

Abstract
Cellular senescence is a stable cell growth arrest. Senescent cells are metabolically active as exemplified by the secretion of inflammatory cytokines, chemokines, and growth factors, which is termed senescence-associated secretory phenotype (SASP). The SASP exerts a range of functions in both normal health and pathology, which is possibly best characterized in cancers and physical aging. Recent studies demonstrated that chromatin is instrumental in regulating the SASP both through nuclear transcription and via the innate immune cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway in the cytoplasm. Here, we will review these regulatory mechanisms with an emphasis on most recent development in the field. We will highlight the challenges and opportunities in developing intervention approaches such as targeting chromatin regulatory mechanisms, to alter the SASP as an emerging approach to combat cancers and achieve healthy aging.
Keywords: Senescence-associated secretory phenotype, chromatin structure, enhancer-promoter interaction, cytoplasmic chromatin, senomorphics
Cellular senescence and the senescence-associated secretory phenotype (SASP)
Cellular senescence was initially characterized as a loss of proliferative capacity after extended culture of human embryonic lung fibroblasts, which is known as replicative senescence (RS) [1]. In addition, senescence can be triggered by several stresses, including activation of oncogenes or DNA damaging cancer therapeutics, known as oncogene-induced senescence (OIS) and therapy-induced senescence (TIS), respectively [2]. Senescent cells also exhibit non-cell autonomous functions exemplified by the secretion of inflammatory cytokines, chemokines, and growth factors, which is called the senescence-associated secretory phenotype (SASP) [3–5]. Senescence plays a critical role in normal development and health (Figure 1). During embryonic development, cells with senescent features are important for body axis patterning, while the associated SASP is implicated in tissue remodeling and clearance of senescent cells through driving macrophage infiltration [6, 7]. In addition, the SASP has been linked to wound healing [8] and tissue homeostasis in mouse models such as liver fibrosis [9]. Age-associated accumulation of senescent cells is thought to contribute to age-related functional decline and pathologies, which was validated by the beneficial effects of senescent cells removal [10].
Figure 1. The multifaceted functions of the senescence-associated secretory phenotype (SASP).
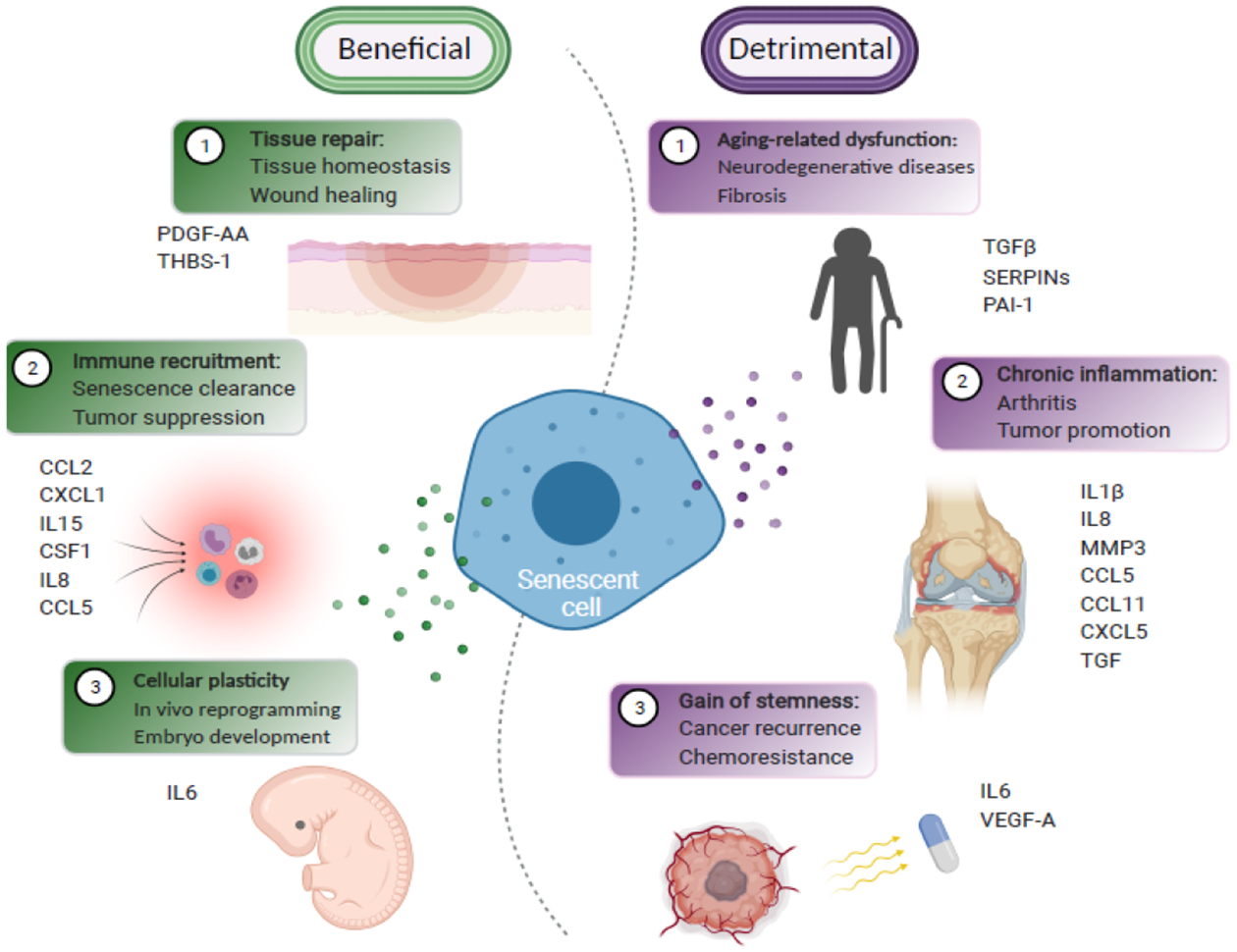
The beneficial (in green) and the detrimental (in purple) roles of the SASP are summarized. The specific SASP factors that function in these processes are indicated. Note that the beneficial and detrimental roles played by the specific SASP factors are context-dependent. For instance, IL6 is critical for senescence-associated cellular reprogramming during embryo development, while contributing to the relapse and cancer stemness during therapy-induced senescence.
Senescence is a programmed developmental mechanism that is thought to have evolved to limit cancer growth [11]. For example, OIS underscores the tumor suppressive role of senescence. The SASP plays a context-dependent role in cancer [2]. The SASP and senescence-associated proliferation arrest cooperate in tumor suppression, by promoting immune clearance of the damaged cells and arresting proliferation of damaged pre-malignant cells [3, 4, 12]. However, as a source of chronic inflammation, the SASP also promotes tissue aging and age-associated diseases including cancer. The SASP is predominantly detrimental in tumors because it promotes several hallmarks of cancer including tumor growth. Consistently, elimination of senescent cells delays tumorigenesis [13]. Consequently, there is currently much effort devoted to developing “senolytics” (see Glossary) that selectively eliminate senescent cells through inducing their apoptosis to promote healthy aging and suppress cancer. However, potential benefits of senolytics are limited by their unwanted side effects such as impairment of tissue repair and immunosurveillance [8, 14, 15].
An alternative to senolytics is senomorphics that specifically inhibit the SASP, without impairing the tumor suppressive proliferation arrest. Compared with senolytics, the SASP-specific senomorphic senescence targeting approach is considered less toxic. Hence, it is important to define the mechanism of SASP regulation because its precise inhibition may be an approach to combat the pro-aging and cancer-promoting effects of senescent cells. Chromatin emerges as a critical integration mechanism underlying the SASP. This article reviews the role of chromatin in regulating the SASP transcriptionally and through the innate immune cGAS-STING pathway. We propose chromatin regulatory mechanisms as emerging targets to alter the SASP.
Senescence-associated changes in chromatin structure
The most visible change in chromatin structure in senescent human cells such as OIS in embryonic lung human diploid fibroblasts (HDF) IMR90 and WI38 is the formation of facultative senescence-associated heterochromatic foci (SAHF) [16]. SAHF are enriched in a variety of heterochromatic markers, such as histone H3 lysine 9 and 27 trimethylation (H3K9me3 and H3K27me3), and the histone H2A variant macroH2A [16, 17]. These epigenetic marks repress the transcription of key proliferation-related genes. Thus, SAHF contribute to the tumor suppressive proliferative arrest of senescent cells [16]. The heterochromatin that is incorporated into the SAHF is not newly formed. Instead, SAHF results from 3D repositioning of existing heterochromatin [18]. Individual chromosomes condensate into a single SAHF focus [19]. The increased density of nuclear pores that control transport between the cytoplasm and nucleus regulates the reorganization of heterochromatin from the nuclear periphery as SAHF [20]. The SASP gene loci such as those of IL1β, IL6 and IL8 are excluded from SAHF [19, 21], suggesting loop-based exclusion of the SASP gene loci as a mechanism of their upregulation in senescent cells.
RS and OIS HDF cells are characterized by large-scale alterations in domains of histone H3 lysine 4 trimethylation (H3K4me3) and H3K27me3-enriched “mesas” and H3K27me3-depleted “canyons” [22]. Mesas form at lamin B1-associated domains (LADs). In contrast, canyons mostly form between LADs and are typically associated with genes- and enhancers-rich genomic regions. Localized H3K27me3 loss in canyons strongly correlates with key senescence-regulatory genes such as SASP genes [22]. This is consistent with the notion that enhancers of the SASP genes often contained H3K27me3 and thus formation of canyons promotes the expression of the SASP genes [22]. However, the underlying mechanism remains to be elucidated. In contrast, inhibition of the H3K4 methyltransferase MLL1 decreases the level of H3K4me3 dramatically over the SASP gene loci and suppresses their expression in both OIS and TIS HDF models [23]. Thus, the chromatin landscape in senescent cells coordinates the formation of SAHF to silence proliferation genes while simultaneously activating the SASP. Of note, the overall chromatin status in senescent cells appears to be more open and accessible [24], which likely facilitates the senescence-maintaining transcriptome such as SASP.
Transcriptional regulation of the SASP by chromatin in nucleus
The genome in the nucleus is hierarchically organized into non-random three dimensions (3D genome). Transcriptionally active regions are segregated from those of inactivated domains. The advancement of techniques such as chromosome conformation capture (3C) and high-resolution microscopy enabled insights into higher-order chromatin organization. Specifically, 3D genome is spatially organized into either active (A) or repressive (B) compartments (multi-megabase), topologically associated domains (TADs) (hundreds-kilobase), and chromatin loops that mediate enhancer-promoter (EP) interaction [25, 26]. Each of the individual structure components is physically separated by boundary regions. Structural maintenance of chromosomes (SMC) components cohesin and CCCTC-binding factor (CTCF) control the organization of chromatin loops and TADs without affecting compartments [27–29]. In contrast, another SMC complex condensin II controls compartment switches [30, 31]. Chromatin loops are dynamically regulated, while cell-type specific TADs are typically stable and associated with transcription and replication [32, 33]. EP interaction allows for access of genomic loci by transcription factors to drive gene expression. While the focus of the current review is on transcriptional underpinning of the SASP, the SASP is also subjected to mRNA translational control downstream of the mammalian target of rapamycin kinase [34, 35].
Transcription factors
The SASP gene transcription is temporally regulated with at least two waves [36, 37], where the first wave of the SASP factors such as TGFβ1 and TGFβ3 is typically immunosuppressive and is controlled by NOTCH1 in OIS HDF cells. In contrast, the second wave of the SASP often consists of inflammatory factors such as IL1α, IL1β, IL-6, IL-8 and CXCL2, which is suppressed by NOTCH1 [36, 37]. Mechanistically, NOTCH1 suppresses the expression of CEBPβ that promotes the transcription of these SASP factors [36, 37]. In addition, late RS or OIS HDF cells are characterized by type I interferon (IFN-I) response to drive a sterile age-associated inflammation [38]. The SASP is regulated through the activation of two main transcription factors: NFκB and CCAAT/enhancer-binding protein β (C/EBPβ) [3, 39–41]. Additionally, the NOTCH1-driven inhibition of the inflammatory SASP is mediated by repression of C/EBPβ transcriptional activity to temporally promote immunosuppressive SASP [36]. Increased stabilization of the transcription factor GATA-binding protein 4 (GATA4) mediates the DNA damage response-induced SASP through NFκB [42]. Further, the janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway have also been involved in transcriptional control of the SASP [43]. In addition to driving cell cycle growth arrest downstream of p16, RB functions downstream of p21 to transcriptionally promote a bioactive secretome across cell types with multiple senescence inducers that partially overlaps with the SASP to place stressed cells under immunosurveillance [44].
Histone variants
MacroH2A isoforms are variants of canonical H2A, which localize to SAHF to stabilize them [17, 19]. Interestingly, macroH2A isoform 1 binds to transcriptionally inactive SASP genes where it likely positions the SASP genes for activation in OIS HDF [45]. Upon senescence induction, macroH2A1 is required for both autocrine and paracrine effects of the SASP. Sustained SASP allows for activation of the ER stress response and DNA damage response, which removes macroH2A1 to prevent the SASP overdrive through elevated ATM activity [45].
Another H2A variant plays a role in the SASP is H2A.J in HDF with multiple senescence inducers [46]. H2A.J is enriched in the chromatin of senescent cells with persistent DNA damage. Depletion of H2A.J strongly inhibits the expression of the SASP genes without affecting the senescence-associated growth arrest. However, genome-wide ChIP-seq analysis revealed that the specificity of H2A.J action cannot be explained by its differential deposition at the SASP genes [46]. This raises the possibility that H2A.J acts in concert with other factors to control the SASP during senescence.
Chromatin architecture proteins
High mobility group proteins are non-histone chromatin-bound proteins that regulate gene transcription by altering chromatin architecture [47, 48]. The high mobility group A (HMGA) proteins increase accessibility of the chromatin to transcription factors in a DNA sequence independent manner [49]. HMGAs promote senescence through SAHF formation in HDF [50]. In addition, HMGAs play a key role in the inflammatory SASP by upregulating NAMPT through regulating enhancer activity in both OIS HDF and TIS ovarian cancer cells [51]. The HMGA1/NAMPT axis regulates inflammatory SASP through increasing NAD+/NADH ratio, which is independent of CEBPβ. NAD+/NADH ratio controls AMPK signaling downstream of HMGA1/NAMPT to govern the strengths of the inflammatory SASP via the p53/p38 MAPK/NFκB pathway. Thus, HMGAs couple the cell growth arrest and the inflammatory SASP during senescence.
In contrast to HMGAs, HMGB1 functions as an extracellular alarmin to activate NFκB and its downstream pro-inflammatory signaling due to its secretion in a p53-dependent manner [52]. Through a distinct mechanism from HMGB1, HMGB2 also plays a pro-inflammatory role during senescence. HMGB2 occupies the loci of key SASP genes, which prevents their incorporation into transcriptionally repressive SAHF regions during OIS in HDF [21]. Thus, HMGB2’s presence creates a permissive chromatin landscape for the expression of the SASP genes. Notably, inhibition of HMGB2 selectively suppresses the SASP without affecting the senescence-associated growth arrest [21].
Promoters and enhancers
Disruptor of telomeric silencing 1-like (DOT1L) is both necessary and sufficient for expression of IL1A, one of the key upstream regulators of other SASP genes, through regulating its promoter activity in OIS of multiple human cell types [53]. A predetermined enhancer landscape that is sequentially employed during the senescence process [54]. Activator protein 1 (AP-1) transcription factor plays a key role in this predetermination by imprinting a prospective senescence enhancer landscape, which foreshadows future transcriptional activities in the absence of traditional enhancer histone modification marks [54]. Indeed, AP-1 is essential for the SASP independent of senescence-associated growth arrest in both OIS HDF and TIS of colorectal cancer cells.
Systematic profiling of enhancer marker H3K27ac in control and OIS HDF revealed the dynamic remodeling of the regulatory enhancer landscape in senescent cells [55]. Specifically, proliferation arrest of senescent cells is associated with loss of narrow typical enhancers adjacent to the promoters of proliferating genes. In contrast, the formation of new super enhancers in senescent cells drives the SASP. A subset of these newly gained SASP-regulating super enhancers are bound by epigenetic regulator BRD4 (Figure 2, Key figure). Consequently, BRD4 inhibition specifically suppressed the SASP without affecting senescence-associated growth arrest, which reduced the immune surveillance both in vitro and in vivo in mouse models [55].
Figure 2. Key Figure. Rewiring of promoters, enhancers and promoter-enhancer loops plays a key role in driving the SASP.
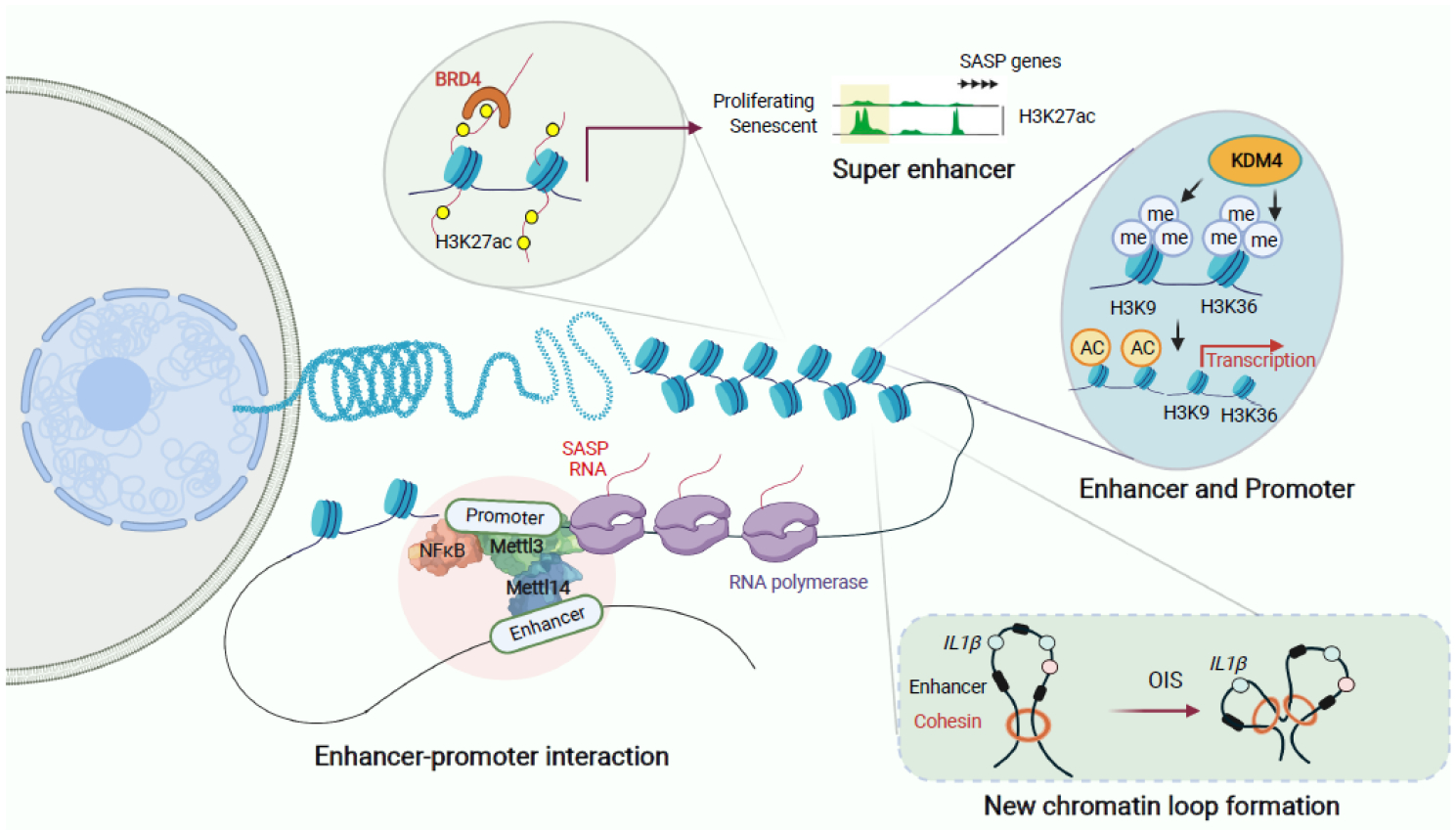
Upper left, global remodeling of the enhancer landscape during senescence. Chromatin reader BRD4 is recruited to the newly formed super enhancers marked by H3K27ac to promote the SASP gene expression. Upper right, histone demethylase KDM4 orchestrates the SASP gene transcription by increasing chromatin openness through demethylating H3K9me3 and H3K36me3 at both distal enhancers and promoters of the SASP genes. Lower left, genome-wide redistribution of the METTL3 and METTL14 complex promotes the SASP gene transcription through mediating enhancer-promoter loop formation. Lower right, cohensin redistribution promotes the SASP through specific enhancer-promoter rewiring and chromatin loop reorganization.
The histone H3-specific demethylase KDM4 diminishes H3K9/H3K36 methylation, which promotes the SASP through altering chromatin accessibility and spatiotemporal reprogramming of the transcriptomic landscape in TIS primary normal human prostate stromal cells [56] (Figure 2, Key figure). Selective targeting of KDM4 suppresses the SASP without affecting senescence-associated growth arrest. Chromatin accessibility changes underlie the observed regulation of the SASP by KDM4, with an increase in accessibility at distal regions of gene promoters. In addition, H3K9me3 and H3K36me3 ChIP-seq analysis indicates that changes of these histone modifications also occur at proximal transcription start sites. Thus, KDM4 likely regulates the SASP through both distal enhancers and promoters [56].
Enhancer-promoter interaction and chromatin loop formation
A recent study revealed that the methyltransferase-like 3 (METTL3) and 14 (METTL14) complex plays a key role in senescence-associated EP contact via chromatin loop formation to promote the SASP in both OIS HDF and TIS ovarian cancer cells [57] (Figure 2, Key figure). METTL3 and METTL14 are core subunits of the methyltransferase complex (MTC) that catalyzes mRNA N6-methyladenosine (m6A) modification [58] (Box 1). Interestingly, genome-wide redistribution of METTL3 and METTL14 promotes the SASP at the transcriptional level independent of its enzymatic function. Specifically, METTL14 is redistributed to the enhancers, while METTL3 is localized to the pre-existing NFκB sites within the promoters of SASP genes during senescence. The METTL3 and METTL14 complex are necessary for SASP without affecting senescence-associated growth arrest. The redistributed METTL3 and METTL14 regulate the SASP gene expression through the formation of chromatin looping via METTL3 and METTL14 interaction in an enzymatic activity-independent manner [57].
BOX 1. MTC and SASP.
The mRNA N6-methyladenosine (m6A) modification plays an important role in the dynamic responses to stresses [93–95]. Notably, the m6A modification is typically coupled with the recruitment of the methyltransferase complex (MTC) to specific chromatin loci to install the m6A modification on their target transcripts. The genome-wide redistribution of METTL3 and METTL14, the core MTC subunits, promotes the SASP transcriptionally in an m6A-independent manner [57]. Specifically, METTL14 is enriched at the enhancers, and METTL3 is enhanced at the NFκB sites within the promoters of the SASP genes during oncogene-induced senescence in IMR90 cells. Inhibition of METTL3 and METTL14 suppresses the SASP without impairing the senescence-associated stable growth arrest. However, the m6A modification on the SASP genes themselves or their chromatin-associated regulatory RNA was not affected. Mechanistically, the redistributed METTL3 and METTL14 allow enhancer and promoter interaction in an enzymatic activity-independent manner to regulate the SASP genes through chromatin looping. Functionally, METTL3 and METTL14 are required for the immune surveillance of senescent cells mediated by the SASP in vivo in an OIS hepatocytes mouse model.
Acute stress-responsive genes are activated by recruitment of transcription factors to pre-existing EP contacts [59–61]. In contrast, OIS in HDF is accompanied by extensive EP rewiring and alteration of chromatin loop structures, which is associated with differential binding of cohesin, but not CTCF [62] (Figure 2, Key figure). Decreased cohesin binding occurs around loop anchors, while de novo cohesin peaks in OIS cells mostly appear at highly active genes with loop domains in a transcription-dependent manner. These de novo cohesin peaks interact with neighboring existing cohesin peaks, suggesting these interactions might mediate local EP contact through the new loop formation. Gene set enrichment analysis using genes involved in differential EP interactions show that transcriptionally upregulated genes are significantly enriched for SASP related “inflammatory” terms. Notably, EP analysis demonstrates a fundamentally distinct difference in the induction of inflammatory cytokines between senescence and acute inflammation [62]. The IL1β expression during OIS is associated with significant EP interaction, but not with TNFα treatment, implying a senescence-specific EP rewiring mechanism to regulate inflammatory SASP. This is entirely different from transcription factor-driven proinflammatory gene expression in response to acute stress inducers. Thus, plasticity associated with the rewiring of EP network and chromatin loop reorganization are additional layers of regulation for inflammatory SASP during senescence as compared with acute stresses or signaling.
TADs and compartments
In RS HDF, Hi-C analysis revealed that compartments and TADs are largely stable compared with either proliferating or quiescent cells [63] (Figure 3). However, there was a significant decrease in long-range interactions (> 2 megabase) and an increase in short-range interactions (< 2 megabase). Additionally, gene expression changes correlated with compartment switching with a higher frequency from B to A and a lower frequency from A to B [63]. In contrast to RS, OIS HDF showed that long-range cross-boundary interactions were significantly gained, but short-range local intra-TAD interactions were reduced [64] (Figure 3). Disruption of TADs does not impact the global organization of A/B compartments, suggesting that organization of TADs and compartments are typically independent of each other. A 4D time course studies using OIS model in HDF showed a massive change in compartmentalization. Specifically, a gain of B-B compartmental interaction leads to SAHF formation in OIS, which is accompanied by a loss of A-B compartmental interaction in OIS. On the contrary, a decrease in RS compartmentalization is characterized by a loss of A-A interactions and a gain of A-B interactions [65]. Notably, SASP genes were shown to be in the stable A compartments in this study. In a OIS HDF model, 3D genome changes become obvious when SAHFs are formed and these changes in 3D genome are stabilized over an extended period [65]. Mechanistically, DMNT1-HMGA2 axis contributes to the differences in 3D genome between RS and OIS, which correlates with differences in the SAHF formation [65].
Figure 3. Three dimensional (3D) genomic control of the SASP.
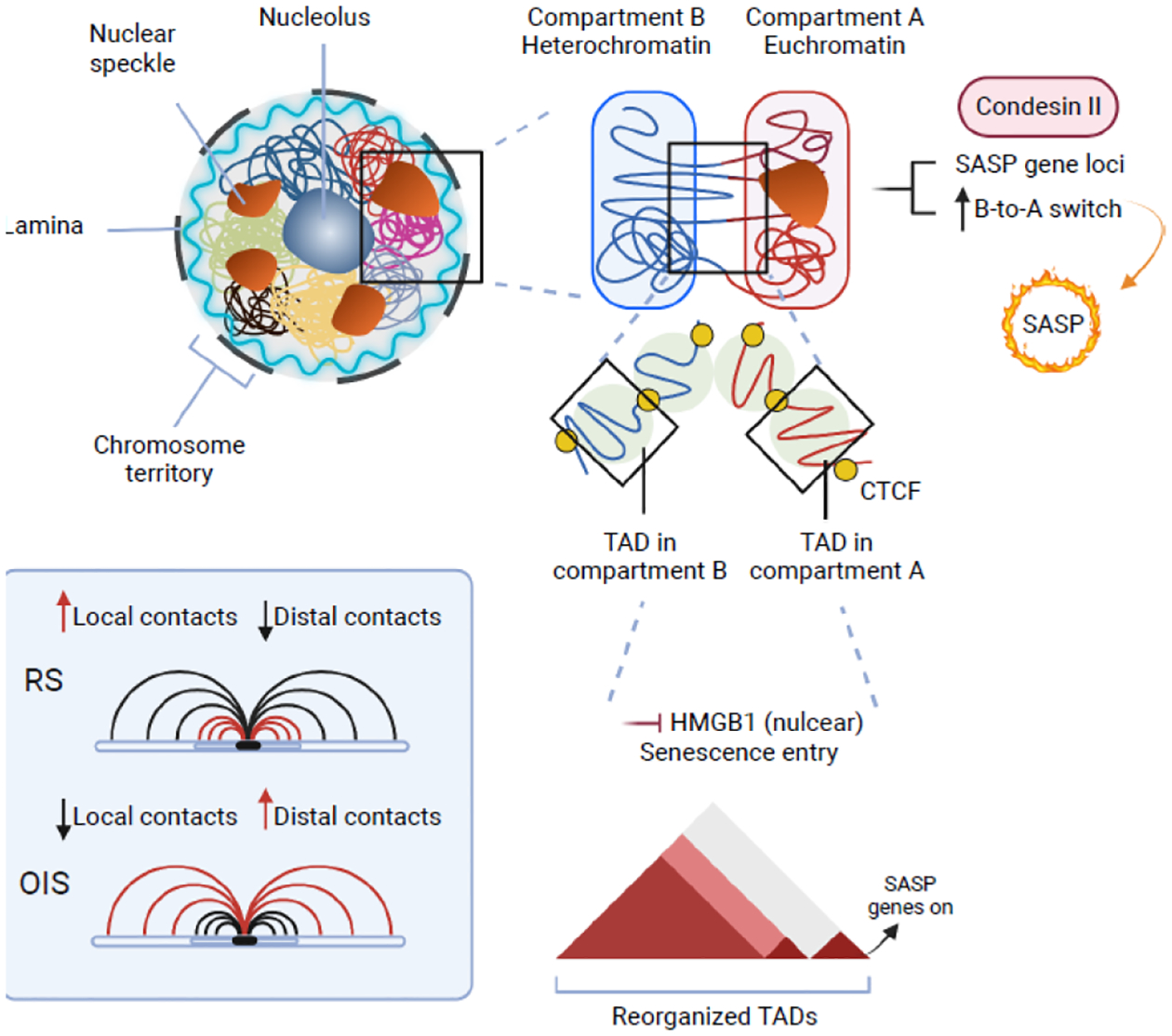
Transcriptionally active and inactive regions of genome form A and B compartments, respectively. Structural maintenance of chromosomes component Condensin II promotes the SASP gene transcription by mediating B-to-A compartmental transition during senescence. Within each compartment, self-interacting domains of genome are organized as topologically associated domains (TADs). Roles of TADs in senescence remain to be fully elucidated. For example, replicative senescence is associated with an increase in short-range intra-TAD interactions, while oncogene-induced senescence is accompanied by a reduction in short-range local intra-TAD interactions. Nuclear HMGB1 regulates the SASP gene transcription by localizing to a subset of TADs boundaries that are enriched in SASP-related genes.
Unlike the well-documented extracellular role of HMGB1 as an inflammatory stimulus, a subset of HMGB1-bound genomic regions mark TAD/loop domain boundaries in RS HDF and primary human umbilical vein endothelial cells (HUVEC) [66] (Figure 3). Domains enriched in HMGB1 binding peaks preferentially associated with SASP genes upon senescence entry or HMGB1 knockdown [66], suggesting that HMGB1 loss affects chromatin topology in a manner relevant to gene expression. Distinct from HMGB1, the loss of HMGB2 does not induce the SASP in RS cells [21, 67]. HMGB1 and HMGB2 control TADs in distinct modes through their non-overlapping genomic binding sites in RS of multiple human cell types: HMGB2 marks the extremities of TADs that shift boundaries upon senescence induction, while HMGB1 is mostly found at invariable TAD/loop domain boundaries enriched for SASP-related genes [66, 67].
Whereas cohesin was reported to orchestrate short-range EP interactions [27, 28], another SMC complex condensin II is implicated in reorganizing compartments and plays a critical role in the formation of TADs and chromosomal territories [30]. Condensin II reinforces the euchromatic A compartments in both OIS and RS HDF [31]. Specifically, condensin II localizes to genes that are highly active in senescent cells and, most notably, SASP and p53 target genes (Figure 3). A function of condensin II-mediated heterochromatic B to euchromatic A compartmental transitions is to enhance the activation of genes within these regions in OIS HDF. Indeed, condensin II is critical for expression of genes at the BA-switching regions compared with other regions.
Cytoplasmic chromatin drives the SASP
The innate immune cGAS-STING pathway is critical for the regulation of SASP [68–72]. cGAS recognizes cytosolic double-strand DNA (dsDNA) in a sequence non-specific manner [73, 74]. The activation of cGAS in senescent cells is mainly due to the induction and accumulation of cytoplasmic chromatin fragments (CCF) caused by nuclear membrane blebbing [68–71] and cDNA generated by reverse transcription of de-repressed retrotransposons [38, 75] (Figure 4).
Figure 4. Cytoplasmic chromatin promotes the SASP through the cGAS-STING innate immune pathway.
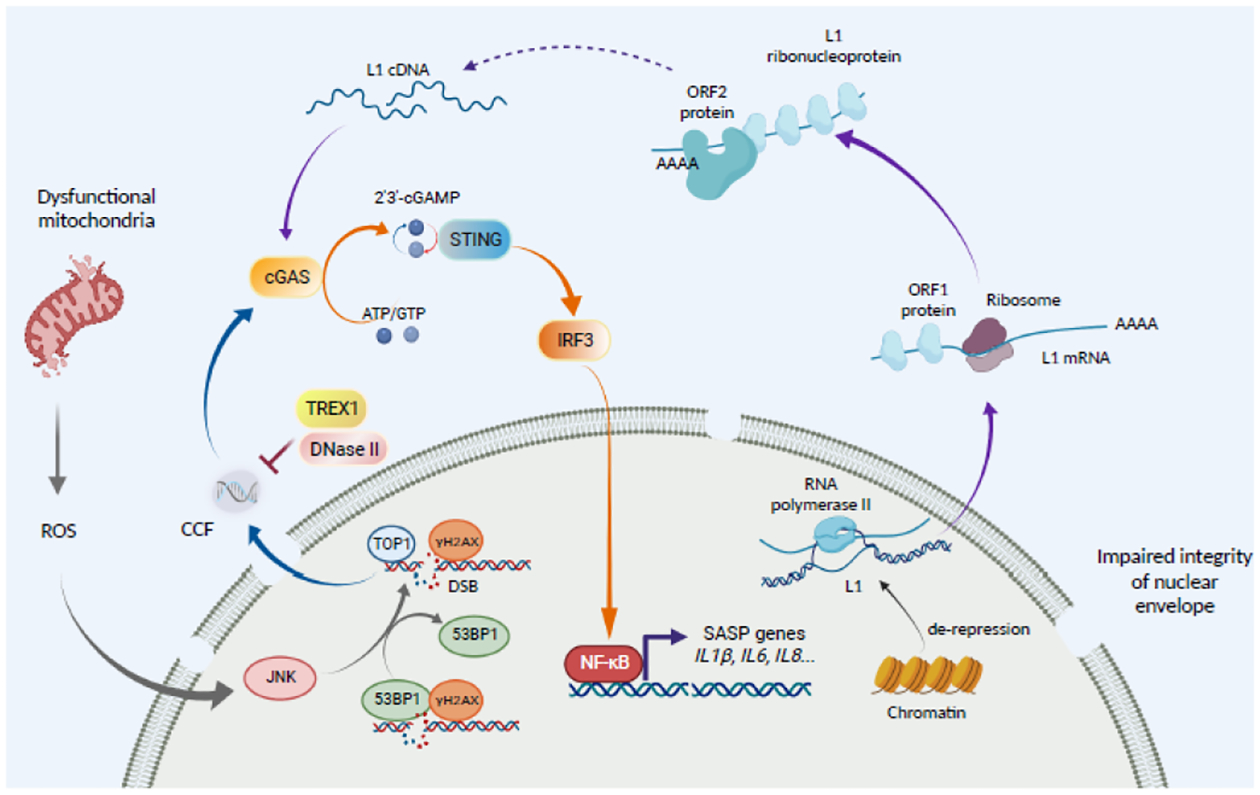
Cytoplasmic chromatin fragments (CCF) formation is initiated by dysfunctional mitochondria that produce excessive reactivated oxygen species (ROS). This activates JNK kinase and detachment of DNA damage repair protein 53BP1 from the DNA damage sites. Topoisomerase I (TOP1) forms a TOP1-DNA covalent cleavage complex to enhance cGAS activity to promote the SASP. Upon activation, cGAS catalyzes adenosine 5’-triphosphate (ATP) and guanosine 5’-triphosphate (GTP) into cyclic GMP-AMP (cGAMP) that binds to and activates STING to recruit tank-binding kinase 1 (TBK1). TBK1 phosphorylates NF-κB to drive the expression of its target SASP genes. CCF formation is negatively regulated by nucleases such as lysosomal nuclease DNase II and cytoplasmic exonuclease TREX1. Another source of cytoplasmic DNA that activates the cGAS-STING pathway is originated from RNA polymerase II-mediated transcription of retrotransposons such as Long Interspersed Element 1 (LINE-1 or L1) during senescence. LINE-1 comprises 17% of human genome. LINE-1 encodes two protein products: the RNA binding protein ORF1 and the endonuclease and reverse transcriptase ORF2. Two possible mechanisms may underlie cytoplasmic L1 cDNA accumulation. High levels of L1 mRNA and protein may result in an increase in cytosolic L1 cDNA through reverse transcription. In addition, high levels of cDNA in the nucleus may fail to re-integrate into the genome, which results in its cytosolic localization through an unknown mechanism. However, the mechanisms by which L1 cDNA is generated from the de-repressed L1 remain unknown.
Cytoplasmic chromatin fragments
During senescence, chromatin undergoes global degeneration and reorganization. The cells lost the integrity of nuclear envelope at a very early stage where the chromatin fragments are pinched off from the nucleus into the cytoplasm to become the CCF [76]. CCF is subjected to autophagosome-lysosome mediated degradation and impairment of autolysome due to downregulation of endonucleases such as DNAase2 and TREX1 which contributes to CCF accumulation in OIS HDF cells [71, 77]. CCF are enriched in DNA damage marker γH2AX, and the repressive histone markers including H3K9me3 and H3K27me3, while devoid of the active chromatin marks [76]. These characteristics suggest that CCF may derive from transcriptionally repressive regions of the genome in senescent cells.
Recently, an upstream signaling event that initiates the formation of CCF have been revealed. Specifically, in an ionizing irradiation (IR)-induced senescent HDF model, the production of reactive oxygen species (ROS) was increased by accumulation of dysfunctional mitochondria [78]. This promotes CCF formation through the activation of c-Jun N-terminal kinase (JNK) via a retrograde signaling pathway. Consistently, mitochondria removal or treatment with either a mitochondria-targeting antioxidant or a JNK inhibitor is sufficient to suppress CCF formation. Notably, JNK interacts with 53BP1, a DNA damage repair protein suppressing the end resection of DNA double strand break (DSB). CCF is negative for 53BP1 and 53BP1 suppresses CCF formation. Knockdown of 53BP1 leads to CCF formation, while its ectopic expression suppresses the CCF formation. However, the exact mechanism by which 53BP1 regulates CCF formation remains unclear (Figure 4). Given the role of 53BP1 in inhibiting the resection of DNA DSB ends [79], it is attempting to speculate that 53BP1 suppresses CCF formation because DNA DSB end resection is required for CCF formation.
CCF accumulate in the cytoplasm to drive the SASP through the cGAS-STING pathway. It was reported that in both OIS and TIS of HDF or ovarian cancer cells, topoisomerase 1-DNA covalent cleavage complex (TOP1cc) are a component of CCF [80]. TOP1cc is both necessary and sufficient for cGAS-mediated CCF recognition and the SASP. Mechanistically, TOP1cc directly interacts with cGAS, which enhances the binding of cGAS to its substrate dsDNA. TOP1cc is stabilized by HMGB2, which is necessary for retaining TOP1cc to CCF. Together, these findings establish a critical role played by HMGB2-TOP1cc that enables CCF recognition by cGAS [80]. They also suggest that TOP1cc modified genomic DNA are selected for enrichment in CCF and, thus, inclusion of genomic DNA in CCF is not random.
Retrotransposons de-repression
In contrast to a potentially passive role displayed by CCF, emerging evidence supports that long-interspersed element 1 (LINE-1, also known as L1) retrotransposable elements become transcriptionally derepressed in RS and OIS HDF and during tissue aging [38, 75]. Cytoplasmic L1 cDNA serves as an important inducer of type I interferon (IFN) signaling through the cGAS-STING pathway to drive chronic age-associated inflammation [38]. These findings suggest that LINE-1 reverse transcription into cDNA and its cytoplasmic localization is a programmed process that is tightly regulated during tissue aging and aberrant activation of this programmed process contributes to cancer [38, 75] (Figure 4).
Notably, in OIS or TIS HDF, CCF-triggered cGAS activation does not induce type I IFN production [68]. This might be due to activation of p38MAPK, which inhibits STING-dependent IFN production [81]. Indeed, p38 inhibition potentiated IFN-β in these cells [68]. In contrast, transcriptionally derepressed L1s activate a type I IFN response in a cGAS-dependent manner [38]. Further, in oxidative stress-induced senescent mouse embryonic fibroblasts, CCF-induced cGAS activation leads to type I IFN production [69]. Therefore, different cell types, senescence inducers and the nature of cytoplasmic DNA may determine the type of cGAS-mediated inflammatory responses.
Harnessing chromatin basis of SASP for intervention
Emerging evidence indicates that targeting senescent cells represents an attractive approach in combating aging and age-associated diseases such as cancer. However, due to the multifaceted roles of senescence, directly eliminating senescent cells by senolytics is often associated with unwanted side effects [8, 14, 15]. Given that the detrimental effect of senescence is typically driven by the SASP, selectively suppressing the SASP while maintaining senescence-associated growth arrest using senomorphics offers an attractive alternative to senolytics.
Chromatin-based control of the SASP in the nuclei offers several avenues to target the SASP. At the transcriptional levels, inhibition of the key SASP-promoting NFκB is an effective approach to inhibit SASP [82]. Epigenetic regulators of the SASP have been explored as senomorphics. BRD4 controls super-enhancer activity of SASP genes and BET inhibitors such as IBET762 are effective in reducing the SASP [55] (Figure 5). In addition, MLL1 inhibition disables the SASP expression by targeting DNA damage pathways [23]. Further, targeting the SASP-promoting pathways downstream of chromatin mechanism has been explored. HMGAs drive inflammatory SASP through upregulating NAMPT [51]. Indeed, selective NAMPT inhibitors such as FK866 dampens the proinflammatory SASP and limits tumor progression in a pancreatic ductal adenocarcinoma mouse model and eliminates ovarian cancer stem cells induced by TIS [51, 83] (Figure 5). Finally, suppressing the EP interaction and the associated loop formation represents a new approach to target the SASP. Inhibition of the METTL3 and METTL14 complex is effective in reducing the SASP in vitro and blunting SASP mediated tumor-promoting and immune surveillance function in vivo in mouse models [57].
Figure 5. Leveraging the SASP for cancer therapy.
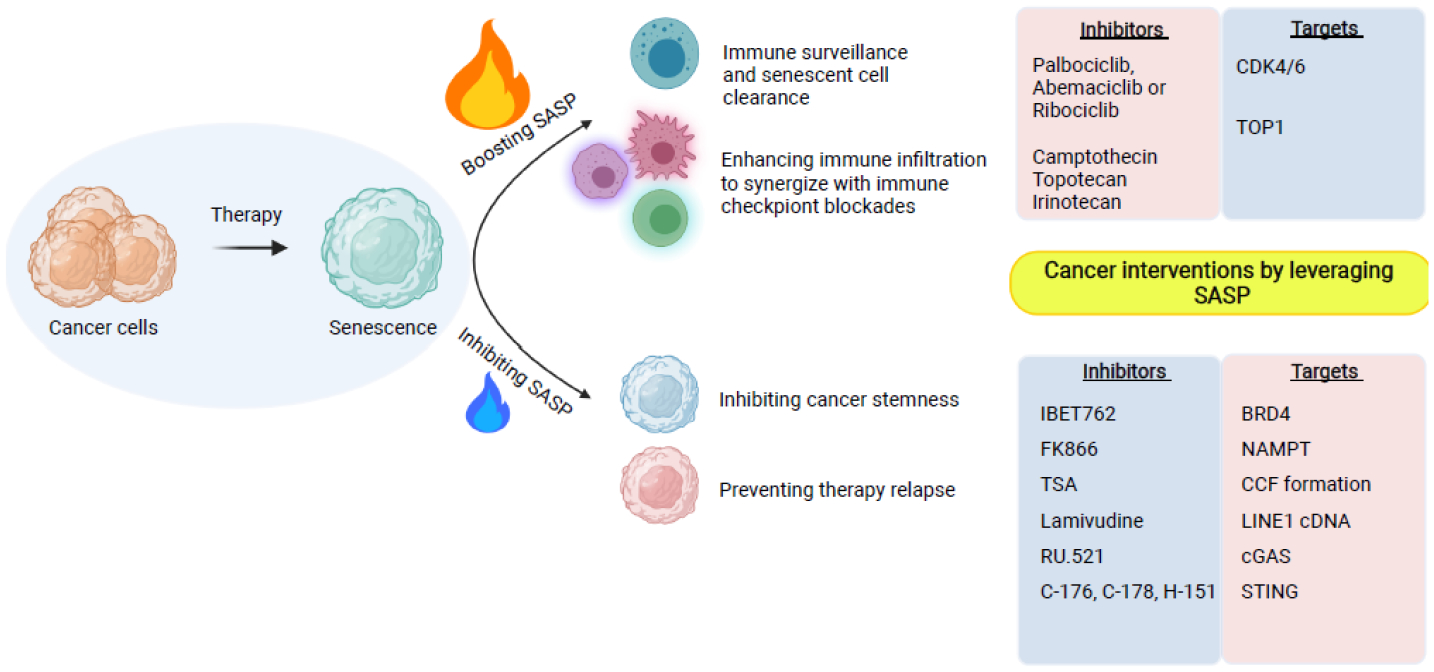
The SASP plays a context-dependent role in senescence-inducing cancer therapy. Boosting the SASP is known to promote immune surveillance and clearance of senescent cells. In addition, the SASP enhances immune infiltration to synergize with cancer immunotherapy such as immune checkpoint blockades. In contrast, inhibiting the SASP is beneficial in reducing cancer stemness and preventing therapy relapse.
The recently discovered cytoplasmic chromatin-mediated activation of the cGAS-STING pathway offers new opportunities to target the SASP. Genetic approaches have validated the role of the cGAS-STING pathway in promoting the SASP and tissue inflammation in vivo [68, 69]. Notably, several inhibitors of cGAS and STING have been developed [84, 85]. In addition to directly targeting the cGAS-STING pathway, approaches that impair its upstream signaling may be equally effective. Nucleotide reverse transcriptase inhibitors such as lamivudine suppress cytoplasmic DNA formation from the L1 retrotransposon, which correlates with a reduction in the late SASP response and an amelioration of several age-associated phenotypes in aged mice [38]. Likewise, low-dose HDAC inhibitors that block the mitochondria-to-nucleus retrograde signaling impairs CCF formation to suppress the SASP [78] (Figure 5).
Inhibition of SASP-associated inflammation is attractive in promoting healthy aging and treating the age-associated diseases. While the net effects of the SASP in cancer might be detrimental, it is important to note that the SASP can be beneficial and, in particular, in the context of cancer immunotherapies such as immune checkpoint blockade (ICB). Indeed, SASP-promoting cGAS is essential for the antitumor effect of ICB [86]. Consistently, cancer therapeutics such as CDK4/6 inhibitor overcomes ICB resistance through inducing proinflammatory SASP and the associated recruitment of immune cells such as effector CD8 T cells [87–89]. Likewise, boosting the SASP by enhancing the cGAS-STING pathway using TOP1 inhibitors sensitizes ovarian tumors to ICBs [90]. It will be interesting to explore the utilization of STING agonists [91] in the context of combining senescence-inducing therapy with immunotherapy. Therefore, caution should be taken when considering targeting the SASP in the context of cancer therapy (Figure 5).
6. Concluding remarks
Chromatin basis of the SASP is in multiple layers and is highly complex. Adding to this complexity, components of the SASP are not universal and often depend on certain stimuli and cell types [92]. Therefore, it will be highly informative to integrate these mechanisms to allow for a better understanding of the relative contributions of various chromatin mechanism in a context and a stimuli-specific manner. Insights gained from these understandings will allow us to leverage the SASP to combat aging and age-associated diseases such as cancer with precision. Many open questions remain (see Outstanding Questions). Cytoplasmic chromatin offers a unique opportunity to gain insights into SASP regulation. L1 cDNA in cytoplasm is linked to late-stage type I IFN chronic inflammation via the cGAS-STING pathway. In addition, CCF activates the cGAS-STING pathway to promote NFκB target genes during acute phase to drive the inflammatory SASP. Understanding the genomic, proteomic and biochemical basis of cytoplasmic chromatin formation and function could offer unique opportunities to not only target the SASP but also selectively eliminate components of the SASP with precision. It is likely that targeting the SASP at different stages of senescence with different agents may allow for harnessing the benefits of senescence while controlling the detrimental effects. Along these lines, temporal and spatial insights into DNA components in the cytoplasmic chromatin remain to be elucidated. Likewise, the nuclear and cytosolic factors in cytoplasmic chromatin that control CCF just began to emerge [80]. These mechanistic insights will allow for targeting these pathways in a context and stage-dependent manner such as differentially targeting nuclear vs. cytoplasmic chromatin. Chromatin basis of the SASP provides exciting opportunities for novel intervention strategies to target the SASP with precision to fight age and age-related pathologies.
Outstanding Questions.
How is the METTL3 and METTL14 redistribution regulated during senescence to drive the SASP via enhancer-promoter interaction?
What are the upstream signaling pathways that control rewiring of enhancer-promoter interaction and chromatin loop reorganization to promote the SASP during senescence?
What is the genomic and proteomic basis of cytoplasmic chromatin fragments?
In addition to nuclear factors, what are the cytoplasmic factors enriched in cytoplasmic chromatin? Can these cytoplasmic factors that selectively localize to cytoplasmic chromatin be targeted to inhibit the SASP?
What is the chromatin basis of context-dependent control of SASP components?
Does the switch of cytoplasmic DNA sources account for acute vs. chronic inflammatory SASP during senescence?
How to leverage chromatin-based mechanisms to target different components of the SASP with precision?
Is it possible to selectively target the SASP by altering high-order chromatin structure?
Highlights.
Two most important characteristics of senescent cells: senescence-associated stable growth arrest and the senescence-associated secretory phenotype are coordinated at the chromatin levels.
High-order chromatin structure controls the SASP transcriptionally in the nucleus of senescent cells.
Rewiring of enhancer and promoter interaction, and reorganization of chromatin loop represent a new layer of the SASP regulation.
Cytoplasmic chromatin promotes the SASP through the cGAS-STING innate immune pathway.
Therapeutic targeting the SASP is an attractive alternative to eliminate senescent cells in combating aging and age-associated diseases such as cancer.
Acknowledgments
We thank Dr. Ken-ichi Noma for his critical reading of the manuscript. The Zhang laboratory is supported by grants from US National Institutes of Health grants (R01CA160331, R01CA163377, R01CA202919, R01CA239128, R01CA243142, P01AG031862, P50CA228991,P30CA010815), US Department of Defense (OC180109 and OC190181), The Honorable Tina Brozman Foundation for Ovarian Cancer Research and The Tina Brozman Ovarian Cancer Research Consortium 2.0, and Ovarian Cancer Research Alliance (Collaborative Research Development Grant #596552).
Glossary
- 3D genome
A hierarchy organizing processes of genomic structures that consist of spatially organized active (A) or repressive (B) compartments, topologically associated domains, and chromatin loops that mediate enhancer-promoter interaction
- Cellular senescence
A state of stable growth arrest triggered by several stress inducers such as critically shortened telomeres, activation of oncogenes and chemotherapeutics
- Chromatin architecture proteins
Non-histone chromatin-bound proteins that regulate gene transcription by altering chromatin architecture
- Cytoplasmic chromatin
DNA and associated proteins localized to cytoplasm that may originate from genomic DNA in the nucleus or cDNA reversely transcribed from de-repressed retrotransponsable elements
- Histone variants
Proteins that substitute for the core canonical histones with specific structural and functional features
- Immune checkpoint blockades
Antibodies or drugs that are designed to interfere with inhibitory pathways that naturally constrain T cell reactivity
- Lamivudine
a nucleoside reverse transcriptase inhibitor
- NAMPT
nicotinamide phosphoribosyltransferase, the rate-limiting enzyme in nicotinamide adenine dinucleotide de novo biosynthesis pathway
- Senescence-associated secretory phenotype (SASP)
Secretion of inflammatory cytokines, immunosuppressive factors, chemokines, and growth factors by senescent cells
- Senescence-associated heterochromatic foci (SAHF)
Facultative heterochromatin domains formed by heterochromatin redistribution and condensation of individual chromosomes that repress proliferation-promoting genes
- Senolytics
Drugs or approaches that eliminate senescent cells inducing them to undergo apoptotic cell death
- Senomorphics
Drugs or approaches that selectively inhibit senescence-associated secretory phenotype without killing senescent cells or affecting senescence-associated growth arrest
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- 1.Hayflick L (1965) The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 37, 614–36. [DOI] [PubMed] [Google Scholar]
- 2.Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuilman T et al. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133 (6), 1019–31. [DOI] [PubMed] [Google Scholar]
- 4.Acosta JC et al. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133 (6), 1006–18. [DOI] [PubMed] [Google Scholar]
- 5.Coppe JP et al. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6 (12), 2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storer M et al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155 (5), 1119–30. [DOI] [PubMed] [Google Scholar]
- 7.Munoz-Espin D et al. (2013) Programmed cell senescence during mammalian embryonic development. Cell 155 (5), 1104–18. [DOI] [PubMed] [Google Scholar]
- 8.Demaria M et al. (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31 (6), 722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krizhanovsky V et al. (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134 (4), 657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Childs BG et al. (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21 (12), 1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collado M and Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10 (1), 51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue W et al. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445 (7128), 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sieben CJ et al. (2018) Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol 28 (9), 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosse L et al. (2020) Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab 32 (1), 87–99 e6. [DOI] [PubMed] [Google Scholar]
- 15.Baker DJ et al. (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479 (7372), 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narita M et al. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113 (6), 703–16. [DOI] [PubMed] [Google Scholar]
- 17.Zhang R et al. (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8 (1), 19–30. [DOI] [PubMed] [Google Scholar]
- 18.Chandra T et al. (2012) Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47 (2), 203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R et al. (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27 (6), 2343–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boumendil C et al. (2019) Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev 33 (3–4), 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aird KM et al. (2016) HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 215 (3), 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah PP et al. (2013) Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27 (16), 1787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capell BC et al. (2016) MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev 30 (3), 321–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH et al. (2020) Heterochromatin: an epigenetic point of view in aging. Exp Mol Med 52 (9), 1466–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belmont AS (2014) Large-scale chromatin organization: the good, the surprising, and the still perplexing. Curr Opin Cell Biol 26, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pueschel R et al. (2016) From single genes to entire genomes: the search for a function of nuclear organization. Development 143 (6), 910–23. [DOI] [PubMed] [Google Scholar]
- 27.Dixon JR et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485 (7398), 376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SS et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159 (7), 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nora EP et al. (2017) Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169 (5), 930–944 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosin LF et al. (2018) Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet 14 (7), e1007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki O et al. (2019) Involvement of condensin in cellular senescence through gene regulation and compartmental reorganization. Nat Commun 10 (1), 5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon JR et al. (2016) Chromatin Domains: The Unit of Chromosome Organization. Mol Cell 62 (5), 668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pope BD et al. (2014) Topologically associating domains are stable units of replication-timing regulation. Nature 515 (7527), 402–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herranz N et al. (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17 (9), 1205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laberge RM et al. (2015) MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17 (8), 1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoare M et al. (2016) NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18 (9), 979–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito Y et al. (2017) Spatial and Temporal Control of Senescence. Trends Cell Biol 27 (11), 820–832. [DOI] [PubMed] [Google Scholar]
- 38.De Cecco M et al. (2019) L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566 (7742), 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chien Y et al. (2011) Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25 (20), 2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y et al. (2019) NF-kappaB signaling in skin aging. Mech Ageing Dev 184, 111160. [DOI] [PubMed] [Google Scholar]
- 41.Chen F et al. (2018) Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat Commun 9 (1), 4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang C et al. (2015) The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349 (6255), aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toso A et al. (2014) Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep 9 (1), 75–89. [DOI] [PubMed] [Google Scholar]
- 44.Sturmlechner I et al. (2021) p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 374 (6567), eabb3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H et al. (2015) MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol Cell 59 (5), 719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Contrepois K et al. (2017) Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun 8, 14995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sumter TF et al. (2016) The High Mobility Group A1 (HMGA1) Transcriptome in Cancer and Development. Curr Mol Med 16 (4), 353–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bianchi ME and Agresti A (2005) HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15 (5), 496–506. [DOI] [PubMed] [Google Scholar]
- 49.Thomas JO (2001) HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans 29 (Pt 4), 395–401. [DOI] [PubMed] [Google Scholar]
- 50.Narita M et al. (2006) A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126 (3), 503–14. [DOI] [PubMed] [Google Scholar]
- 51.Nacarelli T et al. (2019) NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 21 (3), 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davalos AR et al. (2013) p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201 (4), 613–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leon KE et al. (2021) DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J Cell Biol 220 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Zamudio RI et al. (2020) AP-1 imprints a reversible transcriptional programme of senescent cells. Nat Cell Biol 22 (7), 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tasdemir N et al. (2016) BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 6 (6), 612–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang B et al. (2021) KDM4 Orchestrates Epigenomic Remodeling of Senescent Cells and Potentiates the Senescence-Associated Secretory Phenotype. Nat Aging 1 (5), 454–472. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Liu P et al. (2021) m(6)A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. Nat Cell Biol 23 (4), 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yue Y et al. (2015) RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29 (13), 1343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin F et al. (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503 (7475), 290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolovos P et al. (2016) Binding of nuclear factor kappaB to noncanonical consensus sites reveals its multimodal role during the early inflammatory response. Genome Res 26 (11), 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Comoglio F et al. (2018) Thrombopoietin signaling to chromatin elicits rapid and pervasive epigenome remodeling within poised chromatin architectures. Genome Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olan I et al. (2020) Transcription-dependent cohesin repositioning rewires chromatin loops in cellular senescence. Nat Commun 11 (1), 6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Criscione SW et al. (2016) Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv 2 (2), e1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chandra T et al. (2015) Global reorganization of the nuclear landscape in senescent cells. Cell Rep 10 (4), 471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sati S et al. (2020) 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol Cell 78 (3), 522–538 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sofiadis K et al. (2021) HMGB1 coordinates SASP-related chromatin folding and RNA homeostasis on the path to senescence. Mol Syst Biol 17 (6), e9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zirkel A et al. (2018) HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol Cell 70 (4), 730–744 e6. [DOI] [PubMed] [Google Scholar]
- 68.Dou Z et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550 (7676), 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gluck S et al. (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19 (9), 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang H et al. (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114 (23), E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takahashi A et al. (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9 (1), 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller KN et al. (2021) Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184 (22), 5506–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu J et al. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339 (6121), 826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun L et al. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121), 786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simon M et al. (2019) LINE1 Derepression in Aged Wild-Type and SIRT6- Deficient Mice Drives Inflammation. Cell Metab 29 (4), 871–885 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ivanov A et al. (2013) Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202 (1), 129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han X et al. (2020) Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J Biol Chem 295 (14), 4451–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vizioli MG et al. (2020) Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 34 (5–6), 428–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mirman Z and de Lange T (2020) 53BP1: a DSB escort. Genes Dev 34 (1–2), 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao B et al. (2020) Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat Commun 11 (1), 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen Y et al. (2017) p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J Exp Med 214 (4), 991–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laberge RM et al. (2012) Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11 (4), 569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nacarelli T et al. (2020) NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res 80 (4), 890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lama L et al. (2019) Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun 10 (1), 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haag SM et al. (2018) Targeting STING with covalent small-molecule inhibitors. Nature 559 (7713), 269–273. [DOI] [PubMed] [Google Scholar]
- 86.Xiang Y et al. (2017) RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543 (7646), 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jerby-Arnon L et al. (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175 (4), 984–997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wagner V and Gil J (2020) Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 39 (29), 5165–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruscetti M et al. (2020) Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181 (2), 424–441 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hao X et al. (2021) Sensitization of ovarian tumor to immune checkpoint blockade by boosting senescence-associated secretory phenotype. iScience 24 (1), 102016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Amouzegar A et al. (2021) STING Agonists as Cancer Therapeutics. Cancers (Basel) 13 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Basisty N et al. (2020) A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18 (1), e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roundtree IA et al. (2017) Dynamic RNA Modifications in Gene Expression Regulation. Cell 169 (7), 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ries RJ et al. (2019) m(6)A enhances the phase separation potential of mRNA. Nature 571 (7765), 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fu Y and Zhuang X (2020) m(6)A-binding YTHDF proteins promote stress granule formation. Nat Chem Biol 16 (9), 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]