

Abstract
Background and Purpose:
Inwardly rectifying K+ (Kir) channels located on the basolateral membrane of epithelial cells of the distal nephron play a crucial role in K+ handling and blood pressure control, making these channels an attractive target for the treatment of hypertension. The purpose of the present study was to determine how the inhibition of basolateral Kir4.1/Kir5.1 heteromeric K+ channel affects epithelial sodium channel (ENaC)-mediated Na+ transport in the principal cells of cortical collecting duct (CCD).
Experimental Approach:
The effect of fluoxetine, amitriptyline, and recently developed Kir inhibitor, VU0134992, on the activity of Kir4.1, Kir4.1/Kir5.1, and ENaC were tested using electrophysiological approaches in Chinese hamster ovary (CHO) cells transfected with respective channel subunits, cultured polarized epithelial mCCDcl1 cells, and freshly isolated rat and human CCD tubules. To test the effect of pharmacological Kir4.1/Kir5.1 inhibition on electrolyte homeostasis in vivo and corresponding changes in distal tubule transport, Dahl salt-sensitive rats were injected with amitriptyline (15 mg kg−1 day−1) for three days.
Key Results:
We found that inhibition of Kir4.1/Kir5.1, but not Kir4.1 channel, depolarizes cell membrane, induces the elevation of intracellular Ca2+ concentration, and suppresses ENaC activity. Furthermore, we demonstrate that amitriptyline administration leads to a significant drop in plasma K+ level, triggering sodium excretion and diuresis.
Conclusion and Implications:
Present data uncovers a specific role of the Kir4.1/Kir5.1 channel in the modulation of ENaC activity and emphasizes the potential for using Kir4.1/Kir5.1 inhibitors to regulate electrolyte homeostasis and blood pressure.
Keywords: Kcnj10, Kcnj16, hypokalemia, amitriptyline, Kir (IRK) channels, ENaC
1 |. INTRODUCTION
According to the Centers for Disease Control and Prevention and the American Heart Association, nearly one-third of the adult population in the United States has hypertension, which is a leading risk factor for cardiovascular disease, stroke, kidney failure, and all‐cause mortality (Virani et al., 2020). Observational studies and treatment trials provide strong support for a link between excess dietary Na+ intake and the incidence of hypertension. The effect of Na+ on blood pressure is dependent on diet composition, specifically on the ratio of Na+ and K+ ions (Staruschenko, 2018). The beneficial effects of a potassium-enriched diet for blood-pressure control are supported by animal studies and human trials (Dahl et al., 1972; Mente et al., 2014; Tobian, 1991). Understanding the regulatory machinery involved in the interplay between Na+ and K+ homeostasis is a vital milestone to develop an appropriate therapy to prevent hypertension and related cardiovascular and renal disorders.
The aldosterone-sensitive distal nephron (ASDN) plays an essential role in the control of the pressure–natriuresis relationship and long-term regulation of blood pressure. Urinary Na+/water reabsorption and K+ excretion in this region are tightly interconnected and controlled by various channels and transporters expressed in the apical and basolateral membranes of epithelial cells along ASDN. Recent genetic human and animal studies uncovered the critical role of basolateral Kir channels, specifically monomeric Kir4.1 (encoded by Kcnj10 gene) and heteromeric Kir4.1/Kir5.1 (encoded by Kcnj10 and Kcnj16), of the ASDN to control electrolyte homeostasis and blood pressure. Loss-of-function mutations in the genes encoding Kir4.1 are associated with salt-wasting, hypotension, and severe hypokalemia in both human and animal models (Bockenhauer et al., 2009; Cuevas et al., 2017). Recent studies further identified several mutations in KCNJ16 encoding Kir5.1, which result in hypokalemia, salt-wasting, and disturbed acid-base homeostasis (Schlingmann et al., 2021). Deletion of Kcnj16 in Dahl salt-sensitive (SS) rats (SSKcnj16−/−) targeting heteromeric Kir4.1/Kir5.1 channel, a major basolateral Kir channel in ASDN, results in a renal phenotype resembling distinctive features of human and murine loss of function Kir5.1 mutations, including hypokalemia and reduced blood pressure (Manis et al., 2019; Palygin et al., 2017; Schlingmann et al., 2021) providing evidence that modulation of Kir4.1/Kir5.1 channel activity may be a powerful tool for blood pressure and hyperkalemia control in salt-induced hypertension.
The use of genetically modified rodent models provides important insight into the mechanisms of human diseases, but the interpretation of these data is generally complicated due to the possible existence of compensatory mechanisms. The aim of the present study was to dissect the effect of pharmacological inhibition of homomeric Kir4.1 and heteromeric Kir4.1/Kir5.1 channels on the epithelial sodium channel (ENaC) activity in CCD using overexpressed cells, cell culture, and freshly isolated tubules from Dahl SS rat and human kidneys. Using established and newly discovered inhibitors of basolateral Kir channels, amitriptyline, fluoxetine, and VU0134992, we demonstrated that acute inhibition of Kir4.1/Kir5.1 channel but not Kir4.1 induces suppression of ENaC activity in rodent and human CCD principal cells. The suppression of ENaC activity is presumably mediated by the cell depolarization and increase in cytosolic free Ca2+ concentration. We also show that similar to the results obtained in SSKcnj16–/– rats, the injection of SS rats by amitriptyline promotes a significant decline in blood K+ level and induces salt excretion despite the upregulation of renal sodium-chloride co-transporter.
2 |. METHODS
2.1 |. Experimental animals and human kidneys (Compliance with requirements for studies using animals and human tissue)
To fully test our hypothesis, some experiments in our study were performed in whole animals and in the tissue of highly-differentiated cells that contain native Kir channels and retain their physiological function, that cannot be fully replicated by cell lines. All animal studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocol review and approval by the Institutional Care and Use Committee of the Medical College of Wisconsin. Animal studies are reported in compliance with the ARRIVE guidelines and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020; McGrath & Lilley, 2015). The re-derived Rapp Dahl salt-sensitive (SS; RRID:RGD_69369) rat used in our studies (SS/JrHsdMcwi) has been inbred for more than 50 generations at the Medical College of Wisconsin and used to compare the outcomes of pharmacological inhibition of Kir channels with results obtained earlier in the same SS strain with a null mutation of Kcnj16 (Manis et al., 2021; Manis et al., 2019; Palygin et al., 2017). 8-12-week-old male SS rats (TBW approx. 220 g) were used. Animals were kept in standard plastic home cages (Allentown 40 IVC) on hardwood chip bedding. For urine collection, animals were moved into metabolic cages (# 40615; Laboratory Products, one animal per cage). Rats were maintained on a standard 12/12 hrs light-dark cycle with unlimited access to water and rodent chow (0.4% NaCl, 0.36% K, # D113755, Dyets Inc., Bethlehem, PA).
Studies in this manuscript that involve human kidneys are not considered “human subjects research” for the purpose of addressing human subject protection because they are only secondary analysis of coded discarded organs, and none of the investigators can readily ascertain the identity of subjects. MCW IRB Committee reviewed current studies and determined that it does not meet criteria for human subject research since information is not individually identifiable and it does not involve intervention or interaction with living individuals. For experiments with human tissue, human kidneys (no patient identifiers were received) were obtained from an organ procurement company, where they were harvested with the intent to be used for transplantation but were not used and destined to be discarded. The kidneys were stored in a transplant preservation UW solution (ViaSpan) on ice before the isolation of nephron segments by vibrodissociation technique as described (Isaeva et al., 2019; Khedr et al., 2019).
2.2 |. Isolation of renal tubules
Animals were deeply anesthetized with 5% isoflurane in 30% O2/70% N2, and kidneys were harvested. Thereafter rats were humanely euthanized by a thoracotomy. Kidneys were extensively washed in the incubation solution contained (in mM): 119 NaCl, 4.5 KCl, 2 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 5 glucose (pH = 7.35). Both human and rodent kidneys were manually cut into slices (0.5–1 mm thickness) and stored at room temperature in oxygenated incubation solution (95% O2 - 5% CO2) contained (in mM): 119 NaCl, 4.5 KCl, 2 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 5 glucose (pH = 7.35). For renal tubule separation, kidney slices were enzymatically pretreated for 20–25 min at 37°C in physiological saline solution (PSS) of the following composition (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES pH=7.35) containing 0.7 mg ml−1 collagenase type I (Alfa Aesar, Ward Hill, MA) and 4 mg ml−1 of dispase II (Roche Diagnostics, Mannheim, Germany). After enzymatic treatment, slices were extensively washed in PSS and dissociated by applying a local mechanical vibration movement towards the slice surface using a flame-sealed glass micropipette connected to a frequency oscillator (GFG-8250A, Instek).
2.3 |. Chinese hamster ovary (CHO) cell culture and transfection
CHO (RRID:CVCL_0213) cells were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained under standard culture conditions (F12K medium supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin) as previously described (Staruschenko et al., 2005). In transfection experiments, the cells were trypsinized, plated into 35-mm tissue culture dishes with coverslips, and grown for 20–24 hrs prior to transfection with Lipofectamine (Thermo Fisher Scientific). For expression of Kcnj in CHO cells, the cells were transiently cotransfected with green fluorescent protein (eGFP) (0.25 μg of cDNA/35-mm dish) and channel plasmids: Kcnj10 (0.5 μg) for homomer expression or Kcnj10 and Kcnj16 (0.25 μg each) for heteromer expression. Kcnj10 and Kcnj16 plasmids were kindly provided by Dr. Richard Warth from the Institute of Physiology, Regensburg, Germany (Reichold et al., 2010). For expression of ENaC in CHO cells, subunits of α-, β-, γ-ENaC cDNA transfection ratios of 1:1:1 were used (0.3 μg of each cDNA/35-mm dish). To identify transfected cells, 0.5 μg of eGFP was also added to the cDNA mix. After transfection, cells were cultured on coverslips for 24–48 hours, and GFP-positive cells were selected for measurements.
2.4 |. Mouse cortical collecting duct (mCCDcl1) cell culture and short-circuit current (Ieq) measurements
mCCDcl1 cells (Gaeggeler et al., 2005) were provided by Prof. Bernard Rossier (University of Lausanne, Switzerland) and were cultured in 75-cm flasks in Dulbecco’s Minimum Essentials Medium (DMEM/F-12; Cat #11330, Gibco) supplemented with 2% Fetal Bovine Serum (FBS), 5 μg ml−1 insulin, 5 μg ml−1 human apo-Transferrin, 10 ng ml−1 epidermal growth factor (EGF), 1 nM triiodo-L-thyronine Na+ salt, 60 nM Na+ selenite, 50 nM dexamethasone, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin as previously described (Gaeggeler et al., 2005; Pavlov et al., 2014). For patch-clamp studies, cells were seeded into 35 mm cell culture dishes on coverglass chips (5 × 5 mm) and experiments were performed 3–7 days after passage. Transepithelial voltage (Vt) and resistance (Rt) were measured using the Millicell Electrical Resistance System (Millipore) with dual Ag/AgCl pellet electrodes on the cells grow on Transwell permeable supports (Costar) to form polarized monolayers. Intact polarized monolayer formation exhibiting Rt greater than >1000 Ω cm2 and Vt greater than >20 mV was used for experiments (Assmus et al., 2018; Mukherjee et al., 2017). The equivalent short circuit current (Ieq) values were calculated using Ohm’s law. Relative current indicates normalized Ieq values. Apical application of 10 μM amiloride produced near zero drops of Ieq current, demonstrating that the recorded currents were attributable to ENaC. We did not observe significant changes in Rt within 2 hrs following drugs application.
2.5 |. Electrophysiological studies
The single-channel activity was determined in cell-attached patches made under voltage-clamp configuration. Potassium channel and epithelial sodium channel (ENaC) activity were recorded either on CHO cells overexpressing corresponding channels or freshly isolated CCDs. Potassium channel activity was recorded from the basolateral membrane of principal CCD cells. To directly assess ENaC properties, CCDs were split open, and channel activity was recorded from the apical membrane of principal cells.
Data were acquired and subsequently analyzed with Axopatch 200B amplifier interfaced via a Digidata 1440A to a PC running the pClamp 10.6 suite of software (Molecular Devices; RRID:SCR_011323). The recordings were filtered with an 8-pole, low-pass Bessel filter LPF-8 (Warner Inst.) at 0.3 kHz and 0.1 kHz for potassium current and ENaC recordings, correspondently. Events were inspected visually before acceptance to ensure that patches contained activity consistent with the channel of interest and are not contaminated with other conductance. In paired experiments, single channel activity was analyzed over a span of 60–120 s for each experimental condition (over 100 number of open/close events) after a new steady-state was reached in response to a treatment. Automatically detected event histograms fit with single or multiple Gaussian distributions were used to determine unitary current amplitudes (Clampfit 10.3 software). Channel activity and Po were assessed using Clampfit 10.3 software (Molecular Devices). To calculate Po in paired experiments, N was fixed as the greatest number of active channels observed in control or experimental conditions. All electrophysiological experiments were performed at room temperature (22–24 °C). The patch pipettes were pulled with a horizontal puller (Sutter P-97; Sutter Inst.), and their resistances were ranged from 7 to 12 MΩ when filled with corresponding pipette solution. All electrophysiological recordings were performed using PSS as extracellular solution (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35). The single-channel activity of Kir4.1/Kir5.1 and Kir4.1 channels was measured using patch pipettes filled with a solution of the following composition (in mM): 150 KCl, 2 MgCl2, and 10 HEPES (pH 7.35). For the recording of ENaC activity, patch pipettes were filled with a solution of the following composition (in mM): 140 LiCl, 2 MgCl2, and 10 HEPES (pH 7.35).
2.6. Western blotting
Western blotting has been conducted according to BJP Guidelines (Alexander et al., 2018). For the Western blotting analyses, animals were separated into two groups: amitriptyline and control. The first group was treated for three days with i.p. injections of 15 mg kg−1 amitriptyline, while age-matched control rats were treated with vehicle. Kidneys were harvested as it was described in Methods 2.2, flushed with PBS, excised, and cut in 1- to 2-mm slices under a binocular microscope with ×6 magnification. The approximate apical kidney cortex sections were carved and then diced into small pieces with a razor blade. Samples were pulse sonicated in GLB with a protease inhibitor cocktail (Roche) for 10 seconds and spin cleared at 10,000 g for 10 minutes. The resulting supernatant was subjected to PAGE, transferred onto nitrocellulose membrane (Millipore) for probing with antibodies (tNCC # 2430347, Millipore; pNCC # p1311-53 PhosphoSolutions; αENaC # spc-403, γENaC # spc-405 StressMarq), and subsequently visualized by enhanced chemiluminescence (ECL; Amersham Biosciences).
2.7. Confocal microscopy.
For confocal microscopy, mCCDcl1 cells were cultured on glass chips (size 0) to develop monolayer. Cells were loaded either with 2 μl ml−1 FluoVolt™ dye and 10 μl ml−1 PowerLoad Concentrate for membrane potential monitoring, or with 1 μg ml−1, Fluo8HT AM (AAT Bioquest) for Ca2+ imaging studies in the culture media for 20–30 min and incubated in CO2 incubator (37°C). After loading, chips with cells were rinsed twice with PSS and mounted into recording/perfusion chamber (Warner Instruments, Hamden, CT). Confocal imaging recordings were performed in PSS using the Nikon A1-R laser scanning confocal microscope system and analyzed using ImageJ 1.52i software (RRID:SCR_003070), as previously reported (Golosova et al., 2020).
2.7 |. Urine/serum biochemistry measurement
Rats were placed in metabolic cages to acclimate for 24 hrs. Urine was collected over 24 hrs before (two consecutive days of vehicle injection) and after intra-peritoneal (i.p.) administration of amitriptyline (as indicated in 2.6. Western blotting). Blood was collected from the tail vein before animal placement into metabolic cages and 24 hrs after the last injection of amitriptyline. Blood and urine electrolytes, and creatinine levels were analyzed using the ABL800 FLEX analyzer immediately after collection. Creatinine clearance was estimated using the formula: creatinine clearance = (creatinine urine concentration, mmol l−1 × urine flow, ml hr−1)/creatinine serum concentration, mmol l−1.
2.8 |. Drugs
Fluoxetine hydrochloride (# 0927) and Amitriptyline hydrochloride (# 4456/50) were purchased from Tocris, VU0134992 (VU992) was synthesized at Vanderbilt University Medical Center as recently reported (Kharade et al., 2018). All other drugs were purchased from Sigma. All drugs were dissolved in PSS, except VU992, which was initially dissolved in DMSO and then added to aqueous solutions. PSS or DMSO (for VU992) were used as vehicle controls, correspondingly.
2.9 |. Data analysis and statistics
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018; Curtis et al., 2015). The declared group size is the number of independent values, and that statistical analysis was done using these independent values. The statistical analysis was undertaken only for studies where each group size was at least n=5. The experiments on freshly isolated human tissue are subject to availability, so these datasets had limited n per group and were not subjected to statistical analysis. The studies were designed to generate groups of equal size for the small groups (n<20), using randomization and blinded analysis. For group size calculation, we assumed a type I error rate of 0.05 and statistical power of at least 85%, as previously reported with similar experimental designs. Determination or exclusion of outliners within datasets was not applied. We estimated the acute effect of VU992 and amitriptyline on single-channel activity at 5 min following drugs application when the magnitude of the effect has reached its steady-state level.
Data are expressed as the mean ± SE of the mean, and data analysis for all protocols was performed blinded by an independent analyst. Data were tested for normality (Shapiro-Wilk) and equal variance (Levene’s homogeneity test). Paired t-test was used to detect the statistical difference between two variables for the same subject. For more than two groups of variables for the same subject, the repeated measurements (RM) ANOVA was used with corresponding Mauchly’s test of sphericity and Greenhouse-Geisser correction. Unpaired data were analyzed with one-way ANOVA. Tukey or Dunnett multiple-comparisons adjustments were conducted only if the ANOVA F value was significant. P values of < 0.05 (indicated *) were considered significant. The Tukey test was used to compare every mean with every other mean. The Dunnett test was used to assess differences from the control (application of PSS or DMSO as a vehicle control). SigmaPlot 12.5 (RRID:SCR_003210) or OriginPro 9.0 (RRID:SCR_014212) software was used to perform statistical tests.
2.9 |. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3 |. RESULTS
3.1 |. Effect of the antidepressant drugs fluoxetine and amitriptyline on Kir4.1 and Kir4.1/Kir5.1 channels
It was previously reported that the serotonin reuptake blocker fluoxetine effectively inhibits Kir4.1 channel activity in heterologous expression cell systems (Furutani et al., 2009; Kharade et al., 2018; Ohno et al., 2007). Our studies in CHO cells overexpressing Kir4.1 or both Kir4.1 and Kir5.1 subunits confirmed that the application of 100 μM fluoxetine suppresses homomeric Kir4.1 single-channel activity (Figure 1a) but does not affect Kir4.1/Kir5.1 (Figure 1b). It is well established that Kir4.1/Kir5.1 is a major basolateral channel in distal and collecting duct principal cells which has a higher single-channel conductance relative to Kir4.1 homomers, as confirmed by current-voltage relations shown in Figure 1 (21 ± 2 vs 37 ± 2 pS, for homo- and hetereomeric channels correspondingly). Our recent data also revealed that tricyclic antidepressant (TCA) amitriptyline, but not fluoxetine, drastically decreases the macroscopic K+-selective conductance in CCD principal cells (Zaika et al., 2016). Here we demonstrated that the application of amitriptyline (100 μM) in CHO cells overexpressing Kir4.1 and Kir5.1 subunits produces up to a 51% decrease in Kir4.1/Kir5.1 single-channel activity (NPo) (Figure 1b). However, contradictory to previous reports (Kharade et al., 2018; Su et al., 2007), amitriptyline did not affect the homomeric Kir4.1 channel (Figure 1a).
FIGURE 1.
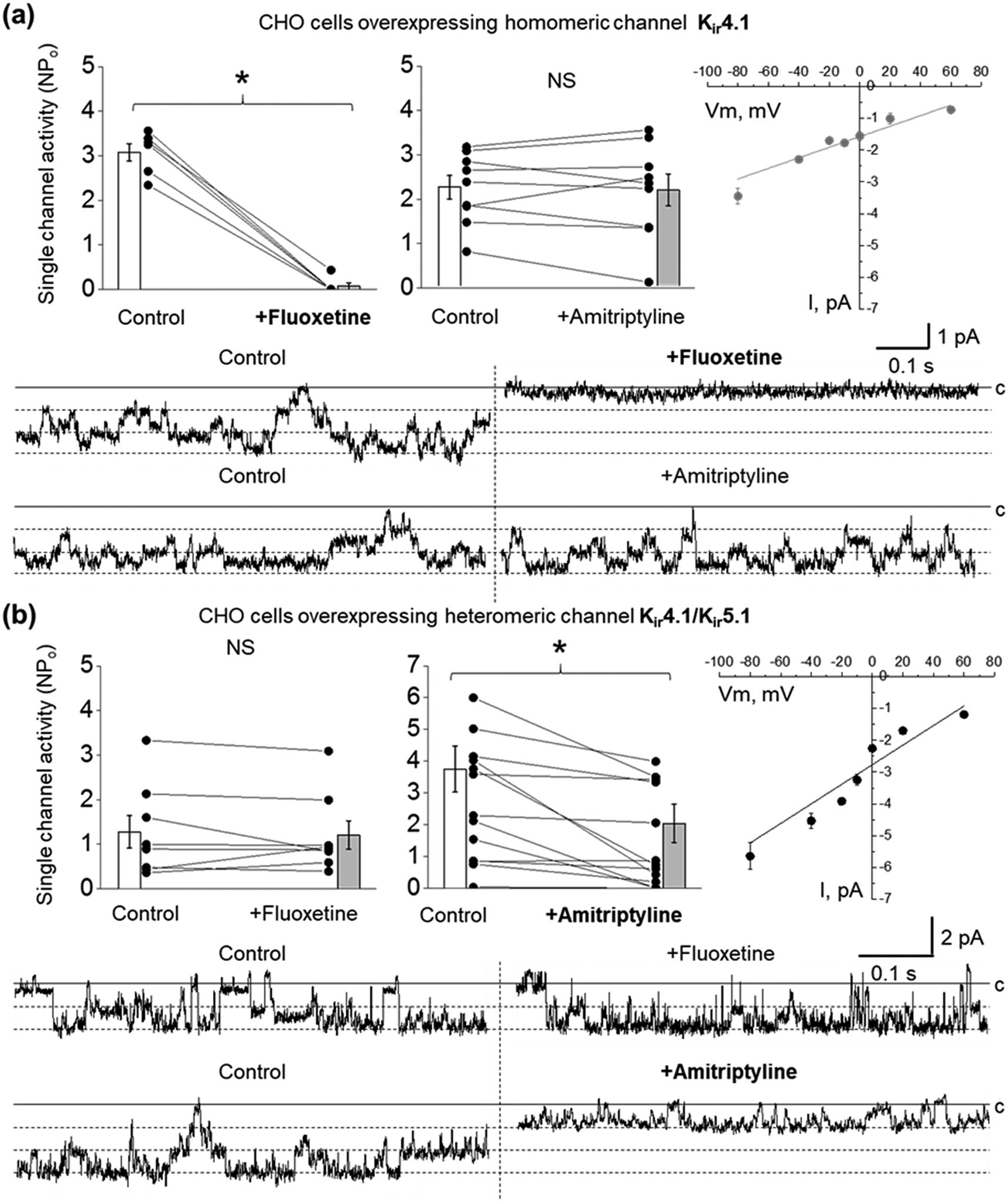
Effect of fluoxetine and amitriptyline on Kir4.1 and Kir4.1/Kir5.1 currents overexpressed in CHO cells. Summary graphs of homomeric Kir4.1 (a) and heteromeric Kir4.1/Kir5.1 (b) single-channel activity (NPo) after application of PSS as vehicle control, 100 μM fluoxetine (Kir4.1, n = 6; Kir4.1/Kir5.1, n = 8) or 100 μM amitriptyline (Kir4.1, n = 9; Kir4.1/Kir5.1, n =13). Paired sample t-test *P < 0.05. Single-channel closed (c) and open states for the representative recorded currents are indicated by solid and dashed lines. Current-voltage relationships of Kir4.1 (n = 6) and heteromeric Kir4.1/Kir5.1 (n = 6) current are shown in the left panels (linear fitting, instrumental weight. Adj. R-Square ≥0.83).
3.2 |. Inhibition of Kir4.1/Kir5.1 but not Kir4.1 channel affects epithelial Na+ transport in cultured mCCDcl1 cells
Next, we examined whether inhibition of Kir4.1 or Kir4.1/Kir5.1 channels would affect apical Na+ transport in polarized mCCDcl1 epithelia. The expression of both Kir4.1 and Kir5.1 subunits in mCCDcl1 cell line was confirmed by immunohistochemical studies (Figure S1). Figure 2a represents the effect of the basolateral application of fluoxetine or amitriptyline on equivalent short-circuit current (Ieq) measured in polarized epithelial mCCDcl1 cells at different time points. We show that the application of 100 μM fluoxetine does not affect Ieq up to 2 hrs after the treatment. However, 50 μM amitriptyline significantly decreases Ieq after application in all time points of the protocol. Importantly, amitriptyline produces a similar effect on Ieq when applied either to the basolateral or apical side of the epithelia (Figure 2b). Amitriptyline is a highly lipophilic molecule (Gupta et al., 1999), and like other TCA is characterized by rapid permeabilization due to affinity for cell membranes, enabling access to K+ channel from the cytosol. The suppressive effect of amitriptyline on Ieq was dose-dependent with an IC50 of 15.0 μM (Figure 2c). To eliminate the possibility that the suppressive effect of amitriptyline on Ieq may be related to the inhibition of ROMK channels, we conducted studies with a specific blocker of ROMK, VU591 (Fodstad et al., 2009; Kharade et al., 2018). Our data demonstrates that the apical application of 1 μM VU591 does not affect the amiloride-sensitive Ieq in polarized mCCDcl1 epithelia (Figure S2). We further supported our observation that basolateral K+ conductance can modulate Na+ transport using a recently developed Kir channel blocker, VU0134992 (VU992) (Kharade et al., 2018). It was shown that this inhibitor is 9-fold more selective for homomeric Kir4.1 (IC50 value of 0.97 μM) over heteromeric Kir4.1/Kir5.1 (IC50 = 9 μM) channels. Figure 2d shows that basolateral application of VU992 in concentrations specific for Kir4.1 (3 μM) has no effect on Ieq. However, higher concentrations of VU992 (30 and 300 μM) which are able to block both homo and heteromeric channels, significantly reduced Na+ transport in polarized epithelia. The effect of high VU992 concentrations was similar to amitriptyline (Figures 2e). Furthermore, we confirmed these observations by analyzing the effect of amitriptyline and high concentrations of VU992 on ENaC activity in mCCDcl1 cells. Figure 3 shows that acute application of either amitriptyline or 30 μM VU992 significantly reduces ENaC single-channel activity. The amplitude of single ENaC current was not affected neither after amitriptyline or VU992 application (0.59 ± 0.08 vs. 0.54 ± 0.07 (n = 7) and 0.82 ± 0.05 vs. 0.65 ± 0.06 (n = 6), pA for control vs. amitriptyline or VU992-treated, correspondingly). Moreover, preincubation of mCCDcl1 cells in amitriptyline for more than 2 hrs resulted in decreased ENaC activity (NPo control: 1.03 ± 0.20 (n=28) vs amitriptyline-treated 0.11 ± 0.04 (n=8), paired sample t-test *P < 0.05). To address the possibility that amitriptyline affects Na+ transport by direct inhibition of ENaC, we examined the effect of 100 μM amitriptyline or 30 μM VU992 on the single-channel activity of ENaC overexpressed in CHO cells. Our data demonstrates that both drugs did not affect ENaC activity (Figure S3).
FIGURE 2.
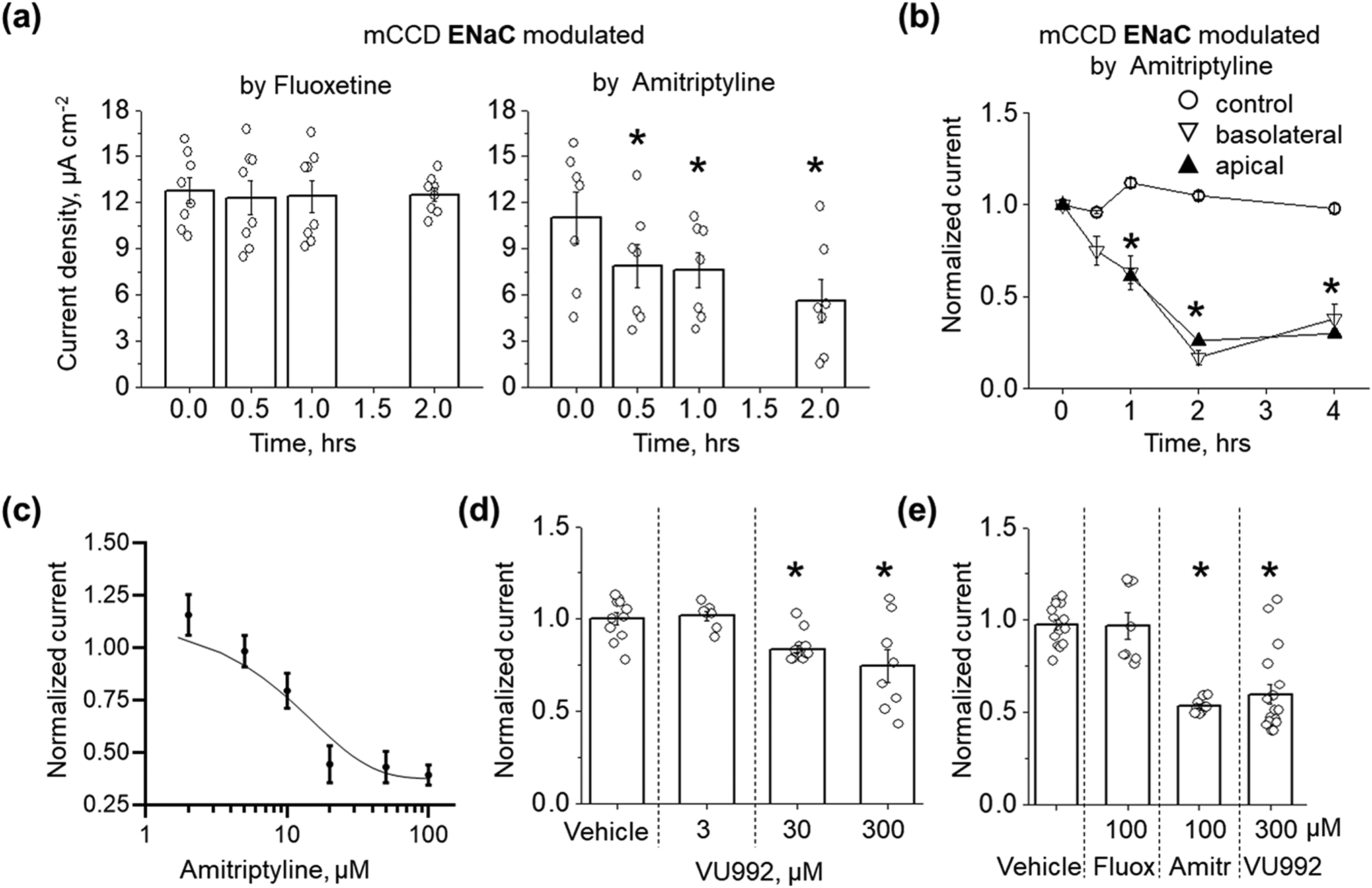
Effect of potassium channel inhibitors on Ieq in mCCDcl1 cells. (a) Effect of basolateral application of 100 μM fluoxetine (n = 8) and 50 μM amitriptyline (n = 7) on Ieq measured at different time points. One Way RM ANOVA with a Greenhouse-Geisser correction (F(1.81, 10.87) = 14.03, P < 0.002), post hoc test using Dunnett correction *P < 0.05 compared to starting point. (b) Effect of apical or basolateral application of 50 μM amitriptyline on amiloride-sensitive Na+ epithelial transport in mCCDcl1 cells (n = 5 for each group, One Way ANOVA, Dunnett post-hoc test *P < 0.05 compared to control). (c) Concentration-dependence of amitriptyline effect on Ieq (n = 6 per point). (d) Modulation of Ieq by different concentrations of VU992 (vehicle, n = 11; 3 μM, n = 7; 30 μM, n = 8; 300 μM, n = 8). (e) Summary graph for the ENaC current modulation (Ieq) by Kir channels blockers (vehicle, n = 14; fluoxetine, n = 8; amitriptyline, n = 9; VU992, n = 17). Ieq for (d) and (e) recorded 2 hrs after drugs or vehicle application (One Way ANOVA, Dunnett post-hoc test, *P < 0.05 compared to vehicle).
FIGURE 3.
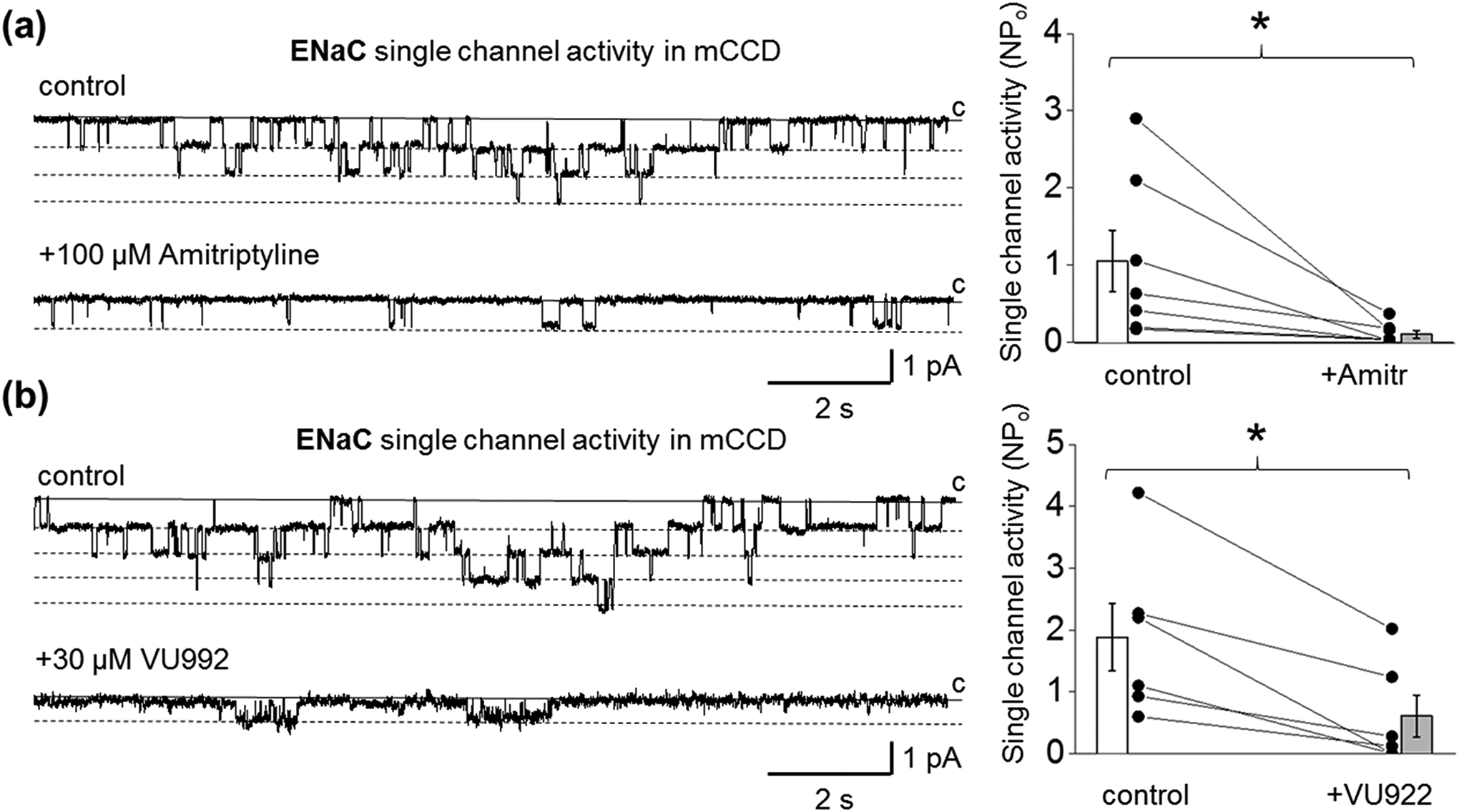
Acute inhibition of Kir4.1/Kir5.1 channel suppresses ENaC single-channel activity in mCCDcl1 cells. Representative traces of ENaC activity recorded in cell-attached mode after application of PSS or DMSO as vehicle control or application of 100 μM amitriptyline (a) or 30 μM VU992 (b). Conformational closed (c) and open states are indicated by solid and dashed lines. Patches were clamped to -Vp = −60 mV. Corresponding summary graphs show that application of 100 μM amitriptyline (n = 7) or 30 μM VU992 (n = 6) induced a decrease in ENaC single-channel activity. Paired sample t-test *P < 0.05.
3.3. Inhibition of Kir4.1/Kir5.1 channel induced cellular membrane depolarization and intracellular Ca2+ transient in cultured mCCDcl1 cells.
It is well established that basolateral Kir channels are involved in the maintenance of a negative resting membrane potential in epithelial cells (Zaika et al., 2016). To evaluate whether inhibition of Kir4.1/Kir5.1 channel will affect epithelial cell membrane potential, cultured mCCDcl1 cells were loaded with voltage-sensitive fluorescent probe FluoVolt™, which increases its emission upon membrane depolarization. Figures 4a and 4b show that application either 100 μM amitriptyline or 30 μM VU992 induced substantial depolarization of membrane potential in these cells. We also examined the effect of different inhibitors of Kir channels on intracellular Ca2+ transient in cultured mCCDcl1 cells loaded with Fluo8 AM dye. We observed the fast and substantial increase in Fluo8 AM fluorescence intensity following the application of 30 μM VU992, which accounts for the elevation of the intracellular Ca2+ concentration (Figure 4c). Summary data reveal that the application of 100 μM amitriptyline or 30 μM VU992 produces a significant increase of intracellular Ca2+ level in mCCDcl1 cells. Application of 100 μM fluoxetine or 3 μM VU992 did not affect Ca2+ transient (Figure 4d).
FIGURE 4.
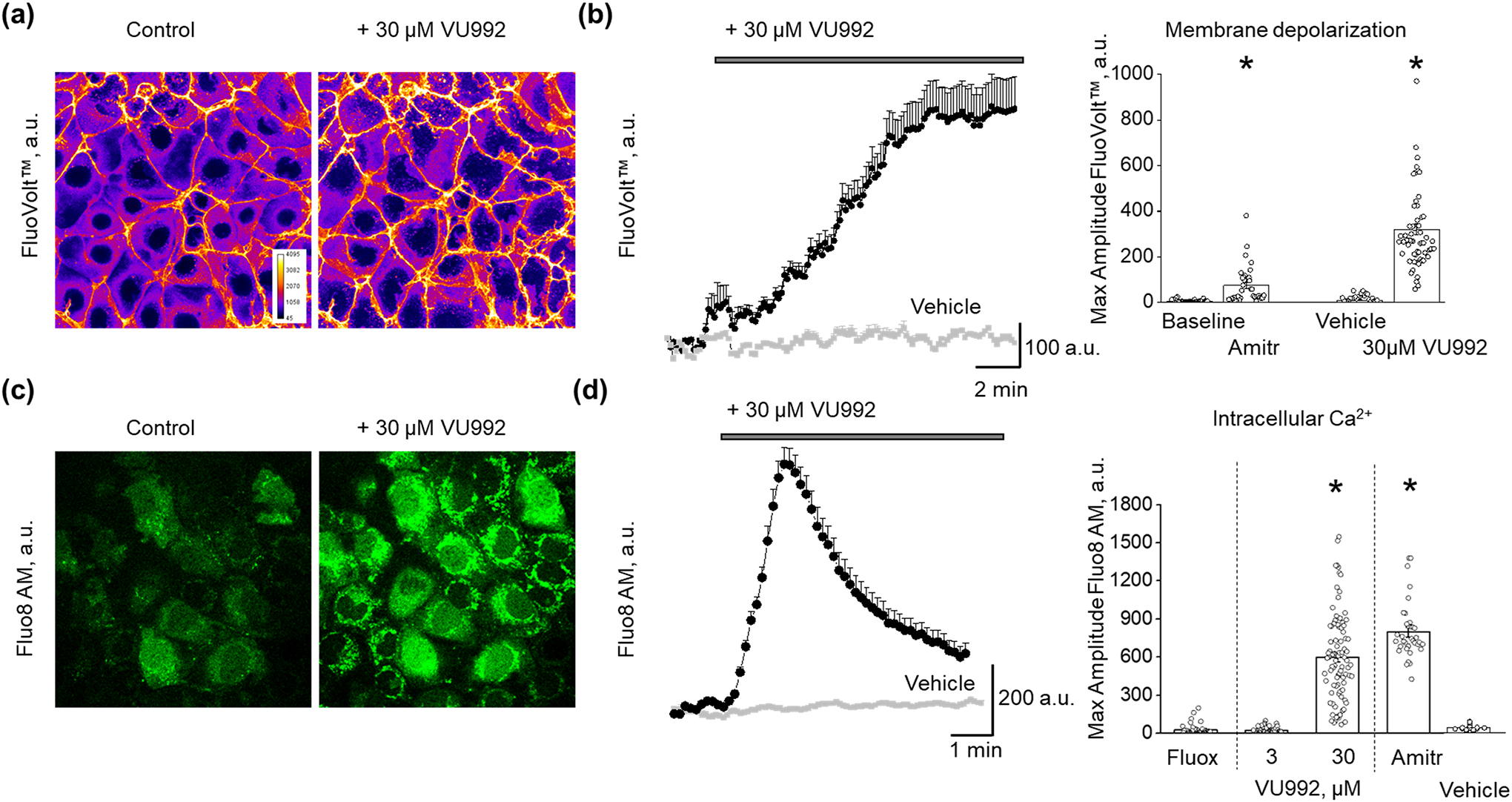
Effect of Kir4.1 and Kir4.1/Kir5.1 channels inhibitors on the membrane potential and intracellular Ca2+ concentrations in mCCDcl1 cells. (a) Representative confocal images of mCCDcl1 cells loaded with membrane potential dye (FluoVolt) acquired before and after application of VU992 with the corresponding graph plotting the time course of changes in the FluoVolt fluorescence intensity pooled from six cells. (b) Summary graph of the peak FluoVolt fluorescence intensity following application of amitriptyline (n = 27) or VU992 (n = 66). PSS or DMSO as a vehicle control shown in grey. One Way RM ANOVA, *P < 0.05. (c) Confocal images of Fluo8HT-stained mCCDcl1 cells acquired before and after application of VU992 with the corresponding graph plotting the time course of VU992-induced intracellular calcium transient pooled from ten cells. (d) Summary graph for the peak Fluo8HT fluorescence intensity following application of fluoxetine (n = 40), 3 μM VU992 (n = 40), 30 μM VU992 (n = 95), and amitriptyline (n = 28). PSS or DMSO as a vehicle control shown in grey. One Way ANOVA, Tukey post-hoc test, *P < 0.05 compared to vehicle, fluoxetine, and 3 μM VU992. Scale bar is 20 μm.
3.4 |. Inhibition of basolateral Kir4.1/Kir5.1 channel suppresses ENaC activity in principal cells of freshly isolated CCD from both human and rodent kidneys
Kir4.1/Kir5.1 channels are highly expressed in the basolateral membranes of the distal convoluted tubule (DCT) and CCD principal cells and are essential contributors to potassium transport in the distal nephron. The inhibition of Kir4.1/Kir5.1 single-channel activity by amitriptyline in the principal cells of freshly isolated CCD from Dahl SS rats is shown in Figure 5a. Using a split-open configuration, we performed patch-clamp recordings on the apical side of principal cells (microphotograph Figure 5b) and directly detected changes in ENaC activity during the modulation of basolateral K+ conductance in freshly isolated CCDs. Figure 5b demonstrates the significant decrease of ENaC activity after adding amitriptyline to the bath solution. The application of amitriptyline leads to a significant reduction of Kir4.1/Kir5.1 channel amplitude but does not modulate the amplitude of ENaC (1.89 ± 0.21 vs. 1.14 ± 0.26 (n = 8), paired sample t-test *P < 0.05 and 0.70 ±0.10 vs. 0.66 ± 0.11 (n = 9) pA for Kir4.1/Kir5.1 and ENaC correspondingly; data are shown for control vs. amitriptyline-treated tubules).
FIGURE 5.
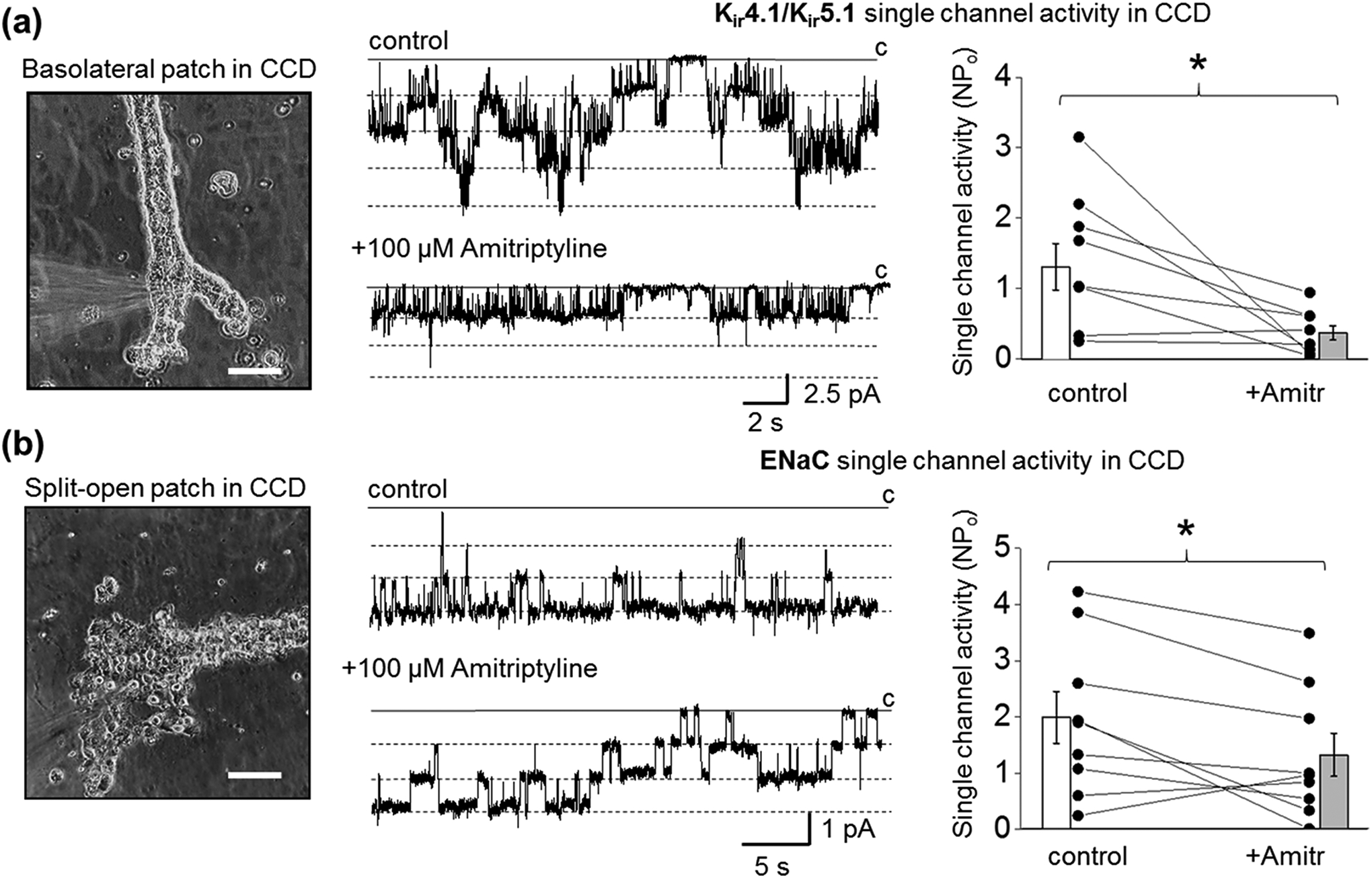
Effect of amitriptyline on Kir4.1/Kir5.1 and ENaC single-channel activity in principal cells of freshly isolated rat CCD. Representative patch-clamp configurations (glass pipette forming a giga-seal contact) and corresponding recordings of basolateral Kir4.1/Kir5.1 (a) and apical (split-open tubule) ENaC (b) single-channel activity recorded from the CCD principal cells after application of PSS as a vehicle control or application of amitriptyline. Scale bars are 100 μm. Patches were clamped to -Vp = −60 mV. Summary graphs of changes in channel’s activity for Kir4.1/Kir5.1 (n = 8) and ENaC (n = 9) channels are shown on the right. Paired sample t-test *P < 0.05.
In order to further extend this line of investigation and the feasibility of clinical use, we used freshly isolated tubules from human kidneys to interrogate Kir4.1/Kir5.1-based control of sodium transport in the collecting duct. Figure 6a represents a microphotograph of split-open (apical) and basolateral electrophysiological configurations to measure single-channel activity in human principal cells of the CCD. As shown in Figure 6b, application of amitriptyline and VU992 resulted in the inhibition of human Kir4.1/Kir5.1 single-channel activity (average amplitude of 1.82 ± 0.29 pA in control, 1.48 ± 0.12 pA after 5 min treatment with 100 μM amitriptyline, and 0.88 ± 0.06 pA after subsequent VU992 application at holding potential −60 mV). Figure 6c represents the effect of Kir4.1/Kir5.1 channel inhibition on ENaC-mediated Na+ transport. These electrophysiological recordings represent the first pharmacological analysis of ENaC single-channel activity in a human kidney. Summary data pulled from four recordings of ENaC single-channel activity in CCDs show that the application of 100 μM amitriptyline produces a substantial decrease in ENaC NPo but does not affect its unitary current amplitude (0.51 ± 0.09 vs. 0.47 ± 0.09 pA, control vs. amitriptyline correspondingly). The application of VU992 produced a similar effect on ENaC activity. Despite the low number of samples and exploratory analyses of the human data set, the observed behavior is consistent with our experiments in polarized epithelia and rat isolated tubules.
FIGURE 6.
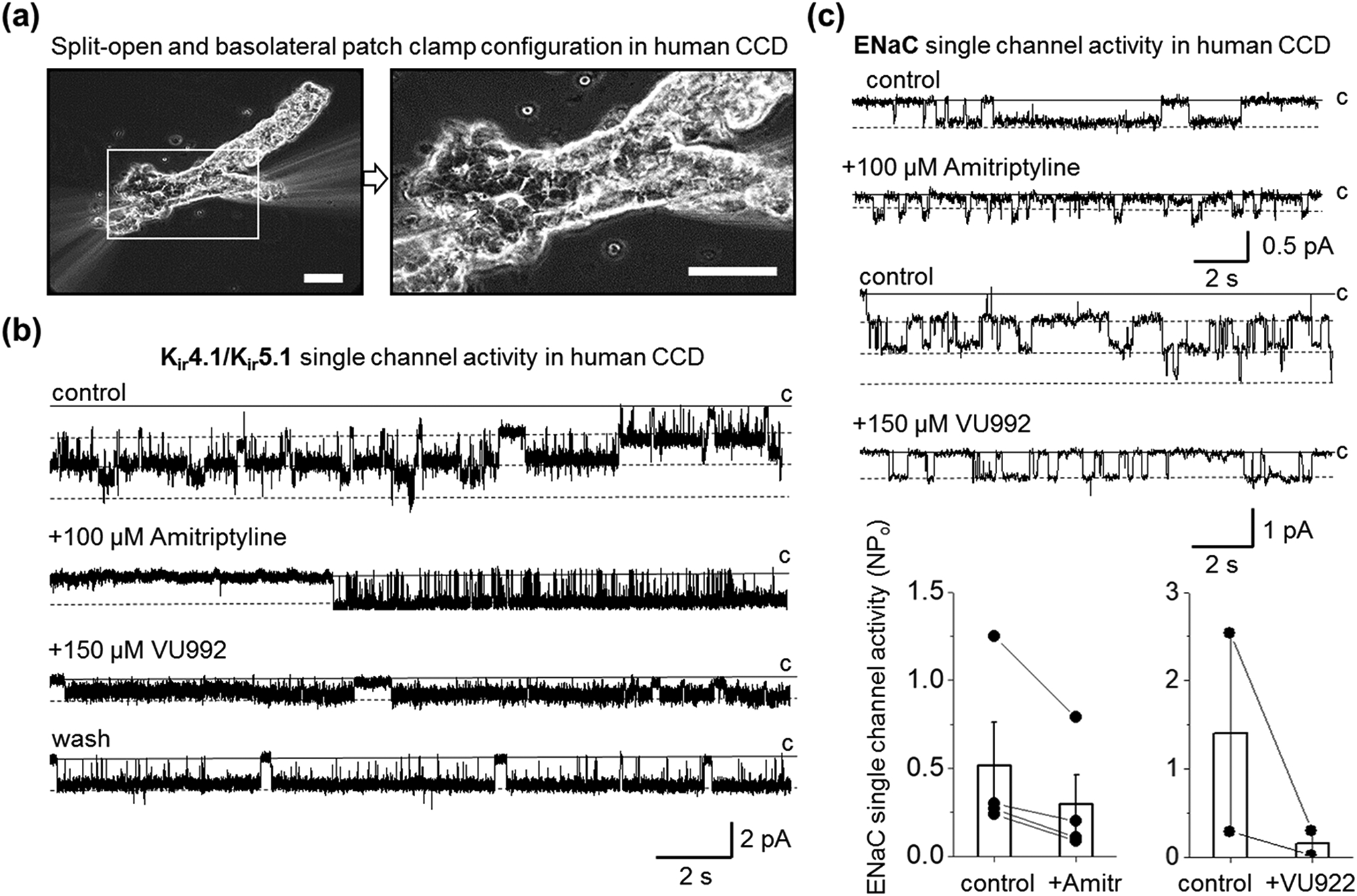
Inhibition of basolateral Kir4.1/Kir5.1 channel modulates ENaC activity in human CCD principal cells. (a) Representative image of patch-clamp configuration on apical (split-open) or basolateral parts of freshly isolated human CCD. Scale bars are 40 μm. (b) Representative current traces monitoring basolateral Kir4.1/Kir5.1 single-channel activity in cell-attached mode after application of PSS or DMSO as vehicle control or amitriptyline (100 μM) and subsequent addition of VU992 (150 μM). The patch was clamped to -Vp = −60 mV. The single-channel conductance of the recorded channel was 54 pS. Note that the effect of the application of VU992 was reversible (wash). (c) Representative current traces monitoring ENaC single-channel activity after application of PSS or DMSO as a vehicle control or amitriptyline (upper panel; -Vp = −40 mV) or VU992 (bottom; -Vp = −60 mV) applications. A solid line denotes a non-conducting closed state. Dash lines indicate open states. Summary graphs of changes in ENaC activity in response to amitriptyline (n = 4) and VU992 (n = 2) application are also shown.
3.5 |. Effect of pharmacological inhibition of Kir4.1/Kir5.1 channel by amitriptyline on K+ and Na+ homeostasis in SS rats
As was previously described in the Dahl SS rats with the functional knockout of Kcnj16 (SSKcnj16–/–), the deletion of Kir4.1/Kir5.1 channels is characterized by elevated excretion of electrolytes, hypokalemia, increased expression of sodium transporters in the ASDN (ENaC, NCC, and NKCC, including active phosphorylated forms), and normal blood sodium levels (Palygin et al., 2017). To evaluate the pharmacological effect of amitriptyline on Kir4.1/Kir5.1 channel activity in an animal model to compare with published data in the knockout, we used 10–12-week-old SS rats injected three consecutive days with amitriptyline. The analyses of blood electrolytes shown in Figure 7a demonstrate that amitriptyline administration led to a significant drop in plasma K+ (4.2 ± 0.1 vs. 3.4 ± 0.1 mmol l−1, control vs. injection day 3) but did not affect plasma Na+ levels after three days of injection. Moreover, both K+ and Na+ excretion was increased (Figure 7b). This increase in urinary electrolytes was accompanied by elevated diuresis (Figure 7c), but we did not detect significant creatinine clearance changes (108.2 ± 11.5 vs. 119.4 ± 11.4 ml h−1, control vs. injection day 3 correspondingly). These data indicate that pharmacological inhibition of Kir4.1/Kir5.1 channel may directly modulate potassium homeostasis, diuresis, and sodium excretion. Figure 8 demonstrates the upregulation of both the total and phosphorylated (active) forms of NCC in renal cortical tissue of rats treated for three days with amitriptyline compared to control. These data are consistent with findings in the SSKcnj16–/– rat, which indicate that the blockade of Kir4.1/Kir5.1 channel produces a salt-wasting phenotype despite the increased compensatory levels of the NCC transporter in ADSN (Palygin et al., 2017). The Western blots show an increase in ENaC α-subunit expression, without any changes in cleaved and noncleaved forms of γ-ENaC (Figure 8), similar to the reported γ-ENaC expression for the collecting duct system-specific Kir4.1 knockout mouse (Penton et al., 2020).
FIGURE 7.
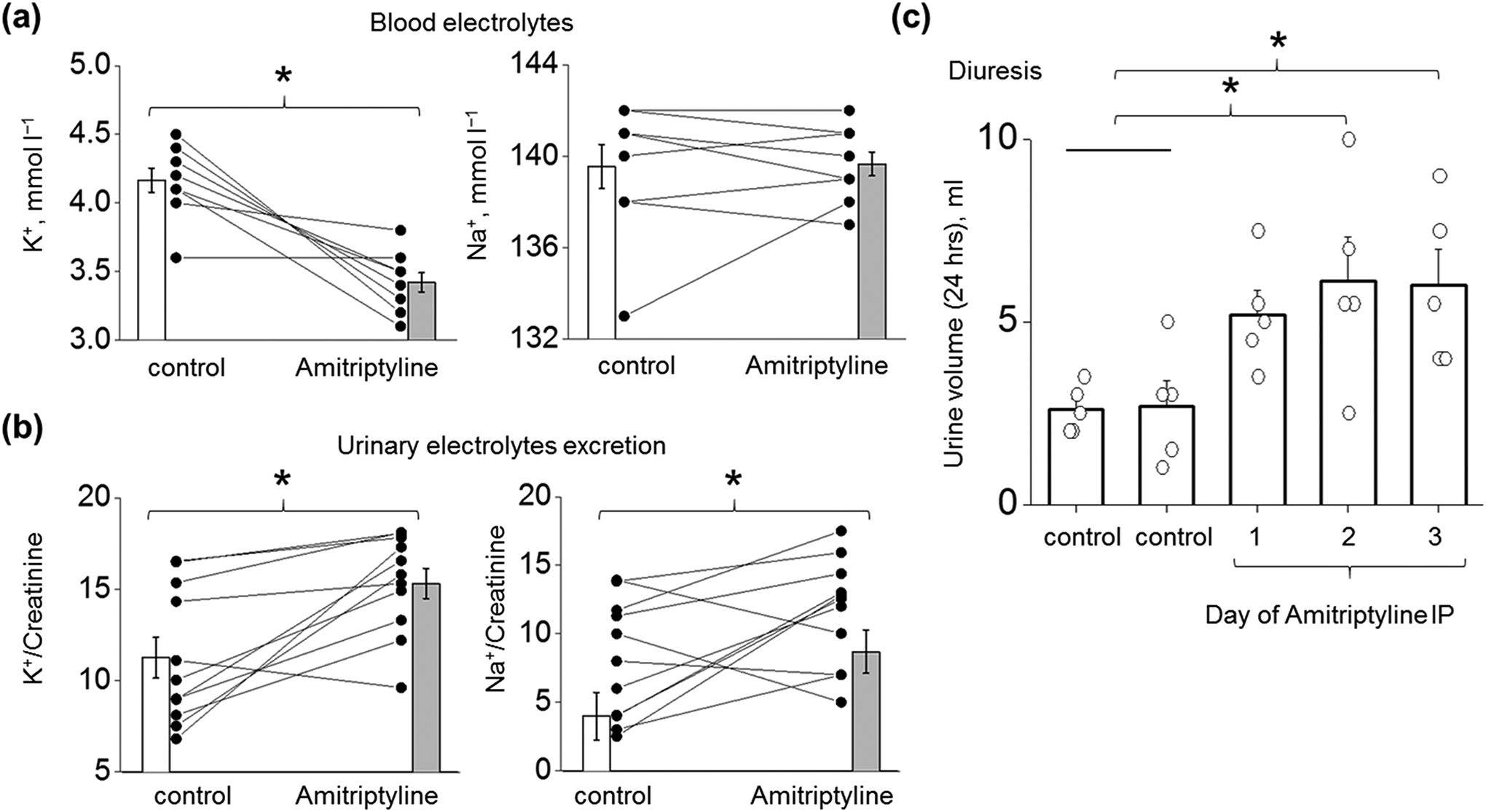
Effect of amitriptyline on sodium/potassium homeostasis in SS rats. (a, b) Biochemical analyses of K+ and Na+ in blood (n = 9 for both groups) and urine samples (n = 11 for both groups) collected from SS rat in control (vehicle injection) and after 3 days of consecutive IP injections of amitriptyline (15 mg kg−1 day−1). Concentrations of electrolytes in urine were normalized to creatinine (24 hrs collections). Paired sample t-test *P < 0.05. (c) Daily urine output in rat injected with vehicle (double control, two or one days before the drug injection) and 15 mg kg−1 amitriptyline (n = 5, One Way RM ANOVA with a Greenhouse-Geisser correction (F(1.56, 6.24) = 5.43, P < 0.05), post hoc test using Dunnett correction *P < 0.05 compared to control).
FIGURE 8.
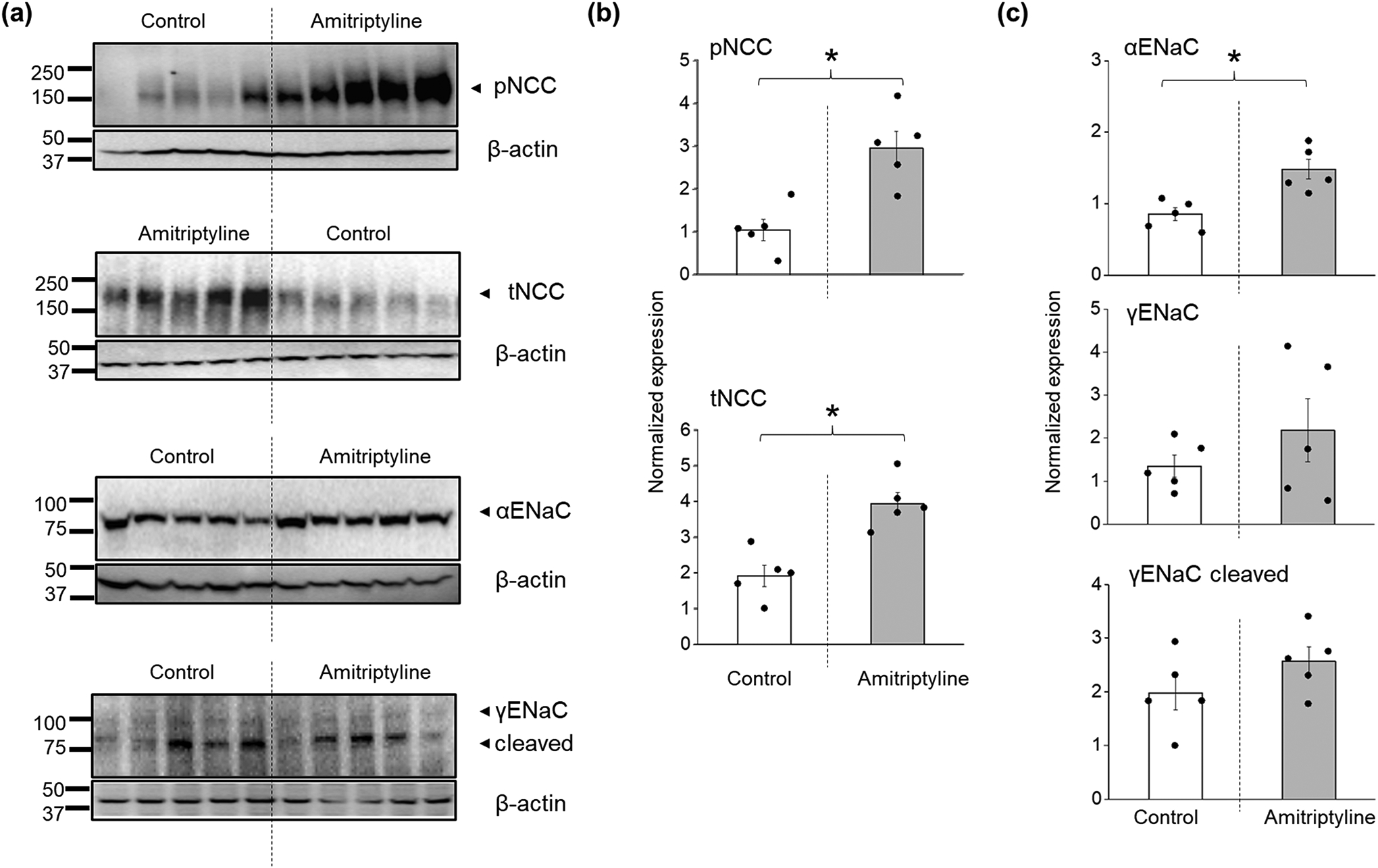
Differences in NCC and ENaC expression in SS rats treated with amitriptyline. (a) Western blotting analysis of tNCC, pNCC, αENaC, cleaved and noncleaved γENaC subunits from the kidney cortex lysates of SS rats under control or after 3 days of i.p. injection with amitriptyline. (b, c) Summary graphs showing the average relative density of the bands (normalized to loading controls) in the studied groups (n = 5 per each group, ANOVA, *P < 0.05).
4 |. DISCUSSION
The key findings of the present study are: 1) inhibition of Kir4.1/Kir5.1, but not a Kir4.1 channels decrease ENaC activity; these effects are most likely mediated by induced depolarization of membrane potential and increase in intracellular Ca2+ level; 2) acute inhibition of inwardly rectifying K+ channel conductance by amitriptyline or high concentrations of VU992 promotes the decrease in ENaC activity in principal cells in rat and human CCD; 3) in vivo amitriptyline treatment recapitulates the effect of Kir5.1 deletion in SS rats on electrolyte homeostasis and expression of total and phosphorylated NCC in the distal nephron.
Although the essential role of Kir4.1-composed K+ channels for the regulation of K+ handling and electrolyte homeostasis in the kidney is well established in knockout rodent models (Manis et al., 2019; Penton et al., 2020; Su et al., 2016), the exact physiological mechanisms as well as the role of ENaC, BK-channels, Ca2+ entry, or Mg2+ transport in this processes still require further investigation. The difference between models with deletion of Kir4.1 or Kir5.1 subunit should be noted. Deletion of Kir4.1 eliminates the activity of both channels, downregulates NCC expression, and induces alkalosis (Scholl et al., 2009). In contrast, deletion of Kir5.1 removes only heteromeric Kir4.1/Kir5.1 channels, upregulates Kir4.1 and NCC expression (Palygin et al., 2017), induces metabolic acidosis (Paulais et al., 2011; Puissant et al., 2019), triggers hyperactivity of the RAAS system and preserves the ability to regulate basolateral K+ transport and survivability on a high K+ diet (Manis et al., 2019). On the other hand, deletion of both subunits is characterized by severe hypokalemia and upregulation of ENaC expression in the kidney (Manis et al., 2019; Wu et al., 2020a). Pharmacological inhibition of the channels could result in different outcomes compared to knockout animal studies. For example, it was reported that inhibition of NCC transport activity by pharmacological means does not increase Na+ reabsorption through ENaC (Hunter et al., 2014). Here we used available pharmacological tools to exclude the discrepancy of the different genetic models and establish the primary basolateral Kir4.1/Kir5.1 channel’s (Wu et al., 2020b) role in regulating ENaC in CCD.
Studies in heterologous expression systems reported that antidepressant TCA drugs act as reversible blockers of Kir4.1 channels (Furutani et al., 2009; Ohno et al., 2007). Our data shows that 100 μM fluoxetine effectively inhibits single-channel Kir4.1 activity but does not affect heteromeric channel activity in CHO cells. Amitriptyline (100 μM) displays an acute inhibitory effect on Kir4.1/Kir5.1 current but has no effect on Kir4.1. Interestingly, in previous studies, the inhibitory effect of amitriptyline on Kir4.1 was demonstrated using patch-clamp studies and thallium (Tl+) flux assay (Kharade et al., 2018; Su et al., 2007). It was shown that the inhibitory action of tricyclic antidepressants on Kir channels is dependent on extracellular concentrations of K+ with a more robust effect in the conditions of smaller potassium gradient. So, we suggest that in our configuration (with 150 mM of K+ in pipette solution), the inhibitory effect of amitriptyline on both homomeric and heteromeric channels maybe be blunted. In our ex vivo study, amitriptyline induced a significant suppression of NPo as well as a decrease in unitary Kir4.1/Kir5.1 current amplitude recorded from rat principal CCD cells, indicating that inhibition of this basolateral channel will result in membrane depolarization. Our data with membrane voltage dye FluoVolt™ in cultured epithelial mCCDcl1 cells supports this assumption. Similar inhibitory effects of amitriptyline or high concentrations of VU992 were observed in human CCD principal cells. Our data demonstrating the important role of basolateral K+ conductance in the regulation of membrane potential support previous findinds on mice Kcnj10 knockout model and C57BL/6J mice (Zaika et al., 2016; Zhang et al., 2014). Taken together, our studies demonstrate that amitriptyline predominantly affects Kir4.1/Kir5.1 channel rather than homomeric Kir4.1 channel activity and could be a valuable pharmacological tool to better understand the physiological role of these channels.
Using an electrophysiological approach, we show that acute application of amitriptyline and VU992 in concentrations that produce an inhibitory effect on heteromeric Kir4.1/Kir5.1 channel results in substantial suppression of the Ieq measured in polarized epithelial mCCDcl1 cells. The inhibition of basolateral K+ conductance reduces single-channel ENaC channel activity in mCCDcl1 and principal cells of freshly isolated rodent and human CCD tubules. The reduction of ENaC activity is not attributed to the direct effect of amitriptyline or VU992 since they do not cause any change in Na+ conductance in CHO cells overexpressing ENaC (Figure S3). Also, the suppressive effect of amitriptyline on amiloride-sensitive Ieq is not likely related to unspecific inhibition of ROMK, since ROMK blockade by VU591 does not affect amiloride-sensitive Ieq in polarized epithelial cortical collecting principal mCCDcl1 cells (Figure S2). These data is in agreement with the previous in vivo study with ROMK inhibitor and benzamil, suggesting that ROMK inhibition does not abolish ENaC-mediated Na+ transport in the collecting duct (Kharade et al., 2016). Interestingly, neither fluoxetine nor VU992 at concentrations specific for Kir4.1 channel affected ENaC activity measured with Ieq. This implies that the homomeric Kir4.1 channel likely plays only a minor role in controlling apical Na+ transport in CCD.
Our study shows that acute inhibition of Kir4.1/Kir5.1 channel induced membrane depolarization, supporting the established role of these channels in the control of membrane potential (Zaika et al., 2016; Zhang et al., 2014). Indeed, classic physiology studies indicate the direct relationship between the activity of Na+/K+ ATPase and potassium conductance on the basolateral membrane (Kubota et al., 1983; Lapointe et al., 1990; Matsumura et al., 1984). We also show that pharmacological inhibition of Kir4.1/Kir5.1 channel promotes the increase in intracellular Ca2+ concentration (Figure 4). Although the exact mechanism involved in the regulation of intracellular Ca2+ by the block of basolateral Kir channel activity needs further in-depth investigation, the inhibitory effect of intracellular Ca2+ increase on ENaC activity is well documented (Gu, 2008; Ishikawa et al., 1998; Ismailov et al., 1997; Palmer & Frindt, 1987). It was proposed that Ca2+ could act directly on the channel pore or through activation of intracellular secondary messengers such as calmodulin, protein kinase C and prostaglandin (Gu, 2008; Palmer & Frindt, 1987). We suggest that the reduction of electrochemical gradient triggered by the depolarization of the basolateral membrane and/or the increase in intracellular Ca2+ could explain the modulatory effect of Kir4.1/Kir5.1 inhibition on ENaC activity. In agreement with this statement, a recent electrophysiological study in mice indicates that the deletion of Kir5.1 decreased ENaC currents in the late distal convoluted tubule, and this effect is even more prominent under high salt conditions (Duan et al., 2021). Furthermore, the increase in intracellular Ca2+ level in response to inhibition of basolateral K+ conductance could also mediate kaliuresis observed in rats exposed to amitriptyline. In this condition, a large conductance, voltage- and Ca2+-gated K+ BK channel presented along the apical side of the distal nephron become activated and may be primarily responsible for renal K+ excretion.
A common side effect of amitriptyline overdose is orthostatic hypotension, metabolic acidosis, hypokalemia (Glassman & Preud’homme, 1993; Kerr et al., 2001; Scalco et al., 2000), which surprisingly mimics phenotypic features of SSKcnj16−/− rats. Although the effect of downregulation of basolateral K+ conductance on blood pressure and salt-wasting phenotypes can vary across models, hypokalemia is always a distinctive feature of this pathological condition. This emphasizes the central role of Kir channels to control plasma K+ levels. In our in vivo study, i.p. injections of amitriptyline for a few days significantly shifted blood potassium concentrations towards hypokalemic values. The SS rats also exhibited elevated K+ and Na+ excretion. The Western blots shows an increase in αENaC expression but no changes in γENaC or it cleaved form by amitriptyline treatment (Figure 8). We also observed a substantial increase in the expression of total and phosphorylated forms of NCC. Likewise, the stimulation of DCT function and increased NCC expression was previously reported in rat and mice models with knockout of Kcnj16 (Palygin et al., 2017; Paulais et al., 2011). These data allow us to presume that amitriptyline treatment in Dahl SS rats recapitulates the renal phenotype developed after genetic ablation of Kcnj16, but not Kcnj10 knockout. In the future, the development of highly specific Kir4.1/Kir5.1 pharmacological tools will allow us to explore in more detail the ways in which basolateral K+ conductance contributes to the regulation of Na+ transport in the kidney.
In summary, our study demonstrates the essential role of renal Kir4.1/Kir5.1 channel not only in the control of plasma K+ but also on the regulation of ENaC-mediated sodium reabsorption. Therefore, future studies are critical to elucidate the physiological effects of Kir4.1/Kir5.1 inhibition in vivo in this and other models, to more fully define the mechanistic actions and potential for therapeutic use.
Supplementary Material
BULLET POINT SUMMARY:
What is already known
Inwardly rectifying K+ (Kir) channels of the distal nephron, specifically Kir4.1 and Kir4.1/Kir5.1, play a critical role in determining blood K+ level.
What this study adds
Our study reveals that inhibition of Kir4.1/Kir5.1 channel, but not Kir4.1 suppresses ENaC activity.
Inhibition of renal Kir4.1/Kir5.1 reduces blood potassium level, promotes diuresis and natriuresis.
What is the clinical significance?
Inhibition of Kir4.1/Kir5.1 channel affects ENaC activity and increases natriuresis, which potentially can help to lower blood pressure and reduce the risk of cardiovascular disease.
This study identifies Kir4.1/Kir5.1 channel as a new potential therapeutic target for the control of electrolyte homeostasis and hyperkalemia.
Acknowledgments:
This research was supported by the National Institutes of Health grants R56 DK121750 and R01 DK126720 (to OP), R35 HL135749 (to ASt), R01 DK120821 (to JSD and ASt), T32 HL134643 CVC A.O. Smith Fellowship (to CAK), Department of Veteran Affairs grant I01 BX004024 (to ASt), endowed funds from the SC SmartState Centers of Excellence (to OP), and RSF №19-14-00114 (to ASh). The authors would like to acknowledge Ashraf El-Meanawy (Department of Nephrology, MCW) for work with human tissues and Nicholas J. Burgraff for help with in vivo animal studies.
Declaration of transparency and scientific rigour:
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Natural Products Research, Design and Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ABBREVIATIONS:
- ASDN
aldosterone-sensitive distal nephron
- CCD
cortical collecting duct
- CD
collecting ducts
- CHO
Chinese hamster ovary
- CNS
central nervous system
- CNT
connecting tubule
- DCT
distal convoluted tubule
- ENaC
epithelial Na+ channel
- HCTZ
hydrochlorothiazide
- Ieq
equivalent short-circuit current
- Kir
inwardly rectifying K+
- ROMK
renal outer medullary potassium channel (Kir1.1)
- NCC
thiazide-sensitive Na+-Cl− cotransporter
- TCA
tricyclic antidepressant
- RAAS
renin-angiotensin-aldosterone system
Footnotes
Conflict of interest disclosure: The authors declare no competing interests.
Data availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alexander SP, Mathie A, Peters JA, Veale EL, Striessnig J, Kelly E, et al. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Ion channels. Br J Pharmacol 178 Suppl 1: S157–S245. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Roberts RE, Broughton BRS, Sobey CG, George CH, Stanford SC, et al. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. Br J Pharmacol 175: 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmus AM, Mansley MK, Mullins LJ, Peter A, & Mullins JJ (2018). mCCDcl1 cells show plasticity consistent with the ability to transition between principal and intercalated cells. Am J Physiol Renal Physiol 314: F820–F831. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. The New England journal of medicine 360: 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, et al. (2017). Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J Am Soc Nephrol 28: 1814–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, et al. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl LK, Leitl G, & Heine M (1972). Influence of dietary potassium and sodium/potassium molar ratios on the development of salt hypertension. J Exp Med 136: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan XP, Wu P, Zhang DD, Gao ZX, Xiao Y, Ray EC, et al. (2021). Deletion of Kir5.1 abolishes the effect of high Na(+) intake on Kir4.1 and Na(+)-Cl(-) cotransporter. Am J Physiol Renal Physiol 320: F1045–F1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodstad H, Gonzalez-Rodriguez E, Bron S, Gaeggeler H, Guisan B, Rossier BC, et al. (2009). Effects of mineralocorticoid and K+ concentration on K+ secretion and ROMK channel expression in a mouse cortical collecting duct cell line. Am J Physiol Renal Physiol 296: F966–975. [DOI] [PubMed] [Google Scholar]
- Furutani K, Ohno Y, Inanobe A, Hibino H, & Kurachi Y (2009). Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol 75: 1287–1295. [DOI] [PubMed] [Google Scholar]
- Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, et al. (2005). Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol 16: 878–891. [DOI] [PubMed] [Google Scholar]
- Glassman AH, & Preud’homme XA (1993). Review of the cardiovascular effects of heterocyclic antidepressants. J Clin Psychiatry 54 Suppl: 16–22. [PubMed] [Google Scholar]
- Golosova D, Palygin O, Bohovyk R, Klemens CA, Levchenko V, Spires DR, et al. (2020). Role of opioid signaling in kidney damage during the development of salt-induced hypertension. Life Sci Alliance 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y (2008). Effects of [Ca2+]i and pH on epithelial Na+ channel activity of cultured mouse cortical collecting ducts. J Exp Biol 211: 3167–3173. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Shah JC, & Hwang SS (1999). Pharmacokinetic and pharmacodynamic characterization of OROS and immediate-release amitriptyline. Br J Clin Pharmacol 48: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RW, Craigie E, Homer NZ, Mullins JJ, & Bailey MA (2014). Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 306: F457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaeva E, Fedoriuk M, Bohovyk R, Klemens CA, Khedr S, Golosova D, et al. (2019). Vibrodissociation method for isolation of defined nephron segments from human and rodent kidneys. Am J Physiol Renal Physiol 317: F1398–F1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Marunaka Y, & Rotin D (1998). Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+. J Gen Physiol 111: 825–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismailov II, Berdiev BK, Shlyonsky VG, & Benos DJ (1997). Mechanosensitivity of an epithelial Na+ channel in planar lipid bilayers: release from Ca2+ block. Biophys J 72: 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr GW, McGuffie AC, & Wilkie S (2001). Tricyclic antidepressant overdose: a review. Emerg Med J 18: 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade SV, Flores D, Lindsley CW, Satlin LM, & Denton JS (2016). ROMK inhibitor actions in the nephron probed with diuretics. Am J Physiol Renal Physiol 310: F732–F737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, et al. (2018). Discovery, Characterization, and Effects on Renal Fluid and Electrolyte Excretion of the Kir4.1 Potassium Channel Pore Blocker, VU0134992. Mol Pharmacol 94: 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khedr S, Palygin O, Pavlov TS, Blass G, Levchenko V, Alsheikh A, et al. (2019). Increased ENaC activity during kidney preservation in Wisconsin solution. BMC Nephrol 20: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Biagi BA, & Giebisch G (1983). Intracellular potassium activity measurements in single proximal tubules of Necturus kidney. J Membr Biol 73: 51–60. [DOI] [PubMed] [Google Scholar]
- Lapointe JY, Garneau L, Bell PD, & Cardinal J (1990). Membrane crosstalk in the mammalian proximal tubule during alterations in transepithelial sodium transport. Am J Physiol 258: F339–345. [DOI] [PubMed] [Google Scholar]
- Lilley E, Stanford SC, Kendall DE, Alexander SPH, Cirino G, Docherty JR, et al. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. Br J Pharmacol 177: 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis AD, Palygin O, Isaeva E, Levchenko V, LaViolette PS, Pavlov TS, et al. (2021). Kcnj16 knockout produces audiogenic seizures in the Dahl salt-sensitive rat. JCI Insight 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis AD, Palygin O, Khedr S, Levchenko V, Hodges MR, & Staruschenko A (2019). Relationship between the renin-angiotensin-aldosterone system and renal Kir5.1 channels. Clin Sci (Lond) 133: 2449–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Cohen B, Guggino WB, & Giebisch G (1984). Electrical effects of potassium and bicarbonate on proximal tubule cells of Necturus. J Membr Biol 79: 145–152. [DOI] [PubMed] [Google Scholar]
- McGrath JC, & Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, et al. (2014). Association of urinary sodium and potassium excretion with blood pressure. The New England journal of medicine 371: 601–611. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Wang Z, Kinlough CL, Poland PA, Marciszyn AL, Montalbetti N, et al. (2017). Specific Palmitoyltransferases Associate with and Activate the Epithelial Sodium Channel. J Biol Chem 292: 4152–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Hibino H, Lossin C, Inanobe A, & Kurachi Y (2007). Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51. [DOI] [PubMed] [Google Scholar]
- Palmer LG, & Frindt G (1987). Effects of cell Ca and pH on Na channels from rat cortical collecting tubule. Am J Physiol 253: F333–339. [DOI] [PubMed] [Google Scholar]
- Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, et al. (2017). Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2: e92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, et al. (2011). Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proceedings of the National Academy of Sciences of the United States of America 108: 10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, Levchenko V, & Staruschenko A (2014). Role of Rho GDP dissociation inhibitor alpha in control of epithelial sodium channel (ENaC)-mediated sodium reabsorption. J Biol Chem 289: 28651–28659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penton D, Vohra T, Banki E, Wengi A, Weigert M, Forst AL, et al. (2020). Collecting system-specific deletion of Kcnj10 predisposes for thiazide- and low-potassium diet-induced hypokalemia. Kidney Int 97: 1208–1218. [DOI] [PubMed] [Google Scholar]
- Puissant MM, Muere C, Levchenko V, Manis AD, Martino P, Forster HV, et al. (2019). Genetic mutation of Kcnj16 identifies Kir5.1-containing channels as key regulators of acute and chronic pH homeostasis. FASEB J 33: 5067–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, et al. (2010). KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proceedings of the National Academy of Sciences of the United States of America 107: 14490–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalco MZ, de Almeida OP, Hachul DT, Castel S, Serro-Azul J, & Wajngarten M (2000). Comparison of risk of orthostatic hypotension in elderly depressed hypertensive women treated with nortriptyline and thiazides versus elderly depressed normotensive women treated with nortriptyline. Am J Cardiol 85: 1156–1158, A1159. [DOI] [PubMed] [Google Scholar]
- Schlingmann KP, Renigunta A, Hoorn EJ, Forst AL, Renigunta V, Atanasov V, et al. (2021). Defects in KCNJ16 Cause a Novel Tubulopathy with Hypokalemia, Salt Wasting, Disturbed Acid-Base Homeostasis, and Sensorineural Deafness. J Am Soc Nephrol 32: 1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, et al. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proceedings of the National Academy of Sciences of the United States of America 106: 5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A (2018). Beneficial effects of high potassium: contribution of renal basolateral K+ channels. Hypertension 71: 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Pochynyuk O, & Stockand JD (2005). Regulation of epithelial Na+ channel activity by conserved serine/threonine switches within sorting signals. Journal of Biological Chemistry 280: 39161–39167. [DOI] [PubMed] [Google Scholar]
- Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, & Kurachi Y (2007). Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther 320: 573–580. [DOI] [PubMed] [Google Scholar]
- Su XT, Zhang C, Wang L, Gu R, Lin DH, & Wang WH (2016). Disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. Am J Physiol Renal Physiol 310: F985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobian L (1991). Salt and hypertension. Lessons from animal models that relate to human hypertension. Hypertension 17: I52–58. [DOI] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. (2020). Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 141: e139–e596. [DOI] [PubMed] [Google Scholar]
- Wu P, Gao ZX, Zhang DD, Duan XP, Terker AS, Lin DH, et al. (2020a). Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice. J Am Heart Assoc 9: e014996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Su XT, Gao ZX, Zhang DD, Duan XP, Xiao Y, et al. (2020b). Renal Tubule Nedd4–2 Deficiency Stimulates Kir4.1/Kir5.1 and Thiazide-Sensitive NaCl Cotransporter in Distal Convoluted Tubule. J Am Soc Nephrol 31: 1226–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Palygin O, Tomilin V, Mamenko M, Staruschenko A, & Pochynyuk O (2016). Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am J Physiol Renal Physiol 310: F311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, et al. (2014). KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proceedings of the National Academy of Sciences of the United States of America 111: 11864–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.