chromatin accessibility40. When actin-mediated decreases in chromatin accessibility are inhibited, myofibroblasts do not persist with time and instead remain transient. Interestingly, a recent study found that actin polymerization reduced pluripotency, or plasticity, in induced pluripotent stem cells by reducing chromatin accessibility63. It is worth noting that the role of actin on chromatin is probably cell-type dependent. Mesenchymal stem cells, which also show a persistent-like phenotype with extended exposure to stiffness, maintain high levels of chromatin accessibility compared with soft hydrogel controls50. Together, these findings suggest that the actin cytoskeleton is crucial for modulating chromatin accessibility and subsequently determines cell-fate decisions.
The actin cytoskeleton exerts forces on the nucleus across the LINC complex, which can regulate chromatin organization. The LINC complex is physically attached to nuclear lamins, which are intermediate filaments that contribute to nuclear stiffness and integrity but are also important for chromatin regulation and spatial organization42,64. Recent studies have demonstrated that the nuclear lamina and the LINC complex are mechanoresponsive65,66 and critical for cell fate and behaviour during mechanical loading67. Importantly, lamin AC can control fibrotic programming in mouse embryonic fibroblasts68 and the LINC complex can mediate dermal myofibroblast activation69, suggesting that nuclear dynamics are important for fibrosis progression. We show that persistent myofibroblasts have increased tension on the LINC complex and that myofibroblast persistence is prevented by disabling or reducing forces on the LINC complex. Moreover, we show that lamin AC is dysregulated with myofibroblast persistence, which is also seen in myofibroblasts from fibrotic human valves. These results indicate that stiffness-dependent myofibroblast persistence is encoded through nuclear mechanosensing. High nuclear forces in persistent myofibroblasts drive increased HDAC activity, which is known to decrease chromatin accessibility70; however, precisely how these LINC-dependent forces drive HDAC activity is unclear. A recent study suggests that forces propagate through the actin cytoskeleton to the LINC complex and via lamina—chromatin interactions, directly stretching chromatin to increase transcription38. Others have hypothesized that nuclear tension can deform the nuclear cytoskeleton (for example, lamina, actin and so on), enabling the release or sequestration of epigenetic remodellers71,72. For instance, a recent study found that polymerization of nuclear actin can increase the nuclear HDAC activity in HeLa cells73. We posit that increased nuclear forces increase HDAC activity directly, as we do not observe increased HDAC activity in the absence of force propagation across the LINC complex. Interestingly, MSCs display the opposite trend: HDAC activity decreases with increased nuclear tension74, which highlights the cell-type-dependent mechanisms likely to be governing mechanical memory. Our ATAC-seq analysis suggests that the high HDAC activity of persistent myofibroblasts reduces chromatin accessibility genome-wide rather than regulating specific genetic loci. Aberrant HDAC activity has been linked to multiple fibrotic diseases, including cardiac and pulmonary fibrosis75–77. Notably, the global chromatin structure in diseased fibrotic human valve myofibroblasts is also less accessible compared with those in healthy valves, possibly indicating the existence of transient myofibroblasts that are activated during normal healing processes in healthy tissue and persistent myofibroblasts populating diseased valves. However, it should be noted that our study is limited to only one healthy patient sample and seven diseased patient samples; thus, our findings require further validation. Future studies should focus on additional measurements and continued development of precision biomaterials to translate these in vitro findings to the human valve disease78.
Tissue fibrosis and the corresponding myofibroblast persistence typically develops over the course of months or years and in a microenvironment with a complex milieu of factors—including fluxes of cytokines and growth factors, recurring injury and repair, and other sources of stress2,79–81. Our in vitro model of persistence represents a simplified system that enables direct evaluation of the interaction between mechanical cues and chromatin accessibility in the absence of other complexities. Although limited by the short time course available with primary cell culture, our findings provide a foundation for understanding chromatin remodelling that leads to the early stages of myofibroblast persistence. Here we used porcine aortic valvular fibroblasts, a dynamic cell population that is constantly under sustained mechanical and biochemical stresses, experiencing thousands of cycles of fibroblast activation and myofibroblast deactivation during a lifetime. We propose that such a highly dynamic fibroblast population represents a generalized example for fibrotic activation but we do not exclude tissue-specific mechanisms controlling persistence. Similar to fibroblasts from other tissues, aortic valvular fibroblasts activate to myofibroblasts in response to stiffness and are themselves stiffer when activated82–84. Future studies should investigate whether myofibroblasts from other tissues demonstrate similar mechanisms of persistence. This study establishes that chromatin remodelling is needed for myofibroblast persistence; however, future investigations are needed to identify the specific proteins and/or signalling pathways affected by the remodelled chromatin.
In summary, this work establishes that acquisition of myofibroblast persistence relies on chromatin remodelling mediated by nuclear mechanosensing of cytoskeletal forces and that a chromatin reset is necessary to restore the ability of myofibroblasts to adapt to the extracellular environment. These findings emphasize the fundamental role of nuclear mechanosensing in determining the epigenetic state of persistent myofibroblasts.
Methods
Synthesis of hydrogel components.
PEGdiPDA was synthesized and characterized as previously described85. The adhesive peptide Ac-OOGRGDSG (acrylamide diethylene glycol-diethylene glycol-glycine-arginine-glycine-aspartic acid-serine-glycine) was synthesized via the 9-fluorenylmethyloxycarbonyl (Fmoc)/1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium 3-oxid hexafluorophosphate (HATU) coupling strategy on RINK amide MBHA resin (0.250 mmol) using a Protein Technologies Tribute automated peptide synthesis machine. The peptide was cleaved from the resin using Reagent B, precipitated in ice-cold diethyl ether and purified via reverse-phase automated flash chromatography (Biotage Isolera) in 0.1% trifluoroacetic acid in an acetonitrile:water gradient. Lyophilization afforded Ac-OOGRGDSG as a white power (0.087 g; 0.117 mmol; 47%).
Characterization of the hydrogel and tissue mechanical properties.
A shear rheometer (ARES; TA) was used to measure the modulus of the PEGdiPDA hydrogel before and after degradation. Briefly, optically thin PEGdiPDA hydrogels (thickness of 50 μm) were polymerized in situ between an 8-mm-diameter tool. Network evolution was tracked using a dynamic time sweep (γ = 1%; ω = 1 rad s−1; determined to be in the linear regime for this material) until the G′ reached a plateau. Degradation by light irradiation (λ = 365 μm; I0 = 10 mW cm−2; Omnicure 1000, LumenDynamics) started after the plateau and was monitored using the same dynamic time-sweep parameters. For measurements of the stiffness of porcine aortic valves, leaflets were isolated from whole hearts of adult male and female pigs provided by Hormel Inc. An 8-mm punch was used to cut out a cylindrical portion of the aortic valve and trimmed to ensure an even height of <1 mm. A shear rheometer (ARES; TA) was used to measure the shear G′ of the aortic valve samples and sandpaper was used between the parallel plate geometry to prevent tissue slippage.
Fabrication of photo-softening hydrogels for cell seeding.
The preparation of PEGdiPDA photo-softening hydrogels was adapted from previously described protocols20. PEGdiPDA was co-polymerized with PEG monoacrylate (Mn ~ 400 Da; Monomer-Polymer and Dajac Laboratories) and acryl-OOGRGDSG in PBS via redox-initiated free-radical polymerization. Gel solutions were prepared with 7.0% (wt) PEGdiPDA, 6.8% (wt) PEG monoacrylate, 5 mM acryl-OOGRGDSG, 0.2 M ammonium persulfate and 0.1 M tetramethylethylenediamine. Gels were formed on acrylated cover glass with a diameter of 12, 15 or 25 mm and a thickness of 100 μm. The gels were rinsed in PBS before cell seeding. Soft hydrogels were prepared by irradiating the initial photo-softening hydrogels with ultraviolet light (λ = 365 nm; I0 = 10 mW cm−2) for 6 min.
Mechanical dosing on photo-softening hydrogels.
The hydrogels were softened in situ on the indicated days after seeding with ultraviolet light (λ = 365 nm; I0 = 10 mW cm−2) for 6 min in PBS, after which the PBS was replaced with growth medium. Valvular fibroblasts were cultured for an additional 2 d on the softened hydrogels before analysis. Culture conditions that were ‘dosed’ (softened) are indicated in green, whereas static culture conditions are indicated in red (stiff) or blue (soft). Transient myofibroblasts are cells that were cultured on stiff hydrogels for 1 or 3 d, and persistent myofibroblasts are cells that were cultured on stiff hydrogels for seven or more days.
Fibroblast isolation and culture.
Fibroblasts from the aortic valve leaflets of both male and female pigs, provided by Hormel, were harvested as previously described24. For ATAC-seq, only male fibroblasts were used. The fibroblast expansion medium consisted of Media 199 (Life Technologies), 15% fetal bovine serum (FBS; Life Technologies), 1% penicillin—streptomycin and 0.5 μg ml−1 fungizone. Cells were cultured at 37 °C and 5% CO2 on tissue-culture polystyrene for expansion before experiments (approximately 3 d). Fibroblasts cultures at 70–80% confluency were harvested using trypsin (Life Technologies) and counted using an automated haemocytometer. The fibroblasts were seeded in the growth area of the PEG hydrogels at a density of 20,000 cells cm−2 in Media 199 supplemented with 1% serum (FBS), 1% penicillin—streptomycin and 0.5 μg ml−1 fungizone.
Immunocytochemistry.
Fibroblasts cultured on photo-softening hydrogels were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 1 h (Sigma-Aldrich) and blocked with 5% (wt) BSA for 1 h (Sigma-Aldrich). The samples were incubated overnight with primary antibodies against αSMA (1:1,000; Abcam, ab7817) or lamin AC (1:1,000; Abcam, ab190380) at 4 °C. The primary antibodies were removed by rinsing in PBS three times. Subsequently, the samples were incubated with the secondary antibodies phalloidin—TRITC (1:300; Sigma-Aldrich, P1951), goat anti-mouse Alexa Fluor 488 (1:500; Life Technologies) or goat anti-rabbit Alexa Fluor 647 (1:500; Life Technologies); HCS CellMask (1:5,000; Life Technologies) or DAPI (1 μg ml−1; Sigma-Aldrich) for 1 h at room temperature. After 1 h, the secondary antibody solution was removed and the samples were rinsed three times in PBS.
Quantification of myofibroblast activation, persistence, cell area, nuclear area, nuclear roundness and cytoskeletal-component expression.
Immunostained samples were imaged with a high-content confocal microscope using a ×20 objective (Operetta, PerkinElmer). Myofibroblasts were defined as cells with αSMA staining organized into stress fibres, and the percentage of myofibroblasts was calculated as: (number of myofibroblasts ÷ total number of cells) × 100%. At least five fields of view were quantified per gel (with at least ten, but fewer than 200, cells per field of view) and at least three gel replicates were performed for each condition. Representative images of each sample were captured with a laser scanning confocal microscope (LSM 710 NLO; Carl Zeiss AG) using a ×20 water objective and a maximum intensity projection of a Z-stack. The image analysis software ImageJ (FIJI) was used to crop images to dimensions that enabled easy visualization. Myofibroblast persistence was calculated by normalizing the per cent activated for the treatment conditions (stiff-to-soft) to the per cent activated determined for the untreated stiff and soft controls, where the mean of the stiff controls was set as the upper limit (100%) and the mean of the soft controls was set as the lower limit (0%). The cell area was quantified with CellMask to define the cytoplasm boundary and determined using the Harmony Image Analysis Software (PerkinElmer). The nuclear area and roundness were quantified with DAPI to define the nuclear boundary and determined using the Harmony Image Analysis Software (PerkinElmer). The cell intensity of the cytoskeletal components (vimentin, f-actin and microtubules) was determined using the Harmony Image Analysis Software (PerkinElmer) using CellMask to define the cell boundaries. Correlative analyses were performed using GraphPad Prism 8 using the average values from each time point (that is, 3, 5, 7 and 9 d).
Protein isolation and quantification.
Cells were washed three times with PBS and lysed in radioimmunoprecipitation assay (RIPA; ThermoFisher, cat. no. 89900) buffer supplemented with 1% protease and phosphatase inhibitor cocktail (ThermoFisher, cat. no. 78440). The lysates were subsequently combined with 6×Laemmli sample buffer and heated to 95 °C for 5 min. The cell lysates were separated on a pre-cast 4–12% gradient in running buffer. The proteins were transferred to either a nitrocellulose or PVDF membrane in transfer buffer for 90 min at 0.4 A and 130 V at 4 °C. The membranes were probed for total protein with the REVERT total protein stain kit (LI-COR Biosciences, cat. no. 926-11010) and imaged. Subsequently, the membranes were blocked in TBST and 5% skim milk powder at room temperature for 1 h and incubated overnight in anti-RNA polymerase II (1:1,000; rabbit; Abcam, cat. no. ab5131); anti-lamin AC (1:1,000; mouse; Abcam, ab190380); anti-fibronectin (1:1,000; mouse; Abcam, ab6328); anti-phosphorylated-cofilin (1:1,000; Cell Signaling, cat. no. 3313); anti-vinculin (1:5,000; Sigma-Aldrich, V9131); anti-phosphorylated erzin, radixin and moesin (1:1,000, Cell Signaling cat. no.3141); anti-cofilin (1:1,000; Abcam, ab11062) or anti-αSMA (1:1,000; mouse; Abcam, ab7817) antibody in blocking solution at 4 °C. The membranes were incubated with secondary goat anti-rabbit or mouse horseradish peroxidase-conjugated antibody for 1 h at room temperature. The chemiluminescence signal was detected using Pierce ECL Plus solution (ThermoFisher Scientific) and an ImageQuant LAS 4000 detector. The captured images were quantified in ImageJ (FIJI) and the intensity values were normalized to the total protein stain or RNA polymerase II.
CCP image acquisition and analysis.
Nuclei were visualized by DAPI staining and imaged with a laser scanning confocal microscope (LSM 710 NLO; Carl Zeiss AG) using a ×20 water objective with a ×3 zoom. At least five fields of view were imaged for each hydrogel. For analysis, individual nuclei for each field of view were isolated by cropping the image using FIJI so that there was only one nucleus per image. To calculate the CCP, a gradient-based Sobel edge-detection algorithm was employed using MATLAB to measure the edge density of individual nuclei, as previously described28. The MATLAB script was adapted to analyse 18-bit images taken with a confocal microscope.
Analysis of human aortic valve tissue.
Frozen tissue sections from the aortic valve were obtained from OriGene. Aortic valve tissue was obtained from both male and female patients (eight patients in total; Supplementary Fig. 4). Only one healthy patient sample was available. Frozen tissue sections were fixed in 4% paraformaldehyde for 30 min, permeabilized for 30 min (0.1% Triton X-100 in PBS) and blocked for 1 h (5% BSA in PBS). The sections were incubated overnight with primary antibodies (anti-αSMA, 1:500 (ab190380) and anti-laminA/C, 1:300 (ab7817)) in 5% BSA in PBS and subsequently washed with PBS. The sections were incubated with secondary antibodies—goat anti-IgG2 mouse Alexa Fluor 488 (1:500; Life Technologies, A-21141) and goat anti-IgG1 mouse Alexa Fluor 647 (1:500; Life Technologies A21240)—and DAPI (1 μg ml−1; Sigma-Aldrich) for 1 h at room temperature and then washed three times in PBS. The sections were imaged using a laser scanning confocal microscope (LSM 710 NLO; Carl Zeiss AG).
Inhibitor treatments.
For TSA dosing, aortic fibroblasts were treated with 75, 150 or 300 nM TSA diluted in DMSO (Sigma-Aldrich, cat. no. T8552). For garcinol dosing, the cells were treated with 1, 5 or 10 μM garcinol diluted in DMSO (Enzo Life Sciences, cat. no. BML-GR343-0010). For cytochalasin D dosing, the cells were treated with 2, 20 or 200 nM diluted in DMSO (Sigma-Aldrich, cat. no. C8273). For Y27632 dosing, the cells were treated with 0.5, 1, 5 or 10 μM Y27632 diluted in DMSO (Abcam, cat. no. ab120129). For withaferin A dosing, the cells were treated with 50 nM or 1 μM withaferin A diluted in DMSO (Santa Cruz Biotechnology, cat. no. sc-200381). For nocodazole dosing, the cells were treated with 10 nM, 100 nM, 1 μM or 10 μM nocodazole diluted in DMSO (Sigma-Aldrich, cat. no. M1404). After the dosing experiments, the final concentrations were determined for experiments as follows: TSA (150 nM), garcinol (5 μM), cytochalasin D (20 nM), Y27632 (5 μM), witheferin A (50 nM) and nocodazole (10 nM).
HDAC activity assay.
The HDAC activity was measured using an in situ histone deacetylase (HDAC) activity fluorometric assay kit (Sigma-Aldrich, EPI003). Briefly, cells cultured on 12-mm hydrogels were incubated with Deacetylated Substrate for 3 h. The supernatant was collected and mixed with developer for 30 min. The HDAC activity was measured by a plate reader reading fluorescence with excitation and emission wavelengths of 368 and 442 nm, respectively. After supernatant removal, the cells were washed with PBS and then lysed and incubated for 5 min in 200 μl RIPA Buffer (Fisher, 89901). The RIPA cell lysates were measured for double-stranded DNA, corresponding to the total cell number, using a Quant-iT PicoGreen dsDNA assay kit (Fisher, P7589). Calculations using standard curves were performed in Microsoft Excel. The HDAC activity values were then normalized to the cell number using the double-stranded DNA content.
RNA isolation and quantitative real-time PCR.
Quantitative real-time PCR was used to quantify the expression levels of HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, HDAC6, HDAC9, HDAC10, Sirt1 and Sirt2 messenger RNA relative to the reference gene RPL30. RNA was isolated from cells cultured (25 mm2) for 3 or 9 d after seeding on stiff or soft hydrogels using an RNeasy mini kit (Qiagen, cat. no. 74104). The RNA quantity and purity were measured via spectrophotometry (ND-1000; NanoDrop). Complimentary DNA was synthesized from total RNA using an iScript Synthesis kit (Bio-Rad, cat. no. 1708841), and the relative mRNA expression levels were measured via quantitative real-time PCR using SYBR Green reagents (Bio-Rad, cat. no. 1708884) on an iCycler (Bio-Rad) and normalized to RPL30 for three technical replicates per condition. The primer sequences are listed in Supplementary Table 1.
Quantification of the nuclear peripheral enrichment of lamin AC.
Immunostained images were captured with a laser scanning confocal microscope (LSM 710 NLO; Carl Zeiss AG) using a ×20 H20 objective with Nyquist sampling. The image analysis software ImageJ (FIJI) was used to quantify the nuclear peripheral enrichment: with single Z-plane nuclei images, an intensity profile was measured across the major axis of the nucleus, starting from the centre to the edge. Using Microsoft Excel, the intensity profile was binned so that 50 intensity points were calculated per nuclei. The average intensity of the nuclear centre (IC) was determined by averaging the first 10% of the intensity points, and the average intensity of the nuclear periphery (IP) was determined by averaging the last 10% of the intensity points. The IP / IC ratio was calculated for each nucleus. At least five fields of view per hydrogel, with three hydrogels per condition, were imaged and subsequently quantified.
Plasmid amplification and isolation.
The mCherry control vector and DNKASH were a gift from J. Lammerding, pcDNA Nesprin-HL (Addgene plasmid no. 68128; http://n2t.net/addgene:68128; RRID:Addgene_68128) and pcDNA Nesprin-TS (Addgene plasmid no. 68127; http://n2t.net/addgene:68127; RRID:Addgene_68127) were a gift from D. Conway, and mTFP1-Lifeact-7 was a gift from R. Campbell and M. Davidson (Addgene plasmid no. 54749; http://n2t.net/addgene:54749; RRID:Addgene_54749).
Plasmid amplification was performed according to an established protocol45. Briefly, bacteria obtained from Addgene were streaked onto an LB plate with the appropriate antibiotic. DNA plasmids were amplified and isolated using 150 ml LB medium containing cultured bacteria with Plasmid Midi-Prep (Qiagen, cat. no. 12145).
Cell transfection.
Cells were cultured on glass coverslips or hydrogels using the indicated growth medium and time lines. The cells were transfected with the Nesprin-TS, Nesprin-HL and mTFP1-Lifeact plasmids using Lipofectamine 3000 (ThermoFisher, cat. no. L3000008) as per the manufacturer’s instructions 24 h before imaging. The transfection cocktails of Lipofectamine reagents were optimized to minimize cell stress.
Image acquisition of the FRET Nesprin-2G-tension sensor.
Images were captured according to a previously established protocol45. Briefly, phenol red-containing medium was replaced with FluoroBrite DMEM medium (ThermoFisher, cat. no. A1896701) supplemented with 1% FBS and 1% penicillin—streptomycin before imaging. Cells were imaged using a spinning-disk confocal (Nikon Ti-E) microscope with a ×60 water objective. The images were captured with lasers to excite mTFP1 (458 nm) and yellow (515 nm). Cells that expressed the Nesprin-TS at a high enough level to be readily visualized and imaged were selected for imaging; however, cells in which the expression levels were too high were avoided45. In addition, cells that expressed mTFP through the entire nucleus were excluded and only cells expressing the sensor in a discernible nuclear ring were imaged. Cells transfected with mTFP1 and a low force control (Nesprin-HL) were used as controls to confirm the sensor and analysis of FRET was working.
NFRET Nesprin-2G-tension sensor image quantification.
Cells that expressed relatively equal amounts of sensor, as suggested by an established method86, were selected for imaging. Images were quantified using the ImageJ (FIJI) PixFRET plug-in to account for spectral bleed-throughs87. PixFRET calculated the spectral-bleed-through correction using donor (458) and acceptor (515) images from control mTFP1 images, using the Donor Model. The spectral-bleed-through ratios were assumed to be constant. To account for hydrogel background, the background intensities were retrieved from the Nesprin-TS images. The parameters used for PixFRET were: Gaussian blur, 1.0; threshold, 0.1; and output, FRET/Donor. The output NFRET image from PixFRET was thresholded to remove negative NFRET pixels and a nuclear membrane mask was overlaid using the acceptor channel. The reported NFRET values are the raw intensity values that were calculated within the nuclear mask and normalized to the nuclear perimeter. At least six cells across at least three hydrogels were quantified per condition. The cell images used for all of the values presented in a figure were acquired during the same imaging session, as absolute values cannot be compared across imaging sessions. For the purpose of clarity, the NFRET values were inverted and then normalized to the left-most condition to show the relative tension across conditions.
Lentiviral production and transduction.
The DNKASH construct was expressed in freshly isolated primary valve fibroblasts (P0) by transduction with a vector based on replication-deficient human immunodeficiency virus. Lentiviral particles were produced by co-transfecting the DNKASH or mCherry control plasmids with packaging delta 8.9 and VSVG-envelope plasmids at a 1.7:2.1:1 ratio into HEK293T cells (ATCC) using Lipofectamine 3000 transfection reagents (Invitrogen). The virus-containing medium was collected 2 d after transfection, centrifuged at 2,000 r.p.m. for 10 min and 0.45-μm filtered. The medium was applied to primary aortic valve fibroblasts, isolated 1 d previously, at a 1:1 ratio. It was verified that >90% cells expressed control or DNKASH through expression of nuclear mCherry (Supplementary Fig. 15). Only cells expressing nuclear mCherry were analysed for CCP, cell area, nuclear roundness and persistence. For the HDAC activity assay, all cells (expressing mCherry or not) were analysed, but visual confirmation was done so that >90% of cells expressed mCherry.
Statistical analysis and illustrations.
Data are shown as the mean ± s.e.m., unless otherwise indicated. GraphPad Prism 8 was used for all statistical analyses and figure creation. Significance was claimed at *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 using a Student’s t-test, or a one-way or two-way ANOVA with Tukey’s or Bonferroni’s post tests for multiple comparisons, unless otherwise stated. Figures 1c, 2f, 4a,b,e,g,h,k, 5a,b,f, 6d,g, 7a,d and 8 created with BioRender.com.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We acknowledge the support of the National Institutes of Health (grant no. R01 HL132353). C.J.W. was supported by the National Science Foundation (training grant no. IGERT 1144807), a National Institutes of Health Predoctoral Fellowship (grant no. F31HL142223) and a Department of Education GAANN Biomaterials Fellowship. A.R.K. was supported by the National Institutes of Health (grant no. R21 AR067469). C.C. was supported by a Human Frontiers Science Program fellowship (grant no. LT001449/2017-L) and American Heart Association postdoctoral fellowship (grant no. 20POST3521111). J.C.G. was supported by a postdoctoral fellowship from the National Institutes of Health (grant no. T32 HL007822-20). B.A.A. acknowledges funding from the National Institutes of Health (grant no. K99 HL148542) and the Burroughs Welcome Fund Postdoctoral Enrichment Program. L.A.L. was supported by the National Institutes of Health (grant nos RHL117138-05 and R01 GM29090). R.D.D. was supported by the National Institutes of Health (grant no. RO1 GM125871). We thank the BioFrontiers Institute Next-Gen Sequencing Core Facility, which performed the Illumina sequencing and library construction. The imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core. Laser scanning confocal microscopy was performed on a Nikon A1R microscope supported by the NIST-CU Cooperative Agreement award number 70NANB15H226. Spinning disc confocal microscopy was performed on a Nikon Ti-E microscope supported by the BioFrontiers Institute and the Howard Hughes Medical Institute. We acknowledge the BioFrontiers Computing Core at the University of Colorado Boulder for providing high-performance computing resources (grant no. NIH 1S10OD012300) supported by BioFrontiers’ IT. C.J.W. thanks T. Ceccato, A. G. Rodriguez, M. Schroeder and D. Batan for their support and feedback on project ideas; and I. Marozas for help with plasmid acquisition and advice.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00709-w.
Data availability
The authors declare that the main data supporting the results in this study are available within the paper and its Supplementary Information. The ATAC-seq data are available through the Gene Expression Omnibus under the accession code GSE167892. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available from the corresponding authors on reasonable request.
References
- 1.Rockey DC, Darwin Bell P & Hill JA Fibrosis-a common pathway to organ injury and failure. N. Engl. J. Med 372, 1138–1149 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Wynn TA Cellular and molecular mechanisms of fibrosis. J. Pathol 214, 199–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells RG Tissue mechanics and fibrosis. Biochim. Biophys. Acta 1832, 884–890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van De Water L, Varney S & Tomasek JJ Mechanoregulation of the myofibroblast in wound contraction, scarring, and fibrosis: opportunities for new therapeutic intervention. Adv. Wound Care 2, 122–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kis K, Liu X & Hagood JS Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med 13, e27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinz B et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol 180, 1340–1355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C & Ogawa R Fibroproliferative disorders and their mechanobiology. Connect. Tissue Res 53, 187–196 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Balestrini JL, Chaudhry S, Sarrazy V, Koehler A & Hinz B The mechanical memory of lung myofibroblasts. Integr. Biol 4, 410–421 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Caliari SR et al. Gradually softening hydrogels for modeling hepatic stellate cell behavior during fibrosis regression. Integr. Biol 8, 720–728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lunyak VV & Rosenfeld MG Epigenetic regulation of stem cell fate. Hum. Mol. Genet 17, R28–R36 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Perino M & Veenstra J Chromatin control of developmental dynamics and plasticity. Dev. Cell 38, 610–620 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Hu B, Gharaee-Kermani M, Wu Z & Phan SH Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am. J. Pathol 177, 21–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duong TE & Hagood JS Epigenetic regulation of myofibroblast phenotypes in fibrosis. Curr. Pathobiol. Rep 6, 79–96 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Tang CB & Kilian KA Matrix mechanics influence fibroblast—myofibroblast transition by directing the localization of histone deacetylase 4. Cell. Mol. Bioeng 10, 405–415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang SK et al. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 4, e621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bechtel W et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med 16, 544–550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caliari SR & Burdick JA A practical guide to hydrogels for cell culture. Nat. Methods 13, 405–414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tibbitt MW & Anseth KS Hydrogels as extracellular matrix mimics for 3D cell. Culture 103, 655–663 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kloxin AM, Kasko AM, Salinas CN & Anseth KS Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 324, 59–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Tibbitt MW, Langer SJ, Leinwand LA & Anseth KS Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc. Natl Acad. Sci. USA 110, 19336–19341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma H, Killaars AR, DelRio FW, Yang C & Anseth KS Myofibroblastic activation of valvular interstitial cells is modulated by spatial variations in matrix elasticity and its organization. Biomaterials 131, 131–144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong DY, Ranganath T & Kasko AM Low-dose, long-wave UV light does not affect gene expression of human mesenchymal stem cells. PLoS ONE 10, e0139307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruskowitz ER & Deforest CA Proteome-wide analysis of cellular response to ultraviolet light for biomaterial synthesis and modification. ACS Biomater. Sci. Eng 5, 2111–2116 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez AG, Schroeder ME, Walker CJ & Anseth KS FGF-2 inhibits contractile properties of valvular interstitial cell myofibroblasts encapsulated in 3D MMP-degradable hydrogels. APL Bioeng. 2, 046104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinz B et al. The myofibroblast: one function, multiple origins. Am. J. Pathol 170, 1807–1816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu AC, Joag VR & Gotlieb AI The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol 171, 1407–1418 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li CX et al. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater 16, 379–389 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Irianto J et al. Osmotic challenge drives rapid and reversible chromatin condensation in chondrocytes. Biophys. J 104, 759–769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K et al. The overlooked fact: fundamental need for spike-in control for virtually all genome-wide analyses. Mol. Cell. Biol 36, 662–667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egan B et al. An alternative approach to ChIP-Seq normalization enables detection of genome-wide changes in histone H3 lysine 27 trimethylation upon EZH2 inhibition. PLoS ONE 11, e0166438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landt SG et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 10.1101/gr.136184.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubin JD et al. Transcription factor enrichment analysis (TFEA): quantifying the activity of hundreds of transcription factors from a single experiment. Preprint at bioRxiv 10.1101/2020.01.25.919738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toth KF Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci 117, 4277–4287 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Szczesny SE & Mauck RL The nuclear option: evidence implicating the cell nucleus in mechanotransduction. J. Biomech. Eng 139, 021006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Downing TL et al. Biophysical regulation of epigenetic state and cell reprogramming. Nat. Mater 12, 1154–1162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho S, Irianto J & Discher DE Mechanosensing by the nucleus: from pathways to scaling relationships. J. Cell Biol 216, 305–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajik A et al. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater 15, 1287–1296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramdas NM & Shivashankar GV Cytoskeletal control of nuclear morphology and chromatin organization. J. Mol. Biol 427, 695–706 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Keeling MC, Flores LR, Dodhy AH, Murray ER & Gavara N Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci. Rep 7, 5219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swift J et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naetar N, Ferraioli S & Foisner R Lamins in the nuclear interior—life outside the lamina. J. Cell Biol 130, 2087–2096 (2017). [DOI] [PubMed] [Google Scholar]
- 43.González-Cruz RD, Dahl KN & Darling EM The emerging role of Lamin C as an important LMNA isoform in mechanophenotype. Front. Cell Dev. Biol 6, 151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirby TJ & Lammerding J Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol 20, 373–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arsenovic PT et al. Nesprin-2G, a component of the nuclear LINC Complex, is subject to myosin-dependent tension. Biophys. J 110, 34–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lombardi ML et al. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem 286, 26743–26753 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crowder SW, Leonardo V, Whittaker T, Papathanasiou P & Stevens MM Material cues as potent regulators of epigenetics and stem cell function. Cell Stem Cell 18, 39–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stowers RS et al. Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat. Biomed. Eng 3, 1009–1019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang C, Tibbitt MW, Basta L & Anseth KS Mechanical memory and dosing influence stem cell fate. Nat. Mater 13, 645–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Killaars AR et al. Extended exposure to stiff microenvironments leads to persistent chromatin remodeling in human mesenchymal stem cells. Adv. Sci 6, 1801483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heo SJ et al. Biophysical regulation of chromatin architecture instills a mechanical memory in mesenchymal stem cells. Sci. Rep 5, 16895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerardo H et al. Soft culture substrates favor stem-like cellular phenotype and facilitate reprogramming of human mesenchymal stem/stromal cells (hMSCs) through mechanotransduction. Sci. Rep 9, 9086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monzack EL & Masters KS Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J. Heart Valve Dis 20, 449–463 (2011). [PMC free article] [PubMed] [Google Scholar]
- 54.Schaefer A et al. Analysis of fibrosis in control or pressure overloaded rat hearts after mechanical unloading by heterotopic heart transplantation. Sci. Rep 9, 5710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Chen B, Liu T & Chen X Reversal of myofibroblast differentiation: a review. Eur. J. Pharm 734, 83–90 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Turner NA & Porter KE Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 6, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramos C et al. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am. J. Respir. Cell Mol. Biol 24, 591–598 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Hinz B & Lagares D Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat. Rev. Rheumatol 16, 11–31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hecker L et al. Reversal of persistent fibrosis in aging by targeting Nox4–Nrf2 redox imbalance. Sci. Transl. Med 6, 231ra47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanders YY et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 1, 8–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez-Segura A, Nehme J & Demaria M Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Cho S, Irianto J & Discher DE Mechanosensing by the nucleus: from pathways to scaling relationships. J. Cell Biol 216, 305–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu X et al. MKL1-actin pathway restricts chromatin accessibility and prevents mature pluripotency activation. Nat. Commun 10, 1695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dechat T et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swift J & Discher DE The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci 127, 3005–3015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buxboim A, Ivanovska IL & Discher DE Matrix elasticity, cytoskeletal forces and physics of the nucleus: how deeply do cells ‘feel’ outside and in? J. Cell Sci 123, 297–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bouzid T et al. The LINC complex, mechanotransduction, and mesenchymal stem cell function and fate. J. Biol. Eng 13, 68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho CY, Jaalouk DE, Vartiainen MK & Lammerding J Lamin A/C and emerin regulate MKL1–SRF activity by modulating actin dynamics. Nature 497, 507–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rashmi RN et al. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus 3, 172–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Görisch SM, Wachsmuth M, Tóth KF, Lichter P & Rippe K Histone acetylation increases chromatin accessibility. J. Cell Sci 118, 5825–5834 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Milon BC et al. Role of histone deacetylases in gene regulation at nuclear lamina. PLoS ONE 7, e49692 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mattioli E et al. Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell 17, e12824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serebryannyy LA, Cruz CM & De Lanerolle P A role for nuclear actin in HDAC 1 and 2 regulation. Sci. Rep 6, 28460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Killaars AR, Walker CJ & Anseth KS Nuclear mechanosensing controls MSC osteogenic potential through HDAC epigenetic remodeling. Proc. Natl Acad. Sci. USA 117, 21258–21266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lyu X, Hu M, Peng J, Zhang X & Sanders YY HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis 10, 2040622319862697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Renaud L et al. HDACs regulate miR-133a expression in pressure overload-induced cardiac fibrosis. Circ. Heart Fail 8, 1094–1104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saito S et al. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFβ–PI3K–Akt pathway. PLoS ONE 12, e0186615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aguado BA, Grim JC, Rosales AM, Watson-Capps JJ & Anseth KS Engineering precision biomaterials for personalized medicine. Sci. Transl. Med 10, eaam8645 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aguado BA et al. Transcatheter aortic valve replacements alter circulating serum factors to mediate myofibroblast deactivation. Sci. Transl. Med 11, eaav3233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ceccato TL et al. Defining the cardiac fibroblast secretome in a fibrotic microenvironment. J. Am. Heart Assoc 10.1161/jaha.120.017025 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grim JC et al. Secreted factors from proinflammatory macrophages promote an osteoblast-like phenotype in valvular interstitial cells. Arterioscler. Thromb. Vasc. Biol 10.1161/ATVBAHA.120.315261 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu H, Sun Y & Simmons CA Determination of local and global elastic moduli of valve interstitial cells cultured on soft substrates. J. Biomech 46, 1967–1971 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Wyss K et al. The elastic properties of valve interstitial cells undergoing pathological differentiation. J. Biomech 45, 882–887 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Pho M et al. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am. J. Physiol. Heart Circ. Physiol 294, H1767–78 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Kloxin AM, Tibbitt MW & Anseth KS Synthesis of photodegradable hydrogels as dynamically tunable cell culture platforms. Nat. Protoc 5, 1867–1887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arsenovic PT & Conway DE in The LINC Complex (Methods in Molecular Biology series) Vol. 1840, 59–71 (Humana Press, 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feige JN, Sage D, Wahli W, Desvergne B & Gelman L PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc. Res. Tech 68, 51–58 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the main data supporting the results in this study are available within the paper and its Supplementary Information. The ATAC-seq data are available through the Gene Expression Omnibus under the accession code GSE167892. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available from the corresponding authors on reasonable request.