

Abstract
A cDNA clone encoding a quinone reductase (QR) from the white rot basidiomycete Phanerochaete chrysosporium was isolated and sequenced. The cDNA consisted of 1,007 nucleotides and a poly(A) tail and encoded a deduced protein containing 271 amino acids. The experimentally determined eight-amino-acid N-terminal sequence of the purified QR protein from P. chrysosporium matched amino acids 72 to 79 of the predicted translation product of the cDNA. The Mr of the predicted translation product, beginning with Pro-72, was essentially identical to the experimentally determined Mr of one monomer of the QR dimer, and this finding suggested that QR is synthesized as a proenzyme. The results of in vitro transcription-translation experiments suggested that QR is synthesized as a proenzyme with a 71-amino-acid leader sequence. This leader sequence contains two potential KEX2 cleavage sites and numerous potential cleavage sites for dipeptidyl aminopeptidase. The QR activity in cultures of P. chrysosporium increased following the addition of 2-dimethoxybenzoquinone, vanillic acid, or several other aromatic compounds. An immunoblot analysis indicated that induction resulted in an increase in the amount of QR protein, and a Northern blot analysis indicated that this regulation occurs at the level of the qr mRNA.
The wood-rotting basidiomycete fungus Phanerochaete chrysosporium degrades polymeric lignin (12, 21, 30) and various aromatic pollutants, including chlorinated phenols, dioxins, and polyaromatic hydrocarbons (9, 23, 26, 40). A wide variety of oxidized metabolic intermediates are generated during the degradation of lignin, lignin model compounds, and aromatic pollutants by this organism. These intermediates include substituted quinones, hydroquinones, benzaldehydes, benzoic acids, and ring-opened fragments (12, 15, 27, 30), which are metabolized further by intracellular processes (7, 42, 55). The extracellular peroxidases that are involved in the initial oxidative steps of lignin and pollutant degradation are well-characterized (15, 21, 27, 30); however, much less is known about the intracellular enzymes involved in the further degradation of monomeric intermediates, such as quinones, hydroquinones, and benzaldehydes. Recent work, including the elucidation of metabolic pathways for the degradation of several aromatic pollutants by P. chrysosporium (53, 54), has suggested that intracellular enzymes are involved in the reduction of quinones. Since benzoquinones are generated by the peroxidase-catalyzed oxidation of lignin and appear to be key intermediates in the degradation of aromatic compounds by P. chrysosporium (29, 42, 44, 51, 53, 54), we have been examining the reduction of benzoquinones by this organism.
The reduction of methoxylated, lignin-derived quinones by P. chrysosporium appears to be catalyzed by an intracellular quinone reductase (QR) or QRs (6, 7, 11, 13), and we have purified and partially characterized one intracellular, NAD(P)H-dependent QR from this organism (6, 7). The soluble protein is a 44-kDa dimer with two similar 22-kDa subunits and appears to contain two flavin mononucleotide (FMN) moieties per dimer. A variety of methoxylated quinones and other electron acceptors serve as substrates for this enzyme (6, 7). The stoichiometry of NADH oxidation to 2,6-dimethoxy-1,4-benzoquinone (DMBQ) reduction is 1:1, and the enzyme apparently catalyzes the reduction of quinones to hydroquinones via a ping-pong, steady-state, kinetic mechanism (6). NADH and NADPH are equally efficient as electron donors, and the enzyme is competitively inhibited, with respect to NADH, by both dicoumarol and Cibacron Blue (6). The latter properties are similar to properties reported for the mammalian QR DT-diaphorase (16, 17, 37).
Here, we describe the isolation and characterization of a cDNA clone encoding the P. chrysosporium QR, as well as studies on the regulation of QR synthesis.
MATERIALS AND METHODS
Culture conditions.
Stock cultures of P. chrysosporium OGC101 (21), a derivative of strain BKM-F-1767 (unpublished data), were maintained on malt extract-yeast extract-Vogel medium slants as previously described (22). Stationary cultures were grown from conidial inocula either in 25 ml of medium in 250-ml flasks or in 50 ml of medium in 500-ml flasks for 2 days at 37°C (7, 42). The medium used has been described previously (31) and was supplemented with 2% glucose, 10 mM dimethyl succinate (pH 4.5), and either 12 mM (high-nitrogen [HN] medium) or 1.2 mM (low-nitrogen medium) ammonium tartrate as the nitrogen source. The 48-h stationary cultures were homogenized for 20 s in a Waring blender, and 100-ml portions of the homogenates were transferred to 250-ml flasks and grown at 28°C on a rotary shaker (150 rpm). After 48 h, inducers were added, and the cells were harvested at the times indicated below.
Isolation of the qr cDNA clone.
A λgt10 cDNA library was constructed from P. chrysosporium mRNA as described previously for a λgt11 library (38). Cultures were grown in 1 liter of low-nitrogen medium as described above. Five-day-old cells were harvested by filtration, and RNA was isolated as described below and previously (8). Poly(A) RNA was isolated by passing the total RNA over oligo(dT)-cellulose (2). cDNA was synthesized as described previously (24, 38), and EcoRI adapter molecules were ligated to cDNA products (38, 43). Finally, cDNA products were ligated to λGT10 arms, packaged in vitro, and amplified as described previously (38, 43). N-terminal sequencing of purified P. chrysosporium QR protein (7) was carried out by D. McMillen, Biotechnology Laboratory, Institute of Molecular Biology, University of Oregon. A fully degenerate 24-mer oligonucleotide, corresponding to the first eight N-terminal amino acids of QR (PKVAIIIY), was synthesized at the Oregon Regional Primate Research Center, Beaverton. The oligonucleotide was end labeled with [γ-32P]dATP (Andotek) by using T4 polynucleotide kinase (New England Biolabs) and was used to screen plaque lifts of the library by standard methods (43). Positive plaques were purified, and the DNA was isolated with a Lambda Midi Kit (Qiagen) and digested with EcoRI to release the cDNA insert. The cDNA insert was subcloned into the plasmid vector Bluescript SKII+ (Stratagene) to generate pcQR1. Plasmid DNA was purified and sequenced in both directions with an Applied Biosystems Inc. model 373 stretch automated sequencer by using primer walking (47). A nucleotide sequence analysis was performed with MacVector 6.0 (Oxford, Molecular).
In vitro transcription and translation.
pQRT1 was generated by isolating the 856-bp HincII-XbaI fragment from pcQR1 and ligating it into HincII-XbaI-restricted pBluescript SKII− (Stratagene). Thus, in pQRT1 the most 5′ ATG of the qr cDNA was 78 bp downstream of the start of the Bluescript T7 promoter. pQRT2 was constructed by isolating the 662-bp PstI-XbaI fragment from pcQR1 and ligating it into PstI-XbaI-digested pBluescript SKII− (Fig. 1). Thus, in pQRT2 the downstream ATG of the qr cDNA was 130 bp downstream of the T7 promoter. The plasmid DNAs used for transcription-translation reactions were purified by using a Plasmid Midi Kit (Qiagen).
FIG. 1.

Nucleotide sequence of the P. chrysosporium qr cDNA. The amino acid sequence of the predicted translation product is shown below the nucleotide sequence. The qr coding region is flanked by a 59-bp 5′ noncoding region and a 132-bp 3′ noncoding region, excluding the poly(A) tail. The initiation codon is followed by an apparent 71-amino-acid leader sequence, which is underlined. The experimentally determined N-terminal amino acid sequence of the isolated QR protein is overlined. The two KEX2 cleavage sites in the leader sequence are enclosed in boxes. The dots indicate the potential glycosylation sites at residues 11 and 223.
Transcription-translation reactions were performed by using the TNT Coupled Reticulocyte Lysate System (Promega). Coupled transcription-translation reaction mixtures (25 μl) contained plasmid DNA (2 μg) and translation grade l-[35S]methionine (4 μl; specific activity, 1,175 Ci/mmol; New England Nuclear). Following incubation at 30°C for 90 to 120 min, 5 μl of the reaction mixture was added to 20 μl of the protein sample buffer, and 10 μl was electrophoresed in a sodium dodecyl sulfate–12.5% polyacrylamide gel electrophoresis (SDS-PAGE) gel (33) that included broad-range, prestained, molecular weight markers (Bio-Rad). The gels obtained were fixed, vacuum dried, and visualized by fluorography.
Chemicals.
The compounds used in induction experiments were obtained commercially and used directly. 2-Methoxybenzoquinone (MBQ) and DMBQ were prepared by silver oxide oxidation of the respective hydroquinones (25).
Enzyme extracts.
Cells were harvested by vacuum filtration through Miracloth (Calbiochem), washed with ice-cold distilled water, and stored at −80°C. Frozen cells (1 g) were broken by grinding in a mortar and pestle with sand. Subsequently, extraction buffer (50 mM sodium phosphate [pH 7.0] containing 1 mM EDTA and 0.004% phenylmethylsulfonyl fluoride) was added with stirring. The homogenate was centrifuged at 17,300 × g for 20 min, and the resulting supernatant was centrifuged at 105,000 × g for 30 min.
Enzyme assays.
The QR activity in the 105,000-×-g supernatant was measured by monitoring the oxidation of NADH at 340 nm. The standard reaction mixtures (1 ml) contained 50 mM sodium citrate (pH 6.0), 100 μM DMBQ, and enzyme (10 to 100 μl). Reactions were initiated by adding 200 μM NADH. Assays were carried out at room temperature by using a Shimadzu model UV260 spectrophotometer. Protein concentrations were determined by the bicinchoninic acid method (45) by using bovine serum albumin as the standard.
Immunoblot analysis.
SDS-PAGE (Mini-PROTEAN; Bio-Rad) of the 105,000-×-g supernatant was performed by using a 12% polyacrylamide resolving gel as described previously (33). Electrophoretic transfers to nitrocellulose were carried out as described previously (49) by using a transfer apparatus (Bio-Rad). Rabbit polyclonal antibody was raised against purified QR as previously described (7). Nitrocellulose transfers were incubated with the rabbit antibody and then with goat anti-rabbit immunoglobulin G conjugated to alkaline phosphatase. Alkaline phosphatase activity was detected by the 5-bromo-4-chloro-3-indolyl phosphate nitroblue tetrazolium assay (5).
RNA preparation and Northern blot hybridization.
Cells from induced and uninduced cultures were filtered through Miracloth, washed with cold distilled water, frozen in liquid N2, and stored at −80°C. Total RNA was isolated by breaking the cells in 1.5-ml Sarstedt tubes containing 1 g of glass beads in the presence of 1 ml of TRI reagent (Molecular Research Center, Inc.) as previously described (8). After spectral quantitation, 20 μg of the RNA was denatured in 2.2 M formaldehyde containing 50% formamide for 15 min at 68°C and electrophoresed in a denaturing gel (0.22 M formaldehyde, 1% agarose) in buffer containing 0.22 M formaldehyde, as described previously (50). The RNA was transferred to a Magna NT membrane (Micron Separations, Inc.). pcQR1 and the P. chrysosporium glyceraldehyde-P-dehydrogenase (gpd) gene (35) were used as templates for randomly primed synthesis (18) of [α-32P]dCTP-labeled probes with a multiprime DNA labeling kit (Amersham). Northern blot hybridizations were performed as previously described (8).
Nucleotide sequence accession number. The cDNA sequence reported in this paper has been submitted to the GenBank library under the accession no. AF106939.
RESULTS AND DISCUSSION
During degradation of polymeric lignin and aromatic pollutants by P. chrysosporium, substituted quinones are generated and reduced to hydroquinones, which undergo further metabolism (6, 7, 11, 13). Thus, the quinones and their corresponding hydroquinones are key intermediates in the degradative process (11, 29, 44, 51, 53, 54). Previously, we described purification and the reaction mechanism of an intracellular QR from P. chrysosporium, which is expressed during both primary and secondary metabolic growth (7). This enzyme reduces methoxyquinones and other substrates to their hydroquinones (6, 7). In order to better understand the mechanism and role of this enzyme in lignin and pollutant degradation, we cloned the qr cDNA from P. chrysosporium and studied the regulation of QR expression.
cDNA sequence.
A degenerate oligonucleotide that was based on the experimentally determined N-terminal sequence of purified QR was used to probe a P. chrysosporium λgt10 cDNA library. The sequence of the cDNA derived from a positive plaque and its predicted translation product are shown in Fig. 1. The cDNA consists of 1,007 nucleotides and a poly(A) tail. The 816-bp open reading frame encodes a 271-amino-acid protein and a TAG stop codon. The coding region is flanked by a 59-bp 5′ noncoding region and a 132-bp noncoding region between the stop codon and the poly(A) sequence. The experimentally determined N-terminal sequence of QR, consisting of the first eight amino acids of the mature protein (PKVAIIIY), corresponds to the sequence of the predicted translation product of the cDNA from Pro-72 to Tyr-79 immediately following Met-71, indicating that this cDNA encodes the QR protein isolated (Fig. 1).
The molecular mass of the purified QR monomer, as determined by SDS-PAGE, is ∼21.4 kDa (7). The calculated molecular mass of the predicted translation product, beginning at the first Met encoded by the ATG at nucleotide 60 of the cDNA, is 28,278 Da; this value is 1.32-fold greater than the molecular mass of the isolated protein. In contrast, the calculated Mr of the predicted translation product of the cDNA, beginning with Pro-72, is 21,236, which is more than 99% of the Mr of the QR monomer, as determined by SDS-PAGE. These results suggest that the mature QR protein may be derived from a precursor containing an N-terminal 71-amino-acid leader sequence. The putative leader sequence contains two potential KEX2 cleavage sites, each consisting of a dibasic pair of amino acids (20), as well as numerous -X-Pro and -X-Ala sequences, which are potential dipeptidyl aminopeptidase cleavage sites (34) (Fig. 1). Both KEX2 and dipeptidyl aminopeptidase are known to be leader-processing enzymes (20, 34).
Table 1 shows the predicted amino acid compositions of the putative mature protein with an Mr of 21,236 and the putative leader peptide with an Mr of 7,060. Ala (25.4%) and Pro (26.8%) comprise 52.2% of the amino acids in the putative leader. The Ala and Pro residues occur as -X-Ala and -X-Pro sequences, which are known to be cleavage sites for dipeptidyl aminopeptidase (34). Ala (12.4%) and Gly (11.9%) are the most abundant amino acids in the putative mature protein. The preponderance of Asp plus Glu (8.46%) over Lys plus Arg (6.96%) in the predicted mature protein suggests a deduced pI of 5.76 (Table 1).
TABLE 1.
Amino acid compositions of the mature deduced QR protein and the leader peptide
| Amino acid(s) | Mature proteina
|
Leader peptideb
|
||
|---|---|---|---|---|
| No. | % | No. | % | |
| Nonpolar amino acids | ||||
| A | 25 | 12.44 | 18 | 25.35 |
| V | 11 | 5.47 | 2 | 2.82 |
| L | 14 | 6.97 | 1 | 1.41 |
| I | 13 | 6.47 | 1 | 1.41 |
| P | 15 | 7.46 | 19 | 26.76 |
| M | 2 | 1.00 | 2 | 2.82 |
| F | 11 | 5.47 | 1 | 1.41 |
| W | 5 | 2.49 | 0 | 0.00 |
| Polar amino acids | ||||
| G | 24 | 11.94 | 1 | 1.41 |
| S | 13 | 6.47 | 3 | 4.23 |
| T | 14 | 6.97 | 5 | 7.04 |
| C | 0 | 0.00 | 1 | 1.41 |
| Y | 8 | 3.98 | 0 | 0.00 |
| N | 4 | 1.99 | 2 | 2.82 |
| Q | 6 | 2.99 | 2 | 2.82 |
| Acidic amino acids | ||||
| D | 3 | 1.49 | 2 | 2.82 |
| E | 14 | 6.97 | 5 | 7.04 |
| Basic amino acids | ||||
| K | 11 | 5.47 | 5 | 7.04 |
| R | 3 | 1.49 | 1 | 1.41 |
| H | 4 | 1.99 | 0 | 0.00 |
| Total | 200 | 71 | ||
The calculated molecular weight is 21,235.62, and the estimated pI is 5.76.
The calculated molecular weight is 7,060.37, and the estimated pI is 4.95.
The cDNA corresponding to the isolated QR protein, beginning at Pro-72, has a G+C content of 65%, and the putative leader sequence has a G+C content of 60.7%, compared with a G+C content of 59% for the total P. chrysosporium genome (39). The 3′ and 5′ noncoding regions of the qr cDNA have G+C contents of 45 and 59%, respectively. A genomic Southern blot in which the qr cDNA was used as a probe (data not shown) indicated that a single copy of the qr gene is present in the P. chrysosporium genome.
Translation start site.
In order to determine experimentally whether translation of qr begins at Met-1 or Met-71, coupled transcription-translation of the cDNA was carried out by using 35S-labeled Met, and the product was analyzed by SDS-PAGE and fluorography. When pQRT1 (in which the most 5′ ATG encoding Met-1 is located 78 bp downstream of the T7 promoter) was used in the transcription-translation experiment, an abundant 35S-labeled translation product was obtained (Fig. 2). This product had a molecular mass of ∼27 kDa, which is within 5% of the calculated molecular mass of the larger predicted translation product. In contrast, when pQRT2 (in which the second ATG encoding Met-71 is located 130 bp downstream of the T7 promoter) was used in the transcription-translation experiment, no significant translation products were observed. Likewise, significant translation products were not observed in the control that lacked DNA (Fig. 2). These results indicate that translation can be initiated from the first ATG, although further work is needed to prove that translation cannot be initiated from the second ATG. The results also suggest that translation of the qr gene probably begins at the first ATG, which is located at nucleotide 60 in the cDNA sequence, and that QR is synthesized as a proenzyme with a 71-amino-acid leader sequence that is removed by enzymatic processing. The results further suggest that the putative KEX2 and/or dipeptidyl aminopeptidase sites observed in the sequence may be authentic cleavage sites. QR has been isolated as a cytosolic protein (7); however, very few cytosolic proteins contain leader sequences. There is a potential N-glycosylation site, conforming to the general rule Asn-X-Thr/Ser (32), beginning at amino acid 11 of the leader sequence (Fig. 1). If this site is glycosylated, then the proenzyme may be membrane bound via the leader sequence and the mature protein may be released to the cytosol or to an internal compartment during processing. There is also a putative N-glycosylation site at Asn-223. While no obvious membrane-spanning region is found in the leader sequence, there is a free cysteine at position 3 in the leader sequence, which may be a potential site for palmitoylation of this protein. Both G proteins (36) and Src proteins (41) are palmitoylated at a site near the N terminus.
FIG. 2.

SDS-PAGE of the in vitro transcription-translation products obtained with the qr cDNA. Reactions and electrophoresis were performed as described in the text. Lane 1, control reaction mixture lacking DNA; lane 2, reaction mixture containing pQRT1, which included the upstream ATG of pcQR1; lane 3, reaction mixture containing pQRT2, which included only the downstream ATG of pcQR1. The prestained molecular mass markers used were carbonic anhydrase (34.8 kDa), soybean trypsin inhibitor (28.3 kDa), and lysozyme (20.4 kDa).
Induction of QR activity.
Table 2 shows the effects on QR activity resulting from the addition of a wide variety of aromatic compounds to 3-day-old HN agitated cultures of P. chrysosporium. The most effective inducers of QR activity, vanillic acid and ferulic acid, are products of degradation of guaiacyl lignin by white rot fungi (28). The fact that QR activity was induced by ferulic and vanillic acids suggests that both of these compounds also are metabolized by P. chrysosporium during primary growth. Several quinones and hydroquinones also are effective inducers of QR activity. Veratric acid, which differs from vanillic acid in methylation at position 4, is a much weaker inducer than vanillic acid. Vanillin is a weaker inducer than vanillic acid, and vanillyl alcohol is not an effective inducer. All compounds that exhibit the 3-methoxy-4-hydroxy substitution pattern except vanillyl alcohol are effective inducers. These compounds may be good inducers in themselves, or they may be metabolized to methoxyquinones, which are effective inducers. The compounds listed in Table 2 which are not effective inducers of QR activity may not be converted readily to methoxyquinones. Most chlorophenols and nitroaromatic compounds are not effective inducers of QR activity; two exceptions are 4-chlorophenol and 2-nitrophenol (Table 2). 1,4-Benzoquinone is produced during metabolism of 4-nitrophenol by Moraxella sp., which also produces an inducible QR (46). Since the oxidized and reduced forms of various quinoid compounds are equally effective as inducers and since these forms probably undergo rapid interconversion, either one or both forms might be recognized by the regulatory system.
TABLE 2.
Induction of QR by various aromatic compoundsa
| Inducer | Concn (mM) | Induction (fold)b |
|---|---|---|
| Quinones | ||
| p-Benzoquinone | 0.2 | 12 |
| MBQ | 0.2 | 12 |
| DMBQ | 0.2 | 12 |
| 1,4-Naphthaquinone | 0.2 | 10 |
| 3,5-Di-tert-buryl-1,2-orthoquinone | 0.2 | 1.3 |
| 2-Chloroquinone | 0.2 | 2.2 |
| Hydroquinones | ||
| p-Hydroquinone | 0.2 | 6.8 |
| 2-Methoxyhydroquinone | 0.2 | 11 |
| 2,6-Dimethoxyhydroquinone | 0.2 | 11 |
| Aromatic acids | ||
| Benzoic acid | 2.0 | 1 |
| p-Hydroxybenzoic acid | 2.0 | 8.8 |
| Vanillic acid | 2.0 | 25 |
| 3,4-Dihydroxybenzoic acid | 2.0 | 1.4 |
| 3,5-Dihydroxybenzoic acid | 2.0 | 1 |
| Syringic acid | 2.0 | 1.3 |
| Ferulic acid | 2.0 | 28 |
| Veratic acid | 2.0 | 4.0 |
| Aromatic aldehyde | ||
| Vanillyl aldehyde | 2.0 | 15 |
| Aromatic alcohols | ||
| Veratryl alcohol | 2.0 | 1 |
| Vanillyl alcohol | 2.0 | 1 |
| Phenols | ||
| Phenol | 0.2 | 1 |
| 4-Chlorophenol | 0.2 | 7.3 |
| 2,4-Dichlorophenol | 0.2 | 1 |
| 2,6-Dichlorophenol | 0.2 | 2.2 |
| 2,6-Dichlorophenol | 0.2 | 2.0 |
| 3,4-Dichlorophenol | 0.2 | 1.9 |
| 3,5-Dichlorophenol | 0.2 | 1 |
| 2,4,5-Trichlorophenol | 0.2 | 1.9 |
| 2,4,6-Trichlorophenol | 0.2 | 1 |
| Pentachlorophenol | 0.2 | 1 |
| Nitroaromatic compounds | ||
| Nitrobenzene | 0.2 | 1 |
| 2-Nitrophenol | 0.2 | 5.7 |
| 4-Nitrophenol | 0.2 | 1.8 |
| 2,4-Dinitrobenzoic acid | 0.2 | 1 |
| 3,5-Dinitrobenzoic acid | 0.2 | 1 |
| 2,4-Dinitrotoluene | 0.2 | 1 |
| 2,6-Dinitrotoluene | 0.2 | 1 |
| 4-Nitrocatechol | 0.2 | 1 |
| 2,4,6-Trinitrotoluene | 0.2 | 2.2 |
Cells were grown for 2 days in HN medium, and then compounds were added as described in the text; 16 h after the compounds were added, cells were harvested and broken, and the enzyme was assayed as described in the test.
The level of induction was determined by dividing the specific activity of an induced culture by the specific activity of the uninduced control.
A time course for the increase in cytosolic QR activity following addition of the inducers vanillic acid (2 mM) and MBQ (0.2 mM) to 3-day-old HN agitated cultures of P. chrysosporium is shown in Fig. 3. An increase in QR activity was observed within 2 h following addition of either compound. Maximum activity with vanillic acid was obtained after 12 h, followed by a decrease after 16 h. The level of induction was approximately 25-fold after 12 h. Addition of MBQ to HN cultures resulted in a more rapid increase in QR activity; maximal activity was attained after 4 h, followed by a slow decline and the activity leveled off after 12 h. A small but measurable amount of activity was present in uninduced cultures (Fig. 3). Addition of either vanillic acid or MBQ to cell extracts had no effect on the QR activity of the extracts (data not shown). While QR is expressed during secondary metabolic growth as well as primary metabolic growth (7), induction of expression by vanillic acid or MBQ appears to be greater during primary metabolic growth (7). However, the amount of QR expressed during secondary metabolic growth is probably sufficient to reduce any quinone generated during lignin degradation (7). At any rate, qr cDNA was isolated from a library prepared from uninduced secondary metabolic cells, demonstrating that the qr gene is expressed under these conditions.
FIG. 3.

Time course for induction of QR activity. Vanillic acid (■), MBQ (▴), or nothing (•) was added to 2-day-old HN cultures, as described in the text. Induced and uninduced cells were harvested at the times indicated, and the QR activity in the supernatants of crude extracts was assayed as described in the text.
The effect of vanillic acid concentration on the induction of QR activity is shown in Fig. 4. Various concentrations of vanillic acid were added to 3-day-old HN agitated cultures of P. chrysosporium, and intracellular QR activity was measured after 16 h. A small increase in activity was observed with as little as 0.1 mM vanillic acid, and the activity increased with increasing vanillic acid concentrations up to 2 mM. Vanillic acid concentrations greater than 2 mM were toxic to the cells, as determined by visual observation of the cultures. Induction by MBQ also was concentration dependent in the range from 0.01 to 0.2 mM (data not shown). Concentrations of MBQ greater than 0.2 mM were toxic to the cells.
FIG. 4.

Induction of QR activity with different concentrations of vanillic acid. Two-day-old HN cultures were induced with the indicated concentrations of vanillic acid. The cells were harvested after 16 h and broken, and QR activity was measured as described in the text.
The rapid induction of QR activity by vanillic acid and MBQ suggests that QR plays a role in the metabolism of these compounds. The first step in the catabolism of vanillic acid is an oxidative decarboxylation by an intracellular vanillate hydroxylase to 2-methoxyhydroquinone (10, 55). 2-Methoxyhydroquinone undergoes autooxidation, or possibly enzymatic oxidation, yielding MBQ. MBQ also has been identified as a product of the oxidation of veratryl alcohol (44) and lignin model dimers (51). Thus, formation of MBQ during lignin and pollutant degradation may regulate QR expression. The fact that induction of QR is more rapid in the presence of MBQ than in the presence of vanillic acid (Fig. 3) and the lower MBQ concentration that is required for induction suggest that MBQ might be the primary inducer and that vanillic acid functions as a source of MBQ. Thus, the difference in response times may reflect the time required to convert vanillic acid to the quinone; i.e., induction may be faster with compounds that do not require metabolic conversion.
The observation that MBQ is toxic to P. chrysosporium at low concentrations suggests that quinones may induce QR by triggering a general oxidative stress response. Indeed, it has been suggested that induction of certain enzymes by quinones may indicate that there is a toxic response (14, 48). However, no data on a general oxidative stress response in P. chrysosporium are available. Furthermore, the oxidative stress agents paraquat and H2O2 do not induce QR activity (data not shown), suggesting that QR induction is specific for quinones rather than part of a general oxidative stress response.
Western blot analysis.
An immunoblot analysis of QR from control and induced cultures was used to determine whether the increase in QR activity in induced cultures was due to an increase in the amount of QR protein or activation of preexisting enzyme. Figure 5 shows the concentration-dependent increase in the amount of QR protein that occurred 16 h after 0.5 or 2.0 mM vanillic acid was added. The results suggest that induction of QR occurs at the level of protein expression rather than via stabilization or activation of preformed enzyme.
FIG. 5.

Immunoblot analysis of QR from uninduced and vanillic acid-induced cells. Two-day-old HN cultures were not induced (lane 1) or were induced with 0.5 mM (lane 2) or 2.0 mM (lane 3) vanillic acid and then were harvested after 16 h, as described in the text. Portions (50 μg) of the supernatant proteins from crude cell extracts were loaded and electrophoresed on SDS-PAGE gels, transferred to nitrocellulose, and immunodetected, as described in the text.
Northern blot analysis.
Northern blot analysis was used to determine whether the regulation of QR expression occurs at the level of RNA. Figure 6 shows a Northern blot of RNA extracted from uninduced HN cells and from HN cells induced with vanillic acid (2 mM) or MBQ (0.2 mM). A large increase in the amount of qr mRNA was observed 3 h after induction with either vanillic acid or MBQ. In contrast, the gpd transcript was not affected by these additions. This confirms the results shown in Fig. 5 and suggests that QR activity is regulated by both vanillic acid and MBQ or their metabolites at the level of mRNA, probably at the level of transcription. The fact that regulation of the qr mRNA by vanillic acid and MBQ corresponds to induction of QR activity indicates that the cloned cDNA encodes the QR protein isolated from P. chrysosporium. The mechanism of QR induction in P. chrysosporium may be similar to the mechanism of DT-diaphorase induction in mammalian cells, which is transcriptional (14, 48).
FIG. 6.

Northern blot of P. chrysosporium RNA probed with the qr cDNA and the gpd gene. Two-day-old HN cultures were not induced (lane 1) or were induced with vanillic acid (2 mM) (lane 2) or MBQ (0.2 mM) (lane 3), as described in the text. After 3 h, RNA were isolated from induced and uninduced cultures, electrophoresed, and transferred to membranes, as described in the text. The blots were probed with randomly primed 32P-labeled pcQR1, as described in the text. Then the blots were stripped and reprobed with 32P-labeled gpd.
The regulation of qr mRNA by MBQ, as well as by vanillic and ferulic acids, suggests that QR plays an important role in the metabolism of aromatic compounds by P. chrysosporium and probably other wood-rotting fungi. Further studies on the regulation of QR expression in P. chrysosporium are planned. By using an RNA ladder (Gibco BRL), the size of the qr message was determined to be 1 kb, which is similar to the size of the 1,007-bp qr cDNA without the poly(A) tail.
A computer search of the GenBank database for similar sequences revealed that the qr cDNA sequence is very similar to the sequences of a Schizosaccharomyces pombe gene encoding brefeldin A resistance (60% identity and 74% overall similarity) (52) and a gene encoding a minor allergen from Alternaria sp. (63% identity and 74% overall similarity) (1) (Fig. 7). The qr cDNA sequence is also similar to the YCR4C (YCR042) gene sequence of Saccharomyces cerevisiae, which encodes a protein whose function is not known (63% identity and 92% overall similarity) (4). Brefeldin A is a fungal metabolite which causes disassembly of the Golgi apparatus (19). The structure of brefeldin A suggests that it is formed by cyclic esterification of an unsaturated fatty acid. In addition, this molecule contains a conjugated ene-one functional group. Given the similarity between the two protein sequences, we propose that the brefeldin A resistance protein may be an FMN-containing reductase which reduces the ene-one to an alcohol, rendering brefeldin A inactive. QR is also similar to the isoflavone reductase gene of higher plants (3), which suggests that all of these genes may be members of an FMN-containing reductase gene family.
FIG. 7.
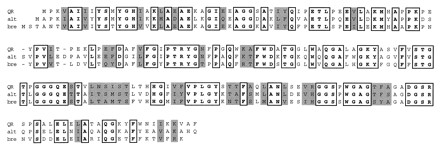
Clustal alignment of deduced protein sequences derived from the QR gene (this work), the brefeldin A resistance gene (bre) from S. pombe (52), and a gene (alt) encoding a minor allergen from Alternaria alternata (1). Identical portions of the sequences (bold) are enclosed in boxes; shading indicates sequence similarity.
ACKNOWLEDGMENTS
This work was supported by grant 96125:JVZ:11/21/96 from the M. J. Murdock Charitable Trust and by grant DE-FG-96ER20235 from the U.S. Department of Energy to M.H.G.
REFERENCES
- 1.Achatz G, Oberkofler H, Lechenauer E, Simon B, Unger A, Kandler D, Ebner C, Prillinger H, Kraft D, Breitenbach M. Molecular cloning of major and minor allergens of Alternaria alternata and Cladosporium herbarum. Mol Immunol. 1995;32:213–227. doi: 10.1016/0161-5890(94)00108-d. [DOI] [PubMed] [Google Scholar]
- 2.Aviv H, Leder P. Purification of biologically active globulin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc Natl Acad Sci USA. 1972;69:1408–1412. doi: 10.1073/pnas.69.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babiychuk E, Kushnir S, Belles-Boix E, Van Montagu M, Inzé D. Arabidopsis thaliana NADPH oxidoreductase homologs confer tolerance of yeasts toward the thiol-oxidizing drug diamide. J Biol Chem. 1995;270:26224–26231. doi: 10.1074/jbc.270.44.26224. [DOI] [PubMed] [Google Scholar]
- 4.Biteau N, Fremaux C, Hebrand S, Menara A, Aigle M, Crouzet M. The complete sequence of a 10.8 kb fragment to the right of the chromosome III centromere of Saccharomyces cerevisiae. Yeast. 1992;8:61–70. doi: 10.1002/yea.320080107. [DOI] [PubMed] [Google Scholar]
- 5.Blake M S, Johnston K H, Russell-Jones G J, Gotschlich E C. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal Biochem. 1984;136:175–179. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- 6.Brock B J, Gold M H. 1,4-Benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium: spectral and kinetic analysis. Arch Biochem Biophys. 1996;331:31–40. doi: 10.1006/abbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 7.Brock B J, Rieble S, Gold M H. Purification and characterization of a 1,4-benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol. 1995;61:3076–3081. doi: 10.1128/aem.61.8.3076-3081.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown J A, Li D, Alic M, Gold M H. Heat shock induction of manganese peroxidase gene transcription in Phanerochaete chrysosporium. Appl Environ Microbiol. 1993;59:4295–4299. doi: 10.1128/aem.59.12.4295-4299.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bumpus J A, Aust S D. Biodegradation of environmental pollutants by the white rot fungus Phanerochaete chrysosporium: involvement of the lignin-degrading system. Bioessays. 1987;6:166–170. [Google Scholar]
- 10.Buswell J A, Ander P, Petterson B, Eriksson K-E. Oxidative decarboxylation of vanillic acid by Sporotrichum pulverulentum. FEBS Lett. 1979;103:98–101. doi: 10.1016/0014-5793(79)81258-2. [DOI] [PubMed] [Google Scholar]
- 11.Buswell J A, Hamp S, Eriksson K E. Intracellular quinone reduction in Sporotrichum pulverulentum by a NAD(P)H:quinone oxidoreductase. FEBS Lett. 1979;108:229–232. doi: 10.1016/0014-5793(79)81216-8. [DOI] [PubMed] [Google Scholar]
- 12.Buswell J A, Odier E. Lignin degradation. Crit Rev Biotechnol. 1987;6:1–60. [Google Scholar]
- 13.Constam D, Muheim A, Zimmermann W, Fiechter A. Purification and characterization of an intracellular NADH:quinone oxidoreductase from Phanerochaete chrysosporium. J Gen Microbiol. 1991;137:2209–2214. [Google Scholar]
- 14.De Long M J, Prochaska H J, Talalay P. Induction of NAD(P)H:quinone reductase in murine hepatoma cells by phenolic antioxidants, azo dyes, and other chemoprotectors: a model system for the study of anticarcinogens. Proc Natl Acad Sci USA. 1986;83:787–791. doi: 10.1073/pnas.83.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eriksson K E, Blanchette R A, Ander P. Biodegradation of lignin. In: Timell T E, editor. Microbial and enzymatic degradation of wood and wood components. Berlin, Germany: Springer Verlag; 1990. pp. 225–333. [Google Scholar]
- 16.Ernster L, Danielson L, Ljunggren M. DT-diaphorase. I. Purification from the soluble fraction of rat liver cytoplasm, and properties. Biochim Biophys Acta. 1962;58:171–188. doi: 10.1016/0006-3002(62)90997-6. [DOI] [PubMed] [Google Scholar]
- 17.Ernster L, Ljunggren M, Danielson L. Purification and some properties of a highly dicoumarol-sensitive liver diaphorase. Biochem Biophys Res Commun. 1960;2:88–92. [Google Scholar]
- 18.Feinberg A P, Vogelstein B. A technique for radolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem. 1988;263:18545–18552. [PubMed] [Google Scholar]
- 20.Fuller R S, Sterne R S, Thorner J. Enzymes required for yeast prohormone processing. Annu Rev Physiol. 1988;50:345–362. doi: 10.1146/annurev.ph.50.030188.002021. [DOI] [PubMed] [Google Scholar]
- 21.Gold M H, Alic M. Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev. 1993;57:605–622. doi: 10.1128/mr.57.3.605-622.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gold M H, Cheng T M. Induction of colonial growth and replica plating of the white rot basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol. 1978;35:1223–1225. doi: 10.1128/aem.35.6.1223-1225.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gold M H, Joshi D K, Valli K, Wariishi H. Degradation of chlorinated phenols and chlorinated dibenzo-p-dioxins by Phanerochaete chrysosporium. In: Hinchee R E, Leeson A, Semprini L, Ong S K, editors. Bioremediation of chlorinated and polycyclic aromatic hydrocarbon compounds. Ann Arbor, Mich: Lewis Publishers; 1994. pp. 231–238. [Google Scholar]
- 24.Gubler U, Hoffman B J. A simple and very efficient method for generating cDNA libraries. Gene. 1983;25:263–269. doi: 10.1016/0378-1119(83)90230-5. [DOI] [PubMed] [Google Scholar]
- 25.Haemmerli S D, Schoemaker H E, Schmidt H W H, Leisola M S A. Oxidation of veratryl alcohol by the lignin peroxidase of Phanerochaete chrysosporium. FEBS Lett. 1987;220:149–154. [Google Scholar]
- 26.Hammel K E. Organopollutant degradation by ligninoloytic fungi. Enzyme Microb Technol. 1989;11:776–777. [Google Scholar]
- 27.Higuchi T. Lignin biochemistry: biosynthesis and biodegradation. Wood Sci Technol. 1990;24:23–63. [Google Scholar]
- 28.Ishikawa H, Schubert W, Nord F F. Investigations on lignins and lignification. XXVII. The enzymic degradation of softwood lignin by white-rot fungi. Arch Biochem Biophys. 1963;100:131–139. doi: 10.1016/0003-9861(63)90043-2. [DOI] [PubMed] [Google Scholar]
- 29.Joshi D K, Gold M H. Degradation of 2,4,5-trichlorophenol by the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol. 1993;59:1779–1785. doi: 10.1128/aem.59.6.1779-1785.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirk T K, Farrell R L. Enzymatic “combustion”: the microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 31.Kirk T K, Schultz E, Connors W J, Lorenze L F, Zeikus J G. Influence of culture parameters on lignin metabolism by Phanerochaete chrysosporium. Arch Microbiol. 1978;117:277–285. [Google Scholar]
- 32.Kornfield R, Kornfield S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Matoba S, Ogrydziak D M. A novel location for dipeptidyl aminopeptidase processing sites in the alkaline extracellular protease of Yarrowia lipolytica. J Biol Chem. 1989;264:6037–6043. [PubMed] [Google Scholar]
- 35.Mayfield M B, Kishi K, Alic M, Gold M H. Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol. 1994;60:4303–4309. doi: 10.1128/aem.60.12.4303-4309.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parenti M, Vigano M A, Newman C M, Milligan G, Magee A I. A novel N-terminal motif for palmitoylation of G-protein alpha subunits. Biochem J. 1993;291:349–353. doi: 10.1042/bj2910349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prestera T, Prochaska H J, Talalay P. Inhibition of NAD(P)H:(quinone-acceptor) oxidoreductase by Cibacron Blue and related anthraquinone dyes: a structure-activity study. Biochemistry. 1992;31:824–833. doi: 10.1021/bi00118a027. [DOI] [PubMed] [Google Scholar]
- 38.Pribnow D, Mayfield M B, Nipper V J, Brown J A, Gold M H. Characterization of a cDNA encoding a manganese peroxidase, from the lignin-degrading basidiomycete Phanerochate chrysosporium. J Biol Chem. 1989;264:5036–5040. [PubMed] [Google Scholar]
- 39.Raeder U, Broda P. Comparison of the lignin-degrading white rot fungi Phanerochaete chrysosporium and Sporotrichum pulverulentum at the DNA level. Curr Genet. 1984;8:499–506. doi: 10.1007/BF00410436. [DOI] [PubMed] [Google Scholar]
- 40.Reddy G V B, Joshi D K, Gold M H. Degradation of chlorophenoxy acetic acids by the lignin-degrading fungus Dichomitus squalens. Microbiology. 1997;143:2353–2360. doi: 10.1099/00221287-143-7-2353. [DOI] [PubMed] [Google Scholar]
- 41.Resh M D. Interaction of tyrosine kinase oncoproteins with cellular membranes. Biochim Biophys Acta. 1993;1155:307–322. doi: 10.1016/0304-419x(93)90012-2. [DOI] [PubMed] [Google Scholar]
- 42.Rieble S, Joshi D K, Gold M H. Purification and characterization of a 1,2,4-trihydroxybenzene 1,2-dioxygenase from the basidiomycete Phanerochaete chrysosporium. J Bacteriol. 1994;176:4838–4844. doi: 10.1128/jb.176.16.4838-4844.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Press; 1989. [Google Scholar]
- 44.Schmidt H W H, Haemmerli S D, Schoemaker H E, Leisola M S A. Oxidative degradation of 3,4-dimethoxybenzyl alcohol and its methyl ether by the lignin peroxidase of Phanerochaete chrysosporium. Biochemistry. 1989;28:1776–1783. [Google Scholar]
- 45.Smith P K, Krohn R I, Hermanson G T. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 46.Spain J C, Wyss O, Gibson D T. Enzymatic oxidation of p-nitrophenol. Biochem Biophys Res Commun. 1979;88:634–641. doi: 10.1016/0006-291x(79)92095-3. [DOI] [PubMed] [Google Scholar]
- 47.Strauss E C, Kobori J A, Siu G, Hood L E. Specific-primer-directed DNA sequencing. Anal Biochem. 1986;154:353–360. doi: 10.1016/0003-2697(86)90536-1. [DOI] [PubMed] [Google Scholar]
- 48.Talalay P, De Long M J, Prochaska H J. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci USA. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsang S S, Yin X, Guzzo-Arkuran C, Jones V S, Davison A J. Loss of resolution in gel electrophoresis of RNA: a problem associated with the presence of formaldehyde gradients. BioTechniques. 1993;14:2–3. [PubMed] [Google Scholar]
- 51.Tuor U, Wariishi H, Schoemaker H E, Gold M H. Oxidation of phenolic arylglycerol-β-aryl ether lignin model compounds by manganese peroxidase from Phanerochaete chrysosporium: oxidative cleavage of an α-carbonyl model compound. Biochemistry. 1992;31:4986–4995. doi: 10.1021/bi00136a011. [DOI] [PubMed] [Google Scholar]
- 52.Turi T G, Webster P, Rose J K. Brefeldin A sensitivity and resistance in Schizosaccharomyces pombe. Isolation of multiple genes conferring resistance. J Biol Chem. 1994;269:24229–24236. [PubMed] [Google Scholar]
- 53.Valli K, Gold M H. Degradation of 2,4-dichlorophenol by the lignin-degrading fungus Phanerochaete chrysosporium. J Bacteriol. 1991;173:345–352. doi: 10.1128/jb.173.1.345-352.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valli K, Wariishi H, Gold M H. Degradation of 2,7-dichlorodibenzo-p-dioxin by the lignin-degrading basidiomycete Phanerochaete chrysosporium. J Bacteriol. 1992;174:2131–2137. doi: 10.1128/jb.174.7.2131-2137.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yajima Y, Enoki A, Mayfield M B, Gold M H. Vanillate hydroxylase from the white rot basidiomycete Phanerochaete chrysosporium. Arch Microbiol. 1979;123:319–321. [Google Scholar]