

Abstract
BACKGROUND
The addition of rituximab to combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), or R-CHOP, has significantly improved the survival of patients with diffuse large-B-cell lymphoma. Whether gene-expression signatures correlate with survival after treatment of diffuse large-B-cell lymphoma is unclear.
METHODS
We profiled gene expression in pretreatment biopsy specimens from 181 patients with diffuse large-B-cell lymphoma who received CHOP and 233 patients with this disease who received R-CHOP. A multivariate gene-expression–based survival-predictor model derived from a training group was tested in a validation group.
RESULTS
A multivariate model created from three gene-expression signatures — termed “germinal-center B-cell,” “stromal-1,” and “stromal-2” — predicted survival both in patients who received CHOP and patients who received R-CHOP. The prognostically favorable stromal-1 signature reflected extracellular-matrix deposition and histiocytic infiltration. By contrast, the prognostically unfavorable stromal-2 signature reflected tumor blood-vessel density.
CONCLUSIONS
Survival after treatment of diffuse large-B-cell lymphoma is influenced by differences in immune cells, fibrosis, and angiogenesis in the tumor microenvironment.
Although diffuse large-B-cell lymphoma is curable with anthracycline-based chemotherapy regimens such as a combination of cyclophosphamide, doxorubicin, vincristne, and prednisone (CHOP),1 the additionof rituximab immunotherapy (R-CHOP) has improved overall survival among patients with diffuse large-B-cell lymphoma by 10 to 15%.2 Diffuse large-B-cell lymphoma is a molecularly heterogeneous disease,3 and it is unclear whether rituximab preferentially improves the outcome in certain subgroups of patients.
Gene-expression profiling has identified two biologically and clinically distinct molecular subtypes of diffuse large-B-cell lymphoma.4,5 The germinal-center B-cell–like diffuse large-B-cell lymphoma subtype probably arises from normal germinal-center B cells, whereas the activated B-cell–like subtype may arise from a post-germinal-center B cell that is blocked during plasmacytic differentiation. Many oncogenic mechanisms distinguish these subtypes: germinal-center B-cell–like diffuse large-B-cell lymphomas have recurrent t(14;18) translocations, whereas activated B-cell–like diffuse large-B-cell lymphomas have recurrent trisomy 3 and deletion of the inhibitor of kinase 4A–alternative reading frame (INK4A/ARF) locus as well as constitutive activation of the antiapoptotic nuclear factor (NF)-κB signaling pathway. 4,6–10 With CHOP-like chemotherapy, the 5-year overall survival rates among patients with germinal-center B-cell–like diffuse large-B-cell lymphoma and those with activated B-cell–like diffuse large-B-cell lymphoma were 60% and 30%, respectively.11
A different analytic approach identified four gene-expression signatures that reflected distinct biologic attributes of diffuse large-B-cell lymphoma tumors that were associated with survival among patients who received CHOP.4 A “germinal-center B-cell” signature was associated with a favorable prognosis and paralleled the distinction between activated B-cell–like and germinal-center B-cell–like diffuse large-B-cell lymphoma. The “proliferation” signature was associated with a poor prognosis and included MYC and its target genes. The “major histocompatibility complex (MHC) class II” signature was silenced in the malignant cells in a subgroup of patients with diffuse large-B-cell lymphoma; this event was associated with inferior survival.4,12 A fourth prognostic signature, termed “lymph node,” was associated with a favorable prognosis and included components of the extracellular matrix, suggesting that it may reflect the nature of the tumor microenvironment. These signatures predicted survival in a statistically independent fashion, indicating that multiple biologic variables dictate the response to CHOP chemotherapy in diffuse large-B-cell lymphoma.
To evaluate the biologic basis of survival after therapy for diffuse large-B-cell lymphoma, we profiled gene expression in pretreatment biopsy samples obtained from patients treated with CHOP or R-CHOP. We used these data to search for gene-expression signatures of different aspects of tumor biology that were associated with survival.
METHODS
STUDY POPULATIONS
Pretreatment tumor-biopsy specimens and clinical data were obtained from 414 patients with newly diagnosed diffuse large-B-cell lymphoma who were treated at 10 institutions in North America and Europe and studied according to a protocol approved by the institutional review board of the National Cancer Institute. Among these patients, a CHOP training group consisted of 181 patients, previously described,4 who were treated with anthracycline-based combinations, most often CHOP or similar regimens. The other 233 patients constituted an R-CHOP cohort that received similar chemotherapy plus rituximab. The median follow-up for the R-CHOP cohort was 2.1 years; the median follow-up for survivors was 2.8 years. A panel of expert hematopathologists confirmed the diagnosis of diffuse large-B-cell lymphoma using current World Health Organization criteria. We also analyzed data from a second cohort of 177 patients who received CHOP; these data were previously reported by the Molecular Mechanisms in Malignant Lymphomas Network Project (MMMLNP).13
GENE-EXPRESSION PROFILING
Gene-expression profiling was performed with the use of Affymetrix U133 plus 2.0 microarrays (data available at www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=rhojvaiwkcsaihq&acc=GSE10846, accession number GSE10846). Cell suspensions from three biopsy specimens were separated by means of flow cytometry into a CD19+ malignant subpopulation and a CD19− nonmalignant subpopulation. Gene-expression profiling was performed after two rounds of linear amplification from total RNA.14 After normalization to a median signal of 500, provided in the Affymetrix Microarray Suite software, version 5.0 (MAS5.0), we selected genes that had a signal value greater than 128 in either the CD19+ or CD19− fractions in at least two of the sorted samples. Detailed statistical and experimental methods are described in the Supplementary Appendix, available with the full text of this article at www.nejm.org.
STATISTICAL ANALYSIS
All aspects of identification of the gene-expression signatures and development of the survival model were based solely on the data from the CHOP training group and are outlined in detail in the Supplementary Appendix. No previous survival analysis or subgroup analysis was performed with the validation groups (i.e., the MMMLNP CHOP and the R-CHOP cohorts). A Cox model was used to identify genes associated with survival and to build multivariate survival models. The models and their associated scaling coefficients were fixed, based on the CHOP training group, and then evaluated in the validation groups. All reported P values are two-sided, except those in the validation groups, which are one-sided P values in the direction of the observed effect on the training group. P values reported for survival associations were based on single-hypothesis testing, except those for testing of multivariate models involving the germinal-center B-cell, stromal-1, proliferation, and MHC class II signatures in the R-CHOP cohort, which were not adjusted for multiple testing.
To discover new signatures associated with survival, we selected individual genes with expression patterns that contributed significantly (P<0.01) to the survival association in the CHOP training group, in a model containing that gene and the germinal-center B-cell and stromal-1 signatures. We organized these genes by hierarchical clustering according to their expression levels in the CHOP training group, and we identified five clusters of coordinately expressed genes (r>0.6). For each of these five candidate signatures, we averaged the expression levels of the component genes and tested whether the average for the signature added to the predictive significance of the bivariate survival model for the CHOP training group. One signature was clearly superior to the others with respect to its predictive contribution to the survival model and was therefore chosen for further analysis. This signature also added to the predictive significance of the bivariate model for the R-CHOP cohort (P = 0.001) and for the MMMLNP CHOP cohort (P = 0.011) (Fig. 8B and 8C in the Supplementary Appendix). In these survival models, this new signature was associated with reduced survival, whereas the stromal-1 signature was associated with increased survival, even though these two signatures were correlated with one another (r>0.8). Therefore, to refine this new signature, we identified genes that were more closely correlated with it than with the stromal-1 signature (P<0.02) in the CHOP training group, and we organized these genes into three signatures by hierarchical clustering, as described above. The signature that most improved the survival model (stromal-2) was chosen for subsequent analyses.
RESULTS
MULTIVARIATE MODEL OF SURVIVAL
We profiled gene expression in 414 pretreatment biopsy samples from patients with newly diagnosed diffuse large-B-cell lymphoma, including a training group of 181 patients treated with CHOP or CHOP-like chemotherapy alone and a validation group of 233 patients treated with similar chemotherapy plus rituximab. In this R-CHOP cohort (Table 1), patients with germinal-center B-cell–like diffuse large-B-cell lymphoma had higher overall and progression-free survival rates than those with activated B-cell–like diffuse large-B-cell lymphoma (Fig. 1A). Three gene-expression signatures that predicted survival in the CHOP training group4 — termed germinal-center B-cell, lymph-node, and proliferation — were significantly associated with the outcome in a second cohort of CHOP-treated patients (from the MMMLNP)13 and in the R-CHOP cohort (Fig. 1 and 2 in the Supplementary Appendix). In contrast, the MHC class II signature was not associated with survival in the R-CHOP cohort (Fig. 1, 2, and 3 in the Supplementary Appendix). From these four signatures, an optimal survival model for R-CHOP combined the germinal-center B-cell and lymph-node signatures (Fig. 4A in the Supplementary Appendix). Since this latter signature reflects the tumor microenvironment (see below), we refer to it as “stromal-1.”
Table 1.
Clinical Characteristics of Patients with Diffuse Large-B-Cell Lymphoma Treated with R-CHOP.*
| Characteristic | All Subtypes (N = 233) | Germinal Center B-Cell–like Subtype (N = 107) | Activated B-Cell–like Subtype (N = 93) | Unclassified Subtype (N = 33) | P Value† |
|---|---|---|---|---|---|
| no./total no. (%) | |||||
| Age >60 yr | 122/233 (52) | 50/107 (47) | 59/93 (63) | 13/33 (39) | 0.023 |
| Ann Arbor stage >II‡ | 121/226 (54) | 49/103 (48) | 56/91 (62) | 16/32 (50) | 0.061 |
| Lactate dehydrogenase >ULN | 93/192 (48) | 38/89 (43) | 44/76 (58) | 11/27 (41) | 0.062 |
| ≥2 Extranodal sites | 30/204 (15) | 13/92 (14) | 13/84 (15) | 4/28 (14) | 0.834 |
| ECOG performance status >1§ | 52/210 (25) | 17/98 (17) | 27/82 (33) | 8/30 (27) | 0.023 |
| IPI score¶ | <0.001 | ||||
| 0 or 1 | 75/182 (41) | 47/85 (55) | 15/71 (21) | 13/26 (50) | |
| 2 or 3 | 83/182 (46) | 28/85 (33) | 45/71 (63) | 10/26 (38) | |
| 4 or 5 | 24/182 (13) | 10/85 (12) | 11/71 (15) | 3/26 (12) | |
R-CHOP denotes rituximab plus combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone, and ULN upper limit of the normal range.
P values are for the comparison of activated B-cell–like and germinal-center B-cell–like diffuse large-B-cell lymphomas.
The Ann Arbor staging system ranges from I to IV, with a higher stage indicating more widespread disease.
The Eastern Cooperative Oncology Group (ECOG) performance score ranges from 0 to 3, with a higher score indicating greater impairment.
The International Prognostic Index (IPI) score ranges from 0 to 5, with 0 indicating the absence of all prognostic factors and 5 indicating the presence of all prognostic factors.
Figure 1. Gene-Expression Predictors of Survival among Patients with Diffuse Large-B-Cell Lymphoma Treated with R-CHOP.
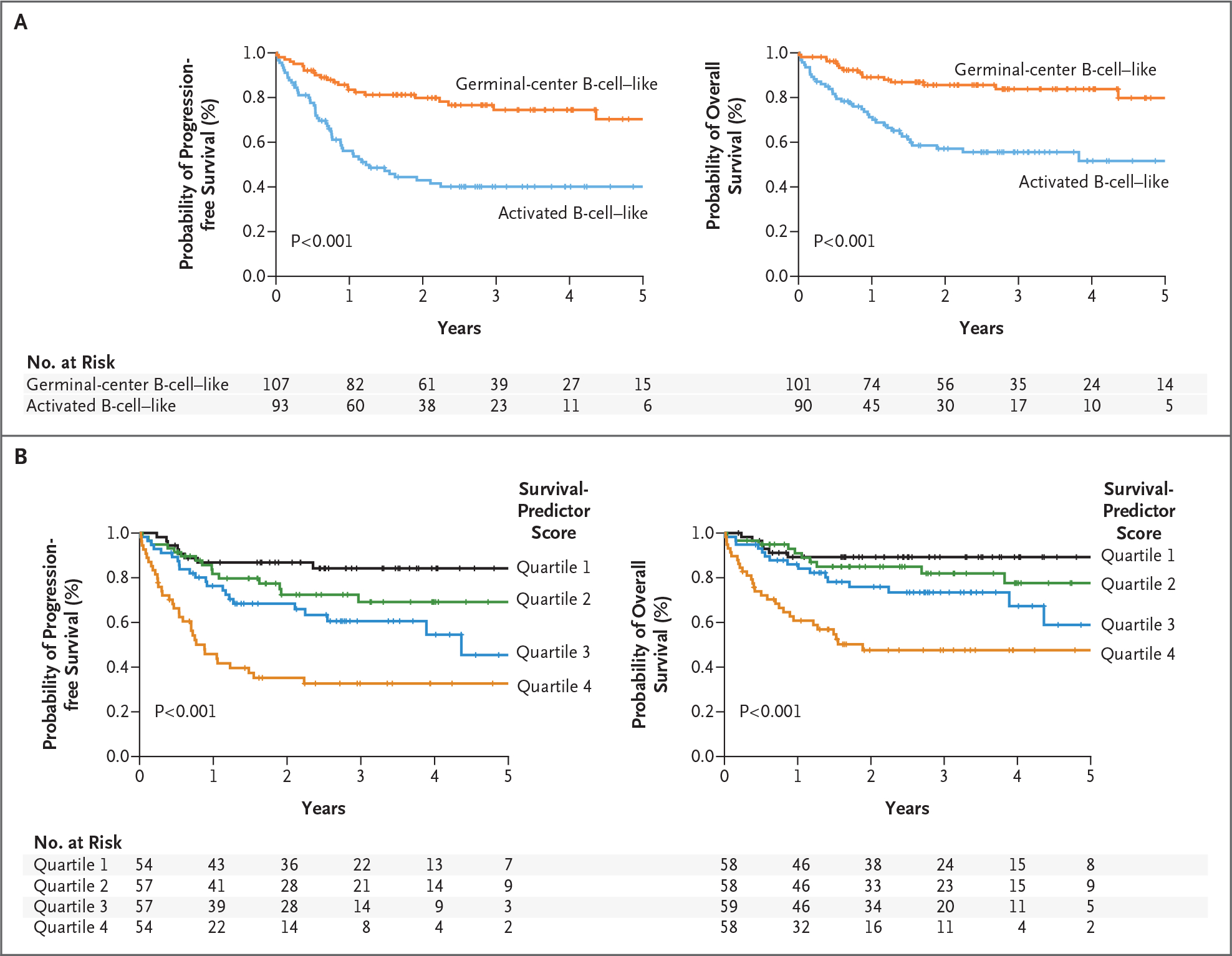
Kaplan–Meier estimates of progression-free and overall survival are shown. Panel A shows that patients with germinal-center B-cell–like diffuse large-B-cell lymphoma had a higher probability of progression-free survival (left) and overall survival (right) than patients with activated B-cell–like diffuse large-B-cell lymphoma. Panel B shows a gene-expression–based predictor of survival among patients with diffuse large-B-cell lymphoma treated with R-CHOP. Kaplan–Meier estimates of progression-free survival (left) and overall survival (right) are based on a multivariate model derived from the germinal-center B-cell, stromal-1, and stromal-2 gene-expression signatures. Survival-predictor scores derived from this model were used to rank the cases of lymphoma, which were then divided into quartile groups as indicated. R-CHOP denotes rituximab plus combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone.
We next discovered a new signature that added to the prognostic significance of the bivariate model for the CHOP training group; we call this signature “stromal-2” by virtue of its association with the tumor microenvironment. The stromal-2 signature added to the predictive significance of the survival model for the R-CHOP cohort (P<0.001) and for the MMMLNP CHOP cohort ( P = 0.002).13 The resulting trivariate model was highly associated with overall and 3-year progression-free survival as a continuous variable in the R-CHOP cohort (P<0.001). Each signature added to the predictive significance of the model, with the germinal-center B-cell and stromal-1 signatures associated with increased survival and the stromal-2 signature associated with reduced survival (Table 2 in the Supplementary Appendix). The survival-predictor score generated by this model was associated with an increase in the relative risk of death of 2.76 (95% confidence interval, 1.90 to 3.90) per unit increment of the score, which varied over 3.58 units and had a standard deviation of 0.76 in the R-CHOP cohort. Model scores were used to divide the R-CHOP cohort into quartiles of 3-year overall survival rates of 89%, 82%, 74%, and 48% and 3-year progression-free survival rates of 84%, 69%, 61%, and 33% (Fig. 1B).
When combined with the International Prognostic Index (IPI),15 the gene-expression–based model added to the predictive power of the IPI (P<0.001), and the IPI added to the predictive power of the gene-expression–based model (P = 0.0033), suggesting that survival in diffuse large-B-cell lymphoma is influenced both by clinical variables and by biologic features of the lymphoma (Fig. 4B and 5 in the Supplementary Appendix).
THE BIOLOGIC BASIS FOR PROGNOSTIC SIGNATURES
To assess whether the gene-expression signatures in our survival model were derived from the malignant lymphoma cells or from the host microenvironment, we separated CD19+ malignant cells from CD19− nonmalignant cells in three biopsy samples of diffuse large-B-cell lymphoma by means of flow sorting. As expected, the germinal-center B-cell signature genes were more highly expressed in the malignant than in the nonmalignant fraction (Fig. 2A). By contrast, the stromal-1 and stromal-2 signature genes were more highly expressed in the nonmalignant fraction. Since these two signatures were synergistic in predicting survival, we combined them into a “stromal score” (Fig. 3), high values of which were associated with adverse outcomes. The stromal score was variably present in both germinal-center B-cell–like and activated B-cell–like diffuse large-B-cell lymphoma, suggesting that the stromal signatures represent biologic attributes of the tumor microenvironment that can be acquired during the pathogenesis of both diffuse large-B-cell lymphoma subtypes (Fig. 3).
Figure 2. Cellular Derivation of Prognostic Gene-Expression Signatures in Diffuse Large-B-Cell Lymphoma.
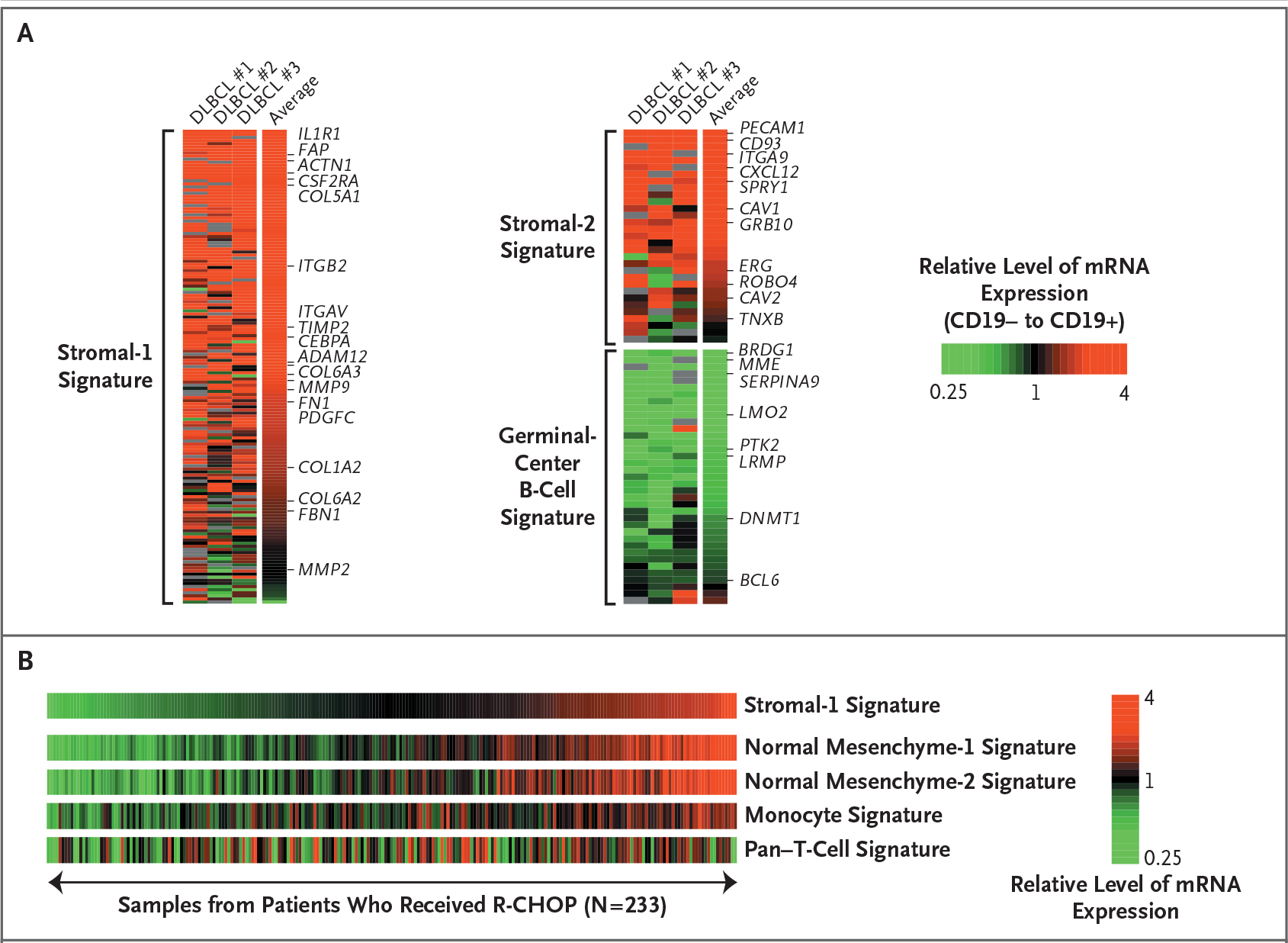
Panel A shows the relative gene expression of the stromal-1, stromal-2, and germinal-center B-cell signatures in CD19+ malignant and CD19− nonmalignant subpopulations of cells isolated from three biopsy specimens from patients with diffuse large-B-cell lymphoma (DLBCL). Stromal-1 and stromal-2 signature genes were more highly expressed in the nonmalignant cells, whereas the germinal-center B-cell signature genes were more highly expressed in the malignant cells. The log2 ratios of gene-expression levels in the CD19− subpopulation to those in the CD19+ subpopulation are depicted according to the color scale shown. Panel B shows the relationship of the stromal-1 signature to gene-expression signatures derived from normal cells. Gene-set enrichment analysis established a relationship between two signatures that are expressed in cells and tissues of mesenchymal origin (normal mesenchyme-1 and mesenchyme-2 signatures) and a monocyte signature, which is expressed more highly in normal blood monocytes than in B cells, T cells, and natural killer cells. No relationship was observed between the stromal-1 signature and a pan–T-cell signature, which is expressed more highly in T cells than in B cells, natural killer cells, and monocytes. The relative levels of gene expression within each sample are depicted according to the color scale shown. R-CHOP denotes rituximab plus combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone.
Figure 3. Expression of Stromal-1 and Stromal-2 Signature Genes in Biopsy Samples from Patients with Diffuse Large-B-Cell Lymphoma.
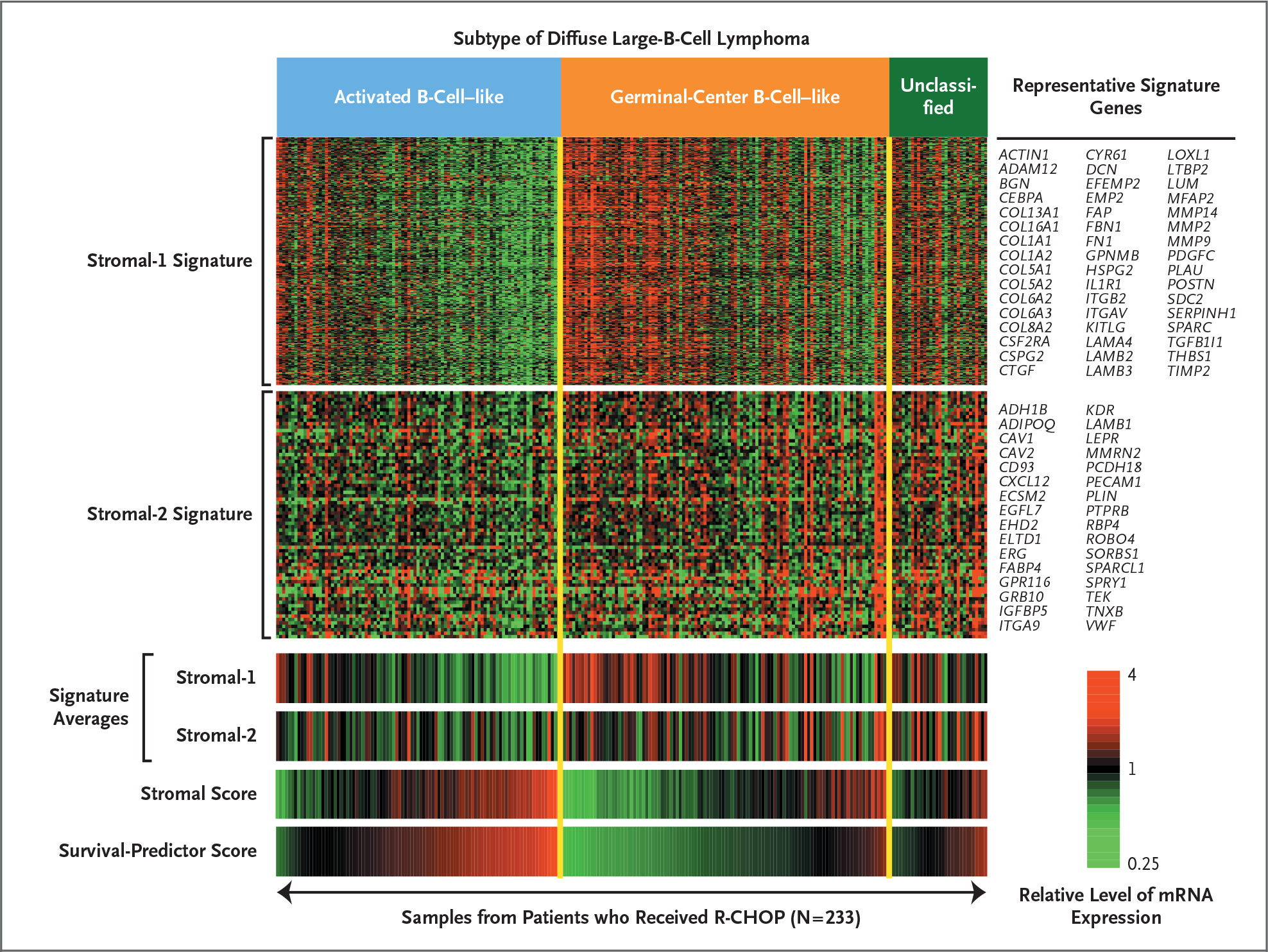
The relative levels of gene expression within each sample are depicted according to the color scale shown, with each row representing a different signature gene and each column representing a different biopsy sample. The stromal-1 and stromal-2 signature averages are shown for each patient. The stromal score is the component of the multivariate survival model contributed by the difference between the stromal-2 and stromal-1 signature averages. The samples were ranked within each diffuse large-B-cell lymphoma subtype according to the survival-predictor score, which was generated by the survival model and incorporates both the stromal score and the germinal-center B-cell signature (not shown). Representative signature genes are shown. R-CHOP denotes rituximab plus combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone.
The genes defining the stromal-1 signature encode components of the extracellular matrix, including fibronectin, osteonectin, various collagen and laminin isoforms, and the antiangiogenic factor thrombospondin (Fig. 3, and Table 3 in the Supplementary Appendix). This signature also encodes modifiers of collagen synthesis (LOXL1 and SERPINH1), proteins that remodel the extracellular matrix (MMP2, MMP9, MMP14, PLAU, and TIMP2), and connective-tissue growth factor (CTGF), a secreted protein that can initiate fibrotic responses.16 In addition, the stromal-1 signature includes genes that are characteristically expressed in cells in the monocytic lineage, such as CEBPA and CSF2RA.
The stromal-1 signature was significantly related to several previously curated gene-expression signatures17 on the basis of gene-set enrichment analysis.18 Two of these signatures include genes that are coordinately expressed in normal mesenchymal tissues but not in hematopoietic subgroups, many of which encode extracellular-matrix proteins (false discovery rate, <0.001) (Fig. 2B, and Fig. 6A in the Supplementary Appendix).19 Also enriched was a “monocyte” signature, comprising genes that are more highly expressed in CD14+ blood monocytes than in B cells, T cells, or natural killer cells (false discovery rate, 0.014) (Fig. 2B, and Fig. 6B in the Supplementary Appendix). By contrast, a pan–T-cell signature was not related to the stromal-1 signature (Fig. 2B, and Fig. 6B in the Supplementary Appendix). These findings suggest that high expression of the stromal-1 signature identifies tumors with vigorous extracellular-matrix deposition and infiltration by cells of the monocytic lineage.
Several stromal-2 signature genes encode wellknown markers of endothelial cells, including von Willebrand factor and CD31 (platelet endothelial-cell adhesion molecule, or PECAM1), as do other genes specifically expressed in endothelium such as EGFL7, MMRN2, GPR116, and SPARCL1 (Table 3 in the Supplementary Appendix). This signature also includes genes encoding key regulators of angiogenesis: kinase-domain-related (KDR) receptor (vascular endothelial growth factor [VEGF] receptor 2); GRB10 (growth factor receptor–bound protein 10), which mediates KDR signaling; integrin alpha 9, which enhances VEGF signaling; TEK (tyrosine kinase, endothelial), the receptor tyrosine kinase for the cytokine angiopoietin; ROBO4, an endothelial-specific molecular guidance molecule that regulates angiogenesis; and ERG (V-ets erythroblastosis virus E26 oncogene homologue gene), a transcription factor required for endothelial-tube formation. The stromal-2 signature genes CAV1, CAV2, and EHD2 encode components of caveolae, which are specialized plasma-membrane structures that are abundant in endothelial cells and are required for angiogenesis.20,21 Although the stromal-2 signature includes a large number of genes expressed in endothelial cells, others are expressed only in adipocytes, including ADIPOQ, FABP4, RBP4, and PLIN.
On immunohistochemical staining, fibronectin, a component of the stromal-1 signature, was prominently localized in fibrous strands running between the malignant cells in biopsy samples obtained from patients with diffuse large-B-cell lymphoma, in keeping with its role in extracellular-matrix formation (Fig. 4A). By contrast, the protein products of three other stromal-1 genes — MMP9, SPARC, and CTGF — were localized primarily in histiocytic-cell infiltrates in the biopsy specimens from patients with diffuse large-B-cell lymphoma (Fig. 4B, 4C, and 4D). On immunofluorescence analysis, SPARC and CTGF colocalized with CD68, a marker for cells in the monocytic lineage (Fig. 4E and 4F). As expected for a stromal-1 gene product, higher SPARC protein levels were associated with increased overall survival (Fig. 4G). Thus, the stromal-1 signature reflects a monocyte-rich host reaction to the lymphoma that is associated with abundant deposition of extracellular matrix.
Figure 4. Immunohistochemical Analysis of Biopsy Specimens from Patients with Diffuse Large-B-Cell Lymphoma.
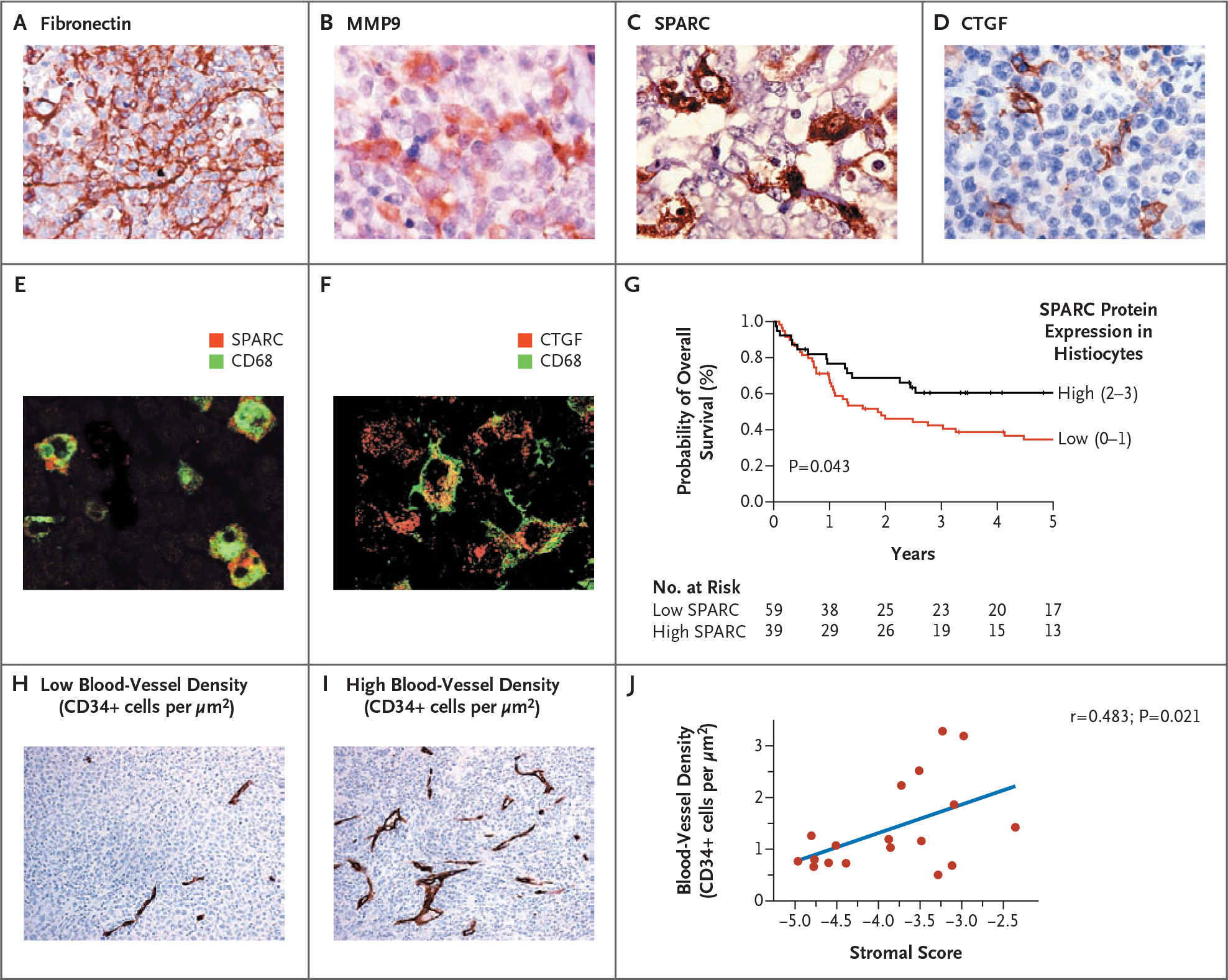
Panel A shows that fibronectin, a stromal-1 signature component, was localized prominently in fibrous strands running between malignant cells in the biopsy specimens. Panels B, C, and D show matrix metalloproteinase 9 (MMP9), secreted protein acidic cysteine-rich (SPARC), and connective-tissue growth factor (CTGF), respectively. Each of these components of the stromal-1 signature was localized primarily in histiocytic cells infiltrating the biopsy specimens. Panels E and F show colocalization of SPARC and CTGF with the myelomonocytic marker CD68. Panel G shows Kaplan–Meier estimates of overall survival according to the level of expression of SPARC. Panels H and I show tumor blood-vessel density according to immunohistochemical analysis of CD34+ endothelial cells in biopsy specimens. Representative cases with low or high blood-vessel density (CD34+ cells per μm2) are shown. Panel J shows the correlation between tumor blood-vessel density and the stromal score.
Finally, we suspected that high relative expression of the stromal-2 signature (i.e., a high stromal score) would reflect high tumor blood-vessel density, given the connection between many stromal-2 signature genes and angiogenesis. Indeed, a quantitative measure of blood-vessel density correlated significantly with the stromal score (r = 0.483, P = 0.021) (Fig. 4H, 4I, and 4J).
DISCUSSION
Biologic variation among diffuse large-B-cell lymphoma tumors, as measured by means of gene-expression signatures, has a consistent relationship to treatment response, regardless of the regimen used. Specifically, the benefit of rituximab immunotherapy combined with chemotherapy was evident in both the activated B-cell–like and germinal-center B-cell–like subtypes of diffuse large-B-cell lymphoma (Fig. 1A, and Fig. 7 in the Supplementary Appendix). Moreover, several other gene-expression signatures that predicted survival among patients who received CHOP chemotherapy retained their prognostic power among patients who received R-CHOP. Hence, future clinical trials in diffuse large-B-cell lymphoma should incorporate quantitative methods to discern these biologic differences so that patient cohorts in different trials can be compared and treatment responses can be related to defined tumor phenotypes.
The survival model includes two gene-expression signatures, stromal-1 and stromal-2, that reflect the character of the nonmalignant cells in diffuse large-B-cell lymphoma. The stromal-1 signature includes genes that are coordinately expressed in many normal mesenchymal tissues, most of which encode proteins that form or modify the extracellular matrix. One stromal-1 signature component, fibronectin, was localized to fibrous strands insinuated between the malignant lymphoma cells, suggesting that this signature reflects the fibrotic nature of many diffuse large-B-cell lymphomas. A key to this fibrotic reaction may be another stromal-1 signature component, CTGF, which participates in many fibrotic responses and diseases. CTGF also promotes tumor growth and metastasis of epithelial cancers.22
Another characteristic feature of tumors with high expression of the stromal-1 signature was infiltration by cells of the myeloid lineage. Various cells in this lineage have been implicated in the pathogenesis of epithelial cancers, including tumor-associated macrophages, myeloid-derived suppressor cells, and Tie2-expressing monocytes.23 In animal models, these cells promote tumor-cell invasion by secreting matrix metalloproteinases such as MMP9 (Fig. 4B), suppress T-cell immune responses, and initiate angiogenesis.
The stromal-2 signature may be an “angiogenic switch” in which the progression of a hyperplastic lesion to a fully malignant tumor is accompanied by new blood-vessel formation.24 We observed that diffuse large-B-cell lymphomas with high relative expression of the stromal-2 signature were associated with increased tumor blood-vessel density and an adverse outcome. Given the complex interplay of cells and cytokines that regulate neoangiogenesis in tumors,23 an understanding of the mechanism of angiogenesis in diffuse large-B-cell lymphoma must await the development of animal models that recapitulate the stromal biology of the human tumors that is revealed by the stromal-1 and stromal-2 signatures. Nonetheless, some aspects of the stromal phenotypes of diffuse large-B-cell lymphoma suggest angiogenic mechanisms. First, the macrophage infiltration in some diffuse large-B-cell lymphomas may confer a predisposition to angiogenesis, since in experimental models, tumor-associated macrophages accumulate before the angiogenic switch and are required for the switch to occur.25 Second, CXC chemokine ligand 12 (CXCL12) (also called stromal-cell–derived factor 1, or SDF-1), a stromal-2 signature component, is a chemokine secreted by either fibroblasts or endothelial cells that can promote angiogenesis by recruiting CXCR4+ endothelial precursor cells from the bone marrow.26 Third, an antagonist of angiogenesis, thrombospondin-2,27 is a stromal-1 signature component, which may explain why tumors with low relative expression of this signature had an elevated blood-vessel density. Finally, the expression of adipocyteassociated genes in diffuse large-B-cell lymphomas with high stromal-2 signature expression may play a role in angiogenesis, since some cells in adipose tissue may have the potential to differentiate into endothelial cells.28 Alternatively, the expression of adipose-associated genes may reflect the recruitment of bone marrow–derived mesenchymal stem cells, which home efficiently to tumors29 and can stabilize newly formed blood vessels.30
The biologic insights gained from our analysis provide a new perspective on current and future clinical trials in diffuse large-B-cell lymphoma. The monoclonal antibody to VEGF, bevacizumab, is currently being investigated in several phase 2 and phase 3 clinical trials involving patients with diffuse large-B-cell lymphoma.31 On the basis of our results, it is possible that only a subgroup of such patients — those with diffuse large-B-cell lymphoma characterized by high relative expression of the stromal-2 signature and increased tumor blood-vessel density — may benefit from this angiogenesis inhibitor. Given the proangiogenic function of SDF-1, small-molecule inhibitors of its receptor, CXCR4, may have activity in diffuse large-B-cell lymphoma.32 The heavy infiltration of some diffuse large-B-cell lymphomas with myeloid-lineage cells raises the possibility that monoclonal antibodies targeting antigens on the myeloid-lineage cells could interfere with trophic interactions between these cells and malignant cells. Antibodies to CTGF have shown activity in preclinical models of cancer33 and might interfere with microenvironmental interactions in diffuse large-B-cell lymphoma. Ultimately, combined treatments that target oncogenic mechanisms in the malignant cell as well as interactions in the tumor microenvironment may prove to be synergistic.
Supplementary Material
Acknowledgments
Supported by grants from the Intramural Research Program of the National Institutes of Health, the National Cancer Institute (NCI), and the Center for Cancer Research; an NCI Strategic Partnering to Evaluate Cancer Signature grant (UO1-CA 114778); and a grant from the German Research Foundation (to Dr. Lenz).
Dr. Vose reports receiving grant support from Genentech; Dr. Greiner, lecture fees from Imedex Future; Dr. Armitage, consulting fees from Ziopharm, L’Oreal, Celgene, Genitope, and Biogen Idec and lecture fees from Genentech; Dr. Gascoyne, consulting fees from Genentech and Roche Canada and lecture fees and grant support from Roche Canada; Dr. Connors, grant support from Roche Canada; Dr. Montserrat, consulting fees from Schering and Hoffmann–La Roche; Dr. Miller, consulting fees and grant support from Genentech and Biogen Idec; Dr. Rimsza, grant support from Ventana Medical Systems; Dr. Cook, consulting fees from Roche Molecular Systems; Dr. Pohlman, consulting fees from Genentech BioOncology and grant support from Genentech BioOncology National Lymphocare Study; Dr. Fisher, consulting fees from Allos Therapeutics, Millennium, Amgen, Pharmion, Celgene, Roche, Genentech, and Seattle Genetics; Dr. Lister, consulting fees from Imedex, Eleos, Educational Concepts, Genentech, and Upside Endeavors and grant support from Millennium; and Dr. Chan, grant support from Roche Molecular Systems. No other potential conflict of interest relevant to this article was reported.
We thank Alexander Kohlmann, Mickey Williams, and Lothar Wieczorek at Roche Molecular Systems for logistical support.
Contributor Information
G. Lenz, Metabolism Branch, National Cancer Institute
G. Wright, Biometric Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute
S.S. Dave, Metabolism Branch, National Cancer Institute
W. Xiao, Bioinformatics and Molecular Analysis Section, Computational Bioscience and Engineering Laboratory, Center for Information Technology — all at the National Institutes of Health, Bethesda, MD
J. Powell, Bioinformatics and Molecular Analysis Section, Bethesda, MD
H. Zhao, Metabolism Branch, National Cancer Institute
W. Xu, Metabolism Branch, National Cancer Institute
B. Tan, Metabolism Branch, National Cancer Institute
N. Goldschmidt, Metabolism Branch, National Cancer Institute
J. Iqbal, University of Nebraska Medical Center, Omaha
J. Vose, University of Nebraska Medical Center, Omaha
M. Bast, University of Nebraska Medical Center, Omaha
K. Fu, University of Nebraska Medical Center, Omaha
D.D. Weisenburger, University of Nebraska Medical Center, Omaha
T.C. Greiner, University of Nebraska Medical Center, Omaha
J.O. Armitage, University of Nebraska Medical Center, Omaha
A. Kyle, British Columbia Cancer Agency, Vancouver, Canada
L. May, British Columbia Cancer Agency, Vancouver, Canada
R.D. Gascoyne, British Columbia Cancer Agency, Vancouver, Canada
J.M. Connors, British Columbia Cancer Agency, Vancouver, Canada
G. Troen, Pathology Clinic, Rikshospitalet University Hospital
H. Holte, Cancer Clinic, Rikshospitalet University Hospital
S. Kvaloy, Cancer Clinic, Rikshospitalet University Hospital
D. Dierickx, University of Leuven, Department of Hematology, Leuven, Belgium
G. Verhoef, University of Leuven, Department of Hematology, Leuven, Belgium
J. Delabie, Pathology Clinic, Rikshospitalet University Hospital
E.B. Smeland, Institute for Cancer Research, Rikshospitalet University Hospital Center for Cancer Biomedicine, Faculty Division of the Norwegian Radium Hospital, University of Oslo — both in Oslo.
P. Jares, Hospital Clinic, University of Barcelona, Barcelona
A. Martinez, Hospital Clinic, University of Barcelona, Barcelona
A. Lopez-Guillermo, Hospital Clinic, University of Barcelona, Barcelona
E. Montserrat, Hospital Clinic, University of Barcelona, Barcelona
E. Campo, Hospital Clinic, University of Barcelona, Barcelona
R.M. Braziel, Oregon Health and Science University, Portland Southwest Oncology Group — both in Tucson.
T.P. Miller, Southwest Oncology Group — both in Tucson University of Arizona Cancer Center — both in Tucson.
L.M. Rimsza, Southwest Oncology Group — both in Tucson University of Arizona Cancer Center — both in Tucson.
J.R. Cook, Southwest Oncology Group — both in Tucson Cleveland Clinic Pathology and Laboratory Medicine Institute — both in Cleveland.
B. Pohlman, Cleveland Clinic Taussig Cancer Institute — both in Cleveland
J. Sweetenham, Cleveland Clinic Taussig Cancer Institute — both in Cleveland
R.R. Tubbs, Southwest Oncology Group — both in Tucson Cleveland Clinic Pathology and Laboratory Medicine Institute — both in Cleveland.
R.I. Fisher, Southwest Oncology Group — both in Tucson James P. Wilmot Cancer Center, University of Rochester School of Medicine, Rochester, NY.
E. Hartmann, Department of Pathology, University of Würzburg, Würzburg, Germany
A. Rosenwald, Department of Pathology, University of Würzburg, Würzburg, Germany
G. Ott, Department of Pathology, University of Würzburg, Würzburg, Germany Department of Clinical Pathology, Robert-Bosch-Krankenhaus, Stuttgart, Germany.
H.-K. Muller-Hermelink, Department of Pathology, University of Würzburg, Würzburg, Germany
D. Wrench, Cancer Research UK, St. Bartholomew’s Hospital, London.
T.A. Lister, Cancer Research UK, St. Bartholomew’s Hospital, London.
E.S. Jaffe, Laboratory of Pathology, Center for Cancer Research, Computational Bioscience and Engineering Laboratory, Center for Information Technology — all at the National Institutes of Health, Bethesda, MD
W.H. Wilson, Metabolism Branch, National Cancer Institute
W.C. Chan, University of Nebraska Medical Center, Omaha
L.M. Staudt, Metabolism Branch, National Cancer Institute
REFERENCES
- 1.Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6. [DOI] [PubMed] [Google Scholar]
- 2.Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42. [DOI] [PubMed] [Google Scholar]
- 3.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol 2005;87:163–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 5.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 6.Bea S, Zettl A, Wright G, et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005;106: 3183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tagawa H, Suguro M, Tsuzuki S, et al. Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood 2005;106: 1770–7. [Erratum, Blood 2006;107:3052.] [DOI] [PubMed] [Google Scholar]
- 8.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2001;194:1861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ngo VN, Davis RE, Lamy L, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006;441:106–10. [DOI] [PubMed] [Google Scholar]
- 10.Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319:1676–9. [DOI] [PubMed] [Google Scholar]
- 11.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A 2003;100:9991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rimsza LM, Roberts RA, Miller TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 2004;103:4251–8. [DOI] [PubMed] [Google Scholar]
- 13.Hummel M, Bentink S, Berger H, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 2006;354:2419–30. [DOI] [PubMed] [Google Scholar]
- 14.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumorinfiltrating immune cells. N Engl J Med 2004;351:2159–69. [DOI] [PubMed] [Google Scholar]
- 15.The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med 1993;329:987–94. [DOI] [PubMed] [Google Scholar]
- 16.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996;107:404–11. [DOI] [PubMed] [Google Scholar]
- 17.Shaffer AL, Wright G, Yang L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev 2006;210:67–85. [DOI] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 2004;101:6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol 2003;23:1161–8. [DOI] [PubMed] [Google Scholar]
- 21.Woodman SE, Ashton AW, Schubert W, et al. Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol 2003;162: 2059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev 2008;19:133–44. [DOI] [PubMed] [Google Scholar]
- 23.Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev 2008;22:559–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996;86: 353–64. [DOI] [PubMed] [Google Scholar]
- 25.Lin EY, Li JF, Gnatovskiy L, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 2006;66:11238–46. [DOI] [PubMed] [Google Scholar]
- 26.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121: 335–48. [DOI] [PubMed] [Google Scholar]
- 27.Kazerounian S, Yee KO, Lawler J. Thrombospondins in cancer. Cell Mol Life Sci 2008;65:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Planat-Benard V, Silvestre JS, Cousin B, et al. Plasticity of human adipose lineage cells toward endothelial cells: physiological and therapeutic perspectives. Circulation 2004;109:656–63. [DOI] [PubMed] [Google Scholar]
- 29.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007;449:557–63. [DOI] [PubMed] [Google Scholar]
- 30.Au P, Tam J, Fukumura D, Jain RK. Bone marrow-derived mesenchymal stem cells facilitate engineering of long-lasting functional vasculature. Blood 2008;111: 4551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganjoo KN, An CS, Robertson MJ, et al. Rituximab, bevacizumab and CHOP (RA-CHOP) in untreated diffuse large B-cell lymphoma: safety, biomarker and pharmacokinetic analysis. Leuk Lymphoma 2006;47:998–1005. [DOI] [PubMed] [Google Scholar]
- 32.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 2007;28:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther 2006;5:1108–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.