

Abstract
Background:
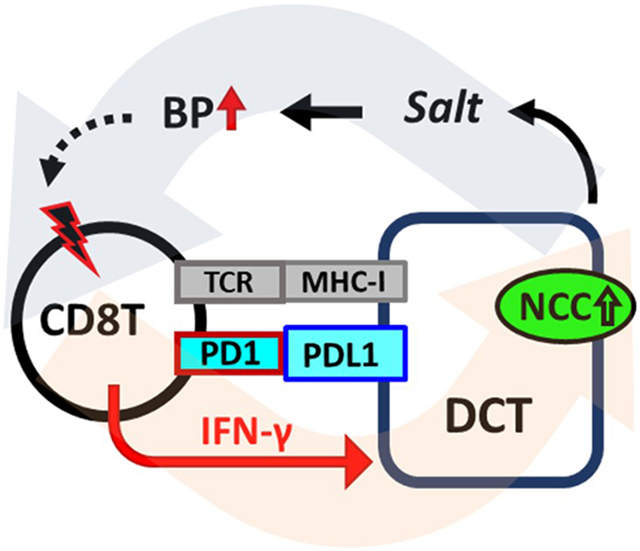
Renal T cells contribute importantly to hypertension, but the underlying mechanism is incompletely understood. We reported that CD8Ts directly stimulate distal convoluted tubule cells (DCTs) to increase sodium chloride co-transporter (NCC) expression and salt reabsorption. However, the mechanistic basis of this pathogenic pathway that promotes hypertension remains to be elucidated.
Methods:
We used mouse models of DOCA+salt (DOCA) treatment and adoptive transfer of CD8+ T cells (CD8T) from hypertensive animals to normotensive animals in in-vivo studies. Co-culture of mouse distal convoluted tubule cells (mDCTs) and CD8Ts was used as in-vitro model to test the effect of CD8T activation in promoting NCC-mediated sodium retention and to identify critical molecular players contributing to the CD8T-DCT interaction. IFNγ-KO mice and mice receiving renal tubule-specific knockdown of PDL1 were used to verify in-vitro findings. Blood pressure was continuously monitored via radio-biotelemetry, and kidney samples were saved at experimental endpoints for analysis.
Results:
We identified critical molecular players and demonstrated their roles in augmenting the CD8T-DCT interaction leading to salt-sensitive hypertension. We found that activated CD8Ts exhibit enhanced interaction with DCTs via IFNγ-induced upregulation of MHC-I and PDL1 in DCTs, thereby stimulating higher expression of NCC in DCTs to cause excessive salt retention and progressive elevation of blood pressure. Eliminating IFNγ or renal tubule-specific knockdown of PDL1 prevented T cell homing into the kidney, thereby attenuating hypertension in two different mouse models.
Conclusion:
Our results identified the role of activated CD8Ts in contributing to increased sodium retention in DCTS through the IFNγ-PDL1 pathway. These findings provide a new mechanism for T cell involvement in the pathogenesis of hypertension and reveal novel therapeutic targets.
Keywords: High Blood Pressure, Hypertension
Graphical Abstract
Introduction:
More than one billion people worldwide are afflicted with hypertension and over half of them exhibit salt sensitivity1, 2, which increases the risk for cardiovascular events and mortality3, 4. Although many drug families are used to treat hypertension, fewer than 50% of patients achieve blood pressure control5-7. Thus, it is important to identify unknown mechanisms involved in the pathogenesis of salt-sensitive hypertension and develop improved medications for its treatment. In recent years, an important role has been proposed for T cells in hypertension8-11. Unfortunately, there are no related treatment options because the mechanisms underlying T cell involvement in blood pressure regulation are largely unknown. Therefore, new research focused on the cellular targets of T cells and their pathogenic signaling pathways are critically needed to improve our understanding of the pathogenesis of hypertension.
A previous study from our group demonstrated a potential mechanism for T cell-mediated sodium handling in the kidney12. We found that CD8+ T cells (CD8Ts) directly interact with renal distal convoluted tubule cells (DCTs), upregulating the expression and activity of the sodium chloride co-transporter (NCC), leading to excessive salt retention12. Interestingly we also observed that CD8Ts isolated from deoxycorticosterone acetate and high salt (DOCA)-treated hypertensive mice more effectively stimulated DCTs to express NCC compared to the same number of CD8Ts isolated from normotensive mice12; however, the mechanisms underlying this observation remain elusive. Recent clinical and pre-clinical studies have indicated that CD8Ts in hypertensive subjects exhibit greater activity, as evidenced by higher production of the cytokine interferon gamma (IFNγ) compared to those isolated from normotensive subjects13, 14. Whether and how this inappropriate activation of CD8Ts contributes to excessive NCC-mediated salt retention are critical questions, that if answered, will not only provide insight into the pathogenesis of salt-sensitive hypertension but also potentially identify new therapeutic targets.
Several reports have shown that the immunosuppressive drug mycophenolate mofetil (MMF), which suppresses T cell activity and disrupts cytokine production, attenuates hypertension in different animal models of the disease15-18. Although the underlying molecular mechanisms are unclear, IFNγ is evidently involved in the upregulation of NCC and the development of hypertension, because deletion of IFNγ blunts NCC upregulation and blood pressure elevation in an angiotensin II infusion mouse model19. These findings led us to hypothesize that IFNγ may be a component of the molecular pathway by which activated CD8Ts induce NCC upregulation in DCTs, thereby contributing to the pathogenesis of excessive salt retention.
In our previous report, we also proposed that an “immunological synapse-like” direct contact between CD8Ts and DCTs is necessary for CD8Ts to stimulate NCC expression and salt retention in DCTs12. However, in addition to the CD8T-specific antigen-presenting molecule MHC-I (major histocompatibility complex class I), a co-signaling molecule is often required to form synapses between CD8T cells and target cells20, 21. The critical co-signaling molecules that mediate the synapse interface between DCTs and CD8Ts in the kidney are unknown. PDL1 (programmed death-ligand 1, also known as CD274 or B7-H1) belonging to the B7 immune regulatory family, is a potent co-signaling molecule that reduces activated CD8T-induced cytotoxicity in target cells22-25. Renal transplant studies reveal that IFNγ produced by activated T cells stimulates the expression of PDL1 in renal tubular epithelial cells. Subsequently PDL1 protects these cells from T cell-induced death by binding to its PD1 receptor on the surface of activated T cells26-28. Despite its protective role in renal transplant, we investigated whether this PDL1-PD1 interaction also may fulfill the requisite co-signaling role for the kidney homing of CD8Ts in hypertension.
To our knowledge, no earlier studies determined the role of IFNγ-mediated renal expression of PDL1 in the development of salt-sensitive hypertension. Thus, in the current study, we propose a novel mechanism contributing to the pathogenesis of hypertension: as a response to renal infiltration of activated T cells, DCTs in the kidney are primed by excessive IFNγ to express a high level of PDL1, which provides co-signaling to its receptor PD1 on CD8Ts, leading to enhanced synapse-like direct interaction between activated CD8Ts and DCTs, pathologically stimulating NCC expression and function, exacerbating salt retention, and elevating blood pressure.
Methods:
Data availability:
All relevant data are available from the authors upon request.
Animals:
The animal use protocol was approved by the Institutional Animal Care and Use Committee at University of Arkansas for Medical Sciences. In the DOCA-salt mouse model, 10~11 weeks old male C57B6 mice were uninephrectomized and randomly assigned to either the sham group, or the DOCA-salt group that received a DOCA pellet subcutaneously followed by 1% NaCl in the drinking water for 3 weeks. The kidneys and splenic T cells were harvested at the end of day 20. In the CD8+ T cell adoptive transfer mouse model, which we described previously12, uninephrectomized 12 weeks old male C57B6 mice were randomly assigned to the sham group that received a saline injection, or assigned to the adoptive transfer group that was injected with fresh isolated CD8+ T cells (from spleens of DOCA-salt mice) via a single tail vein injection at the dose of 1x107 cells/100 μl saline/mouse. High-salt drinking water (1%) was available to sham mice and mice receiving CD8+ T cells as indicated in Figure 7. Blood pressure in this mouse model was measured directly by radio-biotelemetry (DSI). The current study only used male mice because biological sex is a significant factor that affects the regulation of blood pressure and immunity. Specifically, a previous report indicated that adoptive transfer of T cells to T cell deficient mice restores Angiotensin II (AngII)-induced hypertension in male recipient mice but not in female recipients29, suggesting the role of T cells in mediating hypertension is greater in males than females.
Figure 7: Effects of renal tubule-specific knockdown of PDL1 in mice receiving adoptive transfer of CD8Ts.

(a), Effects of renal tubule specific siPDL1 on T cell-accumulation in the kidneys of mice received CD8T-adoptive transfer. Scale=50μm. Data are representative of n>15 images in each group. (b), Intensity quantification of NCC positive segments in stitched images of whole kidney sections from CD8T-adoptive transfer recipient mice treated with or without siPDL1. n=8 mice in each group. (c), Radio-biotelemetry recording of systolic blood pressure in recipient mice of CD8T adoptive transfer with or without injection of nanoparticles containing siPDL1. Blood pressure was recorded for 10 seconds every 15 minutes. n=5 mice for +CD8Ts, n=6 mice for +CD8Ts +siPDL1. Data are means ± sem. Statistical significance was assessed by Mann-Whitney test for a, b, and two-way ANOVA for c.
T cell isolation and analysis:
Mouse spleens or kidneys were dissociated using a GentleMACs tissue dissociator and C-tubes followed the manufacturer’s protocol. Dynabeads T cell isolation kits were used to isolate T cells from splenocytes. The kidney T cell isolation followed a previously reported method10. Briefly, kidney homogenate was filtered using a 70μm sterile filter and leukocytes enriched by centrifugation in 36% and 72% percoll followed by CD45 selection using MagniSort beads. Subtypes of collected T cells were confirmed and analyzed using flow cytometry.
Ex-vivo stimulation:
Intracellular cytokine assays to evaluate cytokine production in CD8T cells used freshly isolated CD8T cells, which were either untreated or stimulated with cell activation cocktail (Bio-Legend #423304, PMA/Ionomycin) in the presence of brefeldin A for 3-4 h at 37°C 30-32.
Renal tubule specific knockdown using nanoparticles:
Ambion in-vivo siRNAs against mouse PDL1 or scramble sequence (as negative control) were purchased from Thermo Fisher and encapsulated into renal tubule-specific nanoparticles at the dose of 5 ng siRNA per 1 mg nanoparticles. Nanoparticles loaded with siPDL1 (or scrambled control) were formulated similarly to a previous description using an emulsion-nanoprecipitation method33-35. Briefly, 100 mg diblock poly (lactic-co-glycolic acid)-carboxy-terminated polyethylene glycol (PLGA-PEG) was dissolved in 2 mL acetonitrile, into which 100 ng of siPDL1 (or scrambled control) in RNase-free water were added dropwise and bath sonicated for 2 minutes. The resulting emulsification was added dropwise to 100 mL water with 100 μL Pluronic F-68 as a surfactant. After stirring for 2 hours, the particle suspension was recovered by centrifugation and immediately washed and centrifuged again. The particles were suspended in 2% sucrose solution and lyophilized to dryness. The hydrodynamic diameter of nanoparticles was evaluated via dynamic light scattering and determined to be 402.5±7.4nm with a polydispersity index (PDI) of 0.34 for siPDL1-nanoparticles and 399.3±16.8nm with a PDI of 0.28 for scrambled control siRNA nanoparticles. Immediately prior to use, nanoparticles were suspended at 16mg/ml in saline and injected into mice at the dose of 1.6mg nanoparticles/mouse via tail vein injection. Our preliminary tests indicate that the treatment with siPDL1 containing nanoparticles does not alter baseline blood pressure or renal T cell infiltration as shown in Figure S1.
Cell culture treatment & harvest:
The mDCT15 cells (referred to in the text simply as mDCTs) and the CD8+T cells (TK1 cells) were introduced in our previous report12. In pre-activation studies, CD8Ts were pre-incubated with cell activation cocktail (PMA/Ionomycine, Biolegend) for 4 hours before being washed and co-cultured with mDCTs. All cells were maintained at 37°C and 5% CO2, and no contaminations were found in cultured cells during our experiments. All treatments of mDCTs were overnight (approximately 12 hours). Co-culture and harvest methods also followed the same protocol as we previously reported12. The quantification of area covered by T cells co-cultured with mDCT cells was estimated by a semi-automated segmentation script using iVision 4.5.4 (BioVision Technologies, Exton, PA). The segmentation was based on intensity threshold, mean intensity, area, and circularity to exclude artifacts. Segmented images were inspected for accuracy and fine adjustments to parameters were made as necessary. Representative images of CD8T-DCT adhesion after co-culture were selected to represent the average of quantification data. Cell co-culture (adhesion) images were acquired using an inverted routine microscopy and Cannon EOS camera with eye-piece adaptor. The scale bars of images were estimated by calculating pixels in an 1mm length image acquired using the same system.
Transfection of siRNAs:
All siRNAs were purchased from ABI. Transfection of siRNAs was accomplished using lipofectamine RNAimax following the manufacturer’s protocol. siRNA-mediated knockdown effects in mDCTs are shown in Figure S2.
Flow cytometry:
For surface staining, cells were stained with antibodies for 30-45 minutes in cell isolation buffer in dark tubes before analysis. For the NCC functional study using CoroNa Green (cell permeable, Molecular Probes), details are described in our previous publication12. Briefly, mDCTs were treated with ouabain, bumetanide and amiloride for 30 minutes at room temperature (RT) followed by loading of CoroNa Green at a concentration of 10 μM for 1 hour at RT. Cells were washed and resuspended in PBS containing the same blockers mentioned above for 45 minutes and analyzed in a BD Accuri C6 flow cytometer immediately. Intracellular cytokine staining of CD8Ts was performed using BD Cytofix/Cytoperm Plus (Cat. 555028) according to manufacturer protocols. In brief, stimulated cells were incubated in the presence of the BD GolgiPlug Protein Transport Inhibitor (with Brefeldin A, 1 microliter/mL cell culture). After 3 hours, cells were harvested and fixed in BD fixation/permeabilization solution, then washed twice in perm/wash buffer and resuspended for intracellular cytokine staining. After 30 minutes of incubation, cells were washed and resuspended in cell isolation buffer for analysis. Flow cytometry data were analyzed using FlowJo software (version v10.7.1). For all flow cytometry studies, cells lacking incubation with fluorescent antibodies (or corona-green) were used as negative controls to determine autofluorescence and gating. Single cells were selected using the FSC-H/FSC-A gating method for all studies and live cells were selected using 7-AAD staining, as shown in Figure S3. Representative flow cytometry images were selected to exemplify quantification data.
Immunostaining:
4% PFA fixed kidneys were processed as 8μm thick paraffin sections in the DNA Damage and Toxicology Core at UAMS. Following antigen retrieval using antigen unmasking solution (Vector), sections were washed and blocked in 5% non-fat milk containing 4% goat serum at RT for 1 hour prior to overnight incubation with primary antibodies. After secondary antibody staining, sections were sealed with ProLong Gold mounting media (Molecular Probes). Images were captured using Keyence XZ-800 microscope. Cell counting was conducted using ImageJ software. Representative images were selected to exemplify the average of quantification data.
Mean intensity quantification of NCC:
All kidney sections in the experimental group and the corresponding controls were stained, imaged, and analyzed under the same conditions. The whole kidney section image was constructed by stitching thousands of images taken on Keyence XZ-800 with a 10x PlanApo NA 0.45 at low power and exposure time to avoid saturation. Intensity cutoffs for tissue and NCC-positive pixels were determined by a blinded investigator and applied to the experimental and control groups uniformly. The mean intensity for each kidney was calculated by (total NCC-positive pixel intensity – mean background tissue intensity x total NCC-positive pixel area) / total tissue area. Image segmentation and analysis were performed using iVision 4.5 (BioVision Technologies).
Western blot:
Detailed methods and materials have been published12. Briefly, bis-tris gel (from Genscript) was used for electrophoresis, and gels were transferred to PVDF membranes on ice. All membranes were blocked in 5% non-fat milk for 1 hour before incubation with primary antibodies overnight. After washing and incubation with HRP conjugated secondary antibodies, western blot images were obtained using a ChemiDoc XRS+ system and analyzed using ImageLab software (BioRad). Representative images were selected that exemplified the average of quantification data.
Statistical analysis:
Data are presented as means ± sem. All statistical analysis were preformed using GraphPad 7.05. Data were tested for normality using D'Agostino & Pearson test (for sample n≥8) or Shapiro-Wilk test (for sample n<8). The threshold for normality tests was set to 0.05, and p>0.05 were considered normally distributed. Unpaired t-tests were used to compare parametric data between two independent groups, whereas Mann-Whitney tests were used to compare non-parametric data between two independent groups. Two-sided p values are indicated in the figures. For multiple comparisons, statistical analysis was carried out by ANOVA followed by Tukey’s or Dunnett’s post hoc tests. P values < 0.05 were considered to be significant, non-significant were labeled as n.s. in figures. No sample size estimate was performed, but sample sizes were selected based on previous experiments. All assays were repeated in independent experiments and displayed figures are representative.
Major Resources:
Please see the Major Resources Table in the Supplemental Materials.
Results:
CD8Ts in hypertensive mice are excessively activated and promote stimulation of DCTs
We reported earlier that CD8Ts increased expression of NCC in mouse DCTs (mDCTs)12. Here we found that compared to Sham CD8Ts (CD8Ts isolated from sham normotensive mice), DOCA CD8Ts (CD8Ts isolated from DOCA hypertensive mice) stimulated higher proportion of mDCTs to exhibit elevated NCC-mediated sodium retention (Figure 1a). NCC-mediated sodium uptake was assessed by the fluorescent intracellular sodium indicator Corona Green as we previously reported12. A comparison between CD8Ts isolated from DOCA hypertensive mice and sham normotensive mice indicated that DOCA CD8Ts exhibited higher activity than sham CD8Ts, as evidenced by higher expression of the cytokines IFNγ and TNFα (Figure 1b). This new finding was supported by cell surface staining with PD1 (Figure S4), a checkpoint molecule that is upregulated in activated CD8Ts36, 37. Higher proportions of PD1-expressing CD8Ts were found in DOCA mice compared to sham mice (Figure S4). To determine whether activation of CD8Ts augments their interaction with DCTs, we pre-activated CD8Ts (act CD8Ts) by stimulating them with PMA and ionomycin for four hours prior to co-culture with mDCTs. After overnight co-culture and washing off non-adherent CD8Ts using PBS, we observed significantly higher numbers of act-CD8Ts adhering to mDCTs compared to naïve CD8Ts (non-act CD8Ts, Figure 1c). Whereas CD8Ts are known to stimulate NCC expression in mDCTs12, we found that CD8T-induced NCC upregulation in mDCTs was remarkably augmented by pre-activating the CD8Ts (Figure 1d). These findings indicate that the higher activity of CD8Ts in salt-sensitive hypertensive subjects may promote inappropriate upregulation of NCC in their kidneys.
Figure 1: Activation of CD8Ts in DOCA mice and impact of activated CD8Ts on DCTs.

(a), Flow cytometry study using the intracellular sodium indicator Corona Green to detect NCC-mediated sodium uptake in mDCTs. Upper population represent DCTs with higher NCC-mediated sodium uptake (number inside the square indicate proportion of cells); lower population represent cells with lower sodium uptake. All studies were performed in the presence of ouabain, bumetanide and amiloride. Data are representative of eight independent tests. Quantifications determined the CD8T-induced Δ proportion of high-sodium retaining mDCTs compared to baseline (mDCTs only, without co-culture). (b), mRNA expression of activation markers IFNγ and TNFα in CD8Ts isolated from spleens of DOCA hypertensive mice and sham normotensive mice. n=4 mice in each group. (c), Effects of pre-activating CD8Ts on their adhesion with DCTs. After overnight co-culture, cells were washed with PBS x 2 times. Scale=30μm. Data are representative of n=7-9 images in each group. (d), Effects of pre-activating CD8Ts on stimulating NCC expression in DCTs. Quantitative western blot data were normalized using GAPDH as a loading control. n=4–5 samples in each group. Data are means ± sem. Statistical significance was assessed by t-test for a, b, c, and ANOVA for d.
IFNγ mediates the stimulatory effect of pre-activated CD8Ts on progressive elevation of blood pressure
One major function of activated T cells is to produce cytokines and chemokines, particularly the cytokines IFNγ and TNFα, which are reported as important contributing factors to the development of hypertension and subsequent kidney injury11, 13, 14, 38, 39. To explore a potential difference in cytokine-producing capacity between sham-CD8Ts and DOCA-CD8Ts, we stimulated these cells ex-vivo and stained for intracellular cytokines using specific antibodies30-32. Results in Figure 2a show that stimulated DOCA CD8Ts produce significantly more IFNγ compared to sham CD8Ts, whereas no difference was found in the production of TNFα between two groups of cells (Figure 2a). These results agree with recent clinical and pre-clinical observations of higher production of IFNγ but not TNFα by CD8Ts obtained from hypertensive patients compared with normotensive individuals13, 14, and support the finding that IFNγ contributes to upregulation of renal NCC and elevated blood pressure in angiotensin II-treated hypertensive mice19. These results led us to speculate a plausible role for IFNγ in augmenting CD8T-DCT interaction and NCC upregulation, thereby exacerbating hypertension. To further explore this hypothesis in in-vitro studies, we introduced neutralizing antibody against IFNγ at 15μg/ml into co-cultures of both types of cells. As expected, pre-activation of CD8Ts-induced additional expression of NCC in DCTs, which was prevented by neutralizing IFNγ in the co-culture environment (Figure 2b). It is worth noting that neutralizing IFNγ only blocked the additional effect of NCC upregulation by pre-activated CD8Ts, without further normalizing the expression of NCC in DCTs to control level (Figure 2b). This result is consistent with our previous report that neutralizing IFNγ does not affect NCC expression in DCTs treated with non-act CD8Ts12. As a corollary, we found that adding recombinant mouse IFNγ (mIFNγ) into the co-culture augmented adherence of DCTs and non-act CD8Ts (Figure S5a) and profoundly enhanced the effect of non-act CD8T-induced NCC upregulation in DCTs (Figure S5b). However, IFNγ per se did not affect NCC expression in DCTs without co-culture (Figure S5b).
Figure 2: Effects of IFNγ deficiency on mDCT cell phenotype, and blood pressure and renal CD8T cell abundance in hypertensive mice.

(a), Flow cytometry was used to determine cytokine production by CD8Ts using intracellular staining of IFNγ and TNFα in PMA (phorbol myristate acetate) & ionomycin-stimulated CD8Ts. Numbers in upper square indicate the percentage of isolated CD8Ts with high production of IFNγ or TNFα. n=6-8 mice each group. (b), Effects of neutralizing IFNγ on NCC expression in DCTs treated with pre-act CD8Ts. Quantitative western blot data were normalized using GAPDH as loading control. n=4 samples in each group. (c), Averaged radio-biotelemetry recording of systolic blood pressure in 4 wildtype mice (WT, red line) and 7 IFNγ-knockout mice (IFNγ-KO, blue line). All mice were given DOCA salt treatment as indicated. Blood pressure was recorded for 10 seconds every 15 minutes. (d), Immunostaining of T cell marker CD3 (brown) in kidneys from DOCA-salt-treated wild type (WT) and IFNγ knockout (KO) mice. Scale-50μm. Data are representative of n>15 images in each group. Data are means ± sem. Statistical significance was assessed by t-test for a, d, one-way ANOVA for b, and two-way ANOVA for c.
Results consistent with the in-vitro findings were obtained from in-vivo studies. We found that knockout of IFNγ (IFNγ-KO) lowers blood pressure in DOCA-salt treated mice (Figure 2c). However, we noticed that this effect only occurred in the later phase of progressive elevation of blood pressure, but not in the early phase of the initial blood pressure rise (Figure 2c). This in-vivo result resembles those in previous studies using DOCA-salt treated thymus-deficient nude mice or DOCA-salt treated mice administered the immunosuppressant MMF18, 40-42, implying a possible contributing role of IFNγ to adaptive immunity-induced suppression of pressure-natriuresis, which leads to progression of hypertension. This concept was supported by our new finding that T cell homing induced by DOCA-salt treatment is attenuated in the kidneys of IFNγ-KO mice compared to the kidneys of DOCA-salt treated wild type mice (Figure 2d). Taken together, these data suggest that IFNγ is a critical factor in mediating activated CD8T-induced interaction between CD8Ts and DCTs, which contributes to excessive NCC upregulation and exacerbates hypertension.
IFNγ primes DCTs to express MHC-I and co-signaling molecule PDL1 via IFNγRs
A large amount of IFNγ is released from activated immune cells and plays an important role in modulating immune responses43-45. In the kidney, one important function of IFNγ is to prime renal tubule cells to express the checkpoint molecule PDL1, which binds to its receptor PD1 on CD8Ts to protect renal tubule cells from CD8T-induced cytotoxicity26-28. This mechanism is common in kidney transplant recipients, a population suffering from a high prevalence of salt-sensitive hypertension46-48. Therefore, we speculated that this protective mechanism of IFNγ may enhance direct contact between DCTs and CD8Ts, suppressing cytotoxic effects, but at the cost of boosting CD8T-induced upregulation of NCC in DCTs. To test this hypothesis, we administered mIFNγ to mDCTs with or without knockdown of IFNγ receptor subunits 1 and 2. Adding IFNγ to mDCTs significantly increased their surface expression of the CD8-specific antigen-presenting molecule MHC-I (Figure 3a, left panel), but not the CD4-specific antigen presenting molecule MHC-II (Figure 3a, right panel), which may result in mDCTs attracting CD8Ts instead of CD4Ts. As expected, knockdown of IFNγ receptor attenuated the IFNγ-induced increase of MHC-I in mDCTs, but this effect in MHC-II was not detectable (Figure 3a). Moreover, IFNγ induced high expression of PDL1 on DCTs at both the mRNA and protein level (Figure 3b&c), which may provide the co-signaling molecule for enhanced interaction between CD8Ts and DCTs. Knockdown of the IFNγ receptor subunits attenuated IFNγ treatment-induced upregulation of MHC-I and PDL1 in DCTs (Figure 3a&c), further confirming that these effects are mediated by the IFNγ signaling pathway.
Figure 3: Effects of IFNγ - IFNγ receptor signaling on priming DCTs.

(a), Flow cytometry study to examine the effects of IFNγ signaling on regulating the surface expression of MHCs on mDCT cells. Groups of cells are indicated by different colors as shown in a. n=5-6 each group. Fold changes of geometric mean fluorescence intensity (MFI) in treated groups are relative to mDCT control cells in each independent test. (b), mRNA expression of PDL1 in untreated mDCTs or mDCTs treated with either IFNγ only, or IFNγ + siIFNγR1&2. n=4 samples each group. (c), Flow cytometry study to examine the effects of IFNγ signaling on regulating the surface expression of PDL1 on mDCTs. Numbers in upper square indicate the proportion of mDCTs with high expression of PDL1. n=5-6 samples each group. Data are means ± sem. Statistical significance was assessed by t-test for a, b, and ANOVA for c.
Increased PDL1 in DCTs is associated with enhanced CD8T-DCT interaction and NCC upregulation
Similar to findings using IFNγ-treated mDCTS (Figure 3), we also observed increased expression of PDL1 in mDCTs treated with pre-activated CD8Ts as determined using flow cytometry (Figure 4a). Next, we introduced the intracellular sodium indicator Corona Green into mDCTs to examine NCC-mediated sodium retention in mDCTs with and without administration of pre-activated CD8Ts. As we expected, mDCTs treated with non-act CD8Ts demonstrated increased sodium retention (Figure 4b) consistent with our previous study12. However, treatment with pre-act CD8Ts resulted in a much greater proportion of mDCTs exhibiting high NCC-mediated sodium retention (Figure 4b). Importantly, most of these pre-act CD8T-induced NCC-hyperactive mDCTs expressed high levels of PDL1 on their surface (Figure 4b), leading us to consider the possibility that NCC hyperactivity in act CD8T-treated DCTs may be related to the surface expression of PDL1 on those DCTs. This association was further tested by adding siRNA to knockdown PDL1 in the mDCTs. We found that knockdown of PDL1 in mDCTs diminished pre-act CD8T-induced enhanced interaction between the two cell types (Figure 4c), and moreover, prevented pre-activation of CD8T-induced additional upregulation of NCC and excessive sodium reabsorption in co-cultured mDCTs (Figure 4d&e). Taken together, these in-vitro studies revealed a critical role for PDL1 in mediating the enhanced interaction between pre-act CD8Ts and DCTs. Next, this mechanism was further evaluated in-vivo to test its role in hypertension.
Figure 4: Impact of PDL1 on DCTs co-cultured with act-CD8Ts.

(a), Expression of PDL1 in mDCTs treated with or without pre-activated CD8Ts. Data are representative of 3 independent experiments with n>6 in each group. (b), Flow cytometry study using intracellular sodium indicator Corona Green to determine NCC-mediated sodium retention in PDL1 positive (top-right) and negative (top-left) mDCTs. n=8 in each group. (c, d & e), Effects of knocking down PDL1 in mDCTs on their adhesion with pre-activated CD8Ts (c, Scale=30μm, data are representative of n>10 images in each group). NCC expression (d, n=4 samples each group) and sodium retention determined by geometric mean fluorescence intensity (MFI) of intracellular sodium indicator Corona Green (e, color defined in the figure, n>15 samples in each group). Data are means ± sem. Statistical significance was assessed by ANOVA for b, d, and t-test for c, e.
Renal tubule specific knockdown of PDL1 ameliorates T cell accumulation in the kidneys of DOCA mice
Numerous earlier studies demonstrated chronic inflammation and cytokine accumulation in the kidneys of hypertensive subjects38, 39, 49, 50. Similarly, we observed higher expression of IFNγ in the kidneys of DOCA mice compared with sham mice (Figure 5a). Moreover, staining the kidney sections with PDL1 specific antibodies revealed that PDL1 is expressed around tubules in the DOCA kidney (Figure 5b) where CD8Ts attach, as demonstrated in our previous report12. An obvious next step was to knockout or block PDL1, which may serve as a potential co-signaling ligand for the CD8Ts. However, knockout of PDL1 or administration of blocking antibodies systemically may cause systemic complications, which could further affect blood pressure, as evidenced by the fact that both hypotension and hypertension are observed as side effects in cancer patients treated with PDL1 inhibiting antibodies51-56. Therefore, we employed renal tubule specific nanoparticles33-35 to introduce siRNAs designed to specifically knockdown PDL1 in renal tubules. Kidney-targeted polymeric mesoscale nanoparticles containing siPDL1 were injected intravenously into mice via tail vein 1 day after the start of DOCA-salt treatment. At the end of the DOCA-salt treatment period, we detected higher levels of PDL1 in the kidneys of DOCA mice, as expected (Figure 5c). In addition, we found that renal tubule-specific nanoparticles+siPDL1 suppressed renal PDL1 expression in DOCA mice through the end of DOCA-salt treatment (18 days after a single injection) without affecting PDL1 expression in other organs (Figure 5c & Figure S6). In the absence of excessive expression of PDL1 in the kidneys, DOCA-salt treatment failed to cause T cell accumulation in the renal cortex (Figure 5d). Quantification of renal accumulation of CD8Ts in mice was performed by counting CD8Ts isolated from kidneys using percoll density gradient centrifugation, CD45+ bead selection and flow cytometry analysis (example shown in Figure 5e left panel). As expected, DOCA-salt treatment led to enhanced infiltration of CD8Ts into the kidneys and this effect was absent after knockdown of PDL1 using specific siRNA (Figure 5e, right panel).
Figure 5: Effects of renal tubule specific knockdown of PDL1 in the kidneys of DOCA mice.

(a & b), The expression of IFNγ (a, mRNA level, n=4 mice each group.) and PDL1 (b, immunostaining, data are representative of n>5 images in each group.) in the kidneys of mice with or without DOCA treatment. (c), Efficacy of siPDL1 delivered by kidney-specific polymeric mesoscale nanoparticles in the kidneys of DOCA mice. n=5-6 mice in each group. (d & e), Renal infiltration of T cells and CD8Ts in DOCA mice with or without targeted siPDL1 as demonstrated by immunostaining of CD3 (d, data are representative of n>10 images in each group) and flow cytometry of CD8+ T cells (e, n=3-4 mice in each group). Left panel indicates gating strategy for CD8 positive cells, right panel shows quantitative numbers of CD8+ T cells from each kidney. Data are means ± sem. Statistical significance was assessed by t-test for a, and ANOVA for c, e.
Knockdown of PDL1 in renal tubules normalizes NCC expression and lowers blood pressure in DOCA mice
We reported earlier that increased infiltration of CD8Ts into the kidney contributes by an unrecognized mechanism to NCC upregulation and excessive salt retention in DOCA mice12. Here we speculate that since renal-specific knockdown of PDL1 prevented CD8T-accumulation in the kidneys, this intervention also would attenuate upregulation of NCC expression in DCTs and consequently attenuate the development of hypertension in these animals. To assess NCC expression in the kidneys of DOCA mice with or without knockdown of PDL1, we immunostained the kidney sections using antibodies against NCC and performed semi-automated quantification analysis of total fluorescence intensities of NCC positive pixels in stitched images of whole kidney sections (Figure 6a). Compared to the kidneys of DOCA mice not receiving nanoparticle treatment, those from mice treated with DOCA plus nanoparticles to knockdown PDL1 expressed significantly less NCC in their kidneys (Figure 6b). Consistently, DOCA treatment-induced hypertension in mice was ameliorated by renal-specific knockdown of PDL1 using nanoparticles (Figure 6c), where nanoparticles containing scrambled siRNA did not affect DOCA treatment-induced elevation of blood pressure in mice (Figure 6c). However, it is worth noting that although knockdown of PDL1 in the kidney almost completely abolished the effect of DOCA-induced accumulation of CD8Ts in the kidney, it did not restore blood pressure of DOCA-treated mice to the baseline level (Figure 6c), indicating possible involvement of other mechanisms in DOCA treatment-induced hypertension.
Figure 6: Effect of renal tubule-specific knockdown of PDL1 on NCC expression and blood pressure in DOCA mice.

(a), Raw fluorescent image of the whole kidney section (left) and intensity-based segmentation (right) of the kidney tissue (blue) and NCC-positive pixels (yellow). Inset: zoomed-in image of the red square. No pixels were saturated, and brightness was adjusted for display purposes only. (b), Intensity quantification of NCC positive segments in stitched images of whole kidney sections from DOCA mice treated with or without siPDL1. n=7-8 mice in each group. (c), Radio-biotelemetry recording of systolic blood pressure in DOCA mice treated with or without nanoparticles containing sham siRNA or siPDL1. Blood pressure was recorded for 10 seconds every 15 minutes. n=6 mice for DOCA only, n=6 mice for DOCA + sham nano, n=10 mice for DOCA + siPDL1 nano. Data are means ± sem. Statistical significance was assessed by Mann-Whitney test for b, and two-way ANOVA for c.
Effects of renal tubule-specific knockdown of PDL1 in mice after adoptive transfer of CD8Ts from DOCA mice
The effects of renal tubule-specific knockdown of PDL1 were further tested in a more direct model of CD8T-induced hypertension, in which mice received adoptive transfer of CD8Ts (1x107/mouse) isolated from DOCA treated hypertensive mice, as demonstrated by us earlier12. Analysis of kidneys from nanoparticle treated CD8T-recipient mice revealed similar results as obtained from the kidneys of nanoparticle treated DOCA mice, namely that kidney-specific knockdown of PDL1 diminished accumulation of CD8Ts around renal tubules (Figure 7a) and attenuated the renal expression of NCC mediated by adoptive transfer of DOCA-CD8Ts (Figure 7b). Mice that received adoptive transfer of DOCA-CD8Ts demonstrated salt-sensitive hypertension (Figure 7c), consistent with our previous report12. Here, we further found that this effect was abolished by renal tubule-specific knockdown of PDL1 (Figure 7c). Interestingly, unlike the endpoint blood pressure in DOCA mice, the blood pressure in adoptive transfer mice with kidney specific knockdown of PDL1 returned to a similar level as their baseline (Figure 7c).
Taken together, our results have demonstrated an important mechanism for the accumulation of CD8Ts in the kidneys of salt-sensitive hypertensive animals, namely that higher production of IFNγ primes DCTs to express MHC-I and PDL1, which enhances the interaction between CD8Ts and DCTs, leading to accentuated upregulation of NCC and consequently, aggravating hypertension.
Discussion:
In recent years, a growing body of evidence has revealed a pivotal role of immune cells, particularly T cells, in the pathogenesis of hypertension8, 10, 11, 16, 57, 58. However, the molecular and cellular mechanisms for T cell contributions to blood pressure elevation remained unrecognized. Earlier we reported one potential molecular event linking T cells to excessive salt retention in the kidney, namely that CD8Ts directly stimulate DCTs to increase NCC expression and salt reabsorption12. In the present study, we further elucidated a mechanism that drives the interaction between DCTs and activated CD8Ts, leading to increased CD8T homing to kidneys and contributing to the pathogenesis of hypertension.
Recent clinical and pre-clinical studies have found inappropriate activation of CD8Ts in hypertensive humans and experimental animals13, 14, and multiple factors may contribute to this inappropriate activation of CD8Ts10, 59-62. For example, it has been reported that a unique TCR subtype of CD8Ts are activated by hypertension-specific neoantigens, including isolevuglandin-modified proteins and heat shock proteins10, 62. Another proposed mechanism is altered gut microbiota, which may play a critical role in altering the activity status of immune cells, as evidenced by high-salt intake altering the microbiome profile, and the imbalance of gut microbiomes stimulating activation of CD8Ts61, 63-65. While ongoing studies are still investigating the causes of the activation of CD8Ts in hypertension, whether and how this activation of CD8Ts contributes to the pathologic CD8T-DCT interaction and participates in the pathogenesis of hypertension are critical questions that need to be addressed. In the current study, we first determined that activation of CD8Ts in salt-sensitive hypertensive animals exhibit a higher ability to interact with DCTs and stimulate NCC upregulation. Moreover, during continued exploration of this new mechanism, we identified key molecules contributing to the direct interaction between activated CD8Ts and DCTs. Eliminating these molecules attenuated the progressive elevation of blood pressure in salt-sensitive hypertensive mice, which further elucidated their important role in the pathogenesis of hypertension.
One important finding of the present study is that the large amount of IFNγ produced by immune cells primes DCTs to accommodate activated CD8Ts; subsequently, the accentuated interaction between CD8Ts and DCTs results in upregulation of NCC and excessive salt retention. In this regard, our findings provide a mechanism for an earlier report that IFNγ increases NCC expression and blood pressure in angiotensin II-induced hypertension19, an effect that we show relies on mediation by activated CD8Ts. We additionally show that knockout of IFNγ ameliorates blood pressure in another classic animal model of hypertension, DOCA-salt treated mice. Interestingly, we noticed that in this same model, IFNγ deficiency only normalized the later phase of progressive blood pressure elevation, which was termed “thymus dependent blood pressure elevation” by an earlier study41. These findings led us to consider the possibility that IFNγ contributes to the impairment of pressure-natriuresis by adaptive immunity and thereby the pathogenesis of salt-sensitive hypertension. Indeed, we found that knockout of IFNγ remarkably reduced CD8T cell accumulation in the kidneys of DOCA-salt treated mice and antibody neutralization of IFNγ diminished the pre-activated CD8T-induced additional upregulation of NCC in DCTs. Collectively, our findings argue for a critical role of IFNγ in activated CD8T-induced upregulation of NCC in DCTs and progression of hypertension.
A second important finding in this study is that we identified a molecular mechanism for IFNγ priming of DCTs, which enhances DCT-interaction with activated CD8Ts. We demonstrated that IFNγ induced higher expression of the CD8-specific antigen-presenter MHC-I in DCTs, but not the CD4-specific antigen MHC-II. This result is consistent with our earlier finding that CD8Ts, but not CD4Ts, play a major role in upregulating NCC in DCTs12. Moreover, we also found that IFNγ stimulates DCTs to express high levels of PDL1, which is a well-known co-signaling molecule required for cells to interact with CD8Ts that express receptor PD122, 23, 55. Some kidney transplantation studies have implied that PDL1 expression in renal tubules may be associated with salt-sensitive hypertension, because high expression levels of PDL1 often are detected on the surface of renal tubular cells in recipients of kidney transplantation26-28. In the same population, the prevalence of salt-sensitive hypertension is extremely high compared to the non-transplant population46-48. We conjecture that this increased expression of PDL1 on DCTs may provide a critical link to augmented cell-cell interaction with activated CD8Ts, resulting in an accentuated homing of CD8Ts to the kidney, higher NCC expression, and excessive salt-retention. It is known that global knockout of PDL1 may cause systemic complications, including diabetes and multi-organ inflammation at an early age, as described by the Jackson Laboratory (www.jax.org/strain/018307). Therefore, we delivered PDL1-specific siRNA directly to renal tubules using renal tubule-specific nanoparticles33-35 to overcome this limitation. Our results confirmed high expression of PDL1 in renal tubules of salt-sensitive hypertension animals, and moreover, renal tubule-specific knockdown of PDL1 abolished kidney accumulation of T cells, reduced renal expression of NCC, and attenuated salt-sensitive hypertension. In addition to identifying a critical role for PDL1 signaling in CD8T homing to kidneys of DOCA mice, the latter findings show the potential of using renal tubule-targeting nanoparticle technology to treat hypertension while minimizing off-target effects.
Although our findings suggest a critical role for PDL1 in mediating the interaction between activated CD8Ts and DCTs, which results in NCC upregulation and development of salt-sensitive hypertension, they do not rule out the possibility of other co-signaling, checkpoint or adhesion molecules participating in this interaction. In fact, diminishing the IFNγ-PDL1 pathway did not block the increase in NCC expression and NCC-mediated sodium retention in mDCTs caused by non-activated CD8Ts (Figures 2&4), suggesting other molecular pathways may promote interaction between CD8Ts and DCTs. Moreover, PDL1 expression in renal tubule cells was anticipated to protect cells from the cytotoxic effects of activated CD8Ts; however, in both cultured DCTs and mouse kidneys with PDL1 knocked down, we observed no obvious cell death (such as loss of cells, damaged renal tubule morphology, etc.) at the end of the treatment period, implying potential involvement of other molecules interfering with downstream cytotoxicity. Hence, we anticipate that future studies will identify additional molecular mechanisms participating in the interaction between immune cells and renal tubule cells.
Finally, our data do not exclude the possibility that other immune cells in addition to CD8Ts also stimulate DCTs and other renal tubule segments by producing IFNγ or other cytokines that contribute to the pathogenesis of hypertension. It is well known that hypertensive kidneys are infiltrated with many types of immune cells16, 38, 50. However, the crosstalk between these immune cells and with renal tubule cells is yet to be identified. One unanticipated finding of our studies was that kidney-specific knockdown of PDL1 in DOCA mice resulted in a more rapid and profound antihypertensive effect compared to the knockout of IFNγ. Because we did not observe siPDL1-induced changes in viability of CD8Ts in our preliminary studies, it is reasonable to consider that other factors in addition to IFNγ may regulate sodium transporter molecules on DCTs or other renal tubule segments. These processes may be clarified by future explorations.
Collectively, our data suggest a novel mechanism by which CD8Ts participate in the pathogenesis of hypertension, namely that activation of CD8Ts magnifies their interaction with DCTs via IFNγ-induced MHC-I and PDL1 expression on DCTs, thereby stimulating enhanced homing of CD8Ts into the kidney and greater NCC expression in the kidney, resulting in excessive salt-retention and salt-sensitive hypertension. This mechanism identified in the current study not only adds to our current understanding of the pathogenesis of hypertension, but also provides insight into new therapeutic targets for antihypertensive medications based on understanding the crosstalk between hypertension and adaptive immunity.
Supplementary Material
Novelty and Significance:
What is known?
Hypertension is a major public health problem, contributing to a plethora of cardiovascular diseases and related death.
The kidney plays a key role in the pathogenesis of hypertension, especially in augmenting salt-sensitivity of hypertension, which increases the risk for cardiovascular events and mortality.
Recently, an important role has been proposed for T cells in hypertension, yet without related treatment options, because the mechanisms underlying T cell involvement are largely unknown.
What new information does this article contribute?
Data from the current study determined a mechanism for T cell homing into the kidney and interacting with kidney cells to promote salt retention and development of hypertension.
The conclusion of this study elucidated immune disorder as a pathogenic event contributing to salt-induced hypertension, justifying a critical need to consider immunological strategies to treat this disease.
We also identified new molecular targets as “new players” contributing to sald-induced hypertension, and provide novel insights for new therapeutic strategies against this form of hypertension, especially those resistant to current treatments.
Our study identified critical molecular players and demonstrated their roles in augmenting the CD8T- distal convoluted tubule cell interaction leading to excessive salt-retention and consequent elevation of blood pressure. These findings provide a new mechanism for T cell involvement in the pathogenesis of salt-induced hypertension.
Acknowledgements:
We would like to thank Drs. Nancy Rusch and Philip Palade in the Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences (UAMS) for contribution to data discussion and editing the manuscript.
Sources of funding
This study was supported by NIH/NHLBI [R01-HL146713] (S. Mu), AHA [15BGIA25730047] (S. Mu), UAMS Medical Research Endowment Awards (S. Mu) and Bronson Foundation Award (S. Mu), as well as the Systems Pharmacology and Toxicology Training Program [Grant T32-GM106999] (L. Benson). L.X.L is supported by NIH/NIAID [R01-AI139124] and NIH/NIGMS [P20 GM103625]. L.H. is supported by NIH/NIGMS [P20 GM103625], ALA [CA-828143], and Sturgis foundation award. D.A.H is supported by NIH/NIDDK [Grant R01 DK114321]. R.M.W is supported by AHA 17POST33650043.
Nonstandard Abbreviations and Acronyms
- CD8T
CD8+ T cells
- DCT
Distal convoluted tubule
- DOCA
Deoxycorticosterone acetate (with high salt intake)
- IFNγ
Interferon gamma
- MHC-I
Major histocompatibility complex class I
- MHC-II
Major histocompatibility complex class II
- MMF
Mycophenolate mofetil
- NCC
Sodium chloride co-transporter
- PMA
Phorbol myristate acetate
- PDL1
Programmed death-ligand 1, also known as CD274 or B7-H1
- PD1
Programmed cell death protein 1, also known as CD279
- TNFα
Tumour necrosis factor alpha
Footnotes
Competing financial interests:
D.A.H. is a co-founder and officer with an equity interest of Goldilocks Therapeutics Inc., Lime Therapeutics, Inc., and Resident Diagnostics, Inc. and a member of the scientific advisory board of Concarlo Holdings LLC, Nanorobotics Inc., and Mediphage Bioceuticals, Inc. R.M.W is a scientific advisor with equity interest in Goldilocks Therapeutics, Inc.
All other authors declare no competing financial interests.
References:
- 1.Sanders PW. Salt-sensitive hypertension: lessons from animal models. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1996;28:775–82. [DOI] [PubMed] [Google Scholar]
- 2.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–90. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, Inenaga T and Kimura G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet (London, England). 1997;350:1734–7. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger MH, Fineberg NS, Fineberg SE and Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–32. [DOI] [PubMed] [Google Scholar]
- 5.Lloyd-Jones DM, Evans JC, Larson MG, O'Donnell CJ, Roccella EJ and Levy D. Differential control of systolic and diastolic blood pressure : factors associated with lack of blood pressure control in the community. Hypertension. 2000;36:594–9. [DOI] [PubMed] [Google Scholar]
- 6.Hajjar I and Kotchen TA. Trends in prevalence, awareness, treatment, and control of hypertension in the United States, 1988-2000. Jama. 2003;290:199–206. [DOI] [PubMed] [Google Scholar]
- 7.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B and Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation. 2008;117:e510–26. [DOI] [PubMed] [Google Scholar]
- 8.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C and Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. The Journal of experimental medicine. 2007;204:2449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tracey KJ. Hypertension: an immune disorder? Immunity. 2014;41:673–4. [DOI] [PubMed] [Google Scholar]
- 10.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y and Harrison DG. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J and Crowley SD. Role of T lymphocytes in hypertension. Current opinion in pharmacology. 2015;21:14–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, Hoover RS, He B and Mu S. CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nature communications. 2017;8:14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC and Park S. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62:126–33. [DOI] [PubMed] [Google Scholar]
- 14.Itani HA, McMaster WG Jr., Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, Konior A, Prejbisz A, Januszewicz A, Norlander AE, Chen W, Bonami RH, Marshall AF, Poffenberger G, Weyand CM, Madhur MS, Moore DJ, Harrison DG and Guzik TJ. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension. 2016;68:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND and Rodríguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. American journal of physiology Renal physiology. 2007;293:F616–23. [DOI] [PubMed] [Google Scholar]
- 16.De Miguel C, Das S, Lund H and Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. American journal of physiology Regulatory, integrative and comparative physiology. 2010;298:R1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor EB and Ryan MJ. Immunosuppression With Mycophenolate Mofetil Attenuates Hypertension in an Experimental Model of Autoimmune Disease. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moes AD, Severs D, Verdonk K, van der Lubbe N, Zietse R, Danser AHJ and Hoorn EJ. Mycophenolate Mofetil Attenuates DOCA-Salt Hypertension: Effects on Vascular Tone. Front Physiol. 2018;9:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG and McDonough AA. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65:569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bromley SK, Iaboni A, Davis SJ, Whitty A, Green JM, Shaw AS, Weiss A and Dustin ML. The immunological synapse and CD28-CD80 interactions. Nature immunology. 2001;2:1159–66. [DOI] [PubMed] [Google Scholar]
- 21.Dustin ML. The immunological synapse. Cancer immunology research. 2014;2:1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riella LV, Paterson AM, Sharpe AH and Chandraker A. Role of the PD-1 pathway in the immune response. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12:2575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M and Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO molecular medicine. 2011;3:581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung K and Choi I. Emerging Co-signaling Networks in T Cell Immune Regulation. Immune network. 2013;13:184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH and Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. The Journal of experimental medicine. 2006;203:883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waeckerle-Men Y, Starke A and Wuthrich RP. PD-L1 partially protects renal tubular epithelial cells from the attack of CD8+ cytotoxic T cells. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2007;22:1527–36. [DOI] [PubMed] [Google Scholar]
- 27.Schoop R, Wahl P, Le Hir M, Heemann U, Wang M and Wuthrich RP. Suppressed T-cell activation by IFN-gamma-induced expression of PD-L1 on renal tubular epithelial cells. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2004;19:2713–20. [DOI] [PubMed] [Google Scholar]
- 28.Ding H, Wu X and Gao W. PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clinical immunology (Orlando, Fla). 2005;115:184–91. [DOI] [PubMed] [Google Scholar]
- 29.Pollow DP, Uhrlaub J, Romero-Aleshire M, Sandberg K, Nikolich-Zugich J, Brooks HL and Hay M. Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension. 2014;64:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li LX and McSorley SJ. B cells enhance antigen-specific CD4 T cell priming and prevent bacteria dissemination following Chlamydia muridarum genital tract infection. PLoS Pathog. 2013;9:e1003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb JR, Milne K and Nelson BH. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer immunology research. 2015;3:926–35. [DOI] [PubMed] [Google Scholar]
- 32.Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, Fischer S, Jackson R, Flavell RA, Wang J, Sanmamed MF, Bosenberg MW and Ring AM. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583:609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams RM, Shah J, Ng BD, Minton DR, Gudas LJ, Park CY and Heller DA. Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Lett. 2015;15:2358–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams RM, Shah J, Tian HS, Chen X, Geissmann F, Jaimes EA and Heller DA. Selective Nanoparticle Targeting of the Renal Tubules. Hypertension. 2018;71:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han SJ, Williams RM, D’Agati V, Jaimes EA, Heller DA and Lee HT. Selective nanoparticle-mediated targeting of renal tubular Toll-like receptor 9 attenuates ischemic acute kidney injury. Kidney international. 2020;98:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riley JL. PD-1 signaling in primary T cells. Immunological reviews. 2009;229:114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA and Ahmed R. Role of PD-1 during effector CD8 T cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:4749–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu X and Crowley SD. Inflammation in Salt-Sensitive Hypertension and Renal Damage. Current hypertension reports. 2018;20:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rucker AJ, Rudemiller NP and Crowley SD. Salt, Hypertension, and Immunity. Annual review of physiology. 2018;80:283–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okuda T and Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Texas reports on biology and medicine. 1967;25:257–64. [PubMed] [Google Scholar]
- 41.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta pathologica et microbiologica Scandinavica Section A, Pathology. 1976;84:523–8. [DOI] [PubMed] [Google Scholar]
- 42.Boesen EI, Williams DL, Pollock JS and Pollock DM. Immunosuppression with mycophenolate mofetil attenuates the development of hypertension and albuminuria in deoxycorticosterone acetate-salt hypertensive rats. Clinical and experimental pharmacology & physiology. 2010;37:1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Maeyer E and De Maeyer-Guignard J. Interferon-gamma. Curr Opin Immunol. 1992;4:321–6. [DOI] [PubMed] [Google Scholar]
- 44.Schroder K, Hertzog PJ, Ravasi T and Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. [DOI] [PubMed] [Google Scholar]
- 45.Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nature reviews Immunology. 2018;18:545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weir MR. Blood pressure management in the kidney transplant recipient. Advances in chronic kidney disease. 2004;11:172–83. [DOI] [PubMed] [Google Scholar]
- 47.Mangray M and Vella JP. Hypertension after kidney transplant. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2011;57:331–41. [DOI] [PubMed] [Google Scholar]
- 48.Hall JE. Renal Dysfunction, Rather Than Nonrenal Vascular Dysfunction, Mediates Salt-Induced Hypertension. Circulation. 2016;133:894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banek CT, Gauthier MM, Van Helden DA, Fink GD and Osborn JW. Renal Inflammation in DOCA-Salt Hypertension. Hypertension. 2019;73:1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rigo D and Orias M. Hypertension and kidney disease progression. Clin Nephrol. 2020;93:103–107. [DOI] [PubMed] [Google Scholar]
- 51.Chen Z, Wang M, Gao S, Guo H, Wang G and Zhou G. [Cardiotoxicity of Anti-PD-L1 Antibody and the Effect of Levothyroxine in Attenuating the Related Mortality in Mice]. Zhongguo Fei Ai Za Zhi. 2021;24:394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu JR, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, Lyon AR, Padera RF, Johnson DB and Moslehi J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovascular research. 2019;115:854–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, Kuntzer T, Michielin O, Peters S, Coukos G, Spertini F, Thompson JA and Obeid M. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–580. [DOI] [PubMed] [Google Scholar]
- 54.Baxi S, Yang A, Gennarelli RL, Khan N, Wang Z, Boyce L and Korenstein D. Immune-related adverse events for anti-PD-1 and anti-PD-L1 drugs: systematic review and meta-analysis. BMJ (Clinical research ed). 2018;360:k793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Philips GK and Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. International immunology. 2015;27:39–46. [DOI] [PubMed] [Google Scholar]
- 56.Gong J, Chehrazi-Raffle A, Reddi S and Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trott DW and Harrison DG. The immune system in hypertension. Advances in physiology education. 2014;38:20–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A and Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haring JS, Badovinac VP and Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. [DOI] [PubMed] [Google Scholar]
- 60.Petrelli A, Mijnheer G, Hoytema van Konijnenburg DP, van der Wal MM, Giovannone B, Mocholi E, Vazirpanah N, Broen JC, Hijnen D, Oldenburg B, Coffer PJ, Vastert SJ, Prakken BJ, Spierings E, Pandit A, Mokry M and van Wijk F. PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. The Journal of clinical investigation. 2018;128:4669–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, Shiota A, Takeshita K, Yasuma-Mitobe K, Riethmacher D, Kaisho T, Norman JM, Mucida D, Suematsu M, Yaguchi T, Bucci V, Inoue T, Kawakami Y, Olle B, Roberts B, Hattori M, Xavier RJ, Atarashi K and Honda K. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600–605. [DOI] [PubMed] [Google Scholar]
- 62.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd and Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bier A, Braun T, Khasbab R, Di Segni A, Grossman E, Haberman Y and Leibowitz A. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferguson JF, Aden LA, Barbaro NR, Van Beusecum JP, Xiao L, Simmons AJ, Warden C, Pasic L, Himmel LE, Washington MK, Revetta FL, Zhao S, Kumaresan S, Scholz MB, Tang Z, Chen G, Reilly MP and Kirabo A. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palmu J, Salosensaari A, Havulinna AS, Cheng S, Inouye M, Jain M, Salido RA, Sanders K, Brennan C, Humphrey GC, Sanders JG, Vartiainen E, Laatikainen T, Jousilahti P, Salomaa V, Knight R, Lahti L and Niiranen TJ. Association Between the Gut Microbiota and Blood Pressure in a Population Cohort of 6953 Individuals. J Am Heart Assoc. 2020;9:e016641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors upon request.
Animals:
The animal use protocol was approved by the Institutional Animal Care and Use Committee at University of Arkansas for Medical Sciences. In the DOCA-salt mouse model, 10~11 weeks old male C57B6 mice were uninephrectomized and randomly assigned to either the sham group, or the DOCA-salt group that received a DOCA pellet subcutaneously followed by 1% NaCl in the drinking water for 3 weeks. The kidneys and splenic T cells were harvested at the end of day 20. In the CD8+ T cell adoptive transfer mouse model, which we described previously12, uninephrectomized 12 weeks old male C57B6 mice were randomly assigned to the sham group that received a saline injection, or assigned to the adoptive transfer group that was injected with fresh isolated CD8+ T cells (from spleens of DOCA-salt mice) via a single tail vein injection at the dose of 1x107 cells/100 μl saline/mouse. High-salt drinking water (1%) was available to sham mice and mice receiving CD8+ T cells as indicated in Figure 7. Blood pressure in this mouse model was measured directly by radio-biotelemetry (DSI). The current study only used male mice because biological sex is a significant factor that affects the regulation of blood pressure and immunity. Specifically, a previous report indicated that adoptive transfer of T cells to T cell deficient mice restores Angiotensin II (AngII)-induced hypertension in male recipient mice but not in female recipients29, suggesting the role of T cells in mediating hypertension is greater in males than females.
Figure 7: Effects of renal tubule-specific knockdown of PDL1 in mice receiving adoptive transfer of CD8Ts.
(a), Effects of renal tubule specific siPDL1 on T cell-accumulation in the kidneys of mice received CD8T-adoptive transfer. Scale=50μm. Data are representative of n>15 images in each group. (b), Intensity quantification of NCC positive segments in stitched images of whole kidney sections from CD8T-adoptive transfer recipient mice treated with or without siPDL1. n=8 mice in each group. (c), Radio-biotelemetry recording of systolic blood pressure in recipient mice of CD8T adoptive transfer with or without injection of nanoparticles containing siPDL1. Blood pressure was recorded for 10 seconds every 15 minutes. n=5 mice for +CD8Ts, n=6 mice for +CD8Ts +siPDL1. Data are means ± sem. Statistical significance was assessed by Mann-Whitney test for a, b, and two-way ANOVA for c.
T cell isolation and analysis:
Mouse spleens or kidneys were dissociated using a GentleMACs tissue dissociator and C-tubes followed the manufacturer’s protocol. Dynabeads T cell isolation kits were used to isolate T cells from splenocytes. The kidney T cell isolation followed a previously reported method10. Briefly, kidney homogenate was filtered using a 70μm sterile filter and leukocytes enriched by centrifugation in 36% and 72% percoll followed by CD45 selection using MagniSort beads. Subtypes of collected T cells were confirmed and analyzed using flow cytometry.
Ex-vivo stimulation:
Intracellular cytokine assays to evaluate cytokine production in CD8T cells used freshly isolated CD8T cells, which were either untreated or stimulated with cell activation cocktail (Bio-Legend #423304, PMA/Ionomycin) in the presence of brefeldin A for 3-4 h at 37°C 30-32.
Renal tubule specific knockdown using nanoparticles:
Ambion in-vivo siRNAs against mouse PDL1 or scramble sequence (as negative control) were purchased from Thermo Fisher and encapsulated into renal tubule-specific nanoparticles at the dose of 5 ng siRNA per 1 mg nanoparticles. Nanoparticles loaded with siPDL1 (or scrambled control) were formulated similarly to a previous description using an emulsion-nanoprecipitation method33-35. Briefly, 100 mg diblock poly (lactic-co-glycolic acid)-carboxy-terminated polyethylene glycol (PLGA-PEG) was dissolved in 2 mL acetonitrile, into which 100 ng of siPDL1 (or scrambled control) in RNase-free water were added dropwise and bath sonicated for 2 minutes. The resulting emulsification was added dropwise to 100 mL water with 100 μL Pluronic F-68 as a surfactant. After stirring for 2 hours, the particle suspension was recovered by centrifugation and immediately washed and centrifuged again. The particles were suspended in 2% sucrose solution and lyophilized to dryness. The hydrodynamic diameter of nanoparticles was evaluated via dynamic light scattering and determined to be 402.5±7.4nm with a polydispersity index (PDI) of 0.34 for siPDL1-nanoparticles and 399.3±16.8nm with a PDI of 0.28 for scrambled control siRNA nanoparticles. Immediately prior to use, nanoparticles were suspended at 16mg/ml in saline and injected into mice at the dose of 1.6mg nanoparticles/mouse via tail vein injection. Our preliminary tests indicate that the treatment with siPDL1 containing nanoparticles does not alter baseline blood pressure or renal T cell infiltration as shown in Figure S1.
Cell culture treatment & harvest:
The mDCT15 cells (referred to in the text simply as mDCTs) and the CD8+T cells (TK1 cells) were introduced in our previous report12. In pre-activation studies, CD8Ts were pre-incubated with cell activation cocktail (PMA/Ionomycine, Biolegend) for 4 hours before being washed and co-cultured with mDCTs. All cells were maintained at 37°C and 5% CO2, and no contaminations were found in cultured cells during our experiments. All treatments of mDCTs were overnight (approximately 12 hours). Co-culture and harvest methods also followed the same protocol as we previously reported12. The quantification of area covered by T cells co-cultured with mDCT cells was estimated by a semi-automated segmentation script using iVision 4.5.4 (BioVision Technologies, Exton, PA). The segmentation was based on intensity threshold, mean intensity, area, and circularity to exclude artifacts. Segmented images were inspected for accuracy and fine adjustments to parameters were made as necessary. Representative images of CD8T-DCT adhesion after co-culture were selected to represent the average of quantification data. Cell co-culture (adhesion) images were acquired using an inverted routine microscopy and Cannon EOS camera with eye-piece adaptor. The scale bars of images were estimated by calculating pixels in an 1mm length image acquired using the same system.
Transfection of siRNAs:
All siRNAs were purchased from ABI. Transfection of siRNAs was accomplished using lipofectamine RNAimax following the manufacturer’s protocol. siRNA-mediated knockdown effects in mDCTs are shown in Figure S2.
Flow cytometry:
For surface staining, cells were stained with antibodies for 30-45 minutes in cell isolation buffer in dark tubes before analysis. For the NCC functional study using CoroNa Green (cell permeable, Molecular Probes), details are described in our previous publication12. Briefly, mDCTs were treated with ouabain, bumetanide and amiloride for 30 minutes at room temperature (RT) followed by loading of CoroNa Green at a concentration of 10 μM for 1 hour at RT. Cells were washed and resuspended in PBS containing the same blockers mentioned above for 45 minutes and analyzed in a BD Accuri C6 flow cytometer immediately. Intracellular cytokine staining of CD8Ts was performed using BD Cytofix/Cytoperm Plus (Cat. 555028) according to manufacturer protocols. In brief, stimulated cells were incubated in the presence of the BD GolgiPlug Protein Transport Inhibitor (with Brefeldin A, 1 microliter/mL cell culture). After 3 hours, cells were harvested and fixed in BD fixation/permeabilization solution, then washed twice in perm/wash buffer and resuspended for intracellular cytokine staining. After 30 minutes of incubation, cells were washed and resuspended in cell isolation buffer for analysis. Flow cytometry data were analyzed using FlowJo software (version v10.7.1). For all flow cytometry studies, cells lacking incubation with fluorescent antibodies (or corona-green) were used as negative controls to determine autofluorescence and gating. Single cells were selected using the FSC-H/FSC-A gating method for all studies and live cells were selected using 7-AAD staining, as shown in Figure S3. Representative flow cytometry images were selected to exemplify quantification data.
Immunostaining:
4% PFA fixed kidneys were processed as 8μm thick paraffin sections in the DNA Damage and Toxicology Core at UAMS. Following antigen retrieval using antigen unmasking solution (Vector), sections were washed and blocked in 5% non-fat milk containing 4% goat serum at RT for 1 hour prior to overnight incubation with primary antibodies. After secondary antibody staining, sections were sealed with ProLong Gold mounting media (Molecular Probes). Images were captured using Keyence XZ-800 microscope. Cell counting was conducted using ImageJ software. Representative images were selected to exemplify the average of quantification data.
Mean intensity quantification of NCC:
All kidney sections in the experimental group and the corresponding controls were stained, imaged, and analyzed under the same conditions. The whole kidney section image was constructed by stitching thousands of images taken on Keyence XZ-800 with a 10x PlanApo NA 0.45 at low power and exposure time to avoid saturation. Intensity cutoffs for tissue and NCC-positive pixels were determined by a blinded investigator and applied to the experimental and control groups uniformly. The mean intensity for each kidney was calculated by (total NCC-positive pixel intensity – mean background tissue intensity x total NCC-positive pixel area) / total tissue area. Image segmentation and analysis were performed using iVision 4.5 (BioVision Technologies).
Western blot:
Detailed methods and materials have been published12. Briefly, bis-tris gel (from Genscript) was used for electrophoresis, and gels were transferred to PVDF membranes on ice. All membranes were blocked in 5% non-fat milk for 1 hour before incubation with primary antibodies overnight. After washing and incubation with HRP conjugated secondary antibodies, western blot images were obtained using a ChemiDoc XRS+ system and analyzed using ImageLab software (BioRad). Representative images were selected that exemplified the average of quantification data.
Statistical analysis:
Data are presented as means ± sem. All statistical analysis were preformed using GraphPad 7.05. Data were tested for normality using D'Agostino & Pearson test (for sample n≥8) or Shapiro-Wilk test (for sample n<8). The threshold for normality tests was set to 0.05, and p>0.05 were considered normally distributed. Unpaired t-tests were used to compare parametric data between two independent groups, whereas Mann-Whitney tests were used to compare non-parametric data between two independent groups. Two-sided p values are indicated in the figures. For multiple comparisons, statistical analysis was carried out by ANOVA followed by Tukey’s or Dunnett’s post hoc tests. P values < 0.05 were considered to be significant, non-significant were labeled as n.s. in figures. No sample size estimate was performed, but sample sizes were selected based on previous experiments. All assays were repeated in independent experiments and displayed figures are representative.
Major Resources:
Please see the Major Resources Table in the Supplemental Materials.