

Abstract
Growth of the musculoskeletal system requires precise coordination between bone, muscle, and tendon during development. Insufficient elongation of the muscle-tendon unit relative to bone growth results in joint contracture, a condition characterized by reduction or complete loss of joint range of motion. Here we establish a novel murine model of joint contracture by targeting Smad4 for deletion in the tendon cell lineage using Scleraxis-Cre (ScxCre). Smad4ScxCre mutants develop a joint contracture shortly after birth. The contracture is stochastic in direction and increases in severity with age. Smad4ScxCre mutant tendons exhibited a stable reduction in cellularity and a progressive reduction in extracellular matrix volume. Collagen fibril diameters were reduced in the Smad4ScxCre mutants, suggesting a role for Smad4 signaling in the regulation of matrix accumulation. Although ScxCre also has sporadic activity in both cartilage and muscle, we demonstrate an essential role for Smad4 loss in tendons for the development of joint contractures. Disrupting the canonical TGFβ-pathway in Smad2;3ScxCre mutants did not result in joint contractures. Conversely, disrupting the BMP pathway by targeting BMP receptors (Alk3ScxCre/Alk6null) recapitulated many features of the Smad4ScxCre contracture phenotype, suggesting that joint contracture in Smad4ScxCre mutants is caused by disruption of BMP signaling. Overall, these results establish a model of murine postnatal joint contracture and a role for BMP signaling in tendon elongation and extracellular matrix accumulation.
Keywords: Tendon, Smad4, BMP, TGFβ, joint contracture
Introduction
The musculoskeletal system provides shape and support to the body and capacity for movement. Precise and highly reproducible connections between muscle and the skeleton account for range and repertoire of movement. Muscle shape and the precise location of tendinous attachments between muscle and bone are established in early stages of limb development (Huang, 2017; Schweitzer et al., 2010). Maintaining the full range of motion during subsequent stages of growth is dependent on exquisite coordination of the rate of growth between skeletal elements and the associated muscles and tendons. When the rate of muscle-tendon growth is not sufficient to accommodate skeletal growth, tension builds across the associated joints eventually resulting in joint contracture, a condition in which the range of motion of affected joints is restricted (Darin et al., 2002; Slakey and Hennrikus, 1996).
Joint contracture may be driven by a primary defect in muscle, bone, connective tissue, or skin, or may be secondary to impaired neurologic function that causes reduced movement or weakness (Rudolph et al., 2011). Notably, contractures can develop due to a wide variety of causes that may be congenital (as in arthrogryposis multiplex congenita and club foot) or acquired (as in cerebral palsy and Duchenne’s muscular dystrophy) (Darin et al., 2002; Hall et al., 1983; Kowalczyk and Feluś, 2016; Slakey and Hennrikus, 1996; Swinyard and Bleck, 1985). The etiology of joint contractures can be attributed to genetic factors or environmental factors, and these conditions are frequently multifactorial (Bevan et al., 2007). Although very little is known about the connection between the etiology and the cellular and molecular mechanisms that account for the emergence of joint contractures, a constant feature of all developmental joint contractures is a shortened muscle-tendon unit (MTU) relative to the growing skeleton.
Tendons are essential musculoskeletal tissues that transmit the force generated by muscle contraction to bone. During tendon development the tenocytes coordinate the production of a massive array of collagen fibrils that occupy most of the volume of mature tendons and serve as the load bearing elements in this tissue. The regulation of tendon growth thus involves coordinated addition of cells and the increase in length and volume of collagen fibrils and overall collagen content. Little, however, is known about the regulation of tendon elongation or the regulation of matrix production. Uncovering the cellular and molecular mechanisms of tendon growth and coordination with muscle and bone length is critical for understanding the initiation and progression of joint contractures. Establishing a genetic model of contracture may also enable mechanistic analyses and the development of novel approaches for therapy.
We have previously shown that TGFβ signaling is essential for tendon formation and maintenance (Pryce et al., 2009; Tan et al., 2020). Canonical signaling in the BMP and TGFβ arms of the TGFβ superfamily is mediated by activation of Smad transcriptional complexes. Activation of pathway-specific receptor-associated Smad proteins is followed by dimerization with a common Smad, Smad4, and translocation of the activated complex to the nucleus (Massagué, 2012). To analyze the role of canonical TGFβ signaling in tendon development, we targeted the Smad4 gene in tendon cells using the ScxCre driver and surprisingly found that Smad4ScxCre manifested acquired postnatal joint contracture. Forelimb contractures were first detected a few days after birth and the severity of the contractures worsened with age. We further found that the phenotype was caused by disruption of BMP signaling and manifested as reduced cellularity and reduced collagen matrix in tendons, suggesting that BMP signaling in tenocytes plays an essential role for tendon growth and coordinated musculoskeletal growth.
Results
Smad4ScxCre mutants develop postnatal contractures
We previously showed that TGFβ signaling is essential for early tendon development (Pryce et al., 2009; Tan et al., 2020). To examine the role of downstream Smad signaling in this process we crossed Smad4 floxed mice (Yu et al., 2002) with ScxCre mice (Blitz et al., 2013) to generate a Smad4ScxCre mutant in which Smad4 activity is eliminated in tenocytes. Loss of the Smad4 protein in the tendons of these mutants was confirmed by western blot and immunohistochemistry (Supplementary Figure 1). To facilitate tendon analysis, the mutant mice also carry the Scleraxis-GFP (ScxGFP) tendon reporter which labels tenocytes and tenocyte progenitors (Pryce et al., 2007). Smad4ScxCre mutant mice were born with no apparent abnormalities (Figure 1 I–J) but within several days after birth developed a severe forelimb contracture (Figure 1A–B). The contracture typically began at or around P5 as an abduction of the wrist resulting in reduced mobility. The forelimb contracture increased in severity rapidly and by P14 most pups presented with additional dorsal or ventral contractures (Figure 1C–D), contractures within the digits, and reduced mobility with contractures even in the hind limbs. The tendons of mutant pups appeared normal at birth but already at the onset of contracture at P5 the tendons appeared thinner, which was exacerbated over time (Figure 1E).
Figure 1: Smad4ScxCre mutants develop contracture shortly after birth.
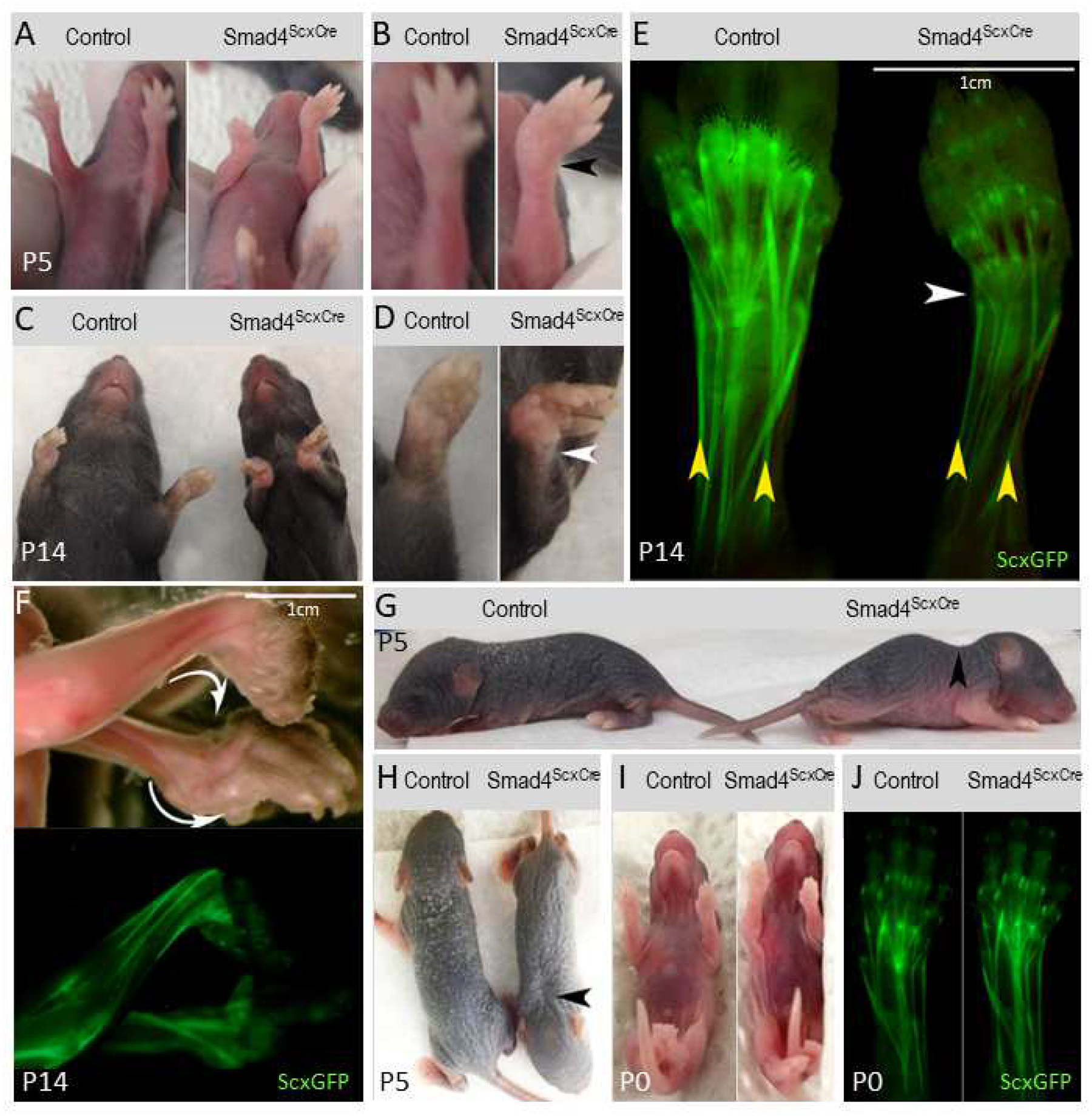
(A) P5 control and Smad4ScxCre mutant mice. (B) Larger image of forelimbs from (A). Mutant displays forelimb abduction (black arrowhead). (C) P14 control and Smad4ScxCre mutant mice. (D) Larger image of left forelimbs from (C). Abduction is more severe by P14 (white arrowhead). (E) ScxGFP-labeled tendons in P5 control and Smad4ScxCre mutant. Mutant tendons appear thinner than control (yellow arrowheads) and the mutant forelimb exhibits abduction at the wrist (white arrowhead). (F) P14 Smad4ScxCre mutant skinned forelimbs. The forelimb contracture in mutant mice can develop as dorsal or ventral contractures even within the same mouse (white arrows). (G and H) P5 control and Smad4ScxCre mutant mice. The mutant displays abnormal cervical curvature (black arrows). (I) P0 Control and Smad4ScxCre mutant mice. (J) ScxGFP-labeled tendons in P0 control and Smad4ScxCre mutant. The forelimb contracture does not manifest in newborn mutant pups.
In human conditions, the dorsal or ventral direction of forelimb contractures involving the wrist is always consistent (Kowalczyk and Feluś, 2016). Surprisingly, in the Smad4ScxCre mutants we found a mixture of both dorsal and ventral contractures and in some mutants, we even found a combination of a dorsal and ventral contracture in the same mouse (Figure 1F). This unique feature of Smad4ScxCre mutant mice likely reflects a stochastic event in the initiation of the contracture and this initial direction is fixed by subsequent growth. While this feature distinguishes the Smad4ScxCre mutants from clinical conditions it may provide clues to the mechanisms of contracture initiation and enhancement.
In addition to limb contracture, the Smad4ScxCre mutant pups also exhibited increased curvature of the cervical spine (Figure 1G–H). Notably, the spinal curvature was not present at birth and appeared in early stages of postnatal development. It is possible that this increased curvature reflects a contracture of the axial skeleton in mutant pups.
Smad4ScxCre mutant tendons exhibit a reduction in volume
A contracture phenotype could be caused by loss of specific tendons that would result in an imbalance in joint movement and prevent recovery of a neutral position. To examine if tendon pattern was affected in Smad4ScxCre mutants, we analyzed the tendon pattern in cross-sections through the forelimb of P5 mutant pups and found that all tendons were present in mutant pups, eliminating the possibility of tendon loss as a primary driver of the contracture phenotype (Figure 2A). While all tendons were accounted for, the tendons of mutant pups were significantly thinner than those of WT littermates (Figure 2B). In mature tendons, the extracellular matrix (consisting of a wide array of collagen fibrils) occupies most of the tendon volume (Kannus, 2000). We therefore determined whether the reduction in tendon cross-sectional area (CSA) was associated with reduction in the number of cells in the tendons and/or a reduction in extracellular matrix. To determine cellular content in tendons, limb cross-sections were stained with DAPI to label nuclei and tendon CSA was measured based on the ScxGFP signal. Because tendon growth may be affected by activity and limb usage was naturally reduced after the onset of the contracture, it was important to determine these parameters at the onset of the limb contracture. In embryonic stages and up to P0, prior to the emergence of contracture, we found no significant difference between control and mutant Flexor Carpi Ulnaris (FCU) tendons in the number of cells per section (counted as number of nuclei per section) (Figure 2C) or in CSA (Figure 2D). However, we found that in mutant pups at P5, when contractures first emerge, there was already a 40% reduction in CSA (Figure 2D) as well as a 30% reduction in the number of tendon cells per section (Figure 2C). Interestingly, the proportional reduction in the number of cells in mutant tendons was stable between P5 and P14 even as tendons continue to grow and lengthen in this interval in both control and mutant pups.
Figure 2: Smad4ScxCre mutant tendons are smaller with fewer cells and smaller collagen fibrils.
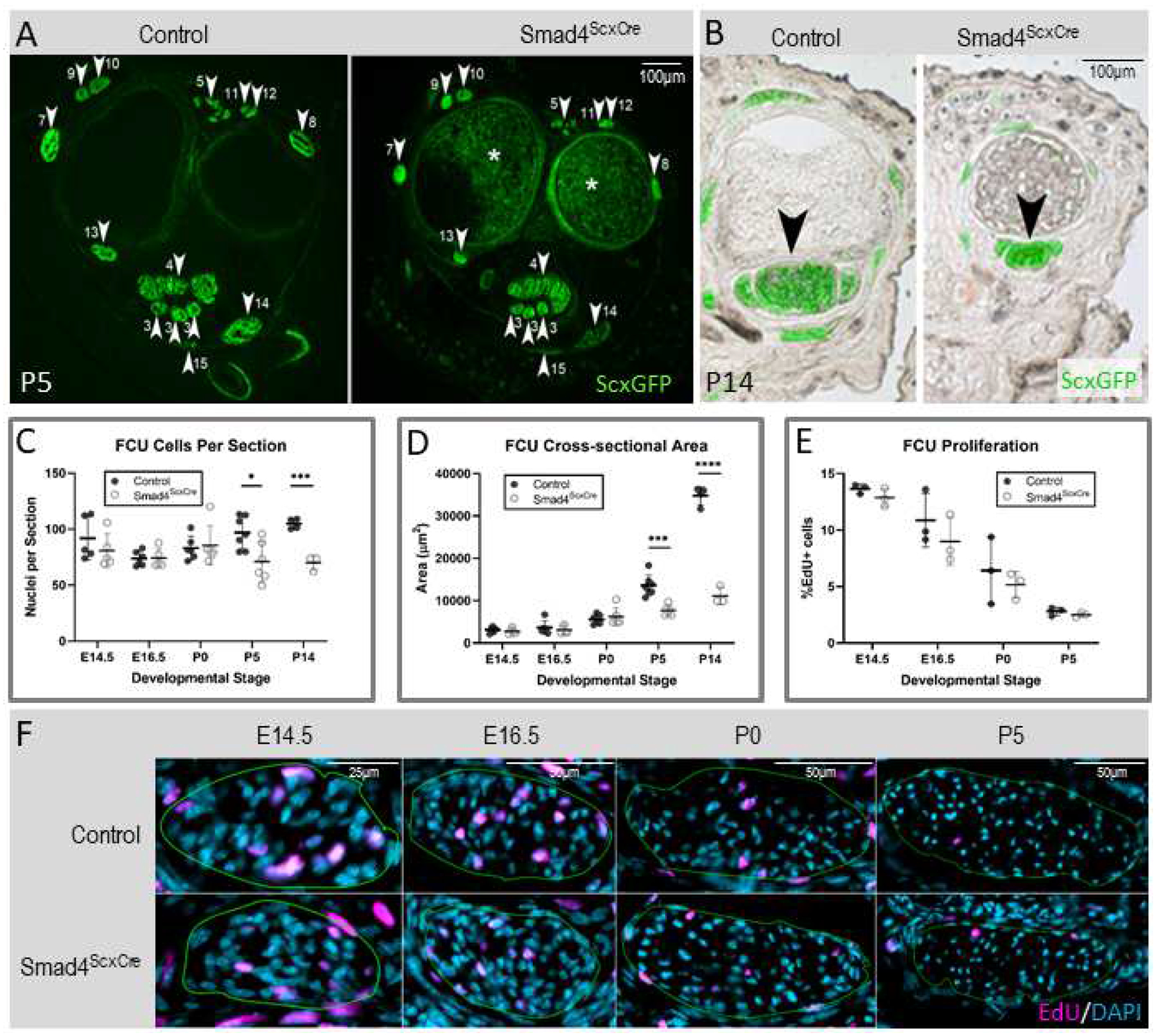
(A) Cross section at wrist levels of forelimbs from P5 control and Smad4ScxCre mutant pups. White arrowheads and associated numbers label tendons as previously published (Watson et al., 2009). * is background signal from bone marrow and is not ScxGFP signal. (B) Cross sections of forelimb digits from P14 control and Smad4ScxCre mutant. Brightfield images were overlaid with ScxGFP signal to label the tendons. Mutant tendons are significantly smaller than controls (black arrowheads). (C,D,E) Quantification of Flexor Carpi Ulnaris (FCU) cells per section (C), cross-sectional area (D), and percentage of EdU+ cells (E) analyzed in cross sections from control and Smad4ScxCre forelimbs at E14.5, E16.5, P0, P5, and P14. Bars represent standard deviation. *p<0.05 **p<0.01 ***p<0.001 ****p<0.0001 (F) Representative images of EdU-stained sections at E14.5, E16.5, P0, and P5. Green outline represents the tendon boundary.
Since the reduction in cell numbers was restricted to the time window between P0–P5 we next focused on this period to identify the cellular process regulated by Smad4 signaling. To determine if tenocyte proliferation is regulated by Smad4 signaling we administered EdU two hours before harvest to label proliferating cells. Surprisingly, we found no difference in proliferation rate between the tenocytes of mutant and control pups at any stage between E14.5 and P5 (Figure 2E–F). An alternative explanation for the sudden drop in relative cell numbers in mutant tendons could be an essential role for Smad4 in tenocyte maintenance and a wave of tenocyte apoptosis in Smad4 mutants. We therefore labeled sections for apoptosis using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. In samples at P0 and P5 from control and mutant tendons apoptotic tenocytes were extremely rare; only one apoptotic tenocyte was found in one control P0 FCU out of thousands of cells analyzed (Supplementary Figure 2C). As a positive control for TUNEL staining we examined hair follicles that predictably cycle and produce apoptotic cells in neonatal development (Magerl et al., 2001). We consistently found TUNEL-labeled hair follicles as expected (Supplementary Figure 2A) but no TUNEL-labeled tenocytes outside of the single P0 positive tenocyte (Supplementary Figure 2B). These results suggest that neither changes in proliferation nor apoptosis are driving the reduction of tendon cells at P5.
The abduction of the paw at P5 occurs consistently towards the ulnar side of the limb. Since the FCU tendon is located on the ulnar side we additionally wanted to examine the cellular dynamics in a tendon at the opposite side of the limb. Analysis of tendon cell dynamics in cross sections can only be performed reliably in tendons that extend in a straight line along the long axis of the limb and thus yield perfect cross sections of the tendons in cross sections of the limb. We therefore chose to analyze the Flexor Carpi Radialis (FCR) and again found no change in cell number (Supplementary Figure 3A) or CSA (Supplementary Figure 3B) from E14.5-P0, with a reduction in cell numbers and CSA in tendons of mutant pups only at P5 (Supplementary Figure 3A–B). In addition, we again found no effect on the rate of proliferation (Supplementary Figure 3C) or on apoptosis in the mutant tendons (data not shown). The reduction in the number of cells in the tendons of neonatal Smad4ScxCre pups is thus not due to effects on tenocyte proliferation or viability and may therefore reflect on a role for Smad4 signaling in cell recruitment to tendons in this time window.
The relative reduction in the number of cells per section remained proportional at P5 and P14 even as the tendon continued to grow. Conversely, the relative reduction of tendon CSA was substantially increased, from 40% reduction at P5 to 70% reduction by P14. As cells comprise only a small portion of the CSA in postnatal stages and cellularity did not continue to decrease between P5 and P14, the enhanced reduction in CSA by P14 is likely a reflection of a direct effect on matrix deposition. These results therefore suggest a possible role for Smad4 signaling in regulation of matrix deposition.
Postnatal loss of Smad4 in tenocytes is not sufficient for the development of limb contractures
ScxCre activity is robust in tendon cells but cre activity is not exclusively restricted to tenocytes and some recombination is also observed in neighboring tissues, e.g. the tendon sheath and other cells that surround the tendon (Supplementary Figure 4). Some ScxCre activity can also be detected in cartilage, especially near the tendon insertion and in the growth plate (Supplementary Figure 4A). Moreover, we also find indications of ScxCre activity in muscle tissue that becomes apparent close to P5 (data not shown). Since ScxCre shows sporadic activity in all of these tissues, it was important to attempt to determine whether limb contractures in Smad4ScxCre mutants were the result of Smad4 loss in tenocytes or whether the recombination in other tissues was also relevant to the development of limb contractures.
ScxCreERT2 is a tamoxifen-inducible Cre driver in which an IRES-CreERT2 cassette was knocked into the 3’UTR of the endogenous Scx gene. Therefore, ScxCreERT2 expression is driven by the endogenous Scx locus and expression of ScxCreERT2 is identical to that of the Scx gene. Induction of ScxCreERT2 in postnatal stages resulted in cre activity that was much more tightly restricted to tenocytes (Supplementary Figure 4B). Smad4ScxCre mutant mice develop contracture only around P5, suggesting an important role for Smad4 signaling in early postnatal development. If Smad4 signaling is indeed required in tendon cells just shortly before the onset of the contracture, inducing Smad4 deletion in tendons at very early postnatal stages should also result in the development of a contracture. To test this hypothesis, we generated Smad4 floxed/ScxCreERT2 mice (Smad4ScxCreERT2) and induced recombination by application of tamoxifen at both P2 and P4, before limb contractures become apparent in the Smad4ScxCre mutants. Surprisingly, the mutant mice did not develop a contracture by P5 or in later stages (Figure 3A–B). Notably, the onset of ScxCre activity in tenocytes is at E13.5. Tenocyte recombination is initially sporadic and gradually becomes more uniform in later stages of embryogenesis. The postnatal emergence of limb contractures in these mutants may therefore be a delayed outcome of this embryonic pattern of loss of Smad4 in tenocytes. However, we could not test this hypothesis at this time since the ScxCreERT2 driver is not efficient in embryonic stages (data not shown).
Figure 3: Smad4ScxCre contracture phenotype is not due to imbalance in muscle or bone growth.
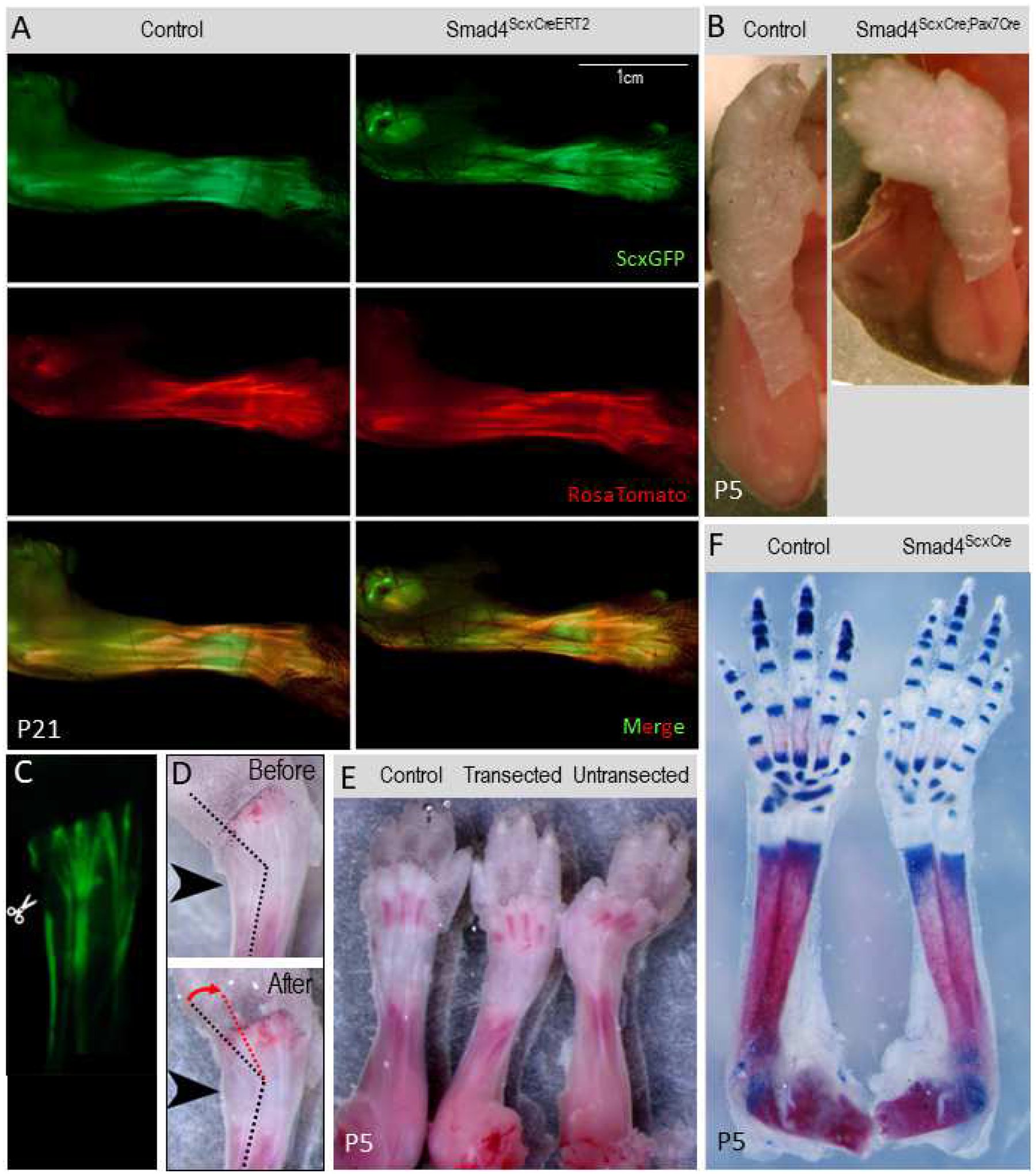
(A) P21 control and inducible Smad4ScxCreERT2 mutant mice. No contracture phenotype is present. (B) P5 Control and Smad4ScxCre;Pax7Cre double-Cre mutant. Limb contracture is present in the double Cre mutant. (C) Example of tendon transection to relieve soft tissue tension. P5 Smad4ScxCre limb was skinned and lateral extensors cut. (D) P5 Smad4ScxCre contracted limb was transected as in (C) and gently straightened with forceps. Wrist angle before transection is indicated by a black dotted line. Change in wrist angle after transection is indicated with red dotted line and an arrow. (E) Skinned P5 limbs from a Control pup, Smad4ScxCre with transection, and Smad4ScxCre with no transection. (F) P5 Control and Smad4ScxCre limb skeletons prepared with Alizarin red (red) and Alcian Blue (blue).
Limb contractures in Smad4ScxCre mutants are not the result of sporadic loss of Smad4 in muscle
ScxCre activity extends beyond the tendons (Supplementary Figure 4). It was therefore also possible that limb contractures may be the result of ScxCre activity in the tissues adjacent to tendons. We recently found that in postnatal stages ScxCre activity may also extend to muscles. Smad4 deletion in muscle leads to a reduction of muscle mass and muscle weakness, however it does not result in limb contractures (Sartori et al., 2013). It is however possible that sporadic recombination in muscles by ScxCre may result in uneven recombination of Smad4 in the muscle and thus an uneven deletion of Smad4 signaling within the muscle and/or between muscle groups. As Smad4 deletion was previously demonstrated to produce a reduction of muscle mass and muscle weakness, such uneven Smad4 signaling in muscles may lead to an imbalance of growth between muscle groups, resulting in an imbalance of strength, that may in turn lead to contracture. To determine if the sporadic activity of ScxCre in muscles may underlie the limb contracture phenotype, we therefore tested whether targeting Smad4 with ScxCre would result in a contracture even on the background of uniform loss of Smad4 in muscles. To achieve this goal, we combined Smad4 floxed mice with ScxCre and Pax7Cre (Keller et al., 2004). Pax7 is an early marker of myoblasts and the Pax7Cre driver leads to uniform recombination in the myogenic lineage by E14.5 (Hutcheson et al., 2009). We therefore generated a double cre knockout mouse, Smad4 floxed/ScxCre/Pax7Cre (Smad4ScxCre/Pax7Cre), in which Smad4 is targeted in both the domain of ScxCre activity and in all muscle cells. If an imbalance in muscle growth due to sporadic ScxCre recombination is driving the Smad4ScxCre contracture phenotype, then a mutant in which Smad4 is evenly deleted in both tendon and muscle should rescue the contracture phenotype. However, if the contracture phenotype remains when Smad4 signaling is complete in both tendon and muscle, then it would suggest an imbalance of Smad4 signaling in the muscle is not driving the Smad4ScxCre contracture phenotype. We found that Smad4ScxCre/Pax7Cre mutants still developed limb contractures shortly after birth, suggesting that the sporadic activity of ScxCre in muscles was not an essential driver of the limb contracture phenotype (Figure 3C).
Smad4ScxCre contracture phenotype is not due to aberrations in skeletal growth
ScxCre also shows sparse recombination in cartilage (Supplementary Figure 4). ScxCre activity in and around the tendon insertion has been reported (Blitz et al., 2009) and some activity can also be detected in other domains in the developing cartilage, notably in some cases even associated with the growth plate (Supplementary Figure 4). Both TGFβ and BMP signaling have been implicated in various aspects of skeletal development and it was directly demonstrated that Smad4 activity is essential for skeletal growth since targeting of Smad4 in chondrocytes using the Col2Cre driver resulted in dwarfism but does not lead to contracture (Chen et al., 2012; Wu et al., 2016; Zhang et al., 2005). Therefore, because Smad4 is essential to normal skeletal growth it is possible that sporadic loss of Smad4 in cartilage by ScxCre activity may result in uneven bone growth and could contribute to the development of joint contractures. To test this hypothesis, we repeated the experimental approach used above by generating a double-cre mutant in which Smad4 will be targeted completely in both tendons and cartilage. To achieve uniform recombination in cartilage we used the Col2Cre driver that shows specific Cre activity in chondrocytes from early stages of skeletal development (Baffi et al., 2006). Unfortunately, in the double Cre experiment targeting Smad4 with both ScxCre and Col2Cre (Smad4ScxCre;Col2Cre) we found that the mutants were embryonic lethal (not shown), precluding the ability to evaluate a postnatal phenotype.
Since we could not determine the role of the cartilage in the Smad4ScxCre joint contracture phenotype using the double cre experiment, we sought alternative approaches to address this question. We first examined mutant limbs in skeletal preps stained for Alcian Blue and Alizarin Red and did not find any gross abnormalities that would indicate aberrant bone or joint development around the wrist (Figure 3F). Next, we performed a functional assay for the possible role of skeletal imbalance in the development of the joint contracture by transecting the tendons to relieve the tension around the contracted joint. If the Smad4ScxCre joint contractures were caused by tension generated by a relatively short muscle-tendon unit (MTU) that cannot accommodate the length of the skeletal elements, transecting the relevant tendons would allow the skeletal elements to spring back to their normal anatomical configuration. Conversely, if the contracture phenotype is caused by an imbalance in bone growth affecting skeletal anatomy around the joint or by disruptions in joint formation, then releasing MTU tension by cutting the tendons would not relieve the contracture phenotype, as the rigid skeleton would maintain the aberrant joint position. Mutant limbs at P5 were skinned and tendons severed at wrist level to release MTU tension (Figure 3C–D). After tendon transection, the wrist angle relaxed and appeared more similar to the control limb (Figure 3E), suggesting that the skeletal tissues did not have a substantial contribution to the development of the joint contracture.
Overall, these results suggest that loss of Smad4 in tendon cells is essential for the contracture phenotype. It is important to note however, that while these experiments prove a direct role for loss of Smad4 activity in tendons in the development of the contracture phenotype, we cannot rule out the possibility that in addition to the loss in tendons, sporadic and regionalized loss of Smad4 in other neighboring tissues may also play a role.
The Smad4ScxCre contracture phenotype does not result from disruption of TGFβ signaling
TGFβ signaling has been implicated in matrix production and tendon development (Havis et al., 2016; Huang et al., 2015; Pryce et al., 2009; Subramanian et al., 2018; Tan et al., 2020) and was therefore the likely pathway implicated in the Smad4ScxCre joint contractures. Canonical signaling of the TGFβ superfamily is mediated by phosphorylation of receptor-associated transcription factors of the Smad family, all of which subsequently dimerize with Smad4 to generate the activated transcription complex. Smad2 and Smad3 are the receptor Smads for the TGFβ proper branch of this signaling pathway and Smad1, Smad5, and Smad8 are associated with BMP signaling (Massagué et al., 2005). Phosphorylated Smad2 is detectable in tendons at P6 (Figure 4A), suggesting the TGFβ pathway is active in tendons near the onset of the contracture phenotype. In addition, we recently reported that targeting TGFβ signaling in tendons by disruption of the type 2 TGFβ receptor (TGFbR2ScxCre) resulted in a severe tendon phenotype involving tenocyte dedifferentiation and the loss of some tendons. The TGFbR2ScxCre mutant pups also developed a neonatal abduction similar to that seen in the Smad4ScxCre mutant (Tan et al., 2020). Importantly, though, the reproducible failure of specific radial extensor tendons was the likely driver of the abduction in the TGFbR2ScxCre mutants but similar tendon damage was not found in the Smad4ScxCre mutants (Figure 1E). To directly test whether joint contracture in the Smad4ScxCre mutants reflects activity of the TGFβ pathway we therefore targeted the Smad2 and Smad3 genes using ScxCre (Li et al., 2008; Liu et al., 2004). Surprisingly, the Smad2;3ScxCre mutant mice did not develop any detectable abnormal tendon phenotype, let alone a joint contracture phenotype (Figure 4B). Canonical TGFβ signaling is therefore unlikely to be the signaling pathway underlying the Smad4ScxCre contracture phenotype.
Figure 4: Disruption of BMP signaling underlies the Smad4ScxCre contracture phenotype.
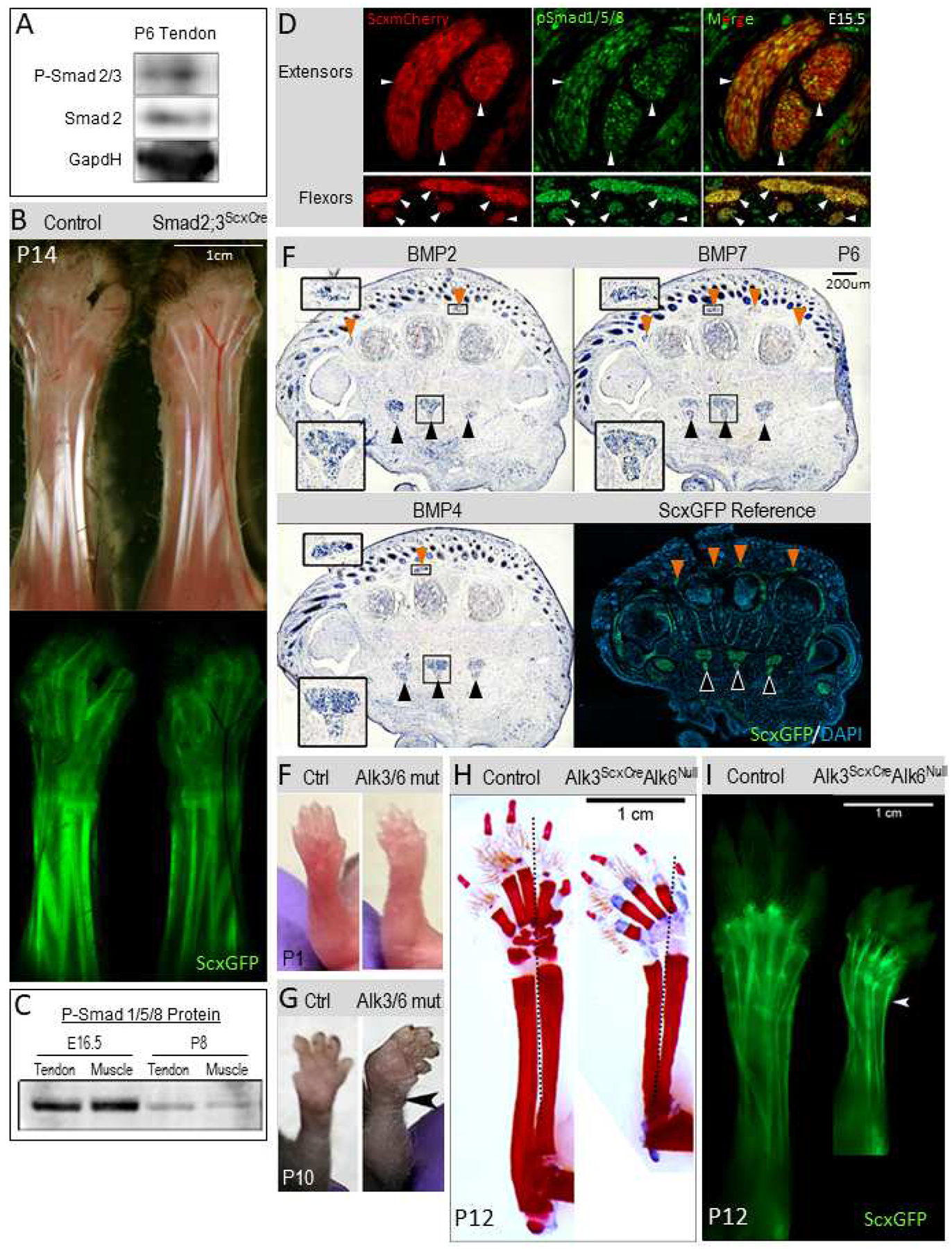
(A) Western blot of protein extract from wild-type tendons detected with anti-pSmad2 antibody shows TGFβ-signaling activation through Smad2 at P6. (B) Brightfield and ScxGFP images of skinned limbs from P14 Smad2;3ScxCre control and mutant mice. No contracture is detected in Smad2;3 mutants. (C) Activation of the BMP pathway through Smad1/5/8 phosphorylation is detectable by western blot in tendons and muscles from E16.5 and P8 limbs. (D) IHC detection of phospho-Smad1/5/8 (green) on cross sections from E15.5 forelimbs. Extensor tendons and flexor tendons were detected using the ScxmCherry tendon reporter (white arrowheads). (E) In-situ hybridization for the BMP ligands BMP2, BMP4, and BMP7 on cross sections from limbs of P6 WT pups. Both extensors (orange arrowheads, upper insets) and flexors (black arrowheads, lower insets) show BMP ligand expression. Insets are zoomed-in regions of the primary image. P6 ScxGFP image is included for anatomical reference, including arrowhead labels (extensors with orange arrowheads and flexors with black arrowheads). (F,G) Disruption of BMP signaling in Alk3ScxCreAlk6Null mutant mice results in wrist abduction. Comparisons of forelimbs from control and Alk3ScxCreAlk6Null mutant pups. (F) Slight wrist abduction can already be detected in mutant forelimbs at P1. (G) The wrist abduction in mutant forelimbs is markedly exaggerated by P10 (black arrowhead). (H) Skeletal preparations of P12 control and Alk3ScxCreAlk6Null mutant mice. Red is mineralized tissue (bone) (Alizarin Red) and blue is cartilage (Alcian Blue). Dotted black line shows center line highlighting the abduction of the mutant forelimb. (I) Tendons, in P12 control and Alk3ScxCreAlk6Null mutant mice, visualized using the ScxGFP reporter. Wrist abduction is visible (white arrowhead).
BMP signaling is activated during early tendon development
The results above suggest that the essential role of Smad4 in tendon growth likely reflects involvement of BMP signaling in this process, however the essential timing and pattern of BMP signaling in tendon was still unknown. Since postnatal deletion of Smad4 using ScxCreERT2 did not result in contracture (Figure 3A), we hypothesized that an earlier loss of Smad4 may be setting the stage for the postnatal emergence of the phenotype. We therefore investigated indications of active BMP signaling from early developmental stages through to the onset of the phenotype in postnatal stages. Interestingly, we detected phosphorylated Smad1/Smad5/Smad8 in both embryonic tendons and in postnatal tendons (Figure 4C–D). Moreover, significant expression of the BMP ligands BMP2, BMP4, and BMP7 was detected in tendons at P6, close to when the contracture develops (Figure 4E). The expression data thus supports the possibility that BMP signaling in tendon cells may play a role in the regulation of tendon elongation.
Disruption of BMP signaling in tendons recapitulates the Smad4ScxCre contracture phenotype
Next, we wanted to determine directly whether BMP signaling was the primary signaling pathway responsible for joint contractures in the Smad4ScxCre mutants. To avoid analysis of a triple mutant for Smads1, 5, and 8 we decided to target receptors for BMP signaling using ScxCre. Receptor specificity in the TGFβ superfamily is complex but the receptors are mostly separate between the TGFβ and BMP pathways. It was previously shown that a double mutant for the type 1 receptors Alk3 and Alk6 was sufficient to almost exclusively target BMP signaling (Yoon et al., 2006, 2005). The specific mutant crosses used a combination of floxed Alk3 and a null mutant for Alk6 (since a conditional mutant for Alk6 does not exist). We therefore generated the Alk3 floxed/Alk6 null/ScxCre mouse (Alk3ScxCre/Alk6null) by crossing the previously-developed Alk3 floxed, Alk6 null, and ScxCre mice (Blitz et al., 2013; Mishina et al., 2002; Yi et al., 2000). Notably, single Alk6null mutants are viable but small, reflecting a role for BMP signaling in skeletal growth (Yi et al., 2000). The Alk3ScxCre/Alk6null mouse mutants were indeed short but also developed joint contractures similar to those found in the Smad4ScxCre mouse shortly after birth (Figure 4F–I). Abduction was seen slightly earlier in the Alk3ScxCre/Alk6null mice than in the Smad4ScxCre mice (Figure 4F) and the contracture remained only in abduction, without additional development of extension or flexion (Figure 4G–I). These results suggest that the integration of BMP signaling by Smad4 is driving the contracture phenotype, pointing to an essential role of BMP signaling in tendon elongation.
Smad4 regulates the rate of collagen growth
Collagen fibrils constitute the major component of the force transmitting apparatus in mature tendons. To sustain these biomechanical functions, the cross-section area of individual collagen fibrils grows dramatically during tendon maturation. At birth, the collagen fibrils in mouse tendons are homogenous consisting of primarily small diameter fibrils with a diameter of approximately 40 nm. At P5 the mode of collagen fibril growth changes and fibril diameters in later stages are heterogeneous with fibril diameters that span 40–300nm (Ansorge et al., 2011; Birk and Brückner, 2010; Moore and Beaux, 1987; Oryan and Shoushtari, 2008). Since emergence of limb contractures in Smad4ScxCre mutants coincided with the shift in the mechanism of collagen fibril growth, we tested whether collagen fibril growth was affected in the mutant pups. Collagen fibril diameters were measured in high magnification transmission electron microscope (TEM) images of tendon cross section from control and mutant littermates at P5 and P14 (Figure 5A–B). In tendons of P14 mutant pups we found that collagen fibril diameters were heterogeneous (Figure 5B), indicating that the mechanistic shift in the growth of collagen fibrils was not disrupted in mutant tendons. Notably however, the distribution of collagen fibril diameters was affected in Smad4ScxCre mutants. At P14, the control Flexor Carpi Ulnaris tendon included fibrils with diameters of up to 170 nm while the diameter of collagen fibrils in the mutant tendons did not exceed 140nm, suggesting a role for Smad4 signaling in postnatal growth of the collagen fibrils in tendons (Figure 5C, Supplementary Figure 5). Interestingly, while CSA of mutant tendons is already reduced at P5, we found that collagen fibril diameter distribution was not affected (data not shown), suggesting that the total number of collagen fibrils may also be affected in the tendons of Smad4ScxCre mutant pups, possibly in correlation with the reduction in the number of cells in these tendons.
Figure 5: Smad4 signaling regulates postnatal growth of collagen fibrils in tendons.
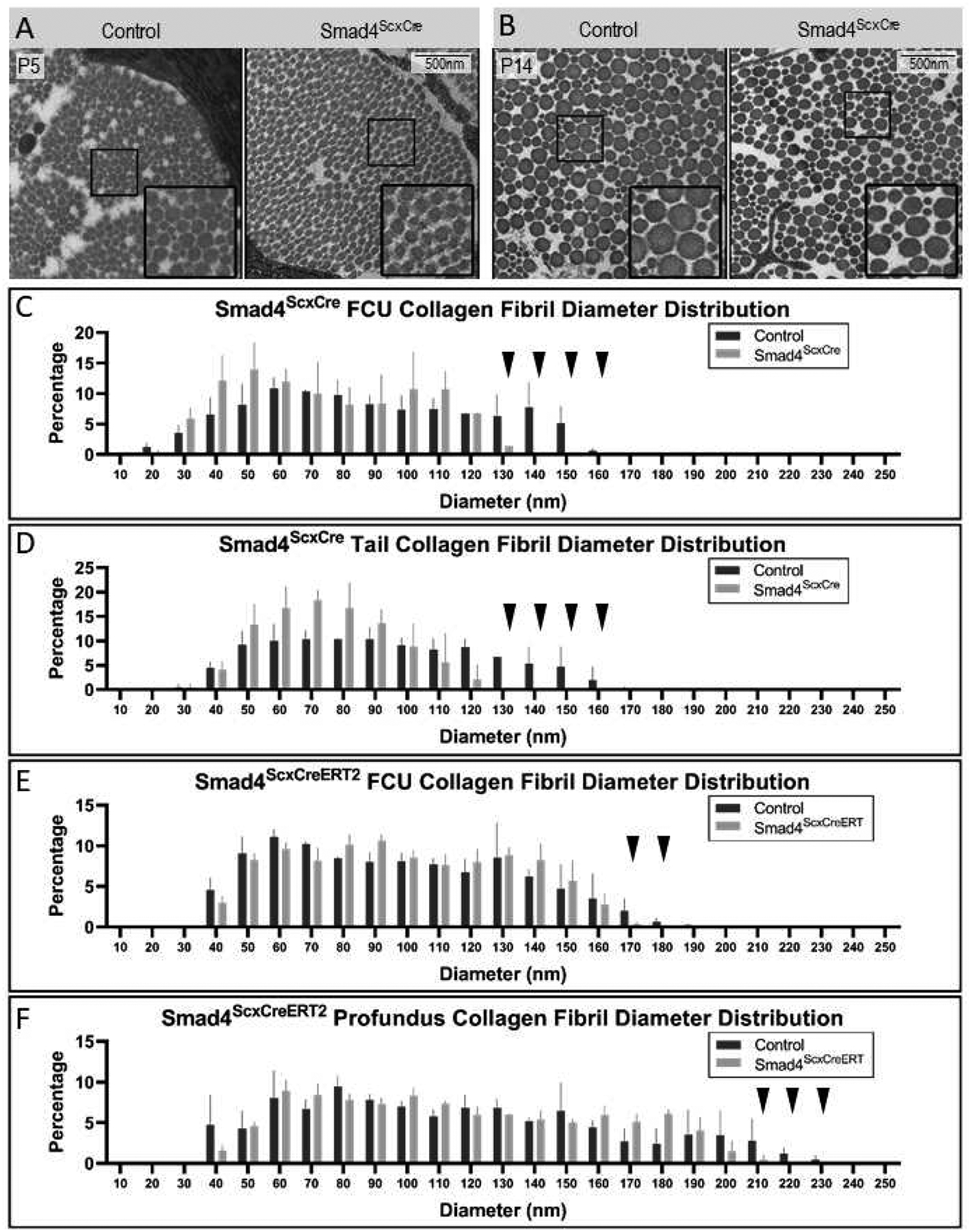
(A,B) Transmission Electron Micrographs of FCU tendons from P5 (A) and P14 (B) control and Smad4ScxCre mutants. Insets are zoomed-in views from the boxed region of the primary image. (C) Quantification of collagen fibril diameters in FCU tendon at P14. Mutant tendons lack highest diameter fibrils (>130nm in diameter, black arrowheads). (D) Distribution of collagen fibril diameter in tail tendons from control and Smad4ScxCre mutant mice at P14. Collagen fibrils with diameters over 130nm are absent or significantly reduced in mutant (black arrowheads). (E,F) Postnatal targeting of Smad4 results in reduced collagen fibril growth. Smad4 was targeted using ScxCreERT2 by administration of tamoxifen at P2 and P4 and control and Smad4ScxCreERT2 mutant mice were harvested at P16. Collagen fibrils were measured in Flexor Carpi Ulnaris (FCU) tendon (E) and the Profundus flexor tendon (F). Collagen fibril diameters are presented as distribution by percentage in control and mutant mice. Mutant displays a reduction of large fibrils over 170nm in diameter in FCU and over 200nm in Profundus (black arrowheads).
Collagen matrix growth is also affected by activity (Kjær et al., 2006; Zhang et al., 2010). Since the limbs of the Smad4ScxCre mutants are contracted at P14, restricted movement of the contracted limb joints may have a direct effect on the growth of the collagen matrix. To more directly determine the role of Smad4 in growth of the collagen matrix we therefore wanted to perform similar measurements in tissues whose movement is not restricted in these mutants. The tail of Smad4ScxCre mutants does not exhibit contractures or movement restrictions. We therefore next performed collagen fibril analysis of mutant tail tendons and again found reduction in the relative proportion of large diameter fibrils in mutant pups (Figure 5D, Supplementary Figure 5). In the tails of control mice at P14, the largest collagen fibrils had a diameter of 160nm, while the largest collagen fibrils in tails of mutant mice had only a diameter of 120nm.
Finally, to test if this role of Smad4 signaling can also be detected in limb tendons, we examined the tendons of postnatal inducible Smad4ScxCreERT2 mutant forelimb tendons, which exhibit loss of Smad4 but do not develop contracture (Figure 3A). Tamoxifen was administered at P2 and P4, and we again found that collagen fibrils with the largest diameters could not be detected at P14 in the Flexor Carpi Ulnaris or the Profundus Flexor tendons of the mutant forelimbs (Figure 5E–F, Supplementary Figure 5). Taken together these data demonstrate a role of Smad4 signaling in the regulation of collagen fibril growth.
Discussion
Joint contractures manifest in a wide range of conditions resulting in mild to severe limitations to joint movement and function. The etiology of joint contractures is complex. A genetic basis has been established for some conditions (e.g. club foot, Duchenne muscular dystrophy), while other conditions were associated with events during pregnancy or birth (e.g. arthrogryposis, cerebral palsy) (Bamshad et al., 2009; Hall, 1985; Hu et al., 2005). Interestingly, the primary effect in most of these conditions is either on muscles (e.g. arthrogryposis, Duchenne muscular dystrophy) or on the nervous system (e.g. cerebral palsy)(Darin et al., 2002; Farmer and James, 2009), but the connection between these primary causes and the subsequent events leading to joint contracture is largely unknown. Treatment for these conditions is focused on relieving the tension on the joints, first by non-invasive approaches such as physical therapy and bracing; if such therapies are not sufficient, then surgical lengthening of the tendons is used (Kowalczyk and Feluś, 2016). Developing mechanistic understanding of the regulation of tendon elongation may thus provide insights both to the etiology of contractures and why tendons do not elongate sufficiently to avoid the development of a contracture. In addition, such a model can aid the development of improvements for the management of these conditions.
We find that disruption of Smad4 signaling in the ScxCre cell lineage resulted in the development of joint contractures in early postnatal stages. A direct disruption of tendon function as a primary cause for contractures has not been identified to date. Therefore, the Smad4ScxCre mutant cannot be considered an animal model for a specific condition of acquired joint contractures. However, it provides a surprising evidence for Smad4 and specifically BMP signaling in the regulation of coordinated tendon elongation. Moreover, as a unique mouse model that develops a joint contracture, it provides insights and opportunity for analysis of the cellular and molecular events that underlie the initiation and progression of joint contractures.
Most bilateral joint contractures result in both limbs exhibiting the same direction of contracture, dorsally or ventrally (Kowalczyk and Feluś, 2016). It is therefore assumed that there is an underlying imbalance of forces around the joint that leads to a consistent direction of the contracture. The forelimb joint contractures in Smad4ScxCre mutants differ in that the contractures can resolve to either dorsal or ventral contracture and in some cases the direction was even different between the two forelimbs of the same animal. We suggest that this unique feature of the Smad4ScxCre phenotype reflects two stages in the development of a joint contracture: initiation and fixation. In the Smad4ScxCre mutant the initiation phase is stochastic between the dorsal or ventral directions. The stochastic nature of joint contractures in the Smad4ScxCre mutant suggests that while the mutation leads to a failure of coordinated growth between the muscle-tendon unit and the skeleton this failure in coordination is not biased in terms of the specific dorsal or ventral direction. However, once a contracture is initiated, the biomechanical conditions around the joint are altered and subsequent progression results in enhancement and fixation of the contracture in each of the affected limbs in the direction determined by the initiation phase. Analysis of the contracted limb at this phase may therefore not reflect the conditions that led to the initiation of the contracture. A unique feature of the Smad4ScxCre phenotype is the defined initiation of contracture around P5. Analysis of the mutant limbs at this phase may therefore help to shed light on the conditions that drive contracture initiation that may provide helpful clues to identify this phase in other cases of contracture during development.
The onset of joint contracture in Smad4ScxCre mutants at P5 was associated with a 30% reduction in the number of cells in the mutant tendons. Interestingly, the reduction in cell numbers of mutant tendons was detected only after P0 and the ratio of 30% less cells in mutant tendons was maintained in subsequent stages, suggesting a narrow time window between P0–P5 in which the loss of Smad4 signaling had an effect on tendon cellularity. Surprisingly, the rate of tenocyte proliferation was not affected in mutant tendons and the loss of Smad4 signaling was also not associated with a wave of cell death. The reduced cellularity in mutant tendons therefore likely reflects a failure of a wave of cell recruitment to tendons in neonates. We recently identified an essential role for tenocyte recruitment during embryonic tendon development (Huang et al. 2019). It is however not known if tendon growth in postnatal stages continues to be dependent on tenocyte recruitment and these results are the first indication that cell recruitment into developing tendons may extend into early neonatal stages. Cell recruitment into tendons in embryos was dependent on Scleraxis activity in the recruited cells (Huang et al. 2019). This is the first indication for a role for Smad4/BMP signaling in tendon cell recruitment, but we cannot determine at this time if Smad4 activity is required in the tendons to initiate cell recruitment or if like Scleraxis it is essential in the recruited cells to ensure recruitment. This observation also raises the question if all cell recruitment to tendons is also dependent on Smad4 signaling or alternatively if these results suggest a unique developmental window between P0–P5 in which Smad4-dependent molecular and/or cellular processes are essential for a neonatal cell recruitment event.
The complex distribution of ScxCre activity precluded a simple attribution of the contracture phenotype to Smad4 activity in tendons. However, the double-Cre experiment combining ScxCre and Pax7Cre and the tendon release experiment established an essential role for Smad4 loss in tenocytes in the development of joint contractures. It should be noted, however, that we cannot exclude at this time the possibility that Smad4 loss in other domains contributed to the development of the full phenotype. Likewise, deleting the BMP receptors Alk6 and Alk3 in tendon recapitulated the Smad4 contracture, suggesting a critical role for BMP signaling in regulation of tendon growth and possibly coordination of growth with other musculoskeletal tissues. However, while BMP receptor deletion recapitulated the timing and development of the joint contracture in Smad4ScxCre mutants, the initial abduction in the forelimbs of these mutants did not progress into a full contracture. The discrepancy between these phenotypes may suggest that additional signaling pathways affected by Smad4 deletion may be contributing to the contracture phenotype. It should however be noted that the BMP receptor mutants were generated as a combination of complete null for Alk6 and a conditional targeting of Alk3. The Alk6 mutation results in various effects on skeletal development, notably stunted growth that may also interfere with the full manifestation of the contracture phenotype in these mutants (Yi et al., 2000).
Most of the volume of mature tendons consists of highly organized collagen fibrils that mediate the mechanical functions of the tendons. Tendon growth therefore combines coordinated addition of tenocytes and collagen matrix. Interestingly we found in the tendons of Smad4ScxCre mutants a reduction in both cellular content and the collagen matrix. Since the tenocytes produce the collagen matrix it may be expected that reduced cell content in tendons may lead to a reduction in matrix content and therefore cross section area of the tendons. However, we found that between P5 and P14 the relative reduction in the number of cells did not change while the relative reduction in cross section area increased significantly. These results therefore suggest that Smad4 signaling regulates both of these components of tendons independently.
Since addition of cells and addition of matrix are both essential for tendon elongation the disruption of these processes may be directly linked to the development of joint contracture. However, contractures were not reported in various mutants that result in disruption and reduction in volume of the collagen matrix. For example, mutants for the homeobox transcription factor Mohawk have a significant reduction in the collagen matrix of their tendons, and mutants in the family of collagen associated small leucine-rich proteoglycans (SLRPs) have collagen fibril with smaller diameters and irregular morphology (Ito et al., 2010; Liu et al., 2010; Zhang et al., 2006). Yet, the Mohawk and SLRP mutations did not result in joint contractures. It is therefore likely that in addition to the specific reduction in cellularity and extracellular matrix, Smad4 signaling is directly involved in the coordinated growth of the skeleton and the soft tissues. The regulation of coordinated growth and of matrix addition by Smad4 signaling may be interrelated, i.e. the regulation of matrix deposition by Smad4 signaling may be the mechanism by which it regulates the coordinated growth of the musculoskeletal components. It should be noted however that we do not have direct evidence for the linkage between these functions and we cannot exclude a possibility that the disruption of coordinated musculoskeletal growth in the Smad4ScxCre mutants may not be associated with the role Smad4 signaling plays in regulated addition of matrix or cells to the growing tendons.
The mechanical properties of tendons are largely determined by the size and composition of the extracellular matrix of the tendon, primarily by a massive array of parallel collagen fibrils, in which collagen I is the major structural protein(Screen et al., 2015). The assembly and growth of the collagen matrix is complex and tightly coordinated process that involves various levels of regulation (Birk and Brückner, 2010; Holmes et al., 2018; Ishikawa et al., 2016; Mouw et al., 2014). The pro-collagens are large polypeptides subjects to numerous posttranslational modifications and a complex process for secretion and incorporation into existing fibrils. At the transcriptional level, the bHLH transcription factor Scleraxis and the homeobox transcription factor Mohawk have been implicated in regulation of the collagen matrix (Espira et al., 2009; Ito et al., 2010; Léjard et al., 2007; Liu et al., 2010). Collagen associated proteins were also associated with collagen deposition and maintenance, notably the family of SLRP proteins and various chaperones (Chen and Birk, 2013; Frolova et al., 2014; Piróg et al., 2013). At even higher levels, mechanical stimulation, such as through exercise, has also been shown to have a direct effect on collagen production and on long term expansion of the collagen matrix (Kjær et al., 2006; Zhang et al., 2010). The deposition and growth of the collagen matrix is thus a highly regulated process, but surprisingly, the role of signaling pathways in this process has not been elucidated to date. Here we found that targeting of Smad4 resulted in significant reduction in the rate of collagen matrix accumulation, suggesting a role for the TGFβ superfamily and, specifically in this case, for BMP signaling in regulation of tendon collagen deposition. Future studies will examine the source and regulation of BMP signaling in growing tendons and the regulatory activity of BMP signaling on collagen production and/or incorporation in fibrils.
Materials and Methods
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Oregon Health & Science University and are consistent with animal care guidelines. Existing mouse lines used in these studies were described previously (See Resource Table). (Pryce et al., 2007)(Keller et al., 2004)(Yu et al., 2002)(Baffi et al., 2006)(Mishina et al., 2002)(Yi et al., 2000)(Howell et al., 2017)(Blitz et al., 2013)(Li et al., 2008; Liu et al., 2004)(Soriano, 1999)(Madisen et al., 2009). For ScxCreERT2 studies, tamoxifen in corn oil was given to neonates by oral gavage (1.25 mg/pup at postnatal days 2 and 4).
Resource Table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (M. musculus) | ScxGFP | (Pryce et al., 2007) | N/A | N/A |
| Genetic reagent (M. musculus) | Pax7Cre | (Keller et al., 2004) | N/A | N/A |
| Genetic reagent (M. musculus) | Smad4 floxed | (Yu et al., 2002) | N/A | N/A |
| Genetic reagent (M. musculus) | Col2Cre | (Baffi et al., 2006) | N/A | N/A |
| Genetic reagent (M. musculus) | Alk3 floxed | (Mishina et al., 2002) | N/A | N/A |
| Genetic reagent (M. musculus) | Alk6 Null | (Yi et al., 2000) | N/A | N/A |
| Genetic reagent (M. musculus) | ScxCreERT2 | (Howell et al., 2017) | N/A | N/A |
| Genetic reagent (M. musculus) | ScxCre | (Blitz et al., 2013) | N/A | N/A |
| Genetic reagent (M. musculus) | Smad3 floxed | (Li et al., 2008; Liu et al., 2004) | N/A | N/A |
| Genetic reagent (M. musculus) | R26R-LacZ reporter | (Soriano, 1999) | N/A | N/A |
| Genetic reagent (M. musculus) | Ai14 Rosa26-TdTomato reporter (RosaT) | (Madisen et al., 2009) | N/A | N/A |
| Genetic reagent (M. musculus) | ScxmCherry | Generated by Ronen Schweitzer | N/A | N/A |
| Antibody | Rabbit anti-phospho Smad2 | Cell Signaling | Cat# 3101 | WB: 1:1000 |
| Antibody | Rabbit anti-Smad2 | Cell Signaling | Cat #5339 | WB: 1:1000 |
| Antibody | Mouse anti-GapdH | Novus | Cat#: NB300-221 | WB: 1:1000 |
| Antibody | Rabbit anti-phospho Smad1/5/8 | Cell Signaling | Cat# 9511 | IF: 1:100 WB: 1:1000 |
| Antibody | Rabbit anti-Smad4 | Abcam | Cat# ab40759 | IF: 1:100 |
| Antibody | Mouse anti-Smad4 | Santa Cruz | Cat# sc-7966 | WB: 1:500 |
| Antibody | Alexa 488 donkey anti-rabbit secondary | Invitrogen Molecular Probes | Cat# A-21206 | IF: 1:1000 |
| Antibody | AlexaFluor647 donkey anti-rabbit secondary | Jackson ImmunoResearch | Cat# 711-607-003 | IF: 1:400 |
| Antibody | HRP goat anti-rabbit secondary | Abcam | Cat# ab6721 | WB: 1:30000 |
| Antibody | HRP goat anti-mouse secondary | Abcam | Cat# ab6789 | WB: 1:30000 |
| Commercial assay or kit | Click-iT EdU kit | Life Technologies | Cat# C10340 | Follow the manufacturer’s instruction |
| Commercial assay or kit | In situ cell death detection kit | Roche | Cat# 12156792910 | Follow the manufacturer’s instruction |
| Other | DAPI stain | Thermo Fisher Scientific | D1306 | 1 μg/ml |
N/A = Not Applicable
Sectioning and Growth Measurements
Sample Preparation: Forelimbs were skinned and fixed in 4% paraformaldehyde overnight at 4°C. Postnatal tissues were decalcified in 5mM EDTA for several days at 4°C following fixation. Limbs were then cryoprotected in 5%−30% graded sucrose/PBS and embedded in Tissue-Tek OCT compound (Sakura Finetek). Alternating transverse cryosections (12 μm) were collected across the length of the limb. Slides were stained and imaged for analysis.
Growth Measurements: The distal insertion of the tendon is determined by the first section with GFP (ScxGFP), and the proximal origin of the tendon body is determined by the first section with muscle attachment. Multiple sections along the length of the tendon were analyzed and averaged to provide a single value per mouse. Cells were quantified by counting DAPI-labeled nuclei. CSA was defined by ScxGFP boundaries in sections.
Immunohistochemistry and Imaging
Immunohistochemistry: Sectioned slides were permeabilized in 0.3% PBS-TritonX for 10 minutes, and blocked in 5% serum for 1 hour at room temperature. Slides were incubated in primary antibody overnight before being incubated in secondary antibody for one hour. DAPI was used to counterstain for cell nuclei. See Resource Table for antibodies used. Fluorescent images of sections were captured using a Zeiss AxioImager and Zeiss Zen software.
Cell Proliferation and Cell Death: For examination of proliferation and cell death, EdU and TUNEL assays were performed using In Situ Cell Death Detection (Roche) and Click-iT EdU (Life Technologies) kits, respectively, following manufacturer’s instructions.
In Situ Hybridization: Standard protocol for in situ hybridization was performed as previously described (Murchison et al., 2007). In situ hybridization images were captured using a Nikon Eclipse E800 compound microscope.
Whole-Mount Imaging: Whole-mount images were captured using a Leica stereomicroscope with fluorescence capabilities (Leica MZFLIII).
Western Blot
Forelimb and hindlimb tendons were dissected, taking care to avoid enthesis and muscle insertions. Tissues were lysed in RIPA buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% SDS, 1.0% NP-40, 0.5% sodium deoxycholate) containing COMPLETE Protease Inhibitor cocktail (Roche). Protein quantification of cell and tissue lysates was performed using the BioRad DC Protein assay. Equal amounts of total protein were prepared in LDS sample buffer (Invitrogen) and 100mM DTT and run on 4–12% NuPAGE Bis-Tris gels (Invitrogen) in NuPAGE MOPS running buffer (Invitrogen). Wet transfer with NuPAGE transfer buffer (Invitrogen)/20% methanol was used to transfer protein onto Millipore Immobilon P (PVDF) membranes. Blots were washed thrice with TBS-0.1%Tween-20 and blocked in 5% serum for 1 hour. Blots were incubated in primary antibody overnight before being incubated in HRP-conjugated secondary antibody for one hour at room temperature. Blots were developed with ECL Substrate (BioRad) and imaged using the ChemiDoc XRS+ System (BioRad) for detection.
Transmission electron microscopy (TEM)
Areas of interest were identified by light microscopy. Ultra-thin sections (~80nm) from the corresponding region were cut on a Leica EM UC7 ultramictrotome (Leica Microsystems) and mounted on formvar coated, copper-palladium 1×2mm slot grids. Samples were stained with saturated uranyl acetate followed by lead citrate and photographed using an AMT 2K × 2K side entry digital camera (AMT, Woburn, MA) mounted on a in a FEI G2 TEM operated at 120KV. For examining overall tendon morphology, an array of overlapping images was collected at 11500x and a montage prepared using ImageJ (National Institutes of Health, Bethesda, MD) to form a (~360-Mb) composite tiff image. For morphometric analysis, fibril diameters were measured from images taken at 50000x using Image J (NIH). A minimum of 1000 fibrils were counted per mouse.
Skeletal Prep
P5 control and mutant limbs were dissected and soft tissue removed. Cartilage and bones were visualized after staining with Alcian blue and Alizarin red S (Sigma) and clarification of soft tissue with KOH (McLeod, 1980).
Surgical transection
Smad4Control and Smad4ScxCre limbs were skinned and imaged. Smad4ScxCre forelimb lateral tendons were severed and wrist was gently repositioned with forceps before being re-imaged.
Statistical analyses
All experiments were performed in triplicate as a minimum and the data shown were chosen as representative of results consistently observed. Results are presented as mean ± standard deviation. Prism 8 (Graphpad Software) was used to visualize quantitative data and perform statistics. Unpaired student’s t-tests were performed to determine significance. Significant differences were detected at p<0.05.
Supplementary Material
Highlights.
Smad4 deletion in tenocytes in mice using Scleraxis-Cre produces a postnatal joint contracture with significantly fewer tenocytes, thinner tendons, and a lack of very large collagen fibrils.
Targeted deletion of BMP signaling, but not TGFb signaling, recapitulates the Smad4 contracture phenotype.
Postnatal inducible Smad4 deletion does not recapitulate the contracture phenotype but does result in a lack of very large collagen fibrils, suggesting Smad4 regulation on matrix is independent of the contracture.
Acknowledgments
This work was funded by Shriners Hospitals for Children [SHC 5410-POR-14].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansorge HL, Hsu JE, Edelstein L, Adams S, Birk DE, Soslowsky LJ, 2011. Recapitulation of the Achilles tendon mechanical properties during neonatal development: a study of differential healing during two stages of development in a mouse model. J Orthop Res Official Publ Orthop Res Soc 30, 448–56. 10.1002/jor.21542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffi MO, Moran MA, Serra R, 2006. Tgfbr2 regulates the maintenance of boundaries in the axial skeleton. Dev Biol 296, 363–374. 10.1016/j.ydbio.2006.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad M, Heest AEV, Pleasure D, 2009. Arthrogryposis. J Bone Jt Surg 91, 40–46. 10.2106/jbjs.i.00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan WP, Hall JG, Bamshad M, Staheli LT, Jaffe KM, Song K, 2007. Arthrogryposis Multiplex Congenita (Amyoplasia). J Pediatr Orthoped 27, 594–600. 10.1097/bpo.0b013e318070cc76 [DOI] [PubMed] [Google Scholar]
- Birk DE, Brückner P, 2010. The Extracellular Matrix: an Overview 77–115. 10.1007/978-3-642-16555-9_3 [DOI] [Google Scholar]
- Blitz E, Sharir A, Akiyama H, Zelzer E, 2013. Tendon-bone attachment unit is formed modularly by a distinct pool of Scx- and Sox9-positive progenitors. Development 140, 2680–2690. 10.1242/dev.093906 [DOI] [PubMed] [Google Scholar]
- Blitz E, Viukov S, Sharir A, Shwartz Y, Galloway JL, Pryce BA, Johnson RL, Tabin CJ, Schweitzer R, Zelzer E, 2009. Bone Ridge Patterning during Musculoskeletal Assembly Is Mediated through SCX Regulation of Bmp4 at the Tendon-Skeleton Junction. Developmental Cell 17. 10.1016/j.devcel.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Deng C, Li Y-P, 2012. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. International Journal of Biological Sciences 8. 10.7150/ijbs.2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Birk DE, 2013. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. Febs J 280, 2120–37. 10.1111/febs.12136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin N, Kimber E, Kroksmark A-K, Tulinius M, 2002. Multiple congenital contractures: Birth prevalence, etiology, and outcome. J Pediatrics 140, 61–67. 10.1067/mpd.2002.121148 [DOI] [PubMed] [Google Scholar]
- Espira L, Lamoureux L, Jones SC, Gerard RD, Dixon IM, Czubryt MP, 2009. The basic helix–loop–helix transcription factor scleraxis regulates fibroblast collagen synthesis. J Mol Cell Cardiol 47, 188–195. 10.1016/j.yjmcc.2009.03.024 [DOI] [PubMed] [Google Scholar]
- Farmer SE, James M, 2009. Contractures in orthopaedic and neurological conditions: a review of causes and treatment. Disabil Rehabil 23, 549–558. 10.1080/09638280010029930 [DOI] [PubMed] [Google Scholar]
- Frolova EG, Drazba J, Krukovets I, Kostenko V, Blech L, Harry C, Vasanji A, Drumm C, Sul P, Jenniskens GJ, Plow EF, Stenina-Adognravi O, 2014. Control of organization and function of muscle and tendon by thrombospondin-4. Matrix Biology J Int Soc Matrix Biology 37, 35–48. 10.1016/j.matbio.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JG, 1985. Genetic aspects of arthrogryposis. Clin Orthop Relat R NA; 44–53. 10.1097/00003086-198504000-00006 [DOI] [PubMed] [Google Scholar]
- Hall JG, Reed SD, Driscoll EP, Opitz JM, 1983. Part I. Amyoplasia: A common, sporadic condition with congenital contractures. Am J Med Genet 15, 571–590. 10.1002/ajmg.1320150407 [DOI] [PubMed] [Google Scholar]
- Havis E, Bonnin M-AA, de Lima JE, Charvet B, Milet C, Duprez D, 2016. TGFβ and FGF promote tendon progenitor fate and act downstream of muscle contraction to regulate tendon differentiation during chick limb development. Development (Cambridge, England). 10.1242/dev.136242 [DOI] [PubMed] [Google Scholar]
- Holmes DF, Lu Y, Starborg T, Kadler KE, 2018. Collagen Fibril Assembly and Function. Current topics in developmental biology 130, 107–142. 10.1016/bs.ctdb.2018.02.004 [DOI] [PubMed] [Google Scholar]
- Howell K, Chien C, Bell R, Laudier D, Tufa SF, Keene DR, Andarawis-Puri N, Huang AH, 2017. Novel Model of Tendon Regeneration Reveals Distinct Cell Mechanisms Underlying Regenerative and Fibrotic Tendon Healing. Sci Rep-uk 7, 45238. 10.1038/srep45238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Nystrom A, Ahmed A, Palmquist M, Dopico R, Mossberg I, Gladitz J, Rayner M, Post J, Ehrlich G, Preston R, 2005. Mapping of an autosomal dominant gene for Dupuytren’s contracture to chromosome 16q in a Swedish family. Clin Genet 68, 424–429. 10.1111/j.1399-0004.2005.00504.x [DOI] [PubMed] [Google Scholar]
- Huang AH, 2017. Coordinated development of the limb musculoskeletal system: Tendon and muscle patterning and integration with the skeleton. Developmental Biology. 10.1016/j.ydbio.2017.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AH, Lu HH, Schweitzer R, 2015. Molecular regulation of tendon cell fate during development. Journal of Orthopaedic Research 33, 800–812. 10.1002/jor.22834 [DOI] [PubMed] [Google Scholar]
- Huang AH, Watson SS, Wang L, Baker BM, Akiyama H, Brigande JV, Schweitzer R, 2019. Requirement for scleraxis in the recruitment of mesenchymal progenitors during embryonic tendon elongation. Development 146, dev182782. 10.1242/dev.182782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson DA, Zhao J, Merrell A, Haldar M, Kardon G, 2009. Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for β-catenin. Genes & Development 23, 997–1013. 10.1101/gad.1769009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Ito S, Nagata K, Sakai LY, Bächinger HP, 2016. Intracellular mechanisms of molecular recognition and sorting for transport of large extracellular matrix molecules. Proc National Acad Sci 113, E6036–E6044. 10.1073/pnas.1609571113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Toriuchi N, Yoshitaka T, Ueno-Kudoh H, Sato T, Yokoyama S, Nishida K, Akimoto T, Takahashi M, Miyaki S, Asahara H, 2010. The Mohawk homeobox gene is a critical regulator of tendon differentiation. Proceedings of the National Academy of Sciences. 10.1073/pnas.1000525107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannus P, 2000. Structure of the tendon connective tissue. Scand J Medicine Sci Sports 10, 312–320. 10.1034/j.1600-0838.2000.010006312.x [DOI] [PubMed] [Google Scholar]
- Keller C, Hansen MS, Coffin CM, Capecchi MR, 2004. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes & Development 18, 2608–2613. 10.1101/gad.1243904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjær M, Magnusson P, Krogsgaard M, Møller JB, Olesen J, Heinemeier K, Hansen M, Haraldsson B, Koskinen S, Esmarck B, Langberg H, 2006. Extracellular matrix adaptation of tendon and skeletal muscle to exercise. J Anat 208, 445–450. 10.1111/j.1469-7580.2006.00549.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk B, Feluś J, 2016. Arthrogryposis: an update on clinical aspects, etiology, and treatment strategies. Archives Medical Sci Ams 12, 10–24. 10.5114/aoms.2016.57578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léjard V, Brideau G, Blais F, Salingcarnboriboon R, Wagner G, Roehrl MHA, Noda M, Duprez D, Houillier P, Rossert J, 2007. Scleraxis and NFATc Regulate the Expression of the Pro-α1(I) Collagen Gene in Tendon Fibroblasts. J Biol Chem 282, 17665–17675. 10.1074/jbc.m610113200 [DOI] [PubMed] [Google Scholar]
- Li Q, Pangas SA, Jorgez CJ, Graff JM, Weinstein M, Matzuk MM, 2008. Redundant Roles of SMAD2 and SMAD3 in Ovarian Granulosa Cells In Vivo. Molecular and Cellular Biology 28, 7001–7011. 10.1128/mcb.00732-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Watson SS, Lan Y, Keene DR, Ovitt CE, Liu H, Schweitzer R, Jiang R, 2010. The Atypical Homeodomain Transcription Factor Mohawk Controls Tendon Morphogenesis. Molecular and Cellular Biology 30, 4797–4807. 10.1128/mcb.00207-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Festing MH, Hester M, Thompson JC, Weinstein M, 2004. Generation of novel conditional and hypomorphic alleles of the Smad2 gene. genesis 40, 118–123. 10.1002/gene.20072 [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh S, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, 2009. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience 13, 133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magerl M, Tobin DJ, Müller-Röver S, Hagen E, Lindner G, McKay IA, Paus R, 2001. Patterns of Proliferation and Apoptosis during Murine Hair Follicle Morphogenesis. J Invest Dermatol 116, 947–955. 10.1046/j.0022-202x.2001.01368.x [DOI] [PubMed] [Google Scholar]
- Massagué J, 2012. TGFβ signalling in context. Nat Rev Mol Cell Bio 13, 616. 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D, 2005. Smad transcription factors. Genes & Development 19. 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- McLeod MJ., 1980. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology, 22, 299–301. 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR, 2002. Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32, 69–72. 10.1002/gene.10038 [DOI] [PubMed] [Google Scholar]
- Moore MJ, & De Beaux A, 1987. A quantitative ultrastructural study of rat tendon from birth to maturity. Journal of anatomy, 153, 163–169. [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Ou G, Weaver VM, 2014. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Bio 15, 771–785. 10.1038/nrm3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison ND, Price BA, Conner DA, Keene DR, Olson EN, Tabin CJ, Schweitzer R, 2007. Regulation of tendon differentiation by scleraxis distinguishes force-transmitting tendons from muscle-anchoring tendons. Development 134, 2697–2708. 10.1242/dev.001933 [DOI] [PubMed] [Google Scholar]
- Oryan A, Shoushtari AH, 2008. Histology and ultrastructure of the developing superficial digital flexor tendon in rabbits. Anatomia Histologia Embryologia 37, 134–40. 10.1111/j.1439-0264.2007.00811.x [DOI] [PubMed] [Google Scholar]
- Piróg KA, Katakura Y, Mironov A, Briggs MD, 2013. Mild Myopathy Is Associated with COMP but Not MATN3 Mutations in Mouse Models of Genetic Skeletal Diseases. Plos One 8, e82412. 10.1371/journal.pone.0082412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce BA, Brent AE, Murchison ND, Tabin CJ, Schweitzer R, 2007. Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Developmental Dynamics 236, 1677–1682. 10.1002/dvdy.21179 [DOI] [PubMed] [Google Scholar]
- Pryce BA, Watson SS, Murchison ND, Staverosky JA, Dünker N, Schweitzer R, 2009. Recruitment and maintenance of tendon progenitors by TGFβ signaling are essential for tendon formation. Development 136, 1351–1361. 10.1242/dev.027342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph C, Rudolph A, Lister G, First L, Gershon A, 2011. Rudolph’s Pediatrics. McGraw-Hill Education. [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H, Sandri M, 2013. BMP signaling controls muscle mass. Nature Genetics 45, 1309–1318. 10.1038/ng.2772 [DOI] [PubMed] [Google Scholar]
- Schweitzer R, Zelzer E, Volk T, 2010. Connecting muscles to tendons: tendons and musculoskeletal development in flies and vertebrates. Development 137, 2807–2817. 10.1242/dev.047498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screen HR, Berk DE, Kadler KE, Ramirez F, Young MF, 2015. Tendon Functional Extracellular Matrix. Journal of Orthopaedic Research 33, 793–799. 10.1002/jor.22818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slakey JB, Hennrikus WL, 1996. Acquired thumb flexion contracture in children: congenital trigger thumb. J Bone Jt Surg Br Volume 78, 481–3. 10.1302/0301-620x.78b3.0780481 [DOI] [PubMed] [Google Scholar]
- Soriano P, 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics 21, 70–71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Kanzaki LF, Galloway JL, Schilling TF, 2018. Mechanical force regulates tendon extracellular matrix organization and tenocyte morphogenesis through TGFbeta signaling. Elife 7, e38069. 10.7554/elife.38069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinyard CA, Bleck EE, 1985. The etiology of arthrogryposis (multiple congenital contracture). Clin Orthop Relat R NA; 15–29. 10.1097/00003086-198504000-00004 [DOI] [PubMed] [Google Scholar]
- Tan G-K, Pryce BA, Stabio A, Brigande JV, Wang C, Xia Z, Tufa SF, Keene DR, Schweitzer R, 2020. TGFβ signaling is critical for maintenance of the tendon cell fate. Elife 9, e52695. 10.7554/elife.52695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SS, Riordan TJ, Pryce BA, Schweitzer R, 2009. Tendons and muscles of the mouse forelimb during embryonic development. Developmental Dynamics 238, 693–700. 10.1002/dvdy.21866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Chen G, Li Y-P, 2016. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4, 16009. 10.1038/boneres.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi S, Daluiski A, Pederson R, Rosen V, Lyons K, 2000. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development (Cambridge, England) 127, 621–30. [DOI] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM, 2005. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. P Natl Acad Sci Usa 102, 5062–5067. 10.1073/pnas.0500031102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM, 2006. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 133, 4667–4678. 10.1242/dev.02680 [DOI] [PubMed] [Google Scholar]
- Yu Y, Yang J, Chapman-Sheath PJ, Walsh WR, 2002. TGF-β, BMPS, and their signal transducing mediators, Smads, in rat fracture healing. J Biomed Mater Res 60, 392–397. 10.1002/jbm.1289 [DOI] [PubMed] [Google Scholar]
- Zhang G, Ezura Y, Chervoneva I, Robinson PS, Beason DP, Carine ET, Soslowsky LJ, Iozzo RV, Birk DE, 2006. Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J Cell Biochem 98, 1436–1449. 10.1002/jcb.20776 [DOI] [PubMed] [Google Scholar]
- Zhang J, Pan T, Liu Y, Wang JH, 2010. Mouse treadmill running enhances tendons by expanding the pool of tendon stem cells (TSCs) and TSC-related cellular production of collagen. J Orthopaed Res 28, 1178–1183. 10.1002/jor.21123 [DOI] [PubMed] [Google Scholar]
- Zhang J, Tan X, Li W, Wang Y, Wang J, Cheng X, Yang X, 2005. Smad4 is required for the normal organization of the cartilage growth plate. Developmental Biology 284. 10.1016/j.ydbio.2005.05.036 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.