

Abstract
Deficiency of MTAP (MTAPdef) mainly occurs because of homozygous loss of chromosome 9p21, which is the most common copy-number loss in metastatic urothelial cancer (mUC). We characterized the clinical and genomic features of MTAPdef mUC in 193 patients treated at MD Anderson Cancer Center (MDACC) and 298 patients from the phase 2 IMvigor210 trial, which investigated atezolizumab in cisplatin-ineligible and platinum-refractory disease. In the MDACC cohort, visceral metastases were significantly more common for MTAPdef (n = 48) than for MTAP-proficient (MTAPprof; n = 145) patients (75% vs 55.2%; p = 0.02). MTAPdef was associated with poor prognosis (median overall survival [mOS] 12.3 vs 20.2 mo; p = 0.007) with an adjusted hazard ratio of 1.93 (95% confidence interval 1.35–2.98). Similarly, IMvigor210 patients with MTAPlo (n = 29) had a higher incidence of visceral metastases than those with MTAPhi tumors (n = 269; 86.2% vs 72.5%; p = 0.021) and worse prognosis (mOS 8.0 vs 11.3 mo; p = 0.042). Hyperplasia-associated genes were more frequently mutated in MTAPdef tumors (FGFR3: 31% vs 8%; PI3KCA: 31% vs 19%), while alterations in dysplasia-associated genes were less common in MTAPdef tumors (TP53: 41% vs 67%; RB1: 0% vs 16%). Our findings support a distinct biology in MTAPdef mUC that is associated with early visceral disease and worse prognosis.
Keywords: Chemotherapy, FGFR, Immunotherapy, MTAP, PIK3CA, Urothelial carcinoma
Patient summary:
We investigated the outcomes for patients with the most common gene loss (MTAP gene) in metastatic cancer of the urinary tract. We found that this loss correlates with worse prognosis and a higher risk of metastasis in internal organs. There seems to be distinct tumor biology for urinary tract cancer with MTAP gene loss and this could be a potential target for treatment.
Located in chromosomal region 9p21, the MTAP gene encodes an enzyme that is essential in the salvage pathway for adenosine and methionine synthesis [1]. Our analysis of focal copy-number losses in The Cancer Genome Atlas muscle-invasive bladder cancer (TCGA-BLCA) cohort (n = 389 patients) previously revealed that 27.2% of cancers exhibited homozygous deletion (HD) of MTAP that was in all cases associated with concurrent HD of CDKN2A, most commonly due to 9p21 loss [2]. We subsequently developed a Clinical Laboratory Improvement Amendments (CLIA)-certified MTAP immunohistochemistry (IHC) test to determine tumor MTAP deficiency (MTAPdef) as a clinically accessible surrogate biomarker to investigate the clinical and biological features of urothelial cancer (UC) with 9p21 loss.
We assessed a total of 212 patients who were treated with standard-of-care chemotherapy and immunotherapy at MD Anderson Cancer Center (MDACC; institutional review board protocol PA17–0577) between November 1997 and April 2018 (Supplementary Fig. 1). CLIA-certified IHC staining for MTAP was carried out using a primary anti-MTAP antibody (1:1200, #11475–1-AP; ProteinTech, Rosemont, IL, USA). Cases with any MTAP cytoplasmic staining were defined as MTAP-proficient (MTAPprof), whereas those with complete loss of MTAP staining were defined as MTAPdef. Somatic alterations in tumor tissue were detected via targeted sequencing; circulating tumor DNA was utilized if tissue was unavailable [3]. Hazard ratios (HRs) and 95% confidence intervals (CIs) were estimated using Cox proportional-hazards regression models to evaluate the association between patient prognostic variables and median overall survival (mOS). All computations were carried out in SAS v9.4 (SAS Institute, Cary, NC).
A total of 193 patients were included in our study after excluding those with undetermined primary tumor, concurrent metastatic disease, or insufficient tissue for MTAP staining (Supplementary Tables 1–3). Median follow-up was 40.6 mo (33.4–44.6). MTAPdef was observed in 24.9% of patients. First-line therapy (Supplementary Table 4) most commonly included chemotherapy (60.6%), followed by immune checkpoint inhibitors (ICIs; 28.5%; Supplementary Fig. 2). Treatment regimens were balanced between the MTAPdef and MTAPprof groups (Supplementary Table 4). mOS was significantly shorter for patients with MTAPdef UC versus MTAPprof UC (12.3 vs 20.2 mo; p = 0.007; Fig. 1A). Similarly, an adverse impact of MTAPdef on progression-free survival (PFS) was noted among the 172 patients who received frontline systemic therapy (4.0 vs 5.5 mo; p = 0.042; Fig. 1B). Further analysis of the frontline ICI and chemotherapy subgroups demonstrated that mOS and PFS in both subgroups were shorter for patients with MTAPdef versus MTAPprof (Supplementary Fig. 3A–D). Since the number of metastatic sites at baseline, baseline visceral metastases, development of subsequent visceral metastases, and Eastern Cooperative Oncology Group performance score ≥1 were all associated with worse OS (Supplementary Table 5), we conducted a multivariable Cox assessment, which showed an independent adverse impact of MTAPdef on OS (HR 1.93, 95% 1.25–2.98; p = 0.003; Supplementary Table 5). Furthermore, patients with MTAPdef mUC had a higher rate of early visceral metastases (75% vs 55.2%; p = 0.02; Fig. 1C, Supplementary Table 6).
Fig. 1 –
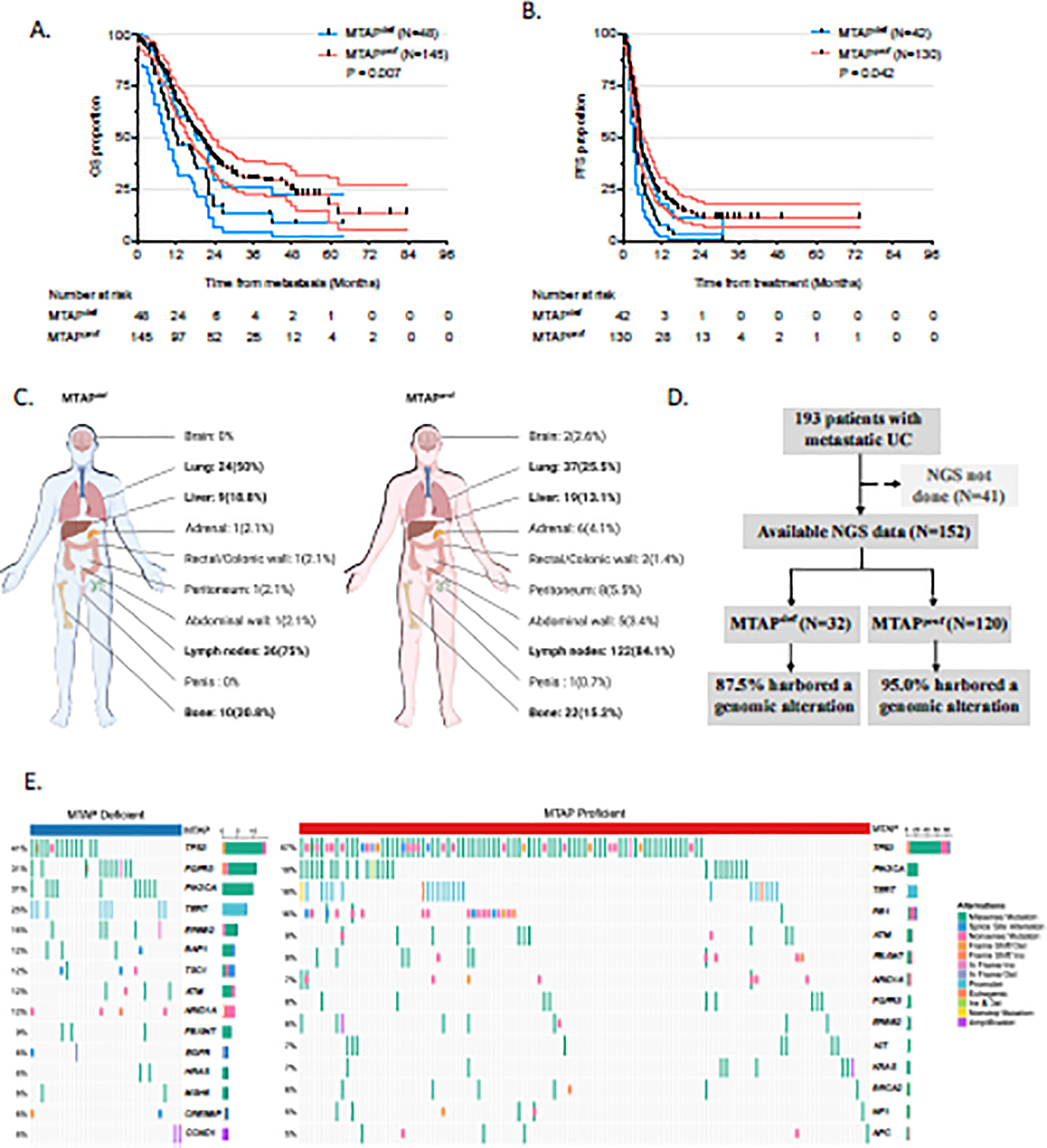
MTAPdef was associated with poor OS and FPS, metastatic status, and genomic alteration. (A) OS for patients stratified by MTAP status. (B) PFS for patients stratified by MTAP status. Only 172 patients who received chemotherapy or immunotherapy as front-line therapy were analyzed for PFS. (C) Proportion of patients by site of metastasis involvement at baseline. Created using a licensed version of biorender.com. (D) CONSORT diagram for patients with available target sequencing data and genomic alterations. (E) Oncoplot showing the most common genomic alterations in our patient cohorts. Patients without genomic alterations and genes with an alteration frequency of <5% in each group were excluded. The right bar plot showed the alteration frequency for each gene. OS = overall survival; PFS = progression-free survival; UC = urothelial cancer; MTAP>def = MTAP-deficient; MTAPprof = MTAP-proficient; NGS = next-generation sequencing.
In total, 152 patients had target sequencing data available (Fig. 1D). The most frequently mutated genes were TP53 (59%), PIK3CA (20%), TERT (18%), FGFR3 (12%), and RB1 (12%; Supplementary Table 7, Supplementary Fig. 4). The dysplasia-associated alterations TP53 and RB1 were detected more often in MTAPprof tumors versus MTAPdef tumors (67% vs 41%; p < 0.001; and 16% vs 0%; p < 0.001), while the hyperplasia-associated alterations FGFR3 and PI3KCA were more common in MTAPdef tumors (31% vs 8%; p < 0.001; and 31% vs 19%; p = 0.05). The majority of FGFR2/3 alterations were functional oncogenic mutations (Supplementary Fig. 5).
Prior efforts highlighted two potential pathways involved in the oncogenesis of early-stage UC: the hyperplasia (FGFR-driven) pathway in low-grade papillary non–muscle-invasive UC (NMIUC) and the dysplasia (TP53- and Rb1-driven) pathway in muscle-invasive UC (MIUC) [4,5]. These studies also suggested that low-grade papillary NMIUCs might progress to MIUCs as a result of CDKN2A loss, a well-known tumor-suppressor gene encoding p16 and p14ARF. MTAP loss in our bladder cancer cohort is a surrogate for 9p21 loss, most commonly encompassing CDKN2A. Our findings of frequent FGFR and PIK3CA alterations among tumors with MTAPdef suggest that these metastatic tumors originating from the hyperplasia pathway have distinct genomic drivers, visceral organ involvement, and clinical prognosis as compared to tumors arising from the dysplasia pathway (Supplementary Fig. 6).
We further assessed clinical and bulk RNA sequencing data for cisplatin-ineligible (cohort 1) and platinum-refractory (cohort 2) patients treated with atezolizumab in the IMvigor210 trial [6]. On the basis of MTAP transcriptional levels, samples were classified into two groups: the bottom 25% was defined as MTAP-low (MTAPlo, n = 75) and the remainder defined as MTAP-high (MTAPhi, n = 223; Fig. 2A). This 25% threshold was chosen to match the prevalence of MTAP deficiency in the TCGA-BLCA and MDACC cohorts. Patients with MTAPlo versus MTAPhi had a higher risk of visceral metastatic disease (82.6% vs 70.8%; p = 0.021; Fig. 2B) and shorter mOS (8.8 vs 12.7 mo; HR 1.4, 95% CI 1.01–1.98; p = 0.029; Fig. 2C,D).
Fig. 2 –
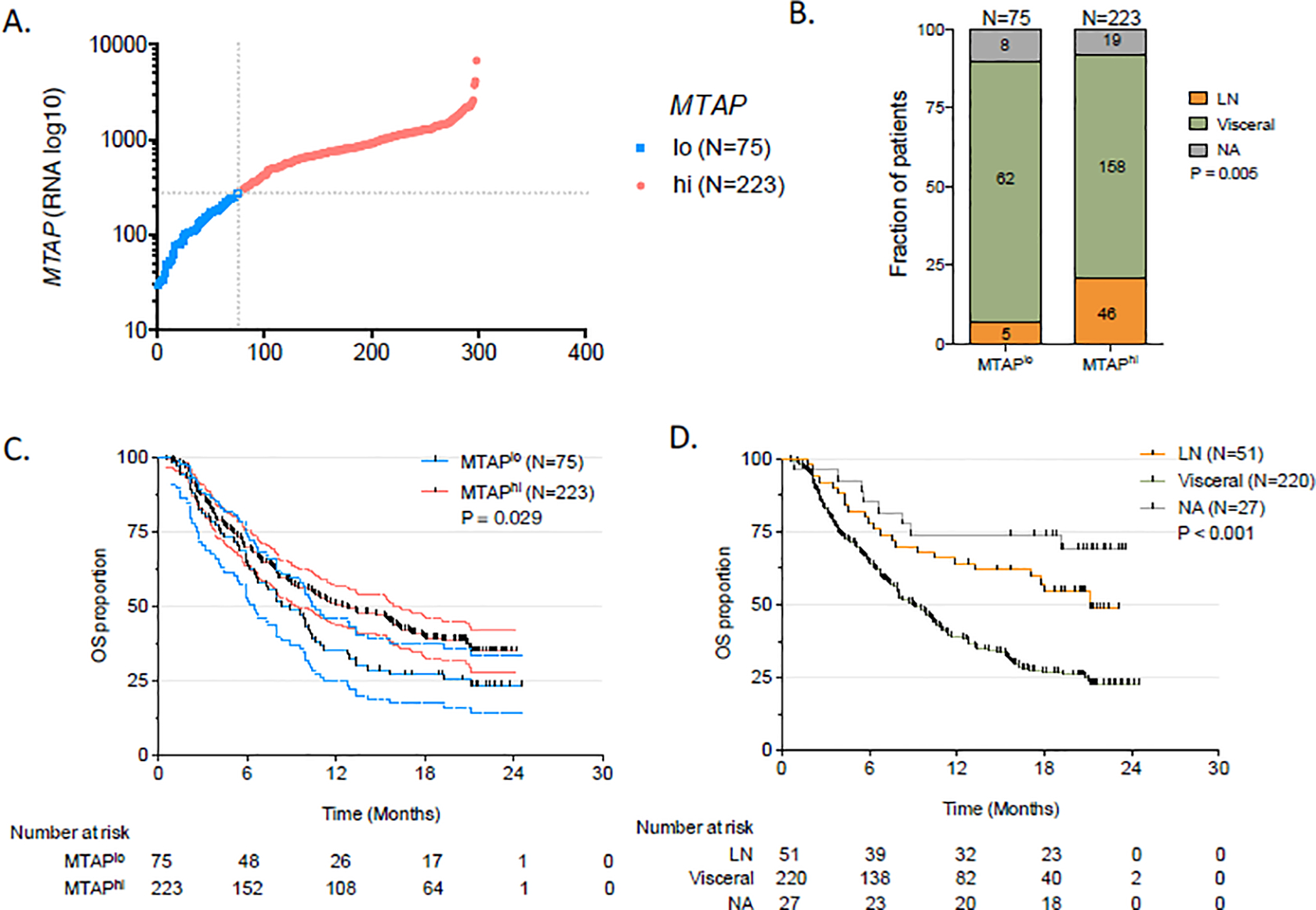
Correlation between MTAP RNA expression, survival, and visceral disease for the validation cohort from the Imvigor210 trial (NCT02951767). (A) MTAP mRNA expression status by number of patients plotted on the x-axis. (B) Metastatic disease in patients with MTAPlo, MTAPhi, and the overall cohort; (C) OS for all patients stratified by MTAP expression. (D) OS for all patients stratified by metastatic sites. OS = overall survival; MTAPlo = MTAP-low; MTAPhi = MTAP-high; LN = lymph node; NA = not available.
The mechanism by which MTAPdef may be associated with visceral metastases is unclear, but could be related to the hyperplasia pathway by which 9p21 loss promotes superficial tumors to acquire muscle invasion [4,5]. MTAP deficiency results in a buildup of its substrate, methylthioadenosine, which inhibits the methylation of STAT1 and, as a result, inhibits interferon signaling pathways [7]. Defects in these pathways have been associated with compromised antitumor immune responses [8]. Furthermore, loss of cell-cycle control due to concomitant CDKN2A HD [9] in this context of immune suppression could explain the early visceral metastases in MTAPdef mUC. Antifolates may be synthetically lethal in MTAPdef mUC via targeting of de novo purine synthesis in the presence of defective salvage synthesis of adenine [10]. We are currently investigating the potential synergy between pemetrexed and avelumab (NCT03744793) along with immune-monitoring studies on pre- and post-treatment MTAPdef tumor tissues.
Our study has the limitations of being a nonrandomized retrospective study from a single institution, with variability in therapy, surveillance, and follow-up times. Furthermore, different methodology (RNA sequencing) was used in the IMvigor210 trial compared to our MDACC cohort (IHC).
In conclusion, our data suggest that MTAPdef in mUC is associated with worse prognosis and a distinct driving biology that may be targetable. These data are hypothesis-generating and provide a basis for future mechanistic studies for the development of effective combination therapies for MTAPdef mUC such as antifolates, cell cycle inhibitors, and/or FGFR inhibitors. Further studies are required to assess the concordance between 9p21 copy-number alterations and MTAP IHC.
Supplementary Material
We investigated the outcomes for patients harboring the most common genomic loss (MTAP loss) in metastatic urothelial cancer. We found that MTAP loss correlates with worse prognosis, a higher risk of visceral involvement, a distinct and potentially targetable tumor biology.
Acknowledgments:
We thank all the patients who participated in this study.
Financial disclosures: Omar Alhalabi certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Jianjun Gao serves as an advisory committee member for CRISPR Therapeutics, Jounce Therapeutics, Polaris, and Seagen, and as a consultant for AstraZeneca, Janssen, Pfizer, and Symphogen. Amishi Y. Shah has received honoraria from Eisai and Oncology Information Group and has received research funding from Bristol-Myers Squibb, Eisai, and EMD Serono. Arlene Siefker-Radtke serves as a consultant for Janssen, Merck, the National Comprehensive Cancer Network, Lilly, Bristol-Myers Squibb, AstraZeneca, BioClin Therapeutics, Bavarian Nordic, Seattle Genetics, Nektar, Genentech, Inovio Pharmaceuticals, and EMD Serono; and has received research funding from the National Institute of Health, the Michael and Sherry Sutton Fund for Urothelial Cancer, Janssen, Takeda, Bristol-Myers Squibb, BioClin Therapeutics, and Nektar. Matthew T. Campbell has served as a consultant or has provided nonbranded educational lectures with honoraria for Pfizer, EMD Serono, AstraZeneca, Eisai, Apricity, Roche, Bristol Myers Squibb, and Merck. Pavlos Msaouel has received honoraria for advisory board participation from Mirati Therapeutics, Exelixis, and Bristol-Myers Squibb, consulting for Axiom Healthcare Strategies, nonbranded educational programs supported by Exelixis and Pfizer; and research funding for clinical trials from Takeda, Bristol-Myers Squibb, Mirati Therapeutics, Gateway for Cancer Research, and the UT MD Anderson Cancer Center. The remaining authors have nothing to disclose.
Funding/Support and role of the sponsor: Jianjun Gao is supported by NCI/NIH grant R01 CA254988–01A1, a Doris Duke Clinical Scientist Development Award (#2018097), an MD Anderson Physician Scientist Award, a Khalifa Physician Scientist Award, an Andrew Sabin Family Foundation Fellows Award, MD Anderson Faculty Scholar Award, the David H. Koch Center for Applied Research of Genitourinary Cancers, the Wendy and Leslie Irvin Barnhart Fund, and the Joan and Herb Kelleher Charitable Foundation. This study was also supported by SMF Core Grant CA016672 (SMF) and the IRG start-up research funds provided to L.W. by MD Anderson Cancer Center. P.M. is supported by a career development award from the American Society of Clinical Oncology, by an MD Anderson Khalifa Scholar Award, and by an MD Anderson Physician-Scientist Award. Omar Alhalabi is supported by a young investigator award from the Conquer Cancer Foundation of the American Society of Clinical Oncology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kamatani N, Carson DA. Dependence of adenine production upon polyamine synthesis in cultured human lymphoblasts. Biochim Biophys Acta 1981;675:344–50. [DOI] [PubMed] [Google Scholar]
- 2.Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017;171:540–56.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson JB, Solano L, Yuki A, et al. Analytical validation of the Oncomine Pan-Cancer Cell-Free Assay in a CLIA- and CAP-regulated laboratory for detection of solid tumor-derived variants in blood plasma. J Clin Oncol 2019;37(15 Suppl):e14614. [Google Scholar]
- 4.Sanli O, Dobruch J, Knowles MA, et al. Bladder cancer. Nat Rev Dis Primers 2017;3:17022. [DOI] [PubMed] [Google Scholar]
- 5.Rebouissou S, Hérault A, Letouzé E, et al. CDKN2A homozygous deletion is associated with muscle invasion in FGFR3-mutated urothelial bladder carcinoma. J Pathol 2012;227:315–24. [DOI] [PubMed] [Google Scholar]
- 6.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mowen KA, Tang J, Zhu W, et al. Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell 2001;104:731–41. [DOI] [PubMed] [Google Scholar]
- 8.Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993;366:704–7. [DOI] [PubMed] [Google Scholar]
- 10.Alhalabi O, Campbell MT, Slack-Tidwell R, et al. A phase II trial to evaluate pemetrexed clinical responses in relation to tumor methylthioadenosine phosphorylase (MTAP) gene status in patients with previously treated metastatic urothelial carcinoma. J Clin Oncol 2019;37(7 Suppl):385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.