

Abstract
This Viewpoint aims to highlight positron emission tomography (PET) research studies that have shaped our understanding of the endocannabinoid system (ECS) through radiopharmaceutical targeting of cannabinoid receptors 1 and 2 (CB1 and CB2), and the enzyme fatty acid amide hydrolase (FAAH), in several brain health illnesses including addiction, schizophrenia, eating disorders and post-traumatic stress disorder. Advances in radiochemistry, including 11C-carbonylation and radiofluorination of non-activated aromatic rings are accelerating our translation of radiotracers with optimal kinetics, bringing us closer to clinical PET research studies to image the enzyme monoacylglycerol lipase (MAGL), and enabling the imaging of unexplored targets in the ECS.
Keywords: PET, CB1, CB2, FAAH, MAGL, endocannabinoid
Graphical Abstract
With the legalization of cannabis and related products emerging internationally, as well as drug development targeting the endocannabinoid system (ECS) in brain health illnesses, it is more timely than ever to increase our understanding of the ECS through molecular imaging. The ECS is a signaling system composed of endogenous lipid-based endocannabinoids, which are retrograde neurotransmitters synthesized in the postsynaptic neuron and released into the synaptic cleft, where they activate presynaptic cannabinoid (CB) receptors to inhibit neurotransmission.1 The ECS is responsible for modulating neurotransmitter release and is involved in maintaining neuronal plasticity, regulating pain, inflammation, emotional processing and behavioural responses. The ECS has been implicated in several brain health illnesses including post-traumatic stress disorder (PTSD), schizophrenia and eating disorders, temporal lobe epilepsy (TLE), and neurodegenerative diseases including Alzheimer’s Disease (AD) and Parkinson’s Disease (PD). Cannabis use disorder is associated with cognitive impairment and has shown psychiatric comorbidity in mood disorders, psychosis and with other substance use disorders (SUDs). Diagnostic and therapeutic efforts are avidly underway to identify potent and selective compounds that target receptors and enzymes in the ECS.
The psychoactive component of cannabis, Δ9–tetrahydrocannabinol (THC), acts on the ECS through the type-1 (CB1) and type-2 (CB2) cannabinoid receptors. The ECS can be pharmacologically probed to modify cannabinoid neurotransmission in several different ways, including direct agonism/antagonism, allosteric modulation of CB receptors, or via enzymes involved in the synthesis and degradation of endogenous cannabinoid ligands. CB1 receptors are primarily expressed on presynaptic GABAergic and glutamatergic neurons, in the central nervous system (CNS), and this receptor is expressed to a lesser extent in peripheral organs. CB1 receptor signaling is involved in motivation, cognition and stress reactivity; its expression is highest within sensory and motor regions, such as the basal ganglia, substantia nigra, globus pallidus, cerebellum and hippocampus. CB2 receptors are primarily expressed on peripheral immune cells and the spleen shows the highest CB2 receptor density, although there is also evidence of expression of this receptor in the CNS. Enzymes of interest in the ECS, are fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), which hydrolyze the major endocannabinoids, anandamide (AEA) and 2-arachidonyl glycerol (2-AG), respectively. FAAH is localized primarily at the post-synaptic terminal where it metabolizes AEA to arachidonic acid, terminating AEA signaling which acts as a partial agonist of CB receptors. MAGL is primarily located in the pre-synaptic axon terminal, where it is co-localized with CB1 receptors. MAGL is responsible for ~85% of 2-AG metabolism in the brain, terminating 2-AG signaling by hydrolysis to arachidonic acid. 2-AG is 100–1000 fold more abundant in the CNS than AEA, and is a full agonist of CB receptors. Other metabolic enzymes involved in synthesis and degradation of AEA and 2-AG include alpha/beta-hydrolase domain containing 6/12 (ABHD6 and ABDH12) which contribute to 2-AG hydrolysis and diacylglycerol lipase (DAGL), which catalyzes the synthesis of 2-AG. Pharmacologically modulating these enzyme targets within the ECS has the capability for analgesic, antihypertensive, antiepileptic, neuroprotective, immunomodulatory and anti-inflammatory effects.
Positron emission tomography (PET) imaging studies of the ECS have been conducted in several brain health illnesses. Herein we aim to further contribute toward the series of Viewpoints and Minireviews: Classics in Neuroimaging, by highlighting PET radiopharmaceuticals used in clinical research studies of CB1 and CB2 receptors, and FAAH. These studies have recently shown differences in target abundance in several neurological and psychiatric disorders including addiction, schizophrenia, eating disorders and post-traumatic stress disorder. We also review the ongoing development towards a PET radiotracer for imaging of MAGL and consider how advances in drug development in the ECS and novel radiochemistry, including 11C-carbonylation and radiofluorination, will impact development of PET radiotracers with optimal brain kinetics and for imaging novel targets in the ECS.
CB1 Receptors:
CB1 receptors are among the most abundant receptors expressed in the human CNS and are the primary receptors involved in the ECS. As such, they have been the most widely studied PET imaging target of the ECS in humans.2 To date, there have been five radiotracers used to image CB1 receptors in humans: [18F]MK-9470, [18F]FMPEP-d2, [11C]OMAR, [11C]MePPEP and [11C]SD5024 (Structures shown in Figure 1).
Figure 1:
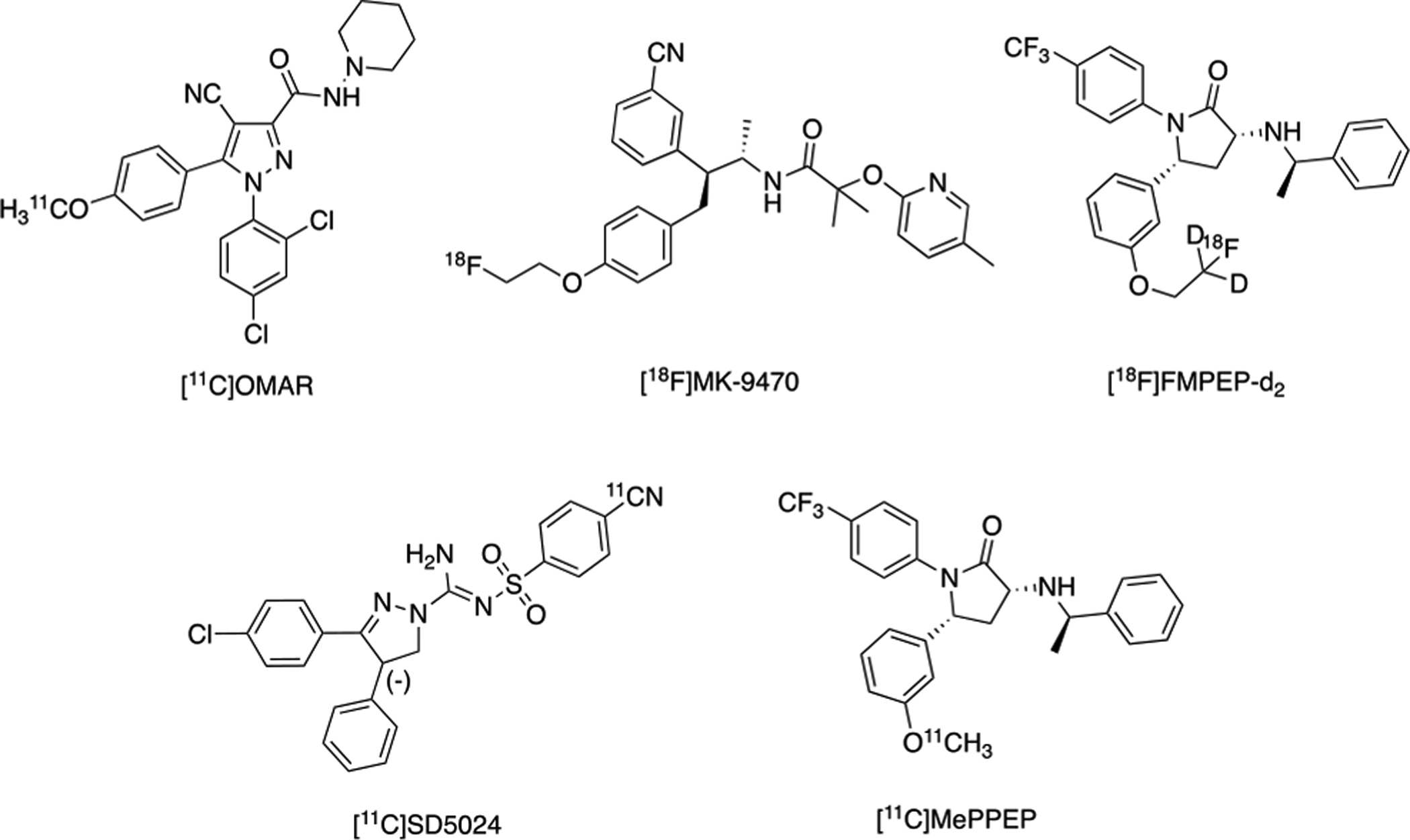
Radiotracers for Human PET Imaging of CB1 Receptors.
[11C]MePPEP and [11C]SD5024 have been translated in humans to evaluate radioligand kinetics using compartmental modeling and test-retest variability. [11C]MePPEP displayed high brain uptake and slow washout in humans, with varying levels of reproducibility using different kinetic models within, and between, studies. [11C]SD5024 showed good precision for measuring CB1 receptors in humans, with moderate brain uptake, low inter-subject variability for distribution volume (VT) and high specific binding. However, [11C]MePPEP and [11C]SD5024 have not yet been used in human studies of psychiatric or neurological disorders. [18F]MK-9470, [18F]FMPEP-d2 and [11C]OMAR are the three radiopharmaceuticals that have been employed in clinical research studies to image patients with brain health illnesses.
The following clinical research studies with [18F]MK-9470 reported receptor availability through modified standardized uptake value (mSUV) or fractional uptake ratio (FUR), whereas [11C]OMAR and [18F]FMPEP-d2 report receptor availability with arterial input functions and reported VT. Figure 2 highlights a study conducted at the National Institute of Mental Health (NIMH) where [18F]FMPEP-d2 uptake is observed in a healthy human brain.
Figure 2. [18F]FMPEP-d2: Antagonist radioligand for cannabinoid CB1 receptors in a healthy human subject.
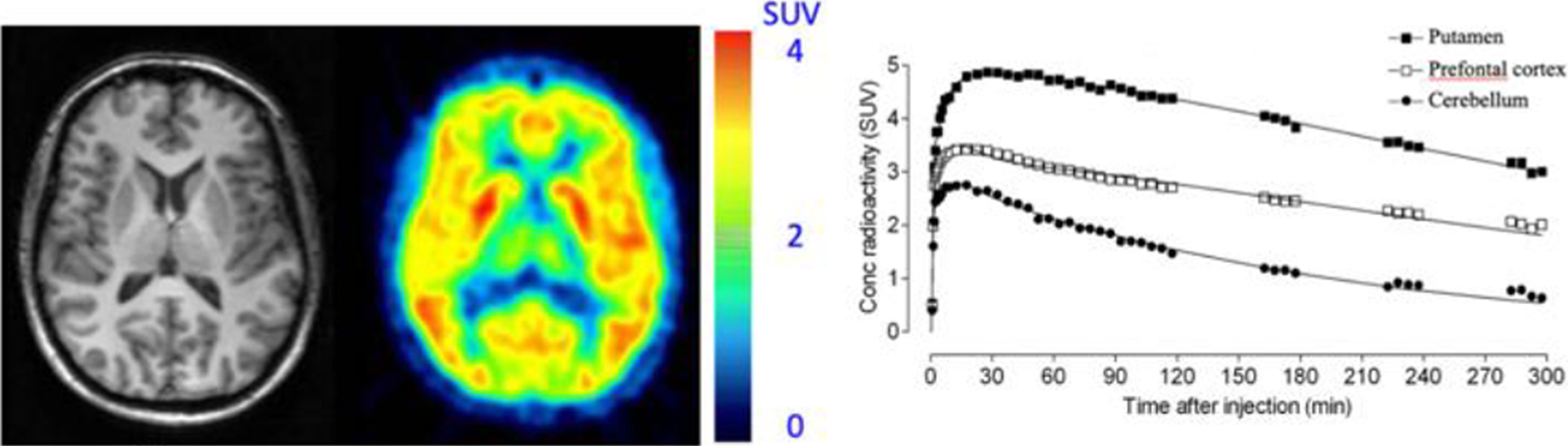
Transaxial section of MRI scan (left) for co-registration, and PET scan (middle) show the distribution of radioactivity in a healthy human brain is consistent with the known widespread distribution of CB1 receptors. Additionally, the regional time activity curves (right) demonstrate the highest uptake in the putamen, followed by the prefrontal cortex and cerebellum, respectively (This research was originally published in JNM. G. E. Terry, J. Hirvone, J.-S. Liow, S. S. Zoghbi, R. Gladding, J. T. Tauscher, J. M. Schaus, L. Phebus, C. C. Felder, C. L. Morse, S. R. Donohue, V. W. Pike, C. Halldin, and R. B. Innis. Imaging and Quantitation of Cannabinoid CB1 Receptors in Human and Monkey Brains Using 18F-Labeled Inverse Agonist Radioligands. J Nucl Med. 2010;51:112–120. © SNMMI).
Researchers at the Katholieke Universiteit (KU) Leuven carried out PET imaging with [18F]MK-9470 in patients suffering from TLE with hippocampal sclerosis and demonstrated increased CB1 receptor availability at the seizure onset zone (ipsilateral temporal lobe) and decreased CB1 receptor availability in the superior insular cortex. Subsequent studies evaluated [18F]MK-9470 in patients suffering from migraines and in patients with PD. In female patients with migraines, CB1 receptor expression was globally increased, in comparison to healthy controls, and in the patients with PD, the authors observed a decrease in CB1 availability in the substantia nigra and an increase in radiotracer uptake in nigrostriatal, mesolimbic and mesocortical dopaminergic projection areas. This study in Parkinsonian patients also found no significant differences in CB1 receptor availability between the PD group with and without levodopa induced dyskinesias (LID) and therefore does not support a role for changes in CB1 expression in LID pathogenesis. CB1 receptors were also imaged using [18F]MK-9470 in patients with AD compared to age-matched controls, where no correlation between amyloid-β pathology and CB1 receptor availability was found.
CB1 receptor availability has also been investigated in SUDs with PET. Researchers at the NIMH evaluated [18F]FMPEP-d2 in cannabis users, to measure receptor availability, where a global decrease in cortical regions, as well as in the hippocampus, was observed in comparison to controls. Further studies using [18F]MK-9470 at KU Leuven and [11C]OMAR at Yale University, were complimentary, also finding a global decrease in receptor availability in cannabis users in comparison to control subjects. Additional studies at Yale investigated [11C]OMAR uptake in individuals with alcohol use disorder to uncover an increase in receptor availability in alcohol-dependent individuals who had been four months abstinent at the time of their PET scans. A following study at the NIMH found the opposite effect when using [18F]FMPEP-d2 to measure receptor availability, where a significant decrease across all brain regions was observed. Patients were scanned without abstinence from alcohol and again after 2–4 weeks abstaining and there were no significant differences in CB1 receptor availability observed. A third study in individuals with alcohol dependence observed a decrease in receptor availability, measured by [18F]MK-9470 at KU Leuven, when compared to controls, across all brain regions, irrespective of an abstinence period. Interestingly, a group of individuals subjected to acute alcohol administration prior to a [18F]MK-9470 PET scan showed a global increase in CB1 receptor availability.
PET imaging has also been used to evaluate CB1 receptor availability in patients suffering from schizophrenia. At Johns Hopkins University, [11C]OMAR was used to show a significant increase of radiotracer uptake in the pons, compared to healthy controls. It can be noted that [11C]OMAR binding was increased in all brain regions studied in the schizophrenia cohort, however statistical significance was not met for any region other than the pons. Another study at Yale using [11C]OMAR displayed the opposite finding; that the combined medicated and unmedicated patients suffering from schizophrenia had significant decreases in CB1 receptor availability compared to controls in all binding regions examined except for the temporal cortex and cerebellum. [18F]MK-9470 has also been used in clinical research studies of schizophrenia at KU Leuven, where it was observed that CB1 receptor availability was increased in both medicated and unmedicated patients, compared to controls.
A noteworthy study was conducted at Yale to investigate CB1 receptor availability in patients with PTSD, where [11C]OMAR was used and showed significantly increased binding in individuals with PTSD compared to healthy and trauma-exposed controls. CB1 receptor availability has also been explored in individuals with eating disorders, using [18F]MK-9470, where a global increase in radiotracer uptake was observed in individuals suffering from anorexia.
The findings in these PET studies assessing CB1 receptor availability in cannabis using individuals are the most consistent with the different radiopharmaceuticals. It has been postulated that the discrepancies in the results in several of the other studies exploring CB1 receptors with PET are attributed to the radiotracers; however, this does not explain the discrepancies using [11C]OMAR in patients with schizophrenia, where radiotracer binding was observed with opposite effects, compared to healthy controls, in two separate studies. There is evidence for genetic factors affecting CB1 availability, including polymorphisms and differences in expression between races, which may also explain the differences in results between studies. When choosing a CB1 radiotracer for use in an imaging program these discrepancies should be taken into consideration, as well as the means of quantitation required for the varying binding mechanisms of each tracer. While [11C]OMAR is the most widely used radiopharmaceutical for imaging CB1 receptors in clinical PET research, it shows peak brain uptake of 1.5–2 SUV, which can make accurate quantitation difficult and while the short half-life of 11C (t1/2 = 20.4 minutes) offers several advantages for testing of paradigms in neuro-PET research, the radionuclide will limit opportunities for multi-center clinical trials. [11C]MePPEP shows higher peak brain uptake of 3–4 SUV but washout from the brain is too slow for precise quantitation, as well as demonstrating high inter-subject variability for VT indicating relatively low precision of the measurement. [18F]MK-9470 has also shown high inter-subject variability for VT in healthy controls and due to the slow kinetics of the radiotracer, challenging quantitation measures may limit its widespread use. In our opinion, [18F]FMPEP-d2 is a highly promising radiopharmaceutical, and has potential for facilitating more accurate quantification of CB1 receptors due to the high brain uptake of ~6 standardized uptake value (SUV), as well as a facilitated measurement of the input function, compared with the other CB1 radiotracers evaluated at the NIMH. [18F]FMPEP-d2 demonstrates high specific binding towards CB1 receptors, displays low inter-subject variability of VT, and moderate test-retest variability. It also has the advantage of being labeled with the radionuclide, 18F (half-life (t1/2 = 109.7 minutes), which enables the option for longer scanning protocols, multi-center trials and widespread distribution. Additional studies with [18F]FMPEP-d2 are warranted for exploring the CB1 receptor in clinical PET research studies.
CB2 Receptors:
CB2 receptors are primarily expressed by the immune system, although they are also expressed within the CNS at very low concentrations. CB2 receptors are upregulated in disorders associated with inflammation, including cancer, pain, osteoporosis and liver diseases.3 CB2 receptors play a role in microglial migration in response to inflammatory stimuli. Evidence for this mechanism comes from immunohistochemistry studies that revealed microglial CB2 receptor expression cluster at amyloid-β plaques in AD brain tissues. There is also an observed upregulation of microglial CB2 receptors in disorders associated with neuroinflammation (AD, multiple sclerosis, human immunodeficiency virus (HIV)-induced encephalitis and Down’s syndrome), providing evidence for CB2 receptors as a PET imaging target of neuroinflammation.3
In vivo evaluation of both fluorine-18 and carbon-11 labeled CB2 receptor agonists have been recently reported. [11C]NE40 was among these reported radioligands and found to be specific for CB2 receptor binding in the spleen and blood of normal rats. Specific and reversible binding of [11C]NE40 was observed in a rat model with local overexpression of human CB2 receptors. [11C]NE40 also exhibited high brain uptake in rhesus monkey. First-in-human PET studies at KU Leuven with [11C]NE40 found lower CB2 receptor availability in AD patients compared to healthy controls, failing to reveal a predicted upregulation in patients with AD. A dual tracer study with [11C]PIB, for imaging amyloid-β, and [11C]NE40, revealed no correlation between amyloid-β binding and CB2 receptor availability, suggesting insufficient affinity and/or selectivity of the tracer for human CB2 receptors. [11C]MA2 and [18F]MA3 were later reported as PET radiotracer candidates; these radiotracers are high affinity analogs of a highly potent N-arylamide oxadiazole CB2 agonist, that demonstrated high brain uptake and efficient clearance in mice. [18F]MA3 did not show specific binding in non-human primate (NHP) brain, and the study concluded that further validation would be required to determine the utility of these radiotracers for imaging CB2 receptors as a biomarker for neuroinflammation.
CB2 receptor radiotracer development has unveiled certain challenges for consideration during these studies. Selectivity against CB1 will play a role in the success of the radioligand. For example, [11C]NE40 demonstrated 100-fold selectivity for CB2 against CB1 receptors; however, since CB1 is one of the most abundant receptors in the human brain, a 100-fold selectivity factor may not be high enough for successful in vivo imaging. Another consideration is that, to our knowledge, the literature is lacking information on CB2 receptor Bmax values in the human brain, so CB2 tracers under development, may not be potent and selective enough for in vivo PET neuroimaging. Radiotracers discussed for PET imaging of CB2 receptors are summarized in Figure 3. Radiotracer development for CB2 receptor agonists and exploration of this receptor in the periphery and in neuroinflammation continue to be actively pursued.3
Figure 3.
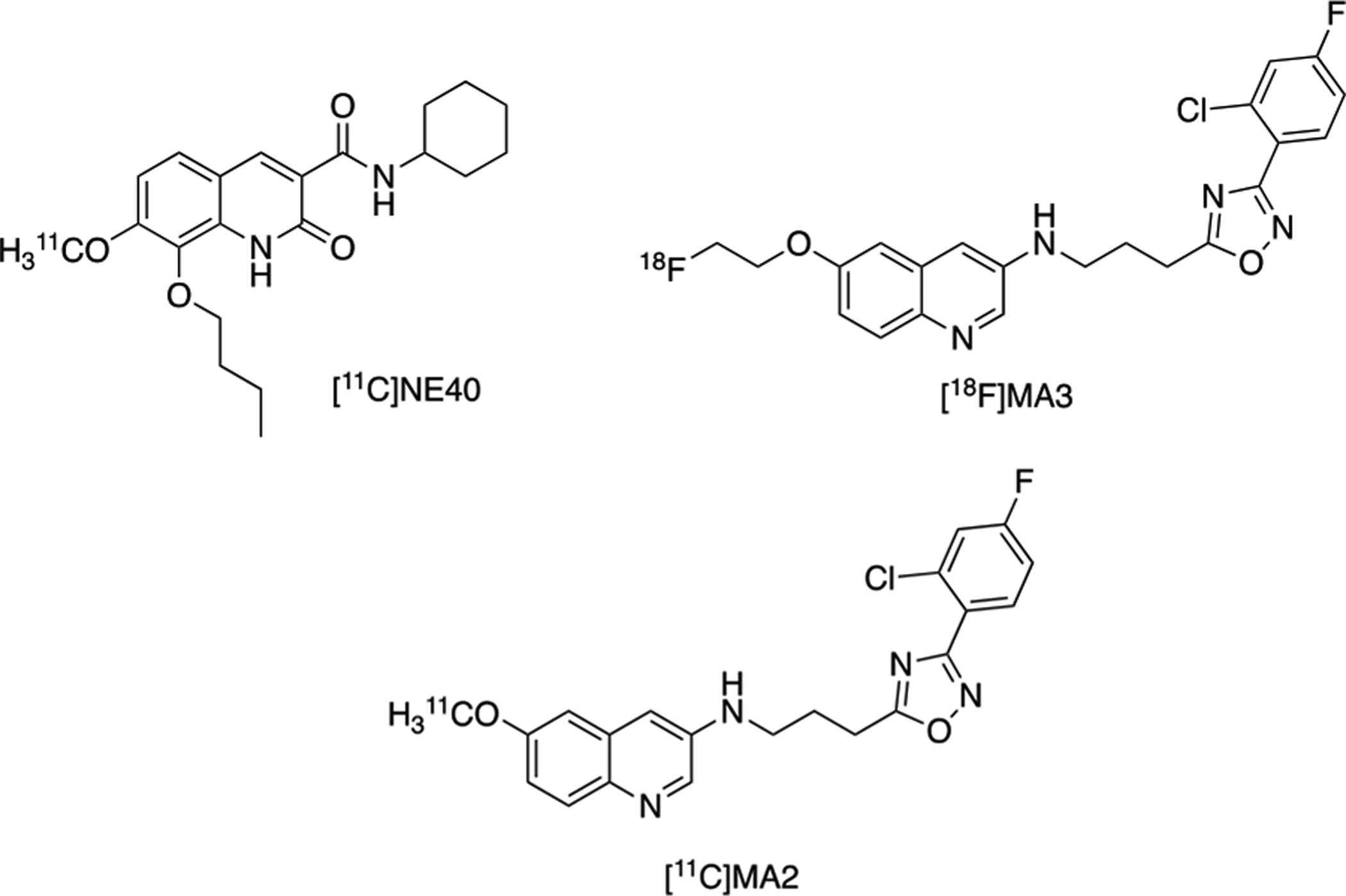
Radiotracers Developed for PET Imaging of CB2 Receptors.
FAAH:
Evidence that FAAH inhibitors resulted in cannabinoid receptor agonist effects, including anandamide-specific behavioral functions and possible regional differences in FAAH inhibitory effects, led to the interest by our laboratories and others to develop PET radiotracers for imaging this enzyme in the CNS.1 Early imaging work included quantifying regional differences of FAAH activity in the rodent CNS using [3H]anandamide, which is enzymatically converted to [3H]arachidonic acid, and is metabolically trapped in membrane phospholipids. Wild-type compared to FAAH knockout mice showed [3H]arachidonic acid binding in a regionally specific manner and correlated with FAAH activity. Subsequently, anandamide analogs which were labeled with iodine-123 as potential metabolic trapping radioligands for in vivo evaluation of brain FAAH. Small molecule radioligand development moved toward synthesizing aryl anandamide analogs, which were also evaluated as metabolic trapping tracers, and two compounds were identified as selective for FAAH over CB1 and CB2 receptors The leads, (named [11C]17 and [11C]18; Figure 4), were radiolabeled with [11C]CH3I, and demonstrated reasonable brain uptake in vivo.
Figure 4.
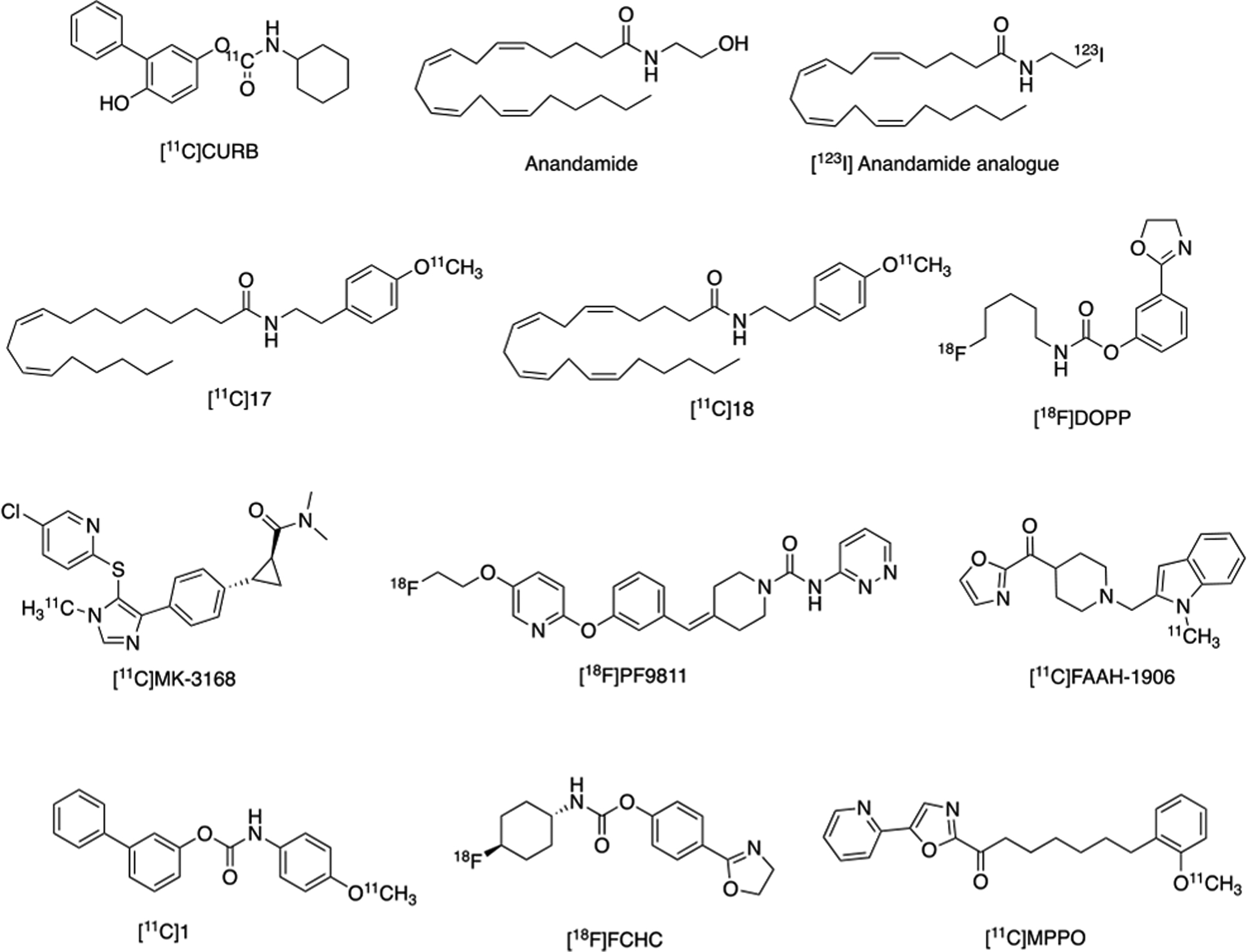
Radiotracers Developed for PET Imaging of FAAH.
Radiotracer development shifted toward analogs of URB597, a potent and selective FAAH inhibitor. Synthesis and evaluation of a carbon-11 labeled analog (biphenyl-3-yl-[11C]-4-methoxyphenylcarbamate ([11C]1; Figure 4)) was found to inhibit FAAH in vitro, displayed high brain uptake, but was the target of non-FAAH mediated metabolism, thereby preventing further translation. Nonetheless, URB597 derivatives continued to serve as a lead scaffold for further radioligand development.
Imaging the enzymes of the ECS, including FAAH and MAGL, require strategic radiotracer design. These enzymes are serine hydrolases which interact with agonists and antagonists through hydrolysis. If hydrolysis is not taken into consideration during radiotracer development, the radiolabel may be cleaved from the target and imaging will be unsuccessful, as was observed during early development of radiolabeled URB597 analogs. It was at this time that [11C]CO2 fixation was developed as facile method of 11C-carbonylation. This methodology is routinely conducted in one-pot and at room temperature, allowing 11C-carbonylation to prepare carbamates, ureas, carboxylic acids, and amides.4 Until [11C]CO2 fixation was adopted for FAAH radiotracer discovery, attempts to radiolabel the carbamate URB597 and its analogs resulted in introduction of the radiolabel at the aromatic position that is released as a radiometabolite once hydrolyzed by FAAH. [11C]CURB was labeled at the 11C-carbonyl moiety via [11C]CO2-fixation using an organic base (BEMP), with cyclohexylamine followed by dehydration with phosphoryl chloride and addition of the respective phenol. [11C]CO2 fixation chemistry was critical to enable the introduction of the radiolabel at a position on the 11C-carbonyl moiety that will not result in brain penetrant radiometabolites and ensures maintenance of the label after enzymatic cleavage by FAAH (Scheme 1).
Scheme 1. Radiosynthesis of 11C]CURB via [11C]CO2-Fixation.
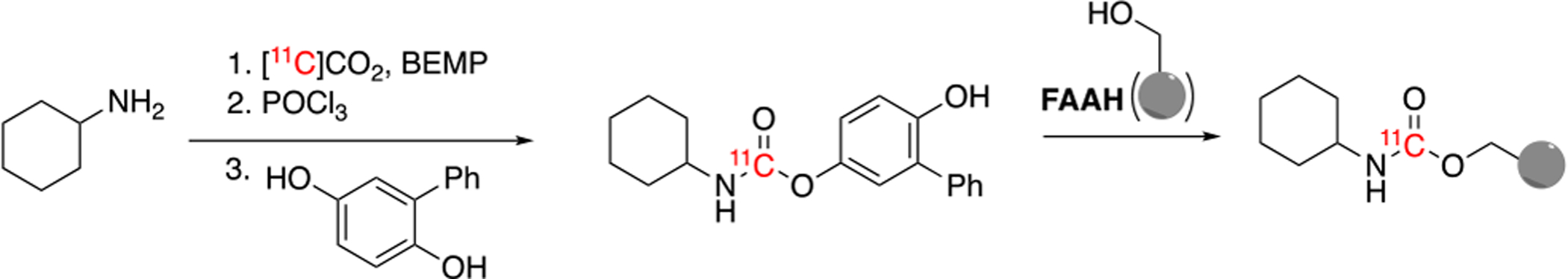
[11C]CO2-fixation chemistry enabled the introduction of carbon-11 radiolabel at the carbonyl position, where the radiolabel is retained on the enzyme, after hydrolysis by FAAH.
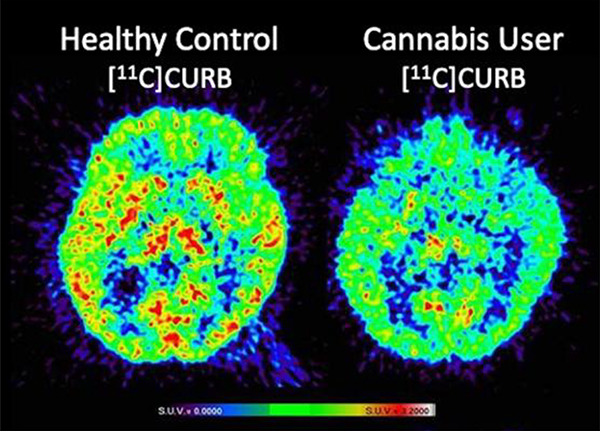
Ultimately this methodology led to the development of [11C]CURB by our laboratories in Toronto and has been successfully translated for human use to map FAAH in the living brain. Imaging with [11C]CURB has demonstrated that the putamen and thalamus have the highest uptake, and the anterior cingulate cortex have the lowest uptake, with widespread and uniform distribution in the grey matter. Specific binding was shown by pretreatment with a the FAAH inhibitor, PF-04457845, which dramatically reduced radiotracer uptake. [11C]CURB quantitation uses an irreversible two-tissue compartment model and the composite parameter λk3 provided the best PET outcome measures. Binding appears irreversible, and λk3 is not dependent on regional cerebral blood flow (Figure 5A). [11C]CURB has been widely used in clinical research for imaging FAAH, including studies of alcohol use disorder, cannabis use disorder and individuals at risk for anxiety and addiction.2 Our laboratories in Toronto conducted a study in individuals with cannabis use disorder, which showed that [11C]CURB brain uptake was reduced in the striatum, thalamus and cortical areas, in comparison to healthy controls (Figure 5B).
Figure 5. Human Neuroimaging of FAAH using [11C]CURB.
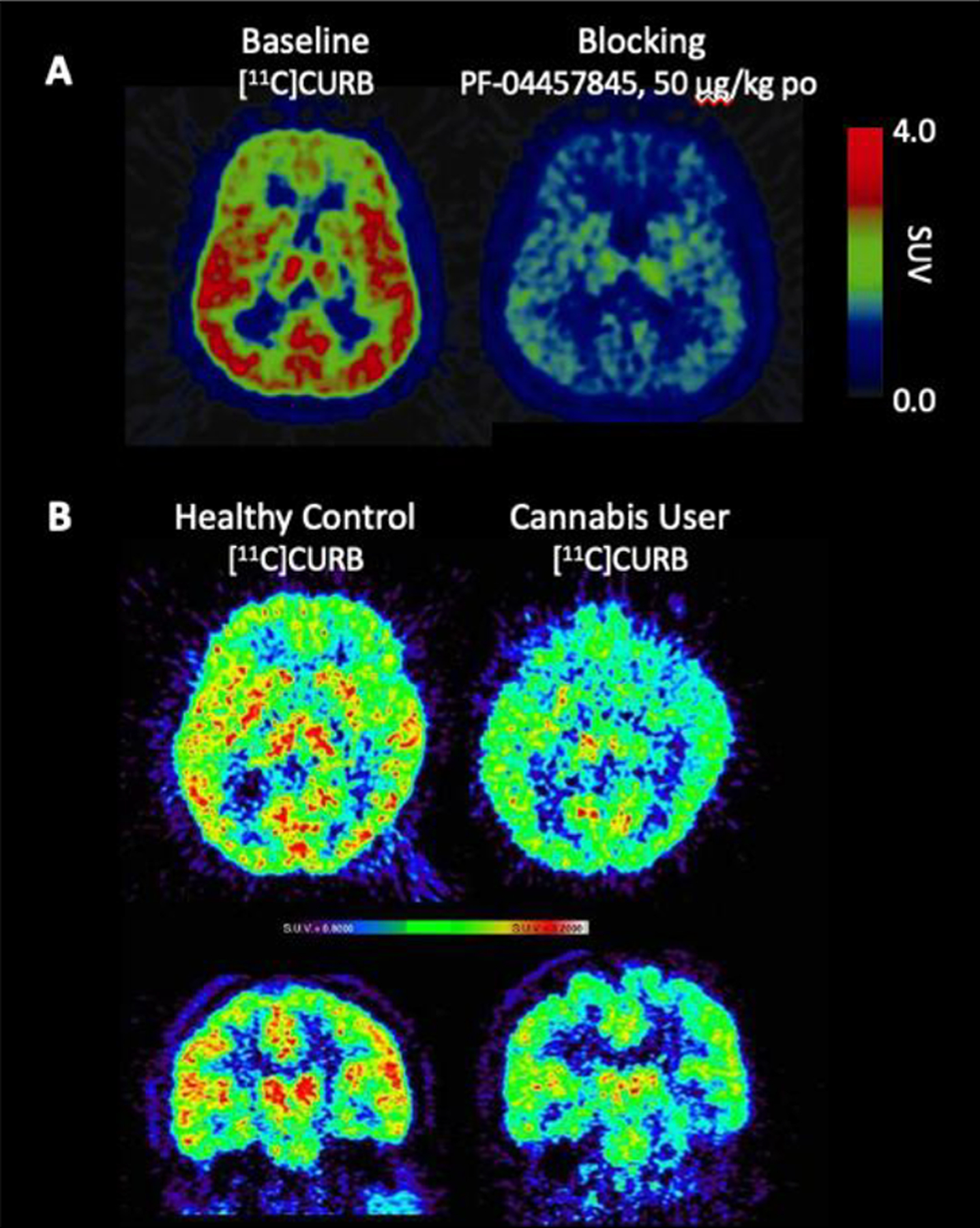
A. Transaxial sections of [11C]CURB-PET at baseline (left) and blocking (right) with oral pre-treatment of a FAAH inhibitor (PF-04457845, 50 μg/kg). B. Transaxial (top) and coronal (bottom) [11C]CURB-PET images in a healthy control showing high uptake in striatum, thalamus and cortical areas (left) compared to a subject meeting criteria for cannabis use disorder scanned after overnight abstinence with visibly lower uptake values in striatum, thalamus and cortical areas (right).
[11C]MK-3168 is a reversible FAAH tracer that was concurrently reported by Merck and has been used in clinical research studies to provide receptor occupancy, has demonstrated good test-retest variability and kinetic modelling has been established in humans. [11C]MK-3168 is the only reversible FAAH radiotracer to have advanced to human PET studies but has only been used in studies of healthy individuals, to our knowledge. Limited access to the precursor and authentic standard compound may have restricted the wider-spread use of this radiotracer. Reversibly binding FAAH radiotracers are still avidly sought and have an advantage for the potential to provide a full quantitative assessment of FAAH distribution, concentration, and activities in brain. For example, our laboratories explored the development of [11C]MPPO and [11C]FAAH-1906 to fill this void, although preliminary PET imaging studies of [11C]MPPO showed low to moderate level of specific binding to FAAH in murine brains. However, the radiosynthesis and preliminary evaluation by PET imaging studies offers a roadmap for the investigation of other FAAH radiotracers, for example, [11C]FAAH-1906 with reversible binding profiles. PF-04457845 was identified as another potent, selective, irreversible FAAH inhibitor and led to our development of [18F]PF-9811, which has been recently evaluated in rats.
We continue to seek new 18F-labeled irreversible FAAH radioligands which led to the multi-step automated radiosynthesis of [18F]DOPP, where it was found to be consistent with FAAH and [11C]CURB distribution, exhibiting high specific binding and brain penetration in NHPs, and we concluded that this ligand was promising for clinical translation. Subsequently, our groups developed [18F]FCHC as an alternative irreversible radiotracer for PET imaging of FAAH. PET imaging and ex vivo biodistribution studies in our laboratories showed that this radiotracer demonstrated a combination of high brain uptake and high specific binding in rats. These properties suggest [18F]FCHC is another promising FAAH radiotracer suitable for neuroimaging and worthy of further evaluation in higher species. The radiotracers developed for PET imaging of FAAH have been summarized in Figure 4.
FAAH inhibitors offer major advances toward therapeutic applications of modifying endocannabinoid signaling. FAAH inhibitors were under investigation in clinical trials, with hopes to advance a therapeutic to aid with neuropathic pain, when in 2016, a Phase I clinical trial of the FAAH inhibitor BIA 10–2474 resulted in one brain dead patient and four others with irreversible brain damage. Leading up to the clinical trial, and following its termination, there had been little information available about the potency of BIA 10–2474 for FAAH in the CNS. Researchers at our site used [18F]DOPP to determine the potency of BIA 10–2474 in rat brain tissue (50–70 μg/kg in various brain regions) in hopes that this information would prove useful for determining what was responsible for the severe adverse events resulting from the trial and inform future dosing regimens based on effective FAAH inhibition in the CNS, rather than maximum tolerated dose, which may have helped prevent the severe adverse events in the BIA 10–2474 trial. This study highlights the importance of PET imaging to determine target engagement during drug development. The tragic conclusion of this trial resulted in termination of other trials involving FAAH inhibitors. We envision that PET will play an important role in the development of next-generation inhibitors of FAAH and other targets in the ECS, to provide insight into clinical trial design and dosing regimens, in hopes to advance safer drugs that retain their efficacy.
The vast majority of FAAH PET imaging studies in clinical research have been conducted using [11C]CURB. To date, no fluorine-18 radioligands have been translated to study FAAH in humans, although both [18F]DOPP and [18F]FCHC show excellent promise. Development of a fluorine-18 labeled FAAH tracer with reversible binding is still an ongoing goal in the field, as such a tool would allow us to access key information from PET quantitation. A reversible fluorine-18 labeled FAAH PET tracer will also allow us to monitor neurological response to therapeutics, in addition to facilitated dissemination and potential for use in multi-center clinical trials.
MAGL:
MAGL PET radiotracers have remained elusive for human translation, despite efforts to achieve this goal by our laboratories and others for nearly a decade.5 Rodent studies involving the genetic and pharmacological blockade of FAAH or MAGL resulted in analgesic, anti-nociceptive and anti-anxiety phenotypes without the negative effects associated with administration of THC. These results spear-headed the development of pharmacological probes of these enzymes. As FAAH PET imaging became more established and clinical research began addressing relevant questions in brain health illnesses, there was an apparent void in the development of a radiotracer for imaging MAGL, partially attributed to lacking the radiochemistry to label promising inhibitors. With regard to MAGL inhibition as a therapy, partial blockade is preferred as chronic, complete blockade has led to reduced pharmacological benefits. Therefore, accurate measurement of enzyme inactivation within the CNS is essential and PET imaging methodology can facilitate therapeutic development. At present, there is no primary literature to disclose any MAGL PET ligand for human studies, although several have shown promise for translation. MAGL PET tracers discussed above are summarized in Figure 6.
Figure 6.
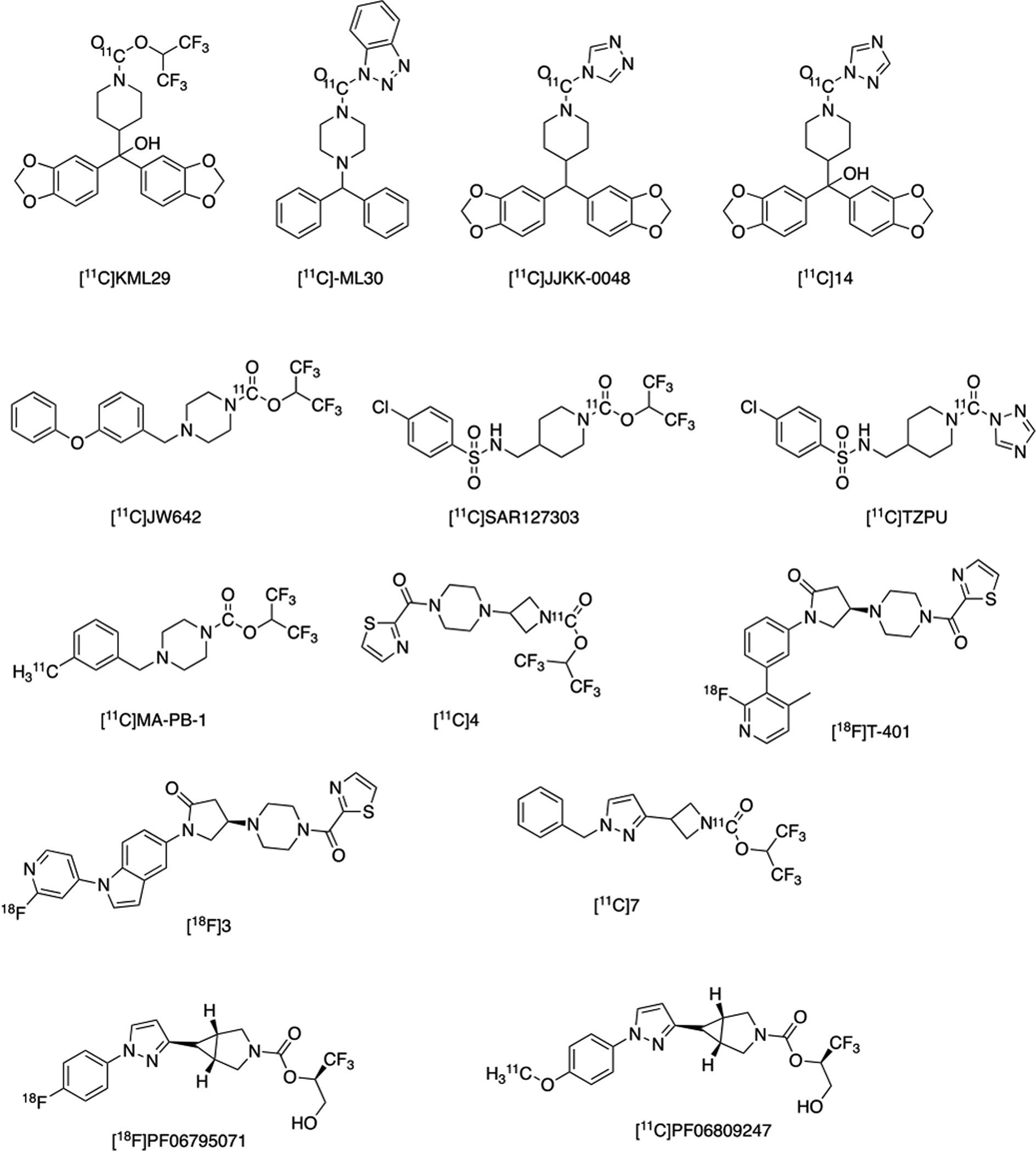
Radiotracers Developed for PET Imaging of MAGL.
The first series of MAGL tracers were labeled with carbon-11, as reported by our group in 2014, but were not translated for human use due to moderate brain uptake, and lack of saturable binding in vivo, which may also be attributed to off-target binding. [11C]TZPU was also developed but deemed unacceptable for further translation due to inadequate brain uptake. [11C]SAR127303 was later reported and prepared by [11C]CO2 fixation. This radioligand was not selective for MAGL, as it also inhibits ABHD6, but was used to image MAGL distribution in the rodent brain for the first time. Radioligand development continued with [11C]MA-PB-1, which was reported and after evaluation in NHP, found to be suitable for in vivo imaging of MAGL. Despite the radiotracers previously reported, several laboratories including ours, continued development of novel imaging agents for MAGL, in attempt to improve upon affinity, selectivity and brain uptake. One of our leads ([11C]4; Figure 6) was reported with picomolar affinity and excellent brain uptake (SUV 2.2). Concurrently, the National Institute of Radiological Sciences (NIRS) developed [11C]7 which has sub-nanomolar affinity for MAGL, selectivity over FAAH and specific for MAGL through structurally dissimilar blocking experiments. Subsequently, researchers at the Karolinksa Institutet synthesized [11C]PF06809247 and demonstrated high MAGL specificity, a low measure of in vitro efflux, good passive permeability, reasonable unbound fraction in the brain.
The first fluorine-18 reversible tracers for MAGL are [18F]3 and [18F]T401, and were also developed and imaged in NHP by the NIRS, demonstrating favorable specificity and kinetics. [18F]PF06795071, an irreversible MAGL inhibitor, was recently reported by our laboratories and prepared through the spirocyclic iodonium ylide (SCIDY) strategy, allowing for radiofluorination of a non-activated aromatic ring, yielding the selective tracer for MAGL imaging.
Radiofluorination using the SCIDY strategy was reported as a method capable of regioselectively introducing [18F]fluoride into non-activated aromatic rings with wide substrate scope under metal-free conditions in high radiochemical yields and has been translated for human use with several PET radiopharmaceuticals.3 The conceptual advantages of incorporation of fluorine-18 into a wide array of non-activated (hetero)arenes makes this methodology suitable for routine radiopharmaceutical production, and this methodology has allowed for the development of [18F]PF06795071 for imaging MAGL in good radiochemical yields, high molar activity and radiochemical purity. NHP imaging has been conducted using the SCIDY strategy to synthesize [18F]PF06795071, demonstrating good brain penetration (Figure 7).
Figure 7. [18F]PF06795071 Baseline PET Imaging in a Rhesus Macaque.
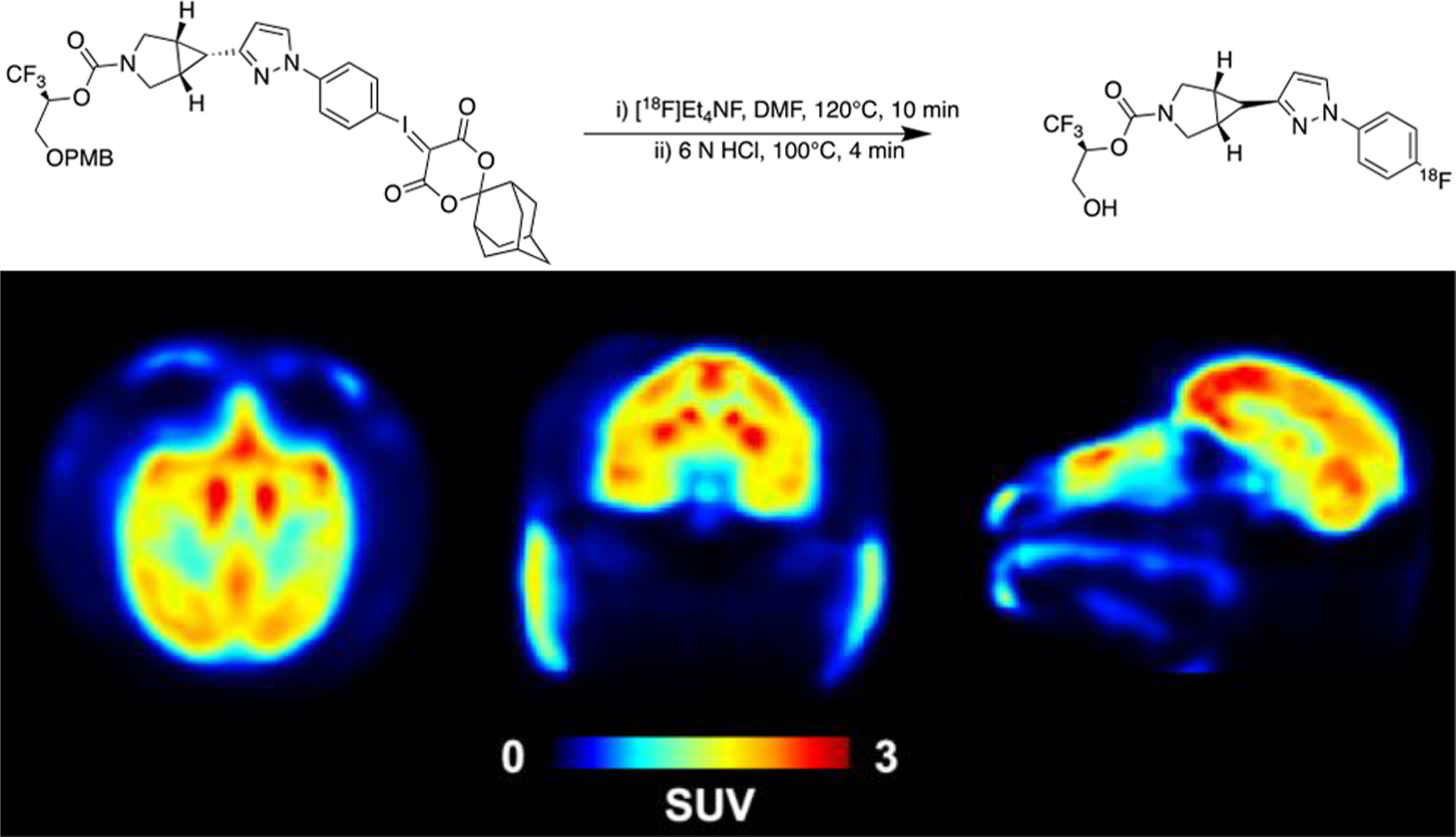
Radiosynthesis of [18F]PF06795071 using the SCIDY strategy was conducted for PET imaging and evaluated in NHP (mean SUV from 60–120 min post-radiotracer injection); transaxial view (left), coronal view (middle), sagittal view (right) depicted. Unpublished results.
Over the past decade it has been challenging to develop a PET radioligand suitable for imaging MAGL in humans. Selectivity over other targets in the ECS and low brain penetration have been two difficulties faced during development and one of the biggest roadblocks has been that the radiochemistry required to access useful MAGL inhibitors for radiolabeling was not readily available for human translation. The recent report using the SCIDY strategy to produce [18F]PF06795071 was an exciting example of new radiochemistry allowing access to a highly selective, potent compound as a promising lead radiotracer for a target that has been historically very difficult to access. Now with access to unique methods for 11C-carbonylation and methodologies to label non-activated aromatic rings readily available and translated for human use we envision that MAGL tracers will find their way to clinical research in the near future.
Concluding Remarks:
Other targets of interest for molecular imaging in the ECS include N-acyl-phosphatidylethanolamine, phospholipase D (NAPE-PLD) and DAGL. These enzymes synthesize endocannabinoids in an “on demand” manner following calcium ion influx in the postsynaptic cell, unlike classical neurotransmitters which are synthesized, stored in vesicles, and released upon signaling.1 The endocannabinoids then diffuse across the synapse in a retrograde manner, acting on presynaptic CB1 receptors to propagate secondary messenger signaling cascades leading to decreased cAMP production, calcium channel closing, and therefore an inhibition of neurotransmitter release. ABHD6 and ABHD12 are two serine hydrolases that compliment MAGL activity, accounting for the remaining ~15% of 2-AG hydrolysis activity. These targets have been unexplored in PET imaging and have potential applications for investigations of neurotransmitter modulation.
While some of the tracers discussed herein have been translated to clinical studies to address several questions surrounding the pathophysiology of the ECS in psychiatric and neurological disorders, there is continued interest to develop new tracers for these targets. Multiple CB1 PET tracers which have advanced to human use will be explored in widespread applications and may be further driven by research into addictions and the effects of recreational and medicinal cannabis use. As legalization of cannabis emerges across the globe research is needed more than ever for a better understanding of how this is affecting our population. Further CB2 receptor radioligands need to be developed for use in neuroinflammation, as the only CB2 radiotracer in humans fail to show a relationship between CB2 receptor availability and neuroinflammation in AD patients compared to healthy controls. Neuroinflammation biomarkers are of major interest in PET imaging, as they provide an opportunity for early diagnosis of neurodegeneration, and CB2 receptors show promise as one of these biomarkers. There is also an opportunity to translate reversible fluorine-18 labeled radiotracers for FAAH and MAGL, and to perform full quantitative assessment of distribution in the brain as well as to monitor neurological response to therapeutics, and to enable widespread use. We envision the translation of the first MAGL tracer to be used in humans in the near future and are encouraged that PET imaging of this enzyme will provide further insights into psychiatric and neurological disorders. Drug development and diagnostic imaging of the ECS for addictions and other brain health illnesses will continue to shape the fields of chemistry and neuroscience.
Funding Sources
N.V. thanks the National Institute on Ageing of the NIH (R01AG054473 and R01AG052414), the Azrieli Foundation, Canada Foundation for Innovation, Ontario Research Fund and the Canada Research Chairs Program for support. S. H. L. thanks the NIH for financial support (DA038000 and DA043507). We also thank Dr. Lu Wang from Jinan University PET Center for helpful discussions.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Horti AG; Raymont V; Terry GE, PET Imaging of Endocannabinoid System. In PET and SPECT of Neurobiological Systems, Dierckx RAJO; Otte A; de Vries EFJ; van Waarde A; Luiten PGM, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2014; pp 249–319. [Google Scholar]
- 2.Sloan ME; Grant CW; Gowin JL; Ramchandani VA; Le Foll B, Endocannabinoid signaling in psychiatric disorders: a review of positron emission tomography studies. Acta Pharmacol. Sin 2019, 40 (3), 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narayanaswami V; Dahl K; Bernard-Gauthier V; Josephson L; Cumming P; Vasdev N (2018) Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: Outlook beyond TSPO. Mol. Imaging, 17, 1536012118792317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang SH and Vasdev N, Total Radiosynthesis: Thinking outside “the box”. Aust. J. Chem 2015, 68 (9), 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimsey NL; Savinainen JR; Attili B; Ahamed M, Regulating membrane lipid levels at the synapse by small-molecule inhibitors of monoacylglycerol lipase: new developments in therapeutic and PET imaging applications. Drug Discovery Today 2020, 25 (2), 330–343. [DOI] [PubMed] [Google Scholar]