

Abstract
Proteolysis targeting chimera (PROTAC) is one of the most frequently used technologies for targeted protein degradation. PROTACs are composed of target protein ligand, E3 ligase ligand and a linker between them. Traditional methods for the development of PROTACs involve step-by-step synthesis and are time consuming. Herein, we report a platform for the rapid synthesis of PROTACs (Rapid-TAC) via a traceless coupling reaction between ortho-phthalaldehyde (OPA) motif on the ligand of targeting protein and an amine fucntional group on the commercially available partial PROTAC library that is composed of different E3 ligase ligands and various types and lengths of linkers. Under our optimized miniaturized conditions, the full PROTACs can be synthesized in a high throughput manner and the products can be directly used for screening without any further manipulations including purification. We demonstrated the utility of this platform by quikcly identifing active degraders for androgen receptor (AR) and BRD4 with DC50 of 41.9 nM and 8.9 nM, respectively. It is expected that this Rapid-TAC platform can be easily extended to many other targets, thus lowering the barrier to access this novel modelity for small molecule drug discovery and faciliate structure activity relationship studies.
Keywords: PROTAC, AR, BRD4, Degradation
Graphic abstract

Water as the only byproduct; Without purification and tested in cell assay
A platform for the rapid synthesis of PROTACs (Rapid-TAC) is reported. The reaction between ortho-phthalaldehyde and amine is the key for the quick generation of a library of PROTACs in DMSO under miniaturized conditions. The products can be used for screening without any purificaiton. The utility of the Rapid-TAC platform is demonstrated by the quick identification of active PROTACs against AR and BRD4
Introduction
Proteolysis targeting chimera (PROTAC), initially introduced twenty years ago [1], represents a revolutionary technology for small molecule drug discovery. PROACs are composed of a ligand for the protein of interest (POI), a ligand for E3 ubiquitin ligase, and a linker between the two ligands. Upon forming the E3-PROTAC-POI ternary complex, the POI is polyubiquitinated and undergoes subsequent proteasomal degradation [2–7]. Traditional small-molecule drugs need to bind to the protein target and change its function. In contrast, PROTAC only needs a binder, which can be non-functional by itself. This opens up the opportunity to target a large number of undrugged proteins that lack functional binding sites for small molecules [8]. In addition, PROTACs can act catalytically to degrade the entire POI via an event-driven mode, which offers great opportunities for achieving the desired pharmacological outcomes. Since its discovery, numerous protein targets in various families have been successfully degraded using the PROTAC strategy. It has been predicted that nearly a dozen of PROTACs will be tested clinically for the treatment of human diseases soon [9].
For the development of PROTACs, synthesis and screening of a library of compounds is still the most viable approach to integrate the three components including the ligand of POI, the ligand of E3 ligase and the linker between them for the generation of active degraders. The length, type, and position of the linker can often significantly affect the activity of the degraders. Various chemical methods have been used to accelerate the development of PROTACs and facilitate the SAR studies, such as coupling one ligand bearing the linker with the other ligand using activated esters [10, 11], click chemistry [12, 13], solid-phase synthesis [14], alkylation reaction [15], Staudinger ligation [16], and multicomponent synthesis [17]. However, the PROTACs need to be synthesized one by one and the final product needs to be purified for further biological evaluations using these methods. It is thus difficult to prepare the library in a high throughput manner. Our group previously reported a strategy for the rapid synthesis of PROTACs (Rapid-TAC) by reacting hydrazide A1 in the POI ligand with aldehyde A2 containing a E3 ligase ligand and various linkers (Figure 1) [18]. The reaction can be carried out under miniaturized conditions for the high throughput synthesis of PROTACs in DMSO solution, which can be directly used for biological testing without further manipulation including purification, because the conversion is generally higher than 80% and the only byproduct is water. Most functional groups in both ligands are also compatible with the conditions.
Figure 1.

Platforms for the rapid synthesis of PROTACs (Rapid-TAC) under miniaturized conditions for direct screening without further manipulations. A) Our previous Rapid-TAC Platform based on hydrazide-aldehyde coupling chemistry. B) Our new Rapid-TAC platform based on OPA-amine coupling chemistry.
However, the acylhydrazone motif in product A3 is hydrolytically labile, which can be a significant issue for assays that require long treatment time and long-term storage after multiple rounds of free-thaw cycles. To solve this problem, we used a two-stage strategy for the development of active and stable PROTACs. In the first stage, we identified active degraders with an appropriate E3 ligase ligand and an optimal linker (e.g., type and length) from a library of compounds A3 using assays that require a relatively short period of time. In the second stage, we replaced the acylhydrazone motif in the linker region by a more stable isostere such as an amide while keeping the rest of the active molecule largely intact. The two-strategy led to the successful development of stable and potent PROTACs for estrogen receptor quickly [18]. However, the change of the acylhydrazone motif to its isosteres can often significantly alter the activity of the degraders and further time-consuming optimization may be required in some cases. In addition, the aldehyde-containing partial PROTAC library A2 is not commercially available and needs to be prepared by step-by-step synthesis.
We herein report our next generation Rapid-TAC platform that can overcome many of the limitations in our previous strategy while keeping most of the advantages for the development of PROTACs. The key to our new platform is the formation of phthalimidine B3 from ortho-phthalaldehyde (OPA) B1 and amine B2, a reaction that has been known for a long time [19]. The phthalimidine motif can be part of the POI ligand or part of the linker. The addition of amine RNH2 to ortho-phthalaldehyde first produces the isoindolinediol intermediate, which can tautomerize to phthalimidine after the elimination of water (Figure S1) [20, 21]. The reaction has been recently optimized for the modification of peptides and protein labeling and profiling under biocompatible conditions [22–25]. We prepared two PROTACs libraries, one for androgen receptor (AR) and the other for bromodomain-containing protein 4 (BRD4), using this highly efficient OPA-amine coupling chemistry from two building blocks B1 and a library of commercially available amine-containing E3 ligase ligands with various linkers. With an appropriate building block B1 in hand, the library can be completed under miniaturized conditions in parallel in a reaction block, which does not require any further manipualtion before the cell-based screening because the only buproduct is water and the conversion is generally very high, a nearly ideal reaction [26].
2. Design of Rapid-TAC platform
We first set out to optimize the reaction conditions to fit the Rapid-TAC platform for a model reaction in Figure 2. The choice of solvent is limited to either water or DMSO to avoid any further manipulation after the synthesis. Although the desired product C3 could be detected under previously reported conditions in PBS (pH 7.4) buffer, a complex mixture was obtained (Figure 2, entry 1). We observed much better results after switching the solvent from PBS buffer to DMSO and the purity of the crude product could reach 84%. We then found that the addition of HOAc additive could significantly improve the conversion and the purity of the product C3 could reach 99%! Because the products will be diluted by buffer before adding to the cells, the HOAc additive is generally inconsequential for the screening.
Figure 2.

Optimization of the OPA-amine coupling reaction for the Rapid-TAC platform. Reaction were conducted with C1 (20 μL of 50 mM DMSO solution), C2 (20 μL of 50 mM DMSO solution), additive, and solvent at 80 °C for 48 h. The purity of C3 directly from the reaction mixture was determined by HPLC.
To this end, we prepared AR ligand 3 bearing a OPA motif by linking part of AR ligand 1 with the carboxylic acid-containing OPA precursor 2 (Figure 3). The benzene ring of the phthalimidine motif is actually part of the AR ligand in this case [27, 28]. For the E3 ligase ligand portion, we used two commercially available partial PROTAC libraries. One of them has 9 members that are composed of a pomalidomide cereblon E3 ligase ligand and various alkyl or polyethylene glycol units (PEG) linkers. The other one has 11 members that are composed of a Von-Hippel-Lindau (VHL) E3 ubiquitin ligase ligand and various alkyl or PEG linkers. These two commercially available partial PROTAC library all contain the NH2 group on the other end of the linker. Each of these building blocks was dissolved in DMSO as a 50 mM stock solution. OPA derivative 3 was also dissolved in DMSO with the same concentration. We next mixed 20 μL of solution 3 with the stock solution of each member of the amine-linker-E3 ligase ligand partial PROTAC library in a 1:1 ratio. The solution was then heated at 80 °C for 24 h. After cooling it to room temperature, to the solution was added another 60 μL of DMSO. The resulting solution was heated at 80 °C for another 24 h. The solution was then cooled down to room temperature and the purity of the product was analyzed by HPLC. Out of the 21 potential AR PROTACs LG-AR-1 to LG-AR-21, we found that the purity ranged from 88% to 98% (Figure 3 and Figure S2). The “linker-less” PROTAC LG-AR-21 bearing a lenalidomide motif was prepared by reacting 3 with 3-aminopiperidine-2,6-dione directly. Without any further manipulation including purification, this PROTAC library can be directly evaluated for their biological activity.
Figure 3.
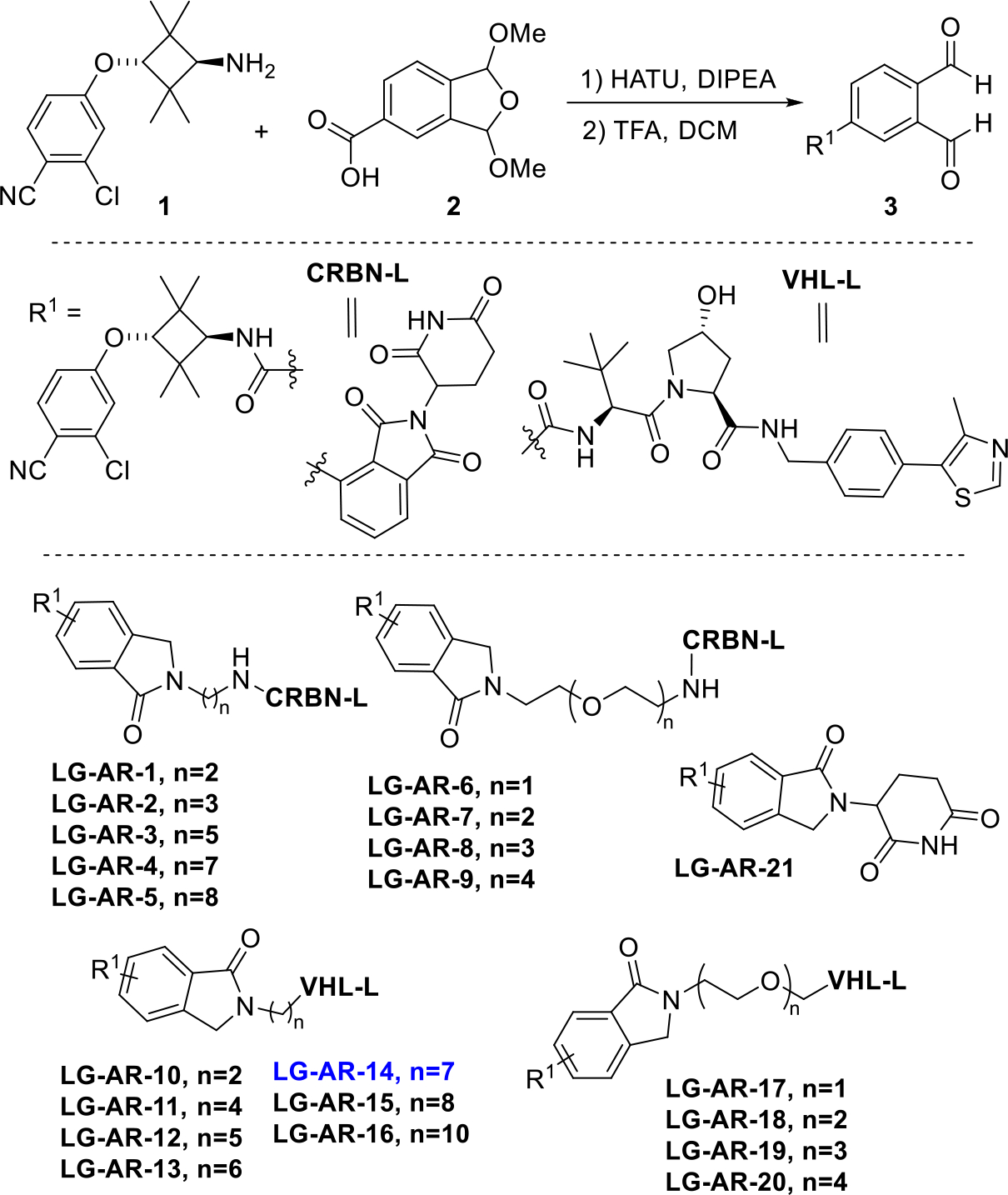
Synthesis of 21 AR PROTACs using the Rapid-TAC platform.
The reaction worked smoothly for all members of the partial PROTAC library bearing a cereblon ligand. For all members of partial PROTAC library that contain a VHL ligand, however, we observed significant amount of byproducts derived from the cleavage of amide bond between the proline residue and the α-tBu glycine residue. We then found that the addition of 2 equivalent of KOAc could suppress this cleavage. Because all of the partial PROTAC library were purchased as their hydrochloride salt, it is likely that the HCl in the system caused the issue and needs to be quenched by KOAc.
3. Result and discussion
3.1. The degradation of AR by Rapid-TAC platform in LNCap cells
Next, we tested the activity of the 21 potential AR PROTACs LG-AR-1 to LG-AR-21 at 1 and 10 μM concentrations in LNCap prostate cancer cells. For the cereblon-recruiting AR PROTACs, no significant degradation was observed (Figure 4). For VHL-recruiting AR PROTACs, many of them showed certain degree of degradation. Not surprisingly, the type and length of linkers played a significant role. Based on the screening result, LG-AR-14 (n=7) containing a linker with seven methylene groups, is the most effective degrader for AR (Figures 4 and S4). Interestingly, LG-AR-14 reduced AR protein by 40% at 10 μM and by 73% at 1 μM, a classical “hook” effect that has been observed for many PROTACs [29].
Figure 4.

A library of 21-members of AR PROTACs was tested by Western blot assay in LNCap cell line for 16 h at 1 μM and 10 μM concentrations.
3.2. Comparison of cyclic amide AR degrader with two isomers of LG-AR-14
The coupling of substituted OPA with amine will always lead to two isomers that are differ from each other on the position of the carbonyl oxygen in phthalimidine product. The difference between the two isomers is so small that we do not expect this difference impacts the activity. In addition, the cyclic amide is generally exposed to the solvent and should not impact the binding to the POI. Nevertheless, we still decided to synthesize the two isomers, LG-AR-14-A and LG-AR-14-B, separately using the standard chemistry and purification procedures to compare their activity (Figure 5A). In addition, we also synthesized and purified compound LG-AR-14-C with a secondary amide, which is closely related LG-AR-14A/B. These three AR PROTACs were evaluated by Western blot assay for their degradation activity for AR. Not surprisingly, the degradation activity of LG-AR-14A and LG-AR-14B is almost identical to LG-AR-14, the mixture generated from our Rapid-TAC platform without any purification (Figure 5B). Interestingly, PROTAC LG-AR-14-C showed slightly lower degradation activity of AR protein than LG-AR-14A and LG-AR-14B, indicating that the cyclic tertiary amide is better than the acyclic secondary amide as part of the linker in this case. Based on these results, it is reasonable to expect that the PROTACs derived from OPA-amine coupling Rapid-TAC platform without purification will have similar activity comparing to the step-by-step synthesized PROTACs with purifications after each step.
Figure 5.

A) The structures of three AR PROTACs based on LG-AR-14. They are synthesized and purified using standard protocols. B) AR PROTACs were tested by Western blot in LNCap cell line for 16 h at 0.1 μM, 1 μM and 10 μM concentrations.
3.3. The characterization of candidate AR PROTACs
We then examined the time course of LG-AR-14-A for the induction of AR degradation in LNCap cell line. Our data showed that LG-AR-14-A effectively reduced the AR protein level within 6 h and it was sufficient to reach the max degradation at 16 h (Figure 6A). We next tested the dose response of LG-AR-14-A in LNCap cell line. Our data showed that LG-AR-14-A induced degradation of AR protein in a dose-dependent manner (Figure 6B and S5). The concentration at which half-maximal degradation occurs (DC50) for LG-AR-14-A was 41.9 nM in the LNCap cell line.
Figure 6.

A) Time course of LG-AR-14-A. LNCap cells were treated with LG-AR-14-A at 100 nM for different time and the results were analysed by Western blot. B) Dose response of LG-AR-14-A. LNCap cells were treated with LG-AR-14-A for 16 h at different concentrations and the results were analysed by Western blot. C) Mechanistic studies of AR degradation induced by LG-AR-14-A in LNCap cells. Cell were co-treated with AR ligand (10 μM), VHL ligand (10 μM), and MLN4924 (0.5 μM) together with LG-AR-14-A (100 nM) for 8 h; D) Cell were co-treated with MG132 (5 μM) for 2 h and then together with LG-AR-14-A (100 nM) for another 6 h.
We next investigated the mechanism of action of LG-AR-14-A in the LNCaP cell line. Our data showed that AR degradation induced by LG-AR-14-A can be effectively abolished by the pretreatment with NEDD8-activating enzyme inhibitor MLN4924, VHL ligand, AR ligand or proteasome inhibitor MG132 (Figure S6 and 6C/D). These results clearly demonstrated that LG-AR-14-A followed the conventional PROTAC mechanism that requires the participation of E3 ligase, AR, neddylation, and proteasome.
3.4. The degradation of BRD4 by Rapid-TAC platform in MV-4-11 cells
To further demonstrate the utility of OPA-amine coupling based Rapid-TAC platform, we prepared a library of potential PROTACs for the second target, BRD4. We first synthesized BRD4 ligand 6 bearing OPA by linking BRD4 ligand 4 derived from JQ1 with the bromide-containing OPA precursor 5 followed by hydrolysis (Figure 7). Previously, the OPA building block contains a carboxylate handle, which can be coupled with an amine-containing ligand of the target. A complementary approach was used for the preparation of key building block 6, which was derived from the cross-coupling reaction between an aryl bromide and a ligand bearing a carboxylate derivative. OPA building blocks 2 and 5 can be used for any POI ligands with an amine or carboxylate handle, respectively. Using the OPA-amine Rapid-TAC platform, we synthesized 21 potential BRD4 PROTACs (LG-BRD-1 to 21) quickly in DMSO solution following the same protocol described previously (Figure 7 and Figure S3). Out of the 21 products, their purity ranged from 78% to 96%.
Figure 7.

Synthesis of 21 BRD4 PROTACs using the Rapid-TAC platform.
We next tested the activity of the 21 potential BRD4 PROTACs in MV-4-11 cell line (Figure 8). Our results showed that there was no obvious degradation for PROTACs LG-BRD-1 to LG-BRD-10 derived by pomalidomide. For PROTACs derived from VHL ligand, LG-BRD-12 with four methylene units in the linker was able to reduce the BRD4 protein significantly at 1 μM as the most potent compound in this series. The classical “hook” effect was observed for some of the PROTACs at 10 μM.
Figure 8.

A library of 21 members of BRD4 PROTACs was tested by Western blot in MV-4-11 cell line for 24 h at 1 μM and 10 μM concentrations.
3.5. Comparison of BRD4 degrader LG-BRD-12 with its isomers
Next, we synthesized the two isomers of BRD4 degrader, LG-BRD-12A and LG-BRD-12B, separately using the standard chemistry and purification procedures to compare their activity (Figure 9A). These two BRD4 PROTACs were evaluated by Western blot assay for their degradation activity for BRD4 at four different concentrations. LG-BRD-12B showed higher degradation activity than LG-BRD-12A (Figure 9B). We then completed a dose response study for compound LG-BRD-12B in MV-4-11 cell line and observed a DC50 of 8.9 nM (Figure 9C and S7). We demonstrated that effective BRD4 degraders could be identified quickly using our OPA-amine coupling reaction based Rapid-TAC platform.
Figure 9.

A) The structures of two AR PROTACs based on LG-BRD-12. They are synthesized and purified using standard protocols. B) BRD4 PROTACs were tested by Western blot in MV-4-11 cell line for 12 h at 100 nM, 30 nM, 10 nM and 3 nM concentrations. C) Dose response of LG-BRD-12B. MV-4-11 cells were treated with LG-BRD-12B for 12 h at different concentrations and the results were analysed by Western blot.
4. Chemistry
To generate the AR PROTACs using the OPA-amine coupling based Rapid-TAC platform, we firstly synthesized the AR ligand based OPA derivative 3. The syntheyic route for 3 was outlined in Scheme 1. Firstly, I-3 was prepared from the reaction of I-1 and I-2, both of which are commercially avaliable. The Boc group in I-3 was removed and then condensed with compound 2, which was prepared following literature [30]. Finally, the protecting group was removed under TFA condition and then the AR ligand based OPA derivative 3 was prepared.
Scheme 1.

The synthetic route of the key OPA derivative 3.
Next, we synthesized LG-AR-14-A and LG-AR-14-B, which are the two isomers of AR degrader LG-AR-14. The synthetic routes are outlined in Schemes 2 and 3. Firstly, commercially avaliable compound I-5 was reacted with two compounds I-6 and I-9, respectively, to afford two intermediates I-7 and I-10. These two compounds were then hydrolyzed to give I-8 and I-11. The desired compounds LG-AR-14-A and LG-AR-14-B were prepared by the condensation of I-3 with the aboved two carboxylic acids.
Scheme 2.

The synthetic route of AR PROTAC LG-AR-14-A.
Scheme 3.
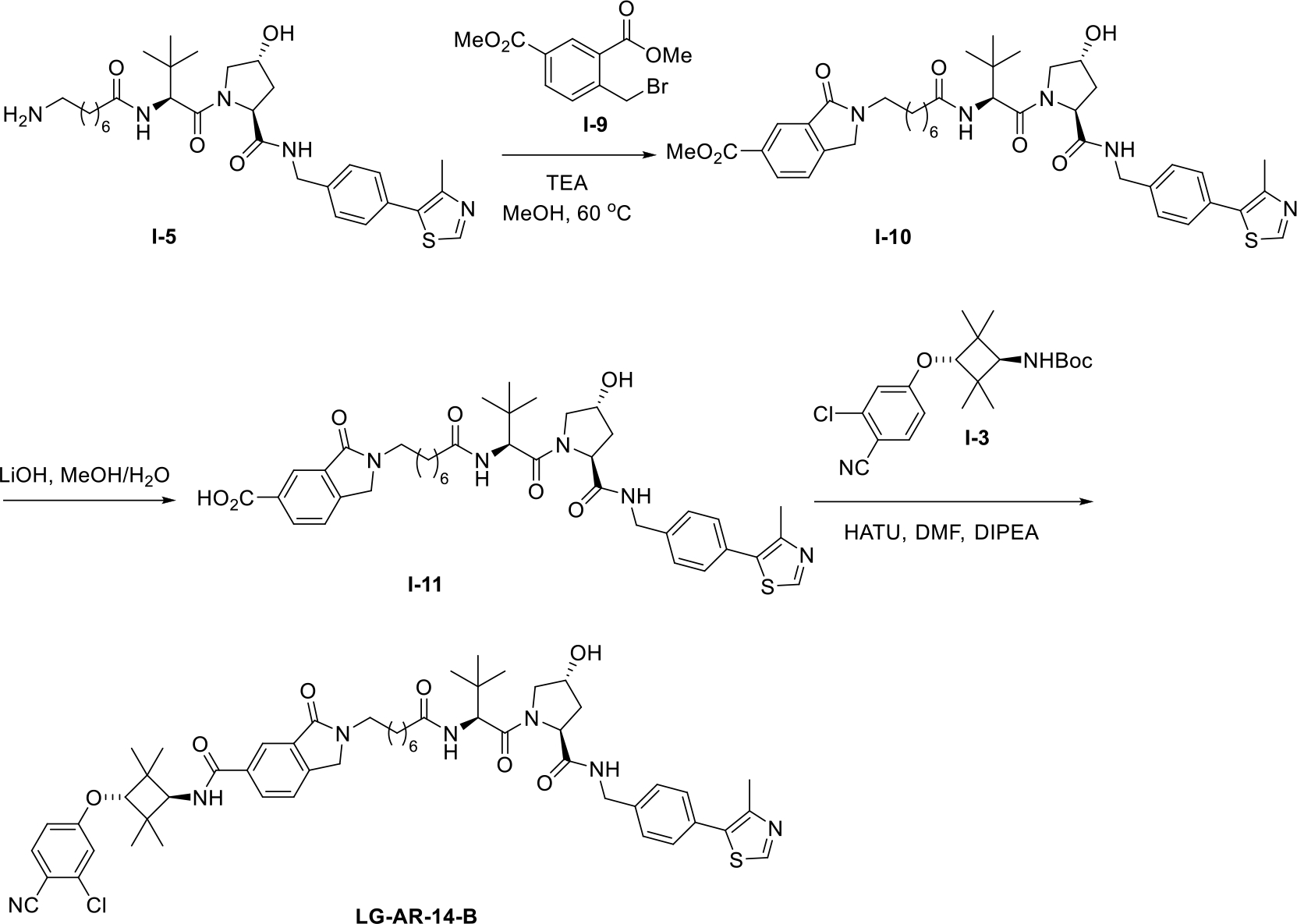
The synthetic route of AR PROTAC LG-AR-14-B.
We also prepared compound LG-AR-14-C with a secondary amide and the synthetic route is shown in Scheme 4. To generate intermediate I-13, compound I-5 was reacted with I-12. After hydrolysis, carboxylic acid I-14 was obtained and then coupled with I-3 to give the AR degrader LG-AR-14-C.
Scheme 4.
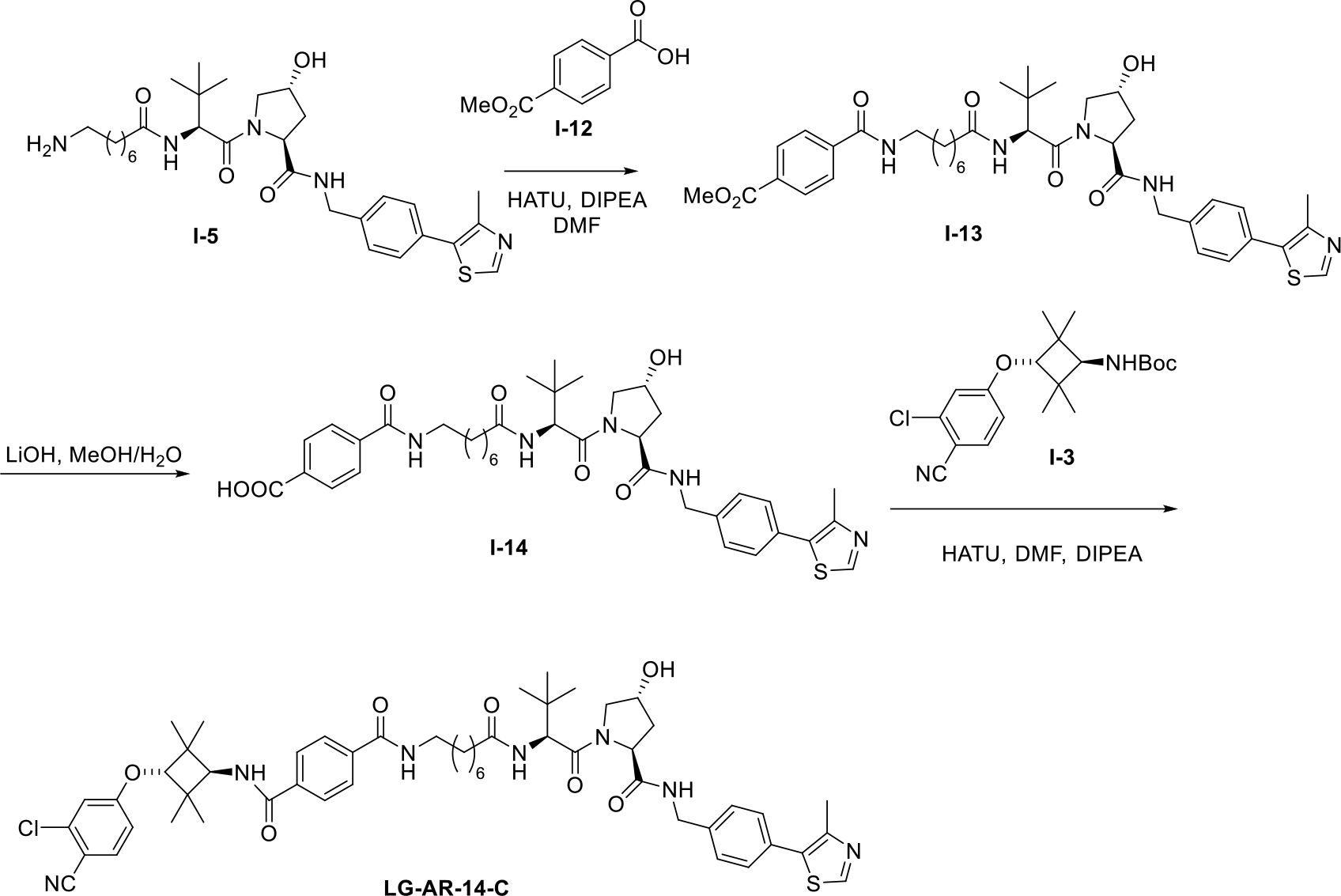
The synthetic route of AR PROTAC LG-AR-14-C.
Furthermore, we prepared BRD4 ligand (JQ1)-OPA derivative 6 and the synthetic route is outlined in Scheme 5. Compound 4, which is the JQ1 primary amide form, was synthesized by the amidation from oxalyl cloride and ammonia. Finally, the OPA derivative 6 was obtained by coupling compound 4 and 5 by the Pd-catalyzed coupling reaction.
Scheme 5.

The synthetic route of key OPA derivative 6.
Next, we synthesized LG-BRD-12A and LG-BRD-12B, which are the two isomers of BRD4 degrader LG-BRD-12. The synthetic routes are outlined in Schemes 6 and 7. Firstly, commercially avaliable compound I-15 was reacted with two compounds I-16 and I-19, respectively, to afford two intermediates I-17 and I-20. These two compounds were then reduced to I-18 and I-21. The desired compounds LG-BRD-12A and LG-BRD-12B were prepared by the amidation of (+)-JQ1-COOH with the aboved two intermediates.
Scheme 6.

The synthetic route of BRD4 PROTAC LG-BRD-12A.
Scheme 7.
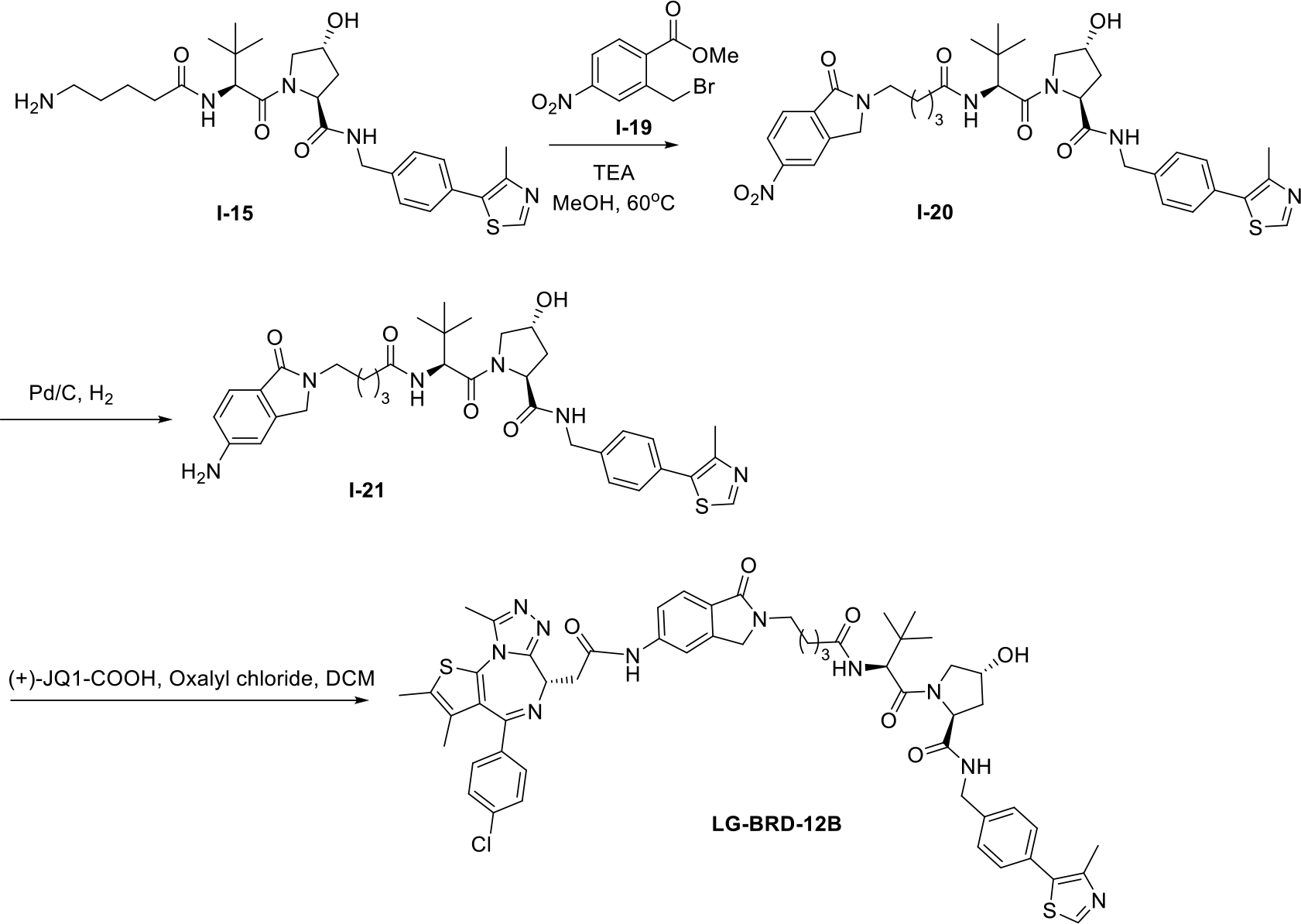
The synthetic route of BRD4 PROTAC LG-BRD-12B.
5. Conclusions
In conclusion, we have developed a novel platform for the rapid synthesis of PROTACs under miniaturized conditions using highly efficient OPA-amine coupling reaction, which produces water as the only byproduct. Using commercially available E3 ligase ligand-linker-NH2 building blocks, this new generation of Rapid-TAC platform can quickly yield a library of stable PROTACs with various linker lengths/types and E3 ligase ligands. The products have sufficient purity and can be directly used for cell-based screening without any further manipulation. The OPA-amine coupling chemistry is compatible with most of the common functional groups. The platform is validated by the quick development of active PROTACs for AR and BRD4. We expect that this new Rapid-TAC platform can be a powerful tool for the development of PROTACs against many other targets. It has the potential to significantly lower the barrier to access PROTAC, a revolutionary technology for small molecule drug discovery, and faciliate the stucture activity relationship studies of PROTACs against various targets.
6. Experimental procedures
6.1. Chemistry General Procedure
Unless otherwise noted, all reagents were purchased from commercial sources and used without further purification. Dry solvents were obtained from a solvent purification system. NMR spectra were recorded on Bruker AV-400 MHz in ppm (δ) downfield of TMS (δ = 0). Signal splitting patterns were described as singlet (s), doublet (d), triplet (t) or multiplet (m), with coupling constants (J) in hertz. High resolution mass spectra (HRMS) were performed by Analytical Instrument Center at the School of Pharmacy on an Electron Spray Injection (ESI) mass spectrometer. The liquid chromatography–mass spectrometry (LC–MS) analysis of final products was processed on an Agilent 1290 Infinity II LC system using a Poroshell 120 EC-C18 column (5 cm × 2.1 mm, 1.9 μm) for chromatographic separation. Agilent 6120 Quadrupole LC/MS with multimode electrospray ionization plus atmospheric pressure chemical ionization was used for detection. The mobile phases were 5.0% methanol and 0.1% formic acid in purified water (A) and 0.1% formic acid in methanol (B). The gradient was held at 5% (0–0.2 min), increased to 100% at 2.5 min, then held at isocratic 100% B for 0.4 min, and then immediately stepped back down to 5% for 0.1 min re-equilibration. The flow rate was set at 0.8 mL/min. The column temperature was set at 40 °C. The purity was determined by high-performance liquid chromatography (HPLC). HPLC was carried out using a Hewlett Packard series 1100 system. Samples were detected using a G1314A VWD detector at two wavelengths (254 nm). Samples were run using an Symmetry C18 3.8 um 4.6×75 mm column and a flow rate of 1.2 mL/min. The eluent consisted of 0.1% formic acid in water (A) and acetonitrile (B) (gradient, from 95% A to 5% A over a period of 7 min or 21 min).
General procedures for the preparation of PROTACs under miniaturized conditions.
Solution X: A 50 mM DMSO stock solution of the OPA building blocks (3 or 6) was prepared by dissolving appropriate amount in DMSO. To this solution was added 10 equivalent of HOAc. Solution Y: A 50 mM DMSO stock solution of each member of the amine-containing partial PROTAC library, which was purchased from either Sigma Aldrich or Tocris Bioscience, was also prepared similarly.
We next mixed 20 μL of DMSO solution X with 20 μL of DMSO solution Y in a vial. The resulting mixture was then heated at 80 °C for 24 h on the reaction block. After cooling it to room temperature, another 60 μL of DMSO was added to the solution. The resulting 10 mM solution of the product was heated at 80 °C for another 24 h. The solution was then cooled down to room temperature and the purity of the product was analyzed by HPLC. Out of the 21 potential AR PROTACs LG-AR-1 to LG-AR-21, we found that the purity ranged from 88% to 98%. Out of the 21 potential BRD4 PROTACs LG-BRD-1 to LG-BRD-21, we found that the purity ranged from 78% to 96%. All Products were directly used for cell-based screening without any further manipulations.
tert-butyl ((1r,3r)-3-(3-chloro-4-cyanophenoxy)-2,2,4,4-tetramethylcyclobutyl)carbamate (I-3)
To a solution of I-2 (243.2 mg, 1 mmol) in DMF was added NaH (28.8, 1.2 mmol) at 0 °C, the reaction mixture was stirred for 20 min. I-1 (243.2 mg, 1 mmol) was added at 0 °C. The reaction mixture was stirred at rt for 4 h. After diluting with ethyl acetate (50 mL) and water (50 mL), the aqueous phase was extracted with ethyl acetate (3 × 50 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the oil was purified by silica column chromatography (eluting with ethyl ether: hexane= 1:4) and the desired fractions were combined and evaporated to yield intermediate I-3 (270 mg, 71%). 1H NMR (400 MHz, CDCl3) δ = 7.53 (d, J=8.7, 1H), 6.92 (d, J=2.4, 1H), 6.76 (dd, J=8.8, 2.4, 1H), 4.73–4.55 (m, 1H), 3.94 (s, 1H), 3.71–3.46 (m, 1H), 1.44 (s, 9H), 1.17 (s, 6H), 1.12 (s, 6H). 13C NMR (101 MHz, CDCl3) δ = 162.8, 155.8, 138.3, 135.1, 116.9, 116.4, 114.2, 105.0, 84.9, 79.6, 59.6, 40.2, 28.4, 23.7, 23.4.
N-((1r,3r)-3-(3-chloro-4-cyanophenoxy)-2,2,4,4-tetramethylcyclobutyl)-3,4-diformylbenzamide (3)
To a solution of I-3 (20 mg, 0.053 mmol) in DCM was added TFA (0.1 mL) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated to give the crude product, which was directly used in the next step. To a solution of 2 (14 mg, 0.063 mmol) in DMF was added HATU (28 mg, 0.074 mmol), DIPEA (27.7 μL, 0.16 mmol) and the above crude product, the reaction mixture was stirred at rt for 4 h. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the oil was purified by silica column chromatography (eluting with ethyl ether: hexane= 1:1) to yield product I-4. To a solution of I-4 in DCM (0.5 mL) was added TFA (0.5 mL), the reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated and the product was purified by reversed-phase HPLC (C18 column, H2O in acetonitrile 20% to 100% over 30 min) to yield product 3 (17.4mg, 75%). 1H NMR (400 MHz, DMSO) δ = 10.53 (s, 0.63H), 10.52 (s, 0.69H), 8.37 (s, 0.8H), 8.31 – 8.17 (m, 1.61H), 8.07 (d, J=7.9, 0.89H), 7.96 – 7.82 (m, 1.77H), 7.47–7.42 (m, 0.24H), 7.22–7.20 (m, 1H), 7.05–6.97 (m, 1H), 6.94–6.77 (m, 0.48H), 6.45–6.38 (m, 0.21H), 6.20–6.11 (m, 0.21H), 4.34 (s, 1H), 4.11 (dd, J=9.2, 4.0, 1H), 1.25 (s, 6H), 1.15 (s, 6H). 13C NMR (101 MHz, DMSO) δ = 193.29, 193.26, 166.49, 163.04, 139.51, 138.42, 137.34, 136.67, 136.56, 133.16, 130.38, 129.83, 117.34, 116.75, 115.16, 104.14, 84.29, 59.15, 58.78, 24.47, 24.45, 23.70, 23.63. HPLC purity: 99% (The mixture of dialdehyde and its additive product with H2O in the HPLC system).
2-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)-1-oxoisoindoline-5-carboxylic acid (I-8)
To a solution of I-5 (182.3 mg, 0.3 mmol) in MeOH was added Triethylamine (166.9 μL, 1.2 mmol) at 0 °C. The reaction mixture was stirred at rt for 5 min. Then I-6 (86.1 mg, 0.3 mmol) was added to the above mixture and the reaction mixture was stirred at 60 °C for 12 h. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with brine and dried over MgSO4. The solvent was evaporated to give the crude product I-7 which was directly used in the next step. To a solution of the above I-7 in MeOH (2 mL) was added LiOH (28.8 mg, 1.2 mmol) in water (0.5 mL). The mixture was stirred at room temperature for 2 h. The solvent was evaporated and acidified by 1N HCl aqueous solution. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with brine and dried over MgSO4. The product was purified by reverse-phase HPLC (C18 column, H2O in acetonitrile 20 to 100% over 30 min) to yield I-8 (75 mg, 34%). 1H NMR (400 MHz, CDCl3/MeOD) δ = 8.82 (s, 1H), 8.20 – 7.95 (m, 2H), 7.73 (d, J=7.8, 1H), 7.65 (s, 1H), 7.42–7.29 (m, 4H), 6.85–6.75 (m, 1H), 4.74 – 4.54 (m, 3H), 4.48 (s, 1H), 4.43 – 4.26 (m, 3H), 4.03 (d, J=11.3, 1H), 3.69 – 3.50 (m, 3H), 2.49 (s, 3H), 2.38 – 2.07 (m, 4H), 1.68–1.47 (m, 4H), 1.37–1.16 (m, 6H), 0.95 (s, 9H). 13C NMR (101 MHz, CDCl3/MeOD) δ = 174.47, 171.60, 171.52, 167.94, 167.89, 151.23, 140.96, 138.68, 136.62, 133.19, 130.08, 129.87, 129.44, 128.07, 124.35, 123.46, 69.84, 59.02, 57.42, 57.33, 57.15, 43.01, 42.66, 36.53, 36.08, 35.26, 28.69, 28.64, 28.14, 26.59, 26.35, 25.37.
N-((1r,3r)-3-(3-chloro-4-cyanophenoxy)-2,2,4,4-tetramethylcyclobutyl)-2-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)-1-oxoisoindoline-5-carboxamide (LG-AR-14-A)
To a solution of I-3 (37.8 mg, 0.1 mmol) in DCM was added TFA (0.5 mL) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated to give the crude product which was directly used in the next step. To a solution of I-8 (73.1 mg, 0.1 mmol) in DMF was added HATU (53.2 mg, 0.14 mmol), DIPEA (52.2 μL, 0.3 mmol) and the above crude intermediate. The reaction mixture was stirred at rt for 4 h. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the oil crude was purified by reverse-phase HPLC (C18, H2O in acetonitrile 20% to 100% over 30 min) to yield product LG-AR-14-A (22.1 mg, 22%). 1H NMR (400 MHz, CDCl3) δ = 9.09 (s, 1H), 7.92 (s, 1H), 7.86 – 7.68 (m, 2H), 7.60–7.51 (m, 2H), 7.46 – 7.31 (m, 4H), 6.99–6.93 (m, 1H), 6.84–6.76 (m, 1H), 6.74–6.65 (m, 1H), 4.79 – 4.29 (m, 7H), 4.23 – 4.00 (m, 3H), 3.75 – 3.50 (m, 3H), 2.53 (s, 3H), 2.40–2.29 (m, 1H), 2.30 – 2.09 (m, 3H), 1.77 – 1.48 (m, 4H), 1.35–1.19 (m, 18H), 0.97 (s, 9H). 13C NMR (101 MHz, CDCl3) δ = 174.89, 171.78, 171.36, 167.98, 167.23, 162.52, 152.22, 145.42, 141.67, 139.36, 138.32, 137.34, 135.46, 135.14, 133.69, 129.42, 128.89, 128.26, 126.34, 123.86, 122.31, 116.78, 116.30, 114.15, 105.16, 84.65, 70.12, 59.14, 58.99, 57.91, 57.11, 50.21, 43.14, 42.67, 40.35, 36.54, 35.91, 35.03, 28.54, 28.43, 28.02, 26.38, 26.35, 25.24, 23.64, 23.50, 14.44. HRMS (ESI): C54H67ClN7O7S [M+H]+; calculated: 992.4511, found: 992.4501. HPLC purity: 99%.
2-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)-3-oxoisoindoline-5-carboxylic acid (I-11)
The procedure for the synthesis compound I-11 is similar to I-8. 1H NMR (400 MHz, CDCl3/MeOD) δ = 8.95 (s, 1H), 8.40 (s, 1H), 8.20–8.15 (m, 1H), 7.75–7.67 (m, 1H), 7.47 (d, J=7.9, 1H), 7.41 – 7.29 (m, 4H), 6.93–6.85 (m, 1H), 4.65 – 4.50 (m, 3H), 4.47 (s, 1H), 4.43 – 4.24 (m, 3H), 3.98 (d, J=11.3, 1H), 3.63–3.59 (m, 1H), 3.55 (t, J=7.2, 2H), 2.47 (s, 3H), 2.30 – 2.08 (m, 4H), 1.66–1.51 (m, 4H), 1.32–1.18 (m, 6H), 0.94 (s, 9H). 13C NMR (101 MHz, CDCl3/MeOD) δ = 174.44, 171.79, 171.71, 171.40, 168.05, 167.84, 151.50, 146.50, 145.60, 138.92, 133.08, 132.84, 130.94, 129.54, 129.35, 128.06, 125.19, 122.87, 69.81, 59.16, 59.11, 57.46, 57.37, 57.10, 43.07, 42.94, 42.50, 36.66, 36.08, 36.03, 35.35, 28.75, 28.64, 28.11, 26.55, 26.30, 25.40, 15.03.
N-((1r,3r)-3-(3-chloro-4-cyanophenoxy)-2,2,4,4-tetramethylcyclobutyl)-2-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)-3-oxoisoindoline-5-carboxamide (LG-AR-14-B)
To a solution of I-3 (37.8 mg, 0.1 mmol) in DCM was added TFA (0.5 mL) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated to give the crude product, which was directly used in the next step. To a solution of I-11 (73.1 mg, 0.1 mmol) in DMF was added HATU (53.2 mg, 0.14 mmol), DIPEA (52.2 μL, 0.3 mmol) and the above crude intermediate. The reaction mixture was stirred at rt for 4 h. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the crude oil was purified by reverse-phase HPLC (C18, H2O in acetonitrile 20% to 100% over 30 min) to yield product LG-AR-14-B (21 mg, 21%). 1H NMR (400 MHz, CDCl3) δ = 9.02 (s, 1H), 8.21 (s, 1H), 8.13 (d, J=7.8, 1H), 7.56 (d, J=8.4, 2H), 7.43–7.30 (m, 5H), 6.98–6.80 (m, 1H), 6.84 – 6.78 (m, 2H), 6.53 (d, J=8.3, 1H), 6.45 (d, J=8.8, 1H), 4.69 (t, J=8.1, 1H), 4.64 – 4.50 (m, 3H), 4.45 (s, 2H), 4.38–4.30 (m, 1H), 4.19 (d, J=8.2, 1H), 4.12–4.03 (m, 2H), 3.68–3.55 (m, 3H), 2.53 (s, 3H), 2.46–2.34 (m, 1H), 2.25–2.10 (m, 3H), 1.71–1.50 (m, 4H), 1.37 – 1.14 (m, 18H), 0.95 (s, 9H). 13C NMR (101 MHz, CDCl3) δ = 174.4, 171.8, 171.1, 168.3, 166.7, 162.6, 151.9, 145.9, 144.4, 139.1, 138.3, 135.1, 134.5, 131.6, 129.5, 128.3, 123.5, 121.1, 116.8, 114.1, 105.1, 84.7, 70.1, 58.9, 57.7, 57.0, 50.3, 43.2, 42.7, 40.4, 36.3, 36.1, 35.1, 28.8, 28.6, 28.1, 26.4, 25.4, 23.7, 23.5, 14.7. HRMS (ESI): C54H67ClN7O7S [M+H]+; calculated: 992.4511, found: 992.4495. HPLC purity: 99%.
methyl 4-((8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)carbamoyl)benzoate (I-13)
To a solution of I-12 (18 mg, 0.1 mmol) in DMF was added HATU (53.2 mg, 0.14 mmol), DIPEA (70 μL, 0.4 mmol) and I-5 (60.8 mg, 0.1 mmol). The reaction mixture was stirred at rt for 4 h. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the oil was purified by silica column chromatography (eluting with DCM: MeOH= 10:1) to yield I-13 (25 mg, 34%). 1H NMR (400 MHz, CDCl3) δ = 8.67 (s, 1H), 8.03 (d, J=8.1, 2H), 7.80 (d, J=8.1, 2H), 7.44 (t, J=5.9, 1H), 7.37–7.28 (m, 4H), 6.76–6.67 (m, 1H), 6.30 (d, J=8.8, 1H), 4.68 (t, J=8.0, 1H), 4.59 – 4.41 (m, 3H), 4.37–4.28 (m, 1H), 4.07–3.99 (m, 1H), 3.91 (s, 3H), 3.63–3.54 (m, 1H), 3.44–3.32 (m, 2H), 2.49 (s, 3H), 2.44–2.35 (m, 1H), 2.22–2.05 (m, 3H), 1.64–1.50 (m, 4H), 1.34 – 1.22 (m, 6H), 0.93 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 173.8, 171.7, 171.0, 166.8, 166.4, 150.4, 138.7, 138.2, 132.5, 130.8, 129.8, 129.5, 128.0, 127.1, 70.0, 58.7, 57.5, 56.8, 55.3, 52.4, 43.3, 43.2, 40.2, 36.3, 36.2, 35.1, 29.7, 29.3, 28.7, 28.6, 26.6, 26.4, 25.2, 18.6, 17.2, 16.0, 12.4.
N1-((1r,3r)-3-(3-chloro-4-cyanophenoxy)-2,2,4,4-tetramethylcyclobutyl)-N4-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctyl)terephthalamide (LG-AR-14-C)
To a solution of the above I-13 (25 mg, 0.034 mmol) in MeOH (2 mL) was added LiOH (3.3 mg, 0.14 mmol) in water (0.1 mL). The mixture was stirred at room temperature for 2 h. The solvent was evaporated and acidified by 1N HCl aqueous solution. The mixture was extracted with ethyl acetate (3 × 10 mL). The solvent was evaporated to give the crude I-14, which was directly used in the next step. To a solution of I-3 (12.9 mg, 0.034 mmol) in DCM was added TFA (0.1 mL) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated to give the crude product, which was directly used in the next step. To a solution of I-14 in DMF was added HATU (18 mg, 0.047 mmol), DIPEA (24 uL, 0.14 mmol) and the above crude intermediate. The reaction mixture was stirred at rt for 4 h. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the crude oil was purified by reverse-phase HPLC (C18, H2O in acetonitrile 20% to 100% over 30 min) to yield LG-AR-14-C (5.4 mg, 16%). 1H NMR (400 MHz, CDCl3) δ = 9.18 (s, 1H), 7.88 – 7.74 (m, 3H), 7.57 (d, J=8.6, 1H), 7.48 – 7.33 (m, 4H), 7.02–6.90 (m, 1H), 6.88–6.77 (m, 2H), 6.39 (d, J=8.1, 1H), 4.73 – 4.52 (m, 3H), 4.41–4.27 (m, 1H), 4.28 – 3.97 (m, 3H), 3.77–3.60 (m, 1H), 3.43 (s, 2H), 2.55 (s, 3H), 2.45–2.11 (m, 4H), 1.74–1.50 (m, 4H), 1.44–1.18 (m, 18H), 0.97 (s, 9H). 13C NMR (101 MHz, CDCl3) δ = 171.4, 167.3, 162.5, 139.4, 138.3, 136.9, 135.1, 129.5, 128.3, 127.5, 127.2, 116.8, 114.1, 105.2, 84.6, 70.8, 59.3, 59.0, 58.3, 57.3, 43.2, 40.3, 35.9, 35.1, 29.1, 28.4, 26.4, 25.1, 23.6, 23.5. HRMS (ESI): C53H67ClN7O7S [M+H]+; calculated: 980.4511, found: 980.4502. LC-MS purity: 99%.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamide (4)
To a solution of (+)-JQ1-CO2H (80 mg, 0.2 mmol) in DCM was added oxalyl chloride (84 μL, 1 mmol) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent was evaporated to give the crude product, which was directly used in the next step. To a solution of the above crude intermediate in DCM was added aq. ammonia solution (2 mL) and then afforded the crude 4 without further purification.
(S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(3,4-diformylphenyl)acetamide (6)
To a solution of 4 (20 mg, 0.05 mmol) in 1,4-dioxane was added 5 (16 mg, 0.06 mmol), Pd(OAc)2 (1.1 mg, 0.005 mmol), Xantphos (2.9 mg, 0.005 mmol) and Cs2CO3 (32.6 mg, 0.1 mmol), The reaction mixture was stirred at 100 °C for 8 h. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with brine and dried over MgSO4. After concentration, the crude oil was dissolved in DCM and added TFA (0.5 mL). The reaction mixture was stirred at rt for 1 h. The solvent and TFA were evaporated and the product was purified by reverse-phase HPLC (C18, H2O in acetonitrile 20% to 100% over 30 min) to yield 6 (11 mg, 20%). 1H NMR (400 MHz, CDCl3) δ = 10.44 (s, 1H), 10.36 (s, 1H), 8.15–8.10 (m, 1H), 8.04–7.97 (m, 1H), 7.85–7.80 (m, 1H), 7.43 (d, J=8.5, 2H), 7.33 (d, J=8.3, 2H), 4.84–4.73 (m, 1H), 4.01–3.92 (m, 1H), 3.81–3.70 (m, 1H), 2.77 (s, 3H), 2.46 (s, 3H), 1.73 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 192.4, 191.2, 169.7, 169.7, 164.6, 155.6, 150.4, 143.8, 143.8, 137.5, 137.3, 136.0, 132.8, 132.8, 131.9, 131.6, 131.3, 131.1, 130.7, 130.0, 128.8, 123.3, 121.4, 54.1, 39.9, 14.5, 13.2, 11.8. LC-MS purity: 99% (The mixture of dialdehyde and its additive product with H2O in the HPLC system).
(2S,4R)-1-((S)-2-(5-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)-1-oxoisoindolin-2-yl)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (LG-BRD-12A)
To a solution of I-15 (158.7 mg, 0.3 mmol) in MeOH was added Triethylamine (166.9 μL, 1.2 mmol) at 0 °C. The reaction mixture was stirred at rt for 5 min. Then I-16 (81.9 mg, 0.3 mmol) was added to the above mixture and the reaction was stirred at 60 °C for 12 h. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were washed with brine and dried over MgSO4. The solvent was evaporated to give the crude product I-17 which was directly used in the next step. I-17 was dissolved in to MeOH and then reduced by H2 with Pd/C. The intermediate I-18 was obtained without further purification and directly used in the next step.
To a solution of (+)-JQ1-CO2H (20 mg, 0.05 mmol) in DCM was added oxalyl chloride (42 μL, 0.5 mmol) at 0 °C. The reaction mixture was stirred at rt for 1 h. The solvent was evaporated to give the crude product. The crude product was then dissolved in DCM and then I-18 (0.05 mmol) and DIPEA were added at 0 °C. The solvent was evaporated and the product was purified by reverse-phase HPLC (C18, H2O in acetonitrile 20% to 100% over 30 min) to yield LG-BRD-12A (14 mg, 27%).1H NMR (500 MHz, DMSO) δ 10.57 (s, 1H), 9.00 (s, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.08 (s, 1H), 7.88 (d, J = 9.3 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.48 (d, J = 8.3 Hz, 2H), 7.45 – 7.36 (m, 6H), 4.64 (t, J = 7.1 Hz, 1H), 4.57–4.50 (m, 1H), 4.45 – 4.39 (m, 4H), 4.37–4.33 (m, 1H), 4.24–4.19 (m, 1H), 3.70 – 3.61 (m, 2H), 3.54–3.49 (m, 4H), 2.62 (s, 3H), 2.44 (s, 3H), 2.42 (s, 3H), 2.33–2.28 (m, 1H), 2.21–2.16 (m, 1H), 2.07 – 1.99 (m, 1H), 1.93–1.89 (m, 1H), 1.63 (s, 3H), 1.59–1.54 (m, 2H), 1.53–1.46 (m, 2H), 0.93 (s, 9H). 13C NMR (126 MHz, DMSO) δ 172.4, 172.3, 170.1, 169.3, 167.6, 163.8, 155.4, 152.0, 150.5, 148.0, 140.0, 139.4, 137.0, 136.7, 135.8, 133.5, 132.7, 131.7, 131.4, 130.6, 130.4, 130.1, 130.0, 129.1, 129.0, 127.9, 124.1, 122.5, 113.3, 69.3, 59.1, 56.8, 56.8, 54.1, 49.4, 42.1, 41.8, 39.0, 38.4, 35.6, 34.8, 27.8, 26.8, 23.2, 16.3, 14.5, 13.1, 11.7. HRMS (ESI): C54H59ClN10NaO6S2 [M+Na]+; calculated: 1065.3647, found: 1065.3642. HPLC purity: 98%.
(2S,4R)-1-((S)-2-(5-(5-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)-1-oxoisoindolin-2-yl)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (LG-BRD-12B)
According to the same synthetic procedure for LG-BRD-12A, compound LG-BRD-12B (7 mg, 14%) was synthesized. 1H NMR (500 MHz, DMSO) δ 10.64 (s, 1H), 8.99 (s, 1H), 8.55 (t, J = 6.1 Hz, 1H), 8.00 (s, 1H), 7.87 (d, J = 9.3 Hz, 1H), 7.61 (d, J = 2.0 Hz, 2H), 7.50 – 7.36 (m, 8H), 4.62 (t, J = 7.1 Hz, 1H), 4.54 (d, J = 9.3 Hz, 1H), 4.45–4.41 (m, 4H), 4.38–3.32 (m, 1H), 4.23–4.18 (m, 1H), 3.69 – 3.46 (m, 6H), 2.61 (s, 3H), 2.44 (s, 3H), 2.42 (s, 3H), 2.33–2.28 (m, 1H), 2.22–2.17 (m, 1H), 2.05–2.01 (m, 1H), 1.91–1.87 (m, 1H), 1.63 (s, 3H), 1.59–1.55 (m, 2H), 1.52–1.46 (m, 2H), 0.93 (s, 9H). 13C NMR (126 MHz, DMSO) δ 172.4, 172.3, 170.1, 169.6, 167.5, 163.8, 158.8, 158.5, 155.4, 151.9, 148.1, 143.4, 142.3, 140.0, 137.1, 135.7, 132.7, 131.3, 130.6, 130.3, 130.0, 129.1, 129.0, 127.9, 127.7, 123.8, 119.0, 113.6, 69.3, 59.1, 56.8, 54.1, 49.7, 42.1, 41.7, 39.1, 38.4, 35.6, 34.8, 27.8, 26.8, 23.2, 16.4, 14.5, 13.1, 11.7. C54H60ClN10O6S2 [M+H]+; calculated: 1043.3827, found: 1043.3834. HPLC purity: 99%.
6.2. Biological Experiments
A. Cell Lines and Materials
LNCap cells were maintained in RPMI supplemented with 10% fetal bovine serum, 1% HEPES, 1% sodium pyruvate and 1% penicillin/streptomycin under 5 % CO2 at 37 °C. MV-4–11 cells were cultured in Iscove’s Modified Dulbecco’s Media (IMDM) supplemented with 10% FBS and 1% penicillin/streptomycin under 5 % CO2 at 37 °C. Androgen receptor (#5153) antibody was obtained from Cell Signaling Technologies. β-Actin (C4) HRP (sc-47778) antibody was obtained from Santa Cruz Biotechnologies. Anti-Brd4 antibody (ab128874) was obtained from Abcam.
B. Western Blotting
Cells were lysed in 1X RIPA lysis buffer containing 25 mM Tris, pH 7−8, 150 mM NaCl, 0.1% (w/v) sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% (v/ v) Triton X-100, Roche protease inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride. Protein samples were adjusted to the equal amount after determining the concentrations by BCA assay and then loaded onto 7.5% SDS−polyacrylamide gel electrophoresis. After transferring, the membrane was first blocked in 5% (w/v) nonfat milk in the TBS-T washing buffer (137 mM NaCl, 20 mM Tris, 0.1% (v/v) Tween) and then incubated with primary antibodies at 4 °C overnight. Next day, the membrane was incubated with secondary HRP-linked antibodies for 1 h followed by acquisition of the immunoblot using ChemiDoc MP Imaging Systems.
C. Cell Viability Assay
LNCap cells were seeded at the density of 2000 cell per well in a 96-well plate. Next day, cells were treated with LG-AR-14-A at indicated concentrations and incubated in regular media for 6 days. Cell viability was measured using AlamarBlue assay by adding 10x AlamarBlue (0.5 mg/mL) to the well and incubated at 37 °C for 2 h. Fluorescent intensity was then determined at 650 nm excitation/680 nm emission and the percentage of cell viability was generated by normalizing to DMSO treated cells.
Supplementary Material
Highlights.
A novel platform for the rapid synthesis of PROTACs under miniaturized conditions.
Libraries of high purity and stable compounds could be prepared quickly.
No further manipulations including purification are necessary.
Active AR and BRD4 degraders were developed using this platform.
Acknowledgements
Support for this research was provided by the University of Wisconsin – Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation through a UW2020 award. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grant R24GM141526.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ, Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. DOI: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Buckley DL, Crews CM, Angew. Chem. Int. Ed. 2014, 53, 2312–2330. DOI: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luh LM, Scheib U, Juenemann K, Wortmann L, Brands M, Cromm PM, Angew. Chem. Int. Ed. 2020, 59, 15448–15466. DOI: 10.1002/anie.202004310;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nalawansha DA, Crews CM, Cell Chem. Biol. 2020, 27, 998–1014. DOI: 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Samarasinghe KTG, Crews CM, Cell Chem. Biol. 2021, 28, 934–951. DOI: 10.1016/j.chembiol.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tomoshige S, Ishikawa M, Angew. Chem. Int. Ed. 2021, 60, 3346–3354. DOI: 10.1002/anie.202004746. [DOI] [PubMed] [Google Scholar]
- [7].Toure M, Crews CM, Angew. Chem. Int. Ed. 2016, 55, 1966–1973. DOI: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- [8].Dang CV, Reddy EP, Shokat KM, Soucek L, Nat. Rev. Cancer 2017, 17, 502–508. DOI: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mullard A, Nat. Rev. Drug Discov. 2021, 20, 247–250. DOI: 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- [10].Papatzimas J, Gorobets E, Brownsey D, Maity R, Bahlis N, Derksen D, Synlett 2017, 28, 2881–2885. DOI: 10.1055/s-0036-1588539. [DOI] [Google Scholar]
- [11].Brownsey DK, Rowley BC, Gorobets E, Gelfand BS, Derksen DJ, Chem. Sci. 2021, 12, 4519–4525. DOI: 10.1039/d0sc05442a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wurz RP, Dellamaggiore K, Dou H, Javier N, Lo M-C, McCarter JD, Mohl D, Sastri C, Lipford JR, Cee VJ, J. Med. Chem. 2018, 61, 453–461. DOI: 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- [13].Liu H, Sun R, Ren C, Qiu X, Yang X, Jiang B, Org. Biomol. Chem. 2021, 19, 166–170. DOI: 10.1039/d0ob02120b. [DOI] [PubMed] [Google Scholar]
- [14].Krajcovicova S, Jorda R, Hendrychova D, Krystof V, Soural M, Chem. Commun. 2019, 55, 929–932. DOI: 10.1039/c8cc08716d. [DOI] [PubMed] [Google Scholar]
- [15].Qiu X, Sun N, Kong Y, Li Y, Yang X, Jiang B, Org. Lett. 2019, 21, 3838–3841. DOI: 10.1021/acs.orglett.9b01326. [DOI] [PubMed] [Google Scholar]
- [16].Bemis TA, La Clair JJ, Burkart MD, Chem. Commun. 2021, 57, 1026–1029. DOI: 10.1039/d0cc05395c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bhela IP, Ranza A, Serafini M, Pirali T, 2021, ChemRxiv preprint. DOI: 10.33774/chemrxiv-2021-440t2-v2. [DOI] [Google Scholar]
- [18].Roberts BL, Ma Z-X, Gao A, Leisten ED, Yin D, Xu W, Tang W, ACS Chem. Biol. 2020, 15, 1487–1496. DOI: 10.1021/acschembio.0c00140. [DOI] [PubMed] [Google Scholar]
- [19].Thiele J, Schneider J, Eur. J. Org. Chem. 1909, 369, 287–299. DOI: 10.1002/jlac.19093690304. [DOI] [Google Scholar]
- [20].Kulla E, Zuman P, Org. Biomol. Chem. 2008, 6, 3771–3780. DOI: 10.1039/b807714m. [DOI] [PubMed] [Google Scholar]
- [21].D’Hollander ACA, Westwood NJ, Tetrahedron 2018, 74, 224–239. DOI: 10.1016/j.tet.2017.11.035. [DOI] [Google Scholar]
- [22].Tung CL, Wong CT, Fung EYM, Li X, Org. Lett. 2016, 18, 2600–2603. DOI: 10.1021/acs.orglett.6b00983. [DOI] [PubMed] [Google Scholar]
- [23].Zhang Y, Zhang Q, Wong CT, Li X, J. Am. Chem. Soc. 2019, 141, 12274–12279. DOI: 10.1021/jacs.9b03623. [DOI] [PubMed] [Google Scholar]
- [24].Zhang Q, Zhang Y, Liu H, Chow HY, Tian R, Eva Fung YM, Li X, Biochemistry 2019, 59, 175–178. DOI: 10.1021/acs.biochem.9b00787. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Q, Zhang Y, Li X, Methods Enzymol. 2020, 639, 237–261. DOI: 10.1016/bs.mie.2020.04.016. [DOI] [PubMed] [Google Scholar]
- [26].Kim Y, Li C-J. Green Synth. Catal. 2020, 1, 1–11. DOI: 10.1016/j.gresc.2020.06.002. [DOI] [Google Scholar]
- [27].Guo C, Linton A, Kephart S, Ornelas M, Pairish M, Gonzalez J, Greasley S, Nagata A, Burke BJ, Edwards M, J. Med. Chem. 2011, 54, 7693–7704. DOI: 10.1021/jm201059s. [DOI] [PubMed] [Google Scholar]
- [28].Han X, Zhao L, Xiang W, Qin C, Miao B, McEachern D, Wang Y, Metwally H, Wang L, Matvekas A, J. Med. Chem. 2021, 64, 12831–12854. DOI: 10.1021/acs.jmedchem.1c00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pettersson M, Crews CM, Drug Discovery Today: Technol. 2019, 31, 15–27. DOI: 10.1016/j.ddtec.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schmidt P, Zhou L, Tishinov K, Zimmermann K, Gillingham D, Angew. Chem. Int. Ed. 2014, 53, 10928–10931. DOI: 10.1002/anie.201406132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.