

Abstract
Microphysiology systems (MPS), also called organs-on-chips and tissue chips, are miniaturized functional units of organs constructed with multiple cell types under a variety of physical and biochemical environmental cues that complement animal models as part of a new paradigm of drug discovery and development. Biomimetic human liver MPS have evolved from simpler 2D cell models, spheroids and organoids to address the increasing need to understand patient-specific mechanisms of complex and rare diseases, the response to therapeutic treatments, and the absorption, distribution, metabolism, excretion and toxicity of potential therapeutics. The parallel development and application of transdisciplinary technologies, including microfluidic devices, bioprinting, engineered matrix materials, defined physiological and pathophysiological media, patient-derived primary cells, and pluripotent stem cells as well as synthetic biology to engineer cell genes and functions, have created the potential to produce patient-specific, biomimetic MPS for detailed mechanistic studies. It is projected that success in the development and maturation of patient-derived MPS with known genotypes and fully matured adult phenotypes will lead to advanced applications in precision medicine. In this Review, we examine human biomimetic liver MPS that are designed to recapitulate the liver acinus structure and functions to enhance our knowledge of the mechanisms of disease progression and of the absorption, distribution, metabolism, excretion and toxicity of therapeutic candidates and drugs as well as to evaluate their mechanisms of action and their application in precision medicine and preclinical trials.
It has been well documented that data from animal models as well as from simple, human, in vitro safety and efficacy experimental models are often not concordant with results from human phase II clinical trials. High failure rates in phase II/III clinical trials (up to 80%) have been primarily ascribed to the genetic and physiological differences between animals and humans as well as to the use of over-simplified in vitro experimental models1–7. The primary challenge in phase II/III clinical trials has been the lack of efficacy from the investigational drug, yet some safety problems have also been identified1,2,8. Liver-related examples include the failure of mouse models to recapitulate aspects of non-alcoholic fatty liver disease (NAFLD) progression that are necessary to predict outcomes in phase II/III clinical trials3,7,9. Other examples include the failure of a promising farnesoid X receptor (FXR) agonist, obeticholic acid, to achieve its efficacy endpoint in a phase III trial for non-alcoholic steatohepatitis (NASH)10 and the failure of the once promising type 2 diabetes drug, fasiglifam, due to idiosyncratic drug-induced liver injury (DILI)11–13, which is not always detected in animal and simple human in vitro models or even in early-stage clinical trials14–17. These late-stage failures cost substantial time and money, potentially exceeding the median cost of a pivotal trial (US$19 million)18 or the cost needed to bring a new drug to market (US$1 billion)19, while also placing hundreds to thousands of patients at risk without public health benefit, making the drug discovery and development process inefficient1,2,20. One promising solution has been the development and application of human microphysiology systems (MPS) of the liver and other organs21–26. MPS, also called organs-on-chips and tissue chips, are miniaturized functional units of organs (for example, liver acinus) constructed from multiple cell types using cell lines, primary cells or pluripotent stem cells (PSCs) from human or animal sources (TABLE 1), constructed either by self-assembly or supervised placement in 3D within a defined structure. MPS recapitulate some key elements of the organ structure and functions, including physical and biochemical microenvironmental cues (for example, microfluidic flow, cell–cell communication and signalling molecules), as well as responsiveness to therapeutics. Both single organ MPS and multiple organ MPS linked by fluidics have been created for high throughput and high content applications21,27. Human biomimetic liver MPS (HBL-MPS) are a more complex type of MPS designed to maximally recapitulate the liver acinus structure and function, including the major cell types as primary or PSC-derived cells (FIG. 1a). They harness defined matrix stiffness and biochemistry, normal and disease-specific media, and physiological microfluidic flow rates and optimally include zonation, vascularization and an ability to image during the experimental time course.
Table 1 |.
Examples of current human liver MPS designs
| MPS | Chip supplier | Design | Contenta | Throughputb | Cells | Key characteristics |
|---|---|---|---|---|---|---|
| vLAMPS56 | Micronit | Two-channel with membrane | High | Med | PHH, LSEC, THP-1, HSC | ECM: collagen and LECM; Material: Glass, PC; physiological zonation |
| Liver-Chip37,200 | Emulate | Two-channel with membrane | High | Med | PHH, LSEC | ECM: Matrigel; Limitation: PDMS materialc and PC; automated platform |
| LAMPS59 | NortisBio | One-chamber | High | Med | PHH, HMVEC or LSEC, THP-1, HSC | ECM: collagen and LECM; Limitation: PDMS materialc; single oxygen zones |
| ExVive201,202 | Organovo | Bioprinting on array of 24-well Transwell membranes | High | Med | PHH, HUVEC, HSC plus KC203 | ECM: Novogel; Material: PS, PC; Limitation: static |
| Organo-Plate50,204 | Mimetas | Array of 96 two-channel chips, phase-guide, rocker-driven flow | Med | High | HepG2, iPSH, HMVEC, THP-1 | ECM: collagen; Material: glass and PS; Limitation: bidirectional perfusion |
| PREDICT96 (REF.79) | Draper | Array of 96 two-channel chips, with membrane, 96 pump array | Med | High | PHH | ECM: collagen, fibronectin; Material: COC, PC |
| LiverChip66,74,109,205 | CNBio Innovations LLC | Array of 12 bioreactors with cells cultured on 3D PC scaffold | Med | Med | PHH, HK and HSC or NPC | ECM: collagen; Material: PS, PU, PC, self-assembly, integral perfusion; Limitation: no imaging until end of study |
| Microliver206,207 | HµRel | Array of four chambers | Med | Med | PHH, NPC | ECM: collagen; Material: PC and elastomer |
| HemoShear Chip65,208 | HemoShear | Two-channel with membrane, cone-plate to induce flow stimulation | Med | Low | PHH, HSC, HM | ECM: collagen; Material: Plastic, PC |
COC, cyclic olefin copolymer; ECM, extracellular matrix; HepG2, an immortalized human hepatoma cell line; HK, human Kupffer cell; HM, human macrophage; HSC, human hepatic stellate cell; HMVEC, human microvascular endothelial cell; HUVEC, human umbilical vein endothelial cell; iPSH, inducible pluripotent stem cell-derived hepatocyte; KC, Kupffer cell; LAMPS, liver acinus microphysiology systems; LECM, liver extracellular matrix; LSEC, human liver sinusoidal endothelial cell; Med, medium; NPC, non-parenchymal cell; PC, polycarbonate; PDMS, polydimethylsiloxane; PHH, primary human hepatocytes; PS, polystyrene; PU, polyurethane; THP-1, immortalized human monocyclic cell line; vLAMPS, vascular liver acinus microphysiology systems.
Content refers to the cell types, structural organization and context of use as a liver biomimetic.
Throughput refers to the number of devices that can be tested in one experiment.
PDMS has high binding affinity to lipophilic compounds.
Fig. 1 |. Human liver acinus structure and organization.
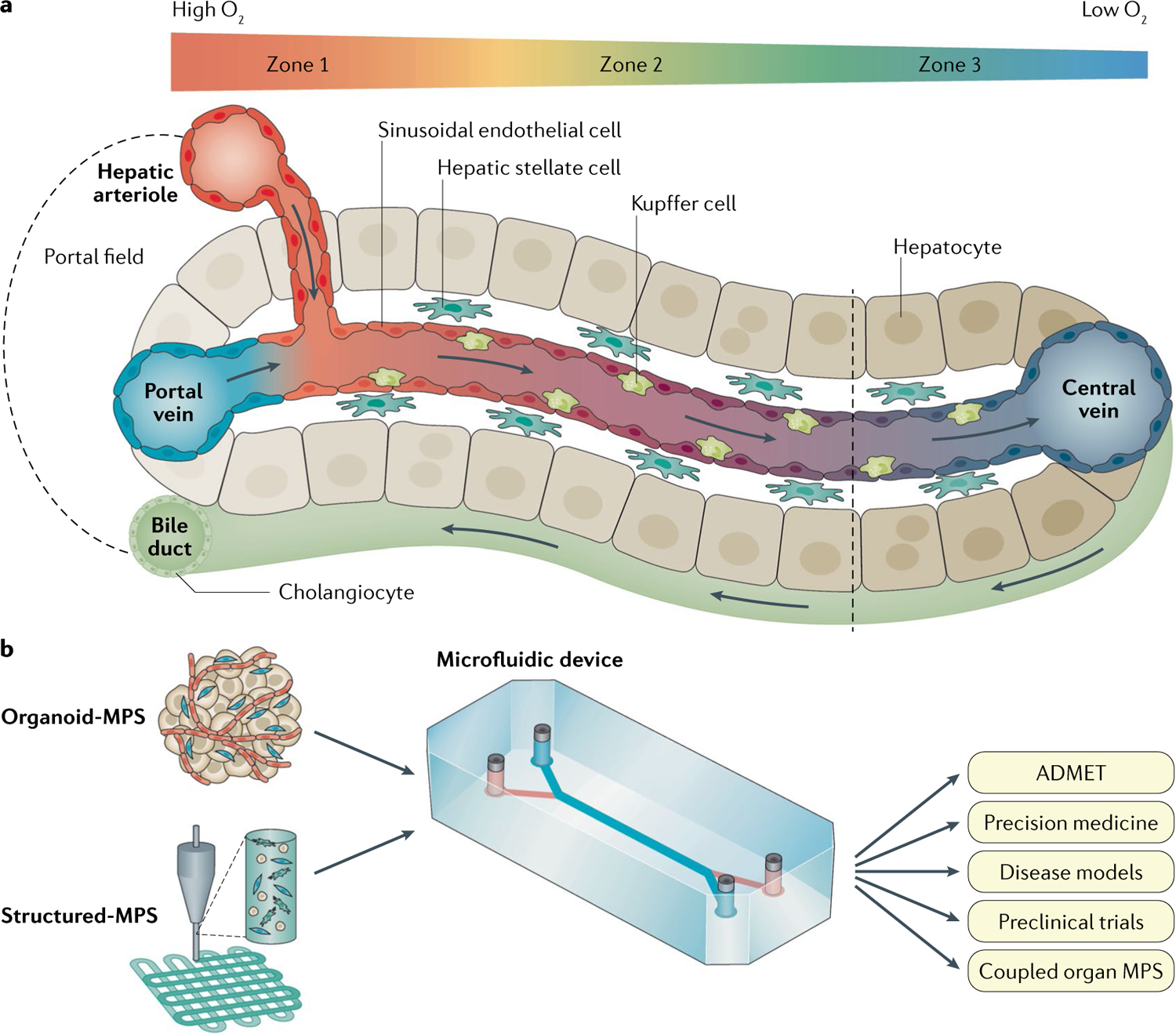
a | The liver sinusoid is created by the combined entry of blood from the nutrient-rich, oxygen-poor portal vein and the oxygen-rich, nutrient-poor hepatic arteriole. The sinusoid is lined with liver sinusoidal endothelial cells that do not form tight junctions, allowing the passage of xenobiotics, nutrients, plasma proteins, lipoproteins, gases, viruses and exosomes across the space of Disse, a thin matrix containing stellate cells, to reach cords of hepatocytes. Resident macrophages (Kupffer cells) are normally in the sinusoid. The sinusoids flow into central veins. The hepatocytes lining the space of Disse are polarized with the basal lateral surface against liver sinusoidal endothelial cells and the apical surfaces forming a network of bile canalicular spaces that link to the cholangiocytes forming the bile ducts that carry the bile produced in the hepatocytes through the biliary track and into the duodenum. A gradient of oxygen tension from high (~15–18% zone 1) to low (~5–6% zone 3) is created along the acinus due to the high rate of oxygen consumption by healthy hepatocytes. Gradients of other factors include the hormones insulin and glucagon, cytokines, and transcriptional regulators. b | The goal of one aspect of developing liver microphysiology systems (MPS) is to fully recapitulate the human liver acinus depicted in panel a. This Review discusses the evolution of the technologies and physiological cues required to make human biomimetic liver MPS based on patient-specific, stem cell-derived organoids (Organoid-MPS) and manually or bioprinted 3D structures of patient-specific stem cells and matrices (Structured-MPS). These next-generation human biomimetic liver MPS have the potential to positively affect patient-specific absorption, distribution, metabolism, excretion and toxicity (ADMET), experimental disease models for therapeutic discovery and development, precision medicine, preclinical trials, and coupled organ MPS. Although not in the scope of this Review, the knowledge gained from Organoid-MPS and Structured-MPS will also lead to the development of advanced human liver tissue derived from stem cells to create regenerative medicine treatments.
MPS can be combined with quantitative systems pharmacology (QSP), an iterative experimental and computational modelling approach that has the potential to improve the efficiency of drug discovery and development8,20,28. Here, data derived from an experimental model is incorporated into a computational model and used to make a prediction that is tested in experimental models. For example, the FDA, in recognition of the potential role of MPS and QSP in enhancing regulatory science and expediting drug discovery and development, is currently evaluating computational clinical pharmacology models to advance model-informed drug development29. A major element of the FDA programme is assessing how MPS can be used to experimentally investigate the mechanisms of adverse drug reactions, provide insights into physiology and disease mechanisms, and to identify relevant endpoints or biomarkers for clinical trial design. The EMA has shown a similar interest in the application of MPS to drug discovery and development, in particular recognizing that MPS are rapidly progressing and represent a promising field for the replacement, reduction and refinement of animal experimental testing (the three ‘Rs’ principle)30. A workshop held in 2017 focused on the challenges and opportunities for the use of MPS and was specifically aimed at “facilitating the regulatory acceptance of innovative non-animal methods in appropriate, defined contexts during the approval of medicines”30.
It has been suggested by the partnership between 23 mostly global pharmaceutical companies and the National Center for Advancing Translational Sciences of the National Institutes of Health that MPS have the potential for driving a paradigm shift in the drug discovery and development pipeline by more closely mimicking human physiology and pathophysiology than current in vitro and in vivo models, leading to improved clinical predictions of disease progression, drug response and pharmacokinetic parameters of absorption, distribution, metabolism, excretion and toxicity (ADMET)23. In this Review, we summarize the evolution, rationale and key characteristics of HBL-MPS, while exploring applications in drug discovery and development. In addition, the further evolution of HBL-MPS is projected and we discuss the potential of patient-specific HBL-MPS in precision medicine and preclinical trials (FIG. 1b).
Evolution of HBL-MPS
MPS have evolved from a long history of valuable animal and human, 2D and 3D cell culture models31–35. The evolution from 2D cell culture to 3D models involved micropatterned cell arrays36, spheroids35,37,38, organoids39–48, and static or fluidic plate-based platforms49,50. A range of MPS designs have been applied to ADMET and liver disease efficacy studies with increasing sophistication over the past decade (TABLE 1). All of these in vitro models have and continue to generate important information about liver physiology and pathophysiology. For instance, pioneering studies have shown that micropatterned co-culture systems of primary human hepatocytes exhibit superior human-specific drug metabolism and enable disease modelling for a number of human liver pathologies51–53. They also provide ease of engineering and facilitate improved physiologically relevant high-throughput screening. It was also demonstrated that controlling cellular positioning via micropatterned cultures can improve the functional maturity of human inducible PSC (iPSC)-derived hepatocytes54. However, human liver MPS have evolved to address the increasing need to understand patient-specific mechanisms of complex and rare diseases as well as ADMET for precision medicine, which was not possible with the simpler models. In practice, the concept of ‘fit-for-purpose’ validation within a clearly defined ‘context of use’23,55 is applied by investigators to select the optimal in vitro experimental model to address specific stages in the drug discovery and development pipeline that require particular measurement and analytical characteristics such as high-throughput screening, high content of mechanistic knowledge or multi-organ physiology. Human liver MPS for high content of mechanistic knowledge (HBL-MPS) are designed to maximally recapitulate the human liver sinusoidal structure and functions, including immune cell infiltration and zonation56 (FIG. 1a,b).
The continued evolution of MPS has been based on the parallel development and application of transdisciplinary MPS technologies, including microfluidic devices, bioprinting, engineered matrix materials, physiological and pathophysiological media, patient-derived primary cells, and PSCs as well as synthetic biology, to engineer cell genes and functions to create the optimal human physiological or pathophysiological experimental platform57 (TABLE 2). Current HBL-MPS already exhibit significant physiological and pathophysiological characteristics and functions of clinical relevance, yielding mechanistic knowledge that is valuable in liver disease therapeutic discovery and ADMET (BOX 1).
Table 2 |.
| Feature | Requirement | Importance |
|---|---|---|
| Matured human iPSC-derived liver cells preferred but primary hepatocytes with non-parenchymal cell lines acceptable62 | Human primary hepatocytes with at least two non-parenchymal cell types | Recapitulates cell communication within the liver acinus and the effect of non-parenchymal cells on disease pathophysiology |
| Microfluidics80 | Controlled media flow through the MPS | Recapitulates the physiological effects of circulation, flow stimulation of cells, and enables sampling and coupling of multi-organ MPS |
| Matrix engineering | Physiological stiffness and biochemistry | For example, hepatocyte gene expression and function is dependent on matrix stiffness210 |
| Fluorescence-based biosensors77,211 | Measurements of cell-specific physiology (for example, apoptosis) | Temporal-spatial measurements to define zone-specific activities during a study |
| Synthetic biology | Cells engineered to control the expression of specific genes | Valuable for modelling specific disease genotypes and phenotypes212 and biosensor expression |
| Carrier protein98 | Physiological levels of proteins (for example, albumin) | Albumin is a carrier protein essential to the transport of hydrophobic or partially hydrophobic molecules |
| Long-term function8,156 | Greater than or equal to 1 month | Many drug exposure effects are chronic, developing over time; enables the mechanistic evaluation of disease progression and therapeutic responses |
| Reproducibility125,186 | Chip-to-chip, day-to-day repeatability | It is critical that procedures are established for consistency and that models demonstrate reproducibility |
| Optimally structured | Enables manual layering or bioprinting101 of matrix and cells | Needed to recapitulate liver acinus structure |
| Amenable to imaging cells8 | Transparent and non-fluorescent | Enables live imaging of morphology, biosensors and other labels throughout the study |
| Low non-specific binding78,213,214 | <50% non-specific binding of lipophilic drugs | Optimizes the testing of diverse therapeutic libraries, avoids loss of hydrophobic biomolecules and ptimizes computational modelling |
| Flow design8,85 | Single pass or recirculation | Single pass flow avoids the depletion of key media components, such as oxygen and glucose, while recirculation enables the concentration of secreted factors and metabolites but also by-products of metabolism |
| In-line sensors81,88,90 | For example, oxygen, glucose, lactate or proteins | Enables real-time, device average monitoring of key biomolecules |
iPSC, inducible pluripotent stem cell; MPS, microphysiological system.
Box 1 |. Measured functions and responses in selected human liver MPS.
Physiology and pathophysiology that can generally be maintained in multicellular microphysiology systems (MPS) for up to 1 month.
Establishment of oxygen zonation and zone-specific hepatocyte functions (for example, CYP450 activities, steatosis, oxidative phosphorylation, glucose metabolism).
Normal and stress-induced secretion from hepatocytes and non-parenchymal cells of key elements of the secretome (for example, albumin, urea, LDH, cytokines, transcription factors, chemokines and exosomes).
Flow-stimulated functions (for example, increased levels of secretome components).
Molecular driver EGF-induced and TGFβ-induced activation of liver sinusoidal endothelial cells lining the vascular channel (sinusoid).
Binding of polymorphonuclear leukocytes to activated liver sinusoidal endothelial cells followed by their infiltration across the space of Disse to the hepatocyte chamber.
Molecular diver-induced and drug-induced activation of hepatic stellate cells (expression of αSMA) in the space of Disse followed by fibrosis (production of collagen), shape change and proliferation.
Molecular driver-induced and drug-induced activation of Kupffer cells in the vascular channel (secretion of TNF).
Insulin regulated glucose uptake and release.
Molecular driver-induced, drug-induced and engineered cell-induced liver disease phenotypes (for example, steatosis, immune infiltration, fibrosis, bile efflux, insulin resistance).
Cell physiology (for example, biosensors of apoptosis and reactive oxygen species).
Inhibition or induction of CYP450 activity (for example, mass spectrometry analysis of metabolic probes).
Mechanisms of toxicity.
Address challenge of drug-induced liver injury.
RNA sequencing comparison of patient data with disease phenotype MPS.
Current HBL-MPS
There has been great progress in the development of HBL-MPS platforms for the recapitulation of the human liver acinus structure and functions58. Initial efforts primarily used human cell lines for hepatocytes and only a limited subset of non-parenchymal cells (NPCs) to avoid some of the cost and technical challenges of using primary cells from a single patient. However, newer human liver MPS have incorporated at least three cell types including combinations of human primary hepatocytes and liver sinusoidal endothelial cells (LSECs), sometimes with well-characterized and functional human cell lines for Kupffer cells and/or hepatic stellate cells (HSCs)56,59. Furthermore, progress in developing partially matured iPSC-derived hepatocytes and NPCs has enabled their use in some MPS46,60,61. With respect to ‘fit-for-purpose’, there are advantages and disadvantages in using cell lines, primary cells and iPSC-derived cells in MPS (TABLE 3).
Table 3 |.
Cell types used to construct MPS liver models89
| Cell type | Cell lines | Primary cells | iPSC-derived cells |
|---|---|---|---|
| All | Unlimited supply | Limited supply, autologous, diseased or healthy, single donor source | Unlimited supply, isogenic, diseased or healthy |
| Hepatocytes | Functional limitsa (for example, HepG2, HepaRG, SV40 transformed hepatocyte cell lines) | Matureb | Partially mature functions215,216 |
| Kupffer cells | Undifferentiated (for example, THP-1, U-937) | Activatedc | Partially mature217 |
| Stellate cells | LX-2 | Activatedc | Partially mature218; activated218 |
| LSEC | Functional limitsa,219,220 (for example, TRP3, TMNK-1) | Matureb,56 | LSEC-like progenitors cells100 |
| Cholangiocytes | Functional limitsa,221 (for example, MMNK) | Matureb,222 | Partially mature223,224 |
iPSC, inducible pluripotent stem cell; LSEC, liver sinusoidal endothelial cell; MPS, microphysiology systems.
Cell possesses limited functions of mature cells
Cell type possesses all adult differentiated functions
Isolation methods fully or partially activate the cell from the basal state. It is also projected that hybrid Structured-MPS and Organoid-MPS will emerge as the field progresses.
Two different approaches have been taken towards developing HBL-MPS platforms: models in which the initial physical and cellular structure mimics the organization of cells in the liver acinus56,59,62, either by design of the device and manual placement of the cells or by 3D bioprinting of cells and matrices to mimic the organization of cells in the liver63,64, and models in which organoids are stimulated or engineered to self-assemble into liver-like structures40,41,65–67. As a result, HBL-MPS are now evolving along two distinct and complementary paths into structured engineered biomimetic MPS (Structured-MPS) and organoid engineered biomimetic MPS (Organoid-MPS), both of which, along with potential combinations, are expected to recapitulate even more clinically relevant characteristics and functions than the current HBL-MPS (Supplementary Table 1). Organoid-MPS and Structured-MPS that contain patient-derived PSCs will enable the highest level of mechanistic investigations in drug discovery and development as well as having applications in precision medicine, including in ‘preclinical trials’, where patient-specific MPS will be used to predict the most appropriate patient cohorts for optimal clinical trial design (FIG. 2).
Fig. 2 |. Illustration of one design of a current HBL-MPS.
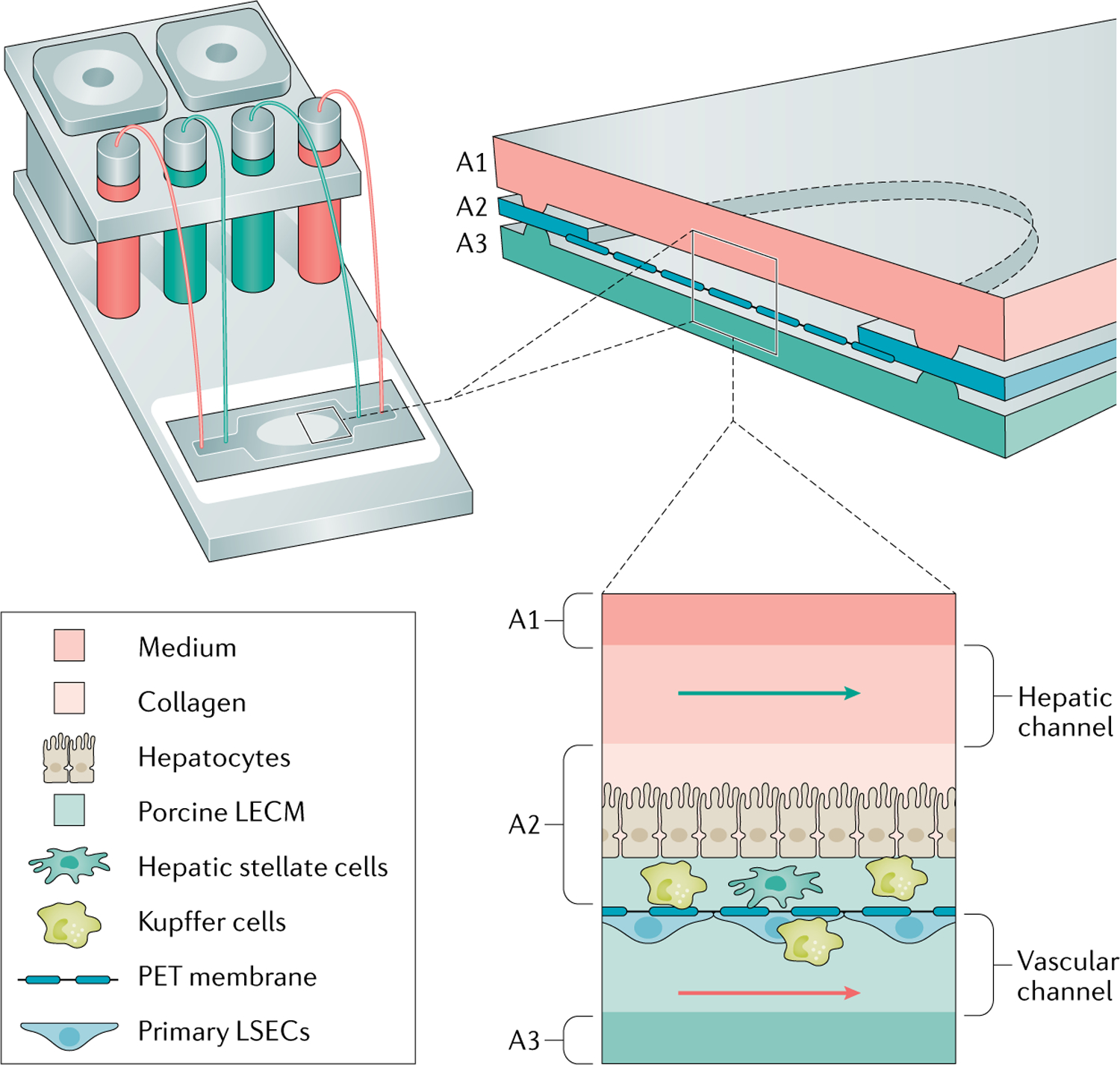
The essential components of a human biomimetic liver microphysiology system (HBL-MPS) are illustrated here as an assembled, self-contained unit with pumps for flow-through or recirculation modes, reservoirs, and a microfluidic device that can be placed in incubators243. In one design, the microfluidic device is a layered assembly with three layers (A1–A3) forming two flow channels separated by a porous membrane filling an opening in the central layer (A2). Cells and extracellular matrix can be layered on the two sides of the membrane to recapitulate the liver acinus as illustrated. Alternatively, cells and matrices can be bioprinted on both sides of the membrane or organoids could be deposited on the membrane. The initial assembly of cells and matrices on two sides of a permeable membrane recapitulates the layered, 3D organization of cells in the liver acinus, which then matures over time as channel flows stimulate cells to establish contacts and communications, while zonation is established56. LECM, liver extracellular matrix; LSEC, human liver sinusoidal endothelial cells; PET, polyethylene terephthalate.
Epithelial liver organoids.
Liver epithelial organoids have been developed to study specific features of hepatic physiology, structure and regenerative function61,68. Despite the limited cell types, constrained access to the sinusoidal lumen and the variability of sizes, these epithelial liver organoids are being successfully used in early drug discovery, screening and safety assessment, in which throughput is often considered more important than mechanistic knowledge early in the pipeline of drug discovery69. Although slightly more difficult to produce than 2D cell models, epithelial liver organoids have a relative ease of production compared to more complex models, show long-term expansion capacity, enable high-throughput measurements and serve as a source of cells68 (Supplementary Table 1).
Epithelial liver organoids have also shown promise for the modelling of some aspects of genetic diseases48,70 and of human liver cancers48,71. However, more advanced liver organoids have been developed from different germ layers, such as endoderm and mesoderm, that recapitulate the diversity of native liver cell types such as HSCs, Kupffer cells and endothelial cells40,41,46,72. The presence of heterotypic cells enables novel translational applications to understand cell–cell interactions in disease modelling, such as steatohepatitis46, which can inform novel drug discovery. Hepato-biliary-pancreatic organoids have also been produced that demonstrate a promising step towards generating experimental multi-organ platforms with the ability to model complex human endoderm organogenesis73.
Designs of current HBL-MPS.
HBL-MPS primarily harness patient-derived human primary liver cells, sometimes with human cell lines for HSCs and Kupffer cells56,59,65,66,74–76 (TABLE 1). Microfluidic devices are constructed of glass, plastic and/or polymers to enable temporal and spatial confocal microscopic interrogation of the distinct 3D cell layers56, a subset of which might contain fluorescence-based protein biosensors of physiological processes such as apoptosis, oxidative stress and insulin resistance77. Although polymers such as polydimethylsiloxane are still widely used, the use of glass or plastic in the microfluidic device minimizes the non-specific binding of hydrophobic molecules8,56,78. In two-channel designs, the hepatic chamber is typically separated from the vascular channel by a porous membrane (presently polyethylene terephthalate (PET)) to permit the communication between compartments of circulatory components such as free fatty acids, gut microbiome products, drugs and cells (including immune cells) as well as secretome and metabolic products. The vascular channel may also be used as a link to other physically coupled organ MPS models56.
In general, current HBL-MPS models are optimally assembled in a microfluidic device that provides connections to microfluidic flow systems and substrates on which to construct the liver tissue, while maintaining the model in a controlled environment (TABLE 2, FIG. 2, Supplementary Table 1). Microfluidic flow (FIG. 2) can be driven by external or integrated mechanical pumps79,80, air pressure81 or gravity50,82–84 in a single pass to provide a constant media composition or with recirculation to enable the accumulation of secreted factors and metabolites as well as of potential toxic by products8,85. The device (FIG. 2) can be one piece or assembled in layers. Organoid-MPS can be encapsulated in a similar microfluidic system31,86,87.
The design of the vascular liver acinus MPS (vLAMPS) model56 serves as an example of combining the options above to create an HBL-MPS. In the vLAMPS model, hepatocytes are placed on top of a thin extracellular matrix containing HSCs, representing the space of Disse, on the hepatic chamber side of the PET membrane (FIG. 2). The microfluidic flow in the hepatic chamber mimics the physiological shear stress on the hepatocytes that optimizes the hepatocyte physiology and interstitial flow that, in turn, optimizes collection of the secretome such as albumin, urea, lactate dehydrogenase (LDH), cytokines and glucose56. LSECs, along with Kupffer cells, line the vascular channel side of the PET membrane with a separate microfluidic flow that creates the shear stress that stimulates the formation and stability of the endothelial cell layer. Oxygen zonation in the vLAMPS model is created by hepatocyte oxygen consumption combined with control of the flow rate along the ‘sinusoid’ to create physiological zone-specific biochemical, metabolic and pathophysiological functions56,58. The vLAMPS model uses in-line sensors for the readout of average glucose or lactate, an approach that is also highly desirable for the readout of oxygen tension and other physiological parameters88–91. HBL-MPS have been successfully applied to pharmacokinetics22,92,93, liver toxicity37,94,95, disease modelling56,65,74 and, when coupled with other organ models, ADMET96–98.
The key characteristics of current HBL-MPS platforms include (1) the ability to experimentally combine, exclude and/or engineer the cell types included in a given study as well as to alter microenvironmental cues, such as matrix stiffness and biochemistry, to define their effect on ADMET and disease models (TABLES 2,3); (2) the ability to test potential therapeutics including small molecules, biologic agents and gene editing in a controlled platform using a vascular delivery mechanism; and (3) the ability to disassemble the platform to extract distinct cell types and matrices for further molecular characterization. The same characteristics are also true for next-generation Structured-MPS and Organoid-MPS (Supplementary Table 1).
Next-generation HBL-MPS
Structured-MPS.
Although HBL-MPS have been shown to provide more physiologically relevant functions than simpler 2D and 3D models, there are still differences between the various models that could affect functions, including the scaling of cells to media99, construction with mixtures of non-isogenic cells, and differences between media and blood composition that could affect the physiological microenvironment. An example of a next-generation HBL-MPS platform is the Structured-MPS that are being engineered within microfluidic devices to optimally recapitulate the liver acinus structure, replacing primary cells predominantly used in the current HBL-MPS with iPSC-derived hepatocytes and NPCs from the same patient100. The iPSC-derived cells are placed in positions that recapitulate hepatic and vascular (sinusoid) channels, linked through a porous membrane that permits the passage of molecules and cells8,56. The bioprinting of cells and matrix materials is also evolving as an important tool to refine the structure and improve the reproducibility and efficiency of Structured-MPS101 (TABLE 2). Structured-MPS will generate patient-specific mechanistic information to drive the selection of molecular targets within computationally identified networks of disease progression, to prioritize ‘hits’ in screening, to identify target engagement, to create precision medicine experimental models of disease progression, to quantify both responses to therapeutics and to ADMET, and to produce platforms for the parallel preclinical testing of multiple drugs to optimize patient selection for each investigational drug trial. Validation of this approach will be necessary and the predictions made in MPS will have to be directly correlated to clinical data obtained in human individuals. Although this approach is not currently used in the clinic, there are major initiatives in place at the National Institutes of Health and the FDA27 to integrate MPS in the drug testing and patient care pipeline.
Organoid-MPS.
Progress in systems and synthetic biology has offered novel genetic approaches to engineer a next-generation HBL-MPS based on organoid technologies (Organoid-MPS)41,72,102–104 (TABLE 2). The opportunity to use genetic manipulation to drive tissue formation has the promise to alleviate the need to have a common culturing media or to sequentially add a cocktail of growth factors to create organoids. Initial studies applying this approach in liver used genetically encoded GATA6 as a regulatory switch to drive the formation of a self-vascularized human fetal liver organoid containing hepatocyte-like, endothelial-like and stellate-like cells from a single population of human iPSCs without the stepwise addition of growth factors41. Organoids with genetically engineered regulatory switches offer multicellular type platforms with control over cell state and maturation in time and space72,102. The integration of systems biology with genetic engineering has further enabled in vitro maturation of the multilineage liver organoids72. These Organoid-MPS that are under development integrate stem cell self-organization, synthetic biology and genetic engineering technologies in microfluidic devices to closely model normal liver physiology and patient-specific disease states as well as ADMET. It is also projected that hybrid Structured-MPS and Organoid-MPS will emerge as the field progresses.
HBL-MPS in ADMET and disease models
The human liver performs a broad range of functions that are integrally coupled with liver structure (BOX 1, FIG. 1a). For example, ammonia detoxification, gluconeogenesis, glycolysis and xenobiotic metabolism predominate in different zones of the liver105. The design and evolution of HBL-MPS has been driven by the need for more physiological and pathophysiological models that recapitulate the structure–function relationships in the human liver, within engineering and biological constraints89,106–108. Although the ideal human biomimetic liver MPS would recapitulate the full range of human liver function, testing, verifying and validating all of those functions together would be a major task. Hence, the present practical approach is to apply ‘fit-for-purpose’ models that are tested and validated for the functionality required in specific applications. A wide range of human liver cell-MPS platforms has been developed50,56,59,62,75,76,109 and the variety of applications continues to expand (TABLE 1). Many of these platforms meet the optimal physiological and device requirements for an optimal HBL-MPS (TABLE 2), consistent with their intended applications. Ultimately, we expect that Structured-MPS and/or Organoid-MPS will be fully developed and tested over a sufficiently broad range of functions such that they will become the standard for patient-specific, mechanistic human liver ADMET studies, disease modelling, precision medicine and clinical trial design.
Of the key structural and functional features required for an HBL-MPS, bile ducts, a crucial toxicological and therapeutic target in cholangiopathies, have not yet been integrated into an HBL-MPS. However, efforts are underway to incorporate these cells as the next step in the evolution of HBL-MPS. Bile ducts are required to create a more complete liver acinus that will enable, for example, the evaluation of the cholestatic component of liver injury from drugs such as troglitazone (withdrawn), leflunomide (black box warning for liver injury) and the combination amoxicillin-clavulanate drug augmentin (one of the most common medications inducing cholestatic DILI)22,110. As a step in this evolution, bile duct organoids developed from human PSC-derived cholangiocytes have been used to model cholangiopathies and to screen for and identify potentially therapeutic compounds111 as well as to bioengineer bile ducts that were used for experimental reconstructive surgery in mice112,113. Another bile duct model, an in vitro mouse cholangiocyte bile duct MPS model, designed with access to the apical and basal sides, has demonstrated barrier function, transport across the barrier and an apical surface that is more resistant to the bile acid glycochenodeoxycholic acid than the basal surface114. These demonstrations of in vitro biliary functions suggest the potential of integration of cholangiocytes and bile ducts into current HBL-MPS and ultimately into Structured-MPS and Organoid-MPS.
Although HBL-MPS can be constructed using some patient PSC-derived cells54,74,75,115, there are still many challenges to producing MPS in which all cells are derived from a single donor and in which all cells exhibit a fully mature phenotype and genotype (TABLE 4, Supplementary Table 2). Culturing partially matured cells in the more physiological environment of the HBL-MPS is expected to contribute to the final maturation of the cells, as they integrate a multicellular, multi-dimensional tissue microenvironment. Full maturation of the differentiated liver cells remains a major challenge to producing next-generation Organoid-MPS and/or Structured-MPS (TABLE 4).
Table 4 |.
Challenges in producing iPSC-derived cells for HBL-MPS
| iPSC-derived cells | Status | Importance |
|---|---|---|
| Hepatocyte-like cells | Maturation protocols are needed to produce the full hepatic metabolic profile | Necessary to mimic all aspects of human liver disease and function; immature iPSC-derived hepatocytes typically have less metabolic capacity and consume only 25% of the oxygen89 |
| Cholangiocyte-like cells | Cholangiocytes that closely resemble the in vivo transcriptional and functional features have been generated but require further testing and validation223,224 | Enables various applications, such as developmental studies, disease modelling, therapeutic target validation and drug screening |
| LSEC-like cells | No protocols have yet been reported for the generation of LSECs | LSECs have unique properties that affect endocytosis, permeability, vascular tone, portal pressure, regeneration and inflammation225–227 |
| VECs | VECs have been generated and shown to have basic functionality but need further testing in MPS228 | Involved in immune, haematological and transport processes |
| Hepatic stellate-like cells | A robust protocol has been generated100,218 but cells need further testing in MPS | Stellate cells play a key role in the initiation, progression and regression of liver fibrosis146,229 |
| Macrophage-like (Kupffer) cells | Human iPSC-derived macrophages perform immunological functions with similar transcriptome profiles as primary cells217 | Scavenger and phagocytic functions to remove protein complexes, small particles, senescent cells and debris143,230 |
| Adaptive immune cells | Several reports of iPSC-derived antigen-specific cytotoxic T lymphocytes and NK cells with high proliferative capacity and expression of central memory T cell markers231 but with immature phenotype | Adaptive immune response to mimic inflammatory response, adoptive immunotherapy, etc. |
HBL-MPS, human biomimetic liver microphysiology system; iPSC, inducible pluripotent stem cell; LSEC, liver sinusoidal endothelial cell; NK, natural killer; VEC, vascular endothelial cell.
Current HBL-MPS ADMET models.
Although the pharmaceutical industry has developed panels of assays to predict ADMET endpoints and human pharmacokinetics, current in vivo models lack species specificity and traditional in vitro models lack organ physiology97. In the liver, a key ADMET parameter is clearance, which is important in the determination of optimal dose, route of administration and drug formulation, collectively forming the basis for risk management in clinical trials. Primary hepatocytes have been the gold standard for measuring clearance but, in simple in vitro models, hepatocytes rapidly lose metabolic function94,116,117. HBL-MPS provide a more physiological and pathophysiological environment, with demonstrated stabilities of 2–4 weeks62,76,118, enabling more accurate predictions of pharmacokinetic models. Furthermore, HBL-MPS can be linked to other organ MPS to construct a more sophisticated multi-organ or even a whole body ADMET model (discussed later)97. HBL-MPS have the potential to experimentally model the key parameters required to build reliable pharmacokinetic and pharmacodynamic models36,63,96,97,119–121.
During drug development, toxicology studies are performed to identify and deprioritize compounds having either on-target or off-target toxic liabilities. Although HBL-MPS models are anticipated to be superior with respect to translatability, ethical considerations and cost compared to current in vivo protocols, acceptance by drug and environmental regulators as well as by the pharmaceutical, chemical and cosmetic industries requires evidence-based results demonstrating that the models are better than, or at least as good as, current animal models21–23. Comparative studies are being pursued within the pharmaceutical industry and regulatory agencies.
DILI is a major reason for the termination of drug development projects. ‘Black box warnings’ and, particularly, the withdrawal of marketed drugs are clear indications that the existing in vitro models and preclinical animal models are not always adequate to detect DILI. HBL-MPS with a broad range of mechanistic metrics have been shown to provide reliable indications of potential liver toxicity and, in some cases, provide a path to mitigate toxicity59,62,94,95,122–124. In one liver toxicity study, a species-specific biomimetic MPS comprised of four liver cell types from rat, dog or humans measured multiple phenotypes, including hepatocellular injury, steatosis, cholestasis and fibrosis, leading to mechanistic profiles that were found to be concordant with some species-specific mechanisms for known drugs75. Multispecies studies such as this might also be useful in developing computational models that improve the translation of preclinical animal study results to human clinical trials. For example, in one study, 14 drugs with varying degrees of liver toxicity were mechanistically profiled in the liver acinus MPS59,125. The profiles were compared with the normalized adverse event frequencies (number of liver-specific adverse events per number of prescriptions) in the Microphysiology Systems Database (MPS-Db)125,126. The rank order of the mechanistic profiles showed excellent concordance with the rank order of the normalized adverse event frequencies from the MPS-Db125. The value of the HBL-MPS for mechanistic analysis was further demonstrated in a study comparing the mechanistic profiles of two antiviral drugs with different DILI phenotypes39,124. Evaluating liver toxicity and DILI in HBL-MPS constructed with PSC-derived cells from patients of varying health (for example, those at different disease stages or those with the presence of comorbidities) as well as varying genetic backgrounds has the potential advantage of identifying patient-specific toxic liabilities127,128.
A review of the performance of a wide range of in vitro liver experimental models, including current HBL-MPS, for the identification of liver toxicity, concluded that the development of MPS has enhanced the general prediction capability but that adoption of liver MPS requires the implementation of standards and data from a set of prototypical liver toxins with diverse mechanisms123. TABLE 5 outlines some suggested standard drugs that could be evaluated in liver MPS for pharmacokinetics, known liver toxicity and DILI38. Collectively, a set of standards for a wide range of genetic backgrounds and liver diseases would provide a useful test set to evaluate model performance and to direct the development of the optimal Structured-MPS and Organoid-MPS. When choosing and applying a human liver MPS, it is important to consider the potential effect of microenvironmental conditions on liver toxicity. For example, toxicity can be associated with an immune cell response62,94,129 or be zone specific56,59,130. Oxygen zonation is a key feature of the liver acinus, which induces a metabolic gradient that can affect drug toxicity56,58,59,89,131,132. Although a critical microenvironmental factor in the liver, oxygen tension and other gradients are often overlooked in MPS models, highlighting the need for integrated oxygen sensors in MPS models56,59,90,133.
Table 5 |.
Standard drugs to evaluate liver pharmacokinetics and DILI in MPS
| Purpose of testing | Drugs/disease drivers | Key MPS metrics |
|---|---|---|
| Pharmacokinetics | Phenacetin, testosterone, tolbutamide, mephenytoin, dextromethorphan, chlorzoxazone | Key cytochrome P450 activities: CYP1A2, CYP3A4, CYP2C9, CYP2C18, CYP2D6, CYP2E1 (REF.241) |
| Classic liver drug toxicity123 | Acetaminophen, troglitazone, amiodarone | ↑ROS, ↑apoptosis, ↑pro-inflammatory cytokines, ↑LDH, ↓functions (albumin, urea)37,59,62 |
| Idiosyncratic DILI242 | Nefazodone, trovafloxacin, sitaxentan | Liver drug toxicity indicators yet to be identified |
DILI, drug-induced liver disease; LDH, lactate dehydrogenase; MPS, microphysiology system; ROS, reactive oxygen species.
HBL-MPS for NAFLD and metabolic syndrome models.
Multiple organs, including the liver, pancreatic islets, heart, adipose tissue, intestines and striated muscle, are involved in the metabolic syndrome that can lead to serious illness and death. Hepatic insulin resistance is a common patient characteristic in NAFLD and our current understanding is that NAFLD represents a hepatic manifestation of the metabolic syndrome7,134. The liver-specific pathophysiology of metabolic syndrome can be induced in stand-alone HBL-MPS and/or through coupling with organ models (as described later) and analysed using well-established phenotypic, genomic and functional metrics (Supplementary Table 3).
NAFLD is a spectrum of pathological conditions that can progress from non-alcoholic fatty liver to NASH and then cirrhosis, a major risk factor for hepatocellular carcinoma and/or liver failure135. In 2020, the term metabolic (dysfunction) associated fatty liver disease (MAFLD) has been recommended to replace NAFLD to more accurately characterize this heterogeneous disease that can include multiple components of the metabolic syndrome136. Currently, no approved therapies for NAFLD or MAFLD exist based on the use of simple in vitro experimental models and animal models3,137. The progression of NAFLD involves the interplay of genetic and environmental factors with complex mechanisms involved in immune and inflammatory responses, oxidative stress, autophagy, DNA damage, and communication between multiple cell types and factors from other organs138–141. The disease state can be induced in HBL-MPS using patient-derived primary disease cells, iPSC-derived cells, molecular drivers of disease (for example, free fatty acids (FFAs), glucose, cytokines, immune cells and cancer cells)65,66,142 and/or by coupling key organ MPS (for example, intestine, adipose tissue and liver)66. HBL-MPS disease models have already made important early contributions to the exploration of potential therapeutic targets and phenotypic profiles, not only in hepatocytes but also in the NPCs and infiltrating immune cells3,65,66,143–146. In one HBL-MPS that incorporates hepatic sinusoidal flow, transport and lipotoxic molecular drivers, 0.5 μM obeticholic acid promoted a healthy lipidomic signature, reducing inflammatory and fibrotic secreted factors in agreement with early clinical trial data, but also showed an increase in ApoB secretion, suggesting a potential adverse effect on lipoprotein metabolism65.
In a second HBL-MPS NASH model, primary human hepatocytes, Kupffer cells and HSCs were co-cultured in physiological ratios in a perfused MPS66. These microtissues displayed a NASH-like phenotype in response to FFA in the media, including hepatic fat accumulation, the production of inflammatory cytokines and the expression of profibrotic markers, which was enhanced with the addition of lipopolysaccharide66. This study further demonstrated some attenuation of these responses following treatment with obeticholic acid, a NAFLD drug candidate that showed some efficacy in clinical trials but was recently rejected for NASH treatment by the FDA due to the risk of side effects10. The NASH phenotype was enhanced when the model was constructed with HSCs carrying the major genetic variant associated with NAFLD progression, the I148M PNPLA3 mutation, which induced a substantial increase in IL-6 secretion66. This HBL-MPS NASH model was evaluated for cytokine production and gene expression but lacked LSECs and image-based spatial relationships and, therefore, some key signalling functions were missing. Deeper profiling of the disease genotypes and phenotypes is required to further evaluate drug safety and efficacy. However, patient-specific Structured-MPS and Organoid-MPS based on patient-derived PSCs are also needed to fully define the mechanisms responsible for this complex disease progression and the patient specific responses, which will lead to precision medicine strategies for drug discovery and development.
In a third study, 11 different healthy and diseased PSC lines were used to construct an organoid-based HBL-MPS composed of hepatocyte-like, HSC-like and Kupffer cell-like cells that exhibited transcriptomic and functional resemblance to in vivo-derived tissues46. FFA treatment induced key features of NAFLD progression in the organoids, including steatosis, inflammation and fibrosis. Interestingly, organoids from patients with genetic dysfunction of lysosomal acid lipase phenocopied severe steatohepatitis and were rescued by an FXR agonist46. In another human liver MPS, immune cell infiltration and control of oxygen zonation were shown to recapitulate functional zonation in the liver. The liver microenvironment had a substantial effect on NAFLD-associated phenotypes, including steatosis, emphasizing the need to control oxygen zonation in HBL-MPS56.
HBL-MPS for human cancer models.
Cancer mortality is often a result of metastases and the liver is a common metastasis site, with the formation of a pro-metastatic niche further promoting the recruitment of metastatic cells and regulating their evolution147–150. A better understanding of the role of the microenvironment in liver cancers, both primary and metastases, is required since it is critical to identifying the biomarkers linked to metastatic disease and the targetable tumour dependencies that can inform novel therapeutic strategies150,151, for example, stromal cell modulation in solid tumours152. The deciphering this coevolution of tumours and their microenvironments is aided by HBL–MPS and, in the future, Structured-MPS and/or Organoid-MPS, as these latter models will enable iPSC-derived cells and microenvironmental components to be further controlled to recapitulate, for example, the emergence of clinically relevant drug resistance.
HBL-MPS offer important opportunities to understand the liver as a cancer niche and to develop therapeutic strategies. In one study, a metastatic breast cancer niche was developed in an HBL-MPS, where it was observed that breast cancer cells engineered to express different oestrogen receptor-α (ERα) mutations that are selected in the clinic during oestrogen deprivation therapy and found to be enriched in metastases that include liver, exhibited different oxygen-dependent growth and drug resistance phenotypes that varied with the tumour microenvironment150. In another metastatic breast cancer liver niche HBL-MPS153, an analysis of the effect of microenvironmental factors on the dormancy and emergence of breast cancer cells and of their relationship to drug efficacy identified potential blood protein clinical biomarkers that could be used to identify the metastatic stage109,154. Using the same model, it was further demonstrated that statins might be an effective long-term treatment to attenuate the outgrowth of breast cancer metastases in the liver155.
The liver in coupled organ MPS
Key advances have been made in constructing coupled human organ MPS27,85,156,157. These multi-organ MPS are valuable for integrating the effect of multiple organs on toxicology, pharmacokinetics, pharmacodynamics, drug efficacy, and disease initiation and progression by enabling factors from one organ MPS to be delivered to another organ MPS as occurs in vivo (Supplementary Figure 1).
Although most multi-organ MPS for ADMET and for liver diseases include a liver, the liver component in most published multi-organ systems varies from simple 2D monolayer hepatocyte cell lines, to co-cultured spheroids and to primary hepatocytes with one or two NPCs96,98,158,159. Most of the early multi-organ MPS included the liver primarily as a compound metabolism engine83–85,160–163. However, successes in evaluating pharmacokinetics in multi-organ systems based on HBL-MPS have been reported, including the permeability and metabolic clearance of diclofenac and hydrocortisone in a liver–intestine MPS164; the organ-specific metabolism of vitamin D3, trimethylamine and terfenadine in a functionally coupled MPS model of the jejunum intestine, liver, vascularized kidney proximal tubule and blood–brain barrier98; diclofenac and tolcapone metabolism through a seven organ MPS model96; and diclofenac metabolism in another seven coupled organs, including MPS models for the intestine, liver and endometrium and simple 2D models of the heart, brain, lung and rat pancreatic islets165.
To date, there are very limited examples of multi-organ MPS disease models that recapitulate the structural organization of the cells in the liver creating an HBL-MPS. A liver MPS can include different sources of liver cells, including animals, humans and cell lines, and consist of hepatocytes alone or with some number of NPCs in plate and/or microfluidic platforms. By contrast, HBL-MPS exhibit more requirements, including multiple human cells in a 3D structure with many liver acinus functions and under flow stimulation. TABLE 2 lists many of the human biomimetic liver requirements that have been implemented or are under development. One example of the use of an HBL-MPS in a multi-organ MPS disease model is the study of fluorescent human colon cancer cells migrating from a hydrogel-fabricated gut to a downstream liver MPS modelling a common route of metastasis156. In another example, a type 2 diabetes mellitus MPS model has been reported that coupled human pancreatic islet tissues with human liver spheroids consisting of HepaRG cells (a carcinoma cell line for hepatocytes) and primary human HSCs163. Although not involving an HBL-MPS for liver as defined here (TABLE 2), this study demonstrated that the insulin released from the pancreatic islet chip promoted glucose uptake by the liver spheroid chip163. The extension of this type of multi-organ model to involve HBL-MPS with a human biomimetic pancreatic islet MPS would enable more detailed mechanistic studies on the crosstalk between the liver and pancreatic islets.
There are also the issues of ‘context of use’ and ‘fit-for-purpose’ for multi-organ MPS. By practical necessity, most of the multi-organ MPS simplify one or more of the organ MPS components and create limited functionality (fit-for-purpose) in some of the organs (for example, including the liver as only a metabolic engine) to meet the experimental design (context of use). One important solution is to create multi-organ biomimetic MPS with the minimum number of component organs to gain the mechanistic capability while harnessing multi-organ communication (for example, liver–pancreatic islets and intestine–liver–kidney). Alternatively, for liver diseases, the application of molecular drivers into the vascular channel of HBL-MPSs and next-generation Structured-MPS and Organoid-MPS can simplify the development of liver disease models without physically coupling to other organ MPS56. The challenges in linking multiple organ MPS are identical to those for HBL-MPS (TABLE 2) with the added complexity of the physiological scaling between the various organ MPS as well as fluid volumes and fluid flow rates within each organ MPS85,99,108,157.
HBL-MPS for precision medicine
Chronic, complex and heterogeneous liver-involved diseases, such as NAFLD3,7, type 2 diabetes mellitus166, liver metastases and rare congenital diseases caused by a defect in a single enzyme or transport protein167, impose distinct challenges for therapeutic development and the optimization of clinical trial design. The implementation of a QSP platform8,20 can address these challenges by revealing the pathogenic mechanisms in individual patients who, in turn, inform the rational development of biomarkers, novel drugs and drug repurposing, therapeutic combinations, and the optimization of clinical trial design. Intrinsic to QSP is the integration of advanced technologies that support the steps required for precision medicine8 (FIGS 1b,3). Here, we discuss the prospective use of Structured-MPS and/or Organoid-MPS as a component of QSP for experimental and computational modelling of NAFLD or MAFLD and two liver-related rare diseases, phenylketonuria and α1-antitrypsin deficiency (AATD). Clinically relevant biomarkers are critical in clinical trials and Structured-MPS and Organoid-MPS have the potential to identify both genetic and secreted molecules as potential biomarkers in preclinical trials (Supplementary Table 4).
Fig. 3 |. Organoid-MPS and Structured-MPS are platforms for advancing precision medicine.
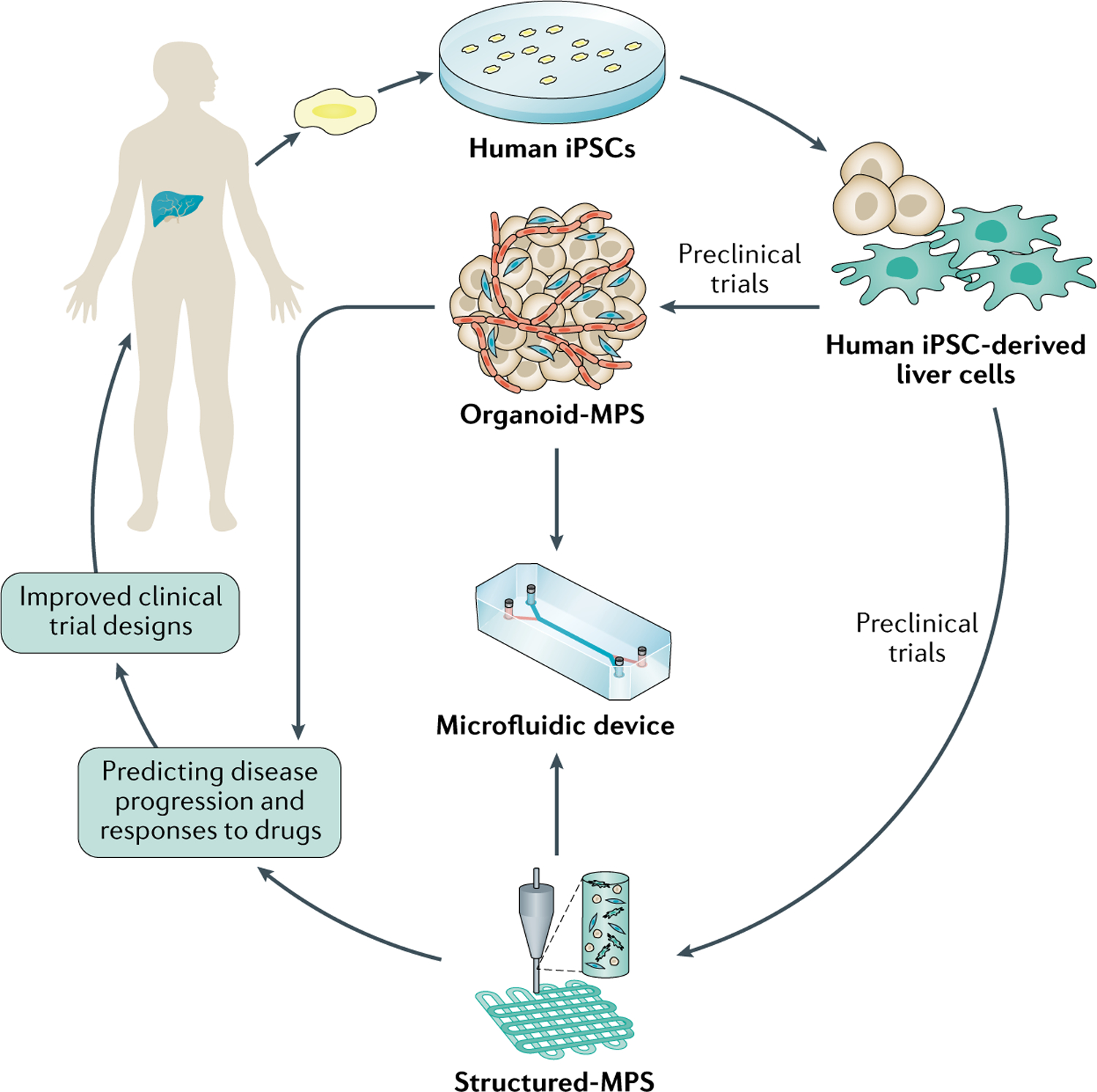
Organoid engineered biomimetic microphysiology systems (Organoid-MPS) and/or structured engineered biomimetic microphysiology systems (Structured-MPS) are dynamic devices capturing key aspects of the spatiotemporal changes in disease phenotypes and genotypes to identify biomarkers and predict both disease progression and response to drugs in individual patients. Organoid-MPS and/or Structured-MPS-driven preclinical study design could address the confounding effect of patient heterogeneity, informing a clinical trial design with more optimal patient selection, reducing the diversity of pharmacological responses and thereby increasing the probability of drug approvals. iPSC, inducible pluripotent stem cell.
Application of HBL-MPS to NAFLD therapies.
Despite an alarmingly high disease prevalence168 as well as an associated and increasing economic169 and health-care burden170–172, to date, no effective therapy for NAFLD has been approved3,7,141. About 40% of undiagnosed patients with NAFLD progress over decades to cirrhosis141, whereas, by contrast, approximately 20% of patients progress over months to years to an advanced fibrotic stage173,174. This heterogeneity intrinsic to NAFLD reflects the diverse but convergent effects and interplay between the environment, microbiome, metabolism, comorbidities and genetic factors141,175. Thus, it is expected that the pathogenic drivers and their relative contribution to disease progression would vary among individual patients3,7,176. A corollary of this diversity in disease mechanisms is that pharmacodynamic markers of drug target engagement would not necessarily correlate with predictive markers of clinical benefit. Thus, although evidence from clinical trials, which includes decreased liver steatosis, stiffness and plasma alanine aminotransaminase levels177, suggests that features of NAFLD are pharmacologically responsive, only 20–40% of patients in these trials have shown benefit from a single therapy, precluding regulatory approval3,7,10,176. Appropriately, the field is moving towards combination therapy178. However, combination therapy per se might not be sufficient for improving clinical trial design and might not demonstrate sufficient benefit for approval. Disease sub-classification that could elucidate the genetic and other factors mechanistically linked to the pathophysiology and thereby lead to a more accurate prediction of disease progression and drug response in individual patients will also be required3,175. To this end, next-generation Structured-MPS and Organoid-MPS represent a novel approach to precision medicine for NAFLD.
HBL-MPS as a precision medicine platform.
Traditional plasma-derived static markers, such as cytokine profiles and extracellular matrix remodelling indicators, along with consensus molecular signatures and advanced imaging modalities have value for the clinical diagnosis and staging of NAFLD3,7. Studies65,66 support the tenable hypothesis that patient-specific Organoid-MPS and Structured-MPS can serve in a complementary manner to these traditional markers to capture, on a weekly timescale, key aspects of the spatiotemporal dynamics of the disease phenotypes (Supplementary Table 3) that could more efficiently predict disease progression. We envisage that, for patients identified with hepatic steatosis through non-invasive imaging, patient-specific Structured-MPS and/or Organoid-MPS based on iPSCs could be implemented to predict rates of progression and enable the early identification of patients (~20%) who are at high risk of rapidly progressing to advanced fibrosis; the early detection of NASH in these patients is critical. For example, compared to the incidence in other liver diseases, a large percentage (35–50%) of hepatocellular carcinomas that arise in NASH occur before patients develop cirrhosis and routine cancer screening is conducted141,172,179,180. Consequently, these tumours tend to be larger and less amenable to therapies than those with other aetiologies141,180.
The next-generation Structured-MPS and/or Organoid-MPS derived from patient cells are inherently non-invasive as the patient-derived cells can be generated from blood cells or superficial skin cell collection. In addition, they are amenable to the controlled variation of experimental parameters (for example, time and pathogenic drivers) and therefore complementary to patient-derived, tissue-based ex vivo studies. In conjunction with the extensive use of diverse imaging modalities, metabolomics and a wide array of real-time cell function readouts (Supplementary Table 3), MPS-derived information can be potentially correlated with disease progression to enable the identification and validation of non-invasive prognostic markers3,66,123,181–185 (Supplementary Table 3). It is anticipated that this cross-validation of accumulating Structured-MPS and Organoid-MPS and clinical data, managed through the MPS-Db125,126,186, will lead to a refined sub-classification of complex heterogeneous diseases such as NAFLD or MAFLD, with important implications for clinical trial design and drug development.
Patient-specific Structured-MPS and/or Organoid-MPS based on patient-specific iPSCs187,188 could be used to address the conundrum of high-risk patients being enrolled in large prolonged studies with a likelihood of failure, while simultaneously being disqualified from other potentially beneficial studies or treatments. The individual patient-specific MPS can be ‘enrolled’ in a preclinical trial for parallel testing of multiple single drugs and/or combinations27. The cohort of patients whose corresponding MPS indicate a therapeutic response to the same investigational treatment could then be enrolled in an actual trial. This MPS-driven preclinical trial could optimize patient selection, leading to an improved clinical trial design and reduce the diversity of pharmacological responses and the effect of confounding disease heterogeneity, thereby increasing the probability of drug approval. Given the participation of many groups worldwide making steady technical progress towards meeting this objective, we project that MPS-driven preclinical trials will achieve clinically significant validation over the next 5 years to support its use as an integral component for clinical trial design. However, there are important functions of adult, PSC-derived Structured-MPS and Organoid-MPS that must be demonstrated for these to be considered physiologically and clinically relevant (Supplementary Table 2).
HBL-MPS for effective drug combinations.
Coupling this precision medicine approach to a more comprehensive and unbiased QSP strategy could take advantage of extensive molecular and cellular datasets available from each Structured-MPS or Organoid-MPS and leverage systems biology to predict novel combinations3,8. For example, consistent changes in the liver transcriptomic pathways associated with NASH and their response to one drug could also uncover the unaffected pathways that would benefit from a second complementary drug3. Furthermore, as was found for Huntington disease189, connecting the canonical drug mode of action to the identification of effective drug combinations could identify emergent disease-specific pathway crosstalk that could in turn lead to novel therapeutic strategies.
The preclinical testing of multiple drugs using these patient-specific HBL-MPS enables drugs to be studied and compared under treatment-naive conditions, thereby circumventing the confounding challenges intrinsic in adaptive clinical trial design, particularly those involving cancer patients190. The potential of Structured-MPS or Organoid-MPS to recapitulate critical aspects of the tumour microenvironment150 will facilitate the identification of those clinically relevant mechanisms of drug resistance that are likely to emerge from a given treatment regimen, thereby proactively informing a more durable and robust therapeutic strategy (TABLE 6). However, the clinical validation of these MPS platforms to accurately recapitulate and thereby predict drug resistance in individual patients will be required to gain support from both the broader translational research community and the regulatory agencies.
Table 6 |.
Challenges presented by complex heterogeneous diseases or rare diseases for MPS
| Challenge | Approach using patient-derived HBL-MPS | Refs |
|---|---|---|
| Validation of predictive biomarkers when disease mechanism, risk and rate of progression display diversity among individuals having different clinical manifestations of the ‘same’ disease | Individualized MPS is itself a dynamic predictive marker of disease progression, recapitulating critical aspects of the complex spatiotemporal interplay of genetic and environmental factors for each patient | 65,66,232,233 |
| Invasive histology-based markers not necessarily reflecting disease progression | Comprehensive unbiased identification of non-invasive markers mechanistically linked to disease progression | 66,234 |
| Predominant enrolment of patients with intermediate or advanced stage of disease (for example, fibrosis for NAFLD) | Enable enrolment of patients predicted to be at high risk (based on genomic and/or MPS analysis) but at earlier clinical stage of the disease; support FDA subpart H pathway | 29 |
| Prolonged trials prevent patients from receiving drugs likely to be more efficacious | Preclinical trial-on-a-chip enables testing of multiple drugs in parallel on same patient-derived MPS model, predicting optimal drug and response for individual patients | 199,235 |
| Given the diversity of pathogenic drivers among individual patients, empirically identifying optimally effective drug combinations is not feasible | Provides a comprehensive systems-oriented framework for predicting repurposed and novel drugs and combinations | 236–238 |
| Complex tissue architecture, microenvironments and inter-organ homeostatic mechanisms may not be recapitulated in animal models | HBL-MPS can capture nuances of innovative therapeutic strategies and organ crosstalk and predicts clinically relevant resistance | 239,240 |
| Complex diseases initiate and progress over years to decades | Time of disease progression can be compressed with the use of specific molecular drivers or gene manipulations | 65,66 |
HBL-MPS, human biomimetic liver microphysiology system; MPS, microphysiology system; NAFLD, non-alcoholic fatty liver disease.
HBL-MPS for congenital liver diseases.
For the diverse class of congenital liver diseases resulting from errors of metabolism167, compensating for loss of function or modulating pathogenic gains of function have, in general, proven challenging (TABLE 6). Although individually rare, monogenic liver diseases (for example, phenylketonuria, Wilson disease, AATD, tyrosinaemia, hereditary haemochromatosis or glycogen storage disease), each caused by a single distinct gene mutation, collectively affect 1% of births191. Monogenic diseases encompass a diverse set of mutations but can be grouped according to whether the mutation results in damage to the liver parenchyma or to specific liver expression that has extrahepatic effects. In many cases, the only therapeutic option is transplantation of the liver and potentially of other organs in the case of extrahepatic damage191. In one study, organoids derived from patients with AATD or Alagille syndrome were shown to mirror the in vivo pathology, demonstrating the potential use of HBL-MPS for experimental disease modelling, ADMET studies and gene therapy for monogenic diseases68.
As an example, for phenylketonuria, a genetic disease characterized by the inability of the liver to metabolize phenylalanine, resulting in elevated plasma levels that lead to neurotoxicity, a synthetic biology approach involving the colonization of the gut with Escherichia coli Nissle (SYNB1618) engineered to metabolize toxic levels of phenylalanine was demonstrated in mice and cynomolgus monkeys192. This approach is currently being tested in the clinic (NCT03516487). We envisage that patient-derived Structured-MPS and/or Organoid-MPS coupling the gut and the liver will facilitate the testing of novel strategies in a human system, expediting the optimization and clinical development of synthetic biologic agents such as SYNB1618 (REF.192) for other diseases in this class. Another example is the case of AATD, where mutations in the gene encoding α1-antitrypsin (AAT) result in its aggregation, impaired secretion, or function and in subsequent hepatocyte damage193. Approaches that reduce AAT expression through RNAi194 or promote its autophagic degradation with small molecules such as carbamazepine195 are in clinical trials (for example, NCT02363946 or NCT01379469). An alternative approach involves the development of small molecule chaperones (for example, VX-864; NCT04474197) that cannot only correct mutant AAT misfolding196,but also preserve its function as a protease regulator. Although the development of these ‘correctors’ has advantages, its mechanistic nuances require optimization in patient-relevant and even in patient-specific systems that recapitulate the critical aspects of the complex liver architecture and function, in both normal and AAT-deficient disease states. iPSC-derived hepatocytes from patients with AATD have been demonstrated to model personalized variations in liver disease197. In this regard, we anticipate an important role for patient-specific Structured-MPS and/or Organoid-MPS in facilitating preclinical development and informing optimal clinical trial design with more precise and timely predictions of disease susceptibility. Thus, the portability of Structured-MPS or Organoid-MPS models is expected to help address the logistical challenge of conducting preclinical and clinical studies across multiple centres, facilitating the execution and statistical analysis of mechanistic studies, especially for rare diseases198,199.
Conclusions
Various designs of human in vitro liver experimental models serve ‘fit-for-purpose’ applications in basic biomedical research and in the drug discovery and development pipeline to address specific ‘contexts of use’ such as the need for throughput of experimentation, content of information, functional biological complexity and/or the clinically relevant recapitulation of disease progression. HBL-MPS have evolved from simpler 2D and 3D models to recapitulate several key elements of the liver acinus structure and functions, including physical and biochemical environmental cues, to maximize the physiological or pathophysiological relevance required to understand the mechanisms of disease progression, identify the biomarkers linked to these mechanisms, and to predict drug response and ADMET. There are multiple designs of liver MPS to serve ‘fit-for-purpose’ applications but we have focused on HBL-MPS, which are designed to optimally recapitulate the liver acinus either as stand-alone liver models or coupled with other organs. Current HBL-MPS models have demonstrated the ability to explore the multicellular and temporal-spatial physiological and pathophysiological heterogeneity within the liver acinus using primary liver cells, sometimes with one or two well-characterized human cell lines. Important mechanisms of disease progression associated with NAFLD, type 2 diabetes and liver metastasis have been explored with these MPS models. The critical step and current challenge to fully realizing the potential power of HBL-MPS is the construction of the MPS with fully matured liver acinus cells derived from patient-specific iPSCs. The latter either undergo self-organization and differentiation as Organoid-MPS or are differentiated into distinct autologous liver cell types and then carefully positioned or bioprinted as Structured-MPS within microfluidic devices. Success here will enable the use of Organoid-MPS and/or Structured-MPS in applications for precision medicine. For example, preclinical trials using patient-specific MPS for parallel drug testing have the potential to predict clinical response, thereby enabling the optimized selection of patient cohorts for clinical trials by addressing heterogeneity in patient populations.
Although MPS made f rom multilineage patient-specific iPSC-derived cells are likely to have a major effect on the future of precision medicine, substantial hurdles remain. For example, the efficiency of lineage differentiation is still variable between donor lines or individual experiments and could lead to varying populations of multiple cell types that arise spontaneously. Nevertheless, given the rate of technical advances and the development of well-designed clinical validation studies, we believe that patient-specific MPS will become an integral component of translational research and precision medicine.
Supplementary Material
Absorption, distribution, metabolism, excretion and toxicity (ADMET).
Studies conducted during the drug discovery, lead optimization and preclinical development phases to provide information for characterization and ranking of compounds based on their properties and to predict their fate after administration into the human body.
Micropatterned cell arrays
Methodologies, often based on nanofabrication, to fix one or more cell types on a substrate with precisely controlled spatial distributions.
Spheroids
In vitro 3D spherical aggregates of cells of either a single cell type or a combination of cells generated by a variety of 3D culturing methods.
Organoids
3D multicellular systems produced primarily from patient-specific stem cells and their progenies via in situ differentiation, cell sorting and self-organization processes.
Plate-based platforms
Platforms designed around microplate standards from the Society of Biomolecular Sciences, available in 6–1,536-well formats.
Fit-for-purpose
A drug development tool that has been accepted for use in a specific application based on thorough evaluation of the information provided.
Synthetic biology
An interdisciplinary area of science focused on the (re) design and construction of biological systems in a bottom-up fashion, often through the engineering of well-characterized genetic components, modules and devices to attain new functions or to correct dysregulated ones.
Secretome
A set of proteins expressed by cells (organs) and secreted into the extracellular space, including cytokines, growth factors, extracellular matrix proteins mediating autocrine, paracrine, endocrine (via circulation) and/or exocrine (via ducts) physiological regulation or pathophysiological dysregulation.
Clearance
The collection of processes by which the body removes a drug, generally categorized as metabolism or elimination.
Pharmacokinetic models
Quantitative models that predict how an organism influences the absorption, distribution, metabolism and excretion of a drug.
Pharmacodynamic models
A quantitative integration of pharmacokinetics, pharmacological systems and (patho-) physiological processes to understand the intensity and time course of drug effects on the body.
Key points.
Liver in vitro experimental models have a long history involving the use of 2D and 3D models that continue to have valuable roles in our understanding of liver physiology and pathophysiology.
Human microphysiology systems (MPS) have evolved from simple cell-based experimental models and have the potential to meet the need for human experimental models for basic biomedical research and the development of therapeutics.
Human biomimetic liver MPS (HBL-MPS) aim to improve the efficiency of developing biomarkers, repurposed drugs and novel therapeutics by maximally recapitulating the structure and functions of the liver acinus.
HBL-MPS are evolving based either on liver organoids derived from patient cells that self-assemble and differentiate or on the directed assembly or bioprinting of matrix materials and cells into microfluidic devices.
Organoid-derived MPS and structured MPS are next-generation HBL-MPS that are projected to enable applications of precision medicine, including preclinical trials, either as stand-alone liver models or as coupled, multi-organ MPS.
Footnotes
Competing interests
A.S.-G. is co-founder and D.L.T. is an adviser for Von Baer Wolff Inc., a company focused on biofabrication of autologous human hepatocytes using stem cell technology and genetic reprogramming to overcome liver failure. Their interests are managed by the Conflict of Interest Office at the University of Pittsburgh, USA, in accordance with their policies. The other authors declare no competing interests.
Peer review information
Nature Reviews Gastroenterology & Hepatology thanks J. Hickman, Y. Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41575-020-00386-1.
Related links
U.s. National Library of Medicine ClinicalTrials.gov: https://clinicaltrials.gov/
References
- 1.Alex A, Harris CJ, Keighley WW & Smith DA In Attrition in the Pharmaceutical Industry: Reasons, Implications, and Pathways Forward (eds Alex A, Harris CJ & Smith DA) 106–127 (Wiley, 2015). [Google Scholar]
- 2.Arrowsmith J & Miller P Trial watch: phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov 12, 569 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL, Neuschwander-Tetri BA, Rinella M & Sanyal AJ Mechanisms of NAFLD development and therapeutic strategies. Nat. Med 24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gribkoff VK & Kaczmarek LK The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 120, 11–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jardim DL, Groves ES, Breitfeld PP & Kurzrock R Factors associated with failure of oncology drugs in late-stage clinical development: a systematic review. Cancer Treat. Rev 52, 12–21 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Parasrampuria DA, Benet LZ & Sharma A Why drugs fail in late stages of development: case study analyses from the last decade and recommendations. AAPS J 20, 46 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Sanyal AJ Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 16, 377–386 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Taylor DL et al. Harnessing human microphysiology systems as key experimental models for quantitative systems pharmacology. Handb. Exp. Pharmacol 260, 327–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eslam M & George J Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol 17, 40–52 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Mullard A FDA rejects NASH drug. Nat. Rev. Drug Discov 19, 501 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Roussel R, Steg PG, Mohammedi K, Marre M & Potier L Prevention of cardiovascular disease through reduction of glycaemic exposure in type 2 diabetes: a perspective on glucose-lowering interventions. Diabetes Obes. Metab 20, 238–244 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Menon V et al. Fasiglifam-induced liver injury in patients with type 2 diabetes: results of a randomized controlled cardiovascular outcomes safety trial. Diabetes Care 41, 2603–2609 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Mosedale M & Watkins PB Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin. Pharmacol. Ther 101, 469–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartung T A toxicology for the 21st century - mapping the road ahead. Toxicol. Sci 109, 18–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sultana J, Cutroneo P & Trifiro G Clinical and economic burden of adverse drug reactions. J. Pharmacol. Pharmacother 4, S73–S77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernal W Acute liver failure: review and update. Int. Anesthesiol. Clin 55, 92–106 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Vernetti L et al. in Drug Efficacy, Safety, and Biologics Discovery: Emerging Technologies and Tools (eds Ekins S & Xu JJ) Ch. 3, 53–73 (Wiley & Sons, 2009). [Google Scholar]
- 18.Moore TJ, Zhang H, Anderson G & Alexander GC Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015–2016. JAMA Intern. Med 178, 1451–1457 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wouters OJ, McKee M & Luyten J Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 323, 844–853 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stern AM, Schurdak ME, Bahar I, Berg JM & Taylor DL A perspective on implementing a quantitative systems pharmacology platform for drug discovery and the advancement of personalized medicine. J. Biomol. Screen 21, 521–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Low LA & Tagle DA Tissue chips - innovative tools for drug development and disease modeling. Lab Chip 17, 3026–3036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watson DE, Hunziker R & Wikswo JP Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp. Biol. Med 242, 1559–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewart L et al. Navigating tissue chips from development to dissemination: a pharmaceutical industry perspective. Exp. Biol. Med 242, 1579–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatia SN & Ingber DE Microfluidic organs-on-chips. Nat. Biotechnol 32, 760–772 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ronaldson-Bouchard K & Vunjak-Novakovic G Organs-on-a-chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22, 310–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Low LA, Mummery C, Berridge BR, Austin CP & Tagle DA Organs-on-chips: into the next decade. Nat. Rev. Drug Discov 10.1038/s41573-020-0079-3 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Tagle DA The NIH microphysiological systems program: developing in vitro tools for safety and efficacy in drug development. Curr. Opin. Pharmacol 48, 146–154 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Sorger PK et al. Quantitative and Systems Pharmacology in the Post-genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms (NIH, 2011). [Google Scholar]
- 29.Isoherranen N, Madabushi R & Huang S-M Emerging role of organ-on-a-chip technologies in quantitative clinical pharmacology evaluation. Clin. Transl. Sci 12, 113–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.European Medicines Agency. Meeting Report: First EMA workshop on non-animal approaches in support of medicinal product development – challenges and opportunities for use of micro-physiological systems (EMA/CHMP/SWP/250438/2018) (European Medicines Agency, 2018). [Google Scholar]
- 31.Fang Y & Eglen RM Three-dimensional cell cultures in drug discovery and development. SLAS Discov 22, 456–472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shamir ER & Ewald AJ Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat. Rev. Mol. Cell Biol 15, 647–664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simian M & Bissell MJ Organoids: a historical perspective of thinking in three dimensions. J. Cell Biol 216, 31–40 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhushan A et al. Towards a three-dimensional microfluidic liver platform for predicting drug efficacy and toxicity in humans. Stem Cell Res. Ther 4 (Suppl. 1), S16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma LD et al. Design and fabrication of a liver-on-a-chip platform for convenient, highly efficient, and safe in situ perfusion culture of 3D hepatic spheroids. Lab Chip 18, 2547–2562 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Underhill GH & Khetani SR Advances in engineered human liver platforms for drug metabolism studies. Drug Metab. Dispos 46, 1626–1637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster AJ et al. Integrated in vitro models for hepatic safety and metabolism: evaluation of a human liverchip and liver spheroid. Arch. Toxicol 10.1007/s00204-019-02427-4 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Proctor WR et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol 91, 2849–2863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y et al. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 18, 3606–3616 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Takebe T et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Guye P et al. Genetically engineering self-organization of human pluripotent stem cells into a liver bud-like tissue using Gata6. Nat. Commun 7, 10243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takebe T, Zhang B & Radisic M Synergistic engineering: organoids meet organs-on-a-chip. Cell Stem Cell 21, 297–300 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Ho BX, Pek NMQ & Soh BS Disease modeling using 3D organoids derived from human induced pluripotent stem cells. Int. J. Mol. Sci 19 10.3390/ijms19040936 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fatehullah A, Tan SH & Barker N Organoids as an in vitro model of human development and disease. Nat. Cell Biol 18, 246–254 (2016). [DOI] [PubMed] [Google Scholar]
- 45.May S, Evans S & Parry L Organoids, organs-on-chips and other systems, and microbiota. Emerg. Top. Life Sci 1, 385–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouchi R et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab 30, 374–384.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lancaster MA & Huch M Disease modelling in human organoids. Dis. Model. Mech 12, dmm039347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prior N, Inacio P & Huch M Liver organoids: from basic research to therapeutic applications. Gut 68, 2228–2237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prestigiacomo V, Weston A, Messner S, Lampart F & Suter-Dick L Pro-fibrotic compounds induce stellate cell activation, ECM-remodelling and Nrf2 activation in a human 3D-multicellular model of liver fibrosis. PLoS ONE 12, e0179995 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang M, Neuzil P, Volk T, Manz A & Kleber A On-chip three-dimensional cell culture in phaseguides improves hepatocyte functions in vitro. Biomicrofluidics 9, 034113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khetani SR & Bhatia SN Microscale culture of human liver cells for drug development. Nat. Biotechnol 26, 120–126 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Davidson MD, Lehrer M & Khetani SR Hormone and drug-mediated modulation of glucose metabolism in a microscale model of the human liver. Tissue Eng. Part C Methods 21, 716–725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidson MD, Kukla DA & Khetani SR Microengineered cultures containing human hepatic stellate cells and hepatocytes for drug development. Integr. Biol 9, 662–677 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Berger DR, Ware BR, Davidson MD, Allsup SR & Khetani SR Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell-cell interactions in vitro. Hepatology 61, 1370–1381 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Avila AM et al. An FDA/CDER perspective on nonclinical testing strategies: classical toxicology approaches and new approach methodologies (NAMs). Regul. Toxicol. Pharmacol 114, 104662 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Li X, George SM, Vernetti L, Gough AH & Taylor DL A glass-based, continuously zonated and vascularized human liver acinus microphysiological system (vLAMPS) designed for experimental modeling of diseases and ADME/TOX. Lab Chip 18, 2614–2631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahadian S et al. Organ-on-a-chip platforms: a convergence of advanced materials, cells, and microscale technologies. Adv. Healthc. Mater 10.1002/adhm.201700506 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Soto-Gutierrez A, Gough A, Vernetti LA, Taylor DL & Monga SP Pre-clinical and clinical investigations of metabolic zonation in liver diseases: the potential of microphysiology systems. Exp. Biol. Med 242, 1605–1616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee-Montiel FT et al. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med 242, 1617–1632 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bin Ramli MN et al. Human pluripotent stem cell-derived organoids as models of liver disease. Gastroenterology 159, 1471–1486.e12 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Sharma A, Sances S, Workman MJ & Svendsen CN Multi-lineage human iPSC-derived platforms for disease modeling and drug discovery. Cell Stem Cell 26, 309–329 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vernetti LA et al. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp. Biol. Med 241, 101–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McAleer CW et al. On the potential of in vitro organchip models to define temporal pharmacokinetic-pharmacodynamic relationships. Sci. Rep 9, 9619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kizawa H, Nagao E, Shimamura M, Zhang G & Torii H Scaffold-free 3D bio-printed human liver tissue stably maintains metabolic functions useful for drug discovery. Biochem. Biophys. Rep 10, 186–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feaver RE et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 1, e90954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kostrzewski T et al. A microphysiological system for studying nonalcoholic steatohepatitis. Hepatol. Commun 4, 77–91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huch M, Knoblich JA, Lutolf MP & Martinez-Arias A The hope and the hype of organoid research. Development 144, 938–941 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Huch M et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saito Y et al. Establishment of patient-derived organoids and drug screening for biliary tract carcinoma. Cell Rep 27, 1265–1276.e1264 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Guan Y et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 10.1172/jci.insight.94954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Broutier L et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Velazquez JJ et al. Synthetic maturation of multilineage human liver organoids via genetically guided engineering. bioRxiv 10.1101/2020.05.10.087445 (2020). [DOI] [Google Scholar]
- 73.Koike H et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature 574, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kostrzewski T et al. Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World J. Gastroenterol 23, 204–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang KJ et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med 10.1126/scitranslmed.aax5516 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Beckwitt CH et al. Liver ‘organ on a chip’. Exp. Cell. Res 363, 15–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Senutovitch N et al. Fluorescent protein biosensors applied to microphysiological systems. Exp. Biol. Med 240, 795–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Auner AW, Tasneem KM, Markov DA, McCawley LJ & Hutson MS Chemical-PDMS binding kinetics and implications for bioavailability in microfluidic devices. Lab Chip 19, 864–874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan K et al. A high-throughput microfluidic microphysiological system (PREDICT-96) to recapitulate hepatocyte function in dynamic, re-circulating flow conditions. Lab Chip 19, 1556–1566 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Wikswo JP et al. Engineering challenges for instrumenting and controlling integrated organ-on-chip systems. IEEE Trans. Biomed. Eng 60, 682–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bavli D et al. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl Acad. Sci. USA 113, E2231–E2240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Esch MB, Ueno H, Applegate DR & Shuler ML Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab Chip 16, 2719–2729 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Miller PG & Shuler ML Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol. Bioeng 113, 2213–2227 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Oleaga C et al. Multi-organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci. Rep 6, 20030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang YI, Carmona C, Hickman JJ & Shuler ML Multiorgan microphysiological systems for drug development: strategies, advances, and challenges. Adv. Healthc. Mater 7, 10.1002/adhm.201701000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Y, Wang L, Guo Y, Zhu Y & Qin J Engineering stem cell-derived 3D brain organoids in a perfusable organ-on-a-chip system. RSC Adv 8, 1677–1685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park SE, Georgescu A & Huh D Organoids-on-a-chip. Science 364, 960–965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang YS et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl Acad. Sci. USA 114, E2293–E2302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ehrlich A, Duche D, Ouedraogo G & Nahmias Y Challenges and opportunities in the design of liver-on-chip microdevices. Annu. Rev. Biomed. Eng 21, 219–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ehrlich A et al. Microphysiological flux balance platform unravels the dynamics of drug induced steatosis. Lab Chip 18, 2510–2522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kilic T, Navaee F, Stradolini F, Renaud P & Carrara S Organs-on-chip monitoring: sensors and other strategies. Microphysiol. Syst 10.21037/mps.2018.01.01 (2018). [DOI] [Google Scholar]
- 92.Sung JH Pharmacokinetic-based multi-organ chip for recapitulating organ interactions. Methods Cell. Biol 146, 183–197 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Sung JH et al. Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med 239, 1225–1239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vernetti LA, Vogt A, Gough A & Taylor DL Evolution of experimental models of the liver to predict human drug hepatotoxicity and efficacy. Clin. Liver Dis 21, 197–214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ewart L et al. Application of microphysiological systems to enhance safety assessment in drug discovery. Annu. Rev. Pharmacol. Toxicol 58, 65–82 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Wang X, Cirit M, Wishnok JS, Griffith LG & Tannenbaum SR Analysis of an integrated human multiorgan microphysiological system for combined tolcapone metabolism and brain metabolomics. Anal. Chem 91, 8667–8675 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Herland A et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng 10.1038/s41551-019-0498-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vernetti L et al. Functional coupling of human microphysiology systems: intestine, liver, kidney proximal tubule, blood-brain barrier and skeletal muscle. Sci. Rep 7, 42296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wikswo JP et al. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip 13, 3496–3511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koui Y et al. An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Rep 9, 490–498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Prendergast ME et al. Microphysiological systems: automated fabrication via extrusion bioprinting. Microphysiol. Syst 10.21037/MPS.2018.03.01 (2018). [DOI] [Google Scholar]
- 102.Velazquez JJ, Su E, Cahan P & Ebrahimkhani MR Programming morphogenesis through systems and synthetic biology. Trends Biotechnol 36, 415–429 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ebrahimkhani MR & Ebisuya M Synthetic developmental biology: build and control multicellular systems. Curr. Opin. Chem. Biol 52, 9–15 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Johnson MB, March AR & Morsut L Engineering multicellular systems: using synthetic biology to control tissue self-organization. Curr. Opin. Biomed. Eng 4, 163–173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Colnot S & Perret C in Molecular Pathology of Liver Diseases (ed. Satdarshan S & Monga P) 7–16 (Springer, 2011). [Google Scholar]
- 106.Marx U et al. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. ALTEX 33, 272–321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baudy AR et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225 (2020). [DOI] [PubMed] [Google Scholar]
- 108.Hughes DJ, Kostrzewski T & Sceats EL Opportunities and challenges in the wider adoption of liver and interconnected microphysiological systems. Exp. Biol. Med 242, 1593–1604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wheeler SE et al. Spontaneous dormancy of metastatic breast cancer cells in an all human liver microphysiologic system. Br. J. Cancer 111, 2342–2350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sundaram V & Björnsson ES Drug-induced cholestasis. Hepatol. Commun 1, 726–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sampaziotis F et al. Directed differentiation of human induced pluripotent stem cells into functional cholangiocyte-like cells. Nat. Protoc 12, 814–827 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sampaziotis F et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat. Med 23, 954–963 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Sampaziotis F Building better bile ducts. Science 359, 1113 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Du Y et al. A bile duct-on-a-chip with organ-level functions. Hepatology 71, 1350–1363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leclerc E et al. Comparison of the transcriptomic profile of hepatic human induced pluripotent stem like cells cultured in plates and in a 3D microscale dynamic environment. Genomics 109, 16–26 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Grant MH et al. Human adult hepatocytes in primary monolayer culture. Maintenance of mixed function oxidase and conjugation pathways of drug metabolism. Biochem. Pharmacol 36, 2311–2316 (1987). [DOI] [PubMed] [Google Scholar]
- 117.Guzelian PS, Bissell DM & Meyer UA Drug metabolism in adult rat hepatocytes in primary monolayer culture. Gastroenterology 72, 1232–1239 (1977). [PubMed] [Google Scholar]
- 118.Long TJ et al. Modeling therapeutic antibody-small molecule drug-drug interactions using a three-dimensional perfusable human liver coculture platform. Drug Metab. Dispos 44, 1940–1948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tsamandouras N et al. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. J. Pharmacol. Exp. Ther 360, 95–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cirit M & Stokes CL Maximizing the impact of microphysiological systems with in vitro-in vivo translation. Lab Chip 18, 1831–1837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shen JX, Youhanna S, Shafagh RZ, Kele J & Lauschke VM Organotypic and microphysiological models of liver, gut and kidney for studies of drug metabolism, pharmacokinetics and toxicity. Chem. Res. Toxicol 33, 38–60 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Truskey GA Human microphysiological systems and organoids as in vitro models for toxicological studies. Front. Public. Health 6, 185–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou Y, Shen JX & Lauschke VM Comprehensive evaluation of organotypic and microphysiological liver models for prediction of drug-induced liver injury. Front. Pharmacol 10, 1093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Terelius Y et al. Transcriptional profiling suggests that Nevirapine and Ritonavir cause drug induced liver injury through distinct mechanisms in primary human hepatocytes. Chem. Biol. Interact 255, 31–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schurdak M et al. Applications of the microphysiology systems database for experimental ADME-Tox and disease models. Lab Chip 20, 1472–1492 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gough A, Vernetti L, Bergenthal L, Shun TY & Taylor DL The microphysiology systems database for analyzing and modeling compound interactions with human and animal organ models. Appl. In Vitro Toxicol 2, 103–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Choudhury Y et al. Patient-specific hepatocyte-like cells derived from induced pluripotent stem cells model pazopanib-mediated hepatotoxicity. Sci. Rep 7, 41238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Koido M et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nat. Med 10.1038/s41591-020-1023-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Adams DH, Ju C, Ramaiah SK, Uetrecht J & Jaeschke H Mechanisms of immune-mediated liver injury. Toxicol. Sci 115, 307–321 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ahn J et al. Human three-dimensional in vitro model of hepatic zonation to predict zonal hepatotoxicity. J. Biol. Eng 13, 22–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tonon F et al. In vitro metabolic zonation through oxygen gradient on a chip. Sci. Rep 9, 13557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Halpern KB et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Prill S et al. Real-time monitoring of oxygen uptake in hepatic bioreactor shows CYP450-independent mitochondrial toxicity of acetaminophen and amiodarone. Arch. Toxicol 90, 1181–1191 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Roden M & Shulman GI The integrative biology of type 2 diabetes. Nature 576, 51–60 (2019). [DOI] [PubMed] [Google Scholar]
- 135.Hardy T, Oakley F, Anstee QM & Day CP Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu. Rev. Pathol 11, 451–496 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Eslam M, Sanyal AJ, George J & International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology, 158, 1999–2014 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Pennisi G et al. Pharmacological therapy of non-alcoholic fatty liver disease: what drugs are available now and future perspectives. Int. J. Environ. Res. Public Health 16, 10.3390/ijerph16224334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Danford CJ, Yao Z-M & Jiang ZG Non-alcoholic fatty liver disease: a narrative review of genetics. J. Biomed. Res 32, 389–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sato K et al. Intercellular communication between hepatic cells in liver diseases. Int. J. Mol. Sci 20 10.3390/ijms20092180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Boeckmans J et al. Human-based systems: mechanistic NASH modelling just around the corner? Pharmacol. Res 134, 257–267 (2018). [DOI] [PubMed] [Google Scholar]
- 141.Anstee QM, Reeves HL, Kotsiliti E, Govaere O & Heikenwalder M From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol 16, 411–428 (2019). [DOI] [PubMed] [Google Scholar]
- 142.Mannaa FA & Abdel-Wahhab KG Physiological potential of cytokines and liver damages. Hepatoma Res 2, 131–143 (2016). [Google Scholar]
- 143.Tacke F Targeting hepatic macrophages to treat liver diseases. J. Hepatol 66, 1300–1312 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Musso G, Cassader M & Gambino R Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov 15, 249–274 (2016). [DOI] [PubMed] [Google Scholar]
- 145.Ezhilarasan D, Sokal E & Najimi M Hepatic fibrosis: it is time to go with hepatic stellate cell-specific therapeutic targets. Hepatobiliary Pancreat. Dis. Int 17, 192–197 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Higashi T, Friedman SL & Hoshida Y Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev 121, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rao SS, Kondapaneni RV & Narkhede AA Bioengineered models to study tumor dormancy. J. Biol. Eng 13, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee JW et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 567, 249–252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wells A, Clark A, Bradshaw A, Ma B & Edington H The great escape: how metastases of melanoma, and other carcinomas, avoid elimination. Exp. Biol. Med 243, 1245–1255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Miedel MT et al. Modeling the effect of the metastatic microenvironment on phenotypes conferred by estrogen receptor mutations using a human liver microphysiological system. Sci. Rep 9, 8341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jia S et al. Clinically observed estrogen receptor alpha mutations within the ligand-binding domain confer distinguishable phenotypes. Oncology 94, 176–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Scherz-Shouval R et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158, 564–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Clark AM et al. A microphysiological system model of therapy for liver micrometastases. Exp. Biol. Med 239, 1170–1179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Clark AM et al. A model of dormant-emergent metastatic breast cancer progression enabling exploration of biomarker signatures. Mol. Cell. Proteomics 17, 619–630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Beckwitt CH et al. Statins attenuate outgrowth of breast cancer metastases. Br. J. Cancer 119, 1094–1105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhao Y, Kankala RK, Wang SB & Chen AZ Multi-organs-on-chips: towards long-term biomedical investigations. Molecules 10.3390/molecules24040675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sung JH et al. Recent advances in body-on-a-chip systems. Anal. Chem 91, 330–351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McAleer CW et al. Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. Sci. Transl. Med 11, eaav1386 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Vunjak-Novakovic G, Bhatia S, Chen C & Hirschi K HeLiVa platform: integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther 4 (Suppl. 1), S8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bricks T et al. Development of a new microfluidic platform integrating co-cultures of intestinal and liver cell lines. Toxicol. In Vitro 28, 885–895 (2014). [DOI] [PubMed] [Google Scholar]
- 161.Bricks T et al. Investigation of omeprazole and phenacetin first-pass metabolism in humans using a microscale bioreactor and pharmacokinetic models. Biopharm. Drug Dispos 36, 275–293 (2015). [DOI] [PubMed] [Google Scholar]
- 162.Maschmeyer I et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 15, 2688–2699 (2015). [DOI] [PubMed] [Google Scholar]
- 163.Bauer S et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: Towards a novel human ex vivo type 2 diabetes model. Sci. Rep 7, 14620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tsamandouras N et al. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J 10.1208/s12248-017-0122-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Edington CD et al. Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep 8, 4530–4530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tilg H, Moschen AR & Roden M NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol 14, 32–42 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Ferreira CR, Cassiman D & Blau N Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases. Mol. Genet. Metab 127, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Younossi ZM, Marchesini G, Pinto-Cortez H & Petta S Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: implications for liver transplantation. Transplantation 103, 22–27 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Younossi ZM, Henry L, Bush H & Mishra A Clinical and economic burden of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Clin. Liver Dis 22, 1–10 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Goldberg D et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology 152, 1090–1099.e1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mikolasevic I et al. Nonalcoholic fatty liver disease and liver transplantation - where do we stand? World J. Gastroenterol 24, 1491–1506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mittal S et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol 14, 124–131.e1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Singh S et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol 13, 643–654.e1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McPherson S et al. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J. Hepatol 62, 1148–1155 (2015). [DOI] [PubMed] [Google Scholar]
- 175.Hoang SA et al. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci. Rep 9, 12541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Younossi ZM et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 68, 361–371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sumida Y & Yoneda M Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol 53, 362–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Polyzos SA, Kountouras J, Anastasiadis S, Doulberis M & Katsinelos P Nonalcoholic fatty liver disease: Is it time for combination treatment and a diabetes-like approach? Hepatology 68, 389 (2018). [DOI] [PubMed] [Google Scholar]
- 179.Dyson J et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol 60, 110–117 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Piscaglia F et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 63, 827–838 (2016). [DOI] [PubMed] [Google Scholar]
- 181.Wong VW, Adams LA, de Lédinghen V, Wong GL & Sookoian S Noninvasive biomarkers in NAFLD and NASH - current progress and future promise. Nat. Rev. Gastroenterol. Hepatol 15, 461–478 (2018). [DOI] [PubMed] [Google Scholar]
- 182.Greene CM et al. α1-antitrypsin deficiency. Nat. Rev. Dis. Primers 2, 16051 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Hazari YM et al. Alpha-1-antitrypsin deficiency: genetic variations, clinical manifestations and therapeutic interventions. Mutat. Res 773, 14–25 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Moat SJ et al. Performance of laboratory tests used to measure blood phenylalanine for the monitoring of patients with phenylketonuria. J. Inherit. Metab. Dis 43, 179–188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Blau N Genetics of phenylketonuria: then and now. Hum. Mutat 37, 508–515 (2016). [DOI] [PubMed] [Google Scholar]
- 186.Bergenthal LM, Shun TY, Vernetti L, Taylor DL & Gough AH The Microphysiology Systems Database http://mps.csb.pitt.edu (2018). [Google Scholar]
- 187.Shi Y, Inoue H, Wu JC & Yamanaka S Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov 16, 115–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Takahashi K & Yamanaka S A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol 17, 183–193 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Pei F et al. Connecting neuronal cell protective pathways and drug combinations in a Huntington’s disease model through the application of quantitative systems pharmacology. Sci. Rep 7, 17803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chow SC & Chang M Adaptive design methods in clinical trials - a review. Orphanet J. Rare Dis 3, 11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fagiuoli S, Daina E, D’Antiga L, Colledan M & Remuzzi G Monogenic diseases that can be cured by liver transplantation. J. Hepatol 59, 595–612 (2013). [DOI] [PubMed] [Google Scholar]
- 192.Isabella VM et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol 36, 857–864 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Ghouse R, Chu A, Wang Y & Perlmutter DH Mysteries of alpha1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease. Dis. Model. Mech 7, 411–419 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Turner AM et al. Hepatic-targeted RNA interference provides robust and persistent knockdown of alpha-1 antitrypsin levels in ZZ patients. J. Hepatol 69, 378–384 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Hidvegi T et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229–232 (2010). [DOI] [PubMed] [Google Scholar]
- 196.Bouchecareilh M, Conkright JJ & Balch WE Proteostasis strategies for restoring alpha1-antitrypsin deficiency. Proc. Am. Thorac. Soc 7, 415–422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tafaleng EN et al. Induced pluripotent stem cells model personalized variations in liver disease resulting from α1-antitrypsin deficiency. Hepatology 62, 147–157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Low LA & Tagle DA Microphysiological systems (tissue chips) and their utility for rare disease research. Adv. Exp. Med. Biol 1031, 405–415 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Blumenrath SH, Lee BY, Low L, Prithviraj R & Tagle D Tackling rare diseases: clinical trials on chips. Exp. Biol. Med 245, 1155–1162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pan G Roles of hepatic drug transporters in drug disposition and liver toxicity. Adv. Exp. Med. Biol 1141, 293–340 (2019). [DOI] [PubMed] [Google Scholar]
- 201.Nguyen DG et al. Bioprinted 3D primary liver tissues allow assessment of organ-level response to clinical drug induced toxicity in vitro. PLoS ONE 11, e0158674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Norona LM, Nguyen DG, Gerber DA, Presnell SC & LeCluyse EL Editor’s highlight: modeling compound-induced fibrogenesis in vitro using three-dimensional bioprinted human liver tissues. Toxicol. Sci 154, 354–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Norona LM et al. Bioprinted liver provides early insight into the role of Kupffer cells in TGF-β1 and methotrexate-induced fibrogenesis. PLoS ONE 14, e0208958 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Trietsch SJ, Israels GD, Joore J, Hankemeier T & Vulto P Microfluidic titer plate for stratified 3D cell culture. Lab Chip 13, 3548–3554 (2013). [DOI] [PubMed] [Google Scholar]
- 205.Domansky K et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 10, 51–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Novik E, Maguire TJ, Chao P, Cheng KC & Yarmush ML A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochem. Pharmacol 79, 1036–1044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chao P, Maguire T, Novik E, Cheng KC & Yarmush ML Evaluation of a microfluidic based cell culture platform with primary human hepatocytes for the prediction of hepatic clearance in human. Biochem. Pharmacol 78, 625–632 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Dash A et al. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am. J. Physiol. Cell Physiol 304, C1053–C1063 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Boeri L et al. Advanced organ-on-a-chip devices to investigate liver multi-organ communication: focus on gut, microbiota and brain. Bioengineering 6, 91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Natarajan V, Berglund EJ, Chen DX & Kidambi S Substrate stiffness regulates primary hepatocyte functions. RSC Adv 5, 80956–80966 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Newman RH & Zhang J The design and application of genetically encodable biosensors based on fluorescent proteins. Methods Mol. Biol 1071, 1–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Collin de l’Hortet A et al. Generation of human fatty livers using custom-engineered induced pluripotent stem cells with modifiable SIRT1 metabolism. Cell Metab 30, 385–401.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Toepke MW & Beebe DJ PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 6, 1484–1486 (2006). [DOI] [PubMed] [Google Scholar]
- 214.Regehr KJ et al. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 9, 2132–2139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Donato MT & Tolosa L Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 106, 15–22 (2019). [DOI] [PubMed] [Google Scholar]
- 216.Rezvani M, Grimm AA & Willenbring H Assessing the therapeutic potential of lab-made hepatocytes. Hepatology 64, 287–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tasnim F et al. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 192, 377–391 (2019). [DOI] [PubMed] [Google Scholar]
- 218.Coll M et al. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell Stem Cell 23, 101–113.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 219.Parent R et al. An immortalized human liver endothelial sinusoidal cell line for the study of the pathobiology of the liver endothelium. Biochem. Biophys. Res. Commun 450, 7–12 (2014). [DOI] [PubMed] [Google Scholar]
- 220.Matsumura T et al. Establishment of an immortalized human-liver endothelial cell line with SV40T and hTERT. Transplantation 77, 1357–1365 (2004). [DOI] [PubMed] [Google Scholar]
- 221.Maruyama M et al. Establishment of a highly differentiated immortalized human cholangiocyte cell line with SV40T and hTERT. Transplantation 77, 446–451 (2004). [DOI] [PubMed] [Google Scholar]
- 222.Tabibian JH et al. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab. Invest 94, 1126–1133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sampaziotis F et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol 33, 845–852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ghanekar A & Kamath BM Cholangiocytes derived from induced pluripotent stem cells for disease modeling. Curr. Opin. Gastroenterol 32, 210–215 (2016). [DOI] [PubMed] [Google Scholar]
- 225.Poisson J et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J. Hepatol 66, 212–227 (2017). [DOI] [PubMed] [Google Scholar]
- 226.Hammoutene A & Rautou PE Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol 70, 1278–1291 (2019). [DOI] [PubMed] [Google Scholar]
- 227.DeLeve LD & Maretti-Mira AC Liver sinusoidal endothelial cell: an update. Semin. Liver Dis 37, 377–387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jang S, Collin de l’Hortet A & Soto-Gutierrez A Induced pluripotent stem cell-derived endothelial cells: overview, current advances, applications, and future directions. Am. J. Pathol 189, 502–512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Li J, Zhao YR & Tian Z Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J. Hepatol 11, 412–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Grunhut J et al. Macrophages in nonalcoholic steatohepatitis: friend or foe? Eur. Med. J. Hepatol 6, 100–109 (2018). [PMC free article] [PubMed] [Google Scholar]
- 231.Nishimura T & Nakauchi H Generation of antigen-specific T cells from human induced pluripotent stem cells. Methods Mol. Biol 1899, 25–40 (2019). [DOI] [PubMed] [Google Scholar]
- 232.Wang G et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med 20, 616–623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Brown JA et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J. Neuroinflammation 13, 306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jenkins RW et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov 8, 196–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Peck RW, Hinojosa CD & Hamilton GA Organs-on-chips in clinical pharmacology: putting the patient into the center of treatment selection and drug development. Clin. Pharmacol. Ther 107, 181–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yi HG et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng 3, 509–519 (2019). [DOI] [PubMed] [Google Scholar]
- 237.Aref AR et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shirure VS et al. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 18, 3687–3702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Miller CP, Shin W, Ahn EH, Kim HJ & Kim DH Engineering microphysiological immune system responses on chips. Trends Biotechnol 38, 857–872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chen WLK et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng 114, 2648–2659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li G, Huang K, Nikolic D & van Breemen RB High-throughput cytochrome P450 cocktail inhibition assay for assessing drug-drug and drug-botanical interactions. Drug Metab. Dispos 43, 1670–1678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Siramshetty VB et al. WITHDRAWN–a resource for withdrawn and discontinued drugs. Nucleic Acids Res 44, D1080–D1086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wikswo JP et al. Integrated human organ-on-chip microphysiolocal systems US patent US 2015/0004077 A1 (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.