

SUMMARY
Bone marrow (BM)-mediated trained innate immunity (TII) is a state of heightened immune responsiveness of hematopoietic stem and progenitor cells (HSPC) and their myeloid progeny. We show here that maladaptive BM-mediated TII underlies inflammatory comorbidities, as exemplified by the periodontitis-arthritis axis. Experimental periodontitis-related systemic inflammation in mice induced epigenetic rewiring of HSPC and led to sustained enhancement of production of myeloid cells with enhanced inflammatory preparedness. The periodontitis-induced trained phenotype was transmissible by BM transplantation to naïve recipients, which exhibited increased inflammatory responsiveness and disease severity when subjected to inflammatory arthritis. IL-1 signaling in HSPC was essential for their maladaptive training by periodontitis. Therefore, maladaptive innate immune training of myelopoiesis underlies inflammatory comorbidities and may be pharmacologically targeted to treat them via a holistic approach.
Graphical Abstract

In Brief
Trained innate immune responses contribute to pathology of a comorbid condition, as seen with arthritis after periodontitis in animal models.
INTRODUCTION
Innate immune cells are now appreciated to retain heterologous memory of earlier microbial or inflammatory challenges that enables them to respond stronger upon future challenge with the same or unrelated stimuli. This state of heightened responsiveness, which is based on epigenetic innate immune memory, is designated as ‘trained innate immunity’ (TII) (Netea et al., 2020; Penkov et al., 2019). We and others have shown that systemic inflammatory stimuli can initiate TII in the mouse and human bone marrow (BM) through long-lasting adaptations in HSPC, associated with a long-term myeloid-bias (‘trained myelopoiesis’) (Chavakis et al., 2019; Cirovic et al., 2020; de Laval et al., 2020; Kalafati et al., 2020; Kaufmann et al., 2018; Mitroulis et al., 2018). Experimental studies show that TII can be protective against infections and tumors (Ciarlo et al., 2020; Kalafati et al., 2020; Moorlag et al., 2020a). However, TII is potentially detrimental, hence maladaptive, in chronic inflammatory pathologies (Chavakis et al., 2019).
By expressing Toll-like receptors and receptors for cytokines and growth factors, HSPC sense peripheral infection/inflammation and adapt via expansion and myeloid skewing (Chavakis et al., 2019). Thus, inflammation-adapted or trained HSPC could act as a central hub that perpetuates inflammation through a positive-feedback loop between the BM and peripheral tissues affected by inflammatory disorders (Chavakis et al., 2019), although experimental evidence for this hypothesis is scarce.
Here we explored the concept that maladaptive TII in BM hematopoietic progenitors may provide a mechanistic link for inflammatory comorbidities, such as the enhanced risk of systemic diseases (e.g., cardiometabolic disease and arthritis) in periodontitis patients (D’Aiuto et al., 2018; Hajishengallis and Chavakis, 2021; Potempa et al., 2017). Periodontitis is a prevalent inflammatory disease of the soft and bone tissues that support the dentition and poses a significant public health burden (Hajishengallis, 2015; Kassebaum et al., 2014; Listl et al., 2015; Peres et al., 2019). Conceivably, the low-grade systemic inflammation caused by periodontitis may contribute to the periodontitis-systemic disease connection (Genco and Sanz, 2020; Hajishengallis and Chavakis, 2021). Indeed, compared to healthy controls, patients with severe periodontitis have elevated inflammatory mediators and neutrophil numbers in the blood (D’Aiuto et al., 2013; Schenkein et al., 2020). Conversely, successful local treatment of periodontitis attenuates systemic inflammatory markers (Bajaj et al., 2018; D’Aiuto et al., 2018; Schenkein et al., 2020). The relationship of periodontitis and linked comorbidities is often bidirectional as systemic diseases can promote susceptibility to periodontitis (Genco and Sanz, 2020; Winning and Linden, 2017). However, there is no documented unifying causal mechanism of how periodontitis affects and is affected by comorbidities.
Since many chronic inflammatory diseases are driven by inflammatory myeloid cells, we hypothesized that inflammation-driven alterations in their BM progenitors, as may occur in the context of maladaptive TII (Chavakis et al., 2019; Netea et al., 2020), could influence distinct inflammatory disorders that emerge as comorbidities. We addressed this hypothesis in the context of the periodontitis-arthritis comorbidity that leads to progressive erosion of cartilage and bone in the joints (Smolen et al., 2018). An epidemiological association between these two inflammatory diseases remains even after adjusting for common risk factors (de Pablo et al., 2008; Scher et al., 2014). Our present findings show that maladaptive BM-mediated TII underlies the comorbid connection between periodontitis and arthritis, as modelled by ligature-induced periodontitis (LIP) and collagen antibody-induced arthritis (CAIA) in mice.
RESULTS
LIP induces a sustained increase in myelopoiesis
We first investigated whether experimental periodontitis induces inflammatory adaptation of HSPC in the BM. To this end, mice were subjected, or not, to LIP by ligature placement around the left and right maxillary second molar teeth. After 7 days, LIP-subjected mice displayed significantly higher frequency and numbers of hematopoietic progenitors (LSK; Lin−cKit+Sca1+) and multipotent progenitors (MPP; CD48+CD150−LSK) in the BM, as compared to unligated controls (Figure S1A). No difference was found in long-term (LT)-HSC (CD48−CD150+LSK), whereas the frequency and numbers of short-term (ST)-HSC (CD48−CD150−LSK) were decreased (Figure S1A). The LIP-induced increase in MPP was associated with an increase in the frequency of the myeloid-biased MPP3 subset (Flt3−CD48+CD150−LSK)(Pietras et al., 2015)(Figure S1B). No significant difference was observed in the lymphoid-biased MPP4 (Flt3+CD48+CD150−LSK) or the erythro-megakaryocytic-biased MPP2 subset (Flt3−CD48+CD150+LSK) (Pietras et al., 2015)(Figure S1B). Although the total numbers of LT-HSC remained unaltered (Figure S1A), LIP led to increased frequencies of CD41+ and CD61+ LT-HSC subpopulations (Figure S1C); CD41+ LT-HSC harbor myeloid-biased cells (Gekas and Graf, 2013) and CD61+ LT-HSC are responsive to inflammatory stimuli (Mann et al., 2018).
Although LIP causes substantial bone loss within a few days, maintaining the ligatures for 21 days mimics the chronic phase of human periodontitis. Mice subjected to LIP for 21 days maintained the changes seen at day 7 (Figure S1), such as, increased frequency and numbers of LSK and MPP (Figure 1A). The elevated frequency of myeloid-biased MPP3 was now associated with a decreased frequency of lymphoid-biased MPP4 in LIP-subjected mice (Figure 1B). There was an even more pronounced difference on day 21 (than on day 7) between LIP-subjected and control mice regarding the elevated frequencies of CD41+ and CD61+ LT-HSC in the former group (Figure 1C). Analysis of myeloid progenitors (MyP; Lin−cKit+Sca1−) revealed that LIP led to elevated absolute numbers of granulocyte macrophage progenitors (GMP; Lin−cKit+Sca1−CD16/32+CD34+) and increased proportion of GMP within the MyP population, associated with a corresponding reduction in the relative abundance of common myeloid progenitors (CMP; Lin−cKit+Sca1−CD16/32−CD34+) (Figure 1D). Consistently, LIP-subjected mice exhibited increased proportion of Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells with corresponding reduction in the proportion of CD19+ B cells and CD3+ T cells, compared to the proportions of these populations in control mice (Figure 1E). Hence, chronic LIP leads to sustained enhancement of myelopoiesis.
Figure 1. LIP causes a sustained increase in myelopoiesis.
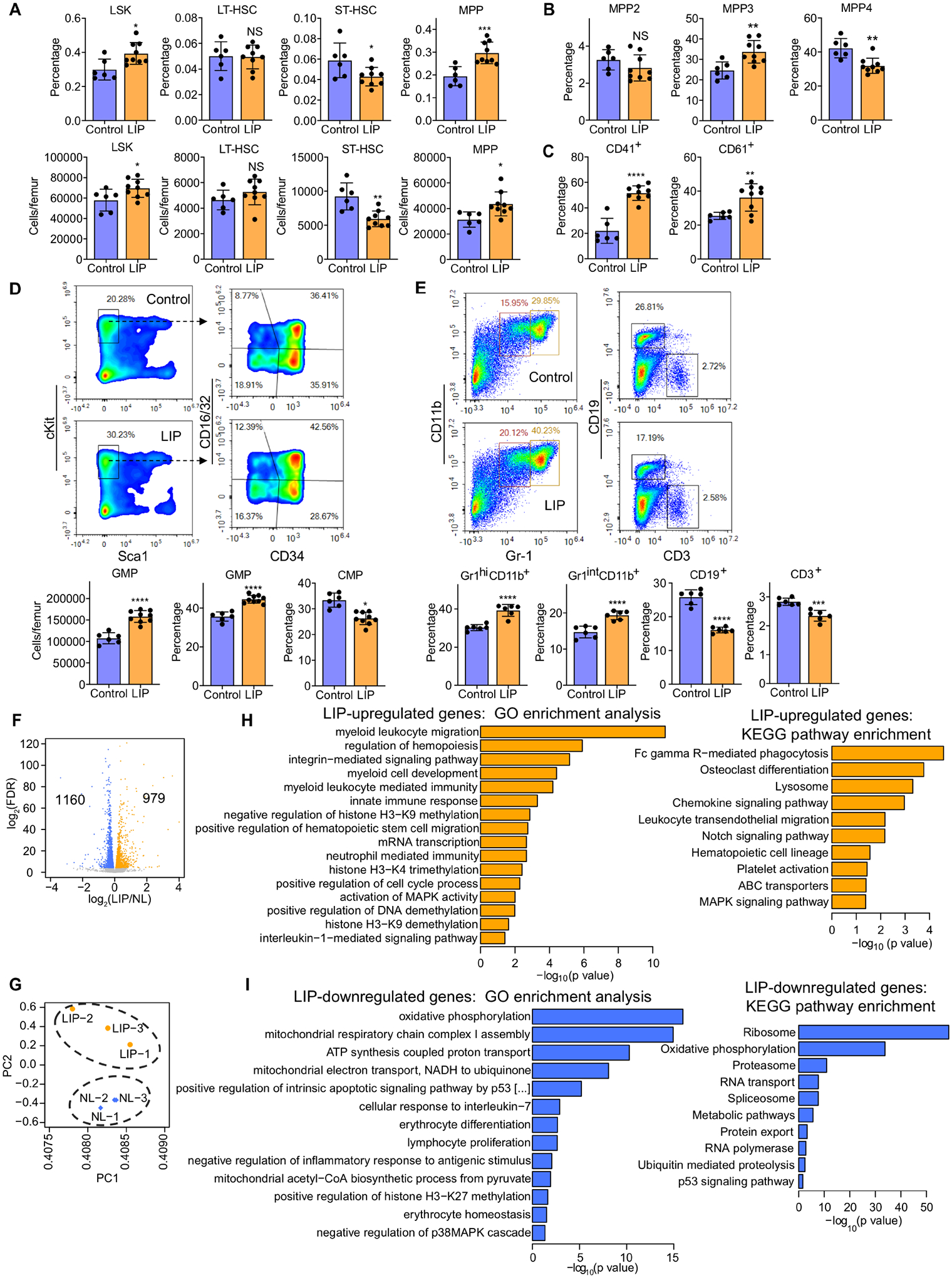
Mice were subjected, or not (control), to LIP followed by BM analysis after 21 days. (A) Frequencies (top) of LSK, LT-HSC, ST-HSC and MPP in total BM cells and absolute cell numbers (bottom) of the same populations. (B) Frequencies of MPP subsets in LSK in the BM of mice. (C) Frequency of CD41+ and CD61+ LT-HSC in total LT-HSC cells in the BM. (D, top) Representative FACS plots for GMP and CMP in the BM; (D, bottom) absolute cell numbers of GMP (left) and frequency within the MyP pool of GMP (middle) and CMP (right) in the BM. (E, top) Representative FACS plots to identify Gr1hiCD11b+ granulocytes, Gr1intCD11b+ myeloid cells, CD19+ B cells and CD3+ T cells and (E, bottom) frequencies of these populations in CD45+ cells in the BM. (F-I) Mice were subjected, or not (control), to LIP and BM cells were harvested on day 7. FACS-sorted LSK were subjected to RNA-sequencing analysis. (F) Differential gene expression in LSK from LIP-subjected mice vs. non-ligated controls. Volcano plot showing the distribution of the adjusted P values (−log2(FDR)) and the fold changes (log2 fold change). Significant changes are shown in blue (down-regulated) or orange (up-regulated) (FDR<0.05), n=3. (G) PCA plot of LSK samples from ligated and non-ligated (NL) groups. Top overrepresented GO terms and KEGG pathways including upregulated (orange) (H) or downregulated (blue) genes (I) in LSK from LIP-subjected mice vs. NL controls. (A-D) Control, n=6 mice/group; LIP, n=9 mice/group and (E) n=6 mice/group. Data are means±SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. control mice; two-tailed Student’s t-test. See Figure S1.
LIP induces transcriptomic changes in hematopoietic progenitors
To study the mechanisms underlying the LIP-induced myeloid-bias in HPSCs, we performed transcriptome analysis of LSK from LIP-subjected and control mice. We identified 2,139 differentially expressed genes of which 979 and 1160 genes were significantly up- or down-regulated, respectively, in LSK from LIP-subjected mice compared to those from control mice (false discovery rate [FDR] <0.05) (Figure 1F). Principal component analysis (PCA) showed clear separation between the two groups (Figure 1G). Gene Ontology (GO) enrichment analysis revealed that several GO terms, including ‘myeloid leukocyte migration’, ‘innate immune response’, ‘neutrophil mediated immunity’, were overrepresented in the significantly up-regulated genes, whereas terms, such as ‘cellular response to interleukin-7’, ‘erythrocyte differentiation’, or ‘lymphocyte proliferation’ were overrepresented in the significantly down-regulated genes (Figure 1H–I). Significant upregulation of myeloid-lineage markers, including Cd68, Csf1r, kit, Ccr2, Csf3r and Mpo (Figure S1D), and downregulation of lymphoid-lineage and erythrocyte markers were observed in LSK of LIP-subjected mice (Figure S1E). Moreover, the GO terms ‘oxidative phosphorylation’ and ‘mitochondrial acetyl-CoA biosynthetic process from pyruvate’ were overrepresented in the significantly down-regulated genes, which was consistent with the KEGG pathway enrichment analysis (Figure 1I). Accordingly, the expression of genes encoding NADH dehydrogenase (e.g., Ndufc2 and Ndufs6) and ATP synthase (e.g., Atp5d and Atp5o) were significantly down-regulated (Figure S1E). The GO term ‘interleukin-1-mediated signaling pathway’ and the KEGG pathway ‘Osteoclast differentiation’ were also significantly overrepresented (Figure 1H and S1). The GO terms ‘negative regulation of histone H3-K9 methylation’ (GO:0051573), ‘histone H3-K4 trimethylation’ (GO:0080182), ‘positive regulation of DNA demethylation’ (GO:1901537) and ‘histone H3-K9 demethylation’ (GO:0033169) were overrepresented in the significantly up-regulated genes, while the GO term ‘positive regulation of histone H3-K27 methylation’ (GO:0061087) was significantly overrepresented in the significantly down-regulated genes (Figure 1H–I). Accordingly, the expression of Kdm1a (Perillo et al., 2008), Kdm3b (Kim et al., 2012) and Pax5 (Johnson et al., 2004) (critical for H3K9 demethylation) as well as Tet3 (Deplus et al., 2013) and Ncoa6 (Qing et al., 2014) (critical for H3K4 methylation) was up-regulated, while the expression of Mtf2 (Perino et al., 2018) and Eed (Margueron et al., 2009) (critical for H3K27 methylation) was down-regulated (Figure S1F).
Ingenuity Pathway Analysis (IPA) for upstream regulators revealed that several myeloid transcription factors were predicted to act as upstream regulators in LSK from LIP-subjected mice compared to those of control mice, including CEBPA, CEBPE, ID1, ID2 and SPI1 (PU.1) (Figure S1G), which mediates IL-1β-induced HSC myeloid differentiation (Pietras et al., 2016). Moreover, IL-1β/IL-1 and growth factors (CSF2 and CSF3) involved in myelopoiesis were also predicted to act on LSK from LIP-subjected mice (Figure S1G). These findings suggest transcriptomic rewiring of BM progenitors associated with inflammatory signaling, such as IL-1.
LIP induces trained innate immunity
We then investigated if hematopoietic progenitors of mice with previous LIP exposure, but without active disease, can elicit enhanced myelopoiesis responses and generate hyper-responsive myeloid cells to a future challenge. Ligature removal abrogates the microbial challenge that drives inflammation, thereby leading to periodontitis resolution (Kourtzelis et al., 2019; Yuh et al., 2020). Mice were thus subjected to LIP for 21 days followed by ligature removal. 14 days later (when local periodontal inflammation was resolved; Figure S2A), analysis of BM myelopoiesis revealed no significant differences between LIP-subjected and control mice (Figure S2B–F), except for slight differences between the two groups regarding the proportion of GMP within the MyP (Figure S2E). Thus, inflammation resolution in the periodontium quantitatively restores phenotypic myelopoiesis, as assessed by flow cytometry, to basal levels.
We next performed scRNA-seq in sorted LSK from mice subjected to 21 days LIP followed by 14 days resolution (‘21dL/14dR’ mice) and naïve controls (rested for 35 days; ‘NL’ mice). Two-dimensional Uniform Manifold Approximation and Projection (UMAP) of 15257 (NL: 7051; 21dL/14dR: 8206) LSK and clustering, partitioned LSK into 8 clusters (C1–C8) (Figure 2A–B). Although most clusters were comparably represented in the 21dL/14dR and NL groups, C7 was modestly enriched with cells from the 21dL/14dR group (Figure 2C). However, C7 comprised a tiny portion of LSK (Figure 2B). GO enrichment analysis of C7 specific markers showed that the GO term ‘leukocyte activation’ and ‘regulation of myeloid cell differentiation’ were in the top 20 significantly enriched GO terms (Figure S2G). There were no significant differences in myeloid marker expression in the overall LSK population from ‘21dL/14dR’ mice compared to those from NL mice (Figure S2H).
Figure 2. LIP-experienced mice display increased myelopoiesis after LPS challenge.
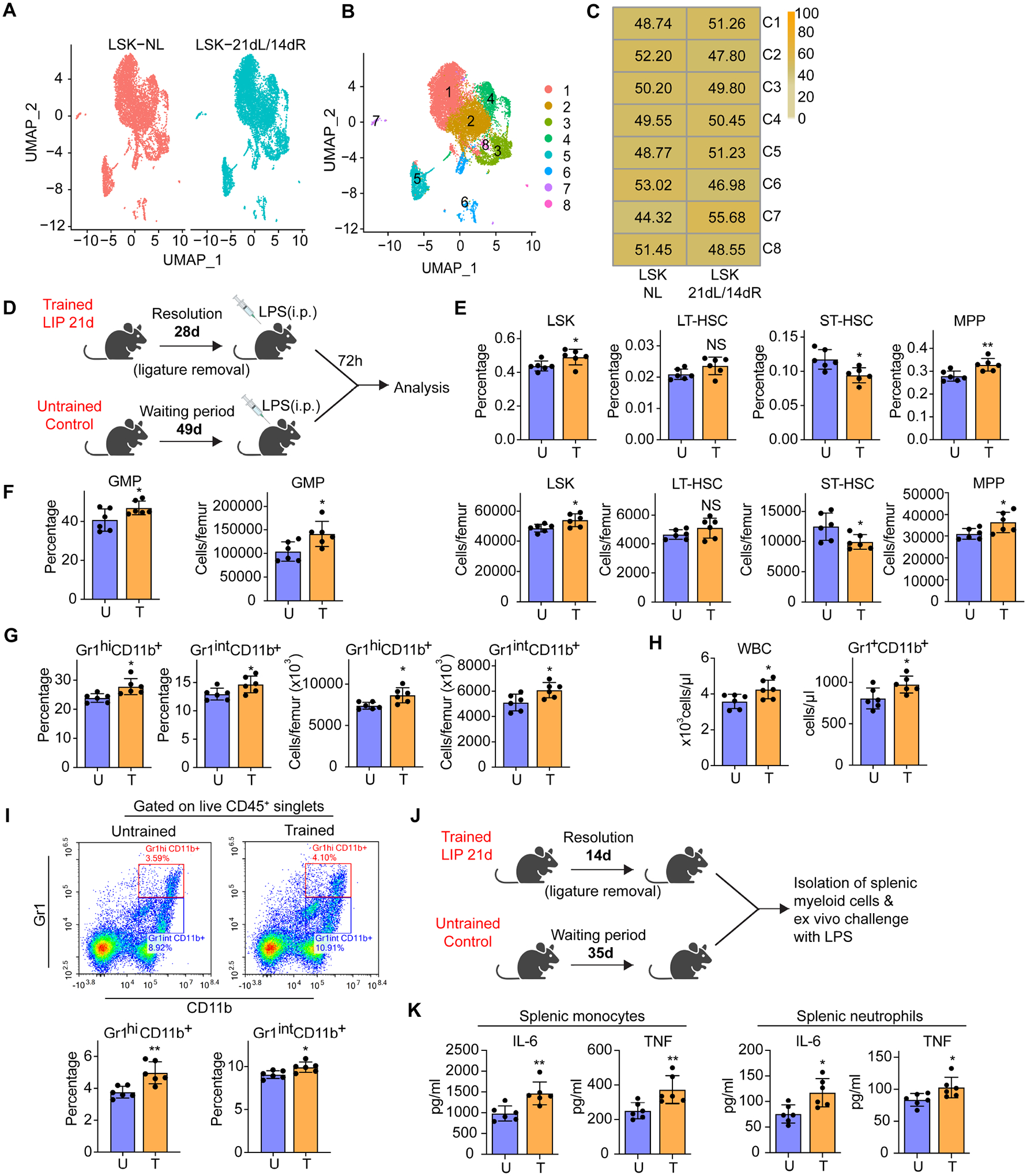
(A-C) BM LSK were sorted from mice subjected to 21dL/14dR or not (NL) and scRNA-seq was performed. (A,B) Two-dimensional UMAP representation of 15257 cells, according to (A) sample origin and (B) results of clustering. (C) Heat map visualization of the distribution of cells within each of the identified clusters, normalized for the number of cells per sample in the dataset. (D) Experimental design. (E) Frequencies (top) of LSK, LT-HSC, ST-HSC and MPP in total BM cells and absolute cell numbers (bottom) of the same populations. (F) Frequency (left) within the MyP pool of GMP and absolute numbers (right) of GMP in the BM. (G) Frequency of Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells in CD45+ cells (left) and cell numbers (right) of the same populations in the BM. (H) Total white blood cell (WBC) count (left) and Gr1+CD11b+ cell counts (right) in the peripheral blood. (I) Representative FACS plots (left) to identify Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells and frequency (right) of the same populations in CD45+ cells in the lungs of mice. (J) Experimental design. (K) Isolated splenic monocytes and neutrophils were re-stimulated ex vivo with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6 and TNF. Data are means±SD (n=6 mice/group) (E-I and K). *P<0.05, **P<0.01, NS, not significant vs. untrained mice; two-tailed Student’s t-test. U, untrained; T, trained. See Figure S2.
We then addressed possible qualitative alterations (associated with trained myelopoiesis) in hematopoietic progenitors of LIP-subjected mice currently under LIP resolution (‘LIP-experienced’ mice). We investigated if the LIP-induced modulation of HSPC could promote a higher response of hematopoietic progenitors to future systemic challenge with LPS (Mitroulis et al., 2017), which would be indicative of inflammatory memory in the BM. To this end, mice, previously subjected to LIP for 21 days followed by ligature removal for 28 days, received a single LPS dose i.p.; BM analysis was performed after 72h (Figure 2D). LIP-experienced mice exhibited a stronger response to LPS than naïve controls, as shown by significantly increased frequency and numbers of the LSK and MPP pools (Figure 2E), GMP (Figure 2F) as well as Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells (Figure 2G). Consistently, the LIP-experienced group showed significantly higher white blood cell counts and Gr1+CD11b+ cells in peripheral blood (Figure 2H). Upon LPS challenge, LIP-experienced mice also exhibited significantly higher frequencies of Gr1hiCD11b+ and Gr1intCD11b+ cells in the lungs (Figure 2I), compared to LPS-challenged controls. Therefore, BM hematopoietic progenitors of LIP-experienced mice maintain a myeloid-differentiation bias and respond with enhanced myelopoiesis to a future systemic challenge. LIP-experienced mice are thereafter referred to as ‘LIP-trained’ mice. Consistent with the notion of LIP-induced inflammatory memory, mature myeloid progeny (spleen-derived neutrophils and monocytes) of LIP-trained mice responded with higher IL-6 and TNF production to secondary (ex vivo) LPS challenge than their counterparts from naïve controls (Figures 2J–K).
LIP induces epigenetic rewiring of BM progenitors
The ability of hematopoietic progenitors and mature myeloid cells from LIP-trained mice to respond stronger to future LPS challenges (Figure 2 D–K) suggested emergence of an epigenetically-based innate immune memory. To establish LIP-induced innate immune memory, we tested if LIP can indeed induce epigenetic rewiring of BM progenitors.
We thus performed single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) analysis of hematopoietic progenitors LSK and GMP in mice that were trained by subjecting them to LIP for 21 days followed by ligature removal for 14 days (‘21dL/14dR’ mice) or were not ligated during the entire period (‘NL’ controls) (Figure 3). Two-dimensional UMAP of 12,758 (NL: 5,341; 21dL/14dR: 7,417) LSK and 25,145 (NL: 12,944; 21dL/14dR: 12,201) GMP (Figure 3A) and clustering, partitioned LSK and GMP into 15 clusters (C1–C15) (Figure 3B). C2, C7, C8, C10 and C12-C14 comprised LSK; C1, C3-C6 comprised GMP, and C9, C11 and C15 included both LSK and GMP (Figure 3C). The LSK cluster C2 displayed enhanced chromatin accessibility in genes indicating myeloid differentiation, Itga2b (Cd41) and Gata2 (Figure S3A). The GMP cluster C3 showed enhanced chromatin accessibility in genes with an inflammatory signature: Csf1r, encodes CD115 and promotes myelopoiesis; Nlrp3, encodes NLR family pyrin domain containing 3 (NLRP3) and acts as an inflammasome component contributing to IL-1β secretion (Figure S3B).
Figure 3. Epigenetic rewiring of trained LSK and GMP.
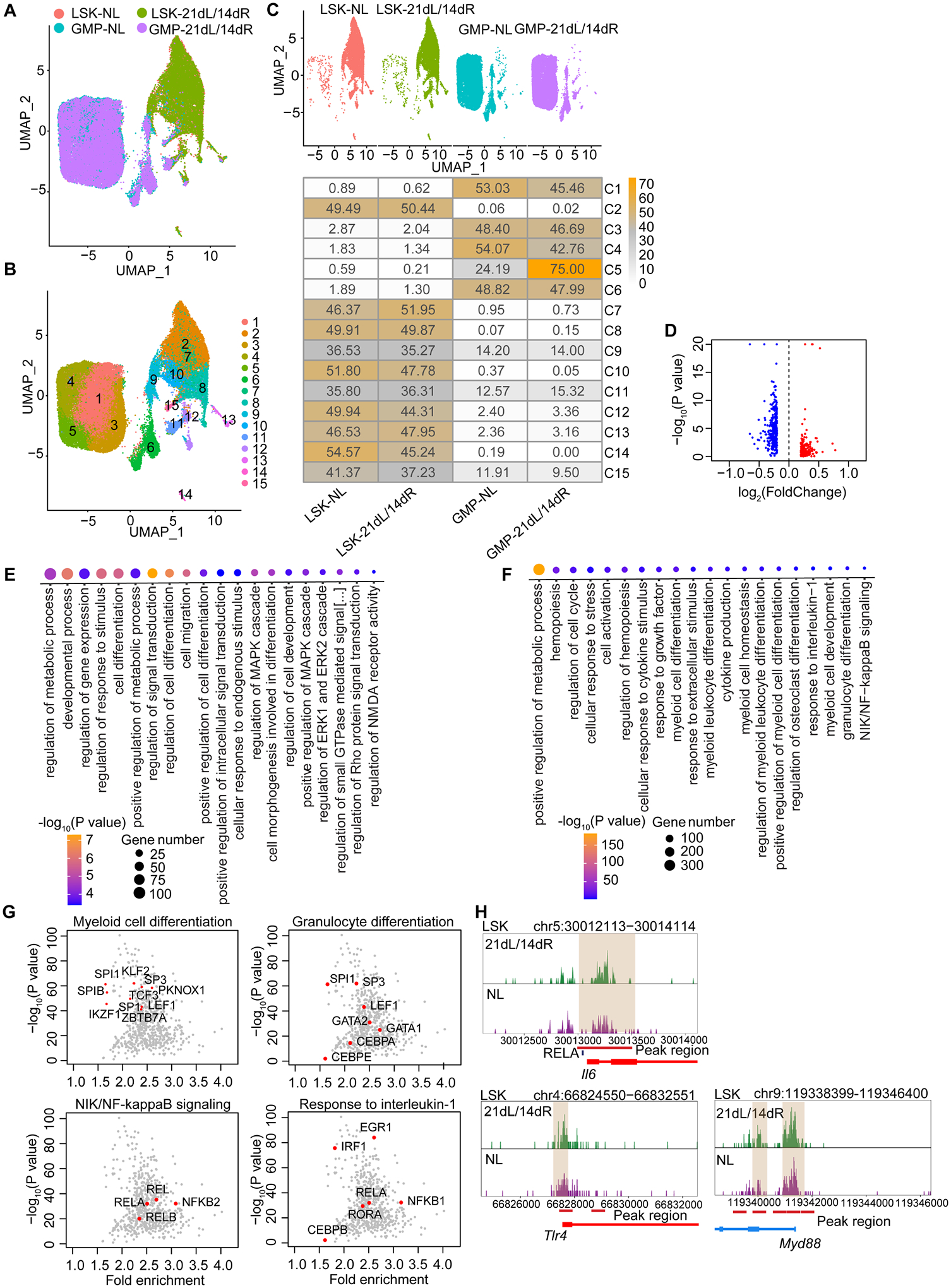
BM LSK and GMP were sorted from mice subjected to 21dL/14dR or not (NL) and scATAC-seq was performed. (A, B) Two-dimensional UMAP representation of 37,903 cells, according to (A) sample origin and (B) results of clustering. (C) UMAP (up) of the distribution of cells from the four different samples (LSK and GMP subjected to 21dL/14dR or not) and heat map visualization (bottom) within each of the identified clusters, normalized for the number of cells per sample in the dataset are shown. (D) Volcano plots displaying differential accessibility analysis results (blue, lower vs. red, greater differential chromatin accessibility) for LSK subjected to 21dL/14dR vs. their counterparts from control mice (abs(Log2FC) ≥ 0.2, P<0.05). (E) GO enrichment results of top 20 significantly enriched GO terms sorted by PANTHER based on genes annotated to regions more accessible due to 21dL/14dR treatment (Bonferroni-corrected, P<0.05). (F) GO enrichment analysis of enriched TF binding motifs in LSK subjected to 21dL/14dR vs. control (NL) group and the top 20 significantly enriched GO terms (Bonferroni-corrected, P<0.05). (G) Visualization of top enriched TF binding motifs in the indicated GO terms for the LIP specifically accessible regions in LSK using the homer TF motif database. (H) Genome browser track showing DAR in the Il6 gene locus and the RELA binding motifs within this region, as well as DARs close to the promoter regions of Tlr4 and Myd88 gene locus. See Figure S3.
In line with a LIP-induced increased myeloid bias in LSK (Figures 1 and S1), differential accessibility analysis revealed differentially accessible regions (DARs) in LSK from ‘21dL/14dR’-trained mice as compared with LSK from ‘NL’ mice (Figure 3D). The top 20 significantly enriched GO terms in LSK, identified on the basis of genes annotated to regions more accessible due to LIP-induced training, revealed terms such as ‘regulation of cell differentiation’, ‘regulation of metabolic process’ and ‘cell migration’ (Figure 3E), consistent with the LSK RNA-seq results (Figure 1H) and with previous findings implicating metabolic reprogramming in TII (Hajishengallis et al., 2021). GO enrichment analysis of the enriched transcription factor motifs in LSK revealed that the GO terms ‘Myeloid cell differentiation’, ‘Granulocyte differentiation’, ‘NIK/NF-kappaB signaling’, ‘Response to interleukin-1’ were significantly enriched by LIP-induced TII (Figure 3F). The top enriched transcription factor motifs in the GO term ‘Myeloid cell differentiation’ includes SPI1 (PU.1), KLF2 (Kruppel-like transcription factor 2, a potent regulator of myeloid cell activation) (Mahabeleshwar et al., 2011) and the GO term ‘Granulocyte differentiation’ includes the transcription factor motifs CEBPA, CEBPE, SP3, SPI1 and GATA2 (Figure 3G). The GO term ‘NIK/NF-kappaB signaling’ includes REL, RELA, RELB and NFKB2, indicating that the NF-κB binding motifs are more accessible in LSK from ‘21dL/14dR’ mice than in their counterparts from NL mice (Figure 3G); in the GO term ‘Response to interleukin-1’, the enrichment of transcription factor motifs IRF1, CEBPB and EGR1 implied the activation of IL-1 signaling pathway (Figure 3G), consistent with the transcriptomic analysis of LSK (Figure 1 F–I and Figure S1 D–G). A genome browser track displays a DAR in the Il6 gene locus and the RELA binding motifs within this region (Figure 3H, top), as well as DARs near the promoter regions of Tlr4 and Myd88 gene locus in LSK from ‘21dL/14dR’ (trained) and control (NL) mice (Figure 3H, bottom).
Differentiation trajectories analysis showed that C4 and C5 existed at the end of the pseudotime trajectories from LSK to GMP (Figure S3C), suggesting that they consist of the most differentiated myeloid precursors within GMP, in which C5 was significantly enriched by LIP-induced TII (Figure 3C). GO enrichment analysis of C5-specific markers revealed that the GO terms ‘Myeloid cell differentiation’, ‘Positive regulation of ROS metabolic process’, ‘Regulation of phagocytosis’ and ‘Positive regulation of MAPK cascade’ were significantly enriched by LIP-induced TII (Figure S3D). C5 also showed enhanced chromatin accessibility in Tlr4, as well as Camk1d, which encodes calcium/calmodulin-dependent protein kinase 1D that promotes ROS production and phagocytosis by granulocytes (Verploegen et al., 2005); Itga1, encoding CD49a, which promotes neutrophil migration (Ridger et al., 2001); Map2k4, encoding MAP2K4, which phosphorylates MAP kinases MAPK8/JNK1, MAPK9/JNK2, and MAPK14/p38 in response to environmental stresses or stimuli (Figure S3E).
Therefore, scATAC-seq suggests that LIP-induced training is associated with a sustained myeloid lineage bias in LSK, allowing for an enhanced myelopoiesis response upon secondary stimulation, as well as pro-inflammatory signatures both in LSK and GMP. Moreover, the results of the scATAC-seq analysis reveal that, upon LIP resolution, HSPC retain an epigenetic myeloid-differentiation bias.
LIP-induced trained myelopoiesis underlies the periodontitis-arthritis comorbidity
We next determined whether the above-documented inflammatory memory renders LIP-trained mice more susceptible to arthritis, as modelled by CAIA (Khachigian, 2006). To this end, mice subjected to LIP for 21 days followed by 14 days of ligature removal were challenged with CAIA. Mice without previous exposure to LIP were also challenged with CAIA and served as controls (Figure S4A). LIP-trained mice were more susceptible to CAIA than untrained controls, as evidenced by significantly higher clinical arthritis score and ankle joint thickness (Figure S4B). Whether this increased susceptibility to arthritis is mediated by an inflammatory LIP-BM axis was addressed by BM transplantation (BMT) experiments from LIP-trained mice to naïve recipients.
To this end, BM cells were isolated from CD45.2+ mice, which were either trained by subjecting them to 21-day LIP followed by 14-day resolution (‘21dL/14dR’ mice), or were left untrained, and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. 12 weeks post-BMT, groups of recipient CD45.1+ mice were subjected to LIP (for 5 days) or CAIA (14 days) (Figure 4A). CD45.1+ mice transplanted with BM cells from ‘21dL/14dR’ mice developed increased bone loss (Figure 4B) and inflammatory gene expression (Figure 4C), as well as higher abundance of monocytes and neutrophils and total CD45+ immune cells in the gingiva (Figure 4D), compared to CD45.1+ mice that received BM cells from untrained CD45.2+ mice. Similarly, upon induction of CAIA in CD45.1+ recipient mice, we observed more severe arthritis in mice transplanted with BM cells from 21dL/14dR mice than in mice transplanted with BM cells from untrained controls, as shown by increased clinical arthritis score and ankle joint thickness (Figure 4E) and aggravated histopathology in ankle joints (Figure 4F). Moreover, compared to the control group, CD45.1+ mice receiving BM cells from ‘21dL/14dR’ CD45.2+ mice showed higher abundance of monocytes and neutrophils and total CD45+ leukocytes in the synovium (Figure 4G). Mature splenic neutrophils and monocytes from CD45.1+ mice receiving BM cells from 21dL/14dR CD45.2+ donor mice responded with significantly higher secretion of IL-6 and TNF to secondary LPS challenge than their counterparts from CD45.1+ mice receiving BM cells from untrained CD45.2+ donors (Figures S4C–D). Thus, LIP-induced maladaptive TII confers increased susceptibility to not only periodontitis but also a comorbid condition, namely arthritis.
Figure 4. LIP-induced trained myelopoiesis contributes to the periodontitis-arthritis comorbidity.

(A) CD45.2+ mice were either trained (T) by LIP for 21 days followed by 14-day resolution or left untrained (U). BM cells were isolated from trained and untrained mice and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. At 12 weeks post-BMT, groups of recipient CD45.1+ mice were subjected to LIP for 5 days (B-D) or CAIA for 14 days (E-G). (B) Bone loss, (C) relative gingival mRNA expression of indicated cytokines and (D) FACS analysis of gingival monocytes (live CD45+CD11c−CD11b+Ly6G−Ly6C+), neutrophils (live CD45+CD11c−CD11b+Ly6G+Ly6C−) and total CD45+ cells in recipient CD45.1+ mice subjected to 5-day LIP. (E) Clinical arthritis score (left) and hind ankle joint thickness (right), (F) representative images of H&E (left) and Safranin-O staining (right) of tissue sections from knee joints harvested on day 7 (scale bars 500μm) and (G) quantification of monocytes (live CD45+CD11c−CD11b+Ly6G−Ly6C+), neutrophils (live CD45+CD11c−CD11b+Ly6G+Ly6C−) and total CD45+ cells in the synovium of knee joints harvested on day 7. (H) 100 LT-HSC sorted from CD45.2+ mice subjected, or not (control), to LIP for 21 days followed by 14-day resolution– were co-transplanted with 5×105 CD45.1+ BM competitors. (I) WBC count, donor-derived percentage, and lineage output (% of indicated cell types in donor-derived cells) in peripheral blood of mice receiving CD45.2+ LT-HSC. Data are means±SD (B and C, n=5 mice/group; D, G and I, n=6 mice/group; E n=7 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice. Two-tailed Student’s t-test (B,C,D,G) except for Rankl in panel C (two-tailed Mann-Whitney U test); two-way repeated measures ANOVA and Sidak’s multiple comparisons test (E,I). W, weeks. See Figure S4.
To exclude the possibility that the differences in the abundance of myeloid cells infiltrating the synovium of recipient groups were due to potential differences in the reconstitution potential of HSPC from the ‘21dL/14dR’ donors vs. those from untrained donors, we performed analysis of BM and peripheral chimerism in recipient mice. Using the CD45.1/CD45.2 congenic system (Figure 4H), 100 LT-HSC isolated from ‘21dL/14dR’ or control CD45.2+ mice were transplanted together with 5×105 CD45.1+ competitors to lethally irradiated C57BL/6.SJL CD45.1+ mice. At 16 weeks post-BMT, no differences were observed in the frequencies of HSPC derived from ‘21dL/14dR’ or control donors (Figures S4E–F). Peripheral blood analysis at 4–16 weeks post-BMT showed no difference in white blood cell counts and in the percentages of ‘21dL/14dR’ or control donor-derived cells (Figure 4I). However, LT-HSC from ‘21dL/14dR’ mice gave rise to elevated proportion of Gr1+CD11b+ myeloid cells with corresponding decrease in the proportion of CD19+ B cells and CD3+ T cells, as compared to LT-HSC from control mice (Figure 4I). Thus, although there was no difference in the overall reconstitution potential, there was an inherent myeloid-differentiation bias of the LT-HSC from the ‘21dL/14dR’ group (vs. those of controls), resulting from LIP-induced trained myelopoiesis in donor mice.
CAIA-induced trained myelopoiesis underlies the arthritis-periodontitis comorbidity
To demonstrate the bidirectional association of periodontitis with arthritis, as suggested by observations in humans (Lee and Choi, 2020; Mikuls et al., 2014; Potempa et al., 2017; Rodríguez-Lozano et al., 2019), we tested whether CAIA causes maladaptive training of HSPC leading to enhanced susceptibility to periodontitis. We first showed that CAIA significantly increased the frequency and numbers of LSK, LT-HSC and MPP (vs. controls; Figure S5A), as well as the frequency of myeloid-biased MPP3 (Figure S5B) and CD41+ LT-HSC (Figure S5C). These changes were associated with decreased frequency of erythro-megakaryocytic-biased MPP2 and lymphoid-biased MPP4 (Figure S5B), thus signifying a myelopoiesis bias. Indeed, CAIA also resulted in increased numbers and frequency of GMP (Figure S5D) and of Gr1hiCD11b+ granulocytes in the BM (Figure S5E).
We next performed BMT experiments to test if CAIA-induced trained myelopoiesis underlies the comorbid connection between arthritis and periodontitis. To this end, BM cells were isolated from CD45.2+ mice 36 days after CAIA. This interval was selected based on observations that 22 days were required for resolution of arthritis (Figure S5F) and another 14 days were required for phenotypic restoration of myelopoiesis to steady-state levels (Figure S5G). BM cells from CAIA-experienced or untreated control CD45.2+ mice were transferred to lethally irradiated CD45.1+ recipients. 12 weeks post-BMT, groups of CD45.1+ recipient mice were subjected to LIP or CAIA (Figure 5A). CD45.1+ mice receiving BM cells from CAIA-subjected CD45.2+ mice developed increased periodontal bone loss (Figure 5B) and inflammatory gene expression (Figure 5C) and higher gingival abundance of monocytes, neutrophils and total CD45+ immune cells (Figure 5D), compared to CD45.1+ mice receiving BM cells from control CD45.2+ mice. Moreover, mice that received BM cells from CAIA-trained mice also developed more severe arthritis (Figure 5E), increased infiltration of monocytes, neutrophils and total leukocytes in the joints (Figure 5F), and aggravated histopathology of ankle joints (Figure 5G), compared to the recipients of BM cells from untrained controls. Mature splenic neutrophils and monocytes from CD45.1+ mice receiving BM cells from CAIA-trained CD45.2+ mice responded with significantly higher production of IL-6 and TNF to secondary LPS challenge than their counterparts from CD45.1+ mice receiving BM cells from untrained CD45.2+ mice (Figures S5H–I). Therefore, LIP-induced (or CAIA-induced) inflammatory maladaptive training of BM progenitors confers, in a bidirectional fashion, increased susceptibility to inflammatory comorbid conditions, namely periodontitis and arthritis.
Figure 5. CAIA– or LIP–induced trained myelopoiesis contributes to the bidirectional arthritis-periodontitis comorbidity.

(A) CD45.2+ mice were either trained (T) by subjecting them to CAIA as indicated, or were left untrained (U; naïve controls). BM cells were isolated from trained and untrained mice and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. At 12 weeks post-BMT, groups of recipient CD45.1+ mice were subjected to LIP for 5 days (B-D) or CAIA for 14 days (E-G). (B) Bone loss, (C) relative gingival mRNA expression of indicated cytokines and (D) FACS analysis of gingival monocytes, neutrophils and total CD45+ cells in recipient CD45.1+ mice subjected to 5-day LIP. (E) Clinical arthritis score (left) and hind ankle joint thickness (right), (F) quantification of monocytes, neutrophils and total CD45+ cells in the synovium of knee joints harvested on day 7, and (G) representative images of H&E (left) and Safranin-O staining (right) of tissue sections from knee joints harvested on day 7 (scale bars 500μm). (H) CD45.1+ mice were transplanted with 200 CD45.2+ LT-HSC from LIP-trained or untrained control mice together with 8×105 CD45.1+ BM competitor cells (left). Frequency of donor-derived cells in the BM of recipient mice (chimerism) 12 weeks post-BMT (right). (I-L) 12 weeks post-BMT, additional recipient CD45.1+ mice were subjected to CAIA. (I) Clinical arthritis score (left) and hind ankle joint thickness (right). (J) Quantification of total monocytes, neutrophils, and CD45+ leukocytes and (K) percentages of CD45.2+ cells in monocytes, neutrophils and CD45+ cells (top panel) and absolute numbers of CD45.2+ monocytes, neutrophils and total CD45.2+ cells (bottom panel) in the synovium of knee joints harvested on day 7. (L) Isolated splenic monocytes and neutrophils were stimulated with LPS (10 ng/ml) for 24h and IL-6 and TNF concentration in the supernatant was measured. Data are means±SD (B-D, F, I-K n=6 mice/group; E, n=7 mice/group; H right, L, n=4 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice. Two-tailed Student’s t-test (B-D,F,H,J-L); two-way repeated measures ANOVA and Sidak’s multiple comparisons test (E,I). See Figures S5 and S6.
Maladaptively trained LT-HSC link comorbid inflammatory conditions
To obtain evidence that maladaptive TII is initiated at the level of LT-HSC, we performed competitive BMT with the CD45.1/CD45.2 congenic system and sorted LT-HSC from LIP (‘21dL/14dR’)-trained mice and untrained donors. Chimerism analysis in the BM at 12 weeks post-BMT showed no significant differences in the frequencies of LIP-trained or untrained control donor-derived cells (Figure 5H). 12 weeks post-BMT, additional recipient CD45.1+ mice were subjected to CAIA. Compared to mice transplanted with LT-HSC from untrained CD45.2+ mice (‘untrained’ LT-HSC), mice receiving LT-HSC from trained CD45.2+ mice (‘trained’ LT-HSC) exhibited more severe arthritis (Figure 5I), higher abundance of monocytes, neutrophils and total CD45+ leukocytes in the synovium (Figure 5J) as well as increased percentage and counts of CD45.2+ monocytes, neutrophils and leukocytes (Figure 5K). Mature splenic neutrophils and monocytes from mice that received trained LT-HSC responded with significantly higher secretion of IL-6 and TNF to secondary LPS challenge than their counterparts from mice transplanted with untrained LT-HSC (Figure 5L). Thus, periodontitis-trained LT-HSC lead to exacerbated inflammation and disease in transplanted mice in the context of a comorbid condition.
To determine if LIP-induced maladaptive TII was restricted to myeloid-biased LT-HSC, we performed a similar competitive BMT using sorted CD41+ (myeloid-biased) or CD41− (non-biased) LT-HSC from LIP-trained or untrained CD45.2+ mice (Figure S6A). Peripheral blood analysis in mice receiving CD41+ LT-HSC showed no difference in white blood cell counts and percentage of LIP-trained or untrained donor-derived cells; however, LT-HSC from LIP-trained mice gave rise to elevated proportion of Gr1+CD11b+ myeloid cells with corresponding decrease in the proportion of CD19+ B cells and CD3+ T cells, relative to LT-HSC from control donors (Figure S6B). No differences were seen in the frequencies of HSPC in the BM derived from LIP-trained or control donors (Figure S6D), suggesting comparable engraftment. Similar observations were made in mice transplanted with CD41− LT-HSC (Figure S6 C,E). These findings reveal an inherent myeloid bias in both CD41+ and CD41− LT-HSC from LIP-trained donors leading to higher myelopoiesis in recipient mice. Consistently, when recipient mice were subjected to CAIA, those transplanted with trained LT-HSC, regardless of CD41 phenotype, had exacerbated arthritis (Figure S6F–G) and joint inflammation (Figure S6H–I). Mature splenic myeloid cells from mice that received trained CD41+ or CD41− LT-HSC responded with higher secretion of IL-6 and TNF to secondary LPS challenge than their counterparts from mice transplanted with untrained LT-HSC (Figures S6J,K). Thus, LIP can induce myeloid-differentiation bias and a maladaptive trained phenotype even in LT-HSC that are not originally myeloid biased, at least phenotypically, based on CD41 expression.
IL-1-signaling in HSPC mediates LIP-induced maladaptive training of myelopoiesis
To better understand how LIP modulates HSPC for enhanced myelopoiesis, we analyzed the BM extracellular fluid for cytokines implicated in the inflammatory modulation of HSPC (Chavakis et al., 2019), collected 14 days post-LIP (between 7 and 21 days when significant changes in HSPC modulation have occurred; Figures 1 and S1). We detected significantly increased concentrations of IL-1β and G-CSF (but not of other cytokines tested) in the BM extracellular fluid in LIP-subjected mice relative to controls (Figure 6A). Parallel analysis of the same cytokines in the serum of LIP-subjected mice indicated elevated concentration of G-CSF but not of IL-1β relative to controls (Figure 6B). Analysis of cytokines upregulated in periodontitis (Dutzan et al., 2018; Shin et al., 2015) revealed high abundance of IL-1β and G-CSF in the periodontal tissues of LIP-subjected mice, although the local induction of G-CSF (6.8-fold vs. control) was more pronounced than that of IL-1β (2.3-fold vs. control) (Figure 6C). These data suggested that LIP-induced serum G-CSF might access the BM, where it might induce IL-1β production.
Figure 6. Analysis of proinflammatory mediators in LIP-trained BM.
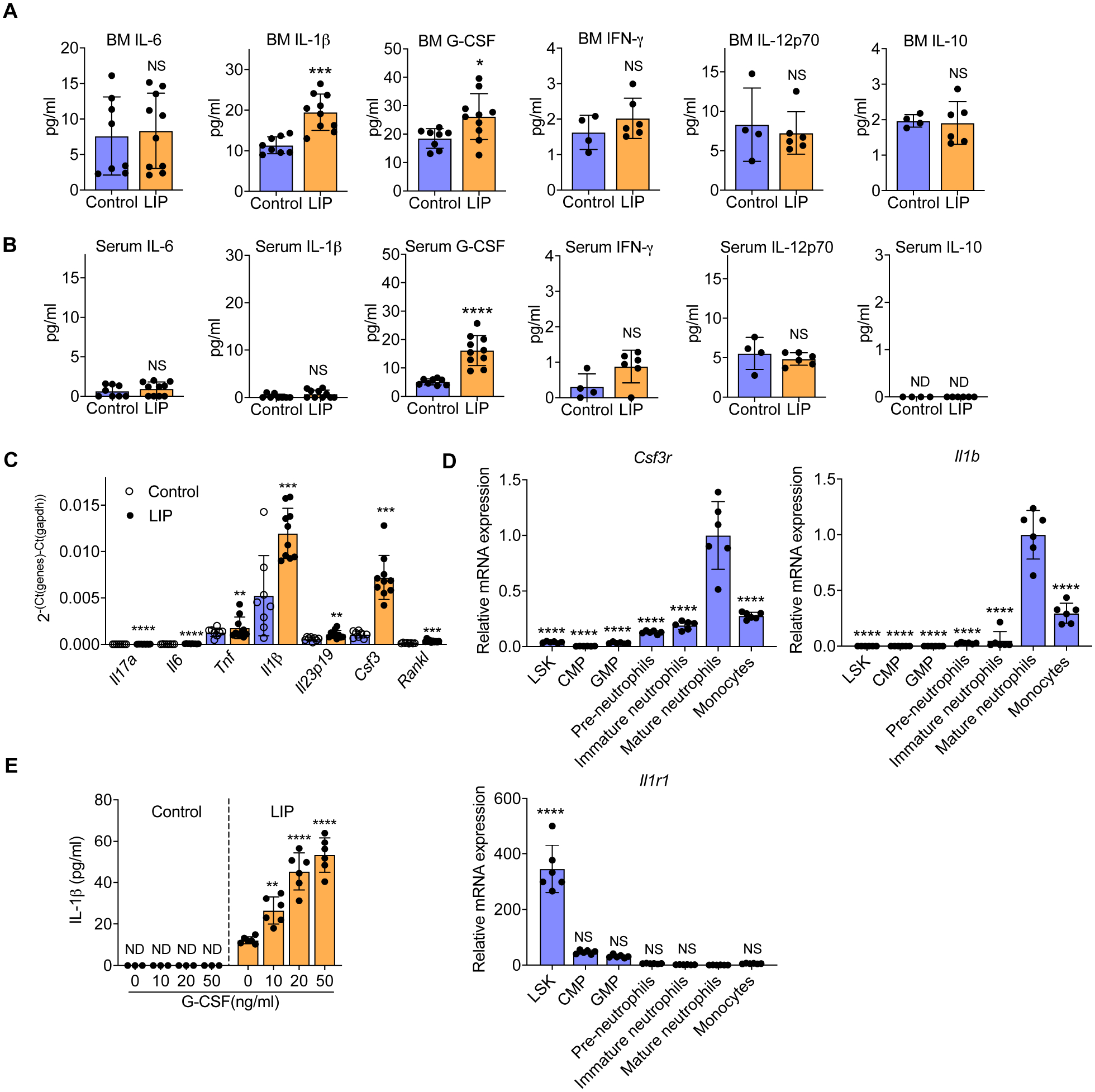
(A-E) Mice were subjected, or not (control) to LIP. After 14 days, indicated cytokines were analysed in BM extracellular fluid (A) and serum (B) (n=4–10 mice/group). (C) After 21 days, relative mRNA expression (normalized to Gapdh) of indicated cytokines was analysed in the gingiva (n=8–10 mice/group). (D,E) After 14 days, BM cells were harvested and the indicated cell types were FACS sorted (Gating strategies of LSK, CMP and GMP in Figures 1 and S1; gating strategies for pre-neutrophils, CD11b+CD115−Gr-1+cKit+CXCR4+; immature neutrophils, CD11b+CD115−Gr-1+cKit−CXCR4−Ly6G+CXCR2−; mature neutrophils, CD11b+CD115−Gr-1+cKit−CXCR4−Ly6G+CXCR2+; monocytes, CD11b+Ly6G−Ly6C+) and examined for Csf3r, Il1b and Il1r1 expression (D). Mature neutrophils isolated from the BM of LIP-trained or untrained controls were stimulated with recombinant mouse G-CSF for 24h and IL-1β was measured in culture supernatants (n=6 mice/group) (E). Data are means±SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. control (A-C, E) or mature neutrophils (D); two-tailed Student’s t-test (A-C), except for IL-12p70 in panel A (two-tailed Mann-Whitney U test); one-way ANOVA with Dunnett’s multiple-comparisons test (D,E).
To test this notion, HSPC, MyP and distinct types of myeloid cells were sorted from the BM of mice subjected to LIP for 14 days. Analysis of G-CSF receptor (colony-stimulating factor 3 receptor; Csfr3) mRNA expression showed that mature neutrophils expressed the highest levels of Csf3r (Figure 6D). Analysis of Il1b expression in the same cell populations revealed that mature neutrophils were also a major source of Il1b (Figure 6D). Thus, mature neutrophils might be the cells that secrete IL-1β upon G-CSF stimulation in the BM. In support of this notion, mature neutrophils isolated from the BM of LIP-subjected (but not control) mice responded to recombinant G-CSF stimulation with dose-dependent increase of IL-1β production (Figure 6E). To identify possible target cells of IL-1β in the BM, we examined the same cell populations from the BM of LIP-subjected mice and found that LSK had the most prominent Il1r expression (Figure 6D). Thus, IL-1β secretion in the BM might link LIP-induced inflammation and activation of HSPC during trained myelopoiesis. Consistently, IL-1, IL-1β and the myeloid transcription factor SPI1 (PU.1) were predicted as activated upstream regulators in LSK of LIP-subjected mice (Figure S1G). Moreover, the TF binding motifs enrichment analysis of the scATAC-seq using LSK revealed that the DNA binding sites of TFs responsive to IL-1 signaling pathway were significantly enriched in the LIP group compared to control (Figure 3F,G).
To directly link IL-1 signaling to LIP-induced maladaptive trained myelopoiesis, we used mice with inducible deletion of IL-1 receptor specifically in HSPC, generated by breeding HSC-SCL-Cre-ERT mice (Schoedel et al., 2016) with Il1r1fl/fl mice (HSC-SCL-Cre-ERT/Il1r1fl/fl), hereafter designated Il1r1HSPC-KO mice. Efficient Il1r1 deletion was achieved by administering tamoxifen by oral gavage, as described (Schoedel et al., 2016) and detailed in Methods. After induction of deletion, Il1r1HSPC-KO and littermate controls with intact IL-1R expression in HSPC were subjected to 21-day LIP. The HSPC-specific deletion of IL-1R resulted in reduced LIP-induced myelopoiesis, as evidenced by decreased frequencies of myeloid-biased HSPC subsets (MPP3 and CD41+ LT-HSC), GMP, Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells (Figure S7A).
Using Il1r1HSPC-KO mice and BMT, we next tested the hypothesis that IL-1-signaling in HSPC mediates LIP-induced trained myelopoiesis and increased inflammatory disease activity. To this end, groups of CD45.2+ Il1r1HSPC-KO and littermate controls with intact IL-1R expression in HSPC were trained by subjecting them to 21-day LIP and 14-day resolution and then used as donors for BMT to groups of lethally irradiated congenic B6.SJL (CD45.1+) mice (Figure 7A). 12 weeks post-BMT, the recipient mice were euthanized for BM and peripheral chimerism analysis or were subjected to LIP for 5 days.
Figure 7. IL-1-signaling in HSPC mediates LIP-induced maladaptive training of myelopoiesis.
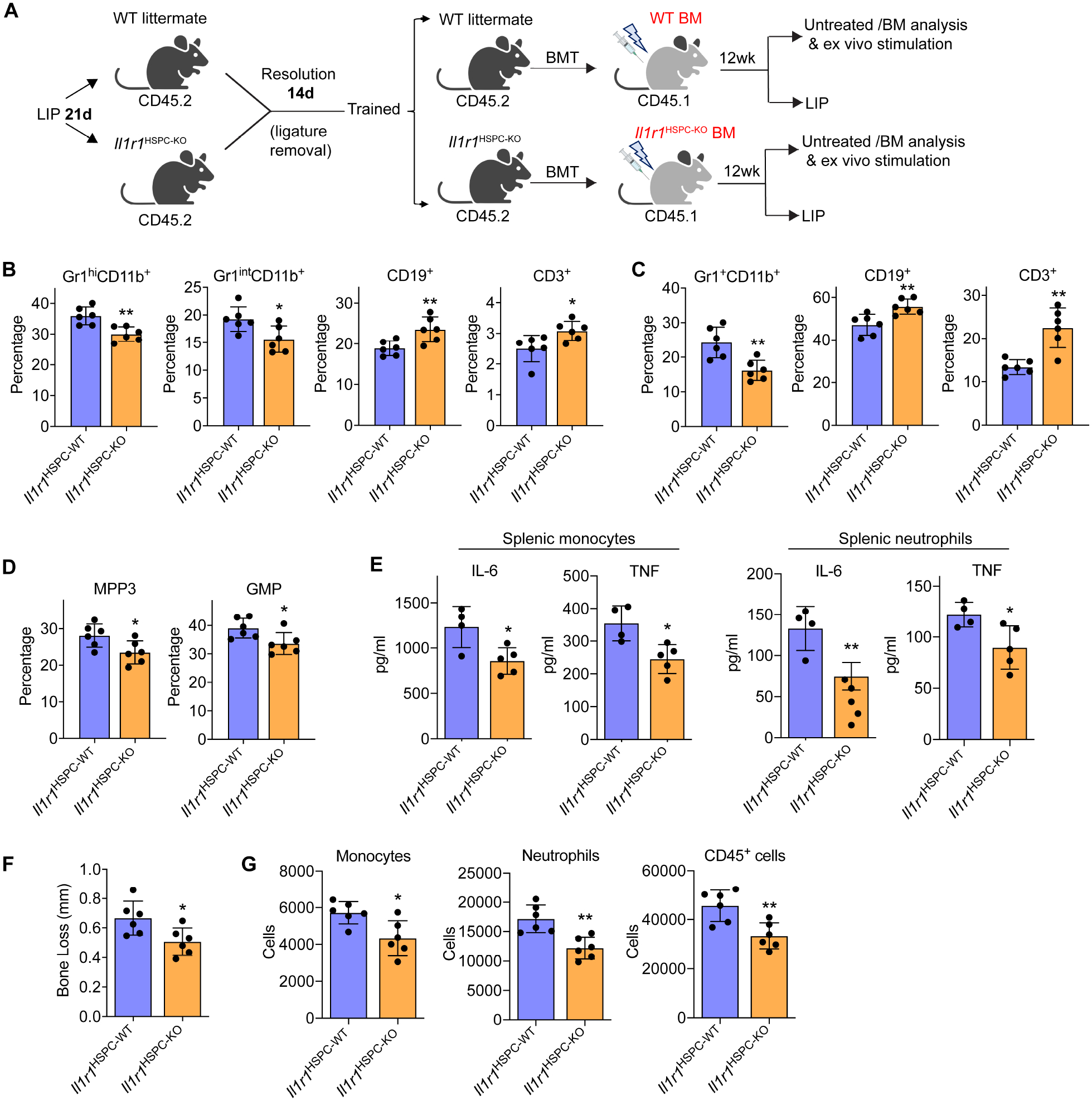
(A) CD45.2+ Il1r1HSPC-KO and littermate controls with intact HSPC IL-1R expression were trained (21-day LIP and 14-day resolution) and used as donors for BMT to lethally irradiated congenic B6.SJL (CD45.1+) mice. (B-D) 12 weeks post-BMT, the frequencies of indicated myeloid cells and lymphocytes in CD45+ cells in BM (B) and peripheral blood (C) of recipients, and the frequencies of MPP3 (in LSK) and GMP (in MyP) (D) were determined by FACS. (E) Splenic monocytes (left) and neutrophils (right) isolated from recipient mice were stimulated with LPS (10ng/ml) for 24h and IL-6 and TNF concentrations in the supernatant were measured. (F,G) The recipient mice were subjected to 5-day LIP, as shown in (A) and assayed for bone loss (F) and abundance of gingival monocytes, neutrophils and total CD45+ cells by FACS (G). Data are means±SD (n=4–6 mice/group). *P<0.05, **P<0.01, NS, not significant vs. WT littermate controls; two-tailed Student’s t-test. T, Trained; U, Untrained. See Figure S7.
At 12 weeks post-BMT, CD45.1+ recipients (without any further challenge) that had received Il1r1HSPC-KO BM cells from LIP-trained donor mice exhibited reduced proportion of Gr1+CD11b+ myeloid cells with corresponding increase in the proportion of CD19+ B cells and CD3+ T cells in the BM (Figure 7B) and peripheral blood (Figure 7C), as compared to mice that received WT BM cells. Consistently, analysis of MPP subsets and downstream progenitors in the BM showed that CD45.1+ mice receiving Il1r1HSPC-KO BM cells had reduced frequency of the myeloid-biased MPP3 and GMP (Figure 7D). These findings indicate a reduced myeloid bias in HPSC due to IL1-R1 deficiency, especially, as the two groups of recipient mice displayed similar frequencies of donor-derived HSPC (Figure S7B). Mature splenic neutrophils and monocytes isolated from CD45.1+ mice transplanted with BM cells from Il1r1HSPC-KO mice responded with significantly lower production of IL-6 and TNF to secondary LPS challenge than their counterparts from CD45.1+ mice that received BM cells from littermate controls bearing IL1-R1-sufficient HSPC (Figure 7E). These data suggest a critical role for HSPC-specific IL-1R signaling in LIP-induced trained myelopoiesis, associated with a transmissible myeloid-differentiation bias and production of mature myeloid cells with increased inflammatory responsiveness.
Consistent with the demonstrated trained phenotype, LIP-subjected recipient CD45.1+ mice that received Il1r1HSPC-KO BM cells from LIP-trained donor mice showed significantly decreased bone loss (Figure 7F) and reduced infiltration of the gingival tissue with monocytes, neutrophils and total leukocytes, relative to control CD45.1+ mice that received BM cells from LIP-trained WT donor mice (Figure 7G). The decreased bone loss could not be attributed to lack of IL-1R signaling in myeloid cells in the gingival tissue since even mice with global deletion of IL-1R were equally susceptible to LIP as WT littermates (Figure S7C), as shown earlier (Dutzan et al., 2018). Therefore, IL-1R signaling acts on HSPC to mediate LIP-induced maladaptive training of myelopoiesis, which exacerbates periodontal inflammation and bone loss.
DISCUSSION
We demonstrated that maladaptive training of myelopoiesis underlies the emergence of inflammatory comorbidities, exemplified here with the periodontitis-arthritis axis. Experimental periodontitis-associated systemic inflammation induced long-lasting myeloid-differentiation bias in HSPC that was retained predominantly at the epigenetic level, indicating prolonged readiness for myelopoiesis induction upon future challenges. The periodontitis-induced trained phenotype was transmissible by transplantation of sorted LT-HSC to naïve recipients, which displayed increased severity of arthritis upon CAIA challenge. In line with the bidirectional association of periodontitis and rheumatoid arthritis (Fuggle et al., 2016; Potempa et al., 2017), CAIA induced alterations to BM HSPC towards a maladaptive inflammatory phenotype, which exacerbated experimental periodontitis in transplanted mice. Thus, induction of central BM-mediated TII due to an inflammatory disease increases susceptibility to another inflammatory condition (comorbidity).
Our present study results are consistent with recent clinical imaging studies based on 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT) (Fifer et al., 2011; Ishai et al., 2019; Van Dyke et al., 2021). The use of 18F-FDG-PET/CT revealed a correlation between metabolic activity within the periodontal tissue (surrogate of periodontal inflammation) and hematopoietic tissue activity (reflecting stimulated hematopoiesis) (Ishai et al., 2019). Although this clinical study is correlative, it suggests an inflammatory periodontitis-BM axis, resembling the one described in our preclinical model.
Similar to the hyper-responsive phenotype of myeloid cells from trained mice, upon ex vivo stimulation, peripheral blood neutrophils or monocytes from periodontitis patients respond with higher inflammatory cytokine production than the same cells from healthy controls; this hyper-responsiveness often persists post-therapy for at least 2 months (Ling et al., 2015; Radvar et al., 2008). This apparent trained phenotype could predispose periodontitis patients to inflammatory comorbidities. As human periodontitis influences hematopoietic tissue activity (Ishai et al., 2019), the hyper-responsiveness of peripheral myeloid cells in periodontitis might result from epigenetically imprinted immune memory in inflammation-adapted BM HSPC, a concept that we and others have recently described in mice (de Laval et al., 2020; Kalafati et al., 2020; Kaufmann et al., 2018)(and this study) and humans (Cirovic et al., 2020; Moorlag et al., 2020b).
As with periodontitis patients, a subset of rheumatoid arthritis patients under clinical remission display elevated BM metabolic activity (by 18F-FDG-PET/CT) and a proinflammatory phenotype of circulating monocytes (Bernelot Moens et al., 2016). These findings imply that remission of arthritis does not necessarily reduce the patients’ risk of a comorbid condition. A common underlying pathophysiology, involving inflammatory memory in the BM that sustains trained myelopoiesis, might thus be an overlooked factor contributing to the connection of distinct comorbid inflammatory disorders.
Studies in rodents have shown that experimental periodontitis promotes experimental arthritis and vice-versa; in these studies, the same animals were subjected to both disease models (Cantley et al., 2011; Flak et al., 2019; Ramamurthy et al., 2005; Sato et al., 2017). This approach would not allow dissecting BM-dependent mechanisms from other mechanisms contributing to the periodontitis-arthritis relationship, such as, the ability of certain periodontal pathogens to cause breakdown of immune tolerance to citrullinated epitopes, leading to the generation of arthritogenic antibodies (Konig et al., 2016; Maresz et al., 2013; Potempa et al., 2017). However, such non-mutually exclusive mechanisms do not explain the bidirectional association of periodontitis and arthritis, in contrast to the mechanism reported here.
Enduring epigenetic modifications that unfold chromatin and render promoter and enhancer regions accessible to transcription factors, constitute a major pillar of the TII concept (Fanucchi et al., 2021). In this study, TII could be transferred via BMT from trained donors to naïve recipient mice, whose mature myeloid cells –12 weeks post-BMT– displayed enhanced inflammatory responsiveness. Although the transmission of the trained state by BMT could involve concurrent transmission of epigenetic modifications, this has not been formally shown in the literature. Indirectly, however, several studies have suggested a key role of epigenetic memory in TII induction and maintenance. LT-HSC retain epigenetic memory of previous inflammatory challenge and this underlies their sustained myeloid bias, i.e., persistent changes in the accessibility of specific myeloid lineage enhancers, which augments the responsiveness of the respective immune genes to secondary stimuli (de Laval et al., 2020). It is currently thought that, unlike repressive modifications (in particular DNA methylation), accessibility of enhancers and active histone modifications are not self-maintained in mammals; indeed, studies in dividing cell populations showing long-term and mitotically inheritable epigenetic changes have so far been attributed to DNA methylation changes and not histone modifications (Sun and Barreiro, 2020). Future research in dividing cells may clarify whether histone modifications are also epigenetically transmissible. In this regard, certain chromatin accessibility changes are transmitted from HSPC to progeny cells along differentiation trajectories (Buenrostro et al., 2018). Moreover, epigenetic changes in the form of histone modifications were shown to be passed on to the mouse offspring (Siklenka et al., 2015). Transgenerational transmission of various immune traits including TII, attributable to sustained epigenetic memory, has been shown recently in mice (Bomans et al., 2018; Katzmarski et al., 2021; Lim et al., 2021) and humans (Berendsen et al., 2021; Gee et al., 2021).
Based on our findings, an inflammatory disease could modulate TII in the BM in a manner that not only aggravates the pre-existing disease but can also increase susceptibility to a distinct inflammatory condition. Such unified conceptual framework could also provide a platform for therapeutic interventions targeting inflammatory comorbidities. Our data suggest that systemic inhibition of IL-1-induced signaling may potentially block the maladaptive training of BM progenitors and thereby disrupt a common mechanism for inflammatory comorbidities. In this regard, it might be argued that the successful application of IL-1β blockade in the CANTOS trial for the treatment of atherosclerosis (Ridker et al., 2017) might, in part, have resulted from inhibition of TII in the BM. In conclusion, our findings establish the principle that maladaptive innate immune training of myelopoiesis underlies inflammatory comorbidities paving the way for their treatment in a holistic manner.
LIMITATIONS OF THE STUDY
Our findings have linked experimental periodontitis to maladaptive BM-mediated TII, which underlies inflammatory comorbidities. Nevertheless, our study has several limitations. Despite initial evidence for an inflammatory periodontitis-BM axis in humans and for a trained phenotype of myeloid cells from periodontitis patients, formal evidence for periodontitis-induced maladaptive TII in humans may require studying hematopoietic stem cell transplant (HSCT) recipients. Thus, future investigations of the transmissibility of inflammatory memory to HSCT recipients from donors with or without periodontitis, would confer increased clinical relevance to our study. Such study would prompt clinicians to take inflammatory memory into consideration when selecting appropriate donors for BMT. In most of the experiments (and all functional assays), LIP lasted for 21 days to mimic chronic periodontitis in humans, although shorter durations (e.g., 7 days) were also used to determine early effects of LIP on modulating myelopoiesis. We cannot rule out the possibility that epigenetic changes seen after 21 days of LIP and 14 days resolution might differ from those occurring at earlier timepoints and whether such earlier changes might also contribute to a trained phenotype. Our findings that a maladaptive trained phenotype was transmissible via BMT in a HSPC-specific and IL-1-dependent manner, indicated that IL-1 is critical for induction of a long-lasting trained phenotype with detrimental consequences in inflammatory disease. We have not determined if IL-1 signaling shapes the epigenetically imprinted inflammatory memory associated with a myeloid-differentiation bias in trained LT-HSC; this question should be studied in the future. Moreover, we cannot exclude the possibility that IL-1β may cooperate (or even synergize) with other as yet unidentified inflammatory factors, thus warranting additional mechanistic investigations. Despite the de facto transmission of the TII phenotype via HSC transplantation, the underlying molecular epigenetic mechanisms remain incompletely understood. Finally, at this point, we do not know how long a maladaptive trained phenotype would last, a question that must be carefully addressed in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact George Hajishengallis (geoh@upenn.edu).
Materials Availability
This study did not generate any unique reagents.
Data and Code Availability
Data are available upon request to the Lead Contact. Sequencing data are available at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE180032.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 and congenic C57BL/6.SJL CD45.1+ male mice (B6.SJL-PtprcaPepcb/BoyJ) were purchased from the Jackson Laboratory. Mice with tamoxifen-inducible deletion of the IL-1 receptor specifically in HSPC, designated HSC-SCL-Cre-ERT/Il1r1fl/fl (hereafter referred to as Il1r1HSPC-KO mice) were generated by crossing using Il1r1fl/fl mice (Stock # 028398; Jackson Laboratory) and HSC-SCL-Cre-ERT mice (Gothert et al., 2005; Schoedel et al., 2016) (donated by Dr. Joachim R. Göthert, University Hospital Essen). To induce Il1r1 deletion, Il1r1HSPC-KO mice were administered tamoxifen (0.3 mg/g body weight by oral gavage) two times within 72h, followed by feeding the mice with tamoxifen-containing (0.5 mg/g) diet for 18 days. Mice were maintained in individually ventilated cages under specific pathogen-free conditions on a standard 12-h light/dark cycle. Food and water were provided ad libitum. The mice were 8- to 10-week-old at the start of the experiments. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and were performed in compliance with institutional, state, and federal policies.
METHOD DETAILS
Ligature-induced periodontitis
Ligature-induced periodontitis (LIP) simulates human periodontitis by generating a local biofilm-retentive milieu leading to inflammation and bone loss (Abe and Hajishengallis, 2013; Dutzan et al., 2018; Kitamoto et al., 2020; Kourtzelis et al., 2019; Tsukasaki et al., 2018). To study the effect of experimental periodontitis on BM hematopoietic progenitor cells, LIP was performed in mice as previously described (Abe and Hajishengallis, 2013). Briefly, 5–0 silk ligatures were tied around the left and right maxillary second molar teeth for time intervals specified in the figure legends (up to 21 days). The same teeth were left unligated in control mice. In some experiments, the placement of ligatures was followed by their removal (for 14 days in most experiments) to enable inflammation resolution (Kourtzelis et al., 2019; Li et al., 2020) prior to BM analysis. In a subset of these experiments, mice during resolution were subjected to a secondary challenge, namely Escherichia coli O111:B4 LPS (InVivogen) which was injected i.p. at 1.5 mg/kg body weight, modeling bacteremia. The mice were euthanized 72h after the LPS injection for analysis. To determine ligature-induced bone loss, the 5-day standard LIP model was used, in which only the left maxillary second molar was ligated, whereas the contralateral (right) molar tooth was left unligated to serve as baseline control for measuring bone loss as we previously described (Abe and Hajishengallis, 2013; Dutzan et al., 2018; Kourtzelis et al., 2019).
Collagen antibody-induced arthritis
Collagen antibody-induced arthritis was induced in mice by i.v. (retro-orbital) injection of 1.5 mg arthritogenic monoclonal antibodies (5-clone collagen antibody cocktail; Chondrex) (Khachigian, 2006; Wang et al., 2021). Three days later, mice were injected i.p. with 50 μg of LPS. Clinical symptoms of arthritis were daily evaluated visually for each paw using a semiquantitative scoring system graded on a scale of 0–4 per paw (Khachigian, 2006) by a blinded procedure: 0 for normal; 1 for mild redness, slight swelling of ankle or wrist; 2 for moderate swelling of ankle or wrist; 3 for severe swelling, including some digits, ankle and foot; 4 for maximally inflamed joint. The clinical score for each mouse was the sum of the 4 paw scores for a maximum score of 16. Hind ankle joint thickness was measured by using J 15 pocket dial thickness gauge (Käfer).
Cells preparations and sample collection
For BM single-cell suspension preparation, femoral bones of C57BL/6 mice were flushed with ice-cold PBS (Gibco) supplemented with 5% FBS (Gibco). Cells were forced through 70-μm nylon cell strainer to get single-cell suspension for further flow cytometric analysis and FACS cell sorting. To isolate BM mature neutrophils, BM cells were flushed from femurs of C57BL/6 mice subjected to LIP for 14 days. Upon lysis of red blood cells with ACK Lysing Buffer (Gibco), the cells were centrifuged in 62% Percoll gradient (GE Healthcare). The sharp interface atop the 62% Percoll (containing immature cells and non-granulocytic lineages) was carefully removed and discarded, whereas the pellet (mature neutrophils) was collected. To collect BM extracellular fluid, mice femurs were flushed with 500 μl ice-cold PBS (Gibco) and the supernatant was harvested after centrifugation at 500 × g for 5 min at 4°C. Serum was collected after retrobulbar bleeding. Specifically, collected whole blood was left undisturbed at room temperature for 30 mins and the clot was removed by centrifugation at 2,000 × g for 10 minutes at 4°C followed by collection of the supernatant serum.
Mice subjected to LIP and a secondary challenge with LPS (see above) were euthanized and lung lobes were dissected after tracheal and intracardial perfusion with cold PBS. Lung tissues were then cut into 1-mm2 pieces with scissor and digested in freshly prepared digestion medium consisting of 2 mg/ml collagenase IV (Invitrogen) in RPMI 1640 medium (Invitrogen) supplemented with 1% penicillin-streptomycin (Invitrogen). The digestion was performed at 37°C with shaking at 100 rpm for 1h and was stopped by addition of EDTA (Invitrogen) at a final concentration of 5mM. Cells were forced through 100-μm nylon cell strainer and subjected to 40% Percoll density gradient centrifugation (GE Healthcare) to remove debris. The cell pellet was resuspended in ACK Lysing Buffer (Gibco) for lysis of red blood cells. Cells were washed and resuspended for immunofluorescence staining and flow cytometric analysis.
To isolate splenic neutrophils and monocytes, splenocytes were incubated with biotinylated anti-mouse Ly6G antibody (clone 1A8; Biolegend) followed by anti-biotin microbeads from Miltenyi Biotec. Neutrophils were positively selected using LS columns on the magnetic field of QuadroMACS™ Separator according to the manufacturer’s instructions (Miltenyi Biotec). For isolating splenic monocytes, neutrophils were first removed by negative selection for Ly6G+ cells and then monocytes were obtained by positive selection for Ly6C+ (clone HK1.4; Biolegend) cells as we previously described (Kalafati et al., 2020).
Flow cytometry and sorting
Flow cytometric analysis was performed on a NovoCyte flow cytometer (ACEA Biosciences). For cell surface phenotypic analysis, a lineage (Lin) cocktail, including the following monoclonal antibodies was used: CD3e (clone 145–2C11), CD11b (clone M1/70), Gr1 (clone RB6–8C5), B220 (clone RA3–6B2) and TER119 (clone TER-119). Other antibody reagents used in experiments included anti-Sca1 (clone E13–161.7), anti-cKit (clone 2B8), anti-CD135 (clone A2F10), anti-CD48 (clone HM48–1), anti-CD150 (clone TC15–12F12.2), anti-CD41 (clone MWReg30), anti-CD61 (clone 2C9.G2 [HMβ3–1]), anti-CD16/CD32 (clone 93), anti-CD34 (clone HM34), anti-CD45.1 (clone A20), anti-CD45.2 (clone 104), anti-CD3 (clone 17A2), anti-CD19 (clone 6D5), anti-CD11b (clone M1/70), anti-Gr1 (clone RB6–8C5), anti-CD11c (clone N418), anti-Ly6C (clone HK1.4), anti-Ly6G (clone 1A8), anti-CD115 (clone AFS98), anti-CXCR2 (clone SA045E1) and anti-CXCR4 (clone L276F12) were used. Data were analyzed with NovoExpress® software (ACEA Biosciences). Gating strategies for hematopoietic stem and progenitor cells were as follows: LSK, Lin−Sca-1+cKit+; LS−K (MyP), Lin−Sca-1−cKit+; LT-HSC, CD48−CD150+LSK; CD41−LT-HSC, CD48−CD150+CD41−LSK; CD41+ LT-HSC, CD48−CD150+CD41+LSK; ST-HSC, CD48−CD150−LSK; MPP, CD48+CD150−LSK; MPP2, Flt3−CD48+CD150+LSK; MPP3, Flt3−CD48+CD150−LSK; MPP4, Flt3+CD48+CD150−LSK; GMP, Lin−Sca-1−cKit+CD16/32+CD34+; CMP, Lin−Sca-1−cKit+CD16/32−CD34+. Gating strategies for neutrophils and monocytes in mouse gingiva and knee joints were as follows: neutrophils, live CD45+CD11c−CD11b+Ly6G+Ly6C−; monocytes, live CD45+CD11c−CD11b+Ly6G−Ly6C+. For BM cell sorting, the following gating strategies were used: for pre-neutrophils, CD11b+CD115−Gr-1+cKit+CXCR4+; immature neutrophils, CD11b+CD115−Gr-1+cKit−CXCR4−Ly6G+CXCR2−; mature neutrophils, CD11b+CD115−Gr-1+cKit−CXCR4−Ly6G+CXCR2+; monocytes, CD11b+Ly6G−Ly6C+. Cells sorting was performed on a FACSARIA™ instrument (Becton Dickinson Immunocytometry Systems, USA).
Bone marrow transplantation
To generate BM chimeras, a total of 2×106 BM cells from C57BL/6 (CD45.2+) mice, or from CD45.2+ Il1r1HSPC-KO and littermate controls were transplanted (via a single retro-orbital injection) into lethally (9.5 Gy) irradiated B6/SJL (CD45.1+) mice. Twelve weeks after BM transplantation, the recipient mice were subjected to treatments and/or analyses as described in Results and Figure legends. The CD45.1/CD45.2 congenic system was also used in competitive BM chimeras, e.g., to assess lineage output of LT-HSC in transplanted mice. Sorted LT-HSC (100 cells per recipient) isolated from the BM of CD45.2+ mice (which were subjected to 21-day LIP and 14-day resolution or were left untreated for 35 days) were retro-orbitally transferred into lethally (9.5 Gy) irradiated naïve B6/SJL (CD45.1) recipients along with 5×105 CD45.1+ competitor cells. The percentage of different CD45.2+ cell populations was assessed at 4, 8, 12 and 16 weeks post transplantation in the blood of recipient mice, and at 16 weeks post transplantation (end point) in the BM of recipient mice.
In additional competitive BM chimera experiments, sorted LT-HSC (total or further sorted into CD41+ LT-HSC and CD41− LT-HSC) isolated from the BM of CD45.2+ mice (which were subjected to 21-day LIP and 14-day resolution or were left untreated for 35 days) were retro-orbitally transferred (200 cells per recipient) into lethally (9.5 Gy) irradiated naïve B6/SJL (CD45.1) recipients along with 8×105 CD45.1+ competitor cells. In all BMT experiments, irradiated recipient mice were kept on antibiotics-containing water for 2 weeks after irradiation. Complete blood count (CBC) test was performed using Sysmex XT-2000iV Hematology Analyzer.
Immunoassays
The concentrations of mouse IL-6, IL-1β, IFN-γ, IL-12p70 and IL-10 in BM fluid and serum were measured using mouse ELISA kits (Invitrogen) according to the manufacturer’s instructions. Mouse G-CSF was measured using ELISA kit from RayBiotech, according to the manufacturer’s instructions. For in vitro stimulation of mature neutrophils from the BM, isolated mature neutrophils were stimulated with different concentration of mouse recombinant G-CSF (R&D Systems) for 24h. The supernatant was collected for measuring IL-1β concentration using mouse ELISA kit (Invitrogen). For in vitro re-stimulation of splenic monocytes and neutrophils with LPS, isolated splenic monocytes and neutrophils were seeded into 24-wells plates and stimulated with 10ng/ml of E. coli O111:B4 LPS (InVivogen) for 24h. The supernatant was collected for measuring IL-6 and TNF concentration using mouse ELISA kit (Invitrogen).
Quantitative real-time PCR
Total cellular RNA was isolated from mouse tissues and sorted BM cells using Trizol (Invitrogen). For real-time PCR, 500 ng of total RNA was reverse-transcribed using High-Capacity RNA-to-cDNA Kit (Applied Biosystems) and real-time PCR with cDNA was performed using the Applied Biosystems 7500 Fast Real-Time PCR System, according to the manufacturer’s protocol (Applied Biosystems). TaqMan probes and gene-specific primers for detection and quantification of murine genes investigated in this study were purchased from Thermo-Fisher Scientific (Supplemental Table 1). Data were analyzed using the comparative (ΔΔCt) method and were normalized to Gapdh mRNA. In LIP experiments assessing gingival cytokine mRNA expression in ligated sites, the data are shown as fold change relative to the contralateral unligated control sites (baseline), set as 1.
Histological analysis
Harvested joints were fixed in 4% buffered formaldehyde. The bones were subsequently decalcified, embedded in Optimal Cutting Temperature (OCT) media and sectioned at 10-μm thickness for hematoxylin and eosin (H&E) or Safranin-O staining (ScienCell) (Wang et al., 2021).
Bulk RNA sequencing
LSK from the BM of mice subjected to 7-day LIP or unligated control mice were sorted using a FACSAria™ sorter. Three mice were pooled for each replicate. Total RNA was extracted by Trizol (Invitrogen) and DNA was removed by using TURBO DNA-free Kit (Invitrogen). PolyA-selected mRNA libraries were generated following the manufacturer’s protocols (BGI). Samples were sequenced on BGISEQ platform to generate 100 bp paired-end reads with an average depth of 45.0 M reads per sample. Clean reads were mapped to the mouse genome (Ensembl assembly GRCm38) using Spliced Transcripts Alignment to a Reference (STAR) (Dobin et al., 2013) with default settings after filtering low-quality, adaptor-polluted and high content of unknown base (N) reads. The average mapping ratio with reference genome was 96.46%, the average mapping ratio with gene was 80.83%. A total of 16,403 genes were detected. Most transcripts were completely covered, and reads were evenly distributed throughout the transcript.
Single-cell RNA sequencing
LSK from the BM of mice which were subjected to 21-day LIP and 14-day resolution, or were left untreated for 35 days, were sorted using a FACSAria™ sorter. The cells were resuspended in 1X PBS containing 0.04% BSA (400 μg/ml) and processed with Chromium Next GEM Single Cell 3’ Kit v3.1, 16 rxns (10x Genomics) at the CAG Sequencing Core of Children’s Hospital of Philadelphia, USA. Cell suspensions contained more than 90% viable cells as determined by microscopy. The cell numbers were adjusted to 700–1200 cells per μl and added to 10x Chromium RT mix to achieve loading target numbers of around 20000 cells. cDNA synthesis was performed per the manufacturer’s instructions, and library preparation and sequencing (Novaseq SP reagent kit; 100 cycles) were performed using the Novaseq 6000 platform (Illumina) per the manufacturer’s instructions.
Single-cell ATAC sequencing
Mouse LSK and GMP were sorted as described above. Nuclei isolation for single-cell ATAC sequencing was performed according to the “Nuclei Isolation for Single Cell ATAC Sequencing demonstrated protocol” (10x Genomics). Briefly, after sorting, cells were centrifuged at 300 rcf for 5 min at 4°C and washed once with PBS with 0.04% BSA. One hundred μL of chilled Lysis buffer (10mM Tris-HCl (pH 7.4), 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 Substrate, 0.01% Digitonin, 1% BSA in nuclease free water) was added to the pellet and cells were incubated on ice for 2 min for LSK and 3 min for GMP. Cells were washed once with 1 ml of wash buffer (10mM Tris-HCl (pH 7.4), 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 1% BSA in nuclease free water) and concentrated by centrifugation at 500 rcf for 5 min at 4°C. Nuclei in pellet were suspended in 1x Nuclei Buffer (10x Genomics); nuclei quality and concentration were determined using a microscope and a Countess II FL Automated Cell Counter (Thermo Fisher). Nuclei suspension was then diluted and used according to the Chromium Single Cell ATAC Reagent Kits protocol (10x Genomics) Briefly, nuclei suspension was mixed with the tagmentation mix and incubated for 1 hr at 37°C. After mixing with a barcoding mix, the nuclei were loaded into a 10x chip H together with barcoded beads and partitioning oil (Chromium Next GEM Chip H Single Cell Kit v1.1, Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1, 10x Genomics) and encapsulated using the Chromium controller (10x Genomics). The Gel Bead-In EMulsions (GEMs) was transferred into a PCR tube and amplified for 12 cycles in a thermocycler. The barcoded DNA was purified and subjected to an index PCR for 11 cycles. The library amplification was assessed using fragment analyzer (Agilent, NGS High Sensitivity Fragment Analysis Kit) and sequenced on an Illumina NovaSeq 6000 system using S1 Reagent Kit v1.5 (100 cycles) in PE mode (50×8×16×50 read lengths), at a median depth of 7500 fragments/cell.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatic analysis of bulk RNA-seq
Reads count in each gene were calculated by htseq-count package. Subsequently, differential gene expression was analyzed using DEGseq2 (Love et al., 2014) and significantly differentially expressed genes (DEGs) were defined by FDR-adjusted P value < 0.05 in the LIP-subjected group relative to their expression in the control group. Significantly up- or down- regulated DEGs were subjected to Gene Ontology (GO) enrichment analyses using PANTHER (Protein ANalysis THrough Evolutionary Relationships) (http://www.pantherdb.org) and KEGG pathway analysis (https://www.genome.jp/kegg/kaas/) with default background and default threshold. Significantly enriched Biological Process GO terms and KEGG pathways were defined by FDR-adjusted P value <0.05. DEGs were also subjected to upstream regulator analysis using Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City www.qiagen.com/ingenuity). Top significantly activated transcription factors and upstream regulators are shown. Volcano plot, principal component analysis (PCA) plot, bar plots and heat maps were generated by customized R script. Overlapping genes, e.g., Csfr1 and Itgb3, in heatmaps of two different GO terms are shown in both terms.
Single-cell RNA sequencing analysis
Cell Ranger 3.0.2 (https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome) was used to process raw paired-end sequencing data. Briefly, Cell Ranger mkfastq pipeline was used to demultiplex sample index reads to generate FASTQ files for each sequencing library, and then raw reads were aligned to the UCSC mouse reference genome (mm10) using STAR aligner with default parameters. Subsequently, data filtering, normalization, scaling, and Principal component analysis (PCA) were performed using the R package Seurat version 4.0.3 (Stuart et al., 2019) and dimensionality reduction and clustering were further done by Uniform Manifold Approximation and Projection (UMAP) analysis. Low-quality cells and possible cell doublets were filtered out using the following criteria: (i) number of detected genes between 200 and 2500, and (ii) percentage of UMIs derived from mitochondrial genes below 5%. The UMI counts were normalized by library size factors. As a result, Cell Ranger recovered total 7051 (NL) and 8206 (LSK-21dL/14dR) LSK. Genes differentially expressed across clusters were identified using likelihood ratio test. GO enrichment analysis of cluster 7-specific markers were performed using PANTHER. Heat maps and bubble plots were generated by customized R script.
Single cell ATAC sequencing analysis
Cell Ranger ATAC 1.2.0 (https://support.10xgenomics.com/single-cell-atac/software/overview/welcome) was used to process raw sequencing data. Cell Ranger ATAC’s pipelines was used to align reads, generate peak matrix with single-cell accessibility counts and fragment file with all unique fragments across all single cells, using ‘refdata-cellranger-atac-mm10-1.2.0’ from the 10X Genomics website (https://support.10xgenomics.com/single-cell-atac/software/downloads/) as reference genome file. The following analysis of the scATAC-seq data was performed using the R package Seurat and Signac (Stuart et al., 2019; Stuart et al., 2020). Briefly, a chromatin accessibility matrix was created by merging fragment files from 4 samples and filtering out peaks with peak width larger than 10000 bp or smaller than 500 bp. Next, Uniform Manifold Approximation and Projection (UMAP) based on latent semantic indexing (LSI) was generated to visualize the data structure in the two-dimensional space (designated LSK-NL, LSK-21dL/14dR, GMP-NL and GMP-21dL/14dR). Smart local moving (SLM) approach implemented in the R package Seurat (Stuart et al., 2019) was used to cluster the single-cell accessibility profiles. A logistic regression framework was used for differentially accessible regions (DAR) analysis (abs(Log2FC) ≥ 0.2, P < 0.05). GO enrichment analysis of the treatment-specific DAR of LSK were performed using PANTHER (Protein ANalysis THrough Evolutionary Relationships) (http://www.pantherdb.org). Additionally, a gene activity matrix was created to quantify the activity of each gene by assessing the chromatin accessibility using Signac package. For motif enrichment analysis, an ArchR project was created using ArchR v1.0.1 (Granja et al., 2021), and peak regions were annotated with homer motif set. Moreover, the peakAnnoEnrichment function implemented in ArchR was used to identify the enriched transcription factor binding motifs based on the treatment-specific DARs in LSK with FDR<0.01 and log2FC ≥ 1, followed by GO enrichment analysis using PANTHER. A cellular trajectory was constructed spanning from LSK over cluster C9 to GMP using Monocle 3 and Cicero tool (Cao et al., 2019; Pliner et al., 2018), based on the chromatin accessibility.
Statistical analysis
After confirming normality, data were analyzed with two-tailed unpaired Student’s t test (comparison of only two groups) or one-way ANOVA followed by Dunnett’s multiple-comparisons test (when comparing more than two groups). In a few instances (comparison of only two groups) where data did not follow normal distribution, the non-parametric two-tailed Mann-Whitney U-test was used. Two-way repeated measures ANOVA and Sidak’s multiple-comparisons test was used to analyze data in repeated-measures designs. All statistical analyses were performed using GraphPad Prism software (version 8.4.3; GraphPad Inc). P values <0.05 were considered to be statistically significant.
Supplementary Material
Figure S1. LIP activates bone marrow and enhances myelopoiesis (Related to Figure 1). Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) followed by BM analysis after 7 days. (A, left) Representative FACS plots for identification of hematopoietic stem and progenitor cells. After gating for Lin− cells, LSK were identified as cKit+Sca1+ cells. LSK subpopulations were further characterized as MPP (CD48+CD150−LSK), ST-HSC (CD48−CD150−LSK), and LT-HSC (CD48−CD150+LSK) cells. (A, right) Frequencies of LSK, LT-HSC, ST-HSC and MPP in total BM cells (top) and absolute cell numbers (bottom) of the same populations. (B, top) Representative FACS plots of MPP subsets. After gating for LSK, MPP4 were defined as CD48+Flt3+CD150−LSK, MPP3 as CD48+Flt3−CD150−LSK, and MPP2 cells as CD48+Flt3−CD150+LSK. (B, bottom) Frequencies of MPP subsets in LSK in the BM. (C, left) Representative FACS plots for the identification of CD41+ and CD61+ LT-HSC cells and (right) frequency of the same populations in total LT-HSC cells in the BM of mice. (D-G) Groups of mice were subjected, or not (control, NL), to ligature-induced periodontitis (LIP) and BM cells were harvested on day 7. FACS-sorted LSK were subjected to RNA-sequencing and transcriptome analysis. (D) Heatmaps of myeloid lineage, osteoclast differentiation. (E) Heatmaps of erythroid, lymphoid lineage and oxidative phosphorylation related genes. (F) Heatmap of significantly regulated epigenetic regulators by LIP. (G) Significant upstream transcription factors (left) and upstream regulators (right) predicted by Ingenuity Pathway Analysis (IPA). Data in (A-C) are means ± SD (n=6 mice/group). *P<0.05, **P<0.01, NS, not significant vs. control mice; two-tailed Student’s t-test.
Figure S2. Inflammation resolution in the periodontium phenotypically restores myelopoiesis to steady-state levels in the BM (Related to Figure 2). Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) for 21 days and subsequently the ligatures were removed (to enable inflammation resolution) for 14 days, at which time BM analysis was performed. (A) Relative mRNA expression of indicated cytokines in the gingival tissue of mice after 14 days resolution. (B) Frequencies in total BM cells (top) and absolute cell numbers (bottom) of LSK, LT-HSC, ST-HSC and MPP. (C) Frequencies of MPP subsets in LSK in the BM. (D) Frequencies of CD41+ LT-HSC in total LT-HSC cells in the BM. (E) Frequency within the MyP pool of GMP and CMP (left and middle, respectively) and cell numbers of GMP (right) in the BM. (F) Frequency of Gr1hiCD11b+ granulocytes, Gr1intCD11b+ myeloid cells, and CD19+ B cells in CD45+ cells in the BM. (G-H) BM LSK were sorted from mice that were subjected to 21dL/14dR or not (NL) and scRNA-seq was performed. (G) GO enrichment results of top 20 significantly enriched GO terms sorted by PANTHER on the basis of marker genes specific for cluster 7 shown in Figure 2A–C (Bonferroni-corrected, P<0.05). (H) Two-dimensional UMAP showing the expression of myeloid marker genes (gray denotes minimal expression, purple intermediate and blue high). Data are means ± SD (n=6 mice/group) (A-F). *P<0.05, NS, not significant vs. control mice; two-tailed Student’s t-test.
Figure S3. Single-cell ATACseq analysis (Related to Figure 3). (A) Violin plots showing gene activity scores of Itga2b and Gata2 across cluster 2. (B) Violin plots showing gene activity scores of Csf1r and Nlrp3 across cluster 3. (C) A cellular trajectory spanning from LSK to GMP. (D) Bubble plot of GO enrichment analysis of the cluster 5-specific marker genes. (E) Violin plots showing gene activity scores of Tlr4, Camk1d, Itga1 and Map2k4 across cluster 5.
Figure S4. LIP-subjected mice are more susceptible to CAIA than non-ligated control mice (Related to Figure 4). (A) Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) for 21 days followed by a 14-day resolution. Mice were then subjected to CAIA model and monitored for 14 days. (B) Clinical arthritis score and hind ankle joint thickness were measured daily. (C) CD45.2+ mice were either trained (T) by subjecting them to 21-day LIP followed by 14-day resolution (‘21dL/14dR’-trainedmice), or were left untrained (U; naïve controls). BM cells were isolated from trained and untrained mice and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. Twelve weeks after receiving BM transplantation from trained or untrained donor mice, splenic monocytes and neutrophils were isolated. (D) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6 and TNF concentration by ELISA. (E-F) Frequency of donor-derived cells in the BM of recipient mice (E) and frequency of donor-derived (CD45.2+) LSK, LT-HSC, ST-HSC and MPP cells in respective populations in the BM of recipient mice (F) at 16 weeks post-BMT, as described in Figure 4H. Data are means ± SD (n = 6 recipient mice/group). P* <0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice; two-way repeated measures ANOVA and Sidak’s multiple comparisons test (B); two-tailed Student’s t-test (D,E,F). T, Trained; U, Untrained.
Figure S5. CAIA activates bone marrow and enhances myelopoiesis (Related to Figure 5). (A-F) mice were subjected, or not (control), to CAIA and BM analysis was performed after 14 days. (A) Frequencies (top) of LSK, LT-HSC, ST-HSC and MPP in total BM cells and absolute cell numbers (bottom) of the same populations. (B) Frequency of MPP subsets in LSK in the BM. (C) Frequency of CD41+ LT-HSC in total LT-HSC cells in the BM. (D) Absolute cell numbers of GMP (left) and frequency within the MyP pool of GMP (right). (E) Absolute cell numbers and frequency of Gr1hiCD11b+ granulocytes in CD45+ cells in the BM. (F,G) mice were subjected, or not (control), to CAIA and monitored daily for ankle joint thickness (F). BM analysis was performed on day 36, i.e., 14 days after complete arthritis resolution. (G) Frequencies of LSK, LT-HSC, ST-HSC and MPP cells in total BM cells. (H) CD45.2+ mice were either trained (T) by subjecting them to CAIA (disease activity returned to baseline on day 22 and the mice were rested post-resolution for 14 days; total 36 days), or were left untrained (U; naïve controls). 36 days post-CAIA, BM cells were isolated and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. Twelve weeks after receiving BM transplantation from donor mice, splenic monocytes and neutrophils from recipient mice were isolated. (I) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6, and TNF concentration by ELISA. Data are means ± SD (n=6 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. control or untrained mice; two-tailed Student’s t-test. T, Trained; U, Untrained.
Figure S6. Effects of competitive BM transplantation using sorted CD41+ and CD41− LT-HSC. (Related to Figure 5). (A) CD45.1+ mice were transplanted with 200 CD41+ or CD41− LT-HSC sorted from LIP-trained or untrained CD45.2+ mice together with 8×105 CD45.1+ BM competitor cells. (B-C) WBC count, donor-derived percentage, and lineage output (% of indicated cell types in donor-derived cells at the indicated time points) in the peripheral blood of mice transplanted with CD41+ (B) or CD41− (C) LT-HSC. (D-E) Frequency of donor-derived cells in the BM of recipient mice and frequency of donor-derived (CD45.2+) LSK, LT-HSC, ST-HSC and MPP cells in respective populations in the BM of mice transplanted with CD41+ (D) or CD41− (E) LT-HSC, at 12 weeks post-BMT. (F-I) 12 weeks post-BMT, the recipient CD45.1+ mice were subjected to CAIA. Clinical arthritis score (left) and hind ankle joint thickness (right) in mice transplanted with CD41+ (F) or CD41− (G) LT-HSC. (H-I) Cell suspensions prepared from the synovium of knee joints (harvested on day 7) were stained for live/dead, CD45, CD45.1, CD45.2, CD11b, CD11c, Ly6C and Ly6G followed by FACS to identify and quantify monocytes (live CD45+CD11c−CD11b+Ly6G−Ly6C+), neutrophils (live CD45+CD11c−CD11b+Ly6G+Ly6C−), and CD45+ or CD45.2 + leukocytes. Total numbers of monocytes, neutrophils and CD45+ cells (top), percentages of CD45.2+ cells in monocytes, neutrophils and CD45+ cells (middle), and absolute numbers of CD45.2+ monocytes, neutrophils and total CD45.2+ cells (bottom) in mice transplanted with CD41+ (H) or CD41− (I) LT-HSC. (J,K) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6 and TNF concentration by ELISA in mice transplanted with CD41+ (J) or CD41− (K) LT-HSC. Data are means ± SD (B,C,F-I, n=6 mice/group; D,E,J,K n=4 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice. Two-tailed Student’s t-test (D,E,H-K) except for panel I, 2nd row, 1st left (two-tailed Mann-Whitney U test); two-way repeated measures ANOVA and Sidak’s multiple comparisons test (B,C,F,G). T, Trained; U, Untrained.
Figure S7. Role of IL-1-signaling in LIP and LIP-induced BM myelopoiesis. (Related to Figure 7). (A) Il1r1HSPC-KO mice and WT littermate controls were subjected to tamoxifen treatment to induce Il1r1 deletion in HSPC. Mice were then subjected to ligature-induced periodontitis (LIP) for 21 days. BM cells were harvested and the frequencies of LSK, ST-HSC, MPP in total BM cells, CD41+ cells in total LT-HSC, MPP3 in LSK, GMP in MyP pool, Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells in CD45+ cells, were analyzed by FACS. (B) CD45.2 Il1r1HSPC-KO and littermate controls with intact HSPC IL-1R expression were trained by subjecting them to 21-day LIP and 14-day resolution and then used as donors for BMT to groups of lethally irradiated congenic B6.SJL (CD45.1+) mice. Frequency of donor-derived LSK, LT-HSC, ST-HSC and MPP cells in CD45.2+ BM cells of recipient mice at 12 weeks post-BMT. (C) Mice with global IL-1R deletion (Il1r1KO) mice and WT littermate controls were subjected to 5-day ligature-induced periodontitis and bone loss was measured. Data are means ± SD (A, n=5 mice/group; B,C, n=6 mice/group). *P<0.05, **P<0.01, ***P<0.001, NS, not significant vs. WT littermate control mice; two-tailed Student’s t-test.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse Lineage Cocktail | Biolegend | Cat#133301 |
| Biotin anti-mouse Lineage Panel | Biolegend | Cat#133307 |
| Rat anti-mouse cKit (CD117) | Biolegend | Cat#105808; RRID: AB_313217 |
| Rat anti-mouse Sca1 (Ly6-A/E) | Biolegend | Cat#122514; RRID: AB_756199 |
| Armenian Hamster anti-mouse CD48 | Biolegend | Cat#103426; RRID: AB_10612755 |
| Rat anti-mouse CD150 | Biolegend | Cat#115922; RRID: AB_2303663 |
| Rat anti-mouse CD16/CD32 | Biolegend | Cat#101324; RRID: AB_1877267 |
| Armenian Hamster anti-mouse CD34 | Biolegend | Cat#128612; RRID: AB_10553896 |
| Rat anti-mouse CD135 | Biolegend | Cat#135310, 135315; RRID: AB_2107050, AB_2571919 |
| Rat anti-mouse CD41 | Biolegend | Cat#133912; RRID: AB_2650893 |
| Armenian Hamster anti-mouse CD61 | Biolegend | Cat#104316; RRID: AB_2561734 |
| Rat anti-mouse CD11b | BD Pharmingen™ | Cat# 552850; RRID: AB_394491 |
| Rat anti-mouse Gr-1(Ly-6G/C) | eBioscience™ | Cat#48-5931-82; RRID: AB_1548788 |
| Rat anti-mouse CD45 | Biolegend | Cat#103132; RRID: AB_893340 |
| Rat anti-mouse CD19 | Biolegend | Cat#115508; RRID: AB_313643 |
| Rat anti-mouse CD3 | Biolegend | Cat#100236; RRID: AB_2561456 |
| Mouse anti-mouse CD45.1 | Biolegend | Cat#110728; RRID: AB_893346 |
| Mouse anti-mouse CD45.2 | Biolegend | Cat#109828; RRID: AB_893350 |
| Rat anti-mouse Ly6G | Biolegend | Cat#127614, 127604; RRID: AB_2227348, AB_1186108 |
| Rat anti-mouse Ly6C | Biolegend | Cat#128008, 128004; RRID: AB_1186132, AB_1236553 |
| Armenian Hamster anti-mouse CD11c | Biolegend | Cat#117306; RRID: AB_313775 |
| Rat anti-mouse CD115 | Biolegend | Cat#135526; RRID: AB_2566462 |
| Rat anti-mouse CXCR4 | Biolegend | Cat#146508; RRID: AB_2562785 |
| Rat anti-mouse CXCR2 | Biolegend | Cat#149610; RRID: AB_2565690 |
| Arthritogenic monoclonal antibodies (5-clone collagen antibody cocktail) | Chondrex | Cat#53010 |
| Chemicals, Enzymes, Reagents and recombinant proteins | ||
| Collagenase IV | Gibco | Cat#17104019; CAS: 9001-12-1 |
| ACK Lysing Buffer | Gibco | Cat#A1049201 |
| Percoll gradient | GE Healthcare | Cat#17-0891-01 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Tamoxifen-containing (0.5 mg/g) diet | Envigo | Cat#TD.130857 |
| Nonidet P40 Substrate | Sigma-Aldrich | Cat#74385 |
| Nuclei Buffer | 10x Genomics | Cat#2000153 |
| Streptavidin microBeads | Miltenyi Biotec | Cat# 130-048-101 |
| Anti-biotin microbeads | Miltenyi Biotec | Cat# 130-090-485 |
| LPS-EB (LPS from E. coliO111:B4) | Invivogen | Cat# tlrl-eblps |
| DAPI (4’,6-Diamidino-2-Phenylindole,Dihydrochloride) | ThermoFisher Scientific | Cat# D1306 |
| TRIzol™ Reagent | Invitrogen | Cat# 15596026 |
| Mouse recombinant G-CSF | R&D Systems | Cat# 414-CS-005/CF |
| Critical commercial assays | ||
| IL-6 mouse ELISA kit | Invitrogen | Cat# 88-7064-88 |
| IL-1β mouse ELISA kit | Invitrogen | Cat# 88-7013-88 |
| IFN-γ mouse ELISA kit | Invitrogen | Cat# 88-7314-88 |
| IL-12p70 mouse ELISA kit | Invitrogen | Cat# 88-7121-88 |
| IL-10 mouse ELISA kit | Invitrogen | Cat# 88-7105-88 |
| Mouse G-CSF ELISA | RayBiotech | Cat# ELM-GCSF-1 |
| High-Capacity RNA-to-cDNA Kit | Applied Biosystems | Cat# 4387406 |
| TURBO DNA-free Kit | Invitrogen | Cat# AM1907 |
| Chromium Next GEM Chip H Single Cell Kit v1.1 | 10x Genomics | Cat#1000162 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10x Genomics | Cat#1000176 |
| NGS High Sensitivity Fragment Analysis Kit | Agilent | Cat#DNF-474 |
| NovaSeq 6000 SP Reagent Kit v1.5 (100 cycles) | Illumina | Cat#20028401 |
| Chromium Next GEM Single Cell 3ʹ Kit v3.1, 16 rxns | 10x Genomics | Cat# PN-1000268 |
| ATAC-Seq Kit | Active Motif | Cat#53150 |
| Deposited data | ||
| RNA sequencing data | This paper | GEO: GSE180002 |
| Single cell ATAC sequencing data | This paper | GEO: GSE180025 |
| Single cell RNA sequencing data | This paper | GEO: GSE196808 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | Stock#000664 |
| Mouse: C57BL/6-CD45.1 B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Stock#002014 |
| Mouse: B6.129(Cg)-Il1r1tm1.1Rbl/J (Il1r1fl/fl mice) | The Jackson Laboratory | Stock#028398 |
| Mouse: HSC-SCL-Cre-ERT | Donated by Dr. Joachim R. Göthert, University Hospital Essen | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | Graphpad Software | N/A |
| STAR | (Dobin et al., 2013) | http://code.google.com/p/rna-star/ |
| Ingenuity Pathway Analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| NovoExpress software | ACEA Biosciences | https://www.aceabio.com |
| Cell Ranger ATAC version 1.2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-atac/software/overview/welcome |
| R package ArchR v1.0.1 | (Granja et al., 2021) | https://github.com/GreenleafLab/ArchR/releases/tag/v1.0.1 |
| R package Seurat | (Stuart et al., 2019) | https://github.com/satijalab/seurat/releases/tag/v3.0.0 |
| R package Signac | (Stuart et al., 2020) | https://github.com/timoast/signac/releases/tag/1.2.1 |
| Monocle 3 v1.0.0 | (Cao et al., 2019) | https://github.com/cole-trapnell-lab/monocle3/releases/tag/1.0.0 |
| Cicero v1.4 | (Pliner et al., 2018) | https://github.com/stjude/CICERO/releases/tag/v1.4.0 |
Highlights.
Experimental periodontitis (EP) induces maladaptive trained myelopoiesis
EP-induced trained phenotype is transmissible by bone marrow transplantation
IL-1 signaling in hematopoietic progenitors mediates maladaptive training by EP
Maladaptively trained myelopoiesis links the periodontitis-arthritis comorbidity
Acknowledgements
This work was supported by NIH grants (DE029436 and DE031206 to GH; DE028561 to GH and TC), and the Deutsche Forschungsgemeinschaft (SFB-TR 127, A3 and SFB1181, C7 to TC). T.C. is also supported by the British Heart Foundation Centre for Research Excellence at The University of Edinburgh (RE/18/5/34216) and the ‘Sonderzuweisung zur Unterstützung profilbestimmender Struktureinheiten 2021’ by the SMWK. MGN is supported by a Spinoza grant of the Netherlands Organization for Scientific Research and an ERC Advanced Grant (#833247). We thank Sylvia Grossklaus (Institute for Clinical Chemistry and Laboratory Medicine, Technische Universität Dresden) for technical assistance. The graphical abstract was created using BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
MGN is the scientific founder and member of Scientific Advisory Board of Trained Therapeutics and Discovery (TTxD). MGN has two patents: US18/61935 (Targeted nanoimmunotherapy to increase trained immunity) and US18/61939 (Targeted nanoimmunotherapy for inhibition of trained immunity). The other authors declare no interest.
References
- Abe T, and Hajishengallis G (2013). Optimization of the ligature-induced periodontitis model in mice. J Immunol Meth 394, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj JS, Matin P, White MB, Fagan A, Deeb JG, Acharya C, Dalmet SS, Sikaroodi M, Gillevet PM, and Sahingur SE (2018). Periodontal therapy favorably modulates the oral-gut-hepatic axis in cirrhosis. Am J Physiol Gastrointest Liver Physiol 315, G824–G837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen MLT, Schaltz-Buchholzer F, Bles P, Biering-Sørensen S, Jensen KJ, Monteiro I, Silva I, Aaby P, and Benn CS (2021). Parental Bacillus Calmette-Guérin vaccine scars decrease infant mortality in the first six weeks of life: A retrospective cohort study. EClinicalMedicine 39, 101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernelot Moens SJ, van der Valk FM, Strang AC, Kroon J, Smits LP, Kneepkens EL, Verberne HJ, van Buul JD, Nurmohamed MT, and Stroes ES (2016). Unexpected arterial wall and cellular inflammation in patients with rheumatoid arthritis in remission using biological therapy: a cross-sectional study. Arthritis Res Ther 18, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomans K, Schenz J, Tamulyte S, Schaack D, Weigand MA, and Uhle F (2018). Paternal sepsis induces alterations of the sperm methylome and dampens offspring immune responses—an animal study. Clin Epig 10, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548.e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley MD, Haynes DR, Marino V, and Bartold PM (2011). Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J Clin Periodontol 38, 532–541. [DOI] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavakis T, Mitroulis I, and Hajishengallis G (2019). Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat Immunol 20, 802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarlo E, Heinonen T, Théroude C, Asgari F, Le Roy D, Netea MG, and Roger T (2020). Trained Immunity Confers Broad-Spectrum Protection Against Bacterial Infections. J Infect Dis 222, 1869–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden WJFM, Bremmers MEJ, van Crevel R, Händler K, Picelli S, et al. (2020). BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe 28, 322–334.e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aiuto F, Gkranias N, Bhowruth D, Khan T, Orlandi M, Suvan J, Masi S, Tsakos G, Hurel S, Hingorani AD, et al. (2018). Systemic effects of periodontitis treatment in patients with type 2 diabetes: a 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol 6, 954–965. [DOI] [PubMed] [Google Scholar]
- D’Aiuto F, Orlandi M, and Gunsolley JC (2013). Evidence that periodontal treatment improves biomarkers and CVD outcomes. J Clin Periodontol 40 Suppl 14, S85–105. [DOI] [PubMed] [Google Scholar]
- de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, Huber C, Gimenez G, Matcovitch-Natan O, Reinhardt S, David E, et al. (2020). C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 26, P657–674.E658. [DOI] [PubMed] [Google Scholar]
- de Pablo P, Dietrich T, and McAlindon TE (2008). Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol 35, 70–76. [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, et al. (2018). A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med 10, eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucchi S, Dominguez-Andres J, Joosten LAB, Netea MG, and Mhlanga MM (2021). The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 54, 32–43. [DOI] [PubMed] [Google Scholar]
- Fifer KM, Qadir S, Subramanian S, Vijayakumar J, Figueroa AL, Truong QA, Hoffman U, Brady TJ, and Tawakol A (2011). Positron Emission Tomography Measurement of Periodontal 18F-Fluorodeoxyglucose Uptake Is Associated With Histologically Determined Carotid Plaque Inflammation. J Amer Coll Cardiol 57, 971–976. [DOI] [PubMed] [Google Scholar]
- Flak MB, Colas RA, Muñoz-Atienza E, Curtis MA, Dalli J, and Pitzalis C (2019). Inflammatory arthritis disrupts gut resolution mechanisms, promoting barrier breakdown by Porphyromonas gingivalis. JCI Insight 4, e125191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuggle NR, Smith TO, Kaul A, and Sofat N (2016). Hand to Mouth: A Systematic Review and Meta-Analysis of the Association between Rheumatoid Arthritis and Periodontitis. Front Immunol 7, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S, Chandiramani M, Seow J, Pollock E, Modestini C, Das A, Tree T, Doores KJ, Tribe RM, and Gibbons DL (2021). The legacy of maternal SARS-CoV-2 infection on the immunology of the neonate. Nat Immunol 22, 1490–1502. [DOI] [PubMed] [Google Scholar]
- Gekas C, and Graf T (2013). CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121, 4463–4472. [DOI] [PubMed] [Google Scholar]
- Genco RJ, and Sanz M (2020). Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol 2000 83, 7–13. [DOI] [PubMed] [Google Scholar]
- Gothert JR, Gustin SE, Hall MA, Green AR, Gottgens B, Izon DJ, and Begley CG (2005). In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood 105, 2724–2732. [DOI] [PubMed] [Google Scholar]
- Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G (2015). Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, and Chavakis T (2021). Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol 21 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Li X, and Chavakis T (2021). Immunometabolic control of hematopoiesis. Mol Aspects Med 77, 100923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishai A, Osborne MT, El Kholy K, Takx RAP, Ali A, Yuan N, Hsue P, Van Dyke TE, and Tawakol A (2019). Periodontal Disease Associates With Arterial Inflammation Via Potentiation of a Hematopoietic-Arterial Axis. JACC Cardiovasc Imaging 12, 2271–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Pflugh DL, Yu D, Hesslein DG, Lin KI, Bothwell AL, Thomas-Tikhonenko A, Schatz DG, and Calame K (2004). B cell-specific loss of histone 3 lysine 9 methylation in the V(H) locus depends on Pax5. Nat Immunol 5, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, Hagag E, Sinha A, Has C, Dietz S, et al. (2020). Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 183, 771–785.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, and Marcenes W (2014). Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res 93, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmarski N, Domínguez-Andrés J, Cirovic B, Renieris G, Ciarlo E, Le Roy D, Lepikhov K, Kattler K, Gasparoni G, Händler K, et al. (2021). Transmission of trained immunity and heterologous resistance to infections across generations. Nat Immunol 22, 1382–1390. [DOI] [PubMed] [Google Scholar]
- Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier JC, et al. (2018). BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190 e119. [DOI] [PubMed] [Google Scholar]
- Khachigian LM (2006). Collagen antibody-induced arthritis. Nat Protoc 1, 2512–2516. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim KB, Eom GH, Choe N, Kee HJ, Son HJ, Oh ST, Kim DW, Pak JH, Baek HJ, et al. (2012). KDM3B is the H3K9 demethylase involved in transcriptional activation of lmo2 in leukemia. Mol Cell Biol 32, 2917–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG III, Hayashi A, Imai J, Sugihara K, Miyoshi M, Brazil JC, Kuffa P, et al. (2020). The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 182, 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, Rosen A, Nigrovic PA, Sokolove J, Giles JT, et al. (2016). Aggregatibacter actinomycetemcomitans–induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med 8, 369ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, Grzybek M, von Renesse J, Czogalla A, Troullinaki M, et al. (2019). DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol 20, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, and Choi YY (2020). Rheumatoid arthritis and periodontitis in adults: using the Korean National Health Insurance Service – National Sample Cohort. J Periodontol 91, 1186–1193. [DOI] [PubMed] [Google Scholar]
- Li X, Colamatteo A, Kalafati L, Kajikawa T, Wang H, Lim J-H, Bdeir K, Chung K-J, Yu X, Fusco C, et al. (2020). The DEL-1/β3 integrin axis promotes regulatory T cell responses during inflammation resolution. J Clin Invest 130 6261–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AI, McFadden T, Link VM, Han S-J, Karlsson R-M, Stacy A, Farley TK, Lima-Junior DS, Harrison OJ, Desai JV, et al. (2021). Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 373, eabf3002. [DOI] [PubMed] [Google Scholar]
- Ling MR, Chapple IL, and Matthews JB (2015). Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun 21, 714–725. [DOI] [PubMed] [Google Scholar]
- Listl S, Galloway J, Mossey PA, and Marcenes W (2015). Global Economic Impact of Dental Diseases. J Dent Res 94, 1355–1361. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Kawanami D, Sharma N, Takami Y, Zhou G, Shi H, Nayak L, Jeyaraj D, Grealy R, White M, et al. (2011). The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity 34, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Mehta A, de Boer CG, Kowalczyk MS, Lee K, Haldeman P, Rogel N, Knecht AR, Farouq D, Regev A, et al. (2018). Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep 25, 2992–3005.e2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, Gawron K, Mizgalska D, Marcinska KA, Benedyk M, et al. (2013). Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog 9, e1003627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, Markt J, McGowan D, Kerr GS, Redman RS, et al. (2014). Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol 66, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroulis I, Chen L-S, Singh RP, Kourtzelis I, Economopoulou M, Kajikawa T, Troullinaki M, Ziogas A, Ruppova K, Hosur K, et al. (2017). Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J Clin Invest 127, 3624–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161 e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, Divangahi M, and Netea MG (2020a). β-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep 31, 107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorlag SJCFM, Rodriguez-Rosales YA, Gillard J, Fanucchi S, Theunissen K, Novakovic B, de Bont CM, Negishi Y, Fok ET, Kalafati L, et al. (2020b). BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep 33, 108387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. (2020). Defining trained immunity and its role in health and disease. Nat Rev Immunol 20, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkov S, Mitroulis I, Hajishengallis G, and Chavakis T (2019). Immunometabolic Crosstalk: An Ancestral Principle of Trained Immunity? Trends Immunol 40, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres MA, Macpherson LMD, Weyant RJ, Daly B, Venturelli R, Mathur MR, Listl S, Celeste RK, Guarnizo-Herreño CC, Kearns C, et al. (2019). Oral diseases: a global public health challenge. The Lancet 394, 249–260. [DOI] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, and Avvedimento EV (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. [DOI] [PubMed] [Google Scholar]
- Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H, van Heeringen SJ, and Veenstra GJC (2018). MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat Genet 50, 1002–1010. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 71, 858–871.e858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potempa J, Mydel P, and Koziel J (2017). The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol 13, 606–620. [DOI] [PubMed] [Google Scholar]
- Qing Y, Yin F, Wang W, Zheng Y, Guo P, Schozer F, Deng H, and Pan D (2014). The Hippo effector Yorkie activates transcription by interacting with a histone methyltransferase complex through Ncoa6. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radvar M, Tavakkol-Afshari J, Bajestan MN, Naseh MR, and Arab HR (2008). The effect of periodontal treatment on IL-6 production of peripheral blood monocytes in aggressive periodontitis and chronic periodontitis patients. Iran J Immunol 5, 100–106. [DOI] [PubMed] [Google Scholar]
- Ramamurthy NS, Greenwald RA, Celiker MY, and Shi EY (2005). Experimental Arthritis in Rats Induces Biomarkers of Periodontitis Which Are Ameliorated by Gene Therapy With Tissue Inhibitor of Matrix Metalloproteinases. J Periodontol 76, 229–233. [DOI] [PubMed] [Google Scholar]
- Ridger VC, Wagner BE, Wallace WA, and Hellewell PG (2001). Differential effects of CD18, CD29, and CD49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J Immunol 166, 3484–3490. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Lozano B, González-Febles J, Garnier-Rodríguez JL, Dadlani S, Bustabad-Reyes S, Sanz M, Sánchez-Alonso F, Sánchez-Piedra C, González-Dávila E, and Díaz-González F (2019). Association between severity of periodontitis and clinical activity in rheumatoid arthritis patients: a case–control study. Arthritis Res Ther 21, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, Nakajima T, Kondo N, Endo N, Yamamoto R, et al. (2017). Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep 7, 6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein HA, Papapanou PN, Genco R, and Sanz M (2020). Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000 83, 90–106. [DOI] [PubMed] [Google Scholar]
- Scher JU, Bretz WA, and Abramson SB (2014). Periodontal disease and subgingival microbiota as contributors for rheumatoid arthritis pathogenesis: modifiable risk factors? Curr Opin Rheumatol 26, 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoedel KB, Morcos MNF, Zerjatke T, Roeder I, Grinenko T, Voehringer D, Göthert JR, Waskow C, Roers A, and Gerbaulet A (2016). The bulk of the hematopoietic stem cell population is dispensable for murine steady-state and stress hematopoiesis. Blood 128, 2285–2296. [DOI] [PubMed] [Google Scholar]
- Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, Mitroulis I, Chavakis T, and Hajishengallis G (2015). DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med 7, 307ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siklenka K, Erkek S, Godmann M, Lambrot R, McGraw S, Lafleur C, Cohen T, Xia J, Suderman M, Hallett M, et al. (2015). Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350, aab2006. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, et al. (2018). Rheumatoid arthritis. Nat Rev Dis Primers 4, 18001. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Lareau C, and Satija R (2020). Multimodal single-cell chromatin analysis with Signac. bioRxiv, 2020.2011.2009.373613. [Google Scholar]
- Sun S, and Barreiro LB (2020). The epigenetically-encoded memory of the innate immune system. Curr Opin Immunol 65, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, Iwakura Y, Nakashima T, Okamoto K, and Takayanagi H (2018). Host defense against oral microbiota by bone-damaging T cells. Nat Commun 9, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke TE, Kholy KE, Ishai A, Takx RAP, Mezue K, Abohashem SM, Ali A, Yuan N, Hsue P, Osborne MT, et al. (2021). Inflammation of the periodontium associates with risk of future cardiovascular events. J Periodontol 92, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verploegen S, Ulfman L, van Deutekom HW, van Aalst C, Honing H, Lammers JW, Koenderman L, and Coffer PJ (2005). Characterization of the role of CaMKI-like kinase (CKLiK) in human granulocyte function. Blood 106, 1076–1083. [DOI] [PubMed] [Google Scholar]
- Wang H, Li X, Kajikawa T, Shin J, Lim JH, Kourtzelis I, Nagai K, Korostoff JM, Grossklaus S, Naumann R, et al. (2021). Stromal cell-derived DEL-1 inhibits Tfh cell activation and inflammatory arthritis. J Clin Invest 131, e150578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winning L, and Linden GJ (2017). Periodontitis and Systemic Disease: Association or Causality? Curr Oral Health Rep 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh DY, Maekawa T, Li X, Kajikawa T, Bdeir K, Chavakis T, and Hajishengallis G (2020). The secreted protein DEL-1 activates a β3 integrin-FAK-ERK1/2-RUNX2 pathway and promotes osteogenic differentiation and bone regeneration. J Biol Chem 295, 7261–7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LIP activates bone marrow and enhances myelopoiesis (Related to Figure 1). Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) followed by BM analysis after 7 days. (A, left) Representative FACS plots for identification of hematopoietic stem and progenitor cells. After gating for Lin− cells, LSK were identified as cKit+Sca1+ cells. LSK subpopulations were further characterized as MPP (CD48+CD150−LSK), ST-HSC (CD48−CD150−LSK), and LT-HSC (CD48−CD150+LSK) cells. (A, right) Frequencies of LSK, LT-HSC, ST-HSC and MPP in total BM cells (top) and absolute cell numbers (bottom) of the same populations. (B, top) Representative FACS plots of MPP subsets. After gating for LSK, MPP4 were defined as CD48+Flt3+CD150−LSK, MPP3 as CD48+Flt3−CD150−LSK, and MPP2 cells as CD48+Flt3−CD150+LSK. (B, bottom) Frequencies of MPP subsets in LSK in the BM. (C, left) Representative FACS plots for the identification of CD41+ and CD61+ LT-HSC cells and (right) frequency of the same populations in total LT-HSC cells in the BM of mice. (D-G) Groups of mice were subjected, or not (control, NL), to ligature-induced periodontitis (LIP) and BM cells were harvested on day 7. FACS-sorted LSK were subjected to RNA-sequencing and transcriptome analysis. (D) Heatmaps of myeloid lineage, osteoclast differentiation. (E) Heatmaps of erythroid, lymphoid lineage and oxidative phosphorylation related genes. (F) Heatmap of significantly regulated epigenetic regulators by LIP. (G) Significant upstream transcription factors (left) and upstream regulators (right) predicted by Ingenuity Pathway Analysis (IPA). Data in (A-C) are means ± SD (n=6 mice/group). *P<0.05, **P<0.01, NS, not significant vs. control mice; two-tailed Student’s t-test.
Figure S2. Inflammation resolution in the periodontium phenotypically restores myelopoiesis to steady-state levels in the BM (Related to Figure 2). Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) for 21 days and subsequently the ligatures were removed (to enable inflammation resolution) for 14 days, at which time BM analysis was performed. (A) Relative mRNA expression of indicated cytokines in the gingival tissue of mice after 14 days resolution. (B) Frequencies in total BM cells (top) and absolute cell numbers (bottom) of LSK, LT-HSC, ST-HSC and MPP. (C) Frequencies of MPP subsets in LSK in the BM. (D) Frequencies of CD41+ LT-HSC in total LT-HSC cells in the BM. (E) Frequency within the MyP pool of GMP and CMP (left and middle, respectively) and cell numbers of GMP (right) in the BM. (F) Frequency of Gr1hiCD11b+ granulocytes, Gr1intCD11b+ myeloid cells, and CD19+ B cells in CD45+ cells in the BM. (G-H) BM LSK were sorted from mice that were subjected to 21dL/14dR or not (NL) and scRNA-seq was performed. (G) GO enrichment results of top 20 significantly enriched GO terms sorted by PANTHER on the basis of marker genes specific for cluster 7 shown in Figure 2A–C (Bonferroni-corrected, P<0.05). (H) Two-dimensional UMAP showing the expression of myeloid marker genes (gray denotes minimal expression, purple intermediate and blue high). Data are means ± SD (n=6 mice/group) (A-F). *P<0.05, NS, not significant vs. control mice; two-tailed Student’s t-test.
Figure S3. Single-cell ATACseq analysis (Related to Figure 3). (A) Violin plots showing gene activity scores of Itga2b and Gata2 across cluster 2. (B) Violin plots showing gene activity scores of Csf1r and Nlrp3 across cluster 3. (C) A cellular trajectory spanning from LSK to GMP. (D) Bubble plot of GO enrichment analysis of the cluster 5-specific marker genes. (E) Violin plots showing gene activity scores of Tlr4, Camk1d, Itga1 and Map2k4 across cluster 5.
Figure S4. LIP-subjected mice are more susceptible to CAIA than non-ligated control mice (Related to Figure 4). (A) Groups of mice were subjected, or not (control), to ligature-induced periodontitis (LIP) for 21 days followed by a 14-day resolution. Mice were then subjected to CAIA model and monitored for 14 days. (B) Clinical arthritis score and hind ankle joint thickness were measured daily. (C) CD45.2+ mice were either trained (T) by subjecting them to 21-day LIP followed by 14-day resolution (‘21dL/14dR’-trainedmice), or were left untrained (U; naïve controls). BM cells were isolated from trained and untrained mice and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. Twelve weeks after receiving BM transplantation from trained or untrained donor mice, splenic monocytes and neutrophils were isolated. (D) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6 and TNF concentration by ELISA. (E-F) Frequency of donor-derived cells in the BM of recipient mice (E) and frequency of donor-derived (CD45.2+) LSK, LT-HSC, ST-HSC and MPP cells in respective populations in the BM of recipient mice (F) at 16 weeks post-BMT, as described in Figure 4H. Data are means ± SD (n = 6 recipient mice/group). P* <0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice; two-way repeated measures ANOVA and Sidak’s multiple comparisons test (B); two-tailed Student’s t-test (D,E,F). T, Trained; U, Untrained.
Figure S5. CAIA activates bone marrow and enhances myelopoiesis (Related to Figure 5). (A-F) mice were subjected, or not (control), to CAIA and BM analysis was performed after 14 days. (A) Frequencies (top) of LSK, LT-HSC, ST-HSC and MPP in total BM cells and absolute cell numbers (bottom) of the same populations. (B) Frequency of MPP subsets in LSK in the BM. (C) Frequency of CD41+ LT-HSC in total LT-HSC cells in the BM. (D) Absolute cell numbers of GMP (left) and frequency within the MyP pool of GMP (right). (E) Absolute cell numbers and frequency of Gr1hiCD11b+ granulocytes in CD45+ cells in the BM. (F,G) mice were subjected, or not (control), to CAIA and monitored daily for ankle joint thickness (F). BM analysis was performed on day 36, i.e., 14 days after complete arthritis resolution. (G) Frequencies of LSK, LT-HSC, ST-HSC and MPP cells in total BM cells. (H) CD45.2+ mice were either trained (T) by subjecting them to CAIA (disease activity returned to baseline on day 22 and the mice were rested post-resolution for 14 days; total 36 days), or were left untrained (U; naïve controls). 36 days post-CAIA, BM cells were isolated and transferred to lethally irradiated congenic B6.SJL CD45.1+ mice. Twelve weeks after receiving BM transplantation from donor mice, splenic monocytes and neutrophils from recipient mice were isolated. (I) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6, and TNF concentration by ELISA. Data are means ± SD (n=6 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. control or untrained mice; two-tailed Student’s t-test. T, Trained; U, Untrained.
Figure S6. Effects of competitive BM transplantation using sorted CD41+ and CD41− LT-HSC. (Related to Figure 5). (A) CD45.1+ mice were transplanted with 200 CD41+ or CD41− LT-HSC sorted from LIP-trained or untrained CD45.2+ mice together with 8×105 CD45.1+ BM competitor cells. (B-C) WBC count, donor-derived percentage, and lineage output (% of indicated cell types in donor-derived cells at the indicated time points) in the peripheral blood of mice transplanted with CD41+ (B) or CD41− (C) LT-HSC. (D-E) Frequency of donor-derived cells in the BM of recipient mice and frequency of donor-derived (CD45.2+) LSK, LT-HSC, ST-HSC and MPP cells in respective populations in the BM of mice transplanted with CD41+ (D) or CD41− (E) LT-HSC, at 12 weeks post-BMT. (F-I) 12 weeks post-BMT, the recipient CD45.1+ mice were subjected to CAIA. Clinical arthritis score (left) and hind ankle joint thickness (right) in mice transplanted with CD41+ (F) or CD41− (G) LT-HSC. (H-I) Cell suspensions prepared from the synovium of knee joints (harvested on day 7) were stained for live/dead, CD45, CD45.1, CD45.2, CD11b, CD11c, Ly6C and Ly6G followed by FACS to identify and quantify monocytes (live CD45+CD11c−CD11b+Ly6G−Ly6C+), neutrophils (live CD45+CD11c−CD11b+Ly6G+Ly6C−), and CD45+ or CD45.2 + leukocytes. Total numbers of monocytes, neutrophils and CD45+ cells (top), percentages of CD45.2+ cells in monocytes, neutrophils and CD45+ cells (middle), and absolute numbers of CD45.2+ monocytes, neutrophils and total CD45.2+ cells (bottom) in mice transplanted with CD41+ (H) or CD41− (I) LT-HSC. (J,K) Isolated splenic monocytes and neutrophils were stimulated with LPS (10ng/ml) for 24h. The supernatant was collected for measuring IL-6 and TNF concentration by ELISA in mice transplanted with CD41+ (J) or CD41− (K) LT-HSC. Data are means ± SD (B,C,F-I, n=6 mice/group; D,E,J,K n=4 mice/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, not significant vs. untrained mice. Two-tailed Student’s t-test (D,E,H-K) except for panel I, 2nd row, 1st left (two-tailed Mann-Whitney U test); two-way repeated measures ANOVA and Sidak’s multiple comparisons test (B,C,F,G). T, Trained; U, Untrained.
Figure S7. Role of IL-1-signaling in LIP and LIP-induced BM myelopoiesis. (Related to Figure 7). (A) Il1r1HSPC-KO mice and WT littermate controls were subjected to tamoxifen treatment to induce Il1r1 deletion in HSPC. Mice were then subjected to ligature-induced periodontitis (LIP) for 21 days. BM cells were harvested and the frequencies of LSK, ST-HSC, MPP in total BM cells, CD41+ cells in total LT-HSC, MPP3 in LSK, GMP in MyP pool, Gr1hiCD11b+ granulocytes and Gr1intCD11b+ myeloid cells in CD45+ cells, were analyzed by FACS. (B) CD45.2 Il1r1HSPC-KO and littermate controls with intact HSPC IL-1R expression were trained by subjecting them to 21-day LIP and 14-day resolution and then used as donors for BMT to groups of lethally irradiated congenic B6.SJL (CD45.1+) mice. Frequency of donor-derived LSK, LT-HSC, ST-HSC and MPP cells in CD45.2+ BM cells of recipient mice at 12 weeks post-BMT. (C) Mice with global IL-1R deletion (Il1r1KO) mice and WT littermate controls were subjected to 5-day ligature-induced periodontitis and bone loss was measured. Data are means ± SD (A, n=5 mice/group; B,C, n=6 mice/group). *P<0.05, **P<0.01, ***P<0.001, NS, not significant vs. WT littermate control mice; two-tailed Student’s t-test.
Data Availability Statement
Data are available upon request to the Lead Contact. Sequencing data are available at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE180032.