

Abstract
A prerequisite to defining the transcriptome-wide functions of RNA modifications is the ability to accurately determine their location. Here, we present N4-acetylcytidine (ac4C) sequencing (ac4C-seq), a protocol for the quantitative single-nucleotide resolution mapping of cytidine acetylation in RNA. This method exploits the kinetically facile chemical reaction of ac4C with sodium cyanoborohydride under acidic conditions to form a reduced nucleobase. RNA is then fragmented, ligated to an adapter at its 3′ end and reverse transcribed to introduce a non-cognate nucleotide at reduced ac4C sites. After adapter ligation, library preparation and high-throughput sequencing, a bioinformatic pipeline enables identification of ac4C positions on the basis of the presence of C→T misincorporations in reduced samples but not in controls. Unlike antibody-based approaches, ac4C-seq identifies specific ac4C residues and reports on their level of modification. The ac4C-seq library preparation protocol can be completed in ~4 d for transcriptome-wide sequencing.
Introduction
N4-acetylcytidine (ac4C) is a modified RNA nucleobase that was first discovered in the 1960s during early efforts to characterize eukaryotic tRNAs1. Subsequent studies found that this RNA modification is conserved in all domains of life2,3, raising the provocative question of its function. In many bacteria, ac4C occurs at the wobble base of elongator tRNAMet and encourages the selective utilization of this tRNA, increasing translational fidelity4. In archaea, ac4C’s rigidified structure was hypothesized to increase the stability of RNA to thermal denaturation as early as the 1990s3, a supposition borne out by recent studies5. In eukaryotes, where ac4C is found in tRNALeu and tRNASer, the combined disruption of ac4C and another tRNA modification (N7-methylguanosine; m7G) destabilizes tRNASer in mutant yeast strains, inhibiting growth6,7. Building on these seminal studies, a breakthrough came in late 2014 and early 2015, when multiple groups concurrently reported that eukaryotic ac4C formation is catalyzed by an orphan GCN5-related N-acetyltransferase enzyme known as Nat10 (Kre33 in yeast)8–10. To direct its activity toward specific targets, Nat10 interacts with an additional adaptor protein—Thumpd1 in humans and Tan1 in yeast—which is required for modification of tRNALeu and tRNASer 6,10. Alternatively, specialized small nucleolar RNAs (snoRNAs) guide Nat10’s acetylation of eukaryotic rRNA11. The unique biochemistry of Nat10/Kre33, together with the facts that cytidine acetyltransferase genes are highly conserved12, essential in eukaryotes13,14 and associated with several diseases including premature aging and cancer15,16, has catalyzed the development of new methods to study ac4C. Here, we describe a detailed protocol for ac4C-seq, a high-throughput sequencing method that enables the quantitative base resolution determination of the location, abundance and dynamics of cytidine acetylation in RNA.
Development of the protocol
Cytidine acetylation does not disrupt canonical base pairing and is therefore undetectable in conventional RNA sequencing protocols17. However, ac4C has two unique chemical properties that together enable its sequencing-based detection. First, N4-acetylation withdraws electron density from cytidine’s pyridine ring, increasing the susceptibility of the modified nucleobase to reduction by hydride donors18. The addition of two equivalents of sodium cyanoborohydride to ac4C results in the formation of a reduced nucleobase, tetrahydro-N4-acetylcytidine (here termed ‘reduced ac4C’). Reduced ac4C can be misread as a ‘U’ rather than a ‘C’ during reverse transcription (RT), causing C→T mutations at sites of ac4C to be observed upon cDNA sequencing19 (Fig. 1). One limitation of this approach is that in addition to ac4C, several other modified nucleobases (m7G, dihydrouridine and N3-methylcytidine) are also susceptible to reduction by hydride donors, meaning the presence of a hydride-dependent misincorporation alone is insufficient to specify a site of cytidine acetylation. To address this, a second distinct chemical feature of ac4C, its hydrolytic lability, can be exploited to chemically deacetylate RNA for control experiments20. Together, these reactions establish a unique chemical signature for ac4C that consists of (i) increased C→T conversion upon reduction by hydride donors, which is (ii) abrogated by pre-treating RNA with mild alkali. Crucial elements of this approach that have been optimized for sensitive and quantitative ac4C detection include reduction chemistry, deacetylation reaction, genetic controls, reverse transcriptase, library preparation and analytical pipeline, all of which are described in detail under Experimental design (Figs. 2 and 3).
Fig. 1 |. Overview of nucleobase reaction chemistry underlying ac4C-seq.
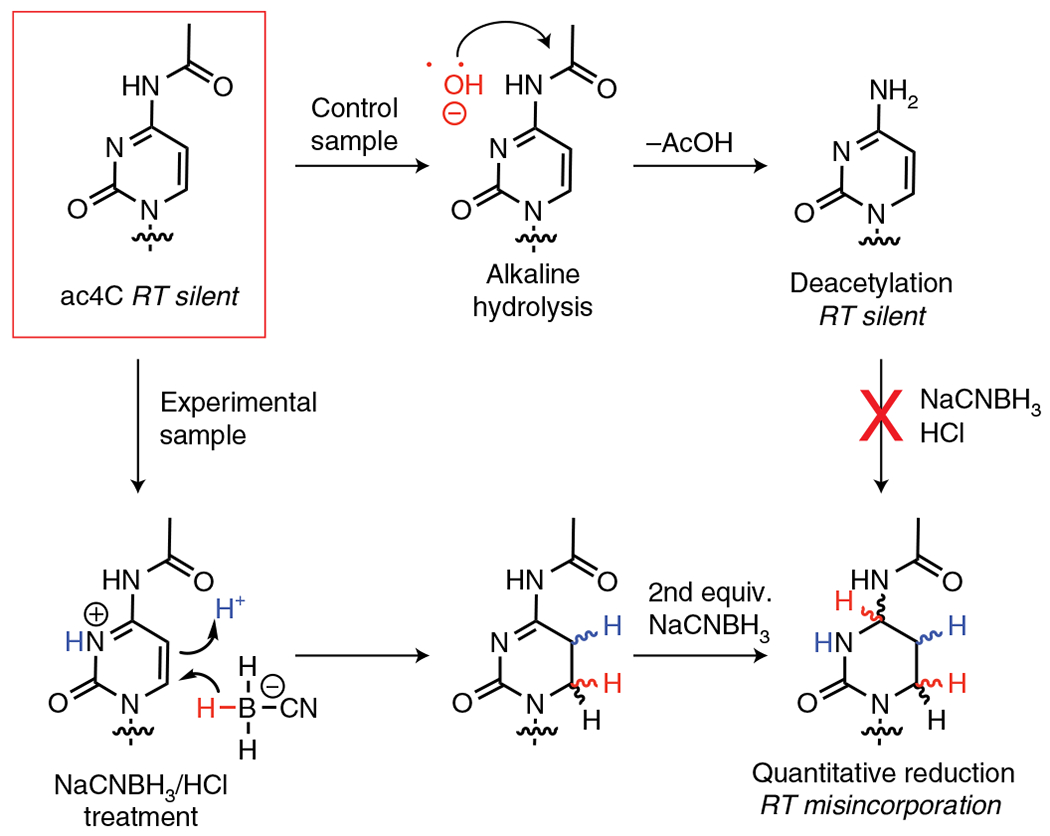
Endogenous ac4C forms base pairs identical to cytidine, rendering it silent in RT and cDNA sequencing experiments (top left). Treatment of ac4C-containing RNA with NaCNBH3 under acidic conditions results in formation of a reduced nucleobase, which is read as a ‘U’ during RT and results in misincorporations that can be detected by cDNA sequencing (bottom right). This chemistry is blocked when ac4C is chemically deacetylated by alkali hydrolysis (top right), which provides a control for specific detection of the acetylated nucleobase. equiv., equivalent.
Fig. 2 |. Experimental workflow for ac4C-seq protocol.
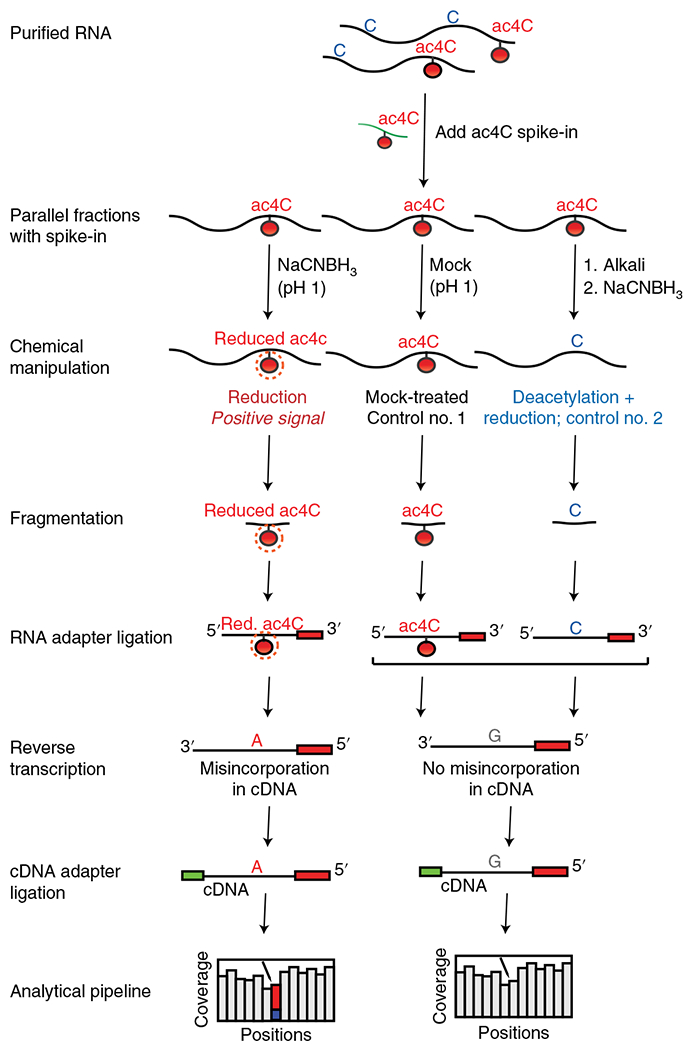
RNA is spiked with a synthetic ac4C RNA spike-in and split into three samples. One experimental sample is treated with NaCNBH3 under acidic conditions (reduction, positive signal), while two control samples are subjected to acidic conditions without reducing agent (mock-treated, control no. 1) and deacetylation followed by NaCNBH3 treatment (deacetylated + reduction, control no. 2). A 3′ adapter is ligated onto fragmented RNA, which is then reverse transcribed, resulting in misincorporation of ‘A’ in cDNA at positions of reduced ac4C. A 3′ adaptor is ligated to facilitate cDNA library construction, with subsequent sequencing and bioinformatic analysis being used to identify ac4C-modified sites. Figure adapted with permission from ref. 5. Red., reduced.
Fig. 3 |. Flowchart illustrating steps involved in ac4C-seq.
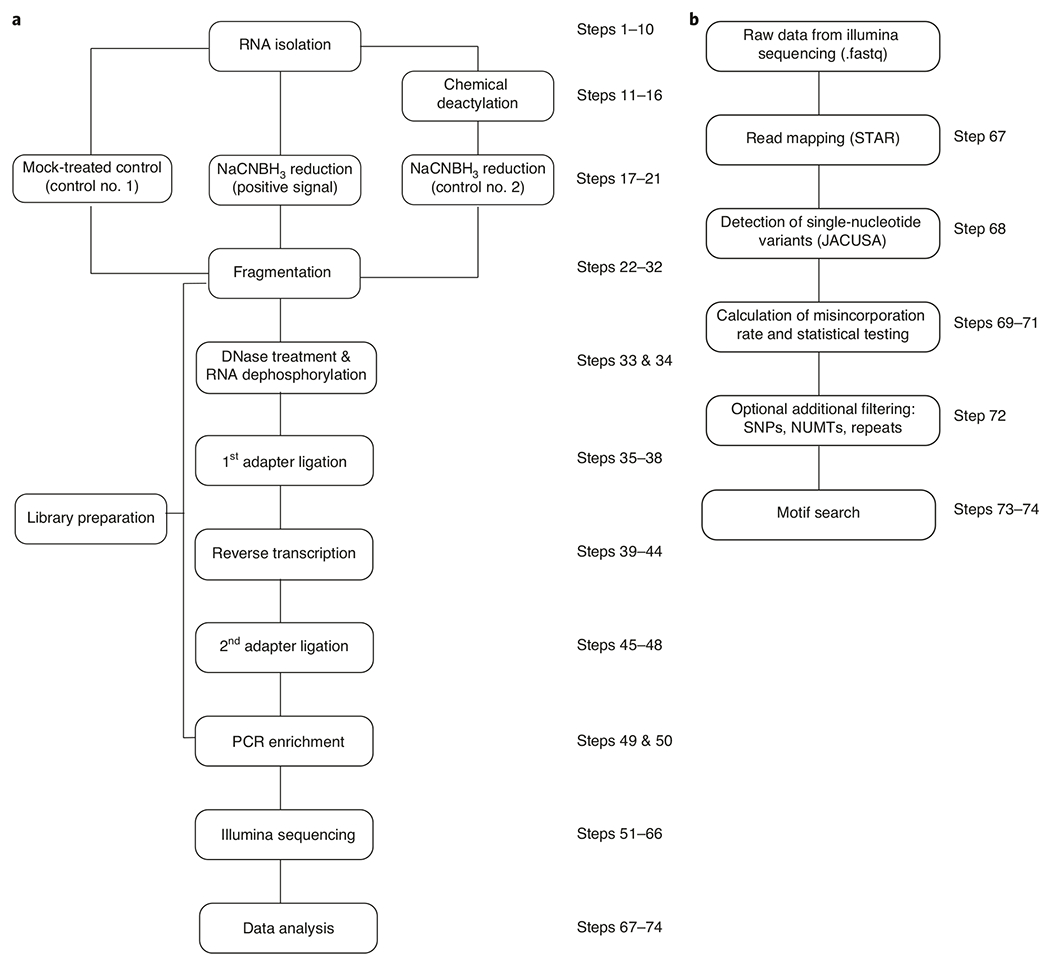
a, First, RNA is isolated, followed by chemical treatment to prepare experimental and control samples. These RNA samples are used to prepare cDNA libraries that undergo next-generation sequencing and are subsequently analyzed by using computational pipelines. b, Data analysis includes mapping of sequenced reads to the appropriate genome, calculating misincorporation rate in treatment and control samples and conducting statistical tests to identify ac4C sites. After additional filtering, motif analysis is applied to strengthen confidence in newly identified candidate ac4C-modified sites. NUMT, nuclear mitochondrial DNA; SNP, single-nucleotide polymorphism.
Applications of the method
In our initial studies, we have applied ac4C-seq to profile cytidine acetylation within human cell lines, fungal model organisms and archaeal extremophiles5. In eukaryotes, ac4C-seq enabled the de novo annotation of all four previously known sites of ac4C in rRNA and tRNA, without requiring a priori knowledge of their location. The quantitative nature of the method also validated these rRNA and tRNA residues as the major targets of Nat10/Kre33-catalyzed cytidine acetylation in eukaryotes, suggesting that additional ac4C sites are either absent or present at very low occupancies under physiological conditions. In addition to providing information on endogenous biology, ac4C-seq can be used to study the biochemistry of Nat10 RNA acetyltransferases. As one example, ac4C-seq has been used to monitor the ability of co-overexpressed human Nat10 and Thumpd1 to acetylate hundreds of mRNA transcripts, providing new insights into the sequence and structural determinants required for productive enzyme–substrate interaction5. The quantitative nature of ac4C-seq has also been powerfully used to study RNA acetylation dynamics within archaeal hyperthermophiles. Here, application of ac4C-seq to archaea of the order Thermococcales led to the discovery of a widespread, temperature-inducible program of cytidine acetylation in rRNA, tRNA and mRNA. This provides a model biological system for studying the physical and phenotypic consequences of cytidine acetylation, aspects of which may be conserved across evolution.
With regard to future applications, it is important to note that in eukaryotes, transcriptome-wide methods may not be necessary to study the dominant ac4C sites found in rRNA and tRNA, although we anticipate that analysis of the dynamics of these sites will benefit from quantitative sequencing. Instead, in these systems, ac4C-seq may find its most powerful application in discovering and validating ac4C sites in viral genomes (evidence for which has been recently reported)21, exploring whether ac4C occurs within mRNA at low stoichiometries in settings marked by Nat10 overexpression such as cancer and studying how ac4C is targeted by protein and snoRNA adapters22–24. In addition, it may be possible to integrate ac4C-seq with other hydride-based sequencing methods to simultaneously profile additional modified nucleobases including m7G25,26, further expanding the scope of this method. Overall, these applications highlight the potential for ac4C-seq to facilitate the high-resolution study of cytidine acetylation, enabling interrogation of its functional role in fundamental biology and disease.
Comparison with other methods
Liquid chromatography coupled to mass spectrometry (LC-MS)
LC-MS has been powerfully applied to study ac4C by several groups, most notably Suzuki and coworkers, who used LC-MS analysis of partially digested tRNA and rRNA fragments as a primary assay to identify ac4C sites and the enzymes responsible for cytidine acetylation in bacteria, yeast and human cell lines8,9,27. In a related method, Sharma et al. used a mung bean nuclease protection assay coupled to HPLC with UV detection to identify ac4C within helix 34 of eukaryotic rRNA, as well as to identify the yeast snoRNAs responsible for guiding rRNA acetylation10,11. Although these methods provide validated methods for discovery, they each require significant technical expertise and suffer from sensitivity limitations due to their lack of signal amplification. This latter limitation may hinder the detection and quantification of ac4C in low-abundance species (i.e., mRNAs) or within RNAs that are only partially modified.
Affinity-based methods
Antibody-based methods have been used to detect ac4C-containing RNAs via dot blot and Northern blotting5,20, as well as to enrich and identify candidate ac4C-containing mRNAs in both human and viral transcriptomes via immunoprecipitation and sequencing21,28. These methods have the advantage of signal amplification, in the forms of chemiluminescent detection for the immunoblotting detection protocols and enrichment for immunoprecipitation-based detection. A major limitation of antibody-based methods is that they cannot detect ac4C at nucleotide resolution or quantify its abundance at putative modification sites. Another limitation of RNA modification immunoprecipitation-sequencing methods is their susceptibility to false positives, which can be caused by even the slightest degree of antibody promiscuity when attempting to enrich a low-abundance modification from an enormous transcriptomic background29–31.
Single-molecule sequencing
A third and emerging method for the analysis of ac4C is the use of single-molecule sequencing technologies32. In one such approach, RNA molecules are pushed by a motor protein through a membrane-bound pore of an Oxford Nanopore Technologies sequencer. This causes a change in the transduction of electric current through the nanopore in a manner that is sequence dependent and also can reflect the presence of minor nucleobases, such as ac4C, that produce basecalling errors at modified sites. Very recently, Grunberger and coworkers applied this technology to the rRNA of archaeal hyperthermophiles and were able to profile 34 sites of cytidine acetylation, many of which have been validated by ac4C-seq5,33. Key advantages of single-molecule platforms are their potential utility in interrogating dense epitranscriptomic landscapes by detecting multiple modifications simultaneously across the entire length of a transcript and the avoidance of biases introduced by RT and PCR amplification during conventional RNA-seq workflows. Current limitations are their relatively low sensitivity, specificity and throughput and challenging analysis of small RNAs. The continued development of these platforms will probably facilitate additional creative applications in the study of ac4C and other RNA modifications.
Experimental design
Preparation of synthetic ac4C spike-in RNA
A valuable element of ac4C-seq is the use of synthetic RNA spike-ins that contain ac4C at a defined position as a control for assessing modified RNA detection5. Because synthetic ac4C RNAs cannot be produced by standard phosphoramidite RNA synthesis, spike-ins are prepared via standard in vitro transcription. These reactions use a T7 RNA polymerase template that encodes a single ‘C’ residue in an AUG-rich sequence and substitutes N4-acetylcytidine triphosphate in the place of cytidine triphosphate19. Preparation of four synthetic RNA spike-ins that contain a single ac4C site in a defined sequence context (ACA, GCA, UCA and ACU) as well as four corresponding cytidine-containing RNAs allows the preparation of a spike-in mixture in which ac4C is present at a defined stoichiometry (10%, 40%, 60% and 100%) within four different sequences, allowing the sensitivity of ac4C detection to be assessed. Because ac4C is a hydrolytically labile RNA modification, synthetic spike-ins should be routinely monitored for degradation via cyanoborohydride-dependent misincorporation assay (see ac4C reduction and mock treatment). Hydrolysis of ac4C within the spike-in would be expected to reduce C→T misincorporations at the known ac4C site. In practice, we have found that aqueous solutions of the synthetic ac4C spike-in RNA mixture are stable for months when stored at −80 °C.
RNA isolation and deacetylation control reaction
RNA to be analyzed by ac4C-seq is prepared from biological samples by standard Trizol extraction and split into three equal fractions, with an aliquot of synthetic ac4C spike-in RNA mixture added to each. One of these fractions is subjected to chemical deacetylation by treatment with mild alkali and is referred to as the ‘chemical deacetylation control’ throughout this protocol. This fraction provides an important specificity control for ac4C detection, because hydrolysis of the N4-acetyl group decreases cyanoborohydride-dependent C→T misincorporations at ac4C sites while leaving other hydride-reducible modifications unaffected. Our group has evaluated several reagents for chemical deacetylation of ac4C20 and favors alkali hydrolysis because of the lack of competing side reactions and known compatibility with next-generation sequencing library preparation. One consideration with chemical deacetylation is that even short exposure to alkali conditions can cause fragmentation of RNA; thus, it is important not to prolong the deacetylation step beyond the recommended duration. Fragmentation in general and in the deacetylation step will affect the final size of the cDNA library and hence will affect the calculation of final dilutions before Illumina sequencing.
ac4C reduction and mock treatment
After addition of spike-ins, RNA is treated with sodium cyanoborohydride in the presence of strong acid to cause the reduction of ac4C-modified sites. This treatment is applied to two fractions (native RNA and the chemical deacetylation control), which are referred to below as the ‘treated’ or ‘reduced’ samples. The remaining (third) RNA sample is subjected to a mock treatment with only strong acid but no reducing agent and serves as the ‘non-treated’ input control. One consideration in choosing an appropriate reduction chemistry is the relative rate of ac4C reduction as compared to ac4C hydrolysis, because the latter reaction erases the signal for cytidine acetylation. As with the chemical deacetylation reaction, our group has evaluated several reactions for the chemical reduction of ac4C and favors our recently reported use of sodium cyanoborohydride in acidic (pH 1) conditions. In model reactions, we have observed that the reduction of ac4C under these conditions proceeds approximately 250-fold faster than an acid-catalyzed hydrolysis reaction5, providing it with excellent signal to noise when integrated with next-generation sequencing workflows.
Choice of reverse transcriptase
Because ac4C-seq detection of cytidine acetylation sites depends on analysis of chemically induced C→T misincorporation sites, the choice of reverse transcriptase is essential34. Different polymerases exhibit different abilities to read through the reduced nucleobase. For locus-specific detection of ac4C sites in rRNA, our group has used the genetically engineered Moloney Murine Leukemia Virus SuperScript III, HIV reverse transcriptase and Type Group II Intron Reverse Transcriptase (TGIRT-III), all of which demonstrate a mixture of read-through and stop activity when transcribing ac4C sites19. For next-generation sequencing analysis of ac4C, we have primarily used TGIRT-III, based on its excellent compatibility with library preparation and misincorporation-based RNA modification detection5,29, but have also obtained highly comparable results by using Moloney Murine Leukemia Virus SuperScript III.
Genetic controls
In addition to spike-ins, which provide a control for monitoring quantitative ac4C reduction, and the deacetylation fraction, which provides evidence that a putative modified site shares the known chemical properties of ac4C (alkaline lability), the use of organismal models in which cytidine acetyltransferase enzymes have been inactivated provides a powerful genetic control for ac4C detection. Almost all known eukaryotic organisms contain a single RNA acetyltransferase gene, which in many cases has been found to be essential. However, in humans it has been shown that genetic targeting of a site that allows only a minor splice isoform to be produced can result in the generation of hypomorphic cell lines in which cytidine acetyltransferases are present at very low levels21,28. Alternatively, in yeast it has been shown that mutation of the acetyltransferase domain is not lethal and can be used to produce RNA that lacks acetylation10. In archaea and bacteria, knockout of cytidine acetyltransferase enzymes is tolerated and results in growth defects only under specific conditions5,27,35. In addition to loss-of-function genetic controls, gain-of-function genetic controls can be used as well. For example, overexpression of Nat10 and Thumpd1 in human cells causes supraphysiological acetylation of hundreds of mRNA sites, which can be attributed directly to the activity of ectopically overexpressed proteins5.
Library preparation and sequencing
The treated RNA samples are used for high-throughput sequencing library construction by using the ac4C-seq protocol detailed in Steps 22–66 of the Procedure below. In short, RNA is first fragmented to produce ~200-bp-long fragments, followed by ligation of an RNA adapter, which is used to prime RT. RT is then performed by using the TGIRT-III enzyme, which results in C→T misincorporation at the position of a reduced ac4C. A second adapter is then ligated to the 3′ end of the single-stranded cDNA product. Importantly, both the RNA and cDNA adapters used are identical across all libraries, minimizing biases. Barcoded primers are then used to amplify the sequencing library via PCR. Libraries are subsequently sequenced on Illumina NextSeq 500 or NovaSeq 6000 platforms, generating short paired-end reads ranging from 25 to 55 bp from each end. Although sequencing in single-end mode is possible, we advise using paired-end mode, which improves the accuracy of sequence alignment and allows better estimation of sample complexity. Of note, fragmentation is not needed when profiling short molecules, such as tRNAs. Although conducting the washing steps with 0.8 volumes of ethanol partially preserves short fragments, further adaptations can be made to the protocol to facilitate analysis of these transcripts.
Data analysis
Detection of ac4C sites is based on identification of a specific C→T misincorporation signature in reduced samples but not in the control samples, as detailed in Steps 67–74 of the Procedure below. In short, raw reads generated via Illumina sequencing are mapped to a custom genome by using the STAR (spliced transcripts alignment to a reference) aligner36, which allows for local alignment (e.g., by activating the ‘soft clipping’ option), followed by detection of single-nucleotide variants by using the JACUSA (JAVA framework for accurate SNV assessment) software in pileup mode37. The JACUSA output is then used to calculate the misincorporation rate of each position by dividing the number of non-cytidine–containing reads by the total number of reads covering that position. Higher misincorporations indicate a higher stoichiometry of ac4C at the given position. Binomial tests of significance can further be conducted to identify putative ac4C sites, whose misincorporation rate in the reduced sample is significantly higher than that of the control samples (chemical and/or genetic controls). The occurrence of the putative ac4C site within a CCG motif, where the middle cytidine is modified, further strengthens confidence in assigned sites because the CCG trinucleotide sequence motif was found to be necessary for targeting by Nat10 and its homologs in eukaryotes and archaea5. In addition, when relevant, putative sites should be assessed post hoc to ensure that they do not originate from misalignaments and misinterpretations related to complexity and redundancy of the genome (e.g., that they do not stem from alignment to repetitive elements, nuclear mitochondrial DNA (NUMTs), paralogs or single-nucleotide polymorphisms (SNPs))34.
Advantages and limitations
The advantages of ac4C-seq relative to related methods are its quantitative nature and nucleotide resolution, which enable monitoring ac4C dynamics and de novo discovery of novel ac4C sites. A limitation that ac4C-seq shares in common with all chemical reaction–based modification sequencing protocols is that its detection limit at any given site is dependent on the stoichiometry of ac4C, the efficiency of the ac4C reduction and the sequencing depth of that site34. Misincorporations caused by quantitative reduction of ac4C by using the protocol described here have been shown to scale linearly with LC-MS measurements of ac4C stoichiometry in single-stranded spike-in RNAs (see Preparation of synthetic ac4C spike-in RNA) and 27 sites of ac4C in eukaryotic and archaeal rRNA5. In addition, ac4C-seq has allowed detection of cytidine acetylation at >200 archaeal sites spread across tRNA, mRNAs and noncoding RNAs (ncRNAs) and within an additional 230 sites that can be induced in eukaryotic mRNA upon Nat10/Thumpd1 or Kre33/Tan1 overexpression5. These observations indicate that cyanoborohydride-mediated reduction of ac4C occurs efficiently across a broad range of RNA contexts to produce a signal that can be robustly detected, and suggest that the sensitivity of ac4C detection by ac4C-seq is likely to primarily depend on stoichiometry and sequencing depth. Deeper sequencing depth of low-stoichiometry sites may be achieved by targeted amplicon sequencing or, theoretically, through pre-enrichment of ac4C-containing RNAs by using commercially available anti-ac4C antibodies20, although the latter approach has not yet been integrated with ac4C-seq.
One limitation of ac4C-seq is that it relies exclusively on detection of C→T misincorporations to identify and quantify ac4C sites, which has the potential to cause an underestimation of modification abundance within RNAs where the reduced ac4C nucleobase causes RT stop events. Thus, the quantitative nature of ac4C-seq may be further improved by combined quantification of both misincorporation and RT stop events38. Although we have successfully applied this protocol to the study of ac4C in archaeal and eukaryotic tRNA, optimized quantitative analysis of tRNA acetylation may further benefit from pre-treatment of RNA with promiscuous demethylase enzymes, which have previously been shown to increase tRNA sequencing depth due to removal of methylated modifications that block RT at those sites39,40, and by replacing the fragmentation step with simple size selection, to limit degradation of these small RNAs.
Finally, it should be noted that the individual chemical treatments used in ac4C-seq can exhibit cross-reactivity with other modified nucleobases. For instance, the alkali conditions used to chemically deacetylate ac4C can also cause Dimroth rearrangements at N1-methyladenosine sites, and cyanoborohydride in strong acid was found to cause a C→T misincorporation at a 5-formylcytidine site in mitochondrial tRNA5. Although ac4C can be distinguished from these nucleobases through additional filters (cyanoborohydride reactivity, occurrence at a 5′-CCG-3′ consensus sequence and sensitivity to alkali treatment), the development of more specific chemical or enzymatic methods may further simplify such analyses.
Materials
Biological materials
Cells and tissues from any organism can be used to isolate RNA for ac4C-seq. Specifically, ac4C-seq has been successfully conducted on RNA from HeLa (CVCL_0030) and HEK-293T (CVCL_0063) human cells. We have also applied ac4C-seq to RNA isolated from Saccharomyces cerevisiae and from archaea of the following strains: Thermococcus kodakarensis strain TS559, Pyrococcus furiosus strain COM1, Thermococcus sp. AM4, Methanocaldococcus jannaschii and Saccharolobus solfataricus. Experiments shown in the Anticipated results section were carried out on HeLa and TS559 cells, as indicated in the figure legend. ! CAUTION Use aseptic techniques and practices to avoid contamination. Human cell lines used should be regularly checked to ensure that they are not infected with mycoplasma.
Reagents
TRIzol reagent (Invitrogen, cat. no. 15-596-018) ! CAUTION TRIzol reagent is a serious health hazard; use personal protective equipment when handling.
Chloroform (Sigma-Aldrich, cat. no. C2432) ! CAUTION Chloroform is a health hazard. Wear personal protective equipment when handling.
Isopropanol (Acros Organics, cat. no. 32727-0010) ! CAUTION Isopropanol is highly flammable; keep away from all sources of ignition.
Ethanol (Fisher Scientific, cat. no. BP2818) ! CAUTION Ethanol is highly flammable; keep away from all sources of ignition.
Nuclease-free ultra-pure water (Invitrogen, cat. no. 10977)
NaCNBH3 (Sigma-Aldrich, cat. no. 156159) ! CAUTION This is a flammable solid and a health hazard. Keep away from sources of ignition and any possible contact with water. Use personal protective equipment when handling, and work in a chemical hood.
Sodium bicarbonate (Sigma-Aldrich, cat. no. S6297)
Sodium acetate (3 M, pH 5.5; Invitrogen, cat. no. AM9740)
Hydrochloric acid solution, BioReagent, suitable for cell culture (1 N; Sigma, cat. no. H9892)
UltraPure 1 M Tris-HCI buffer, pH 8.0 (Invitrogen, cat. no. 15567027)
RNA fragmentation reagents (Ambion, cat. no. AM8740); includes fragmentation reagent and stop solution
Silane beads: Dynabeads MyOne Silane (Thermo Fisher Scientific, cat. no. 37002D)
RLT buffer (RNeasy lysis buffer) (Qiagen, cat. no. 79216)
T4 PNK (includes 10× PNK buffer containing 700 mM Tris-HCl pH 7.6, 100 mM MgCl2, 50 mM DTT; New England Biolabs, cat. no. M0201L)
FastAP thermosensitive alkaline phosphatase (includes 10× Fast AP buffer containing 100 mM Tris-HCl pH 8, 50 mM MgCl2, 1 M KCl, 0.2% (vol/vol) Triton X-100 and 1 mg/ml BSA; Thermo Fisher Scientific, cat. no. EF0654)
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, cat. no. D8418) ! CAUTION DMSO is a flammable liquid; keep away from all sources of ignition.
RNase inhibitor, murine (New England Biolabs, cat. no. M0314)
Turbo DNase (Life Technologies, cat. no. AM2238)
T4 RNA Ligase 1 (single-stranded RNA ligase) high concentration (includes 100 mM ATP, 50% (wt/vol) PEG 800 and 10× T4 RNA ligase reaction buffer; New England Biolabs, cat. no. M0437M)
TGIRT-III RT enzyme (Ingex)
Potassium chloride (Sigma-Aldrich, cat. no. P9333)
50 mM magnesium chloride solution (New England Biolabs, cat. no. B0510A)
dNTP mixture (10 mM solution; Jena Bioscience, cat. no. NU-1006L)
ExoSap-it PCR product cleanup reagent (Affymetrix, cat. no. 78201)
DTT (0.1 M; Invitrogen, cat. no. Y00147)
EDTA disodium salt solution for molecular biology (EDTA, 0.5 M in H2O; Sigma-Aldrich, cat. no. 7889)
Sodium hydroxide solution BioUltra, for molecular biology (NaOH, 10 M in H2O; Sigma, cat. no. 72068)
2× KAPA HiFi HotStart ReadyMix PCR kit (KAPA Biosystems)
2% (wt/vol) E-Gel EX agarose gel (Invitrogen, cat. no. G402002)
100-bp DNA ladder (New England Biolabs, cat. no. N0467S)
Ampure Beads: Ampure XP Agencourt (60 ml; Beckman Coulter, cat. no. A63881)
Qubit RNA HS assay kits (Invitrogen, cat. no. Q32852)
Qubit double-stranded DNA HS assay kit (Invitrogen, cat. no. Q32854)
High Sensitivity RNA ScreenTape analysis (Agilent, cat. no. 5067-5579)
High Sensitivity DNA ScreenTape analysis (Agilent, cat. no. 5067-5584)
RNA adaptor for first ligation: 5′-/5Phos/rArG rArUrC rGrGrA rArGrA rGrCrA rCrArC rGrUrC/3ddC/-3′ (Integrated DNA Technologies, custom order, standard desalting; dissolved in H2O to 10 μM and stored at −20 °C for ≤1 year)
DNA adaptor for second ligation: 5′-/5Phos/AG ATC GGA AGA GCG TCG TGT AG/3ddC/-3′ (Integrated DNA Technologies, custom order; dissolved in H2O to 100 μM and stored at −20 °C for ≤3 years)
Primer for RT: 5′-AGA CGT GTG CTC TTC CG-3′ (Integrated DNA Technologies, custom order; dissolved in H2O to 10 μM and stored at −20 °C for ≤3 years)
Primers for PCR (Integrated DNA Technologies, custom order; primers should be dissolved in H2O to 100 μM and stored at −20 °C for ≤3 years; The working dilution to be used at Step 49 is 25 μM). Full barcoded primer sequences are listed in ref. 41. Sequences of the Read 1 primer (2P_universal) and three possible barcoded Read 2 primers (2P_504, 2P_375 and 2p_630) are provided in Table 1.
Table 1 |.
PCR primers to be used in Step 49
| Name | Sequence (5′-3′) |
|---|---|
| Primer 2P_universal | AATGATACGGCGACCACCGAGATCTACACTCTTTCC CTACACGACGCTCTTCCGATCT |
| 2P_504 | CAAGCAGAAGACGGCATACGAGATCCTGGTAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2P_375 | CAAGCAGAAGACGGCATACGAGATTAAGCATGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2P_630 | CAAGCAGAAGACGGCATACGAGATAGATGTGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
Equipment
Microcentrifuge tubes (1.7 ml; Sorenson Bioscience, cat. no. 11700)
PCR tubes (0.2 ml, flat cap; Corning, cat. no. 3745) or PCR strips (0.2 ml, flat cap; Gunster MB-P08-B)
Refrigerated microcentrifuge (Eppendorf, model no. 5424)
Vortex mixer
Veriti 96-well thermal cycler (Applied Biosystems, cat. no. 4375786)
Agarose gel electrophoresis equipment
E-Gel Safe Imager E-Gel real-time transilluminator (Invitrogen, cat. no. G6500)
NanoDrop 2000 spectrophotometer (Thermo Scientific)
DynaMag-96 Side Magnet (Thermo Fisher Scientific, cat. no. 12331D)
Ice bucket/cold block
Sequencing system (Illumina, NextSeq 500 or NovaSeq 6000)
Desiccator for storing NaCNBH3 stock
Qubit 3.0 fluorometer (Invitrogen, Q33216)
4200 TapeStation system (Agilent, cat. no. G2991A)
Software and data
STAR (version 2.5.3a) for mapping short reads to call modification sites36
Samtools (v.0.1.19) for converting, sorting and indexing the sequence alignment map/binary alignment map (SAM/BAM) mapping result files42 (http://www.htslib.org/)
JACUSA for base calling from BAM files37
R package lme444
Reagent setup
1 M NaCNBH3
In a 1.7-ml tube, dissolve 10 mg of NaCNBH3 in 159.1 μl of H2O. The solution should be made up fresh each time. Keep the tube open to avoid accumulation of gases, which are released upon addition of H2O. Leftover NaCNBH3 dissolved in water should be quenched with an equal volume of 1 M HCl before disposal. ! CAUTION NaCNBH3 is a flammable solid that releases flammable and toxic gases upon reaction with water and/or HCl. Thus, all handling should be carried out in a chemical hood. Refer to the reagent’s safety data sheet for information on disposal.
100 mM sodium bicarbonate pH 9.5
Dissolve 8.401 mg of sodium bicarbonate in 800 μl of nuclease-free H2O. Adjust pH to 9.5 with 5 M NaOH. Bring the final volume to 1 ml with H2O. This solution can be stored at room temperature for ≤1 year.
FNK buffer
Mix 300 μl of 10× PNK buffer, 300 μl of 10× Fast AP buffer and 600 μl of water. This buffer can be stored at −20 °C for ≤1 year.
5× TGIRT-III buffer
Mix reagents to prepare the 5× TGIRT-III buffer with a final composition of 250 mM Tris-HCl (pH 8.3), 375 mM KCl and 15 mM MgCl2. This buffer can be stored at room temperature or −20 °C for ≤1 year.
Procedure
Total RNA isolation ● Timing 3 h
-
1Culture and harvest cells as instructed in option A (mammalian cells) or option B (bacteria, yeast or archaea).
- Mammalian cells
- Cells can be grown in monolayer in culture dishes or flasks to 80% confluency (e.g., 8–10 × 106 cells in a 10-cm2 dish). Remove growth medium, add 2–4 ml of cold PBS and harvest by scraping with a cell scraper. Pellet cells by centrifugation at 400g and 4 °C for 4 min, discard the medium and proceed to TRIzol extraction in Step 2. Alternatively, TRIzol can be directly added to the culture dish after removing the medium to lyse the cells.
- For cells that can be grown in suspension, aim to get 5–10 × 106 cells. Pellet cells by centrifugation at 400g and 4 °C for 4 min and discard the supernatant before proceeding to Step 2.
- Bacteria, yeast and archeal cells
- Cells can be grown in suspension to reach an OD600 of ~0.6–0.8 for bacteria or archaea or an OD600 of 1 for yeast. Pellet cells by centrifugation at 5,000g and 4 °C for 15 min and discard the medium.
-
2
Isolate total RNA from mammalian, yeast, bacterial or archaeal cells by using TRIzol reagent according to the manufacturer’s protocol45,46. Add TRIzol reagent (~1 ml of TRIzol per 5–10 × 106 mammalian or yeast cells or 1 × 107 bacterial or archaeal cells grown in suspension) directly to the cell pellet and ensure complete lysis by vortexing the suspension. Alternatively, hot acid phenol can be used to isolate RNA from yeast47.
-
3
After cell lysis, incubate the samples for 5 min at room temperature.
-
4
Add 0.2 ml of chloroform per 1 ml of TRIzol used and vortex for 15 s.
-
5
Incubate at room temperature for 3 min, and centrifuge at 12,000g and 4 °C for 15 min.
-
6
Transfer the colorless upper aqueous layer to a new tube.
▲ CRITICAL STEP The aqueous phase contains the RNA. Avoid transferring the bottom or the interface layer, which contain DNA.
-
7
To precipitate the RNA, add an equal volume of isopropyl alcohol to the aqueous layer. Mix by vortexing for 15 s, incubate at room temperature for 10 min and centrifuge at 12,000g and 4 °C for 10 min.
-
8
Discard the supernatant and vacuum- or air-dry the pellet for ≤10 min. Take care to avoid over-drying the RNA pellets.
-
9
Resuspend the pellet in 100 μl of nuclease-free water and quantify by using the NanoDrop 2000 spectrophotometer.
-
10
Check RNA size and quality by using an Agilent 4200 TapeStation system or an Agilent 2100 bioanalyzer or by running an aliquot on a 1% (wt/vol) agarose gel electrophoresis with 1× TBE buffer (stained with ethidium bromide).
■ PAUSE POINT Isolated RNA can be stored at −80 °C for ≤1 year.
? TROUBLESHOOTING
Chemical treatment of RNA ● Timing 6 h
▲ CRITICAL RNA from Step 10 is used for reductive treatment with NaCNBH3 as well as for two controls. We recommend starting with 1 μg of RNA per reaction; however, this amount can be as low as 0.2 μg. Synthetic ac4C spike-in RNA mixture can be added to RNA from Step 10 before proceeding to reactions.
▲ CRITICAL Set up three parallel reactions: (i) NaCNBH3 treated (+NaCNBH3), (ii) chemical deacetylation followed by NaCNBH3 treatment (+alkali +NaCNBH3) and (iii) mock-treated control (–NaCNBH3). RNA from Step 10 can be directly used for reactions A and C. For reaction B, RNA from Step 10 should be pre-treated with alkali as described in Steps 11–16.
-
11
Chemical deacetylation. Incubate 1 μg of RNA in 100 μl of 100 mM sodium bicarbonate pH 9.5 at 60 °C for 1 h.
▲ CRITICAL STEP Alkali treatment with sodium bicarbonate promotes substantial conversion (>80%) of ac4C to cytidine while also causing partial degradation of the RNA backbone, as assessed by bioanalyzer or TapeStation (Fig. 4).
-
12
Adjust the volume of the RNA reaction to 200 μl with nuclease-free H2O.
-
13
Add 0.1 volume of 3 M sodium acetate (pH 5.5) and 2.5 volumes of 100% ethanol to the sample. Mix thoroughly and keep at −20 °C for 1 h. Centrifuge at 18,000g and 4 °C for 30 min.
-
14
Carefully remove the supernatant and wash the pellet with 500 μl of ice-cold 70% (vol/vol) ethanol. Centrifuge for an additional 10 min at 18,000g and 4 °C.
-
15
Discard the supernatant without disturbing the pellet and vacuum- or air-dry the pellet for ≤10 min. Take care to avoid over-drying the RNA pellets.
-
16
Resuspend the pellet in nuclease-free water such that the RNA concentration will be >0.2 μg/μl and quantify RNA by using the NanoDrop 2000 spectrophotometer.
-
17Set up three 1.7-ml Eppendorf tubes per sample.
- For +NaCNBH3 RNA, mix 1 μg of RNA (from Step 10) in 80 μl of nuclease-free H2O and 10 μl of 1 M NaCNBH3.
- For +alkali +NaCNBH3 RNA, mix 1 μg of alkali-treated RNA (from Step 16) in 80 μl of nuclease-free H2O and 10 μl of 1 M NaCNBH3.
-
For –NaCNBH3 RNA, use 1 μg of RNA (from Step 10) in 90 μl of nuclease-free H2O.▲ CRITICAL STEP Prepare a fresh solution of NaCNBH3 immediately before using. For the untreated control, NaCNBH3 is replaced with 10 μl of nuclease-free water.
-
18
Add 10 μl of 1 M HCl to all three tubes, vortex briefly, spin down and start the timer.
▲ CRITICAL STEP HCl will initiate the reducing reactions, and hydrogen gas bubbles will be produced as the reaction progresses.
? TROUBLESHOOTING
-
19
Incubate the samples for 20 min at room temperature.
-
20
Add 30 μl of 1 M Tris-HCl (pH 8.0) to each sample to quench the reaction by neutralizing the pH.
-
21
Adjust the reaction volume to 200 μl by adding 70 μl of nuclease-free H2O and purify RNA by ethanol precipitation as described in Steps 13–16.
■ PAUSE POINT Treated RNA can be stored at −80 °C for ≤1 year.
Fig. 4 |. Quality control of RNA and cDNA libraries.
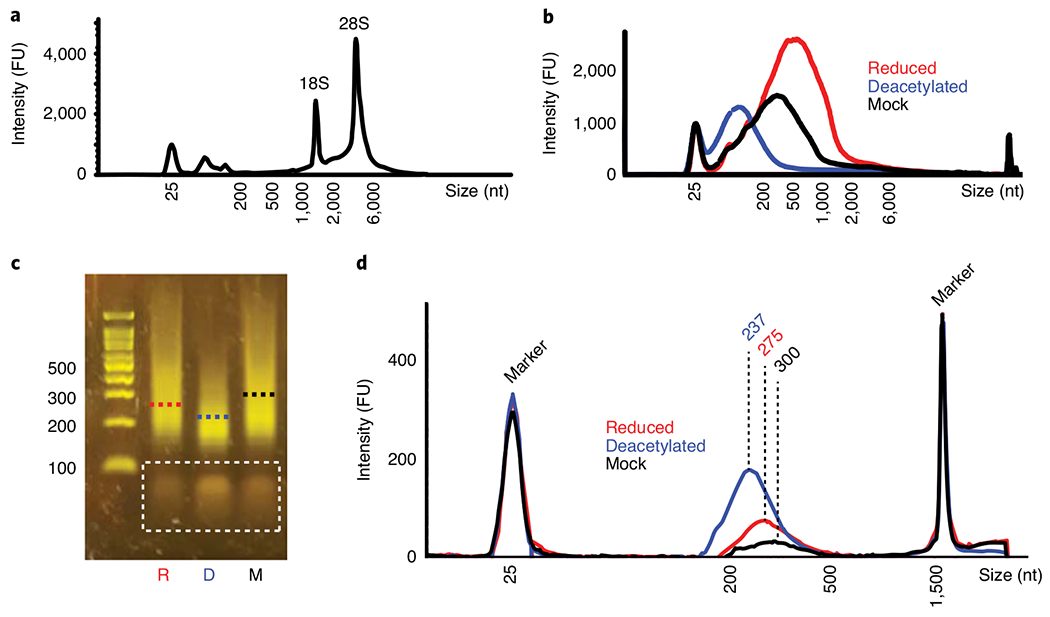
a and b, Typical TapeStation-generated trace of RNA from human HeLa cells after isolation (a, Step 10) or after treatment with NaCNBH3, alkali deacetylation followed by NaCNBH3 or mock treatment (b, Step 21). c and d, Typical cDNA libraries generated from RNA of T. kodakarensis, analyzed by using E-gel (c, Step 53) or TapeStation (d, Step 64). Dashed lines indicate the mean fragment size of each library. Bands in the dashed rectangle represent leftover primers from the PCR reaction (Step 50), which are eliminated upon the final washes on Ampure beads (Step 61). R, reduced; D, deacetylated; M, mock; FU, fluorescence units.
Preparation of cDNA libraries for high-throughput sequencing ● Timing 2 d
▲ CRITICAL The procedures described below entail repeated steps of sample washing, meant to clean the sample from the reagents of the current reaction and resuspend it in an appropriate volume for the next step. Cleanup includes binding the reaction mixture to pre-cleaned silane beads, washing the sample on the beads, air-drying the RNA (while avoiding over-drying the pellets) and eluting it from the beads to a new tube. Although the order of these stages is constant, volumes and elution buffers may vary. Thus, we describe the ‘cleanup’ process in detail in Steps 26–32 below and only mention variations of it in the following wash steps. Pre-cleaning of beads (Steps 26 and 27), when applicable, can be conducted in parallel to the reaction it is meant to follow, and thus save time for the user. Varying the concentration of ethanol at the binding stage (Step 29) is used to optimize retention of RNA/cDNA while disposing of unwanted oligonucleotide used as reagents in the reaction, because a higher concentration of ethanol will enable retention of smaller fragments. Thus, when applicable, it is advised to use either 0.8 volumes of 100% ethanol for the retentions of short fragments below ~200 nt (e.g., tRNAs) or 0.6 volumes of ethanol for longer fragments (e.g., rRNA and mRNA). Specific volumes are indicated when relevant.
RNA fragmentation ● Timing 30 min
-
22
In a 0.2-ml tube, take 50–200 ng of purified RNA from Step 21 and adjust the reaction volume to 18 μl by adding nuclease-free H2O.
-
23
Add 2 μl of RNA fragmentation reagent.
-
24
Heat at 70 °C for 2 min in a thermocycler.
▲ CRITICAL STEP Do not exceed the duration of incubation at 70 °C, because this may result in excessive fragmentation of the RNA.
-
25
Place the sample on ice or a cold block and add 2 μl of fragmentation stop solution.
-
26
To pre-clean silane beads, put 20 μl of beads into a new tube and place on a magnet. Remove the supernatant and add 50 μl of RLT buffer.
-
27
Remove the supernatant and resuspend beads in three times the sample volume (66 μl) of RLT buffer.
-
28
To wash the samples, add the 66 μl of beads to the fragmentation reaction tube from Step 25.
-
29
Add 125 μl of 100% ethanol, mix gently by pipetting up and down 10 times and incubate for 1 min. Place on a magnet and remove the supernatant.
-
30
Add 100 μl of 75% (vol/vol) ethanol, wash by transferring from side to side of the magnet and remove the supernatant.
-
31
Repeat the 75% (vol/vol) ethanol wash in Step 30. Air-dry the beads for 1 min.
-
32
Resuspend the sample in 16.5 μl of nuclease-free H2O, mix well, place on a magnet and elute by transferring 15.5 μl of the sample into a new tube.
DNase treatment and dephosphorylation ● Timing 45 min
-
33To the eluted sample, add the components as outlined in the table below and incubate at 37 °C for 30 min.
Component Amount (μl) Sample from Step 32 15.5 FNK buffer 6 RNase inhibitor 0.5 (20 U) Turbo DNase 1 (2 U) FastAP 3 (3 U) T4 PNK 4 (40 U) -
34
Conduct cleanup as in Steps 26–32 by pre-washing 20 μl of beads, except resuspend beads in 90 μl of RLT. To elute, add 7 μl of nuclease-free H2O to the beads, mix well, place on a magnet and elute by transferring 6 μl of the sample into a new tube.
First adapter ligation to the RNA ● Timing 2 h
-
35
Add 1 μl of 10 μM RNA adapter 5′-/5Phos/rArG rArUrC rGrGrA rArGrA rGrCrA rCrArC rGrUrC/3ddC/-3′ to the eluted RNA.
-
36
Incubate at 70 °C for 2 min and place directly on ice or a cold block.
-
37Combine the following components as outlined in the table below; incubate at 23 °C for 90 min.
Component Amount (μl) Sample from Step 36 7 10× T4 RNA ligase buffer 2 DMSO (100%) 1.8 ATP (100 mM) 0.2 PEG 8000 (50%, wt/vol) 8 RNase inhibitor 0.3 (12 U) T4 RNA ligase 1, high concentration 1.2 (36 U) -
38
Conduct cleanup as in Steps 26–32 by pre-washing 15 μl of silane beads, except resuspend the beads in 60 μl of RLT and add either 64 or 48 μl of 100% ethanol (for short or long RNA, respectively). To elute, add 13.5 μl of nuclease-free H2O to the beads, mix well, place on a magnet and elute by transferring 12.5 μl of the sample into a new tube.
First-strand cDNA synthesis ● Timing 2 h
-
39
Add 1 μl of the primer 5′-AGA CGT GTG CTC TTC CG-3′ (10 μM stock, 10 picomoles). Mix well by pipetting up and down 10 times.
-
40
Heat at 70 °C for 2 min in a thermocycler and transfer directly to a cold block.
-
41On ice, combine the following components as outlined in the table below. Incubate at 50 °C for 60 min and allow the thermocycler to cool down to 4 °C before transferring to ice or a cold block.
Component Amount (μl) Sample from Step 40 13.5 5× TGIRT-III buffer 4.4 100 mM DTT 1 10 mM dNTP mixture 1 RNase inhibitor 0.5 (20 U) TGIRT-III enzyme 0.9 (180 U) ▲ CRITICAL STEP It is recommended that TGIRT-III enzyme be pre-incubated with the reaction mixture for 20–30 min before adding dNTPs to increase RT efficiency.
-
42
Add 3 μl of ExoSap-it and incubate at 37 °C in a thermocycler for 12 min. This step will digest the oligodT and primers from the sample.
-
43
To hydrolyze RNA, add 1 μl of 0.5 M EDTA and 2.5 μl of 1 M NaOH and incubate at 70 °C for 12 min in a thermocycler. Add 2.5 μl of 1 M HCl to neutralize NaOH.
-
44
Conduct cleanup as in Steps 26–32 by pre-washing 12 μl of beads, except resuspend the beads in 90 μl of RLT and add either 96 or 72 μl of 100% ethanol (for short or long RNA, respectively). To elute, add 6.5 μl of nuclease-free H2O to the beads, mix well, place on a magnet and elute by transferring 5.5 μl of the sample into a new tube.
■ PAUSE POINT Treated cDNA can be stored at −80 °C for the next day.
Second adapter ligation to the cDNA ● Timing 15 min + overnight incubation + 30 min
-
45
To the 5.5 μl of cDNA, add 0.5 μl of a 100 μM stock of the DNA adaptor 5′-/5Phos/AG ATC GGA AGA GCG TCG TGT AG/3ddC/-3′.
-
46
Incubate at 75 °C for 3 min and place on ice or a cold block.
-
47On ice, combine the following components as outlined in the table below. Incubate at 23 °C overnight.
Component Amount (μl) Sample from Step 46 6 10× T4 RNA ligase buffer 2 DMSO (100%) 0.8 ATP (100 mM) 0.2 PEG 8000 (50%, wt/vol) 10 T4 RNA ligase 1, high concentration 1.6 (48 U) -
48
Conduct cleanup as in Steps 26–32 by pre-washing 5 μl of beads, except resuspend the beads in 60 μl of RLT and add either 64 or 48 μl of 100% ethanol (for short or long RNA, respectively). To elute, add 23 μl of nuclease-free H2O to the beads, mix well, place on a magnet and elute by transferring 11.5 μl into each of two tubes.
▲ CRITICAL STEP Separation of the eluted cDNA sample into two aliquots in 0.2-ml tubes allows the user to conduct the following PCR enrichment step on half of the material, keeping the other half as a backup for potential troubleshooting. The backup tube should be stored at −20 °C until needed.
■ PAUSE POINT After this step, the protocol can be paused at any point and cDNA kept at −20 °C until further use.
PCR enrichment ● Timing 30 min
-
49To the tube with 11.5 μl of cDNA intended for PCR, add the following components as outlined in the table below.
Component Amount (μl) One aliquot from Step 48 11.5 25 μM Read 1 PCR primer 0.5 25 μM Read 2 PCR primer (a distinct barcoded primer for each sample) 0.5 2× KAPA HiFi HotStart ReadyMix PCR kit 12.5 -
50Set up and run the PCR program as follows:
Stage Cycle number Denature Anneal Extend 1 1 95 °C, 2 min - - 2 2-5 98 °C, 20 s 65 °C, 15 s 72 °C, 15 s 3 6-10 98 °C, 15 s - 72 °C, 15 s 4 11 - - 72 °C, 2 min ▲ CRITICAL STEP Although the number of cycles in stage 3 of the PCR reaction can vary from 5 to 10, it is recommended to keep it to a minimum, to preserve the complexity of the resulting cDNA library.
? TROUBLESHOOTING
Quality control of cDNA libraries and preparation for high-throughput sequencing ● Timing 1–1.5 h
-
51
Place 5 μl of the cDNA library into a new tube, and add 15 μl of nuclease-free H2O. Keep the remaining 20 μl on ice until used in Steps 54–62.
-
52
In a new tube, mix 3 μl of a 100-bp ladder and 17 μl of nuclease-free H2O.
-
53
Run the cDNA library samples alongside the ladder sample on an agarose gel (Invitrogen E-Gel EX agarose gels (2%, G402002) can be used) to check the average size of the libraries, which should appear as 200–300-bp smears (Fig. 4c).
? TROUBLESHOOTING
-
54
Ampure cleanup. To the remaining 20 μl of cDNA library, add 20 μl of nuclease-free H2O.
-
55
Add 40 μl of Ampure beads, mix well by pipetting 10 times and let stand at room temperature for 2 min.
-
56
Place on a magnet until all beads have collected, and remove the supernatant.
-
57
Add 100 μl of 75% (vol/vol) ethanol and move back and forth on the magnet 15–20 times until the beads are no longer clumpy.
-
58
Remove the supernatant carefully and repeat Step 57.
-
59
Remove the supernatant and dry at room temperature on a magnet until beads appear dry (2–3 min).
-
60
Resuspend the sample in 40 μl of nuclease-free H2O, mix well, place on a magnet and transfer the supernatant to a new tube.
-
61
Repeat Steps 55–59.
-
62
Resuspend the sample in 13 μl of nuclease-free H2O, mix well, place on a magnet and transfer the supernatant to a new tube. This is the final cleaned cDNA library to be sequenced.
-
63
Measure library concentration by using a Qubit fluorometer and the Qubit double-stranded DNA HS assay kit. Store the library at −20 °C (expected concentration should be >2 ng/μl).
-
64
(Optional) Perform additional quality control of the libraries by using an Agilent 2100 bioanalyzer or High Sensitivity D1000 ScreenTape on the TapeStation system to check the size of the libraries (Fig. 4d).
-
65Calculate the final dilution of each library for Illumina sequencing. Given the concentration and average length of each resulting library (i.e., sample), calculate the concentration in nM as follows and add H2O to dilute to the desired concentration according to the Illumina sequencing protocol used.
▲ CRITICAL STEP The final dilution of the cDNA library is based on the average size of the library (estimated from Step 53 or 64) and its concentration (taken from Step 63). The amount of material taken from each library depends on experimental needs and the sequencing protocol used. Thus, usually only an aliquot of the final library should be diluted, while the rest can be stored at −20 °C.
Illumina sequencing ● Timing 12 h
-
66
Sequence the generated libraries with the Illumina NextSeq 500 or NovaSeq 6000 platforms by using paired-end reads, ranging from 25 to 55 bp from each end, according to the manufacturer’s instructions.
Data analysis ● Timing ≥1 d
▲ CRITICAL Misincorporation-based mapping of modifications is subject to diverse sources of artifacts, ranging from ones introduced during the RT step, or in the library preparation, or ones due to the analysis, including mismapping of reads, misannotation of genomic regions, or misinterpretation of sources of misincorporations (e.g., due to the presence of a SNP). Thus, it is advised to incorporate computational considerations of misincorporation analysis, the rationale and implementation of which are described by Sas-Chen and Schwartz34. These include performing local alignment, filtering out sites showing positional or directional biases and filtering out known SNPs, NUMTs and sites with multiple annotations (e.g., annotated as part of a tRNA and an mRNA).
Read mapping
Detection of single-nucleotide variants
-
68
Use the BAM files from Step 67 as input to the JACUSA software to detect single nucleotide variant positions37. JACUSA should be applied in pileup mode, and minimal coverage can be limited to reduce computational load (e.g., to detect only positions with coverage >5), to output a tabular format summarizing the abundance of each nucleotide at each position.
Calculation of misincorporation rate and statistical testing
-
69
On the basis of the output of JACUSA, calculate the misincorporation rate of each position by dividing the number of non-cytidine-containing reads by the total number of reads.
-
70A putative ac4C site is defined by having a statistically significant higher misincorporation rate in the NaCNBH3-treated sample compared to the control samples (mock-treated, chemically deacetylated and/or a genetic control), on the basis of the results of binomial testing. Perform testing as described in options A and B, depending on how many replicates per condition are available.
- Multiple replicates per condition
- If multiple replicates for each condition are available, calculate the P value of a generalized linear mixed-effects model (GLMM), by using the ‘glmer’ function from the ‘lme4’ package in R44. For example, in an experiment with two treatment and two control samples with five reads in each, the numbers of cytidines and non-cytidines in each sample are as follows:
sample Cytidine Non-cytidine Treatment_1 4 1 Treatment_2 3 2 Control_1 1 4 Control_2 2 3 - In R, create a dataframe (myData) in which each row represents a single site from the experiment, as in the table below, and conduct the GLMM test as follows: GLMM.test = glmer(isCytidine ~ condition + (1|sample), data = myData, family = ‘binomial’).
condition isCytidine sample Treatment 0 Treatment_1 Treatment 1 Treatment_1 Treatment 1 Treatment_1 Treatment 1 Treatment_1 treatment 1 Treatment_1 Treatment 0 Treatment_2 Treatment 0 Treatment_2 Treatment 1 Treatment_2 Treatment 1 Treatment_2 Treatment 1 Treatment_2 Control 0 Control_1 Control 0 Control_1 Control 0 Control_1 Control 0 Control_1 Control 1 Control_1 Control 0 Control_2 Control 0 Control_2 Control 0 Control_2 Control 1 Control_2 Control 1 Control_2 - To obtain the P value, apply: GLMM.pvalue = coef(summary(GLMM.test))[,’Pr(>|z|)’][2]
- Single replicate per condition
- If a single replicate is available for each condition, calculate the P value of a χ2 test, by using the ‘chisq.test’ function from the ‘stats’ package in R43. For example, in an experiment that has only one replicate per condition (in the treatment sample, 20 cytidines and 80 non-cytidines, and in the control sample, 95 cytidines and 5 non-cytidines), create a matrix (myMatrix) as in the table below, and conduct the chisq.test as follows: CHISQ.pvalue = chisq.test(myMatrix)$p.value
20 80 95 5
▲ CRITICAL STEP To reduce computational load, statistical tests (detailed in Step 70) can be calculated on a subset of sites fulfilling conditions specified in Step 71. Note that the thresholds specified in Step 71 are dependent on the experimental design, namely the type of controls and the type of organism.
-
71For each position, register the following parameters by using the custom code available at https://github.com/SchwartzLab/ac4c-seq and conduct statistical tests only for positions fulfilling all five of the following conditions.
- The base to which cytidine is most frequently converted is a thymidine.
- The number of reads displaying a C→T conversion is greater than MINC2T (MINC2T = 3 in the original study; this can be defined by the user when running the code).
- The misincorporation rate in the NaCNBH3-treated sample is greater than MINmisTreat (MINmisTreat = 2–3% in the original study; this can be defined by the user when running the code).
- The misincorporation rate in control sample(s) is less than MAXmisCont (MAXmisCont = 1–5% in the original study; this can be defined by the user when running the code). Note that deacetylated controls and/or partial knockdown of the acetyltransferase may result in a higher misincorporation rate compared to mock, and thus this threshold might need to be relaxed.
- The difference in the misincorporation rate of the NaCNBH3-treated versus control sample is greater than MINdiff (MINdiff = 2% in the original study; this can be defined by the user when running the code).
Additional filtering
-
72
When applicable, flag sites overlapping with known SNPs, rRNA and tRNA repeats, repetitive regions (such as Alu repeats in primates), mitochondrial pseudogenes that have ‘transferred’ into the nuclear DNA (aka NUMTs) and paralog genes derived from a common ancestor gene through duplication events.
Motif search
-
73
For each putative ac4C site, the custom code extracts the 10 bases upstream and downstream of the site and stores the 21-base sequence in a column named ‘surrSeq’ within the table saved as ‘ac4c_significantSites.txt’. Extract relevant sequences from this column for motif analysis. Note that sequences are extracted on the basis of the user-provided .fasta file. Make sure to use a relevant reference of the genome or transcriptome, according to the guidance provided in Experimental design (e.g., in the case of assessing sites in the coding sequence of intron-containing mRNA, make sure to use a file containing sequences of the mature mRNA).
-
74
Use the 21-base-long sequences as input for multiple sequence alignment via the WebLogo software (https://weblogo.berkeley.edu/logo.cgi).
▲ CRITICAL STEP Experimental results indicate that a CCG motif is necessary for catalysis by the acetyltransferase5. Thus, the presence of a CCG motif in the putative ac4C sites can serve as a quality control of the validity of the sites. The code stores the 3-base motif under the column ‘motif’ within the table saved as ‘ac4c_significantSites.txt’.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 10 | Poor RNA quality and low yield | RNA degradation | Use RNase-free materials and equipment when isolating total RNA |
| 18 | No bubbles are formed after addition of HCl to the NaCNBH3-treated samples | Inactive NaCNBH3, perhaps due to exposure to humidity in the air (if the powder absorbs water from the air, it is less active and its color turns from bright white to pale white) | Prepare a fresh aliquot of NaCNBH3 Make sure that the stock powder is properly desiccated |
| 50 and 53 | No library is seen in the E-gel | The number of PCR cycles in Step 50 was insufficient (this may indicate a low complexity of the library and thus should be taken into account in downstream analysis) | Place the 20 μl of sample from Step 51 back in the thermocycler and conduct two to five additional PCR cycles (stage 3 of Step 50), followed by a final extension (stage 4 of Step 50). Re-run the library on an E-gel. If the library now shows the correct 200-300-bp smear, re-do the PCR reaction on the 11.5 μl left from Step 48, using the newly optimized number of cycles. Continue with the PCR product to Step 51 |
| No library was generated | If no product is seen after increasing the number of PCR cycles, verify the quality of RNA and materials used and re-do library preparation (Steps 22–53) | ||
| 53 | The library contains adaptor dimers (~140-bp peak) or excess primers (70-80 bp) | This may result from insufficient removal of adaptors and primers from prior steps This may indicate low complexity of the generated library and thus may affect the downstream quality of the sequencing output |
Conduct Ampure cleanup as in Steps 54-62, and verify that unwanted bands are removed The presence of adaptor sequences can be detected in the .fastq files generated by the Illumina platform |
Timing
Steps 1–21 (days 1–2), isolation of total RNA and chemical treatment of RNA: 9 h
Steps 22–65 (days 2–3), preparation of cDNA libraries for high-throughput sequencing: 2 d
Step 66 (day 4), Illumina sequencing: 12 h
Steps 67–74 (day 5), data analysis: minimum of 1 d
Anticipated results
RNA isolation throughout the protocol
After RNA isolation using TRIzol (Step 10), >100 μg of RNA is recovered, and RNA size distribution should resemble that shown in Fig. 4a, indicating that rRNA, mRNA and short RNAs were purified with no notable degradation. Ethanol precipitation of RNA after chemical treatments (Step 21) results in recovery of >70% RNA.
ac4C-seq library construction
Distribution of RNA fragment size between reduced, deacetylated and mock-treated samples varies, with deacetylated samples usually exhibiting smaller RNA fragments (Fig. 4b). A library prepared by using ~200 ng of total RNA results in >25 ng of amplified cDNA at a concentration of >2 ng/μl (Step 63). The size of the libraries is 200–300 bp, with the deacetylated control samples usually resulting in smaller cDNA fragments compared to reduced and mock-treated samples (Fig. 4c,d).
Read mapping and misincorporation calculations
The mapping results in BAM files, sorted and indexed via Samtools, which can be used for visual inspection and quality control of the data via the IGV software48. Although ac4C sites in reduced samples should exhibit a mixture of nucleotides (predominantly cytidines and thymidines), control samples will show negligible amounts of non-cytidine incorporation (Fig. 5a). Note that deacetylated controls and partial knockdown of the acetyltransferase may result in some non-cytidine-containing reads, but their percentage should be lower than that in the reduced sample. Furthermore, misincorporations can be seen for additional modifications; however, these signatures are not expected to vary between experimental conditions. Known sites can be used as positive controls during visual inspection of the data via the IGV software and when generating the catalog of statistically significant ac4C sites (Fig. 5b,c).
Fig. 5 |. Anticipated results of ac4C-seq.
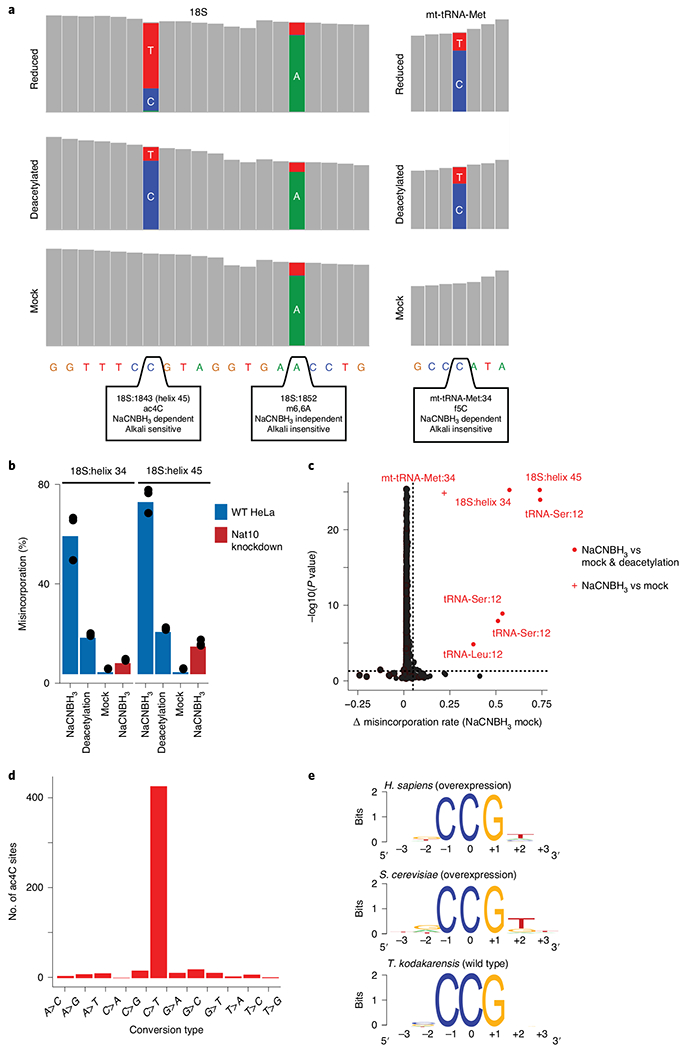
a, Coverage tracks of regions within human rRNA 18S (left) and mitochondrial tRNA methionine (right), as depicted in the IGV browser. Positions with a misincorporation rate >1% are shown in color (blue, cytidine; red, thymidine; green, adenosine; gray, misincorporation <1%). Although both the ac4C in helix 45 of 18S and the f5C in mt-tRNA-Met are dependent on reduction with NaCNBH3 (display a misincorporation upon reduction), only ac4C is sensitive to alkali deacetylation (showing reduced misincorporation in ‘reduced’ versus ‘deacetylated’ conditions). m6,6A in 18S induces a misincorporation in an NaCNBH3- and deacetylation-independent manner and is thus similar in all conditions. b, Misincorporation rates of known sites in 18S in wild-type and NAT10-depleted HeLa cells (bars, mean of 3 biological samples; error bars, s.d.). c, Statistical significance plotted against the difference in misincorporation rates between NaCNBH3- and mock-treated total RNA from HeLa cells. Vertical dashed line, 5%; horizontal dashed line, P = 0.05 (χ2 test). n = 3 biological samples. d, Frequency of the 12 possible misincorporation patterns (y axis) found across all statistically significant sites in T. kodakarensis, showing an enrichment of C→T conversions. e, Sequence motif surrounding the ac4C sites identified in humans, yeast and archaea. Figure adapted with permission from ref. 5. f5C, 5-formylcytidine; mt-tRNA-Met, mitochondrial tRNA methionine; m6,6A, N6,N6-dimethyladenosine; WT, wild type.
ac4C sites and motif identification
Because reduction by hydride donors leads to C→T conversions at ac4C sites, it is expected that putative ac4C sites that passed statistical testing will be enriched in C→T conversions compared to other conversion types. In contrast, control samples are expected to show no enrichment for a specific conversion type. This can be assessed by plotting the number of sites displaying each conversion type in reduced and control samples, as in Fig. 5d.
Furthermore, we previously identified that ac4C occurs within a CCG motif, which is necessary for modification of the middle cytidine. Thus, individual putative ac4C sites, as well as the signature identified via WebLogo (Step 74), should contain a CCG motif (Fig. 5e).
A few few examples of putative ac4C sites identified by using ac4C-seq5 are listed in Table 3.
Table 3 |.
Identified putative ac4C sites
| Species | RNA type | No. of identified ac4C sites |
|---|---|---|
| Yeast (S. cerevisiae) | 18S rRNA | 2 (helix 34 and helix 45) |
| tRNA | 2 (tRNA-Ser and tRNA-Leu) | |
| Mammalian cells (HeLa) | 18S rRNA | 2 (helix 34 and helix 45) |
| tRNA | 2 (tRNA-Ser and tRNA-Leu) | |
| Archaea (T. kodakarensis grown at 85 °C) | rRNA | 173 |
| tRNA | 77 | |
| mRNA | 119 | |
| Ribonuclease P | 19 | |
| Signal recognition particle | 16 |
Acknowledgements
We thank members of the Schwartz and Meier laboratories for many helpful comments. S.S. is funded by the Israel Science Foundation (543165), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 714023) and the Estate of Emile Mimran. S.S. is the incumbent of the Robert Edward and Roselyn Rich Manson Career Development Chair in Perpetuity. J.L.M. is supported by the Intramural Research Program of the National Institutes of Health (NIH), the National Cancer Institute, The Center for Cancer Research (ZIA BC011488-06).
Footnotes
Code availability
The custom code used for the ‘Calculation of misincorporation level and statistical testing’ section is available at https://github.com/SchwartzLab/ac4c-seq.
Competing interests
The authors declare no competing interests.
Data availability
Results depicted in Fig. 5 are based on ac4C-seq data previously deposited in the Gene Expression Omnibus under accession number GSE135826 as part of the publication by Sas-Chen et al.5.
References
- 1.Zachau HG, Dütting D & Feldmann H Nucleotide sequences of two serine-specific transfer ribonucleic acids. Angew. Chem. Int. Ed. Engl 5, 422 (1966). [DOI] [PubMed] [Google Scholar]
- 2.Ohashi Z et al. Characterization of C+ located in the first position of the anticodon of Escherichia coli tRNAMet as N4-acetylcytidine. Biochim. Biophys. Acta 262, 209–213 (1972). [PubMed] [Google Scholar]
- 3.Bruenger E et al. 5S rRNA modification in the hyperthermophilic archaea Sulfolobus solfataricus and Pyrodictium occultum. FASEB J. 7, 196–200 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Stern L & Schulman LH The role of the minor base N4-acetylcytidine in the function of the Escherichia coli noninitiator methionine transfer RNA. J. Biol. Chem 253, 6132–6139 (1978). [PubMed] [Google Scholar]
- 5.Sas-Chen A et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 583, 638–643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansson MJ & Bystrom AS The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA 10, 712–719 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotelawala L, Grayhack EJ & Phizicky EM Identification of yeast tRNA Um(44) 2′-O-Methyltransferase (Trm44) and demonstration of a Trm44 role in sustaining levels of specific tRNASer species. RNA 14, 158–169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S et al. A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem 289, 26201–26212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem 289, 35724–35730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 43, 2242–2258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma S et al. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet. 13, e1006804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas JM, Bryson KM & Meier JL Nucleotide resolution sequencing of N4-acetylcytidine in RNA. Methods Enzymol. 621, 31–51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dempster JM et al. Extracting biological insights from the Project Achilles genome-scale CRISPR screens in cancer cell lines. Preprint at bioRxiv 10.1101/720243 (2019). [DOI] [Google Scholar]
- 15.Larrieu D, Britton S, Demir M, Rodriguez R & Jackson SP Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344, 527–532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschida BR et al. Sleeping Beauty insertional mutagenesis in mice identifies drivers of steatosis-associated hepatic tumors. Cancer Res. 77, 6576–6588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryvkin P et al. HAMR: high-throughput annotation of modified ribonucleotides. RNA 19, 1684–1692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerutti P & Miller N Selective reduction of yeast transfer ribonucleic acid with sodium borohydride. J. Mol. Biol 67, 90260–90264 (1967). [DOI] [PubMed] [Google Scholar]
- 19.Thomas JM et al. A chemical signature for cytidine acetylation in RNA. J. Am. Chem. Soc 140, 12667–12670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinclair WR et al. Profiling cytidine acetylation with specific affinity and reactivity. ACS Chem. Biol 12, 2922–2926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai K et al. Acetylation of cytidine residues boosts HIV-1 gene expression by increasing viral RNA stability. Cell Host Microbe 28, 306–312.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balmus G et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun 9, 1700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q et al. NAT10 is upregulated in hepatocellular carcinoma and enhances mutant p53 activity. BMC Cancer 17, 605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X et al. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 46, 9601–9616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin S et al. Mettl1/Wdr4-mediated mG tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 71, 244–255.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchand V et al. AlkAniline-Seq: profiling of M7 G and M3 C RNA modifications at single nucleotide resolution. Angew. Chem. Int. Ed. Engl 57, 16785–16790 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Ikeuchi Y, Kitahara K & Suzuki T The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J. 27, 2194–2203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arango D et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safra M et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Grozhik AV et al. Antibody cross-reactivity accounts for widespread appearance of m1A in 5′UTRs. Nat. Commun 10, 5126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helm M, Lyko F & Motorin Y Limited antibody specificity compromises epitranscriptomic analyses. Nat. Commun 10, 5669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun 10, 4079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grünberger F et al. Exploring prokaryotic transcription, operon structures, rRNA maturation and modifications using nanopore-based native RNA sequencing. Preprint at bioRxiv 10.1101/2019.12.18.880849 (2019). [DOI] [Google Scholar]
- 34.Sas-Chen A & Schwartz S Misincorporation signatures for detecting modifications in mRNA: not as simple as it sounds. Methods 156, 53–59 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Orita I et al. Random mutagenesis of a hyperthermophilic archaeon identified tRNA modifications associated with cellular hyperthermotolerance. Nucleic Acids Res. 47, 1964–1976 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. J. Bioinform 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piechotta M et al. JACUSA: site-specific identification of RNA editing events from replicate sequencing data. BMC Bioinformatics 18, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sexton AN, Wang PY, Rutenberg-Schoenberg M & Simon MD Interpreting reverse transcriptase termination and mutation events for greater insight into the chemical probing of RNA. Biochemistry 56, 4713–4721 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cozen AE et al. ARM-Seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 12, 879–884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng G et al. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinodoz SA et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H et al. The Sequence Alignment/Map format and SAMtools. J. Bioinform 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2020) Available at https://www.R-project.org/ [Google Scholar]
- 44.Bates D, Mächler M, Bolker B & Walker S Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 45.Chomczynski P A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15, 532–534 (1993). [PubMed] [Google Scholar]
- 46.Hummon AB, Lim SR, Difilippantonio MJ & Ried T Isolation and solubilization of proteins after TRIzol® extraction of RNA and DNA from patient material following prolonged storage. Biotechniques 42, 467–472 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collart MA & Oliviero S Preparation of yeast RNA. Curr. Protoc. Mol. Biol Ch. 13, Unit 13.12 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Robinson J et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Results depicted in Fig. 5 are based on ac4C-seq data previously deposited in the Gene Expression Omnibus under accession number GSE135826 as part of the publication by Sas-Chen et al.5.