

Abstract
Leishmania donovani, the causative organism for visceral leishmaniasis, contains a unique bisubunit DNA–topoisomerase IB (LdTopIB). The catalytically active enzyme is a heterodimer constituted by a large subunit (LdTopIL) containing a non-conserved N-terminal end and the phylogenetically conserved core domain, whereas the small subunit (LdTopIS) harbors the C-terminal domain with the characteristic tyrosine residue in the active site. Site-directed mutagenesis was used to substitute the basic amino acid (Arg-314, Lys-352, Arg-410 and His-453) of the LdTopIL subunit by the neutral amino acid alanine. The expression of these mutants in a topoisomerase-free yeast strain produced inactive proteins. Similarly, when the Tyr-222 from small subunit, involved in DNA cleavage, was substituted by Phe no topoisomerase activity was detected in yeast overexpressing extracts. In addition two substitutions involved in camptothecin inhibition were also analyzed. Asp-353 located in the core domain of the large subunit and Asn-221 which heads Tyr-222 in the small subunit, were replaced by Ala and Ser, respectively. These mutants were insensitive to the inhibitor; despite they displayed significant relaxation activity.
Keywords: Topoisomerase, IB Leishmania, Trypanosomatids, Camptothecin, Site-directed mutagenesis, Tropical diseases
1. Introduction
Leishmania donovani is the etiological agent of visceral leishmaniasis a parasite-borne zoonotic disease transmitted by the bite of female sandflies [1]. Since no effective vaccine has been developed at present, chemotherapy is the unique way to control a disease that may be fatal if left untreated. Most of current pharmacopoeia against visceral leishmaniasis is plenty of undesirable side effects, require parental administration and long-term treatments or is ineffective due to drug resistances [2,3].
DNA topoisomerases (Top) catalyze changes on duplex DNA unwinding during replication, transcription, recombination and DNA repair processes. Type I Top (TopI) are ATP-independent monomeric enzymes introducing transient single-stranded breaks in DNA followed by passage and rejoining. Type II Top (TopII) are multimeric ATP hydrolyzing proteins that generate temporary double-stranded breaks in the double helix, followed by passage and rejoining [4,5].
TopIA are bacterial proteins that bind covalently to the 5′-end of the broken strand thereby allowing single step changes in the linking number of circular DNA. This subfamily includes bacterial TopI and TopIII and reverse gyrases enzymes [6]. TopIB subfamily includes eukaryotic enzymes producing transient covalent bonds at the 3′-end of the scissile DNA strand. Structurally speaking, eukaryotic TopIB are monomeric proteins constituted by four structural and functional domains: a poorly conserved N-terminal containing multiple nuclear localization signals (NLS); the phylogenetically conserved core domain, required for relaxation of supercoiled DNA and the C-terminal end containing a tyrosine residue that cleaves one of the DNA strands, creating a transient covalent phosphodiester bond [7,8]. Core and C-terminal end are connected to each other by a poorly conserved linker region [9].
Several reports describing TopIB in trypanosomatid parasites show remarkable differences between this enzyme and the one from other eukaryotes. Unlike most organisms TopIB from trypanosomes [10] and leishmanias [11] are heterodimers whose corresponding encoding genes are placed in different chromosomes. LdTopIL gene – placed at chromosome 31 – encodes for a 73-kDa subunit containing a short and non-conserved N-terminal end (start-Met-Glu-32) followed by the conserved core region (Gln-33-Ser-456). Beyond this amino acid a long and non-conserved C-terminal end occurs, displaying multiple predictable NLS. On the other hand, LdtopIS gene encodes for the 28 kDa small subunit, which it is located at chromosome 4. This protein contains the catalytic C-terminal domain headed by a non-conserved N-terminal extension (start-Met-Ala-210), which might play role as a putative linker. These striking differences between human and trypanosomatid’s TopIB have been accounted for a potential differentiable target against these organisms [11–13].
Multiple alignment analysis with other TopIB shows that the large LdTopIL subunit contains the expected catalytic tetrad – a series of four basic amino acids interacting with DNA – whereas the small LdTopIS subunit displays the consensus cleaving residue [11]. From this model, Arg-410 and Lys-352 are involved in a “proton relay” mechanism for catalysis similar to that described for vaccinia TopIB where Arg-130 and Lys-167 are involved in the transfer of a proton from the enzyme to DNA 5′-free end [14]. X-ray analysis of a crystalline LdTopIB specimen confirmed this theory, showing the Arg-410 positioned at 3.2 Å from the phenolic oxygen atom of Tyr-222, and playing role in stabilizing the phenolate anion during the attack on the scissile phosphate group. Lys-352 acts as the general acid in the cleavage reaction by donating a proton to the scissile strand, according to the crystallized active site, while His-453 – close to the non-bridging oxygen atom of vanadate – mimics the transient state of enzyme–DNA covalent complex. A water molecule at 2.5 Å from the active Tyr plays role as specific base accepting the proton yielded by the phenolic group of Tyr-222 [15].
Several amino acids have been involved in camptothecin (CPT) resistance. Tamura et al. [16] showed that the unusual resistance of a human acute lymphoblastic leukemia cell line to CPT was due to a substitution of Asp-533 to Gly. On the other hand, changes in the Tyr-neighboring Asn within the conserved “SKxxY” motif, entails CPT resistance with no changes in relaxation activity [17]. In addition, Marquis et al. have shown that the L. donovani CPT-resistant LdRCPT.160 strain is due to two amino acid substitutions within the core domain of the large LdTopIB subunit (Gly-185-Arg and Asp-325-Glu) resulting from two single nucleotide mutations. In addition, authors observed a decrease in DNA relaxation, presumably due to the presence of these mutations [18]. Recently, the authors have undoubtedly shown the relevance of the RPPVV pentapeptide placed at positions 175–180 of the non-conserved N-extension end of LdTopIS small subunit, in CPT resistance by preventing the formation of a stable cleavage complex with DNA [19].
The present paper confirms the role played by the amino acids occurring at the catalytic tetrad of LdTopIB using site-directed mutagenesis. We also report two amino acid changes responsible for the resistance to CPT in the leishmanial enzyme.
2. Methods and materials
2.1. Reagents and culture media
Pyrococcus furiosus (Pfu) and Klenow polymerases and restriction enzymes were procured from Roche (Basel, Switzerland) and Amersham Biosciences. DNA ligase from bacteriophage T4 was obtained from Stratagene (La Jolla, USA). Cell culture media, raffinose, CPT and other chemicals and reagents were purchased from Sigma (St. Louis, USA). Primers for PCR amplification were from Sigma Genosys (Cambridge, UK).
2.2. Yeast and leishmanial strains
The purified proteins were overexpressed from Saccharomyces cerevisiae EKY3 strain [MAT α ura3–52 his3Δ200 leu2 Δ1 trp1 Δ63 top1 Δ::TRP1] whereas MBY3 strain [MAT α ura3–52 his3Δ200 leu2 Δ1 trp1 Δ63, top1 Δ::TRP1 rad52 Δ::LEU2] was transformed with the same gene constructions for the cytotoxic assays in spot test and dose–response. Both strains are deficient in TopI activity [20]. L. donovani LEM75 (Ethiopian) promastigotes were a kind gift from Dr. J.M. Requena (Centro de Biología Molecular “Severo Ochoa”, CSIC Madrid, Spain).
2.3. Cloning of leishmanial DNA topoisomerase I
Cloning of heterodimeric L. donovani DNA topoisomerase I was performed as described previously [11]. LdTop1L (GenBank accession AF303557) gene, was obtained by the screening of a λ-EMBL3 genomic library [21], whereas LdTop1S (GenBank accession AY062908) gene was amplified using primers based on L. major Friedlin genome Project [22] using L. donovani genomic DNA as template.
2.4. Direct mutagenesis in LdTopIB
The procedure to obtain the mutated genes was developed according the QuickChange® method: each gene encoding for the LdTopIL and LdTopIS were mutated separately and subcloned in the pESC URA. The restrictions sites for LdTopIL gene were BamHI and XhoI, whereas NotI and SpeI were those for LdTop1S gene. Fig. 1 describes the distinct mutations performed on LdTopIB subunit.
Fig. 1 –

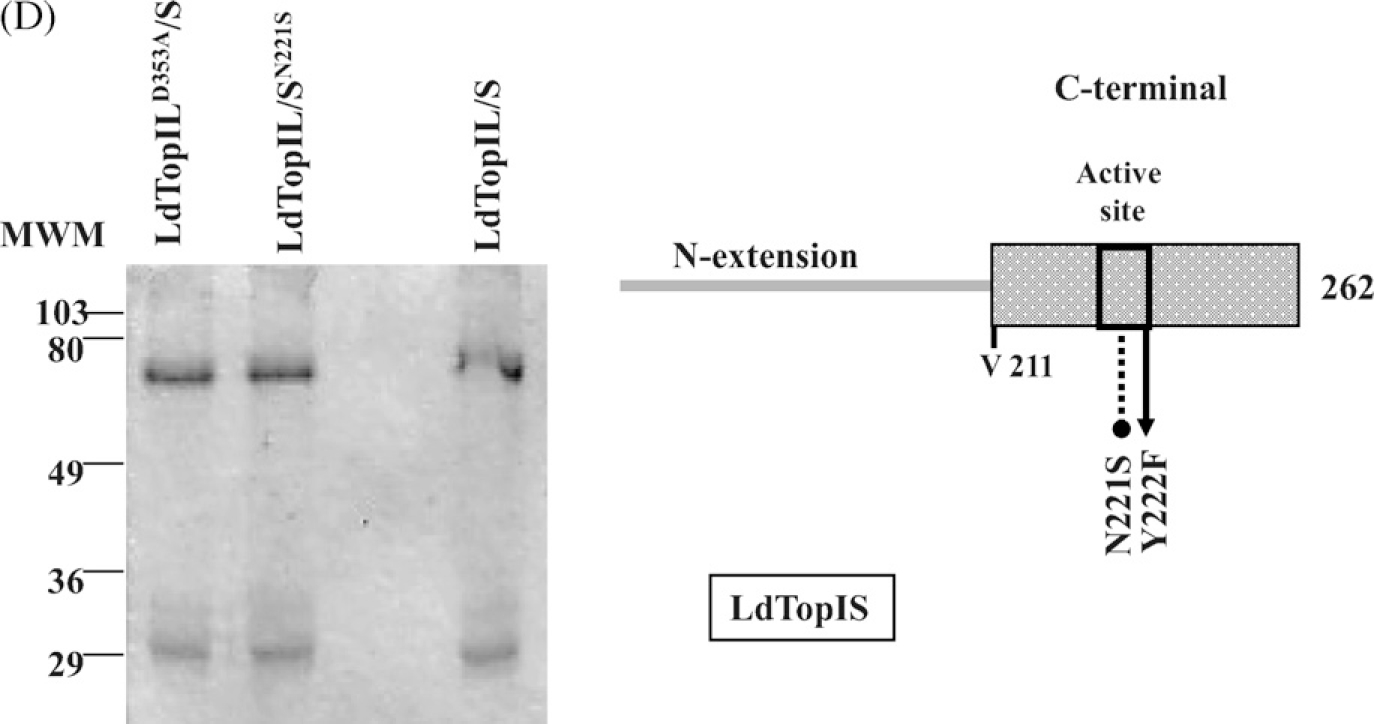
Multiple alignments of partial TopIB sequences showing the representative amino acids of the catalytic tetrad and the active site. (A) Partial alignment of the core region of human (hTopIB), yeast (yTopIB), trypanosomal (LdTopIL) and leishmanial large subunits (TbTopIL). (B) Partial alignment of the C-terminal end of hTopIB, yTopIB and the small subunits of T. brucei (TbTopIS) and L. donovani (LdTopIS). The mutated residues: Arg-314, Lys-352, Asp-353, Arg-410, His-453, Asn-221 and Tyr-222, are highlighted in grey, as well as the homologous residues in the orthologous sequences. (C) Schematic representation of LdTopIB showing the putative functional domains, and the allocation of the different point mutations performed in both subunits: arrows indicate the catalytical residues positions, and the dotted line ended in circle shows the CPT-resistant amino acids substitutions. (D) SDS-PAGE gel showing the purification level for the mutants LdTopILD353A/S y LdTopIL/SN221S and wild-type.
The sequence of the primers used and their codon changes are listed in Table 1. The PCR reaction contained 20 ng of plasmid pSK-LdTopIL or pSK-LdTopIS as template depending on the different mutations, 1 μM of each oligonucleotide, 10 μM dNTPs, 5 μl of 10× Pfu-buffer and 2.5 units of Pfu-polymerase for a total volume of 50 μl. Reactions were carried out in a Mastercycler gradient thermocycler (Eppendorf®) as follows: a 5-min first step at 94 °C, followed by one cycle at 94 °C for 1 min, 55 °C for 1 min and 45 °C for 12 min; this cycle was repeated 5 times, increasing the elongation temperature 5 °C in each cycle until 68 °C were reached, when the last cycle was repeated for 16 times. PCR-products were incubated in presence of DpnI in order to digest the parental DNA template, and used to transform DH5α E. coli; four to six clones were sequenced to assure that site-directed mutation had been introduced accurately. Typically, the efficiency of mutagenesis was around 80%. Mutants were sequenced (Sistemas Genomicos, Spain) in order to confirm the absence of undesirable additional mutations. The genes were subcloned into the BamHI/XhoI sites for the LdTopIL subunit and NotI/SpeI for LdTopIS in the yeast expression vector pESC-URA- under control of promoters GAL1 and GAL10, respectively.
Table 1 –
Sequences of the primers used in this study to mutate LdTopIB
| Mutation | Primers (5′–3′) | |
|---|---|---|
| R314A | FW | GAC CGC CTC GCC CTC GCC GTT GGT AAT GAG AAG |
| RV | CTT CTC ATT ACC AAC GGC GAG GGC GAG GCG GTC | |
| K352A | FW | TTC GAC TTC CTG GGC GCG GAC TCG ATC CGC TAC |
| RV | GTA GCG GAT CGA GTC CGC GCC CAG GAA GTC GAA | |
| R410A | FW | CTG CCA AGG TGT TCG CCA CCT ACA ATG CCT CCA TC |
| RV | GAT GGA GGC ATT GTA GGT GGC GAA CAC CTT GGC AG | |
| H453A | FW | GCC ATT CTG TGC AAC GCC CAG AAG TCC GTC TCG |
| RV | CGA GAC GGA CTT CTG GGC GTT GCA CAG AAT GGC | |
| Y222F | FW | CCA GTA AAA TCA ACT TCA TCG ACC CCC GTA TC |
| RV | GAT ACG GGG GTC GAT GAA GTT GAT TTT ACT GG | |
| D353 | FW | GAC TTC CTG GGC AAG GCC TCG ATC CGC TAC CAG |
| RV | CTG GTA GCG GAT CGA GGC CTT GCC CAG GAA GTC | |
| N221S | FW | GGC ACC AGT AAA ATC AGC TAC ATC GAC CCC CGT |
| RV | ACG GGG GTC GAT GTA GCT GAT TTT ACT GGT GCC |
2.5. Yeast expression system
S. cerevisiae strains EKY3 and MBY3 were transformed with the constructs showed in Fig. 1, using the lithium acetate method [23]. The GAL promoted expression vectors carry the URA3 selection marker and were maintained by selection in synthetic complete (SC) – uracil deficient (URA-) – media. At least four independent clones were selected from each transformation. The independent colonies were incubated overnight in SC URA- 2% dextrose (w/v) for an optimal starting growth. Prior to the 6 h induction with 2% galactose (w/v), each starter culture was incubated in SC URA- 2% raffinose (w/v) medium to eliminate any traces of dextrose that might inhibit the GAL promoter expression. The cells were spun down, washed with cold water to remove traces of media and resuspended in the lysis buffer composed by TEEG buffer (50 mM Tris–HCl pH 7.4, 1 mM EDTA, 1 mM EGTA, 10% glycerol) supplemented with 0.2 M KCl and protease inhibitors cocktail [0.1 mg/ml sodium fluoride, 0.8 mg/ml sodium bisulphite, 2× Complete Mini® (Roche Molecular Biochemicals)]. Cell pellets were prepared for disruption by glass beads [24] with one freeze/thaw cycle at −80 °C, with the purpose of weakening the yeast wall; after lysis with 425–600 μm acid-washed glass beads, the extracts were cleared by centrifugation at 15,000 × g for 30 min at 4 °C.
2.6. Purification of the proteins
Purification of LdTopIB was done according to Knab et al. [17]. Briefly, overexpressing yeast extracts were subjected to double-ammonium sulphate fractionation (35 and 75% saturation) and the supernatant from the second precipitation was loaded onto a phosphocellulose (P-11) column, previously equilibrated as manufacturer indications. The recombinant proteins (wild-type topoisomerase I enzymes and mutated proteins) were eluted at 4 °C with a discontinuous gradient of KCl (0.2, 0.4, 0.6, 0.8 and 1 M) in TEEG buffer, supplemented with 0.1 mg/ml sodium bisulphite, 0.8 mg/ml NaF and the protease inhibitors cocktail. In order to reach an appropriate concentration for the different in vitro assays, the eluate from the P-11 column was concentrated by Microcon YM-30 (Millipore,). When required, the active fractions were loaded onto a phenyl–sepharose column (Sigma–Aldrich, St. Louis, USA) and eluted with a discontinuous inverse gradient of KCl (1, 0.8, 0.6, 0.4 and 0.2 M) and the concentrated as indicated before. Purity of the samples was analyzed by SDS-PAGE. To store, 40% glycerol was added to preserve the activity and keep in a −20 °C freezer. Protein concentration was determined by the Bradford method [25].
2.7. DNA relaxation assays
DNA topoisomerase I activity was assayed by the relaxation of negatively supercoiled plasmid DNA. The reaction mixture in a total volume of 20 μl contained 0.3 μg of supercoiled Rf I ΦX 174 DNA, 10 mM Tris–HCl buffer pH 7.5, 5 mM MgCl2, 0.1 mM EDTA, 15 μg/ml bovine serum albumin, 50 mM KCl and various extracts containing altered proteins or wild-type enzyme, starting with 0.2 μg of the total protein. The reaction mixtures were incubated for 30 min at 37 °C. The enzyme reactions were stopped by the addition of up to 1% SDS – final concentration -and digested by proteinase K 2 mg/ml with 1 h incubation to remove protein bonded to the DNA fragment. The extent of plasmid DNA relaxation was assessed by electrophoresis in a 1% agarose gel in 0.1 M Tris borate EDTA buffer pH 8.0 at 2 V/cm for 14 h. The gels were visualized under UV illumination after being stained with ethidium bromide (0.5 mg/ml) and a posterior electrophoresis in the presence of 0.1 mg/ml ethidium bromide, in order to separate the nicked DNA from the relaxed topoisomers [26].
2.8. DNA cleavage assay
Wild-type and mutant enzyme sensitivity to CPT was determined in DNA cleavage assays. A 282 bp PvuII/HindIII fragment of pBluescript SK(−) phagemid DNA (pSK) DN substrate, [32P]-labeled at a single 3′-end, was prepared as described elsewhere [27]. Equal concentrations of the purified wild and modified leishmanial proteins were incubated with at least 1.5 μl of DNA containing a minimum of 150,000 cpm in 10 mM Tris–HCl buffer pH 7.5, 5 mM MgCl2 mM DTT, 0.1 mM EDTA, 15 μg/ml bovine serum albumin, 50 mM KCl, and the indicated concentrations of CPT in a final 1% DMSO. Following incubation for 30 min at room temperature, the reactions were terminated with final concentration of 1% SDS and afterwards heated to 75 °C for an extra 15 min period. The samples were treated with proteinase K and double-ethanol precipitation prior to electrophoresis in an 8% polyacrylamide/7 M urea gel. The cleavage products were visualized with a PhosphorImager (Molecular Dynamics, Sunnyvale, USA).
2.9. In vivo sensitivity to CPT
To determine cellular sensitivity to CPT, MBY3 yeast strain (ΔtopI; Δrad52) were transformed with vectors carrying the wild-type and different mutations made onto LdTopIL and LdTopIS genes driven by GAL1/GAL10 promoters. Cultures were grown O/N and then they were adjusted to OD595 = 0.3 and serially diluted 10-fold in TE buffer [28]. Five microliters were spotted onto selective media under repressed (2% dextrose) or induced (2% galactose) conditions, in the presence or absence of 15 and 30 μM CPT. All plates contained 0.4% DMSO. Control assays (“mock”) were performed under similar assay conditions with the yeast expressing the pESC-URA- vector without any insert.
For dose–response and time-course experiments, the corresponding yeast strains were grown in SC URA- 2% dextrose culture O/N and diluted 100-fold in SC URA- 2% raffinose in order to eliminate any traces of dextrose that might inhibit the GAL promoters. After 20 h incubation, the cultures were adjusted to OD595 = 0.3 and incubated in SC URA-2% galactose in the presence of increasing amounts of CPT starting from 0 to 10 μM. After 24 h, the cultures were 10-fold serially diluted and 50 μl were platted onto SC URA- 2% dextrose and incubated at 30 °C for 72 h. Viable colonies were counted up and plotted in a logarithmic scale.
3. Results
3.1. L. donovani topoisomerase IB
LdTopIB activity is reconstituted when both large – LdTopIL -and small – LdTopIS – subunits were cloned simultaneously in the biscistronic yeast expression vector pESC-URA- and expressed in a TopIB-deficient yeast system (EKY3) [11]. LdTopIB were purified from overexpressing yeast extracts, using NH4(SO4)2 precipitation and further column elution according to [17]. SDS-PAGE electrophoresis of the purified extracts shows two major bands corresponding to the expected molecular weights of each LdTopIB subunits. This purification protocol was used to obtain pure protein required for further in vitro studies (Fig. 1D).
3.2. Mutations on active-site amino acids
Fig. 1A shows a partial sequence alignment of the LdTopIL core region with the homologous from human (hTopIB), yeast (yTopIB) and African trypanosomes (TbTopIB), showing the high degree of phylogenetic conservation between the amino acids described in the human enzyme (Gln-33-Ser-456) and the ones composing the putative core domain of the other species including the catalytic tetrad [15]. However, the homology at the C-terminal end (Val-457-onwards) is dramatically lost in trypanosomatids. This region of LdTopIL contains a long tail enriched in lysine residues, which lacks the conserved Tyr catalytic residue involved in DNA cleavage.
On the other hand, the first 210 amino acidic residues composing the N-terminal end (start-Met-Ala-210) of the small LdTopIS subunit, shows no homology with any other TopIB described at present. The C-terminal end of this protein (Asn-210-onwards) is an authentic catalytic domain displaying a high homology degree with other eukaryotic catalytic sites. LdTopIS also includes the phylogenetically conserved “SKxxY” motif in which Tyr-222 (Tyr-723 in the human enzyme) plays role in DNA cleavage (Fig. 1B).
As mentioned before, five conserved amino acids are involved in topoisomerization activity of any TopIB. The role of these amino acids in the leishmanial enzyme has been analyzed by site-directed mutagenesis, studying the relaxation activity of the mutated proteins. Based on these analogies, we replaced the residues sited at the large subunit by the neutral amino acid alanine, which is not protonable at physiological pH, preventing the electrostatic contributions in substrate catalysis and hydrogen bonding (mutations LdTopILR314A/S, LdTopILR410A/S, LdTopILH453A/S and LdTopILK352A/S). On the other hand Tyr-222, placed in the small subunit, which aligns with a homologous Tyr at position 723 in the human enzyme, was replaced by Phe, which is sterically homologous but lacks the ability to cleave DNA (mutation LdTopIL/SY222F) (scheme of the Fig. 1C). All these mutations were created in the pSK vector, carried to the yeast biscistronic pESC-URA plasmid, and heterologously expressed in the TopIB-deficient yeast system (EKY3). Fig. 2 compares the residual relaxation activity of these mutants with the one found in the recombinant wild-type enzyme (lanes 4–8 show two-fold dilutions of the purified wild-type LdTopIB) and the commercial calf-thymus enzyme (lane 3) used as positive control. As expected, none of the mutants of the active site created, displayed any significant ability to relax supercoiled circle DNA under standard assay conditions, using undiluted extracts.
Fig. 2 –
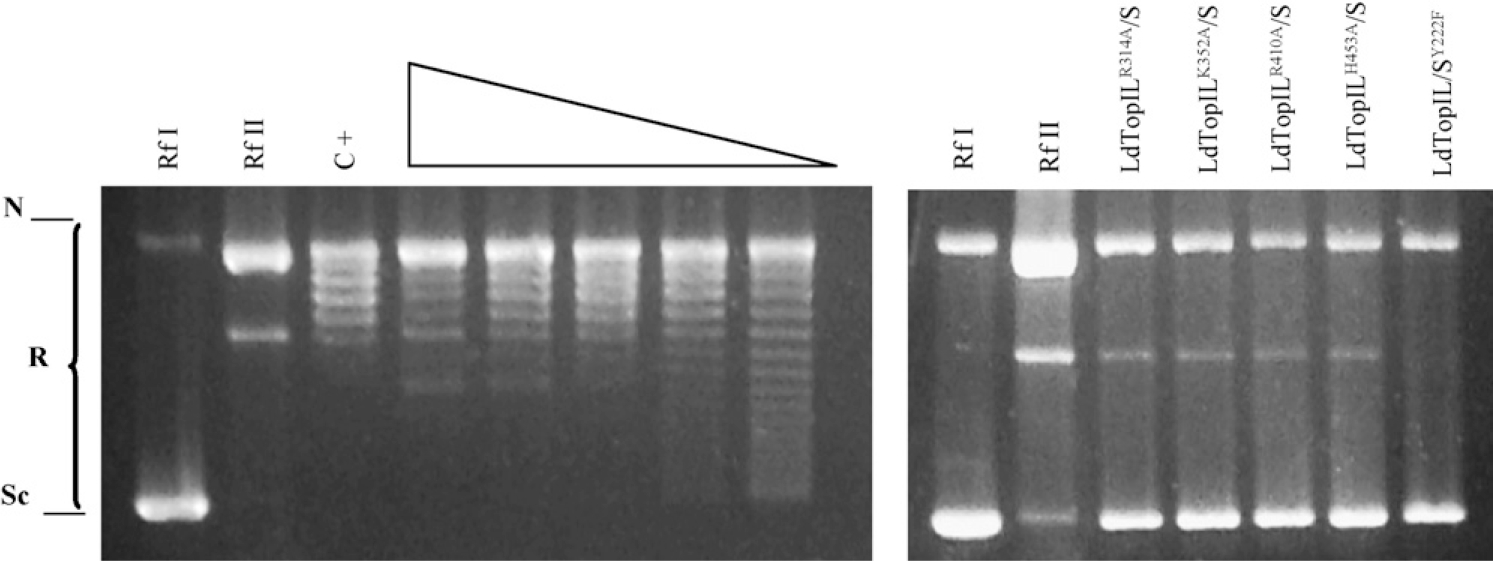
Relaxation activity of yeast extracts expressing different point mutants of the catalytic tetrad and active site of LdTopIB. The relaxation activity of the mutants LdTopILR314A/S, LdTopILK352A/S, LdTopILR410A/S, LdTopILH453A/S and LdTopIL/SY222F (lanes 11–16) is compared with the wild-type (lanes 4–8). In lanes 1, 2, 9 and 10 supercoiled DNA (RfI) and nicked DNA (RfII), respectively, were loaded, and in lane 3, a commercial assay for TopIB was carried out as positive control. Sc and N indicate the position in the electrophoresis agarose gel of supercoiled and nicked DNA each, whereas R specifies the relaxed DNA isoforms.
3.3. D353A and N221S mutants
Pursuant to the crystal structure of hTopIB, Asp-353 placed at large LdTopIL subunit and Asn-221 heading the active Tyr-222 in the “SKxxY” motif of the active site of small LdTopIS subunit (Asp-533 and Asn-722, respectively, in the human enzyme) would be able to bind the DNA molecule and cleave/reseal a single strand of DNA. However, they have lost their sensitivity to the specific poison CPT when substituted by neutral amino acids. Site-directed mutagenesis in both amino acids was carried out to alanine (mutation LdTopILD353A/S and serine (mutation LdTopIL/SN221S), respectively, removing the possibility of interaction with the inhibitor, according with the CPT docking models. Mutants were subcloned into pESC-URA- vector that contained the non-mutated complementary subunit and expressed into EKY3 topoisomerase-deficient yeast system and purified according to the above-mentioned protocol (Fig. 1D). Pictures from Fig. 3 show relaxation assays carried out with different half-dilution extracts either from wild-type or mutants. No differences in relaxation activity were accounted under standard assay conditions.
Fig. 3 –
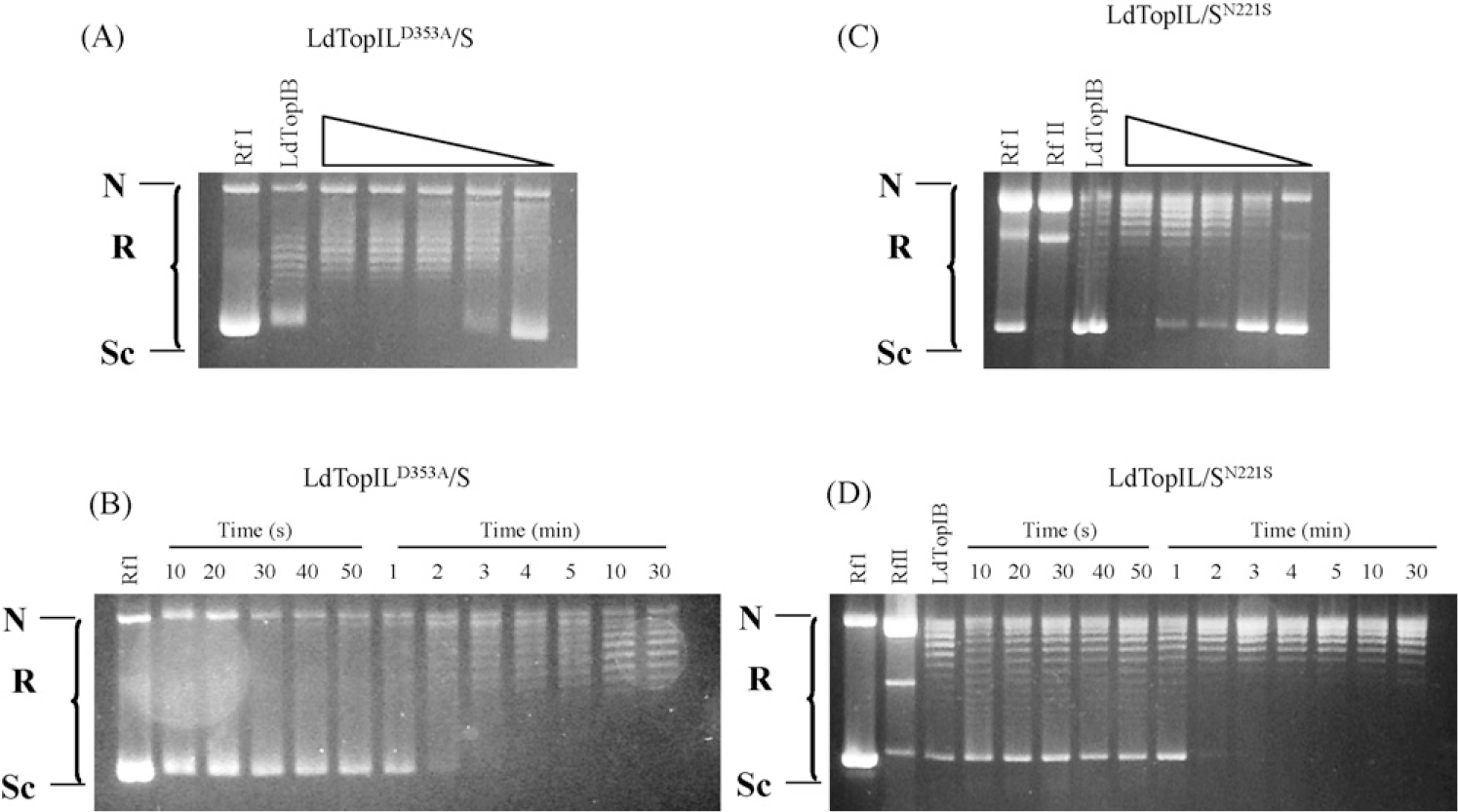
Relaxation activity of purified recombinant LdTopIB point mutants. (A) Fourfold serial dilutions of LdTopILD353A/S mutant were assayed in a plasmid DNA relaxation assay (0.3 μg of supercoiled RfI DNA as substrate) for 30 min at 37 °C (as described under Section 2.7). (B) Equal concentrations of LdTopILD353A/S were incubated stepwise for 10 s to 30 min in a plasmid DNA relaxation assay protein. (C and D) Similar to A and B but using LdTopIL/SN221S mutant.
3.4. Topoisomerase-deficient yeast sensitization to CPT
In spite of the unusual dimeric structure, LdTopIB I is conserved in terms of reaction mechanism and drug sensitivity. The heterologous expression of the wild-type protein in a TopIB-deficient yeast strain (top1Δ), defective in double-strand breaks repair (rad52Δ), MBY3 was able to confer sensitivity to 15 and 30 μM CPT when cultures where induced with 2% galactose (Fig. 4).
Fig. 4 –
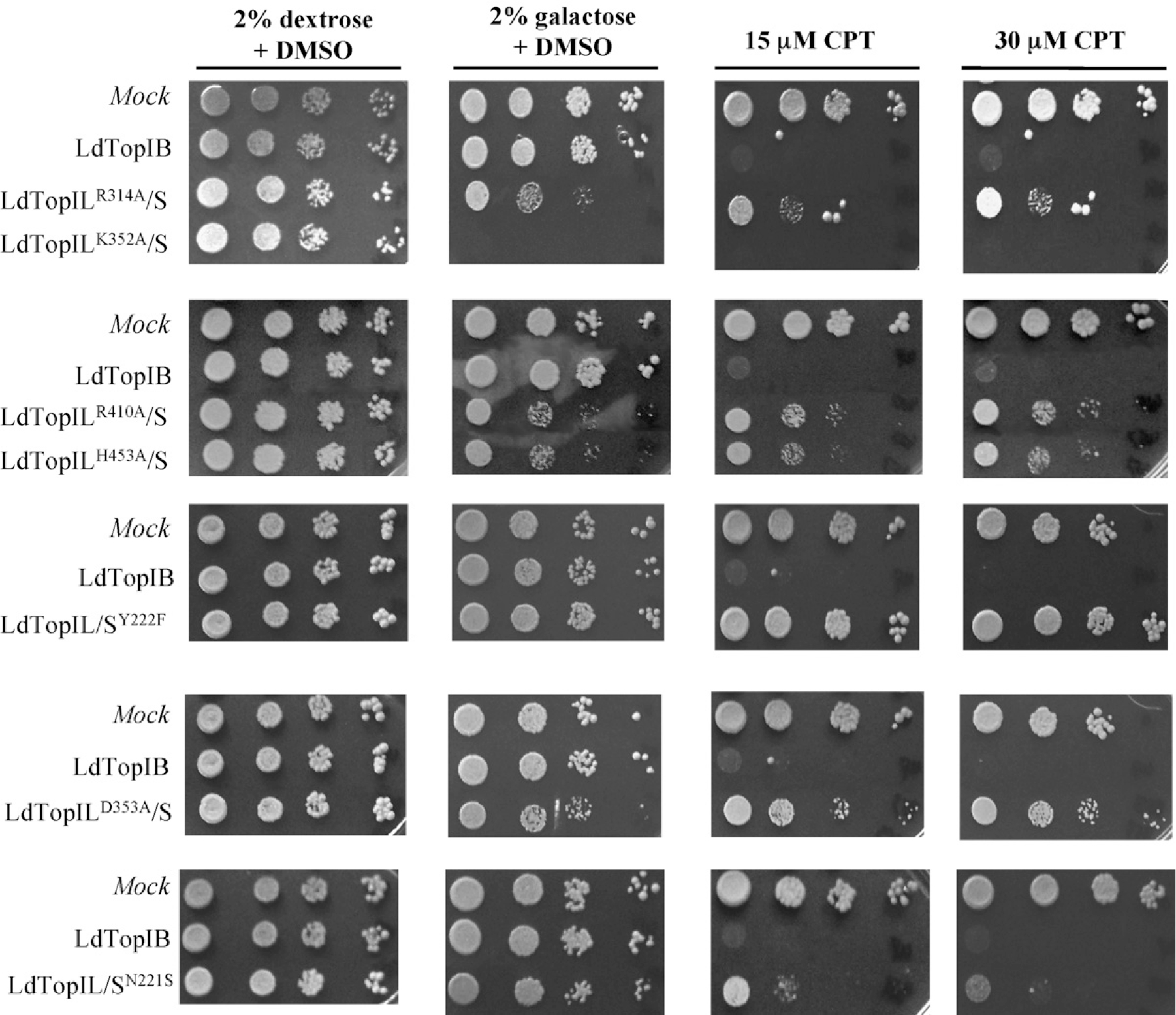
Expression of L. donovani topoisomerase induces sensitivity to CPT-resistant yeast strain MBY3 (rad52Δ). Exponentially growing cells in dextrose, transformed with vectors carrying the leishmanial wild-type and different point mutants, driven by GAL1/GAL10 promoters, were serially 10-fold diluted starting from OD595 = 0.3. Five microliters were spotted on selective media under repressed (2% dextrose) or induced (2% galactose) conditions, in the presence or absence of 15 and 30 μM CPT. Pictures are representative of multiple experiments.
The series of point mutants prepared previously, were transfected into the MBY3· yeast-deficient strain plated and induced with 2% galactose in absence and presence of 15 and 30 μM CPT. Pictures of Fig. 4 show two different patterns after addition of the inducer sugar. The overexpression of the active site mutant LdTopILK352A/S was lethal for the yeast-deficient strain in absence of CPT, whereas the mutants LdTopILD353A/S LdTopIL/SN221S, LdTopILR314A/S, LdTopILR410A/S, LdTopILH453A/S and LdTopIL/SY222F did not produce any deleterious effect in absence or presence of the inhibitor. Unlike LdTopIL/SY222F and LdTopILD353A/S, the mutant LdTopIL/SN221S-induced sensitivity to CPT although to a lesser extent than the wild-type LdTopIB.
Pre-induced 2% galactose MBY-3 cells, expressing the wild-type and LdTopIL/SY222F mutant were incubated with 20 μM CPT for a 2–24 h time span, and the surviving cells were plated in 2% galactose. Fig. 5A shows that the viability of cells expressing LdTopIL/SY222F was unaffected by CPT. In contrast, CPT induced a significant drop in the number of viable cells expressing wild-type LdTopIB, with a apparent decrease of more than 100 times in cell counting within 24 h. Based on this experiment, dose–response curves were carried out with wild-type (positive control) LdTopILD353A/S, LdTopIL/SN221S and LdTopIL/SY222F (negative control), at different concentrations of CPT, showing that LdTopIL/SY222F and LdTopILD353A/S were not sensitive at all to the drug. The IC50 values for CPT were estimated to be 0.24 and 1.82 μM for the wild-type and LdTopIL/SN221S mutant, respectively (Fig. 5B).
Fig. 5 –
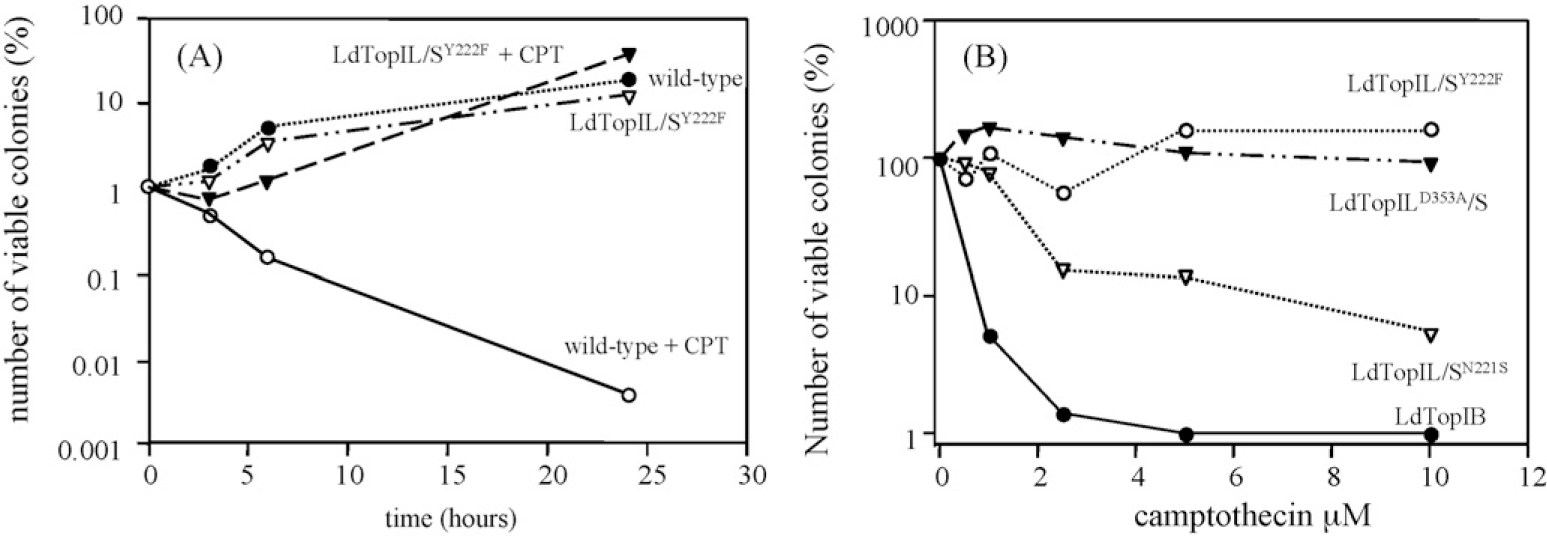
Time-course and dose–response curves of TopIB-transformed yeasts to the TopIB poison CPT. (A) Time-course of CPT lethality to TopIB-transformed yeasts. One suspension culture of LdTopIB and LdTopIL/SY222F were grown in SC URA-2% galactose, as indicated in Section 2. They were exposed to 20 μM of CPT or 1% DMSO; a colony of MBY3 yeast expressing the empty vector (mock) was used as control of drug or dissolvent resistance. Different aliquots were harvested at the indicated times, and platted onto a SC URA-2% dextrose agar plates for recovery: the surveillance data were plotted with SigmaPlot® software. (B) LdTopIB overexpressing MBY3 cells were exposed to different concentrations of CPT for 24 h, after which the cultures were allowed to recover in SC URA-2% dextrose agar plates. The viable colonies were counted up and the percentage of viability was plotted by SigmaPlot® software.
3.5. CPT-resistant D353A substitution does not stabilize DNA–topoisomerase cleavage complexes but N221S does
To further investigate the lack of sensitivity to CPT of LdTopILD353/S and LdTopIL/SN221S, CPT-induced DNA cleavage was examined over a range of protein concentrations. Induction of DNA cleavage complexes in the presence of different protein concentrations (0.1–3 μg of purified protein) of wild-type and the point mutations LdTopILD353A/S and LdTopIL/SN221S was tested in the PvuII/HindIII fragment of pBluescript SK(−) phagemid DNA (pSK). Fig. 6 shows that both wild-type and LdTopIL/SN221S displayed a protein-dependent increase in cleavage complexes formation in the presence of a fixed 100-μM CPT concentration. However, at this concentration CPT was not able to stabilize the cleavage complex with the LdTopILD353A/S substitution at all the protein concentrations assayed.
Fig. 6 –
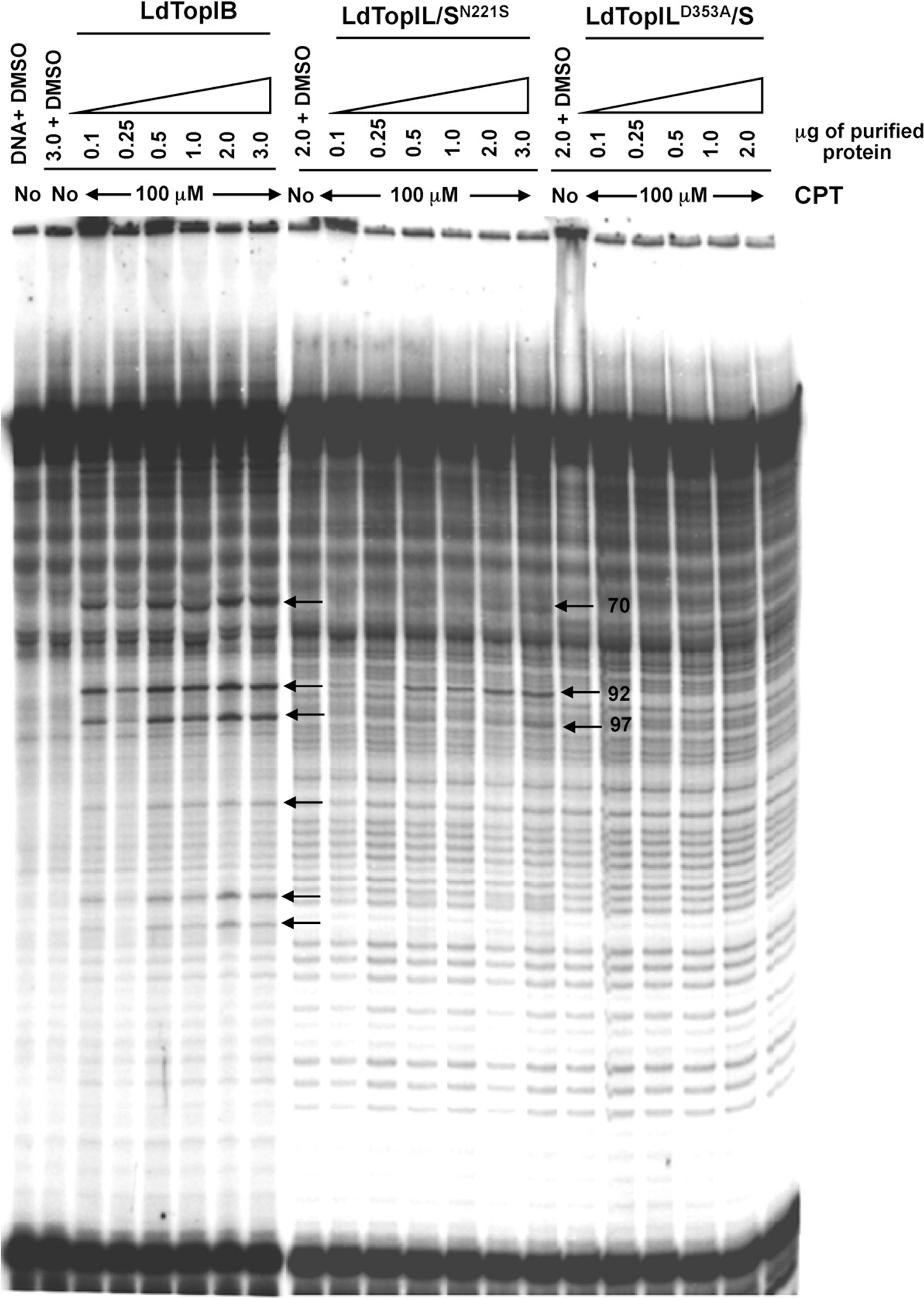
Expression of resistance to stabilization of the cleavage complex by CPT. The gel shows the specific pattern of cleaved bands of LdTopIB when exposed to 100 μM CPT with increasing amounts of protein, in contrast to its mutated counterparts, which exposition to CPT reveal how the drug is unable to stabilize cleavage complex in the presence of mutants LdTopILD353A/S and LdTopIL/SN221S, showing no cleaved bands in the electrophoresis gel except of a unique band for LdTopIL/SN221S.
The cleavage pattern exhibited by LdTopIB over the pSK vector differs slightly to that found for hTopIB, as previously described [19]. LdTopIL/SN221S cleaves DNA at the recognizable 70, 92 and 97 bp bands unlike LdTopILD353A/S mutation, that does not produce any differentiable band from those found in lane 1 lacking enzyme.
4. Discussion
X-ray analysis of a LdTopIB specimen containing parts of two subunits, showed that the catalytic machinery of the leishmanial enzyme was highly conserved when compared with that of the human counterpart, despite the structural differences forced by its bisubunit nature [15]. On the one hand, multiple alignments of the amino acid sequences of LdTopIB with the homologue from human and yeast, allows the unambiguous location of the conserved active tetrad amino acids within the putative core domain of the large LdTopIL subunit. On the other hand, the small LdTopIS subunit retains the Tyr-222 residue involved in the nucleophilic attack to the scissile strand of DNA. Despite these amino acids were pointed as putative parts of the active binding pocket of LdTopIB, their functionality have not been deciphered up to this report. The active site mutants of the large LdTopIL subunit LdTopILR314A/S, LdTopILR410A/S, LdTopILH453A/S were inactive and did not induce cytotoxicity to yeast when expressed. The mutant, including Lys-352 → Ala substitution (LdTopILK352A/S) was performed according to the role played by Lys-167 in vaccinia topoisomerase (vTopIB) [29] and hTopIB (Lys-532) [30]. LdTopILK352A/S contacts with a thymidine nucleotide at the −1 position of the scissile DNA strand and it yields one proton to the 5′-O of the leaving DNA strand. The heterologous expression of LdTopILK352A/S mutan in S. cerevisiae induced cell death, mimicking the effect of CPT. The lethality of this mutant in absence of CPT may be explained by the massive production of double-strand breaks in the overexpressing yeast strain, which lacks an effective rad52 repair system. Eventually, the mutant created substituting Tyr-222 by Phe in the LdTopIS subunit does not have the ability to attack DNA phosphodiester bonds, and thus to establish the cleavage complex lacking topoisomerase activity at all, and does not provide sensitivity to CPT to yeast expressing this mutant.
African trypanosomes and leishmanias are killed by CPT by establishing stable cleavage complexes between TopIB and DNA [31]. Based on the crystal structure of human hTopIB in presence of CPT and DNA, Redinbo et al. [7] have proposed a model of interactions amongst the components of the cleavage complex that can be assumed in trypanosomatid’s (Fig. 7). In this model, all of the amino-acid residues that line the drug-binding pocket are conserved in Leishmania. The side chains of Asp-353 in the large subunit and Asn-221 of the small one (Asp-532 and Asn-722, respectively, in the monomeric hTopIB) can establish hydrogen bonds with the carboxylate form of CPT setting the cleavable complex up, whereas Tyr-222 pro-motes the nucleophilic attack to the scissile strand of DNA [32]. Substitution of Asn and Asp residues by Ala-produced mutant proteins that entirely retain the relaxation activity but are insensitive to CPT prevented the hydrogen bonding with the drug [33].
Fig. 7 –
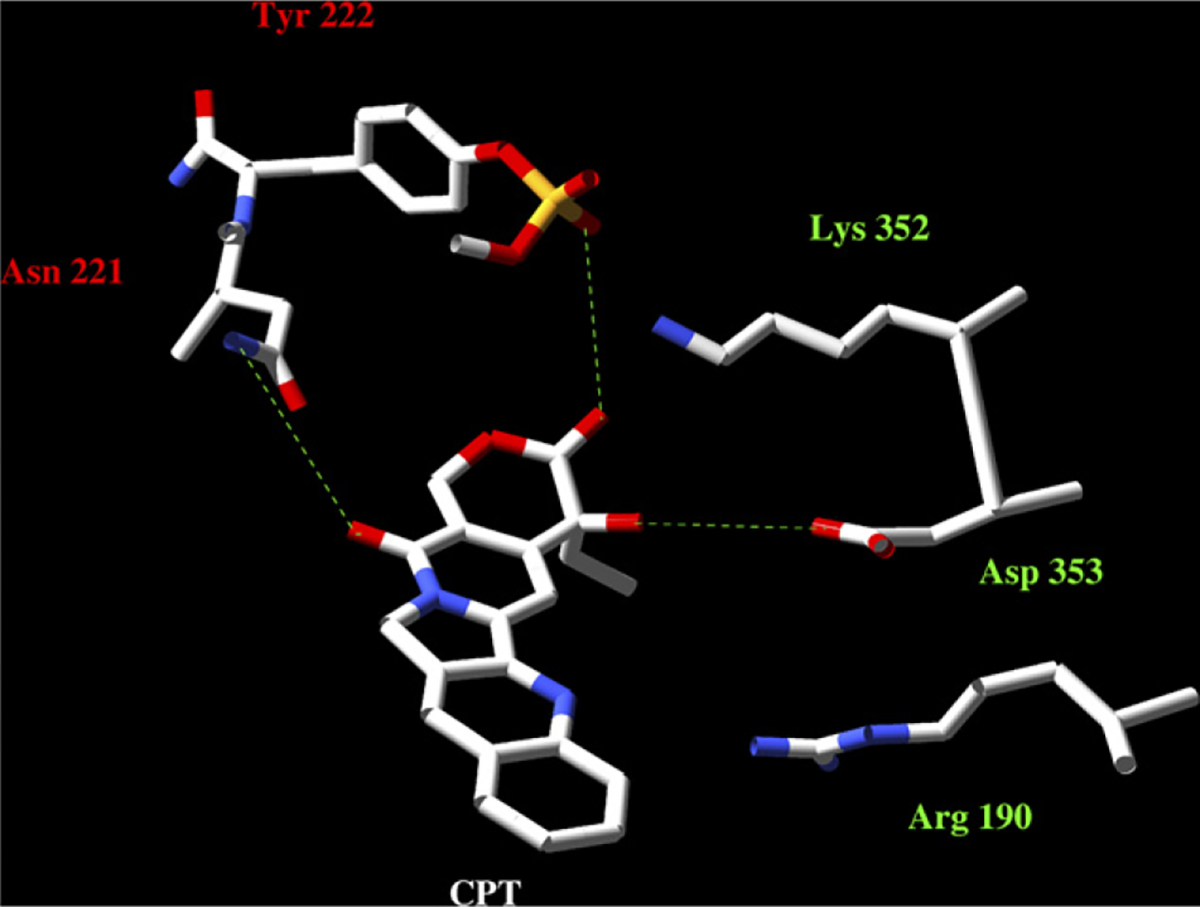
Predicted 3D structure of the CPT binding pocket in LdTopIB. The homology modelling was made using the Swiss pdb database. The PDB ID 1t8i file corresponding to hTopIB − 70 kDa in a non-covalent complex with a 22-mer DNA duplex – was used as template. The final plots were obtained by using the MOLMOL programme. As the image displays, the residues of our study bound to the CPT E-ring (Asp-353) and the C17 (Asn-221); the presence of Arg-190 (Arg-364 in hTopIB) opens the possibility of interaction with the drug [32].
Other amino acids substitutions described in mammalian cells that lead CPT resistance are Glu-418 [34], Ala-653 [35] and Thr-729 [36], despite their position from the cleavage site are not close enough to directly interact with the drug. Interestingly, these amino acids are not conserved in the leishmanial enzyme at all. Thr-729 is substituted by an Ile residue, Glu-418 is changed by an Asp and Ala-653 is placed within the linker domain of the human topoisomerase, which homology is completely lost in Leishmania. The absence of these amino acids can explain in part the reduced sensitivity of the leishmanial enzyme to CPT in in vitro assays. It deserves special attention the phylogenetically conserved Thr-719 that heads the “SKxxY” catalytic motif. Thr-719 contributes in maintaining the stability of the covalent intermediate formed between the enzyme and DNA [37]. In Leishmania Thr-217 is placed in the small LdTopIS subunit and its substitution by Ala produces an unusual accumulation of nicked DNA (data not shown), confirming the previous results obtained by Megonigal et al. [38].
It has been pointed that amino acids included into the linker domain of mammalian topoisomerases can be involved in CPT sensitivity. A partially truncated human topoisomerase lacking the linker domain (topo70ΔL-monomer), conserved the enzymatic activity but lost the sensitivity to CPT. Despite there is not a clear linker domain in the bisubunit TopIB of trypanosomatids, it has been stated that this role can be played by the N-extension end of the small LdTopIS subunit. Sequential deletion experiments demonstrated that the absence of a RPPVV pentapeptide located at positions 175–179 this subunit produces an enzymatic specimen which conserves the relaxation activity but is CPT resistant. An additional RS deletion (residues 180 and 181 of the small subunit) produces an inactive protein [19]. Since the CPT was unable to trap covalent complexes between the truncated R180End protein and DNA, it is likely that a reduced affinity of the enzyme-DNA covalent complex for the drug is probably due to the conformational changes introduced in the protein.
From these results, we conclude that all of the amino acids of the active tetrad are conserved in both monomers of the bisubunit LdTopIB. The CPT resistance provided by D352A and N221A substitutions in combination with the RPPVV motif should be useful to model interactions between the drug and trypanosomatid TopIB/DNA complexes. Further studies on putative amino acids involved in CPT resistance [39] (conserved FxGR motif in the core domain of Leishmania in positions 187–190) are being carrying out, to perform a map of drug interactions that can be of use for testing and developing new topoisomerase cleaving drugs against these organisms.
Acknowledgements
This research was partially supported by MEC (grant AGL2006-07420/GAN) and Instituto de Salud Carlos III (grant PI06302) from Ministerio de Salud y Consumo from the Spanish Kingdom. Rosario Díaz González is granted with a FPI scholarship funded by Ministerio de Educación y Ciencia (MEC).
Abbreviations:
- TopIB
type IB DNA topoisomerase
- TopII
type II DNA topoisomerase
- LdTopIB
Leishmanial DNA topoisomerase
- LdTopIS
small subunit of leishmanial DNA topoisomeraseIB
- LdTopIL
large subunit of leishmanial DNA topoisomerase
- yTopIB
yeast DNA topoisomerase
- CPT
camptothecin
- DMSO
dimethylsulfoxide
REFERENCES
- [1].Croft S, Coombs GH. Leishmaniasis—current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol 2003;19:502–8. [DOI] [PubMed] [Google Scholar]
- [2].Berman J Clinical status of agents being developed for leishmaniasis. Exp Opin Invest Drugs 2005;14:1337–46. [DOI] [PubMed] [Google Scholar]
- [3].Balaña-Fouce R, Cubría JC, Reguera RM, Ordóñez D. The pharmacology of leishmaniasis. Gen Pharmacol 1998;30:435–43. [DOI] [PubMed] [Google Scholar]
- [4].Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001;70:369–413. [DOI] [PubMed] [Google Scholar]
- [5].Corbett K, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct 2004;33:95–118. [DOI] [PubMed] [Google Scholar]
- [6].Wang J DNA topoisomerases. Annu Rev Biochem 1996;65:635–92. [DOI] [PubMed] [Google Scholar]
- [7].Redinbo M, Stewart L, Kuhn P, Champoux JJ, Hol WG. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998;279:1504–13. [DOI] [PubMed] [Google Scholar]
- [8].Stewart L, Ireton GC, Champoux JJ. The domain organization of DNA topoisomerase I. J Biol Chem 1996;271:7602–8. [DOI] [PubMed] [Google Scholar]
- [9].Stewart L, Ireton GC, Champoux JJ. A functional linker in human topoisomerase I is required for maximum sensitivity to camptothecin in a DNA relaxation assay. J Biol Chem 1999;274:32950–6. [DOI] [PubMed] [Google Scholar]
- [10].Bodley A, Chakraborty AK, Xie S, Burri C, Shapiro TA. An unusual type IB topoisomerase from African trypanosomes. Proc Natl Acad Sci USA 2003;100:7539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Villa H, Otero Marcos AR, Reguera RM, Balaña-Fouce R, García-Estrada C, Pérez-Pertejo Y, et al. A novel active DNA topoisomerase I in Leishmania donovani. J Biol Chem 2003;278:3521–6. [DOI] [PubMed] [Google Scholar]
- [12].Reguera R, Redondo CM, Gutiérrez de Prado R, Pérez-Pertejo Y, Balaña-Fouce R. DNA topoisomerase I from parasitic protozoa: a potential target for chemotherapy. Biochim Biophys Acta 2006;1759:117–31. [DOI] [PubMed] [Google Scholar]
- [13].Das BB, Sengupta T, Ganguly A, Majumder HK. Topoisomerases of kinetoplastid parasites: why so fascinating? Mol Microbiol 2006;62:917–27. [DOI] [PubMed] [Google Scholar]
- [14].Krogh B, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol Cell Biol 2000;5:1035–41. [DOI] [PubMed] [Google Scholar]
- [15].Davies D, Mushtaq A, Interthal H, Champoux JJ, Hol WG. The structure of the transition state of the heterodimeric topoisomerase I of Leishmania donovani as a vanadate complex with nicked DNA. J Mol Biol 2006;357:1202–10. [DOI] [PubMed] [Google Scholar]
- [16].Tamura H, Kohchi C, Yamada R, Ikeda T, Koiwai O, Patterson E, et al. Molecular cloning of a cDNA of a camptothecin-resistant human DNA topoisomerase I and identification of mutation sites. Nucl Acids Res 1991;19:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Knab A, Fertala J, Bjornsti MA. Mechanisms of camptothecin resistance in yeast DNA topoisomerase I mutants. J Biol Chem 1993;268:22322–30. [PubMed] [Google Scholar]
- [18].Marquis JF, Hardy I, Olivier M. Topoisomerase I amino acid substitutions, Gly185Arg and Asp325Glu, confer camptothecin resistance in Leishmania donovani. Antimicrob Agents Chemother 2005;49:1441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Díaz-González R, Pérez-Pertejo Y, Redondo CM, Pommier Y, Balaña-Fouce R, Reguera RM. Structural insights on the small subunit of DNA topoisomerase I from the unicellular parasite Leishmania donovani. Biochimie 2007;89:1517–27. [DOI] [PubMed] [Google Scholar]
- [20].Hann CL, Carlberg AL, Bjornsti MA. Intragenic suppressors of mutant DNA topoisomerase I-induced lethality diminish enzyme binding of DNA. J Biol Chem 1998;273:31519–27. [DOI] [PubMed] [Google Scholar]
- [21].Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005;309:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol 1983;153:163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jazwinski SM. Preparation of extracts from yeast. Methods Enzymol 1990;182:154–74. [DOI] [PubMed] [Google Scholar]
- [25].Bradford M A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- [26].Thrash C, Voelkel K, DiNardo S, Sternglanz R. Identification of Saccharomyces cerevisiae mutants deficient in DNA topoisomerase I activity. J Biol Chem 1984;259:1375–7. [PubMed] [Google Scholar]
- [27].Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, et al. Cellular topoisomerase I inhibition and antiproliferative activity by MJ-III-65 (NSC 706744), an indenoisoquinoline topoisomerase I poison. Mol Pharmacol 2005;67:523–30. [DOI] [PubMed] [Google Scholar]
- [28].Woo M, Vance JR, Marcos AR, Bailly C, Bjornsti MA. Active site mutations in DNA topoisomerase I distinguish the cytotoxic activities of camptothecin and the indolocarbazole, rebeccamycin. J Biol Chem 2002;277:3813–22. [DOI] [PubMed] [Google Scholar]
- [29].Wittschieben J, Petersen BO, Shuman S. Replacement of the active site tyrosine of vaccinia DNA topoisomera by glutamate, cysteine or histidine converts the enzyme into a site-specific endonuclease. Nucl Acids Res 1998;26:490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Interthal H, Quigley PM, Hol WG, Champoux JJ. The role of lysine 532 in the catalytic mechanism of human topoisomerase I. J Biol Chem 2004;279:2984–92. [DOI] [PubMed] [Google Scholar]
- [31].Bodley AL, Shapiro TA. Molecular and cytotoxic effects of camptothecin, a topoisomerase I inhibitor, on trypanosomes and Leishmania. Proc Natl Acad Sci USA 1995;92:3726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Staker B, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 2002;99:15387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lynn R, Bjornsti MA, Caron PR, Wang JC. Peptide sequencing and site-directed mutagenesis identify tyrosine-727 as the active site tyrosine of Saccharomyces cerevisiae DNA topoisomerase I. Proc Natl Acad Sci USA 1989;86:3559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fiorani P, Chillemi G, Losasso C, Castelli S, Desideri A. The different cleavage DNA sequence specificity explains the camptothecin resistance of the human topoisomerase I Glu418Lys mutant. Nucl Acids Res 2006;34:5093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fiorani P, Bruselles A, Falconi M, Chillemi G, Desideri A, Benedetti P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J Biol Chem 2003;278:43268–75. [DOI] [PubMed] [Google Scholar]
- [36].Kubota N, Kanzawa F, Nishio K, Takeda Y, Ohmori T, Fujiwara Y, et al. Detection of topoisomerase I gene point mutation in CPT-11 resistant lung cancer cell line. Biochem Biophys Res Commun 1992;188:571–7. [DOI] [PubMed] [Google Scholar]
- [37].Fiorani P, Amatruda JF, Silvestri A, Butler RH, Bjornsti MA, Benedetti P. Domain interactions affecting human DNA topoisomerase I catalysis and camptothecin sensitivity. Mol Pharmacol 1999;56:1105–15. [DOI] [PubMed] [Google Scholar]
- [38].Megonigal M, Fertala J, Bjornsti MA. Alterations in the catalytic activity of yeast DNA topoisomerase I result in cell cycle. J Biol Chem 1997;272:12801–8. [DOI] [PubMed] [Google Scholar]
- [39].Urasaki Y, Laco G, Takebayashi Y, Bailly C, Kohlhagen G, Pommier Y. Use of camptothecin-resistant mammalian cell lines to evaluate the role of topoisomerase I in the antiproliferative activity of the indolocarbazole, NB-506, and its topoisomerase I binding site. Cancer Res 2001;61: 504–8. [PubMed] [Google Scholar]