

Abstract
Abstract
The 5-membered oxadiazole and thiadiazole scaffolds are the most privileged and well-known heterocycles, being a common and essential feature of a variety of natural products and medicinal agents. These scaffolds take up the center position and are the core structural components of numerous drugs that belong to different categories. These include antimicrobial, anti-tubercular, anti-inflammatory, analgesic, antiepileptic, antiviral, and anticancer agents. In this review, we mostly talk about the isomers 1,2,4-oxadiazole and 1,3,4-thiadiazole because they have important pharmacological properties. This is partly because they are chemical and heat resistant, unlike other isomers, and they can be used as bio-isosteric replacements in drug design. We are reviewing the structural modifications of different oxadiazole and thiadiazole derivatives, more specifically, the anti-tubercular and anticancer pharmacological activities reported over the last 5 years, as we have undertaken this as a core area of research. This review article desires to do a thorough study and analysis of the recent progress made in the important biological isomers 1,2,4-oxadiazole and 1,3,4-thiadiazol. This will be a great place to start for future research.
Key points
• Five-membered heterocyclic compound chemistry and biological activity recent survey.
• Synthesis and pharmacological evolution of 1,2,4-oxadiazole and 1,3,4-thiadiazole are discussed in detail.
• The value and significance of heterocyclic compounds in the field of drug designing are highlighted.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00253-022-11969-0.
Keywords: Anticancer, Anti-tubercular, Heterocycles, Oxadiazole, Pharmacological activities, Thiadiazole
Introduction
It ought to be the top priority of researchers to identify and develop new and better pharmaceuticals, pesticides, and insecticides, and to do so, they follow natural models. The pharmaceutical products with biological activity that resemble natural products are called heterocycles (Sompalle and Roopan 2017). Heterocyclic compounds are cyclic compounds in which another atom replaces one or more carbons in the ring (Ram et al. 2019). Thus, the compounds comprising heteroatoms like nitrogen, oxygen, sulfur, phosphorous, silicon, boron, and the like make the heterocyclic compounds one of the most prevalent classes of organic compounds (Roopan and Palaniraja 2016). They are persistent in biology and do play an integral function in medicinal chemistry, particularly in drug design and synthesis. In nature, heterocycles are broadly distributed; many of them exist in the flora regarded as alkaloids, and a few of them are used in historical instances as medicinal agents (Paulo et al. 2018; Cascioferro et al. 2019).
Importance of heterocyclic compounds in medicinal chemistry
Heterocyclic compounds possess a broad spectrum of pharmacological activities, and for this reason, it continues to yield new therapeutic agents. The biological activity exhibited by the heterocycles is owed to their potential to bind with various enzymes either to the active sites or with enzyme pocket structures through a broad range of intra-molecular interactions such as van der Waals and hydrophobic forces, hydrogen bonding, and metallic coordination bonds, making them an important scaffold in medicinal chemistry (Pearce 2017). A massive variety of naturally occurring substances, such as hemoglobin, chlorophyll, pyrimidines, and purine bases and enzyme co-factors, belong to the family of heterocycles, which are integral to dwelling cells (Arunachalapandi and Roopan 2021; Sompalle and Roopan 2016). They are almost critical at every step of many biochemical processes and processes that are important to life. Heterocyclic compounds, especially those containing nitrogen, sulfur, and oxygen heteroatoms, are the most important class of compounds in the pharmaceutical and agrochemical industries, in which heterocycles comprise around 60% of the drug substances. Five-membered nitrogen- and oxygen- or sulfur-containing heterocycles such as oxazolidine, isoxazolidine, oxazole, isoxazole, thiazolidine, isothiazolidine, thiazole, isothiazole, oxadiazole, and thiadiazole are important structural motifs and are present in a vast number of biologically active compounds. These heterocycles make up the core structure of numerous drugs and are therefore of great interest in the pharmaceutical industry (Manjupriya and Roopan 2021; Li et al. 2013).
The following few sections cover the review of the 5-membered oxadiazole and thiadiazole heterocycles as their derivatives continue to gain interest from researchers, being used as bio-isosteric replacements in drug designing and widely studied for their utility in agrochemical and material science (Sauer et al. 2019): Kumari et al. 2020). The main objective behind this review article was not to cover a broad spectrum of topics. Instead, this article focuses more specifically on recent advances in the anti-tubercular and anticancer pharmacological activities of isomers 1,2,4-oxadiazole and 1,3,4-thiadiazole. Because of their high chemical and thermal strength, these two isomers possess the potential to be an essential part of the pharmacological sector. During our literature review, we attempted to cover a large number of related research articles in order to make it as comprehensive as possible. We included the most recent articles to keep it up-to-date. This review article seeks to conduct an in-depth study and analysis and provide a comprehensive report in the form of detailed technical discussion and even information in the form of figures. Overall, this will serve as a great starting point for future research in this sector.
Therapeutic targeting of the hallmarks of tuberculosis (TB)
Tuberculosis, a communicable disease caused by the bacillus Mycobacterium tuberculosis, is one of the top 10 causes of death. At the first-ever UN High Level Meeting in September 2018, at its headquarters in New York, heads of state came together and made strong commitments to end TB. The title of the meeting was “United to End TB: An Urgent Global Response to a Global Epidemic” (WHO, Global Tuberculosis Report 2018).
In the field of anti-TB drug discovery, although many drugs are currently in use, the developments of multidrug-resistant Mycobacterium tuberculosis (MDRTB) and extremely drug-resistant TB (XDRTB) are the two major challenges. Currently, the development of an effective anti-TB drug, which will have a short treatment duration, be simpler, be less toxic, be drug-resistant, and have minimal drug-drug interactions, is the current requirement in the field (Ginsberg 2010).
For TB, the main targets of choice for the researchers are mycobacterial transcriptional repressor (EthR), enoyl-ACP-reductase (InhA), enzyme decaprenylphosphoryl-β-D-ribose 2′epimerase (DprE1), shikimate kinase, alanine racemase, etc. The hallmarks of TB are represented in Figure S1 (Zumla et al. 2014). The current anti-TB-drug pipeline contains new chemical scaffolds and a variety of targets, including the 1,2,4-oxadiazole and 1,3,4-thiadiazole, as both the nuclei are able to coordinate well with these targets.
Therapeutic targeting of the hallmarks of cancer
Researchers have produced a vast quantity of records on proteins and muted genes towards the development of cancer cells. Recently, many environmental aspects that are associated with mutations at a genetic level have been reported. To determine the efficacy of gene expression and defective proteins, as well as to detect novel most cancers biomarkers, a one-of-a-kind molecular method has been used.
It can be beneficial to deal with most cancers and decrease associated complications. Although it appears that many aspects of epigenetic remain unknown, various studies to ascertain the mechanisms and their relationship collectively to the development and spread of quite a number of diseases, mainly most cancers, are continuing. Lack of specificity or low therapeutic index, drug resistance, and poor ADME profile are a few of the challenges in anticancer drug discovery. Hallmarks of cancer are represented in Figure S2 (Hanahan and Weinberg 2011).
Oxadiazole
Introduction to 1,2,4-oxadiazole and its biological importance
As mentioned earlier, growing interest is noted among medicinal chemists for oxadiazole (1) due to its biological activity and being used as a privileged scaffold in drug design with various therapeutic applications (Kumar et al. 2011). While the combination of hydrophilic and electron donor properties makes the oxadiazole ring biologically active, the thermal and chemical resistance provides metabolic stability. In drug discovery and development, 1,2,4-oxadiazoles are widely used as bio-isosteric replacements for ester, amide, and acid compounds to take care of associated metabolism-related liabilities with these functional groups (Saunders et al. 1988; Patani and Voie 1996).
In a few clinical trials, 1,2,4-oxadiazole-based medicines have been found to be effective. The 1,2,4-oxadiazole scaffolds containing marketed drugs are reported in Fig. 1, which underlines their significance in the discipline of medicinal chemistry (Kleeman et al. 2001).
Fig. 1.

Some marketed drug’s containing 1,2,4-oxadiazole
Oxolamine (2) majorly exhibits anti-tussive and anti-inflammatory activity. Irrigor (3) shows anesthetic and vasodilator activity, while Libexin (4) indicates anti-tussive activity. Ataluren (5) is used in the treatment of cystic fibrosis (Bora et al. 2014). The 1,2,4-oxadiazole ring binds and inhibits receptors such as 5-hydroxytryptamine1B/D (5-HT1B/D) (Selkirk et al. 1998) 1,2,4-oxadiazole exhibits an array of pharmacological activities such as anti-inflammatory (Nicolaides et al. 1998), anticancer (Matsumoto et al. 1994; Zhang et al. 2005), anti-TB, anti-tussive, and anti-tubercular (Upare et al. 2019).
Oxadiazoles have also been used within the area of material science due to their inherent characteristics to excellent thermal and chemical stability and high photoluminescence quantum yield. The 1,2,4-oxadiazoles show useful properties in blue phosphorescent devices, solar cells, liquid crystals, fluorogenic chemosensory polymers, organic light-emitting diodes, and heat-resistant polymers (Li et al. 2014; Parra et al. 2006; Guo et al. 2014).
Chemistry
As a five-membered ring heterocycle, oxadiazole consists of oxygen and two nitrogen atoms. Based on the position of these N and O heteroatoms in the ring, it has four isomeric structures as represented in Fig. 2.
Fig. 2.

Four isomers of oxadiazole heterocycle (A–D) and oxadiazole heterocycle reactive site (E)
The stability of these analogues was explained using the free Gibb’s energies. Free Gibb’s energies for analogues 1,3,4; 1,2,3; 1,2,4; and 1,2,5 is 0.0, 21.28, 8.64, and 40.61 kcal/mol, respectively, making 1,3,4 analogues the most stable among them all, which has the smallest amount of free Gibb’s energy. The stability order decreases as 1,3,4 > 1,2,4 > 1,2,3 > 1,2,5; the 1,2,3-oxadiazole isomer is found solely as an unstable diazoketone tautomer due to its cyclic structure instability, making 1,3,4 and 1,2,4 isomers more frequent in medicinal chemistry than the other two isomers. Further UV spectral data of these isomers points out that the 1,2,4-oxadiazole heterocyclic, unlike its aromatic nature, the ring is more heterodiene in character, whilst 1,3,4-oxadiazoles have increased aromaticity, most likely due to their symmetry (Pace and Pierroa 2009). Consequently, these traits differentiate 1,2,4-oxadiazole from its 1,3,4-isomer in reactivity. The experimentally measured bond lengths in X-ray diffraction endorse the planar structure as well as its aromatic character.
It is the most electron-poor azole because it contains two pyridine-type nitrogen atoms and a furan-type oxygen atom. Mono-substituted 1,2,4-oxadiazoles are much less stable than the disubstituted. The latter can even tolerate strong acids such as concentrated sulfuric acid acidic and strong bases. While the 3 and 5 positions are susceptible to creating 3,5-disubstituted 1,2,4-oxadiazoles via nucleophilic attack, they are almost inert towards electrophilic substitution, and, thus, disubstituted are the most commonly synthesized derivatives. The 1,2,4-oxadiazole ring electron-withdrawing effect is effectively exercised via its C-5 position rather than its C-3 position. See Fig. 2E.
Synthesis
With time, synthetic chemists have developed many preparatory methods for 1,2,4-oxadiazole. Tiemann and Kruger report the very first synthetic method in 1884 (Tiemann et al. 1884).
Traditional route
Worldwide, the most common and widely used two synthetic routes by chemists start with readily available nitriles, as shown in Fig. 3: (a) 1,3-dipolar cycloadditions of nitriles to nitrile N-oxides; (b) amidoxime heterocyclization. The amidoxime intermediate is formed when hydroxylamine reacts with nitrile. Acylation of the amidoxime intermediate yields O-acylamidoxime, isolable in some instances, upon cyclization as the ultimate step gives oxadiazole (Paulo et al. 2018).
Fig. 3.
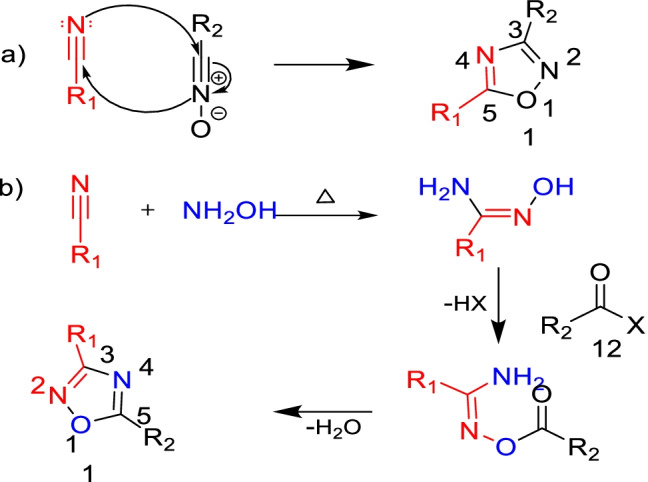
Traditional synthetic routes towards 1,2,4-oxadiazoles: a 1,3-dipolar cycloaddition of nitriles to nitrile N-oxides; b cyclization of amidoxime derivatives
It must be mentioned that when one pursues the 1,3-dipolar cycloaddition route for synthesis, the corresponding substituent (R1) of nitrile acquires the C-5 position of ultimate Oxadiazole, whilst the same acquires the C-3 position when one follows the amidoxime route (Paulo et al. 2018). Most synthetic strategies belong to either one of the two traditional routes. As a variant to the predominant route, while different acylating agents were explored to synthesize the 1,2,4-oxadiazoles, researchers additionally made use of catalyst, coupling agents, or even the preliminary O-acylation of amidoxime to tolerate the reaction conditions, assist in cyclization, and enhance the yield. They also tried some greener methods, such as microwave or ultrasound irradiation. A few of the synthetic approaches developed by researchers are summarized in Fig. 4, and various types of biologically active oxadiazole derivatives are shown in Fig. 5 and Fig. 6.
Fig. 4.

Some synthetic approaches to oxadiazole molecules: 1 Amidoxime reacted with suitably activated acid derivative; 2 amidoxime reacted with amide at high temperature; 3 pyrolysis of amidoxime and their esters; 4 ring transformations; 5 amidoxime reacted with acid anhydrides; 6 oxidation of an imino group with sodium hypochlorite; 7 oxidation of oxadiazoline; 8 addition of benzonitrile oxides to aromatic nitriles
Fig. 5.

List of oxadiazole derivative 1–12 (Upare et al. 2019). Reprinted with permission from Upare et al. 2019,
copyright 2019 Elsevier
Fig. 6.

List of oxadiazole derivative 13–24
Biological activity
Anti-tubercular activity
Villemagne et al. (2020) have worked on fragment-based drug-design and synthesized new oxadiazole compounds as potent EthR inhibitors (Villemagne et al. 2020). As a result of their approach, they successfully synthesized compounds with decreased metabolism and enhanced solubility. The compound 1a (BDM 71,339) showed an EC50 of 0.072 μM, solubility of 9.9 μg.mL−1, and an excellent pharmacokinetic profile with an elimination half-life (t1/2) of 19 min and CL of 69 μL min−1 mg−1.
Parikh et al. (2020) have presented the development of substituted 1,2,4-oxadiazole as a potent anti-TB agent (Parikh et al. 2020). The research group tested these compounds for their in vitro anti-tuberculosis activity towards Mycobacterium tuberculosis (Mtb.) H37Rv, anti-microbial activity, and antimalarial activity. In vitro studies performed at two different concentrations, the % inhibition observed for compound 2a against M. tuberculosis H37Rv is 92% at both concentrations, while the compound 2b shows 96% inhibition at a concentration of 250 μg mL−1 and a 91% inhibition at 100 μg mL−1.
Shruthi et al. (2019) worked on a quinoline scaffold. The 1,2,4-oxadiazole moiety is linked to quinoline with piperazine as a linker (Shruthi et al. 2019). They have generated the structure activity relationship (SAR) via combinatorial synthesis. Among the synthesized compounds, compound 3a shows a minimum inhibitory concentration (MIC) of 0.5 μg mL−1 towards the wild-type, Mycobacterium tuberculosis strain (WT H37Rv). Compound 3a has been found to be active against monoresistant strains of Mtb. The observed activities against this strain of Mtb. are as given in Table S1.
Excellent metabolic stability and bioavailability were noted for the compound 3a, with a long T1/2 (h) of 1.63 h and a comparatively short concentration of a drug in the blood, cerebrospinal fluid, or target organ after a dose was given (Cmax) of 2503.25 ng ml−1. Relatively, compound 3b showed a T1/2 (h) of 1 h, with a Cmax 7442.73 ng ml−1, owing to its less stability in human liver microsomes. Its MIC against Mtb. strain H37Rv is 0.25 g mL−1.Upare et al. (2019) have synthesized the styryl oxadiazoles in good yields from cinnamic acid, wherein the terminal carboxylic group of cinnamic acid was replaced with a bio-isostere 1,2,4-oxadiazole (Upare et al. 2019). The in vitro anti-tubercular activity study against the Mycobacterium tuberculosis (Mtb) H37Ra strain showed varied anti-tubercular profiles. The compound 4a was reported to possess the highest anti-tubercular activity [concentration of drug required for 50% inhibition (IC50) = 0.045 µg/mL] among the synthesized compounds. Computational molecular docking studies on the InhA enzyme corroborated experimentally observed activities.
Desai et al. (2016) have designed molecules based on nicotinamide and pyrazinamide drugs. The synthesized 1,2,4-oxadiazole-5-ones and or -5-thiones are substituted with pyridines and pyrazines at the C3 position. Pivaloyloxymethyl derivatives (5a) of 1,2,4-oxadiazole-5-thiones were made with improved lipophilicity and evaluated for anti-TB activity. Because these compounds have an MIC greater than 50 mg ml−1 against Mtb. H37Rv strain, they may not be considered potent molecules (Desai et al. 2016).
Gold et al. (2016) synthesized a novel analogue of cephalosporins which is selectively active on non-replicating Mtb (Gold et al. 2016). The compound 6a has a MIC of 0.88 g/mL for non-replicating Mtb, a MIC of > 100 g/mL for replicating Mtb, and an LD50 of > 100 g/mL in human liver cancer cell line (HepG2). Jain et al. (2016) have reported a novel hybrid of quinoline and oxadiazole as agents for anti-TB (Jain et al. 2016). They have replaced the isoxazole ring with 1,2,4-oxadiazole as a bio-isosteric replacement. While several molecules tested against the Mtb. H37Rv strain are active, the compound 7a is shown to possess the highest activity, with a MIC of 0.4 µM and a selectivity index of > 610.
Shruthi et al. (2016) have proposed the design strategy and synthesized benzimidazole–oxadiazole hybrid compounds as an anti-tubercular agent (Shruthi et al. 2016). Among the 20 novel compounds tested, compound 8a was found to be 546-fold selective toward Mycobacterium tuberculosis strain H37Rv, with an MIC of 1.6 mg mL−1. Thus, it is twofold superior to the standard drug pyrazinamide and fourfold to isoniazid. Furthermore, the active compounds were tested for toxicity and found to be safe even at high concentrations. Westhuyzen et al. (2015) optimized a hit compound to produce a class of compounds known as pyrrolo[3,4c]pyridine-1,3(2H)diones, which target the mycobacterial respiratory cytochrome bc1 complex, a validated drug target in Mtb (Westhuyzen et al. 2015).. The optimized compound 9a was synthesized from its hit compound wherein the ester functionality was replaced for its metabolic stability with methyl oxadiazole as a bio-isosteric replacement. This modification leads to an improved MIC of 0.065 µM and shows improved metabolic stability, that is, human S9 (% remaining after 40 min) = 97% and good Clin < 11.6 mL/min/mg.
Flipo et al. (2012) worked on EthR inhibitors. Ethionamide is the main second-line drug used in multidrug-resistant tuberculosis (MDR-TB) (Flipo et al. 2012). This molecule has been reported to have side effects. To overcome problems associated with ethionamide, this research group came up with the most active compound, 10a, that showed an IC50 of 400 nM and an EC50 of 60 nM against M. tuberculosis–infected macrophages at nanomolar concentration. They also reported that this compound has good microsomal stability and suitable physicochemical properties. This compound has a solubility of 410 μg mL−1 and Clint (microsomes) of 15 mL/in/mg. The bioavailability of the compound is AUC (mouse): 98.6 µg mL−1 h. Besides, compound 10a (BDM41906) shows a tenfold improved activity of ethionamide and is called “ethionamide boosters.” The molecule is in a phase II clinical trial as an anti-TB agent.
Almansour and co-workers (2012) reported a 1,2,4-oxadiazoles substituted series of molecules (Almansour et al. 2012). The in vitro anti-mycobacterial behavior of this compound 11a against Mtb. is 0.07 μM, and MDR-TB is 0.14 μM.
Anticancer activity
Kala et al. (2020) have worked on the design and synthesis of 1,2,4-oxadiazoles with quinoline derivatives and evaluated anticancer activity against etoposide (Kala et al. 2020). Etoposide is used for the treatment of non-lymphocytic leukemia, lung cancer, lymphoma, testicular cancer, and glioblastoma multiforme. In treating testicular cancer, it is usually given in combination with other drugs such as bleomycin. The compounds 12a,12b, and 12c exhibited excellent anticancer activity when compared with etoposide (Table S2). The designed series can be considered as an extension or a modification of M. srinivasa, S. satyavenia, and B. rambis published work wherein the isoxazole and quinazoline heterocycles are replaced with quinolone heterocycles.
Srinivasa et al. (2019) have presented the design and synthesis of new 1,2,4-oxadiazole-isoxazole linked quinazoline derivatives as anticancer agents (Srinivasa et al. 2019). Among the tested compounds against human are cell lines for breast (MCF-7, MDA MB-231), lung (A549), and prostate cancer (DU-145). Compounds 13a and 13b are the most active in the series. The compound 13a has an IC50 of 0.056 against MCF-7, 0.06 against MDA MB-231, 0.76 against A549, and 0.011 against DU-145. The compound 13b shows an IC50 0.021 against MCF-7, IC50 0.14 against A549, IC50 0.064 against DU-145, IC50 0.19 against MDA MB-231.
Aguiar et al. (2019) have reported the synthesis of O-acylamidoximes adamantyl and 3-Aryl-5-adamantane-1,2,4-oxadiazoles from adamantane carbonyl chloride and variously substituted arylamidoximes. Under microwave irradiation conditions, the O-acylamidoxime intermediate was cyclized (Aguiar et al. 2019). Compounds 14a and 14b were tested for cytotoxic activity against vero cells and anticancer activity against chronic myeloid leukemia (CML) cancer cells. Compound 14a showed a concentration-dependent profile for the CML human immortalized myelogenous leukemia cell line (K562) and acute pro-myelocytic leukemia for human leukemia cell line (HL60).
Sudhakara et al. (2019) worked on the design and synthesized of new derivatives that contain both the 1,2,4-isomer of oxadiazole and the thiadiazole motif as part of a molecule (Sudhakara et al. 2019). These derivatives exhibited moderate antitumor efficacy in four human cancer cell lines tested for antitumor efficacy, namely, Colo205 (colon), MCF-7 (breast), A2780 (ovarian), and A549 (lung), in a colorimetric assay for assessing cell metabolic activity (MTT assay). Among the tested compounds, 15a shows an IC50 of 0.10 µM, 0.24 µM, 0.11 µM, and 0.32 µM for Colo-205, MCF-7, A2780, and A549, respectively.
Reddy et al. (2019) have reported the design and synthesis of a series of 1,2,4-oxadiazole linked imidazopyrazine derivatives (Reddy et al. 2019). Compounds were tested against three human cell lines. The compounds 16a and 16b show excellent cytotoxicity activity and are more potent than adriamycin. The reported IC50 values for compound 16a for cell lines MCF-7, A-549, and A-375 are 0.68 µM, 1.56 µM, and 0.79 µM, respectively. Similarly, for the other compounds, 16b reported IC50 values for cell lines MCF-7, A-549, and A-375 are 0.22 µM, 1.09 µM, and 1.18 µM, respectively.
Yu et al. (2019) identified isatin as 2,3-indolinedione derivatives as a new promising class of anticancer agents (Shengping et al. 2019). They synthesized and tested 18 compounds against mantel cell lymphoma (MCL) cell lines. The most prominent active compound from the 1,2,4-oxadiazole series is compound 17a, which shows IC50 values of 0.4 µM, 0.4 µM, 0.5 µM, 0.7 µM, and 1.5 µM, against MCL cell lines Mino, chronic lymphocytic leukemia cell line (Z138), Maver-1, Jeko-R, and leukemia cell line (Granta-519) respectively.
Cascioferro et al. (2019) reported structural modifications made to nortopsentin, an alkaloid, to obtain the new derivatives wherein the 1,2,4-oxadiazole moiety replaces the central imidazole ring as the main modification and generates the SAR (Cascioferro et al. 2019). The compounds were tested on cancer cell lines such as HCT-116 (human colon cancer cell line), MCF-7 (cervical cancer cell line), and Caco-2. While compound 18a and 18b are the prominent active compounds in the series, and both induce a halt in the cell cycle resting phase (G0)–growth phase (G1) phase, the 18a is 2 to 4 times more active than the 18b, except for the HeLa cell. The SAR studies performed revealed that the halogen atom is crucial for the antiproliferative activity. Table S3 shows the cytotoxic activities measured by the MTT colorimetric assay.
Srinivas et al. (2018) have described the design and synthesis of novel 1,2,4-oxadiazole linked benzimidazoles (Srinivas et al. 2018). Preliminary evaluations of the substances’ antitumor activities against A549 (lung), MCF-7 (breast), and human melanoma cell line (A375) revealed that they were more potent than the doxorubicin drug. The most active compounds, 19a and 19b, have IC50 values of 0.24 M, 0.31 M, 1.67 M, and 1.90 M, 0.17 M, and 1.45 M, respectively, against MCF-7, A549, and A375 cell lines. Chakrapani et al. (2018) and co-workers have described the design and synthesis of a new 1,2,4-oxadiazole linked imidazothiadiazole analogue that is structurally close to levamisole (Chakrapani et al. 2018). Anticancer activity data was generated for these compounds using the MTT assay for three human cell lines: A375, MCF-7, and human renal carcinoma cell line (ACHN) (Table S4). The compounds 20a, 20b, and 20c turned out to be the most prominent compounds with almost similar activity to that of the doxorubicin drug.
A 1,2,4-oxadiazole scaffold has been developed by S. Moniot et al. (2017) as Sirt2 inhibitors. They successfully optimized their previously reported lead compound 3-(4-chlorophenyl)-5-(piperidine-1-ylmethyl)-1,2,4-oxadiazole. For SAR studies, α–tubulin-acetylLys40 peptide was used as the Sirt2 substrate (Moniot et al. 2017). The study revealed that for a molecule to possess Srit2 inhibitory action, the 1,2,4-oxadiazole must have a para substituted phenyl ring in its 3rd position and a cyclic aminomethyl or haloalkyl chain in its 5th position as a crucial substituent. Amongst the compounds tested against leukemia cell lines, 21a and 21b emerged to be the most active in the series; 21a shows an IC50 of 10 µM against Sirt2, while compound 21b shows an IC50 of 1.5 µM (Table S5).
Han et al. (2016) and his co-workers have designed and synthesized a new series of 4-chloro-benzamide derivatives containing substituted 1,2,4-oxadiazole heteroaryl rings as a potent rearranged transfection kinase inhibitor (RET) for cancer therapy (Han et al. 2016). The compound 22a strongly inhibits cell proliferation driven by RET wild-type and gatekeeper mutations. Western blotting analysis revealed that the compound 22a completely inhibits the phosphorylation of the RET enzyme at a 1-µM concentration. The compound 22a shows an IC50 of 1.8 nM against RET, whereas that of Ponatinib shows an IC50 of 0.9 nM.
Cai et al. (2015a, b) post their successful investigation of a few derivatives of Vorinostat as an antitumor agent. The group has explored the optimization of lead compound entinostat, leading to a series of 2-aminobenzamide and hydroxamate derivatives containing 1,2,4-oxadiazole (Cai et al. 2016). The MTT-based assay performed to investigate the in vitro antiproliferative activities of these compounds revealed that the 2-aminobenzamides 23b [R = Phenyl, 4-Me-Phenyl, 4-Cl-phenyl, etc.] are predominantly active towards the histiocytic lymphoma cell line (U937) among the tested human cancer cell lines U937, NCI-H661, A549, HCT116, and MDA-MB-231. Furthermore, the compounds were tested against histone deacetylases (HDAC) 1, 2, and 8 for their inhibitory activities, while the 2-aminobenzamide derivatives 23b are active against HDAC 1, hydroxamate derivatives 23a [R = Phenyl, 4-Me-Phenyl, 4-Cl-Phenyl, etc.] are active against the HDAC 8 and exhibit lower IC50 values compared to suberoylanilide hydroxamic acid (SAHA) and entinostat.
Cai et al. (2015a, b) discussed how they made hydroxamate, 2-aminobenzamide, and trifluoromethyl ketone analogues of 1,2,4-oxadiazoles (Cai et al. 2015a, b). They have replaced the amide functional group of vorinostat with 1,2,4-oxadiazole as a bio-isostere to investigate the effect of replacement on activity. When these compounds were tested against three human cancer cell lines, most of the compounds showed more prominent anticancer activity against human acute myeloid leukemia cell U937 than the other two human lung cancer cell lines A549 andNCI-H661. The compound 24a shows an IC50 of 5.31 μM, 3.09 μM, and 0.29 μM against A549, NCI-H661, and U937 cell lines. The compound 24b shows an IC50 of 9.17 μM, 0.41 μM, and 0.46 μM against A549, NCI-H661 andU937 cell lines, respectively. The compound 24c shows an IC50 of > 100 μM, 18.68 μM, and 3.73 μM against A549, NCI-H661, and U937 cell lines, respectively.
Thiadiazole
Introduction of thiadiazole and its biological importance
Thiadiazole, the most prevalent and indispensable heterocycle, is a five-membered heterocyclic compound, a scaffold that makes an important structure of several naturally occurring as well as medicinal products (Dawood and Farghaly 2017). The thiadiazole moiety acts as a “hydrogen-binding domain” and “two-electron donor system,” which makes the thiadiazole ring biologically active, sulfur atom imparts lipo-solubility, leading to analogues with higher lipophilicity. A lot of people use 1,3,4-thiadiazoles as bio-isosteric replacements for pyrimidine, pyridine azine, oxadiazole, oxazole, thiazole, and benzene in the development of new drugs (Serban et al. 2018). A few 1,3,4-thiadiazole scaffold containing marketed drugs are reported in Fig. 7, which underlines their importance in the field of medicinal chemistry (Serban 2019). Acetazolamide (9) and methazolamide (10) are potent carbonic anhydrase inhibitors, drugs used in the treatment of glaucoma, an eye-related disorder that causes damage to optic nerves. Megazole (11) is an antitrypanosomal agent, a drug used to treat African trypanosomes, also called sleeping sickness. Sulfamethizole (12) is an anti-microbial agent. Cefazolin (13) and cefazedone are used as antibiotics that belong to the cephalosporin family. Azeteta (14) is a phosphorous-containing drug used for the treatment of cancer.
Fig. 7.

Some marketed drugs containing 1,3,4-thiadiazole (Li et al. 2013)
Thus, 1,3,4-thiadiazole exhibits various pharmacological activities such as anti-inflammatory (Amir and Kumar 2007), antihypertensive (Hasui et al. 2011), anti-HIV, antidepressant, anticonvulsant, anticancer (Haider et al. 2015), anti-TB, and anti-microbial agents (Hu et al. 2014).
Chemistry
Thiadiazole is a heterocycle ring with two nitrogen and one sulfur heteroatom. Based on the nitrogen and sulfur heteroatoms’ positions in the ring, it has four isomers as represented in Fig. 8a.
Fig. 8.

a Isomers of thiadiazole based on the position of heteroatoms. b Thiadiazole heterocycle reactive site
1,3,4-Thiadiazole is a conjugated, weekly basic, planner, and electron-deficient ring system. While high aromaticity with the added + I effect of a sulfur atom makes it a weekly basis, the nitrogen atoms electron-withdrawing effect makes it electron deficient. This nature makes the carbon atoms at the C-2 and C-5 positions relatively inert towards electrophilic substitution while more reactive to nucleophilic attack. Substituents at the C-2 or C-5 positions activate the ring, favoring nucleophilic attack on carbon atoms. Although electrophilic attack on sulfur atoms is rare, the ring nitrogen atoms, dependent on the nature of the substituent on the carbon C-2 or C-5 position, do undergo electrophilic attack. See Fig. 8b. The ring is quite stable and can tolerate aqueous acidic solutions, but not the strong basic conditions under which ring cleavage is observed.
Synthesis
Emil Fischer first synthesized 1,3,4-thiadiazole in 1882 (Goerdler et al. 1956). The most common and extensively explored synthetic route is the cyclization of acyl hydrazine, which includes diacylhydrazines and monoacylhydrazines. The other synthetic route to 1,3,4-thiadiazoles makes use of thiohydrazines. From the recent literature, we have summarized a few strategies employed by chemists for the synthesis of 1,3,4-thiadiazoles as shown in Fig. 9 (Al-Omar et al. 2011: Kristinsson and Winkler 1982). The transformation of the 1,3,4-oxadiazole ring is also a route of choice.
Fig. 9.

A few synthetic approaches to thiadiazole molecules with different Acyl hydrazides, diacylhydrazides, thiosemicarbazide, thiocarbazide, thiourea, bisthiourea
Biological Activity
Biologically active 1,3,4-thiadiazole derivatives’ structure is illustrated in Fig. 10.
Fig. 10.

List of thiadiazole derivative 25–41
Anti-tubercular activity
Kumar et al. (2019) have synthesized a series of azetidinone nucleus containing 1,3,4-thiadiazole derivatives from the Schiff base and are reported to exhibit good antibacterial, antifungal, and anti-tubercular activity. To obtain anti-tubercular activity data, the microplate Alamar Blue Assay was performed. The MIC of these derivatives against the Mtb. H37Rv strain ranged from 6 to 25 g mL−1. The compounds 25a, 25b, 25c, and 25d with the substituent in the para position are active and have a MIC close to 6 μg mL−1, while the substituent in the meta position makes them inactive.
Taflan et al. (2019) reported two series of novel imidazo[2,1-b][1,3,4]thiadiazole (ITD) hybrid compounds 26a with excellent anti-tubercular profiles and MICs ranging from 0.24 to 0.49 g mL−1 concentration against Mycobacterium smegmatis organisms (Ebru et al. 2019). At the same time, these compounds were also found to possess antioxidant effects due to the presence of a 3,4-dihydroxy phenolic group C-2 position.
Demirci et al. (2018) have reported the synthesis of novel 1,3,4-thiadiazole-based fluoroquinolone hybrid compounds as anti-tuberculosis agents. All of the norfloxacin-derived compounds have 4-chlorophenyl 27a and 2,4-dichlorophenyl 27b substituents on the 1,3,4-thiadiazole ring and exhibit significant antimicrobial activity against the Mtb. H37Rv strain. MIC was measured using the Broth Microdilution method and was found in the range of 8 to 64 μg.mL−1.
Wadhwa et al. (2017) presented the results of computational studies that revealed the most vital structural requirements of molecules for InhA inhibition. The molecular docking experiment performed on imidazo[2,1-b] [1,3,4] thiadiazole 28a revealed that PRO156, GLN100, TYR158, ALA198, LEU197, LEU218, and MET199 (active site) are the essential binding residues that are responsible for interactions between the InhA enzyme and inhibitors.
As anti-TB agents, presented 5-substituted-2-[(3,5-dinitrobenzyl)-sulfanyl]-1,3,4-oxadiazoles and 1,3,4-thiadiazole derivatives (Karabanovich et al. 2016). They assessed the anti-tubercular profile (in vitro) of the synthesized compounds as well as studied SAR deeply to conclude their earlier reported findings that the dinitrobenzylsulfanyl substitution is crucial for anti-mycobacterial activity. The nitro-substituted 2-alkyl/aryl-5-benzylsulfanyl-1,3,4-thiadiazoles are reported to possess greater activity than the first-line anti-TB drugs, with a measured MIC as low as 0.03 μM for the 29a compound. The compounds were also tested against nontuberculosis mycobacterial species, namely Mycobacterium avium, Czechoslovak National Collection of Type Cultures (CNCTC) My 331/88, Mycobacterium kansasii CNCTC My 235/80, the clinically isolated M. kansasii 6509/96, and six multidrug-resistant clinically isolated strains of M. tuberculosis.
Tatar et al. (2016) have synthesized and reported conjugated thiadiazole-thiourea as anti-tuberculosis agents. The 5-(4-chlorolphenyl and 4-fluorophenyl)-1,3,4-thiadiazole 3-substituted thioureas were identified as the most active as well as selective compounds against the M. tuberculosis H37Rv strain. The MIC values measured for compounds 30a and 30b were 10.96, 11.48 μM, respectively. Molecular docking studies performed on these compounds showed good docking scores for the enzyme InhA.
Tatar et al. (2015) reported another two series of compounds synthesized from L-methionine as anti-TB agents. The synthesized 1,3,4-thiadiazole and 1,2,4-triazole derivatives exhibited anti-tubercular activity (Batt et al. 2015). The 1,3,4-thaidizole with (4-chloro-(3-trifluoromethyl) phenyl] substituted thiourea 31a is the most active compound against the M. tuberculosis H37Rv strain, with a measured MIC of 30.88 μM.
Batt et al. (2015) successfully used the target overexpression approach to screen the lead molecules, identifying GSK710 (32a), one of the series’ analogues, as a strong receptor of enzymatic reactions, Mt-DprE1 IC50 = 54 nM), in M. bovis BCG. This compound showed an eightfold higher MIC relative to the control strain. Furthermore, spontaneous resistant mutant sequencing studies were used to conclude that DprE1 is the target of these analogues, with a single nucleotide change at both positions E221Q and G248 in the enzyme DprE1 gene molecule, in a clinical trial. SAR studies revealed that the modification could be allowable at the phenyl’s terminal ring inclusive of fluorine substitution, pyridine regioisomers, and other functional groups can be substituted for the para position ensuing when solubility and distribution factors improve dramatically, while inhibitory exercise is maintained.
Anticancer activity
Naggar et al. (2019) recently presented design, synthesis, and molecular docking experiments of antimicrobial and anticancer 5-(3,5-dinitrophenyl)-1,3,4-thiadiazole compounds. These compounds were studied in vitro and their activities were compared to those of Doxorubicin, a standard drug. The compounds 33a, 33b, and 33c are the most active compounds and show the following IC50 values against various tumor cell lines (Table S6).
Altıntop et al. (2018) have described the design and synthesis of 1,3,4-thiadiazole derivatives and evaluated their anticancer activity towards chronic myelogenous leukemia cells. The compound 34a was screened against eight kinases and found to inhibit the Bruton’s tyrosine kinase inhibitor (BTK), FYN proto-oncogene (FYNA), LCK, and C-terminal Src kinase (CSK kinases) with the highest selectivity for the Bcr-Abl-positive K562 cell line. With a measured IC50 value of 7.4 µM for Abl kinase protein compound 34a, it turned out to be the most potent of all the compounds. It also had a different kinase inhibitory profile than imatinib. Compound 34a exhibited BTK activity of 30.3 µM, whereas imatinib showed activity of > 100 µM.
Chowrasia et al. (2017) investigated novel fused thiadiazole scaffolds as anticancer agents. The fluorinated analogue 35a is more active than the parent analogue 35b. The antiproliferative activity observed for the most active compound 35a against different cell lines is MCF7 with an IC50 value of 22.1 µM, a cell line derived from the primary osteosarcoma (SaOS-2) IC50 value of 19 µM and a K562 IC50 value of 15 µM. The compound 35b showed significantly low activity against the same cell lines and observed are MCF7 IC50 value of 30.2 µM, SaOS-2, IC50 value of 39 µM, and a K562 IC50 value of 29.4 µM.
Amin et al. (2017) presented the synthesis of various coumarin-thiadiazole-based analogues with potential antitumor activity. The DNA binding assay was performed to assess the antitumor activity of the synthesized compounds. These compounds were tested against a few human cancer cell lines; liver cancer (HepG-2), colorectal cancer (HCT-116), and breast cancer (MCF-7). The measured IC50 values for compound 36a in HepG-2, HCT-116, and MCF-7 (human cancer cell lines) are 266, 238, 398 μg.mL−1, respectively. Similarly, compound 36b shows an IC50 of 114, 30.7, 54.9 μg.mL−1 in HepG-2, HCT-116, and MCF-7, cell lines.
Jakovljevic et al. (2017) developed two series of 1,3,4-thiadiazole amide derivatives containing a substituent catechol moiety as a potential antioxidant and anticancer agents. Among the two 3,4- and 2,3-dihydroxy series of derivatives, the latter showed the best activity. Moreover, the antioxidant properties of these derivatives totally depend on the substituent attached to the amide bond. Thus, adamantane-containing compounds 37a and 37b showed enhanced cytotoxic activity with IC50 values of 7.4 and 7.3 µM, respectively, towards the human acute pro-myelocytic leukemia HL-60 cells and lung carcinoma A549 cells, while they showed decreased toxicity towards normal MRC-5, suggesting a selective nature in action.
Wang et al. (2017) and his group identified [1,2,4]-triazolo-[3,4-b][1,3,4]-thiadiazole a novel scaffold as a disruptor of telomeric silencing 1 (DOT1L) inhibitor that plays a crucial role in cell cycle regulation, transcriptional elongation (Wang et al. 2017). The compound 38a showed strong binding affinity to DOT1L with measured IC50 of 8.3 μM. It has average selectivity over other methyl transferases and non-MLL-rearranged leukemia cell lines.
Kenji et al. (2016) have described the synthesis of nitrogen-containing bisphosphonates. These compounds show antitumor activity in particular in breast cancer and myeloma patients. Due to poor absorption, it has difficulty in cell entry and hence exhibits weak activity; it also causes bone side effects. Hence, research groups have synthesized their Pivaloyloxymethyl esters as a prodrug approach. The compound 39a shows IC50 of 11 µM for U937 cell lines and 8.5 µM IC50 for Cancer cell lines (EJ-1).
Tingting et al. (2016), have synthesized a novel disubstituted 1,3,4-thiadiazole that includes substituted N-heterocylic rings, mainly indole, pyridine, and quinoline, along with the aromatic rings (Tingting et al. 2016), Synthesized compounds were tested against CML cells and breast cancer cells. The compound 40a with indole as a substituent on the thiadiazole is reported to be the most potent compound among all, with a measured IC50 value of 5.9 ± 0.56 μM/L for epithelial, human breast cancer cell lines (MDA-MB231 cells), and a measured IC50 value of 4.2 ± 0.32 μM/L against K562 cell lines. Gossypol is used as the positive control.
Romagnoli et al. (2015) described the synthesis of a hybrid compound imidazo[1,2-b][1,3,4]thiadiazole as a biologically active scaffold with anticancer activity (Romagnoli et al. 2015).The compound 41a shows IC50’s of 0.17, 0.37, 0.41, and 0.67 µM against mouse lymphocytic leukemia cell line (L1210), murine breast cancer cell line (FM3A), lymphoblastic cells that happened to come from a child who had acute lymphoblastic leukemia (CEM), and HeLa cell lines, respectively. The compound 41b shows an IC50 of 0.25, 0.61, 0.83, and 0.87 µM against L1210, FM3A, CEM, and HeLa cell lines, respectively.
Conclusion
It is evident from the plethora that today, the 1,2,4-oxadiazole and 1,3,4-thiadiazole scaffolds are widely explored by researchers in medicinal chemistry, particularly as anti-tubercular and anticancer agents. The appearance of these scaffolds in the number of newly reported molecules also underlines their importance as bio-isosteres. While improving the anticancer and anti-tubercular profiles, while both the positions of these heterocycles were substituted and explored, new scaffolds in combination with other heterocycles are also reported. Although both the scaffolds are tested against various targets for their anti-tubercular and anticancer properties, other potential targets are still to be explored. SAR studies conducted helped in identifying the few structural key requirements. A few of the derivatives showed promising anti-tubercular and anticancer activity. We believe the summarized literature herein provides an overview of anti-tubercular and anticancer activities demonstrated by the 1,2,4-oxadiazoles and 1,3,4- thia diazoles, and helps researchers in rational drug design and development in this area.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Author Dr. Selvaraj Mohana Roopan thanks the VIT top-level management for their encouragement especially during COVID-19 lockdown period.
Author contribution
A.A.U. conceived and designed the study and wrote the initial manuscript. S.M.R. conceived and designed the study and edited the manuscript. All authors read and approved the manuscript.
Declarations
Ethics approval
This article does not contain any studies on human volunteers or animals.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aguiar DF, Dutra LAA, Dantas WM, Carvalho GC, Lemes RP, Pessoa CO, Paier CK, Araujo PL, Araujo ES, Pena LJ, de Oliveira RN. Synthesis, antitumor and cytotoxic activity of new adamantyl o-acylamidoximes and 3-aryl-5-adamantane1,2,4-oxadiazole derivatives. Chem Select. 2019;4:9112–9118. doi: 10.1002/slct.201901285. [DOI] [Google Scholar]
- Almansour AL, Kumar RS, Arumugam N, Sriram DA. Solvent free, four-component synthesis and 1,3-dipolar cycloaddition of 4(H)-pyrans with nitrile oxides: synthesis and discovery of antimycobacterial activity of enantiomerically pure 1,2,4-oxadiazoles. Eur J Med Chem. 2012;53:416–423. doi: 10.1016/j.ejmech.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Al-Omar MA, Amar AEE, Wahab AA. Synthesis and antimicrobial activity of some heterocyclic 2, 6-bis(substituted)-1,3,4-thiadiazolo-, oxadiazolo-, and oxathiazolidino-pyridine derivatives from 2,6-pyridine dicarboxylic acid dihydrazide. J Heterocycl Chem. 2011;48(5):1103–1110. doi: 10.1002/jhet.690. [DOI] [Google Scholar]
- Altıntop MD, Ciftci HI, Radwan MO, Sever B, Kaplancıkl ZA, Ali Taha FS, Koga R, Fujita M, Otsuka M, Özdemir A. Design, synthesis, and biological evaluation of novel 1,3,4-thiadiazole derivatives as potential antitumor agents against chronic myelogenous leukemia, striking effect of nitrothiazole moiety. Molecules. 2018;23(1):59–76. doi: 10.3390/molecules23010059. [DOI] [Google Scholar]
- Amin KM, Taha AM, George RF, Mohamed NM, Elsenduny FF (2017) Synthesis, antitumor activity evaluation, and DNA-binding study of coumarin-based agents. Arch Pharm Chem Life Sci 351(1):1–18. 10.1002/ardp.201700199
- Amir M, Kumar S. Synthesis and evaluation of anti-inflammatory, analgesic ulcerogenic and lipid peroxidation properties of ibuprofen derivatives. Acta Pharm. 2007;57(1):31–45. doi: 10.2478/v10007-007-0003-y. [DOI] [PubMed] [Google Scholar]
- Arunachalapandi M, Roopan SM. Ultrasound/visible light-mediated synthesis of N-heterocycles using g-C3N4/Cu3TiO4 as sonophotocatalyst. Res Chem Intermed. 2021;47:3363–3378. doi: 10.1007/s11164-021-04461-3. [DOI] [Google Scholar]
- Batt SM, Izquierdo MC, Pichel JC, Stubbs CJ, Del Peral LV, rez-Herra E, Dhar N, Mouzon B, Rees M, Hutchinson JP, Young RJ, McKinney JD, Aguirre DB, Ballell L, Besra GS, Argyrou A. Whole cell target engagement identifies novel inhibitors of Mycobacterium tuberculosis Decaprenyl phosphoryl-β-D-ribose Oxidase. ACS Infect. Dis. 2015;1(12):615–626. doi: 10.1021/acsinfecdis.5b00065. [DOI] [PubMed] [Google Scholar]
- Bora RO, Dar B, Pradhan V, Farooqui M. [1, 2, 4]-oxadiazoles: synthesis and biological applications. Mini Rev Med Chem. 2014;13(12):34. doi: 10.2174/1389557514666140329200745. [DOI] [Google Scholar]
- Cai J, Hongtao W, Ho HK, Xiaoqing W, Meng C, Xi Z, Lushen L, Chunlong S, Junqing C, Min J. Discovery and preliminary evaluation of 2-aminobenzamide and hydroxamate derivatives containing 1,2,4-oxadiazole moiety as potent histone deacetylase inhibitors. Eur J Med Chem. 2015;96:1–13. doi: 10.1016/j.ejmech.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Cai J, Wei H, Hong KH, Wu X, Zong X, Cao M, Wang P, Li L, Sun C, Chen B, Zhou G (2015b) Discovery, bioactivity and docking simulation of Vorinostat analogues containing 1,2,4-oxadiazole moiety as potent histone deacetylase inhibitors and antitumor agents. Bioorg Med Chem 23(13):3457–3471. 10.1016/j.bmc.2015.04.028
- Cascioferro S, Attanzio A, Sarno VD, Musella S, Tesoriere L, Cirrincione G, Diana P, Parrino B. New 1,2,4-oxadiazole nortopsentin derivatives with cytotoxic activity. Mar Drugs. 2019;17(1):35. doi: 10.3390/md17010035. [DOI] [Google Scholar]
- Chakrapani B, Ramesh V, Chander RGP, Ramachandran D, Reddy TM, Chakravarthy AK, Sridhar G. Synthesis and anticancer evaluation of 1,2,4-oxadiazole linked imidazothiadiazole derivatives. Russ J Gen Chem. 2018;88(5):1020–1024. doi: 10.1134/S1070363218050304. [DOI] [Google Scholar]
- Chowrasia D, Karthikeyan C, Choure L, Gupta S, Arshad MM, Trivedi P. Synthesis, characterization and anticancer activity of some fluorinated 3,6-diaryl-[1,2,4]triazolo[3,4-b][1,3,4] thiadiazoles. Arab J Chem. 2017;10(2):424–442. doi: 10.1016/j.arabjc.2013.08.026. [DOI] [Google Scholar]
- Dawood KM, Farghaly TA. Thiadiazole inhibitors: a patent review. Expert Opin Ther Pat. 2017;27:477–505 b. doi: 10.1080/13543776.2017.1272575. [DOI] [PubMed] [Google Scholar]
- Demirci A, Karayel KG, Tatar E, Okullu SO, Unubol N, Tasli PN, Kocagoz ZT, Sahin F, Kucukguzel I. Synthesis and evaluation of novel 1,3,4-thiadiazole-fluoroquinolone hybrids as antibacterial, antituberculosis, and anticancer agents. Turk J Chem. 2018;42(3):839–858. doi: 10.3906/kim-1710-35. [DOI] [Google Scholar]
- Desai NC, Somani H, Trivedi A, Bhatt K, Nawale L, Khedkar V, Jha PC, Sarkar D. Synthesis, biological evaluation and molecular docking study of some novel indole and pyridine based 1,3,4-oxadiazole derivatives as potential antitubercular agents. Bioorg Med Chem Lett. 2016;26(7):1776–1783. doi: 10.1016/j.bmcl.2016.02.043. [DOI] [PubMed] [Google Scholar]
- Ebru T, Hacer B, Mehtap E, Pardeshi RK. Novel imidazo[2,1-b][1,3,4]thiadiazole (ITD) hybrid compounds: design, synthesis, efficient antibacterial activity and antioxidant effects. Bioorg Chem. 2019;89:102998–103008. doi: 10.1016/j.bioorg.2019.102998. [DOI] [PubMed] [Google Scholar]
- Flipo M, Desroses M, Guillet NL, Villemagne B, Blondiaux N, Leroux F, Piveteau C, Mathys V, Flament MP, Siepmann J, Villeret V, Wohlkonig A, Wintjens R, Soror SH, Christophe T, Jeon HK, Locht C, Brodin P, Deprez B, Baulard AR, Willand N. Ethionamide boosters 2 Combining bioisosteric replacement and structure-based drug design to solve pharmacokinetic issues in a series of potent 1,2,4-oxadiazole EthR inhibitors. J Med Chem. 2012;55:68–83. doi: 10.1021/jm200825u. [DOI] [PubMed] [Google Scholar]
- Ginsberg A. Drugs in development for tuberculosis. Drugs. 2010;70(17):2201–2214. doi: 10.2165/11538170-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Goerdler J, Ohm J, Tegmeyer O. Darstellung und Eigenschaften des 1.2.4- und des 1.3.4-Thiodiazols. Chem Ber. 1956;89:1534–1543. doi: 10.1002/cber.19560890624. [DOI] [Google Scholar]
- Gold B, Smith R, Nguyen Q, Robert J, Ling Y, Quezada L, Somersan S, Warrier T, Little D, Pingle M, Zhang D, Ballinger E, Zimmerman M, Dartois ´ V, Hanson P, Mitscher LA, Porubsky P, Rogers S, Schoenen FJ, Nathan C, Aube J. Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. J Med Chem. 2016;59(13):6027–6044. doi: 10.1021/acs.jmedchem.5b01833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Hua R, Sui Y, Cao J. Synthesis of 3,5-disubstituted 1,2,4-oxadiazoles and their behavior of liquid crystallines. Tetrahedron Lett. 2014;55(9):1557–1560. doi: 10.1016/j.tetlet.2014.01.066. [DOI] [Google Scholar]
- Haider S, Alam MS, Hamid H. 1,3,4-Thiadiazoles: a potent multi targeted pharmacological scaffold. Eur J Med Chem. 2015;92:156–177. doi: 10.1016/j.ejmech.2014.12.035. [DOI] [PubMed] [Google Scholar]
- Han M, Li S, Ai J, Sheng R, Hu Y, Geng M. Discovery of 4-chloro-3-(5-(pyridin-3-yl)-1,2,4-oxadiazole-3-yl) benzamides as novel RET kinase inhibitors. Bioorg Med Chem Lett. 2016;26(23):5679–5684. doi: 10.1016/j.bmcl.2016.10.061. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hasui T, Matsunaga N, Ora T, Ohyabu N, Nishigaki N, Imura Y, Igata Y, Matsui H, Motoyaji T, Tanaka T, Habuka N, Sogabe S, Ono M, Siedem CS, Tang TP, Gauthier C, De Meese LA, Boyd S, Fukumoto SA. Identification of Benzoxazin-3-one derivatives as novel, potent, and selective nonsteroidal mineralocorticoid receptor antagonists. J Med Chem. 2011;54(24):8616–8631. doi: 10.1021/jm2011645. [DOI] [PubMed] [Google Scholar]
- Hu Y, Li CY, Wang XM, Yang YH, Zhu HL. 1,3,4-thiadiazole: synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem Rev. 2014;114(10):5572–5610. doi: 10.1021/cr400131u. [DOI] [PubMed] [Google Scholar]
- Jain PP, Degani MS, Raju A, Anantram A, Seervi M, Sathaye S, Ray M, Rajan MGR. Identification of a novel class of quinoline–oxadiazole hybrids as anti-tuberculosis agents. Bioorg Med Chem Lett. 2016;26(2):645–649. doi: 10.1016/j.bmcl.2015.11.057. [DOI] [PubMed] [Google Scholar]
- Jakovljevic K, Matic IZ, Stanojkovic T, Krivokuc A, Markovic V, Joksovic MD, Mihailovic N, Niciforovic M, Joksovic L. Synthesis, antioxidant and antiproliferative activities of 1,3,4-thiadiazoles derived from phenolic acids. Bio Org Med Chem Lett. 2017;27(16):3709–3715. doi: 10.1016/j.bmcl.2017.07.003. [DOI] [Google Scholar]
- Kala P, Sharif SK, Krishna CM, Ramachandran D. Design synthesis and anticancer evaluation of 1,2,4-oxadiazole functionalized quinoline derivatives. Med Chem Res. 2020;29(1):136–144. doi: 10.1007/s00044-019-02467-6. [DOI] [Google Scholar]
- Karabanovich G, Zemanova J, Smutny T, Szekely R, Sarkan M, Centarova I, Vocat A, Pavkova I, Conka P, Nemecek J. Development of 3,5-dinitrobenzylsulfanyl-1,3,4-oxadiazoles and thiadiazoles as selective antitubercular agents active against replicating and nonreplicating Mycobacterium tuberculosis. J Med Chem. 2016;59(6):2362–2380. doi: 10.1021/acs.jmedchem.5b00608. [DOI] [PubMed] [Google Scholar]
- Kenji M, Kosuke H, Kaoru H, Masashi I, Haruki O, Nagahiro M, Craig TM, Yoshimasa T. Targeting Cancer Cells with a Bisphosphonate Pro drug. Chem Med Chem. 2016;11(24):2656–2663. doi: 10.1002/cmdc.201600465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeman A, Engels J, Kutscher B, Georg DR (2001) Pharmaceutical substances: syntheses, patents applications, 4th Edition. (2 Volumes) Thieme Verlag: Stuttgart. 2521:599
- Kristinsson H, Winkler T. Synthese von heterocyclen. V. 1, 3, 4‐thiadiazol‐2 (3 H)‐one. Helv Chim Acta. 1982;65:2606. doi: 10.1002/hlca.19820650833. [DOI] [Google Scholar]
- Kumar D, Patel G, Chavers AK, Chang KH, Shah K. Synthesis of novel 1,2, 4-oxadiazoles and analogues as potential anticancer agents. Eur J Med Chem. 2011;46:3085. doi: 10.1016/j.ejmech.2011.03.031. [DOI] [PubMed] [Google Scholar]
- Kumar SA, Furqan DM, Mazahar F, Pardeshi RK. Design and synthesis of biologically active azetidinones nucleus containing 1, 3, 4-thiadiazole derivatives and evaluate their Tuberculosis activity. Chem Biol Interfaces. 2019;9(3):157–162. [Google Scholar]
- Kumari S, Carmona AV, Tiwari AK, Trippier PC (2020) Amide bond bioisosteres: strategies, synthesis, and successes. J Med Chem 63(21):12290–12358. 10.1021/acs.jmedchem.0c00530
- Li Y, Geng J, Liu Y, Yu S, Zhao G. Thiadiazole-a promising structure in medicinal chemistry. Chem Med Chem. 2013;8:27–41. doi: 10.1002/cmdc.201200355. [DOI] [PubMed] [Google Scholar]
- Li Q, Cui LS, Zhong C, Jiang ZQ, Liao LS. Asymmetric design of bipolar host materials with novel 1,2,4- oxadiazole unit in blue phosphorescent device. Org Lett. 2014;16(6):1622–1625. doi: 10.1021/ol5002494. [DOI] [PubMed] [Google Scholar]
- Manjupriya R, Roopan SM. Carbon dots-based catalyst for various organic transformations. J Mater Sci. 2021;56:17369–17410. doi: 10.1007/s10853-021-06354-7. [DOI] [Google Scholar]
- Matsumoto J, Takahashi T, Agata M, Toyofuku H, Sasada N. Study of the biological pharmacology of IFO, a new selective and reversible monoamine oxidase-B inhibitor. Jpn J Pharmacol. 1994;65(1):51–57. doi: 10.1254/jjp.65.51. [DOI] [PubMed] [Google Scholar]
- Moniot S, Forgione M, Alessia L, Gebremedhin S, Hailu S, Nebbioso A, Carafa V, Baratta F, Altucci L, Giacche N, Passeri D, Pellicciari R, Mai A, Steegborn C, Rotili D. Development of 1,2,4-oxadiazoles as potent and selective inhibitors of the human deacetylase sirtuin 2: structure−activity relationship, X ray crystal structure, and anticancer activity. J Med Chem. 2017;60(6):2344–2360. doi: 10.1021/acs.jmedchem.6b01609. [DOI] [PubMed] [Google Scholar]
- Naggar M, Sallam HA, Shaban SS, Azab ME, Abdel-Wahab SS, Amr AG, Al-Omar MA. Design, synthesis, and molecular docking study of novel heterocycles incorporating 1,3,4-thiadiazole moiety as potential antimicrobial and anticancer agents. Molecules. 2019;24(6):1066. doi: 10.3390/molecules24061066. [DOI] [Google Scholar]
- Nicolaides DN, Fylaktakidou KC, Litinas KE, Litina LD. Synthesis and biological evaluation of several coumarin-4-carboxamidoxime and 3-(coumarin-4-yl)-1,2,4-oxadiazole derivatives. Eur J Med Chem. 1998;33(9):715–724. doi: 10.1016/S0223-5234(98)80030-5. [DOI] [Google Scholar]
- Pace A, Pierroa P. The new era of 1,2,4-oxadiazoles. Org Biomol Chem. 2009;7:4337–4348. doi: 10.1039/B908937C. [DOI] [PubMed] [Google Scholar]
- Parikh PH, Timaniya JB, Patel MJ, Patel KP. Design, synthesis, and characterization of novel substituted 1,2,4-oxadiazole and their biological broadcast. Med Chem Res. 2020;29(3):538–548. doi: 10.1007/s00044-020-02505-8. [DOI] [Google Scholar]
- Parra M, Hidalgo P, Carrasco E, Barber J, Silvino L. New 1,2,4- and 1,3,4-oxadiazole materials: synthesis, and mesomorphic and luminescence properties. Liq Cryst. 2006;33(8):875–882. doi: 10.1080/02678290600871614. [DOI] [Google Scholar]
- Patani GA, Voie EJ. Bioisosterism: a rational approach in drug design. Chem Rev. 1996;96:3147. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Paulo SP, Vitor SS, Marco EF. 1,2,4- and 1,3,4-oxadiazoles as scaffolds in the development of antiparasitic agents. J Braz Chem Soc. 2018;29(3):435–456. doi: 10.21577/0103-5053.20170208. [DOI] [Google Scholar]
- Pearce S. The importance of heterocyclic compounds in anti-cancer drug design. Int J Drug Discov. 2017;18(2):66–70. [Google Scholar]
- Ram VJ, Sethi A, Nath M, Pratap R. The chemistry of heterocycles. US: Elsevier Science Publishing Co Inc; 2019. pp. 1–503. [Google Scholar]
- Reddy KT, Sreenivasulu R, Raju RR. Synthesis and biological evaluation of 1,2,4-oxadiazole linked imidazopyrazine derivatives as anticancer agents. J Indian Chem Soc. 2019;96:1–6. [Google Scholar]
- Romagnoli R, Baraldi PG, Prencipe F, Balzarini J, Liekens S, Estévez F. Design, synthesis and antiproliferative activity of novel heterobivalent hybrids based on imidazo[2,1-b][1,3,4]thiadiazole and imidazo[2,1-b][1,3]thiazole scaffolds. Eur J Med Chem. 2015;101:205–217. doi: 10.1016/j.ejmech.2015.06.042. [DOI] [PubMed] [Google Scholar]
- Roopan SM, Palaniraja J. Synthesis of indazoloquinazoline system (microreview) Chem Heterocycl Compd. 2016;52(2):93–95. doi: 10.1007/s10593-016-1838-2. [DOI] [Google Scholar]
- Sauer AC, Wolf L, Quoos N, Rodrigues MB, Braga AL, Rodrigues OED, Dornelles LA. Straightforward and high-yielding synthesis of 1,2,4-oxadiazoles from chiral n-protected α-amino acids and amidoximes in acetone-water: an eco-friendly approach. J Chem. 2019;16:1–9. doi: 10.1155/2019/8589325. [DOI] [Google Scholar]
- Saunders J, MacLeod AM, Merchant K, Showell GA, Snow RJ, Street LJ, Baker R. Ester bio-isosteres: synthesis of oxadiazolyl-1-azabicyclo[2.2.1]heptanes as muscarinic agonists. Chem Soci Chem Commun. 1988;88:1618–1619. doi: 10.1039/C39880001618. [DOI] [Google Scholar]
- Selkirk JV, Scott C, Ho M, Burton MJ, Watson J, Gaster LM, Collin L, Jones BJ, Middlemiss DN, Price GW. SB-224289 a novel selective (human) 5-HT1B receptor antagonist with negative intrinsic activity. J Pharmacol. 1998;125(1):202–208. doi: 10.1038/sj.bjp.0702059. [DOI] [Google Scholar]
- Serban G. Future prospects in the treatment of parasitic diseases: 2-amino-1,3,4-thiadiazoles in leishmaniasis. Molecules. 2019;24(8):1557–1579. doi: 10.3390/molecules24081557. [DOI] [Google Scholar]
- Serban G, Stanasel O, Serban E, Bota S. 2-Amino-1,3,4-thiadiazole as a potential scaffold for promising antimicrobial agents. Drug Des Dev Ther. 2018;12:1545–1566. doi: 10.2147/DDDT.S155958. [DOI] [Google Scholar]
- Shengping Y, Yang L, Zhen Z, Jingya Z, Guisen Z. Design synthesis and biological evaluation of novel 2,3-indolinedione derivatives against mantle cell lymphoma. Bioorg Med Chem Lett. 2019;27(15):3319–3327. doi: 10.1016/j.bmc.2019.06.009. [DOI] [Google Scholar]
- Shruthi N, Poojary B, Kumar V, Hussain MM, Rai VM, Pai VR, Bhata M, Revannasiddappad BC. Novel benzimidazole–oxadiazole hybrid molecules as promising antimicrobial agents. RSC Adv. 2016;6:8303–8316. doi: 10.1039/C5RA23282A. [DOI] [Google Scholar]
- Shruthi TG, Eswaran S, Shivarudraiah P, Narayanan S, Subramanian S. Synthesis, antituberculosis studies and biological evaluation of new quinolone derivatives carrying 1,2,4-oxadiazole moiety. Bioorg Med Chem Lett. 2019;29(1):97–102. doi: 10.1016/j.bmcl.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Sompalle R, Roopan SM. Microwave assisted synthesis of ring junction heterocyclic antioxidants. Res Chem Intermed. 2016;42(6):5353–5366. doi: 10.1007/s11164-015-2371-0. [DOI] [Google Scholar]
- Sompalle R, Roopan SM. Review on Benzothiazoles: synthesis and diverse biological activities. Chem Sci Rev Lett. 2017;2:408–414. [Google Scholar]
- Srinivas M, Satyaveni S, Ram B. Synthesis and Anticancer activity of 1,2,4-oxadiazol linked benzimidazole derivatives. Russ J Gen Chem. 2018;88(12):2653–2657. doi: 10.1134/S1070363218120289. [DOI] [Google Scholar]
- Srinivasa M, Satyavenia S, Ramb B. Design synthesis and biological evaluation of 1,2,4-oxadiazole-isoxazole linked quinazoline derivatives as anticancer agents. Russ J Gen Chem. 2019;89(12):2492–2497. doi: 10.1134/S1070363219120260. [DOI] [Google Scholar]
- Sudhakara DGS, Rao AS, Reddy CVR. Design, synthesis and anticancer activity of 1,2,4-thiadiazole derivatives bearing 1,2,4-oxadiazole. Russ J Gen Chem. 2019;89(8):1696–1701. doi: 10.1134/S1070363219080243. [DOI] [Google Scholar]
- Taflan E, Bayrak H, Er M, Karaoğlu ŞA, Bozdeveci A (2019) Novel imidazo [2, 1-b][1, 3, 4] thiadiazole (ITD) hybrid compounds: Design, synthesis, efficient antibacterial activity and antioxidant effects. Bioorg Chem 89(1):102998
- Tatar E, Kucukguzel SG, Karakus S, De Clercq CE, Andrei G, Snoeck R, Pannecouque C, Oktem OS, Unubol N, Kocagoz T. Synthesis and biological evaluation of some new 1,3,4-thiadiazole and 1,2,4-triazole derivatives from L-methionine as antituberculosis and antiviral agents. Marmara Pharm J. 2015;19(2):88–102. doi: 10.1248/bpb.b15-00698. [DOI] [Google Scholar]
- Tatar E, Karakus S, Kucukguzel SG, Oktem OS, Unubol N, Kocagoz T, De CE, Andrei G, Snoeck R, Pannecouque C. Design, synthesis, and molecular docking studies of a conjugated thiadiazole-thiourea scaffold as antituberculosis agents. Biol Pharm Bull. 2016;39(4):502–515. doi: 10.1248/bpb.b15-00698. [DOI] [PubMed] [Google Scholar]
- Tiemann F, Kruger P, Ueber A. Amidoxime und Azoxime. Dtsch Chem Ges. 1884;17:1685. doi: 10.1002/cber.18840170230. [DOI] [Google Scholar]
- Tingting L, Yichao W, Renshuai L, Lin M, Minyong L, Hao F. Improved antiproliferative activities of a new series of 1,3,4-thiadiazole derivatives against human leukemia and breast cancer cell lines. Chem Res Chin Univ. 2016;32(5):768–774. doi: 10.1007/s40242-016-6159-6. [DOI] [Google Scholar]
- Upare AA, Gadekar PK, Sivaramakrishnan H, Naik N, Khedkar VM, Sarkar D, Choudhari A, Roopan SM. Design, synthesis and biological evaluation of (e)-5-styryl-1,2,4-oxadiazoles as anti-tubercular agents. Bioorg Chem. 2019;86:507–512. doi: 10.1016/j.bioorg.2019.01.054. [DOI] [PubMed] [Google Scholar]
- Villemagne B, Machelart A, Tran NC, Flipo M, Moune M, Leroux F, Piveteau C, Wohlkönig A, Wintjens R, Li X, Gref R, Brodin P, Deprez B, Baulard AR, Willand N. Fragment-based optimized EthR inhibitors with in vivo ethionamide boosting activity. ACS Infect Dis. 2020;6(3):366–378. doi: 10.1021/acsinfecdis.9b00277. [DOI] [PubMed] [Google Scholar]
- Wadhwa P, Bagchi S, Sharma A. In-silico analysis of imidazo[2,1-b][1,3,4]thiadiazole analogs as putative Mycobacterium tuberculosis enoyl reductase inhibitors. Current Drug Therapy. 2017;12(1):46–63. doi: 10.2174/1574885511666160930121123. [DOI] [Google Scholar]
- Wang Y, Li L, Zhang B, Xing J, Chen S, Wan W, Song Y, Jiang H, Luo C, Zheng M. Discovery of novel disruptor of silencing Telomeric 1-Like (DOT1L) inhibitors using a target-specific scoring function for the (S)-Adenosyl-L-methionine (SAM)-Dependent Methyltransferase Family. J Med Chem. 2017;60(5):2026–2036. doi: 10.1021/acs.jmedchem.6b01785. [DOI] [PubMed] [Google Scholar]
- Westhuyzen RV, Winks S, Wilson CR, Boyle GA, Gessner RK, Melo CS, Taylor D, Kock C, Njoroge M, Brunschwig C, Lawrence N, Rao SPS, Sirgel F, Helden PV, Seldon R, Moosa A, Warner DF, Arista L, Manjunatha WH, Smith PW, Street LJ, Chibale K. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: a novel antimycobacterial class targeting mycobacterial respiration. J Med Chem. 2015;58(23):9371–9381. doi: 10.1021/acs.jmedchem.5b01542. [DOI] [PubMed] [Google Scholar]
- WHO, Global Tuberculosis Report (2018)
- Yu S, Liu Y, Zhang Z, Zhang J, Zhao G. Design, synthesis and biological evaluation of novel 2,3-indolinedione derivatives against mantle cell lymphoma. Bioorg Med Chem Lett. 2019;27(15):3319–3327. doi: 10.1016/j.bmc.2019.06.009. [DOI] [Google Scholar]
- Zhang HZ, Kasibhatla S, Kuemmerle J, Kemnitzer W, Mason KO, Qiu L, Crogan-Grundy LC, Tseng B, Drewe J, Cai SXJ. Discovery and structure-activity relationship of 3-Aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers and potential anticancer agents. Med Chem. 2005;48(16):5215–5223. doi: 10.1021/jm050292k. [DOI] [Google Scholar]
- Zumla AI, Gillespie SH, Hoelscher M, Philips PPJ, Cole ST, Abubakar I, McHugh TD, Schito M, Maeurer M, Nunn AJ. New antituberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. Lancet Infect Dis. 2014;14:327–340. doi: 10.1016/S1473-3099(13)70328-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.