

Abstract
Telomeres are chromosome-capping structures that protect the linear genome from DNA damage sensors. However, these structures present obstacles during DNA replication. Incomplete telomere replication accelerates telomere shortening and limits replicative lifespan. Therefore, continued proliferation under conditions of replication stress requires a means of telomere repair, particularly in the absence of telomerase. It was recently revealed that replication stress triggers break-induced replication (BIR) and mitotic DNA synthesis (MiDAS) at mammalian telomeres; however, these mechanisms are error-prone and primarily utilized in tumorigenic contexts. In this review, we discuss the consequences of replication stress at telomeres and how use of available repair pathways contributes to genomic instability. Current research suggests that fragile telomeres are ultimately tumor-suppressive and thus may be better left unrepaired.
Keywords: Telomeres, replication stress, break-induced replication, mitotic DNA synthesis, alternative lengthening of telomeres
Replication Stress at Telomeres
Mammalian chromosomes are capped by telomeres, GGTTAG DNA repeats bound by the shelterin complex (see Glossary) [1]. Telomeres inhibit cellular DNA damage responses (DDR) from recognizing chromosome ends as DNA double-stranded breaks (DSBs) requiring repair. This prevents chromosome end ligation and aberrant recombination that would otherwise generate deleterious chromosomal fusions and rearrangements. By the classic end-replication model, telomeres shorten by 50–200 nucleotides with every round of cell division until they become too short to fully inhibit the DDR, resulting in cell senescence or death [2]. With this, telomere length limits proliferation capacity and provides a formidable obstacle for tumorigenesis. However, it is now evident that stress resulting from, for example, excessive reactive oxygen species (ROS) or ionizing radiation, accelerates telomere shortening and promotes length-independent dysfunction, indicating that telomeres are responsive to the cellular environment and adding a layer of complexity to their proposed tumor suppressive function [3–6].
Ironically, several structural features of telomeres, including bound shelterin, the single-stranded DNA (ssDNA) overhang-obscuring t-loop, and R-loops arising from telomeric repeat-containing RNA (TERRA), present obstacles to replication fork progression with the potential to compromise genome stability [7–9]. Furthermore, the G-rich sequence of the telomere lagging strand is susceptible to oxidative base damage and G quadruplex formation that generate additional barriers to DNA replication (Figure 1) [3, 10]. These obstacles render telomeres vulnerable to replication stress, as demonstrated by the appearance of fragile telomeres analogous to the gaps that appear on metaphase chromosomes at difficult-to-replicate common fragile sites (CFSs) [11]. As observed for CFSs, fragile telomeres are “expressed” in response to replication stress induced by pharmacologic agents such as aphidicolin (APH), as well as overexpression of oncogenes such as cyclin E [11, 12]. Fragile telomere expression is also observed in response to genetic deletion of the shelterin component TRF1, demonstrating the essential role of mammalian shelterin for telomere replication originally reported in budding yeast [11, 13, 14]. While TRF1 does not directly inhibit DDR, it enables efficient telomere replication by recruiting proteins that promote fork progression, remove obstacles, and protect stalled forks from DDR recognition to prevent cell cycle arrest [11, 15–17]. In the absence of excessive replication stress, mammalian telomeres are replicated at similar rates to the rest of the genome, indicating that cells are sufficiently equipped to prevent or quickly restart stalled forks in telomeres under normal circumstances [18, 19].
Figure 1. Replication Fork Obstacles within Telomeres.

Top, telomere replication primarily initiates from origins within the subtelomere and progresses to end of the chromosome. Bottom, the guanine-rich repetitive sequences and secondary structures within telomeres present obstacles to replisome progression during semi-conservative DNA replication, indicated by stars. Various proteins that transiently interact with shelterin as well as DNA and RNA maintenance pathways remove these obstacles to prevent replication fork stalling and collapse [94]. G quadruplexes are unwound by BLM and RTEL1 helicases [11, 44, 95]. Base lesions such as 8-oxoguanine are removed by base excision repair [3]. R-loops are prevented by nonsense mediated decay of TERRA and are dismantled by RNase H1 and the TERRA-interacting proteins NONO and SPFQ [96, 97]. t-loops are dismantled by RTEL1 [95].
However, if the extent of stress is too severe to efficiently restart the replication fork, cleavage of exposed ssDNA may cause the fork to collapse into a DSB. Collapsed forks in other parts of the genome can be compensated by converging forks from adjacent origins. While some instances of replication initiation within telomeres have been reported, telomeres generally lack replication origins, and telomere replication is primarily initiated within the subtelomere to drive unidirectional fork progression to end of the chromosome (Figure 1) [11, 18, 20, 21]. If terminated replication renders the telomere too short to functionally inhibit DDR recognition, it will become a telomere dysfunction-induced focus (TIF) [22]. Five TIFs are sufficient to trigger senescence, therefore telomeres that break under replication stress must be repaired to maintain continuous cell proliferation [23]. Most cancers achieve this by activating the specialized reverse transcriptase telomerase, while telomerase-negative cancers maintain telomere length using the recombination-mediated Alternative Lengthening of Telomeres (ALT) [24, 25].
Although replication stress is a considerable source of telomere dysfunction, the mammalian cellular response to stalled or collapsed replication forks within telomeres is just beginning to be elucidated. Telomere repair in response to replication stress has recently been associated with break-induced replication (BIR) and mitotic DNA synthesis (MiDAS). However, current research suggests that these pathways inaccurately synthesize telomeric repeats, deregulate telomere length, and promote genomic rearrangements, as will be discussed. As such, a repaired telomere may be more detrimental than a broken telomere to genomic stability. Here, we review how mammalian cells respond to DSBs in telomeres arising from collapsed replication forks, how these responses affect telomere integrity, and how they differ between untransformed and cancer cells, with an emphasis on ALT cancers. We also discuss how replication stress may contribute to the proposed tumor-suppressive function of telomeres and address current knowledge gaps in this underappreciated aspect of telomere biology.
Break-induced Replication
The single-ended DSBs that arise from collapsed replication forks cannot be repaired by nonhomologous end-joining (NHEJ) or conventional homology-directed repair (HDR) because chromosome fusion and double Holliday junction formation each require two exposed dsDNA ends (Figure 2A) [26]. Instead, replication is resumed through BIR, an origin-independent replication method that is well-characterized in yeast but has only recently been observed in mammalian models [27]. In yeast, BIR diverges into two mechanisms: Both require Rad52 to promote annealing of a resected 3’ ssDNA to a homologous donor; however, only one type requires Rad51-mediated homology search and strand invasion. The less characterized, Rad51-independent pathway has been proposed to use exposed microhomologies to initiate replication via microhomology-mediated BIR (mmBIR) [28]. In each mechanism, the resected break overhang acts as the primer using the homologous donor as a template, and synthesis proceeds through a migrating D-loop rather than a replication fork (Figure 2A). In contrast to conventional HDR, which uses both ends of the DSB to replicate a relatively small gap of DNA, BIR uses only one end to initiate and sustain long-tract, conservative replication [26].
Figure 2. Consequences of Replication Fork Collapse within Telomeres.

A. Top, Replication fork collapse generates single-ended double-stranded breaks (DSBs) that cannot be repaired by direct ligation or conventional homology-directed repair. In telomeres, failure to restart replication at these break sites results in loss of unreplicated telomeric DNA, culminating in accelerated telomere shortening. Bottom, single-ended DSBs can complete replication through break-induced replication (BIR). Broken telomeres may utilize intra-, inter-, or extra-chromosomal telomeric sequences, potentially though annealing to microhomologies, to initiate long-tract BIR, resulting in conservative replication and unregulated telomere lengthening. B. Induced replication stress inhibits timely replication of fragile sites including CFSs and telomeres, causing cells to enter mitosis with unreplicated DNA and stalled replication fork intermediates. These sites are replicated by MiDAS detectable as mitotic EdU+ foci. MiDAS at CFSs and telomeres share common BIR-associated factors and are both dependent on the SLX4 nuclease scaffold protein, although CFS MiDAS requires the Mus81-EME1 nuclease while telomeric MiDAS does not. Telomeric MiDAS is almost exclusively replicated conservatively, while CFSs appear to partially rely on a semi-conservative replication mechanism.
In yeast, BIR requires Pol32, a non-catalytic subunit of DNA polymerase δ that is nonessential under normal replication conditions. Pol32-mediated replication is error-prone due to its low processivity, which causes frequent base-skipping and template switching that generates mismatches, duplications, and rearrangements [29–31]. This is exacerbated in mmBIR, which permits non-specific template annealing that can generate gross chromosomal rearrangements [32]. This propensity for error, combined with the ability to extend hundreds of kilobases to the end of the chromosome, offers BIR the potential to introduce significant genomic instability, rendering it as a seldom-used “last-resort” response to severe replication stress.
Replication error associated with PolD3, the mammalian ortholog to Pol32, is less characterized. However, depletion of PolD3 decreases copy number alterations in human cancer cells undergoing oncogene-induced replication stress, suggesting that BIR introduces genomic instability in mammals [27]. Furthermore, knockout of Rad52, which is nonessential under normal physiological conditions, decreased tumor size and extended lifespan in cancer-prone APC+/− mice, demonstrating that BIR is ultimately tumorigenic and detrimental to survival [33]. Indeed, PolD3 is frequently overexpressed in cancer cells [34]. Notably, the extent of BIR can be limited by convergence with incoming replication forks from adjacent origins [26, 35]. However, due to their lack of replication origins and repetitive sequence that facilitates non-specific strand annealing and template switching, telomeres are especially vulnerable to the deleterious effects of BIR.
Mitotic DNA Synthesis
MiDAS is detectable as mitotic EdU foci that colocalize with the chromosome gaps formed at CFSs under induced replication stress, indicating that MiDAS restarts replication at stalled forks within fragile loci [36, 37]. As CFSs are among the last loci to be replicated in S phase, MiDAS is thought to be a final effort to resolve stalled fork intermediates and complete replication prior to chromosome segregation (Figure 2B). MiDAS is dependent on PolD3 and Rad52 and often replicates DNA conservatively, strongly suggesting that DNA is replicated in mitosis through a BIR mechanism. Furthermore, MiDAS is Rad51-independent, suggesting that this mechanism uses the more deleterious mmBIR [32, 37, 38]. While observed at low levels in noncancerous, diploid cells, MiDAS is far more active in cancer cells, especially in lines that exhibit aneuploidy [37]. This implies that MiDAS is primarily used by cancer cells to survive under chronic replication stress, rather than as a physiological DNA maintenance mechanism. However, a Rad52-independent form of MiDAS has been reported in noncancerous cells, suggesting that a less deleterious MiDAS mechanism exists for routine maintenance of fragile sites [39]. It should be noted that BIR and MiDAS are not synonymous, as BIR can occur outside of mitosis, and a portion of DNA synthesized by MiDAS is replicated by semiconservative mechanisms that are yet to be characterized [12, 38, 40].
Telomere Synthesis Following Replication Fork Collapse
Evidence of BIR at mammalian telomeres was first observed in human ALT cells, which exhibit spontaneous, conservative telomere synthesis analogous to Rad51-independent BIR [41, 42]. Furthermore, induction of telomeric DSBs with a TRF1-FokI endonuclease fusion protein increased BIR-mediated telomere synthesis, indicating that BIR is triggered in response to broken telomeres [31]. Surprisingly, this effect was observed in both ALT-positive and ALT-negative cell lines, indicating that BIR may be a universal cellular response to telomere damage [41]. Supporting this notion, TRF1-FokI induction in telomerase-positive mouse embryonic fibroblasts (MEFs) triggers HDR-mediated telomere repair that is at least partially attributable to BIR [43, 44]. In fact, the de Lange group recently reported that BIR-mediated telomere synthesis underlies fragile telomere expression in MEFs, suggesting that fragile telomeres are akin to CFSs in that they represent DNA synthesized by BIR following replication fork collapse [44].
While the study from the de Lange group noted BIR at broken telomeres in S phase, the Hickson group reported that replication stress induced by APH or ATR inhibition in human cancer cells activated MiDAS at telomeres that did not correlate with fragile telomere expression [44, 45]. In addition to potential differences between mouse and human models, this discrepancy may in part be due to differing sources of telomere damage: APH and ATR inhibition cause replication fork stalling that generates one-sided DSBs or sister chromatid-linking intermediates, while TRF1-FokI induction generates two-sided DSBs that can be repaired by mechanisms other than BIR, such as conventional HDR or noncanonical-NHEJ [43, 46]. As such, the former would be more challenging to repair and thus more likely to persist past S/G2 into mitosis. Notably, damaged telomeres do not activate the G2/M cell cycle checkpoint, potentially increasing the likelihood of MiDAS [47].
In agreement with the Hickson study, telomeric MiDAS has been observed by others in ALT and telomerase-positive cancer cells in response to pharmacological and genetically induced replication stress (Figure 2B) [12, 48–51]. However, while MiDAS is activated at 80% of CFSs under replication stress, its frequency averages at less than one telomere per cell per cycle, implying that MiDAS may not be the primary pathway for repairing fragile telomeres as it is for CFSs [12, 37, 40]. This could partially be due to the difference in timing between CFS and telomere replication: CFSs are replicated late in S phase, therefore stalled forks within CFSs are less likely to restart before mitosis [36]. In contrast, telomeres are replicated throughout S phase depending on the chromosome-specific subtelomere; thus, earlier-replicating telomeres would be more likely to complete replication in S or G2 before onset of mitosis [18, 52]. Although it has not been determined whether specific late-replicating telomeres are more prone to repair by MiDAS, this could explain the lacking correlation between telomere fragility and MiDAS.
While the contribution of MiDAS remains somewhat unclear, telomere synthesis following induced replication stress is consistently dependent on conservative, PolD3-mediated BIR. In fact, while MiDAS events at CFSs appear to partially rely on a semi-conservative replication mechanism, telomeric MIDAS is ~95% conservative, indicating that telomeres are more reliant on BIR than other types of fragile sites (Figure 2B) [12]. BIR was recently observed at telomeres in conditional TRF1 knockout MEFs, the model in which telomeres were originally characterized as fragile replication sites [11, 53]. Surprisingly, proteomic analysis of telomeric chromatin revealed that TRF1-ablated telomeres were enriched for DNA repair and chromatin-modifying factors commonly associated with ALT. Experimental validation further demonstrated that replication stress following loss of TRF1 triggered several ALT-associated phenotypes, including formation of ALT-associated promyelocytic leukemia bodies (APBs), telomeric sister chromatid exchanges (T-SCEs), and transcription of TERRA [53]. These results contribute to mounting evidence that replication stress at telomeres drives ALT activation during tumorigenesis.
Replication Stress Drives Alternative Lengthening of Telomeres
In the absence of telomerase, 4–11% of tumors rely on ALT to maintain telomeres for extended proliferation [25]. ALT uses inter- and intrachromosomal recombination to elongate telomeres; this manifests several detectable phenotypes, including heterogeneous telomere lengths, extrachromosomal telomeric repeats, APBs, and TERRA [25, 54]. In humans, ALT is not utilized outside of an oncogenic context, rendering it a desirable target for treatment of ALT-positive cancers. However, the molecular understanding of ALT is still largely incomplete, in part because ALT encompasses multiple pathways that can antagonize or compensate for loss of another. For instance, ALT can initiate telomere recombination through either Rad51-dependent or independent homologous strand annealing, with Rad51 depletion enhancing the latter pathway [41, 55]. While both pathways initially appeared to rely on Rad52, a Rad52-independent pathway was recently discovered that is activated after telomere shortening following Rad52 depletion [56]. This adaptability and the late activation of ALT during tumorigenesis suggest that ALT arises and evolves with increasing mutational load [57]. Notably, ALT is activated in mice and human cancer cells following telomerase inhibition and subsequent telomere crisis, further indicating that ALT arises from genomic instability and emphasizing the need for ALT-targeting therapies [12, 58].
While the underlying causes of ALT activation are not fully elucidated, recent evidence indicates that ALT is driven by a combination of chromatin remodeling and replication stress at telomeres. Cytological evidence, including length independent TIFs and fragile telomeres in untreated cells, indicates that telomeres are under chronic replication stress in ALT cells [12, 59]. This stress is at least partially attributable to altered nucleosome assembly, as the most common mutations associated with ALT affect the ATRX/DAXX histone chaperone complex [60, 61]. Additionally, loss of the ASF1 histone chaperone is sufficient to activate ALT in primary cells, and deletion of HIRA, an alternative H3.3 chaperone, selectively kills ATRX-deficient ALT cells, demonstrating that ALT is driven by and dependent on altered chromatin maintenance at telomeres [62, 63]. Chromatin decompaction at ALT telomeres increases transcription of TERRA, which form R-loops within the telomere that block replication fork progression to generate replication stress (Figure 1) [7, 63, 64]. Additionally, ALT telomeres harbor nucleotide variants of the GGTTAG repeat, which disrupt shelterin binding and cause remodeling of telomeric chromatin to a recombination-permissive state that further enhances ALT [65, 66]. These variant repeats are presumed to result from recombination events at subtelomeres, although it is tempting to speculate that they arise due to base misincorporations or microhomology-derived rearrangements attributable to BIR, as has been observed in yeast as well as non-telomeric regions of human cancers [30, 32].
It has been repeatedly demonstrated that pharmacologic or genetically induced replication stress in ALT cells exacerbates ALT-associated phenotypes, including direct detection of BIR-mediated telomere elongation [12, 53, 67–72]. Notably, the BIR mechanism maintained in ALT cells is not identical to the break-induced telomere synthesis response observed immediately following induced telomere damage; the latter response is frequently observed as MiDAS and does not necessitate telomere clustering, while ALT occurs primarily in G2 in telomeres clustered at APBs [40, 56, 70, 73]. However, it was recently reported that “baseline” BIR in ALT cells continuously generates replication stress and promotes telomere clustering at APBs, providing a positive feedback mechanism to sustain ALT throughout progressive cell cycles at the expense of genome stability [70]. This cycle may be triggered by oncogenic replication stress during tumorigenesis complemented by chromatin decompaction to trigger break-induced telomere synthesis in the absence of telomerase (Figure 3). While the cause of activation remains unknown, this recent work strongly indicates that ALT is perpetuated by replication stress.
Figure 3. Cellular Response to Replication Stress at Telomeres.
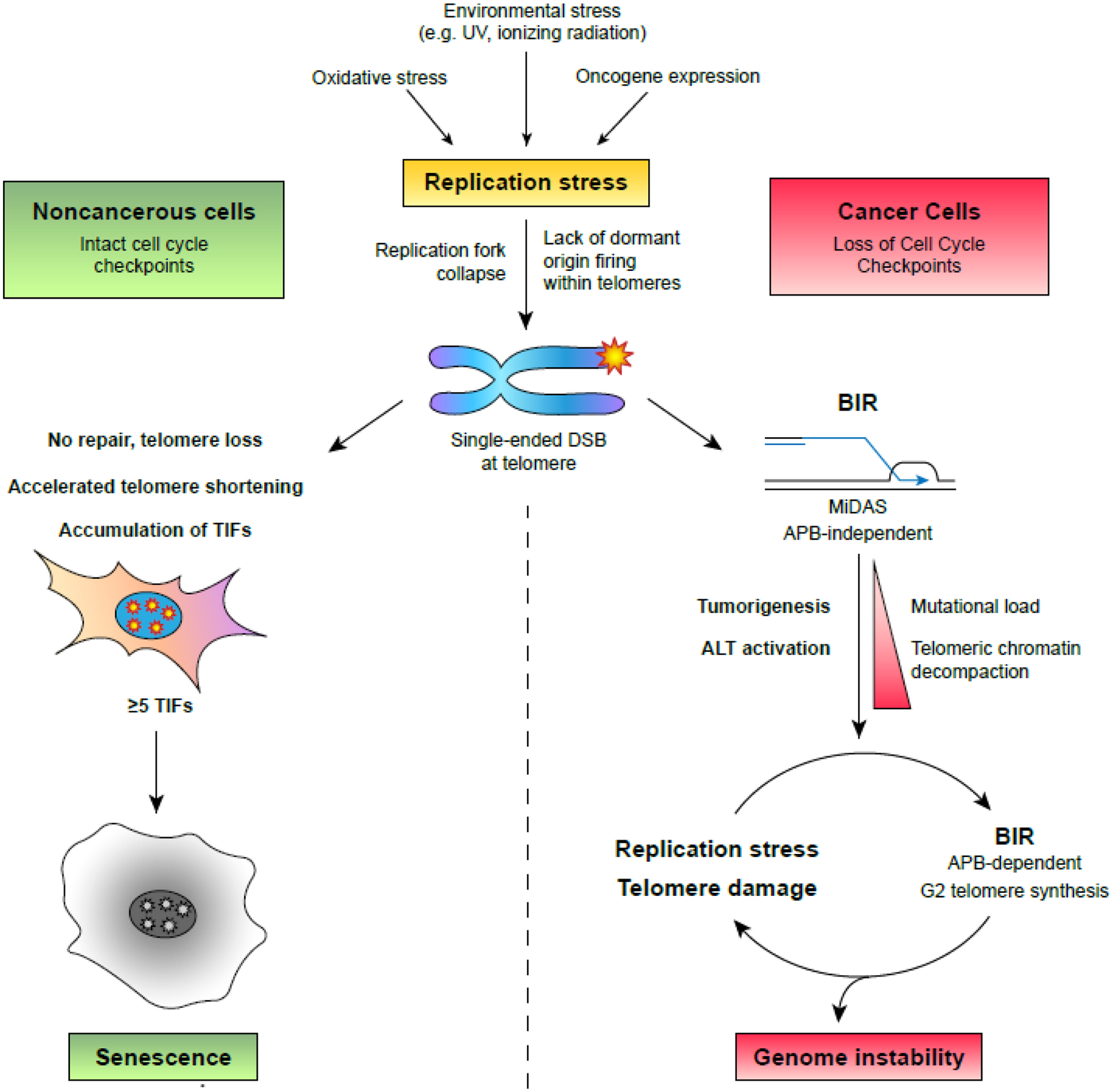
Excessive replication stress causes replication fork stalling and collapse at origin-poor fragile sites such as telomeres. Left, noncancerous cells with normal cell cycle checkpoints do not attempt to repair telomeres that break during replication. This results in telomere loss and accelerated telomere shortening. If the nucleus accumulates five or more telomere dysfunction-induced foci (TIFs), cell cycle checkpoints will be activated to trigger senescence. Right, precancerous or cancerous cells with compromised cell cycle checkpoints restart telomere replication through break-induced replication (BIR) and mitotic DNA synthesis (MiDAS). Over the course of tumorigenesis, reliance on error-prone BIR generates additional replication stress that may contribute to cell transformation, alter telomeric chromatin compaction, and activate alternative lengthening of telomeres (ALT). In ALT+ cells, an altered BIR mechanism that acts in G2 and relies on telomere clustering in PML bodies (APBs) continuously repairs broken telomeres while introducing additional replication stress. This manifests a positive feedback loop that ultimately generates genome instability that threatens the viability of the cell and its host.
Ironically, ALT cells are vulnerable to increased replication stress, suggesting that ALT is regulated to maintain a stress level just above the threshold for survival. For instance, it was revealed that T-SCEs, long considered a marker of ALT, are not indicative of telomere elongation but represent a recombination event prematurely terminated by SLX4-mediated D-loop resolution, which competes with BLM-TOP3A-RMI (BTR)-mediated D-loop migration and dissolution to prevent extensive telomere synthesis [74]. The mismatch repair MutSα complex and translesion Polη also mitigate replication stress and limit BTR-mediated extension at ALT telomeres. This may suggest an increased reliance on ssDNA repair and the ability to bypass DNA lesions during replication to preserve telomere integrity in ALT cells, although MutSα and Polη have been proposed to regulate ALT through less straightforward mechanisms [50, 75]. It is speculated that these antagonistic pathways exist to limit the extent of genome instability generated by BIR-mediated telomere synthesis. This sensitivity presents a promising avenue for ALT-targeting therapies. For instance, some ALT cells are hypersensitive to ATR inhibition, although this is unfortunately not applicable to all ALT cell lines [76, 77]. A more optimistic target is FANCM, a multifunctional protein that alleviates replication stress through replication fork remodeling and R-loop resolution. Multiple groups recently reported that FANCM depletion generates replication stress that is selectively toxic in ALT cells, validating it as a potential therapeutic target [67–69]. A subset of ALT cells lines and tumors were also recently reported to be hypersensitive to poly(ADP-ribose) polymerase (PARP) inhibitors, perhaps due to loss of PAR-mediated HIRA H3.3 deposition [62, 78, 79]. Altogether, ALT epitomizes the risk of repairing telomeres under replication stress and illustrates how genomic instability derived from BIR-mediated telomere synthesis can drive tumorigenesis.
Physiological Responses to Telomere Damage and Replication Stress
While cancer cells readily engage BIR-mediated telomere synthesis following induced telomere damage or replication stress, this is not observed in untransformed human cells [80]. In fact, it was presumed that noncancerous cells were incapable of repairing broken telomeres, as fibroblasts treated with high doses of ionizing radiation fail to repair DSBs that arise in telomeres, which manifest as persistent TIFs and induce senescence [5, 6]. However, fibroblasts treated with lower doses of DNA-damaging agents repair telomeric DSBs via HDR [81]. Although DSBs within telomeres are recognized by DDR, shelterin remains bound and prevents end ligation via NHEJ, the major DSB repair pathway available in G1 [5]. As such, telomeres can only be repaired by HDR following entry into S phase, which is impeded if the extent of damage acquired in G1 is sufficient to trigger the G1/S checkpoint. This clarifies why telomerase expression does not prevent senescence in irradiated fibroblasts, as telomerase activity is also restricted to S phase [5, 6, 82]. Notably, the HDR observed in proliferating fibroblasts was dependent on Rad51 and generated T-SCEs, indicating that the double-ended DSBs arising in G1 are repaired via the canonical “error-free” HDR pathway, rather than BIR [46].
Oxidative stress is a more physiologically relevant source of telomere damage, as excessive ROS accelerates telomere shortening and dysfunction [3]. However, the mechanism underlying this has been elusive because ROS damages multiple cellular components that could indirectly contribute to telomere dysfunction. To address this, chemoptogenetic tools were recently developed that target ROS-induced 8-oxoguanine (8-oxoG) lesions specifically to telomeric DNA (Figure 1) [49, 83, 84]. This revealed a striking difference in the response to telomeric oxidative stress between cancerous and noncancerous cells: While cancer cell lines only exhibited telomere shortening and aberrant mitosis following repeated, long-term induction of telomeric 8-oxoG, fibroblasts and epithelial cells accrued extensive telomere damage and underwent senescence after a single induction. These phenotypes appeared to result from replication stress, as 8-oxoG induction increased fragile telomeres and MiDAS and did not trigger senescence in quiescent cells [48, 49]. Furthermore, noncancerous cells activated ATR while cancer cells did not, indicating that the difference in response between transformed and untransformed cells is due to preservation of active cell cycle checkpoints (Figure 3).
The finding that replication stress restricted to telomeres is sufficient to trigger senescence suggests that telomere fragility could contribute to an antitumor mechanism. Spontaneous TIFs are also observed in precancerous lesions but not in malignant cells from the same tissues, suggesting that TIF accumulation prevents tumorigenesis and must be bypassed during cell transformation to allow continued proliferation [80]. While these TIFs cannot be entirely attributed to replication stress, it is a probable source of damage, as increased ROS and oncogene expression are prominent features of tumorigenesis that generate replication stress. These recent findings indicate that noncancerous cells are less amenable than cancer cells to stress that compromises telomere integrity. This suggests that broken telomeres are not meant to be repaired, but rather serve as sensory signals that trigger cell cycle arrest to prevent DNA replication in a mutagenic environment [85, 86].
Concluding Remarks
Here we’ve reviewed recent insights regarding how unresolved replication stress at telomeres contributes to genomic instability. While it has long been appreciated that accumulated telomere damage correlates with senescence, mitotic crisis, and tumorigenesis, novel experimental strategies capable of targeting DNA damage specifically to telomeres have recently validated that telomere damage is sufficient to generate these phenotypes. Recent findings reveal that replication stress within telomeres triggers BIR-mediated telomere synthesis and MiDAS. These pathways deregulate telomere length and generate errors that exacerbate replication stress; as such, intact cell cycle checkpoints thwart these repair responses. However, several questions remain (see Outstanding Questions). As most of the discussed studies subjected telomeres to severe damage presumably above the determined five TIF threshold for senescence, it is unclear if untransformed cells use BIR or MiDAS to resolve lower levels of stress. Additionally, how the subtelomere impacts the timing or chosen pathway for repair of their corresponding telomere remains undetermined. To address these questions, methods targeting damage at individual telomeres on specific chromosomes will need to be developed.
Outstanding Questions:
Is BIR-mediated telomere synthesis exclusive to cancer cells? If so, are untransformed cells capable of repairing collapsed replication forks in telomeres?
Is the mammalian PolD3 BIR replisome as error-prone as the Pol32 yeast replisome? How does BIR generate telomere replication stress to sustain ALT?
How is BIR-mediated telomere synthesis initiated in the absence of Rad51-mediated strand invasion? Is replication initiated from exposed ssDNA in the stalled replication fork, ssDNA gaps in damaged telomeres, or extrachromosomal telomeric DNA? Does this use mmBIR?
Are late-replicating telomeres more likely to activate MiDAS? How does the subtelomere affect the repair of collapsed replication forks?
What is the semi-conservative mechanism of MiDAS and why is it utilized at CFSs but not telomeres?
What combinations of acquired mutations and stressors initiate ALT?
How does induced BIR in ALT-negative cells differ from baseline BIR in ALT-positive cells? What is the advantage of these mechanistic differences for telomere maintenance?
Which tissues are most susceptible to stress-induced telomere dysfunction? Does this significantly contribute to aging-associated phenotypes?
Why do damaged telomeres localize to the nuclear pore complex? Does this promote or inhibit BIR?
Do cancer-associated POT1 mutations promote tumorigenesis by increasing replication stress? If so, what is the molecular mechanism behind this? Is telomere fragility a significant source of genomic instability in other cancers?
Does replication stress contribute to telomere shortening in telomere biology disorders?
Is active telomerase sufficient to counteract telomere replication stress in highly proliferative cell types such as stem cells?
It was recently reported that human telomeres relocate to the nuclear pore complex (NPC) in response to replication stress, which had previously only been observed in yeast. NPC localization was shown to prevent T-SCE formation, suggesting that NPCs inhibit conventional HDR and cross-over by either preventing collapse of stalled forks or by promoting conservative BIR [51, 87]. It will be interesting to further elucidate this aspect of telomere repair and stress management, as it mirrors the localization of telomeres to APBs, which serve as recombination hubs in ALT cells. Additionally, while the discussed results suggest that untransformed cells are incapable of repairing the single-ended DSBs arising from collapsed replication forks, other measures, such as nonrandom segregation of chromosomes with damaged telomeres, may be used to limit TIF accumulation and extend replicative lifespan [88]. Further studies would be needed to test these possibilities.
Finally, in addition to ALT activation, telomere replication stress may also contribute to earlier stages of tumorigenesis in telomerase-positive cancers. Somatic and germline mutations in POT1, a shelterin component that binds telomeric ssDNA [89], are associated with a range of cancers, yet the molecular basis behind this is unknown [90]. There is strong evidence that these mutations increase replication stress at telomeres, as POT1 dysfunction has been shown to stall replication at telomeres, activate ATR, and trigger MiDAS [51, 91]. However, it has also been argued that POT1 mutations promote tumorigenesis by increasing telomere length, as POT1 negatively regulates telomerase [92, 93]. Further study will be needed to separately assess these functions. Altogether, the discussed results argue for an expanded definition of the “end-replication problem” that includes replication stress as a significant source of telomere attrition and limited cell proliferation. Parallel to telomere shortening, telomere fragility serves as double-edged sword that protects genome stability when physiological checkpoints are intact but paradoxically promotes tumorigenesis should they fail.
Highlights.
Telomeres are prone to replication fork stalling and collapse when subjected to replication stress, and failure to complete telomere replication accelerates telomere shortening and limits replicative lifespan.
Telomere replication after fork collapse can resume via break-induced replication (BIR) and mitotic DNA synthesis (MiDAS), however, recent findings indicate that these pathways are error-prone and associated with genomic instability and tumorigenesis.
BIR and MiDAS at telomeres are more prevalent in cancer cells and are suppressed by cell cycle checkpoints, implicating telomere fragility under replication stress as a tumor-suppressive mechanism.
Further characterization of BIR-mediated telomere synthesis in different cell types should provide insight into the molecular basis of tumorigenesis, particularly regarding activation of alternative lengthening of telomeres (ALT).
Acknowledgments:
While we tried our best to provide an unbiased and comprehensive view of the current status of the telomere replication field of study, we apologize if any studies or citations were omitted due to space constraints. The Nandakumar laboratory is funded by National Institutes of Health grant R01GM120094 (to J.N.) and the American Cancer Society Research Scholar grant RSG-17-037-01-DMC (to J.N.).
Glossary
- 8-oxoguanine (8-oxoG)
Common mutagenic DNA lesion arising from guanine oxidation. Telomeres are enriched for 8-oxoG due to high guanine content
- ALT-associated Promyelocytic Leukemia Bodies (APBs)
PML bodies that aggregate telomeres and recombination proteins serve as hubs for telomere recombination in ALT cells
- Alternative Lengthening of Telomeres (ALT)
Telomerase-independent telomere maintenance mechanism using telomere recombination
- Ataxia telangiectasia and Rad3- related (ATR)
DDR protein kinase that recognizes persistent ssDNA within stalled replication forks. Triggers a signaling cascade that prevents entry into mitosis and stabilizes the stalled fork, resulting in completed replication or permanent cell cycle arrest
- Break-Induced Replication (BIR)
Error-prone HDR mechanism that drives long-tract, conservative DNA synthesis from one-ended DSBs
- Common Fragile Site (CFS)
Large, difficult-to-replicate loci that manifest as DAPI-negative gaps in metaphase chromosomes when cells are subjected to replication stress
- D-loop
Displacement loop, three-strand DNA structure in which one strand of the DNA duplex is displaced by an invading homologous strand
- Fragile Telomeres
Elongated or multi-signal telomere foci visualized by fluorescent in situ hybridization (FISH) on metaphase chromosomes, indicative of replication stress at telomeres
- G-quadruplex
Highly stable secondary structure formed by Hoogsteen hydrogen bonding between 4 guanines. Form on the G-rich telomere lagging strand
- Homology-Directed Repair (HDR)
A DSB repair pathway using homologous DNA as a template for DNA synthesis and repair. Conventional HDR is “error-free”, restricted to S/G2, and requires two DNA ends for double Holliday junction formation
- Microhomology-mediated BIR (mmBIR)
Rad51-independent BIR pathway that uses microhomologies of 1–3 nucleotides to initiate replication. Prone to template switching, generates genomic rearrangements
- Mitotic DNA Synthesis (MiDAS)
HDR-mediated repair mechanism that resolves late replication intermediates and completes replication of fragile sites in prophase of mitosis
- Nonhomologous End-Joining (NHEJ)
An error-prone, cell cycle-independent DSB repair pathway that directly ligates broken DNA ends back together
- R-loop
Three-stranded nucleic acid structure composed of a DNA:RNA hybrid and displaced ssDNA
- Reactive Oxygen Species (ROS)
Reactive oxygen products formed from cellular respiration
- Shelterin
Telomere-capping protein complex consisting of TRF1, TRF2, TIN2, TPP1, RAP1, and POT1 in humans. Inhibits DDR at telomeres, assists telomere replication, and regulates telomere extension by telomerase
- Telomerase
Reverse transcriptase that elongates telomeres using an RNA template. Absent in most somatic cells, catalytic subunit expression is limited to stem cells and germ cells
- Telomere dysfunction-induced foci (TIF)
Colocalization of a DDR marker (typically yH2AX or 53bp1) and a telomere visualized through immunofluorescent staining and FISH. Indicates a structurally compromised telomere. TIFs in the absence of telomere fusions represent “intermediate” state telomeres that inhibit DSB repair but permit DSB recognition by DDR[86]
- Telomeric Repeat-containing RNAs (TERRA)
Noncoding RNA transcribed from telomeres, upregulated with telomeric chromatin decompaction
- Telomeric Sister Chromatid Exchanges (T-SCEs)
Detectable by CO-FISH, indicative of telomere cross-over via HDR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palm W and de Lange T, How Shelterin Protects Mammalian Telomeres. Annual Review of Genetics, 2008. 42(1): p. 301–334. [DOI] [PubMed] [Google Scholar]
- 2.Levy MZ, et al. , Telomere end-replication problem and cell aging. Journal of Molecular Biology, 1992. 225(4): p. 951–960. [DOI] [PubMed] [Google Scholar]
- 3.Barnes RP, Fouquerel E, and Opresko PL, The impact of oxidative DNA damage and stress on telomere homeostasis. Mechanisms of ageing and development, 2019. 177: p. 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson R, et al. , Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. The EMBO journal, 2019. 38(5): p. e100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fumagalli M, et al. , Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature cell biology, 2012. 14(4): p. 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hewitt G, et al. , Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature communications, 2012. 3: p. 708–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandes RV, Feretzaki M, and Lingner J, The makings of TERRA R-loops at chromosome ends. Cell Cycle, 2021. 20(18): p. 1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Higa M, Fujita M, and Yoshida K, DNA Replication Origins and Fork Progression at Mammalian Telomeres. Genes, 2017. 8(4): p. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarek G, et al. , TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Molecular cell, 2015. 57(4): p. 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryan TM, G-Quadruplexes at Telomeres: Friend or Foe? Molecules (Basel, Switzerland), 2020. 25(16): p. 3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sfeir A, et al. , Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell, 2009. 138(1): p. 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min J, Wright WE, and Shay JW, Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Molecular and cellular biology, 2017. 37(20): p. e00226–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martínez P, et al. , Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes & development, 2009. 23(17): p. 2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller KM, Rog O, and Cooper JP, Semi-conservative DNA replication through telomeres requires Taz1. Nature, 2006. 440(7085): p. 824–828. [DOI] [PubMed] [Google Scholar]
- 15.Leman AR, et al. , Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell cycle (Georgetown, Tex.), 2012. 11(12): p. 2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann M, et al. , TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes & development, 2014. 28(22): p. 2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YW, et al. , TRF1 participates in chromosome end protection by averting TRF2-dependent telomeric R loops. Nature structural & molecular biology, 2018. 25(2): p. 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drosopoulos WC, et al. , Human telomeres replicate using chromosome-specific, rather than universal, replication programs. The Journal of cell biology, 2012. 197(2): p. 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart JA, et al. , Human CST promotes telomere duplex replication and general replication restart after fork stalling. The EMBO journal, 2012. 31(17): p. 3537–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alver RC, Chadha GS, and Blow JJ, The contribution of dormant origins to genome stability: From cell biology to human genetics. DNA Repair, 2014. 19: p. 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drosopoulos WC, et al. , TRF2 Mediates Replication Initiation within Human Telomeres to Prevent Telomere Dysfunction. Cell reports, 2020. 33(6): p. 108379–108379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takai H, Smogorzewska A, and de Lange T, DNA Damage Foci at Dysfunctional Telomeres. Current Biology, 2003. 13(17): p. 1549–1556. [DOI] [PubMed] [Google Scholar]
- 23.Kaul Z, et al. , Five dysfunctional telomeres predict onset of senescence in human cells. EMBO reports, 2011. 13(1): p. 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greider CW and Blackburn EH, Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell, 1985. 43(2, Part 1): p. 405–413. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J-M and Zou L, Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell & Bioscience, 2020. 10(1): p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramara J, Osia B, and Malkova A, Break-Induced Replication: The Where, The Why, and The How. Trends in genetics : TIG, 2018. 34(7): p. 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costantino L, et al. , Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science (New York, N.Y.), 2014. 343(6166): p. 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hastings PJ, Ira G, and Lupski JR, A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS genetics, 2009. 5(1): p. e1000327–e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, et al. , Tracking break-induced replication shows that it stalls at roadblocks. Nature, 2021. 590(7847): p. 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kockler ZW, et al. , Repair of DNA Breaks by Break-Induced Replication. Annual Review of Biochemistry, 2021. 90(1): p. 165–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deem A, et al. , Break-induced replication is highly inaccurate. PLoS biology, 2011. 9(2): p. e1000594–e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osia B, et al. , Cancer cells are highly susceptible to accumulation of templated insertions linked to MMBIR. Nucleic Acids Research, 2021. 49(15): p. 8714–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sotiriou SK, et al. , Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Molecular cell, 2016. 64(6): p. 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs J, Cheblal A, and Gasser SM, Underappreciated Roles of DNA Polymerase δ in Replication Stress Survival. Trends in Genetics, 2021. 37(5): p. 476–487. [DOI] [PubMed] [Google Scholar]
- 35.Mayle R, et al. , Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science, 2015. 349(6249): p. 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Özer Ö and Hickson ID, Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open biology, 2018. 8(4): p. 180018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minocherhomji S, et al. , Replication stress activates DNA repair synthesis in mitosis. Nature, 2015. 528(7581): p. 286–290. [DOI] [PubMed] [Google Scholar]
- 38.Bhowmick R, Minocherhomji S, and Hickson ID, RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Molecular Cell, 2016. 64(6): p. 1117–1126. [DOI] [PubMed] [Google Scholar]
- 39.Graber-Feesl CL, et al. , Mitotic DNA Synthesis Is Differentially Regulated between Cancer and Noncancerous Cells. Molecular cancer research : MCR, 2019. 17(8): p. 1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verma P, et al. , RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes & development, 2019. 33(3–4): p. 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dilley RL, et al. , Break-induced telomere synthesis underlies alternative telomere maintenance. Nature, 2016. 539(7627): p. 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roumelioti F-M, et al. , Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO reports, 2016. 17(12): p. 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doksani Y and de Lange T, Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell reports, 2016. 17(6): p. 1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z, et al. , Break-induced replication promotes fragile telomere formation. Genes & development, 2020. 34(19–20): p. 1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Özer Ö, et al. , Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget, 2018. 9(22): p. 15836–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doksani Y, The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes, 2019. 10(4): p. 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cesare AJ, et al. , The telomere deprotection response is functionally distinct from the genomic DNA damage response. Molecular cell, 2013. 51(2): p. 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnes RP, et al. , Telomeric 8-Oxoguanine Drives Rapid Premature Senescence In The Absence Of Telomere Shortening. bioRxiv, 2021: p. 2021.05.05.442662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fouquerel E, et al. , Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Molecular cell, 2019. 75(1): p. 117–130.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Exposito L, et al. , Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase η in the Alternative Lengthening of Telomeres. Cell reports, 2016. 17(7): p. 1858–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pinzaru AM, et al. , Replication stress conferred by POT1 dysfunction promotes telomere relocalization to the nuclear pore. Genes & development, 2020. 34(23–24): p. 1619–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnoult N, et al. , Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS genetics, 2010. 6(4): p. e1000920–e1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porreca RM, et al. , TRF1 averts chromatin remodelling, recombination and replication dependent-break induced replication at mouse telomeres. eLife, 2020. 9: p. e49817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sobinoff AP and Pickett HA, Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends in Genetics, 2017. 33(12): p. 921–932. [DOI] [PubMed] [Google Scholar]
- 55.Cho NW, et al. , Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell, 2014. 159(1): p. 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J-M, et al. , Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell reports, 2019. 26(4): p. 955–968.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JY, et al. , Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clinical cancer research : an official journal of the American Association for Cancer Research, 2017. 23(6): p. 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu J, et al. , Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell, 2012. 148(4): p. 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cesare AJ, et al. , Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nature Structural & Molecular Biology, 2009. 16(12): p. 1244–1251. [DOI] [PubMed] [Google Scholar]
- 60.Schwartzentruber J, et al. , Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 2012. 482(7384): p. 226–231. [DOI] [PubMed] [Google Scholar]
- 61.Dyer MA, et al. , ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harbor perspectives in medicine, 2017. 7(3): p. a026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoang SM, et al. , Regulation of ALT-associated homology-directed repair by polyADP-ribosylation. Nature structural & molecular biology, 2020. 27(12): p. 1152–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Sullivan RJ, et al. , Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nature structural & molecular biology, 2014. 21(2): p. 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li F, et al. , ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. The EMBO journal, 2019. 38(19): p. e96659–e96659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conomos D, et al. , Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. The Journal of cell biology, 2012. 199(6): p. 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conomos D, Reddel RR, and Pickett HA, NuRD–ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nature Structural & Molecular Biology, 2014. 21(9): p. 760–770. [DOI] [PubMed] [Google Scholar]
- 67.Lu R, et al. , The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nature Communications, 2019. 10(1): p. 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan X, et al. , FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Scientific Reports, 2019. 9(1): p. 19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Silva B, et al. , FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nature Communications, 2019. 10(1): p. 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J-M, et al. , Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Molecular Cell, 2021. 81(5): p. 1027–1042.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cox KE, Maréchal A, and Flynn RL, SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell reports, 2016. 14(5): p. 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang T, et al. , Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS genetics, 2019. 15(2): p. e1007925–e1007925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Min J, Wright WE, and Shay JW, Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes & development, 2019. 33(13–14): p. 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sobinoff AP, et al. , BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. The EMBO journal, 2017. 36(19): p. 2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barroso-González J, et al. , Anti-recombination function of MutSα restricts telomere extension by ALT-associated homology-directed repair. Cell reports, 2021. 37(10): p. 110088–110088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deeg KI, et al. , Cancer Cells with Alternative Lengthening of Telomeres Do Not Display a General Hypersensitivity to ATR Inhibition. Frontiers in oncology, 2016. 6: p. 186–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flynn RL, et al. , Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science (New York, N.Y.), 2015. 347(6219): p. 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mukherjee J, et al. , A subset of PARP inhibitors induces lethal telomere fusion in ALT-dependent tumor cells. Science Translational Medicine, 2021. 13(592): p. eabc7211. [DOI] [PubMed] [Google Scholar]
- 79.George SL, et al. , Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine, 2020. 59: p. 102971–102971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suram A, et al. , Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. The EMBO journal, 2012. 31(13): p. 2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mao P, et al. , Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nature communications, 2016. 7: p. 12154–12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tomlinson RL, et al. , Cell cycle-regulated trafficking of human telomerase to telomeres. Molecular biology of the cell, 2006. 17(2): p. 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun L, et al. , Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic acids research, 2015. 43(13): p. 6334–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan J, et al. , An R-loop-initiated CSB–RAD52–POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Research, 2020. 48(3): p. 1285–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.von Zglinicki T, Oxidative stress shortens telomeres. Trends in Biochemical Sciences, 2002. 27(7): p. 339–344. [DOI] [PubMed] [Google Scholar]
- 86.Cesare AJ and Karlseder J, A three-state model of telomere control over human proliferative boundaries. Current opinion in cell biology, 2012. 24(6): p. 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hauer MH and Gasser SM, Chromatin and nucleosome dynamics in DNA damage and repair. Genes & development, 2017. 31(22): p. 2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xing M, et al. , Replication Stress Induces ATR/CHK1-Dependent Nonrandom Segregation of Damaged Chromosomes. Molecular Cell, 2020. 78(4): p. 714–724.e5. [DOI] [PubMed] [Google Scholar]
- 89.Baumann P and Cech Thomas R, Pot1, the Putative Telomere End-Binding Protein in Fission Yeast and Humans. Science, 2001. 292(5519): p. 1171–1175. [DOI] [PubMed] [Google Scholar]
- 90.Wu Y, Poulos RC, and Reddel RR, Role of POT1 in Human Cancer. Cancers, 2020. 12(10): p. 2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pinzaru AM, et al. , Telomere Replication Stress Induced by POT1 Inactivation Accelerates Tumorigenesis. Cell reports, 2016. 15(10): p. 2170–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim W-T, et al. , Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. The EMBO journal, 2021. 40(12): p. e107346–e107346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lei M, et al. , Switching Human Telomerase On and Off with hPOT1 Protein in Vitro*. Journal of Biological Chemistry, 2005. 280(21): p. 20449–20456. [DOI] [PubMed] [Google Scholar]
- 94.Glousker G and Lingner J, Challenging endings: How telomeres prevent fragility. BioEssays, 2021. 43(10): p. 2100157. [DOI] [PubMed] [Google Scholar]
- 95.Vannier J-B, et al. , RTEL1 Dismantles T Loops and Counteracts Telomeric G4-DNA to Maintain Telomere Integrity. Cell, 2012. 149(4): p. 795–806. [DOI] [PubMed] [Google Scholar]
- 96.Arora R, et al. , RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nature communications, 2014. 5: p. 5220–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petti E, et al. , SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nature Communications, 2019. 10(1): p. 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]