

Abstract
Bispecific antibodies (BsAb) refer to a class of biomacromolecules that are capable of binding two antigens or epitopes simultaneously. This can elicit unique biological effects that cannot be achieved with either individual antibody or with two unlinked antibodies. Bispecific antibodies have been used for targeting effector cells to tumor cells, preferential targeting of cells expressing two target biomarkers over cells expressing either target biomarker individually, or to couple two molecular targets on the same cell surface to trigger unique intracellular signaling pathways. Here, we present two related methods that enable direct, rapid assembly of bispecific antibodies from any two “off-the-shelf” Immunoglobulin G (IgG) antibodies, in as little as one day. Both workflows can be summarized into two steps: 1) attach a small photoreactive antibody binding domain (pAbBD) fused to SpyCatcher or SpyTag (peptide-protein partners derived from the S. pyrogenes fibronectin-binding protein FbaB) to each component IgG, respectively; 2) assemble the BsAb through the spontaneous isopeptide bond formation that occurs between SpyTag and SpyCatcher. These approaches enable production of BsAbs from any two IgG molecules without the need to elucidate their amino acid sequences or genetically alter their structure. Binding assays and T cell-mediated cytolysis assays were performed to validate the binding and functional properties of Trastuzumab x Cetuximab BsAb and Cetuximab x OKT3 BsAb, respectively. This approach enables rapid, low-cost production of highly homogenous tetravalent BsAbs in a modular fashion, presenting an opportunity to quickly evaluate antibody pairs in a BsAb format for unique or synergistic functionalities.
Keywords: bispecific antibody, production, conjugation, off-the-shelf
Graphical Abstract
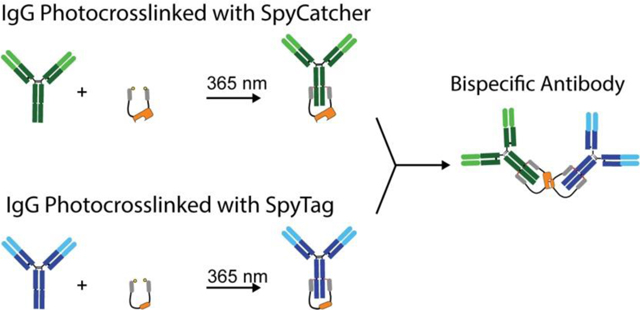
Introduction
Bispecific antibodies (BsAb) refer to a class of biomacromolecules that consist of two unique targeting domains that can simultaneously bind two different epitopes or antigens. The ability to bind to two different targets simultaneously can confer novel functionality to BsAbs. For example, BsAbs can bridge effector cells and target cells.1 In one such case, bispecific T cell engagers (BiTEs), which contain tumor surface antigen-binding domains and T cell-specific CD3-binding domains, recruit T-cells to promote cytolysis of tumor cells2–3. BsAbs can also be used to preferentially bind cells that simultaneously express two molecular targets, compared to cells that express only one, due to increased avidity.4 The ability of BsAbs to bind two different epitopes on a cell surface simultaneously can also interfere with signaling pathways in unique ways that is not possible with either antibody alone or the combination of unlinked antibodies.5 The crosslinking of cell-surface antigens can trigger, redirect, or inhibit cross-talk between signaling pathways.6 The simultaneous binding of two receptors is especially valuable in angiogenesis inhibition and tumor treatments, where the biological signals are often redundant and target cells can evade inhibition of one receptor by upregulating another.7 With several bispecific antibodies, such as emicizumab and blinatumomab, gaining FDA approval and dozens more in clinical trials, BsAbs are garnering growing interest as a therapeutic modality for the treatment of a wide range of diseases such as cancer, hematological disorders, inflammatory disorders, diabetes, and Alzheimer’s disease.8
As of 2019, more than 100 BsAb formats have been developed.8 Structurally, BsAbs are often conceptualized as an assembly of two antigen binding moieties, sometimes with the addition of crystallizable fragments (Fc) and linkers. Together, these features affect the level of production complexity, stability, functional valency, mechanism of action, plasma half-life, and toxicity of BsAb therapeutics. Many different approaches have been employed to generate bispecific antibodies, including chemical conjugation methods, the fusion of antibody fragments (e.g. BiTEs), and genetic engineering of full-length antibodies that enable controlled pairing of two antibodies along their line of symmetry, e.g. “knob-into-hole”.9–10 While each of these approaches can yield functional BsAbs, chemical methods are generally inefficient and/or result in highly heterogeneous products, limiting their adoption, and genetic methods generally require several rounds of recombinant protein engineering in addition to construction and optimization of mammalian cell lines.10–11 The time and cost associated with generating such genetically engineered BsAbs is often only justified with strong evidence supporting the proposed dual-targeting strategy. Therefore, while these platforms are useful in targeting well-studied or predictable biological pathways, there is still a need for an accessible technology that enables the rapid testing of any two antibody pairs for unique or synergistic activity. This is perhaps most easily achieved if BsAbs can be assembled from any pair of “off-the-shelf” Immunoglobulin Gs (IgGs).
Here we present two methods for rapid, in vitro assembly of tetravalent, bispecific antibodies from two full-length IgGs. Both methods use small photoreactive antibody binding domains (pAbBDs)12–13 fused to SpyCatcher (SC) or SpyTag (ST)14 to steer full-length IgG molecules into forming heterodimers (Figure 1). pAbBDs are composed of IgG-binding domains derived from the bacterial protein G12–13 or protein Z15–16 and enable highly-efficient photocrosslinking to the heavy chains of nearly any IgG, from a diverse range of hosts and subclasses. pAbBDs can be expressed as a fusion protein in series with other proteins of interest to label IgGs covalently and in a site-specific manner. Here, we use pAbBDs to label IgGs with either SpyCatcher or Spytag. This protein-peptide binding pair was originally developed from the S. pyrogenes fibronectin binding protein FbaB domain and can spontaneously form a covalent isopeptide bond.14 Therefore, once SpyCatcher and SpyTag are covalently attached to two different antibodies via the pAbBDs, they can drive the formation of a stable BsAb. This eliminates the need for any genetic engineering of the component antibodies and allows the use of any off-the shelf IgG to create BsAbs in a modular fashion. Thus, this represents a powerful new approach to test any combination of antibodies in a BsAb format for unique or synergistic functionalities.
Figure 1.

Schematic representation of the two different schemes used to rapidly produce bispecific antibodies. (A) In scheme 1, each component IgG is first conjugated to either a photoreactive antibody-binding domain fused to SpyCatcher (pAbBD-SC) or SpyTag (pAbBD-ST). Photocrosslinking is performed by exposing the sample to non-damaging 365 nm light for 2 hours. The IgG-SC and IgG-ST conjugates are then combined to form a BsAb via SpyCatcher-SpyTag isopeptide bond formation. (B) In scheme 2, each component IgG is conjugated to either pAbBD-SC-pAbBD or pAbBD-ST-pAbBD. Using a construct with two pAbBDs flanking SC or ST, the IgG is more likely to be labeled with only a single SpyCatcher or SpyTag. Once IgGs are labeled with SpyCatcher or SpyTag, their conjugates are combined to form a BsAb.
Results
BsAb Production Using pAbBD-SpyCatcher and pAbBD-SpyTag (Scheme 1)
The initial approach that was tested to rapidly create bispecific antibodies involved first crosslinking two IgGs to either pAbBD-SC or pAbBD-ST, respectively, and then using the interaction between SpyCatcher and SpyTag to form a bridge between these two antibodies (Figure 1, Scheme 1). As proof of principle, bispecific antibodies were formed from different combinations of the clinical IgG antibodies, Trastuzumab (anti-Her2/neu), Cetuximab (anti-EGFR), and OKT3 (anti-CD3).
Since every IgG molecule has two heavy chains to which pAbBD can crosslink, Scheme 1 yields a heterogenous mixture consisting of uncrosslinked IgG, IgG conjugated to a single pAbBD-SC/ST, and IgG conjugated to two pAbBD-SC/ST, in addition to any unbound pAbBD-SC/ST. On non-reducing SDS-PAGE, this mixture shows up as 3 bands around 150 kDa, representing IgG crosslinked to two, one, and no pAbBD-SC/ST from top down. When reduced, this mixture contains the heavy chain crosslinked to one pAbBD-SC/ST (~60–70 kDa), unlabeled heavy chain (~50 kDa), and unlabeled light chain (~25 kDa) (Figure 2A, B). While unconjugated IgGs will not be crosslinked in the SpyCatcher-SpyTag binding step and can be easily separated from 330 kDa BsAbs with size-exclusion methods, it is imperative to minimize the amount of IgG crosslinked with pAbBD-SC/ST on both heavy chains as it can form chains of 3 or more antibodies and can significantly reduce the yield of the correct BsAb product.
Figure 2. SDS-PAGE gels of precursor IgG conjugates and BsAbs produced via the scheme 1 conjugation method.

(A) SDS-PAGE of Trastuzumab (Tras) before and after photocrosslinking to pAbBD-SC, under reducing (R) and non-reducing (NR) conditions. (B) Protein G resin was used to capture and enrich for IgG conjugated with one or no pAbBD-SC fusion proteins. The fraction of the photocrosslinked sample that was captured by the Protein G resin as well as the flow-thru (i.e. uncaptured) was evaluated by SDS-PAGE. (C) SDS-PAGE of Cetuximab (Cetux) before and after photocrosslinking to pAbBD-ST. (D) SDS-PAGE of the photocrosslinked IgG-ST after capture and enrichment by Protein G resin, as well as the flow-thru (i.e. uncaptured). Photocrosslinking reactions between IgG and pAbBD-SC or pAbBD-ST were performed using non-damaging 365nm light, for 2 hours, at a pAbBD to IgG ratio of 1.2:1. (E) Tras-SC and Cetux-ST were mixed at an equimolar ratio, to generate a Tras x Cetux BsAb, and analyzed by SDS-PAGE before and after purification by size exclusion chromatography (SEC). (F) Cetux-SC and OKT3-ST were mixed at an equimolar ratio, to generate a Cetux x OKT3 BsAb, and analyzed by SDS-PAGE before and after purification by SEC. All gels were stained using SimplyBlue Safe Stain. Protein sizes were confirmed against Novex Sharp Prestained Protein Standard. Each crosslinking experiment was performed on at least three separate occasions. Gel images and quantification are from a representative experiment.
To maximize the relative amount of IgG conjugated to a single pAbBD-SC/ST, the amount of pAbBD-SC/ST was titrated against a fixed amount of IgG. The maximum percentage of IgG-pAbBD-SC/ST achieved was ~40%, at a pAbBD to IgG ratio ~1.2:1 (Figure 2A,C). To further enrich the population of IgGs labeled with a single SpyCatcher or SpyTag, recombinant protein G (pG) Agarose was used. The envisioned pG recovery process relies on the fact that pG Agarose binds IgG at the CH2-CH3 junctions, thus it is unable to bind IgGs crosslinked to two pAbBD-SC/ST. As a result, these constructs are ideally removed in the flow-through during the capture/wash steps. As shown in Figure 2B,D, however, pG Agarose recovery of IgG conjugated to one or no pAbBD proved to be less effective than expected as it also bound to dual-conjugated IgGs, even at a much lower pG Agarose-to-IgG ratio than recommended. We hypothesized that this is because pG Agarose can also bind to the Fab region.17 IgG-SC/ST was also found in the flow-through and was not captured as efficiently as unlabeled IgG. As a result, the pG Agarose recovery step only led to a slight enrichment of IgGs crosslinked to one or no pAbBD-SC/ST.
After pG Agarose recovery, IgG-SC and IgG-ST were incubated at equimolar concentrations at 4°C overnight. BsAbs were formed with an overall efficiency of ~20–40%. The unreacted IgGs and aggregates were removed via size exclusion chromatography (SEC), leading to a final purity of >90% (Figure 2C,D) and an overall yield of ~15%. While Scheme 1 led to the successful creation of BsAb, a notable drawback of this approach was the low efficiency of labeling IgG with a single pAbBD-SC/ST and the low percent of BsAbs recovered after SC-ST complementation. This prompted the invention of scheme 2.
BsAb Production Using pAbBD-SC-pAbBD and pAbBD-ST-pAbBD (Scheme 2)
To overcome the limitations of generating bispecific antibodies with pAbBD-SC/ST, we designed a fusion protein containing SpyCatcher or SpyTag flanked by two pAbBDs (Figure 1, Scheme 2). This method was designed to maximize the chance of crosslinking only one SpyCatcher or SpyTag per IgG by simultaneously occupying both CH2-CH3 junctions with a single construct.
Notably, a potential side-product of Scheme 2 is a homodimer, where two IgG molecules are crosslinked via a single pAbBD-SC-pAbBD or pAbBD-ST-pAbBD construct. To maximize the relative amount of IgG that has been crosslinked by a single pAbBD-SC/ST-pAbBD, the molar ratio of pAbBD-SC/ST-pAbBD to IgG and the IgG concentration were varied (Figure 3). The optimal crosslinking conditions were found to use ~2 equivalent(s) of pAbBD fusion per IgG. IgG concentrations up to 0.3 mg/mL could be utilized. Under these conditions, approximately 70–80% of the IgG was labeled with just one SpyCatcher/SpyTag. For all experiments, photocrosslinking was performed for 2 hours at 4°C. The large size difference between IgG homodimers (~300 kDa) and the desired product, i.e. IgG modified with a single SpyCatcher/SpyTag (~170kDa), enabled further purification using size-exclusion chromatography (SEC) (Figure 4).
Figure 3. SDS-PAGE gels of IgG photocrosslinked with pAbBD-SC/ST-pAbBD at varying SC/ST-to-IgG ratios and IgG concentrations.

Samples containing 0.15 mg/mL IgG (Cetuximab) and varying amounts of (A) pAbBD-SC-pAbBD or (B) pAbBD-ST-pAbBD were exposed to 365nm light for 2 hours and analyzed by SDS-PAGE, under non-reducing conditions. The gels were stained using SimplyBlue Safe Stain. The intensity of each band on the gel was quantified in ImageJ and the percentage of each molecular species was plotted as a function of the (C) pAbBD-SC-pAbBD-to-IgG ratio and (D) pAbBD-ST-pAbBD-to-IgG ratio. Samples at a fixed 2:1 (E) pAbBD-SC-pAbBD-to-IgG and (F) pAbBD-ST-pAbBD-to-IgG ratio, but with increasing concentrations of IgG were also analyzed by non-reducing SDS-PAGE, following photocrosslinking. The intensity of each band on the gel was quantified in ImageJ and the percentage of each molecular species was plotted as a function of IgG concentration for both (G) IgG-SC and (H) IgG-ST. Each crosslinking experiment was performed on at least three separate occasions. Gel images and quantification are from a representative experiment.
Figure 4. Purification of IgG conjugated to a single SpyCatcher/SpyTag by SEC.

(A) SEC trace of Trastuzumab following photocrosslinking with pAbBD-SC-pAbBD. (B) SEC trace of Cetuximab following photocrosslinking with pAbBD-ST-pAbBD. Photocrosslinking reactions between IgG and pAbBD-SC-pAbBD or pAbBD-ST-pAbBD were performed using non-damaging 365nm light, for 2 hours, at a pAbBD-to-IgG ratio of 2:1 and an IgG concentration of 0.3 mg/mL. (C) Non-reducing SDS-PAGE of Trastzumab crosslinked to pAbBD-SC-pAbBD and Cetuximab crosslinked to pAbBD-ST-pAbBD before and after SEC. Protein gels were stained using SimplyBlue Safe Stain. Each experiment was performed on at least three separate occasions. Gel images are from a representative experiment.
After successful isolation of IgG-SC and IgG-ST via SEC, the two conjugates were mixed at equimolar concentrations and incubated at 4°C overnight. BsAbs were formed with an overall SpyCatcher/SpyTag reaction efficiency of ~70%. The unreacted component IgGs were then removed via SEC, leading to a final purity of 90–95% (Figure 5) and an overall yield of 40–50%. With Scheme 2, both the efficiency of labeling IgG with a single SpyCatcher/SpyTag and the efficiency of BsAb formation, owing to SC-ST complementation, was ~2-times more efficient than Scheme 1 and the overall yield was ~3-times higher, with a higher level of purity. Therefore, all subsequent studies were performed using the BsAbs generated using Scheme 2.
Figure 5. SEC purification of BsAbs produced via Scheme 2.

(A) SEC trace of Trastuzumab x Cetuximab BsAb after mixing an equimolar amount of purified Trastuzumab-pAbBD-SC-pAbBD and Cetuximab-pAbBD-SC-pAbBD conjugates. Samples were mixed overnight at 4°C. (B) Non-reducing SDS-PAGE Trastuzumab x Cetuximab BsAb before and after SEC. The gel was stained with SimplyBlue Safe Stain. A representative gel image is shown. Each experiment was performed on at least three separate occasions.
Cell Surface Ligand Binding Assay
Binding assays were performed to survey the binding potential of Trastuzumab x Cetuximab BsAbs (anti-Her2 x anti-EGFR) against HER2/neu- and EGFR-positive cells. BsAb binding was compared to that of monospecific IgGs from which the BsAbs were derived. T6–17 human breast cancer (HER2+/EGFR-) and MDA-MB-468 human breast cancer cell lines (HER2-/EGFR+) were used to evaluate binding to HER2 and EGFR, respectively. The BsAbs exhibited similar dose-dependent binding to both T6–17 and MDA-MB-468 cells as the respective individual parental IgGs (Figure 6A,B). Paired t-tests revealed no significant differences between the EC50 of the Trastuzumab x Cetuximab BsAb and either component IgG (p<0.05).
Figure 6. Functional evaluation of BsAbs produced via Scheme 2.

Dose-dependent binding of Trastuzumab (circle), Cetuximab (square), and Trastuzumab x Cetuximab BsAbs (triangle) to (A) Her2-positive T6-17 cells and (B) EGFR-positive MDA-MB-468 cells. Each data point represents a set of duplicated wells. (C) T cell mediated cytolysis of EGFR-positive MDA-MB-468 cells as a function of Cetuximab x OKT3 BsAb concentration (triangle). A mixture of equivalent concentrations of un-crosslinked Cetuximab and OKT3 at 10 nM (circle) was included as a negative control. (D) Kinetics of T-cell mediated cytolysis of MDA-MB-468 cells in increasing concentrations of Cetuximab x OKT3 BsAbs (triangle) or an equimolar mixture of Cetuximab and OKT3 (circle). For both Cetuximab x OKT3 BsAb treatment and the equimolar Cetuximab, OKT3 mixture treatment, each data point represents a set of duplicated wells.
T cell-Mediated Cytolysis Assay
Cetuximab x OKT3 BsAbs (anti-EGFR x anti-CD3) were tested for their ability to induce T cell-mediated cytolysis of EGFR+ MDA-MB-468 tumor cells. When Cetuximab x OKT3 BsAbs were incubated with target cells in the presence of human T cells expanded in vitro, dose-dependent cytolysis was observed (Figure 6C, D). In comparison, an equimolar mixture of Cetuximab and OKT3 exhibited no cytolysis, in the presence of T cells, at equivalent doses and incubation time. A 10:1 effector-to-target ratio was used in all studies. These results confirm that cytolysis is mediated by the direct linkage between Cetuximab and OKT3. The BsAbs were effectively able to recruit T cells and enable formation of functional immunological synapses between tumor cells and T cells.
Discussion
In recent years, there has been a clear interest in developing new methodologies to rapidly prepare bispecific antibodies;18–20 however, prior attempts that rely on chemical conjugations result in inefficient reactions, low yields, and aggregates.10 Under optimal conditions, the yield of purified BsAbs using traditional chemical conjugation techniques has been reported to be only as high as 15%, and that is with a final purity of just ~70%.21 Genetic engineering approaches produce BsAbs with significantly higher homogeneity and purity (>95%), but it can take weeks or longer to acquire a functional product for testing, owing to complexities associated with expression, folding, and yield.11 Prior knowledge of the antibody sequence is also required in most cases, particularly if antibodies capable of binding specific targets are to be incorporated into the BsAb design. This presents a barrier to exploring and discovering new and unique antibody pairs that may exhibit interesting synergistic functionality.
To accelerate BsAb production, we leveraged photoreactive antibody binding domains that site-specifically label nearly any off-the-shelf IgG with SpyCatcher/SpyTag. The specific interaction between SpyCatcher and SpyTag was then used to drive the formation of a covalent isopeptide bond between the labeled IgGs. This allowed BsAbs to be generated against any two desired targets in less than a day, with high purity and yield. In contrast to genetic engineering approaches, this method does not require prior sequencing of the parental antibodies. Moreover, no recombinant protein engineering of the component IgGs is required and no mammalian/bacterial expression systems are required beyond the production of the pAbBD fusion proteins, which can be prepared in bulk. Two antibodies of choice are simply conjugated with SpyCatcher and SpyTag, respectively, and then combined to make a BsAb. The pAbBD is broadly compatible with antibodies from a wide range of hosts and subclasses, including all mouse, human, and rabbit isotypes.12–13, 15–16 This feature makes this approach highly modular and could enable small panels of BsAbs to be rapidly generated by mixing and matching antibodies that have first been functionalized with SpyCatcher and/or SpyTag.
Between Scheme 1 and Scheme 2, we found Scheme 2 to be superior both in terms of the IgG labeling efficiency with a single SpyCatcher/SpyTag and the efficiency of BsAb formation. While scheme 1 produces 30–40% of IgG labeled with a single SpyCatcher/SpyTag, the overall yield of final BsAb product is only ~15%, with a purity of ~90%. The biggest challenge associated with Scheme 1 was the undesirable formation of IgG modified with two SpyCatcher/SpyTag. This byproduct led to multimeric species composed of more than two IgG molecules (>450 kD) when SpyCatcher- and SpyTag-labeled IgG were combined. These multimeric species were difficult to avoid without significantly reducing the ratio of SpyCatcher/SpyTag-to-IgG, which leads to a large fraction of unlabeled IgG, with no improvement in the yield of IgG-SC/ST. This led us to explore various approaches to minimize the amount of IgG labeled with two SpyCatcher/SpyTag.
Size exclusion chromatography could not be used to separate IgG labeled with two SpyCatcher/SpyTag from IgG labeled with one SpyCatcher/SpyTag, due to the small difference in size. As an alternative approach, we attempted to use Protein G resin to isolate IgG with only a single SpyCatcher/SpyTag, since one heavy chain is still available for capture. Unlabeled IgG is also captured, but it is not able to form BsAbs and can be easily separated from BsAbs on size exclusion chromatography. Since both heavy chains of IgG are blocked when labeled with two pAbBD-SC/ST, we expected that this construct would not be captured by the Protein G resin. However, while a capture step with Protein G resin did lead to some enrichment, there were still significant levels of the double-labeled IgG impurity. We believe that this is because Protein G has some affinity for the Fab domain.17 Perhaps, using an affinity resin that is specific for just the heavy chain could avoid this issue, but this approach was not tested here.
The use of pAbBD-SC/ST-pAbBD overcomes the shortcomings of using pAbBD-SC/ST since the two tethered pAbBD are much more likely to bind the same IgG than two different IgG, due to the proximity effect. This provides a simple way to attach just a single SpyCatcher/SpyTag to each IgG. Initially, we performed a preliminary study using glycine-serine linkers between the two pAbBD domains, with lengths of 20 and 50 amino acids. We found that the shorter linker predominantly led to homodimer formation, while the longer linker labeled both heavy chains on a single IgG much more efficiently, with minimal homodimer formation. Therefore, we prepared SpyCatcher and SpyTag constructs with ~50 amino acids or longer between the two flanking pAbBDs, i.e. pAbBD-(GGS)7-SC/ST-(GGS)4-pAbBD and pAbBD-(GGS)7-SC/ST-(GGS)7-pAbBD. For Cetuximab and Trastuzumab, both designs offered efficient IgG labeling with a single SpyCatcher/SpyTag (>70%). However, for OKT3, the longer fusion protein was needed to achieve comparable labeling efficiency with a single SpyCatcher/SpyTag. In all cases, the fraction of homodimer could be slightly reduced at lower IgG concentrations, but if the concentrations became too low the interaction between SpyCatcher and SpyTag became inefficient when the two IgGs were combined to form BsAbs. Therefore, concentrations of at least 0.1 mg/mL IgG are recommended. Fortunately, the homodimer could be easily removed from IgG labeled with pAbBD-SC/ST-pAbBD by size exclusion chromatography. This led to high purity in IgG labeled with just a single SpyCatcher/SpyTag. Following size-exclusion chromatography and incubation of the single-labeled IgGs at a 1:1 molar ratio, BsAbs are formed with an efficiency of ~70%. The overall yield of the final BsAb product is 40–50% with a ~95% purity.
It is important to point out that the BsAbs formed from two full-length IgG using the approach described in Scheme 2 requires additional development if it is to be adopted as a therapeutic platform. First, the pAbBD, as well as SpyCatcher/SpyTag, are derived from bacterial proteins and thus could be prone to eliciting an immune response, although it should be noted that bacterial antibody binding domain variants have previously been used in clinical trials.22 Secondly, all four of the BsAb’s CH2-CH3 hinges, two on each antibody, are crosslinked with pAbBDs. The CH2-CH3 junction has been shown to play an important role in IgG recycling through binding to the neonatal Fc receptor (FcR).23 Therefore, blocking of these sites could affect the half-life of these BsAbs in vivo. Full-length IgG have also been shown to bind to Fcγ receptors at the hinge proximal end of CH2,24 but this site is not modified in this format. Secondary immune functions such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) are therefore unlikely to be affected in the IgG x IgG format. Finally, photocrosslinking of IgG with pAbBDs requires exposure to non-damaging 365nm light for two hours. While this can be performed at milliliter scales, scaling up for clinical use would require further development of crosslinking instrumentation. With these challenges in mind, the methodologies described here are currently best suited for discovery of novel synergistic targets, probing biological systems and uncovering unique signaling pathways. If desirable, any unique antibody combinations discovered with this approach can be introduced into other bispecific formats for clinical development. Considering the tetravalent nature of the BsAbs described here, it is envisioned that their behavior will be most predictive of other tetravalent BsAb formats such as those with second targeting domains fused to a native IgG antibody, although this would require further studies to be proven.
Conclusion
In summary, we developed two methods for the production of tetravalent, bispecific antibodies from off-the-shelf IgGs in less than a day. Both methods use SpyCatcher and SpyTag, fused to small photoreactive binding domains, to potentiate IgG molecules for BsAb assembly. The Trastuzumab x Cetuximab BsAb binds to each antigen without a significant reduction in affinity compared to each component IgG. The Cetuximab x OKT3 BsAb successfully re-directed and engaged human T cells to facilitate the cytolysis of tumor cells in vitro. Notably, scheme 2 is a rapid, low-cost, and high-yield process that produces highly homogeneous products.
Bispecific antibodies with novel functionalities have largely been developed and tested based on prior knowledge about the potential effects of binding to both molecular targets simultaneously. This approach precludes the discovery of useful and synergistic target pairs, of which the biological mechanisms are yet to be fully understood. The methods presented here enables the use of pre-established, off-the-shelf IgG molecules in BsAb discovery without engineering the IgG architecture using a relatively high-yield, low-cost process. Thus, this represents a powerful new approach to test any combination of antibodies in a BsAb format for unique or synergistic functionalities.
Methods
Cloning, Expression, and Protein Purification
All pAbBD fusion proteins are expressed and purified via the Sortase-Tag Expressed Protein Ligation (STEPL) method.25 The pSTEPL plasmid was first modified to include a His12 tag instead of a His6 tag. Plasmids encoding variants of pAbBD-SpyCatcher/SpyTag and pAbBD-SpyCatcher/SpyTag-pAbBD were cloned via In-Fusion (Takara Bio) reactions of their respective GeneBlock (IDT) into pSTEPL-His12 plasmids. The first generation SpyCatcher (ATHIKFSKRDEDGKELAGATMELRDSSGKTISTWISDGQVKDFYLYPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVN) and SpyTag (AHIVMVDAYKPTK) constructs were used here. For pAbBD-SC/ST, a (GGS)7 linker is used to connect the pAbBD with SC or ST. For pAbBD-SC/ST-pAbBD, the linker on the N-terminus of SC/ST is (GGS)7. For the linker following SC/ST, two versions with either (GGS)4 and (GGS)7 were produced. Sequences are verified via restriction digest and Sanger sequencing.
Plasmids were then co-transformed into T7 Express Competent E. coli cells (New England Biolabs) with pEVOL-pBpF plasmid (Addgene). The cells were spread onto a Luria Broth (LB) agar plate containing ampicillin (100 μg/mL) and chloramphenicol (20 μg/mL) incubated at 37°C. After 16–18 hours, a starter culture is prepared by inoculating bacterial colonies in 3 ml lysogeny broth (LB) supplemented with ampicillin (100 μg/mL) and chloramphenicol (20 μg/mL) and cultured for 16 hours in a shaker (225 rpm) at 37 °C. For expression, the starter cultures were inoculated into Auto Induction Media LB Broth (Formedium) containing glycerol (0.6% v/v), chloramphenicol (20 mg/L), ampicillin (100 mg/L), L-benzoylphenylalanine (BPA, 200 μM, Bachem) and L-arabinose (0.1% w/v) at a 1:1000 ratio. The bacterial culture was then allowed to grow on shaker (225 rpm) at 37 °C for 18 hours.
Cultures were pelleted down, resuspended in 1% OTG in PBS (w/v), and lysed on rotation at 25 °C for 30 min. The lysate was then briefly sonicated on ice and centrifuged at 15,000 g for 20 min at 4 °C. The resulting supernatant was transferred into in a 10 mL Poly-Prep chromatography column (Bio-Rad) and co-incubated with TALON® Superflow Metal Affinity Resin (Takara Bio USA) at room temperature on rotation for 30 min. The resin was then washed with 1 column volume (CV) PBS, 1 CV of 20 mM imidazole in PBS, and 2CV PBS. The resin was then incubated and eluted in 2 mM triglycine and 125 μM CaCl2 in PBS for an hour at 37 °C.
The eluted proteins were concentrated using a 3K MWCO Amicon Ultra Centrifugal Filter (Millipore) and purified via size-exclusion chromatography on a Superdex s30 10/300L column (Cytiva). Total protein concentration was measured with Pierce™ BCA Protein Assay Kit (ThermoFisher) and protein size was verified via SDS-PAGE. Protein gels are stained with SimplyBlue Safe Stain (ThermoFisher). Protein sizes were compared against Novex Sharp Prestained Protein Standards (ThermoFisher). Protein samples were aliquoted, frozen, and stored at −80 °C.
Crosslinking pAbBD Fusion Proteins to IgGs
pAbBD fusion proteins were added to IgGs and the mixture was exposed to 365 nm UV light in ice bath using a UVP CL-1000L UV crosslinker, which houses 5 × 8 watt bulbs. For bispecific antibody production, the crosslinking reactions were set up at an IgG concentration of 0.05–0.3 mg/mL, using a pAbBD-fusion-to-IgG ratio of 1:1 to 32:1 and an irradiation time of 2 hours. The crosslinking mixture was then concentrated with a 50K MWCO Amicon Ultra Centrifugal Filter (Millipore) and purified via size-exclusion chromatography on a Superdex s200 increase 10/300L column (Cytiva). Crosslinking product was analyzed with SDS-PAGE and ImageJ. Total protein concentrations were measured via BCA assays.
Purification of Crosslinked IgG Using Recombinant Protein G Agarose
IgG crosslinked to pAbBD-SpyCatcher/SpyTag was allowed to bind recombinant protein G resin for 30 minutes at room temperature on rotation. The resin was then spun down, washed three times with 50 mM Tris-HCl with 150 mM NaCl. 0.1 M Glycine-HCl (pH=2.7) was added to elute bound protein from spin columns into 1 M Tris-HCl (pH=8), which neutralizes the solution.
Production of BsAb Using Isopeptide Bond Formation
For isopeptide bond formation between SpyCatcher and SpyTag, an equimolar mixture of IgG-SC and IgG-ST in PBS, each at 1 μM (0.17 mg/mL) was incubated at 4°C overnight. Bispecific antibodies were purified via size-exclusion chromatography on a Superdex s200 increase 10/300L column (Cytiva). When necessary, BsAbs were concentrated using a 50K MWCO Amicon Ultra Centrifugal Filter (Millipore).
Fixed-cell Surface Ligand Binding Assay
MDA-MB-468 (EGFR+, HER2-) and T6–17 (HER2-, EGFR+) were each assayed for their respective binding to Cetuximab, Trastuzumab and Trastuzumab x Cetuximab BsAb. For each cell line, cells were seeded in black-wall, transparent-bottom 96-well plates (Corning) at 5,000 cells/well. The cells were incubated at 37°C in a 5% CO2 humidified incubator for 3 days or until 90% confluency. Cells were then fixed in neutral buffered formalin solution (Sigma-Aldrich) for 15 min at 25 °C and washed three times with 0.05% PBST. Plates were stored at 4°C with 100 μL 0.05% PBST in each well and were used within 2 weeks of production.
In each binding assay, cells were first blocked with 10% Normal Goat Serum (Life Technologies) for 15 minutes at 25 °C and washed three times with 0.05% PBST.
Antibodies and bispecific antibodies in blocking buffer (1x PBS with 0.05% Tween 20 and 0.25% BSA) were added in duplicates to cells in a three-fold serial dilution from 100 nM to 0.033 nM and allowed to bind at 25°C for an hour on a horizontal shaker. Cells were then washed three times with 0.05% PBST to remove unbound antibodies. Cells were then incubated in 1:1000 dilution of Goat Anti-Human IgG Fc Secondary Antibody conjugated to PE (eBiosciences) for an hour at 25°C. Cells were washed again with 0.05% PBST three times before measuring fluorescence at 544/585 nm in 0.05% PBST with a Tecan M200 Infinite plate reader.
Duplicate untreated cells remain in 0.05% PBST through binding and washing steps but are detected with the same method. Signals from these untreated cells were designated as background signal. Background signal was subtracted from all raw signals. Signals were then normalized, with 0% designated as the average minimal signals from each cell line and 100% the average maximal signals from each cell line. Normalized data were analyzed and plotted with GraphPad Prism8, both for original data and averaged data. Multiple unpaired t-tests were conducted for all pairings of EC50s (unaveraged) assuming Gaussian distribution (p<0.05).
T cell-Mediated Cytolysis Assay
MDA-MB-468 cells were maintained in DMEM (Corning) supplemented with 10% FBS (Corning) and 1% penicillin/streptomycin (ThermoFisher). Healthy human T cells were obtained from the Human Immunology Core (University of Pennsylvania) and expanded as previously described.26 Briefly, CD4 and CD8 T cells were incubated 1:1 and stimulated with CD3/CD28 Dynabeads (Gibco). Human IL-2 (Gibco) was maintained at a concentration of 50 IU/mL for 10 days. The Dynabeads were removed after 7 days of culturing, and the cells were maintained at 0.5–1 M/mL an additional 7 days. The cells were frozen down using a 1:1 mixture of X-VIVO media (Lonza) and 10% DMSO in FBS and allowed to rest in RPMI media (Corning) supplemented with 10% FBS and 1% penicillin/streptomycin 24 hours before cytolysis assays.
10,000 tumor cells were seeded per well 24 hours prior to adding BsAb treatments and T cells at an E:T ratio of 10:1. Controls included 3-fold serial dilutions starting at 100 nM or 10 nM of the monoclonal antibodies alone (Cetuximab and OKT3 separately) and an equimolar mixture of Cetuximab and OKT3. Tumor cytolysis was tracked up to 72 hrs post-treatment using xCelligence Real-Time Cell Analysis (ACEA Biosciences). Data were analyzed and plotted with GraphPad Prism8.
Acknowledgements
This work was supported in part by the NIH (NCI R01CA241661 and NCI R21CA187657).
Footnotes
Notes
L.M., F.Z., and A.T. have a pending patent on this technology. A.T. is a founder and owns equity in AlphaThera, a biotechnology company that sells antibody conjugation products.
References
- 1.Lum LG; Davol PA, Retargeting T Cells and Immune Effector Cells with Bispecific Antibodies. Cancer Chemother Biol Response Modif 2005, 22, 273–291. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA; Reinhardt C, Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res 2009, 69, 4941–4944. [DOI] [PubMed] [Google Scholar]
- 3.Bargou R; Leo E; Zugmaier G; Klinger M; Goebeler M; Knop S; Noppeney R; Viardot A; Hess G; Schuler M; Einsele H; Brandl C; Wolf A; Kirchinger P; Klappers P; Schmidt M; Riethmuller G; Reinhardt C; Baeuerle PA; Kufer P, Tumor Regression in Cancer Patients by Very Low Doses of a T Cell-Engaging Antibody. Science 2008, 321, 974–977. [DOI] [PubMed] [Google Scholar]
- 4.Mazor Y; Sachsenmeier KF; Yang C; Hansen A; Filderman J; Mulgrew K; Wu H; Dall’Acqua WF, Enhanced Tumor-Targeting Selectivity by Modulating Bispecific Antibody Binding Affinity and Format Valence. Sci Rep 2017, 7, 40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatta P; Whale KD; Sawtell AK; Thompson CL; Rapecki SE; Cook DA; Twomey BM; Mennecozzi M; Starkie LE; Barry EMC; Peters SJ; Kamal AM; Finney HM, Bispecific Antibody Target Pair Discovery by High-Throughput Phenotypic Screening Using in Vitro Combinatorial Fab Libraries. MAbs 2021, 13, 1859049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nie S; Wang Z; Moscoso-Castro M; D’Souza P; Lei C; Xu J; Gu J, Biology Drives the Discovery of Bispecific Antibodies as Innovative Therapeutics. Antib Ther 2020, 3, 18–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kontermann RE, Dual Targeting Strategies with Bispecific Antibodies. MAbs 2012, 4, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labrijn AF; Janmaat ML; Reichert JM; Parren P, Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat Rev Drug Discov 2019, 18, 585–608. [DOI] [PubMed] [Google Scholar]
- 9.Godar M; de Haard H; Blanchetot C; Rasser J, Therapeutic Bispecific Antibody Formats: A Patent Applications Review (1994–2017). Expert Opin Ther Pat 2018, 28, 251–276. [DOI] [PubMed] [Google Scholar]
- 10.Szijj P; Dhudasam V, The Renaissance of Chemically Generated Bispecific Antibodies. Nature Reviews Chemistry 2021, 5, 78–92. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q; Chen Y; Park J; Liu X; Hu Y; Wang T; McFarland K; Betenbaugh MJ, Design and Production of Bispecific Antibodies. Antibodies (Basel) 2019, 8. [DOI] [PMC free article] [PubMed]
- 12.Hui JZ; Tamsen S; Song Y; Tsourkas A, Lasic: Light Activated Site-Specific Conjugation of Native Iggs. Bioconjug Chem 2015, 26, 1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zappala F; Tsourkas A, Site-Specific Photocrosslinking to Immunoglobulin G Using Photoreactive Antibody-Binding Domains. Methods Mol Biol 2019, 2033, 275–286. [DOI] [PubMed] [Google Scholar]
- 14.Zakeri B; Fierer JO; Celik E; Chittock EC; Schwarz-Linek U; Moy VT; Howarth M, Peptide Tag Forming a Rapid Covalent Bond to a Protein, through Engineering a Bacterial Adhesin. Proc Natl Acad Sci U S A 2012, 109, E690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hui JZ; Al Zaki A; Cheng Z; Popik V; Zhang H; Luning Prak ET; Tsourkas A, Facile Method for the Site-Specific, Covalent Attachment of Full-Length Igg onto Nanoparticles. Small 2014, 10, 3354–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui JZ; Tsourkas A, Optimization of Photoactive Protein Z for Fast and Efficient Site-Specific Conjugation of Native Igg. Bioconjug Chem 2014, 25, 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erntell M; Myhre EB; Sjobring U; Bjorck L, Streptococcal Protein G Has Affinity for Both Fab- and Fc-Fragments of Human Igg. Mol Immunol 1988, 25, 121–126. [DOI] [PubMed] [Google Scholar]
- 18.Carlring J; De Leenheer E; Heath AW, A Novel Redox Method for Rapid Production of Functional Bi-Specific Antibodies for Use in Early Pilot Studies. PLoS One 2011, 6, e22533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HS; Dunshee DR; Yee A; Tong RK; Kim I; Farahi F; Hongo JA; Ernst JA; Sonoda J; Spiess C, Tethered-Variable Cl Bispecific Igg: An Antibody Platform for Rapid Bispecific Antibody Screening. Protein Eng Des Sel 2017, 30, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shatz W; Ng D; Dutina G; Wong AW; Dunshee DR; Sonoda J; Shen A; Scheer JM, An Efficient Route to Bispecific Antibody Production Using Single-Reactor Mammalian Co-Culture. MAbs 2016, 8, 1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Q; Zhang H; Wang Y; Ting HH; Yu W; Cao X; Ge W, Purified Anti-Cd3 X Anti-Her2 Bispecific Antibody Potentiates Cytokine-Induced Killer Cells of Poor Spontaneous Cytotoxicity against Breast Cancer Cells. Cell Biosci 2014, 4, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tolmachev V; Orlova A, Affibody Molecules as Targeting Vectors for Pet Imaging. Cancers (Basel) 2020, 12. [DOI] [PMC free article] [PubMed]
- 23.Ying T; Ju TW; Wang Y; Prabakaran P; Dimitrov DS, Interactions of Igg1 Ch2 and Ch3 Domains with Fcrn. Front Immunol 2014, 5, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Taeye SW; Bentlage AEH; Mebius MM; Meesters JI; Lissenberg-Thunnissen S; Falck D; Senard T; Salehi N; Wuhrer M; Schuurman J; Labrijn AF; Rispens T; Vidarsson G, Fcgammar Binding and Adcc Activity of Human Igg Allotypes. Front Immunol 2020, 11, 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warden-Rothman R; Caturegli I; Popik V; Tsourkas A, Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal Chem 2013, 85, 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minutolo NG; Sharma P; Poussin M; Shaw LC; Brown DP; Hollander EE; Smole A; Rodriguez-Garcia A; Hui JZ; Zappala F; Tsourkas A; Powell DJ Jr., Quantitative Control of Gene-Engineered T-Cell Activity through the Covalent Attachment of Targeting Ligands to a Universal Immune Receptor. J Am Chem Soc 2020, 142, 6554–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]