

Abstract
B cells, which are critical for intestinal homeostasis, remain understudied in ulcerative colitis (UC). In this study, we recruited three cohorts of patients with UC (primary cohort, n = 145; validation cohort 1, n = 664; and validation cohort 2, n = 143) to comprehensively define the landscape of B cells during UC-associated intestinal inflammation. Using single-cell RNA sequencing, single-cell IgH gene sequencing and protein-level validation, we mapped the compositional, transcriptional and clonotypic landscape of mucosal and circulating B cells. We found major perturbations within the mucosal B cell compartment, including an expansion of naive B cells and IgG+ plasma cells with curtailed diversity and maturation. Furthermore, we isolated an auto-reactive plasma cell clone targeting integrin αvβ6 from inflamed UC intestines. We also identified a subset of intestinal CXCL13-expressing TFH-like T peripheral helper cells that were associated with the pathogenic B cell response. Finally, across all three cohorts, we confirmed that changes in intestinal humoral immunity are reflected in circulation by the expansion of gut-homing plasmablasts that correlates with disease activity and predicts disease complications. Our data demonstrate a highly dysregulated B cell response in UC and highlight a potential role of B cells in disease pathogenesis.
UC is a chronic inflammatory bowel disease (IBD) characterized by relapsing episodes of inflammation of the colonic mucosa1. In healthy individuals, intestinal B cell responses are dominated by the homeostatic generation of IgA-producing plasma cells (PCs) that promote tolerance at mucosal surfaces through pleotropic effects2–7. Host IgA also shapes the microbial communities of the microbiota by mediating colonization of specific commensals in defined intestinal niches and can select specific bacteria through differential adhesion8,9.
Although less frequent than IgA-producing PCs, IgG-producing PCs have been identified in the healthy intestinal mucosa. Additionally, polyreactive or monoreactive clonotypes recognizing commensals and/or pathogens have been identified10,11. Intestinal IgG is thought to play a protective role against pathogens but can also enhance inflammation in IBD11–13.
T follicular helper (TFH) cells, a subset of CD4+ T cells, which are specialized in the induction of T cell help to B cells14, are increased in circulation of patients with UC15. TFH-like T peripheral helper (TPH) cells can drive pathological B cell responses in the synovium of patients with rheumatoid arthritis16, but they have not been identified in UC.
Given that recent data have suggested involvement of B cells in the pathogenesis of IBD17,18, the goal of the present study was to define the phenotypic and molecular landscape of intestinal and circulating B cells in patients with UC.
Results
Remodeling of the B cell compartment in UC.
To dissect the dynamics of B cells in UC, three distinct cohorts of patients were studied (Extended Data Fig. 1 and Methods). Lamina propria cells were isolated from the left colon of five healthy controls (HCs) and four patients with UC with active disease (Supplementary Table 1), analyzed by single-cell RNA sequencing (scRNA-seq)19 and mapped to 47 previously identified clusters17. Grouping the clusters by expression profile similarities indicated the presence of seven major compartments consisting of stromal/glial cells and six immune cell lineages (Extended Data Fig. 2a), including T cells (CD3D, CD2 and CD7), innate lymphoid cells (CD2, CD7 and no CD3D), B cells (BANK1, CD79B, CD22, MS4A1 and MHC-II genes), PCs (TNFRSF17 and MZB1), mononuclear phagocytes (LYZ and MHC-II genes) and mast cells (TPSAB1) (Extended Data Fig. 2b). Overall, the proportions of these seven cell lineages were similar in HCs and patients with UC (Extended Data Fig. 2c).
B cells and PCs were selected in silico (Methods) and re-clustered using an unsupervised clustering algorithm17. Twenty clusters of B cells (CD19, MS4A1, BANK1 and HLA-DRA) and PCs (SDC1, XBP1, TNFRSF17, PRDM1, MZB1 and SEC11C) with variable cell numbers (276–3,778 cells; Supplementary Table 2), and unique molecular inhibitor (UMI) counts per cell were identified (Extended Data Fig. 3a,b and Supplementary Table 3). By analyzing co-expression patterns of gene modules defined by strong gene-to-gene correlation of expression (Extended Data Fig. 3c and Supplementary Table 4), we identified naive (IGHD, FCER2 and CD72; clusters 8 and 12) and memory (CD27, TNFRSF13B and no expression of IGHD; clusters 2, 7 and 13) B cells (Fig. 1a and Extended Data Fig. 3d), including atypical memory B cells (cluster 13; FCRL5, FCRL4 and DUSP4) (Extended Data Fig. 3d), which are associated with infectious and inflammatory diseases20,21. Cluster 16 was annotated as germinal center (GC)-like B cells based on the expression of well-established GC B cell genes, such as AICDA, BCL6, CD38, FAS and MME (CD10) (Fig. 1a and Extended Data Fig. 3d), as well as the expression of cell cycle genes (STMN1, MKI67, PCNA and TYMS) (Extended Data Fig. 3d). PCs were grouped into IgA (clusters 5, 9, 10, 11, 14, 17, 18 and 19), IgM (cluster 3) and IgG (clusters 6, 15 and 20) PCs based on their differential expression of genes encoding for immunoglobulin isotypes (Fig. 1b). Two PC clusters displayed increased expression of cell cycling genes (clusters 1 and 4) (Fig. 1c) and were annotated as plasmablasts (PBs). Expression analysis of IgA-encoding or IgG-encoding genes at the single-cell level indicated that PB 1 was enriched in IgA, whereas PB 4 segregated into two distinct subgroups with mutually exclusive expression of either IgG or IgA (Fig. 1d,e).
Fig. 1 |. PBs, IgG-secreting PCs and IFN-imprinted naive B cells are expanded in the inflamed colons of patients with UC.
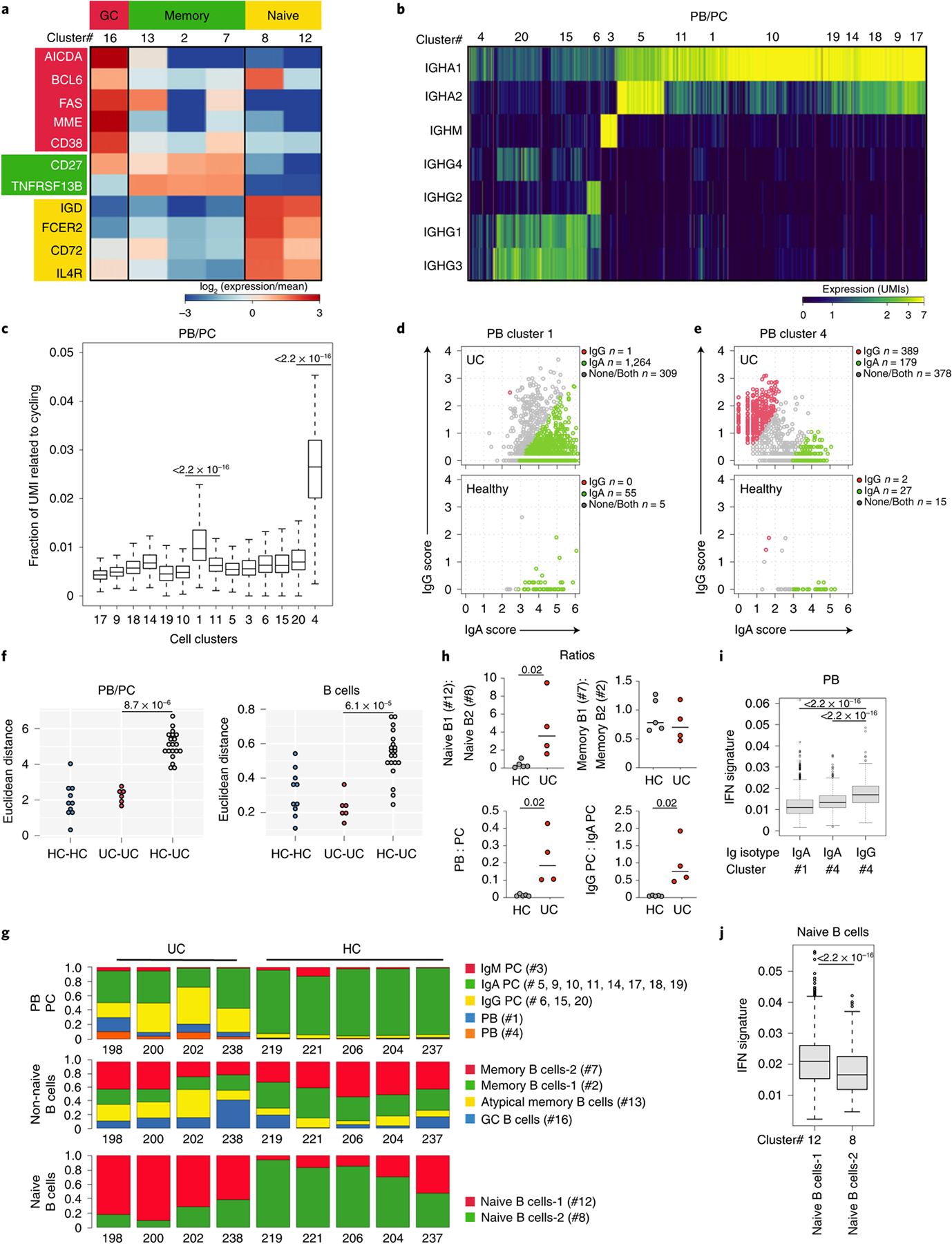
a, Heat map showing relative expression values of genes (rows) enriched in B lymphocyte clusters (columns; total B lymphocytes = 7,621 cells). b, Heat map showing color-coded downsampled UMI counts of immunoglobulin genes in PB (clusters 1 and 4; total PBs = 2,705 cells) and PC clusters (total PCs = 18,720 cells). c, Total UMI counts of the cycling transcriptional programs were calculated for each cell and divided by the total number of UMIs per cell. Bonferonni-adjusted one-sided t-test. t.statistic (cluster 4) = 76.08, t.statistic (cluster 20) = 5.62 and t.statistic (cluster 1) = 31.02. d, e, IgG versus IgA UMI scores per PB single cells in HCs (bottom) and UC samples (top) within cluster 1 (d) and cluster 4 (e). Cells were classified as IgG, IgA or unknown based on a normal mixture model with expected significance at 0.05% per cell, Kruskal–Wallis test. f, Euclidean distance for lineage-normalized cell type frequencies was compared within the indicated groups for PBs/PCs (left) and B lymphocytes (right), Wilcoxon rank-sum test. g, Subtype frequencies of PBs/PCs and non-naive and naive B cells within total respective cell types. h, Dot plots comparing the ratios of naive B cell subsets; memory B cell subsets; PBs and PCs; and PC subsets between HC and UC; two-sided Mann–Whitney U-test. i IFN signature scores between IgG PBs from cluster 4 and IgA PBs from cluster 4 (Kruskal–Wallis chi-squared = 108.63, df = 1, P < 2.2 × 10−16) and between IgG PBs from cluster 4 and IgA PBs from cluster 1 (Kruskal–Wallis chi-squared = 377.15, df = 1, P < 2.2 × 10−16). j, IFN signature scores between naive B cells-2 (cluster 8) and naive B cells-1 (cluster 12) (Kruskal–Wallis chi-squared = 114.25, df = 1, P < 2.2 × 10−16). c, i, j, Supplementary Table 2 shows cell counts per cluster; box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements; and the whiskers represent 1.5 times the interquartile range from the top or bottom of the box.
To assess global compositional changes in HCs and patients with UC while avoiding testing interdependent compositional covariates separately, we calculated the Euclidean distance22 between the cell type frequencies of sample pairs and compared the distributions of distances within and between sample groups. This analysis revealed significant dissimilarities of the PC cell composition between HCs and patients with UC (Wilcoxon rank-sum test of distances of log cell composition vector between UC samples versus distances between UC and HC samples; P = 8.7 × 10−6) (Fig. 1f). In particular, the PB/PC ratio was increased in UC (Mann–Whitney U-test; P = 0.0159), which associated with higher proportions of both IgA+ PB cluster 1 and PB cluster 4 (Fig. 1g, h). Concordant with historical descriptions23, this indicated that, whereas acute IgA responses were also observed in the inflamed intestines, part of the acute B cell response was skewed toward IgG production, as further supported by a marked increase of IgG PC proportions (Fig. 1g,h). IgG production associates with interferon (IFN)-γ-mediated type 1 immunity, although the specific mechanisms remain to be clarified in humans24,25. In UC samples, significant differences were obsereved in IFN-induced gene expression scores between IgG PB cluster 4 and IgA PB cluster 4 (Kruskal–Wallis chi-squared = 108.63, df = 1, P < 2.2 × 10−16) as well as between IgG PB cluster 4 and IgA PB cluster 1 (Kruskal–Wallis chi-squared = 377.15, df = 1, P < 2.2 × 10−16), supporting higher expression of the IFN signature specifically in IgG PBs (Fig. 1i).
The calculation of Euclidean distances within the B cell compartment also revealed major alterations in UC (Euclidean distance Wilcoxon rank-sum test, P = 6.1 × 10−5) (Fig. 1f). There was marked enrichment of naive B cells-1 (cluster 12) versus naive B cells-2 (cluster 8) in UC (Fig. 1g,h). When restricting the comparison to cells from UC samples only, naive B cells-1 displayed a high IFN signature compared to naive B cells-2 (Kruskal–Wallis chi-squared = 114.25, df = 1, P < 2.2 × 10−16; Fig. 1j). Computing the overlap with hallmark gene sets in the Molecular Signature Database (MSigDB)26 of genes differentially expressed between naive B cells-1 and naive B cells-2 (Supplementary Table 5) confirmed type I IFN-α and type II IFN-γ responses as the top two pathways enriched in naive B cells-1 (adjusted P < 10−10), whereas tumor necrosis factor (TNF) signaling, mTORC1 signaling and hypoxia signatures overlapped with genes enriched in naive B cells-2 (adjusted P < 10−10). Atypical memory B cells depend on the transcription factor (TF) T-bet and expand in response to IFN-γ27. Accordingly, they had the highest expression of the IFN-induced gene score (Extended Data Fig. 3e). However, we did not observe a clear expansion of atypical memory B cell in UC inflamed colon.
Overall, these data suggest that a type 1 inflammatory microenvironment contributes to reshape the B cell compartment in UC colons by imprinting naive B cell molecular programs and skewing part of the acute PC response toward IgG production.
Major disruptions of B cell populations in UC.
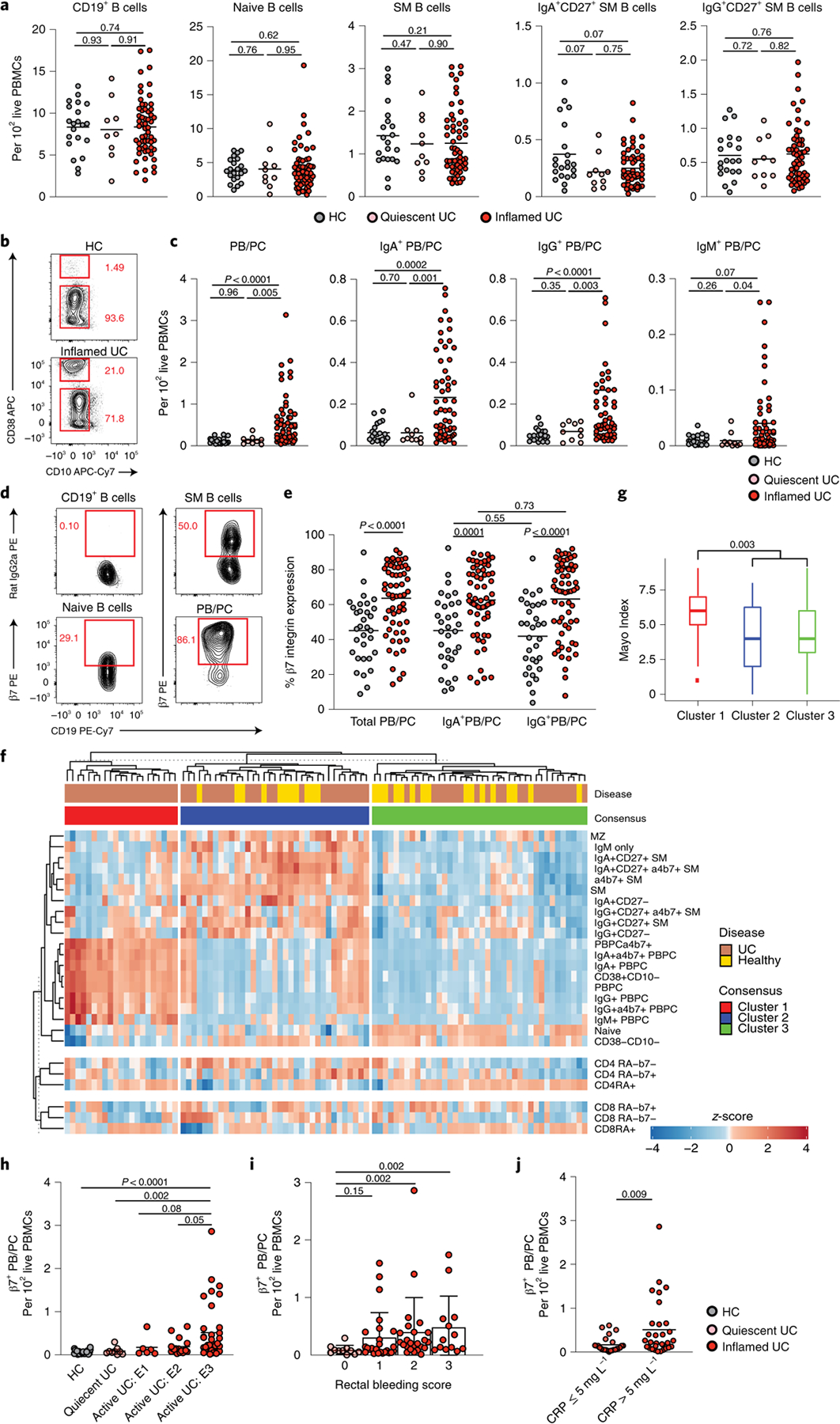
Next, we examined colonic B cell populations in a subset of patients within the primary cohort using multi-parametric flow cytometry (Supplementary Fig. 1). Consistent with scRNA-seq analyses, there was a significant increase in IgG+IgA−IgM−CD38++CD27+ colonic PC and IgG:IgA PC ratios in patients with active UC compared to HCs (Fig. 2a,b), with a significant proportion of actively cycling colonic PCs (Fig. 2c,d). Using published criteria28, we defined short-lived (CD19+CD45+), long-lived (CD19−CD45+) and ultra-long-lived (CD19−CD45−) CD38++CD27+ PCs. Concordant with the PB expansion, we observed a significant increase in the frequency of short-lived PCs and the ratio of short-lived PCs:long-lived PCs in patients with UC with active disease (Fig. 2e,f). Naive B cells were significantly increased in UC inflamed colon as compared to HCs, possibly reflecting the increased proportion of IFN-imprinted naive B cells observed by scRNA-seq analysis (Fig. 2g). In addition, although HCs and patients with UC had similar frequencies of total CD19+ B cells and CD19+CD38−CD10−CD27+IgM−IgD− switched memory (SM) B cells (Fig. 2g), significantly higher frequencies of IgG+ SM B cells and IgG:IgA SM B cell ratios were detected in UC (Fig. 2h,i).
Fig. 2 |. Major disruptions of B cell populations in inflamed colon of patients with UC.
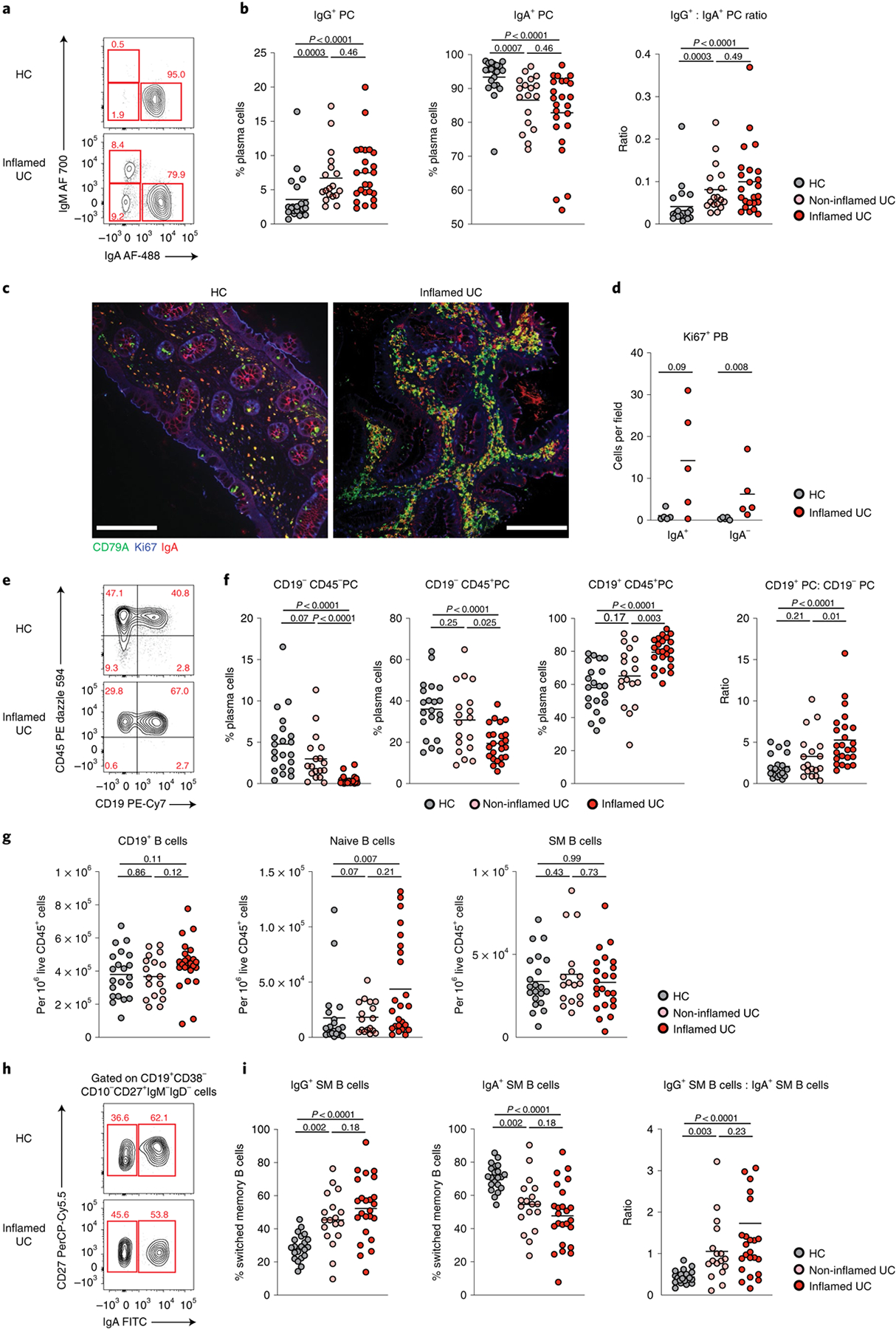
a, Representative flow plots showing the frequency of IgA and IgM expressed on CD27+CD38++ PCs in biopsies of an HC and a patient with UC with inflammation. b, Frequency of IgG (IgA−IgM−) PC, IgA PC and IgG:IgA ratio on colonic PC in HCs (n = 21) and in inflamed (n = 24) and non-inflamed (n = 18) biopsies from patients with UC. Data are shown as individual data and mean, Mann–Whitney test, two-tailed. c, Representative images of colonic biopsies from an HC (left) and a patient with UC with inflammation (right) after immunofluorescence staining with CD79a (green), Ki67 (blue) and IgA (red) (×20 magnification); scale bar, 150 μm. d, Quantification of lamina propria IgA+ (IgA, CD79a and Ki67 triple-positive cells) and IgA− PB (CD79a and Ki67 double-positive cells) in five HCs and five patients with UC with inflammation. Data are shown as individual data and mean, Mann–Whitney test, two-tailed. e, Representative flow plots showing the frequency of short-lived (CD27+CD38++CD19+CD45+), long-lived (CD27+CD38++CD19−CD45+) and ultra-long-lived (CD27+CD38++CD19−CD45−) PCs in colonic biopsies of an HC and a patient with UC with inflammation. f, Frequency of each subset among total PCs and ratio of CD19+CD45+ to CD19−CD45+ PCs in HCs (n = 21) and in inflamed (n = 24) and non-inflamed (n = 18) biopsies from patients with UC. Data are shown as individual data and mean, Mann–Whitney test, two-tailed. g, Frequency of CD19+ B cells and naive (CD45+CD19+CD38−CD10−IgD+IgM+) and SM (CD45+CD19+CD38−CD10−IgD−IgM−) B cells out of live CD45+ lamina propria mononuclear cells in HCs (n = 21) and in inflamed (n = 24) and non-inflamed (n = 18) biopsies from patients with UC. Data are shown as individual data and mean, Mann–Whitney test, two-tailed. h, Representative flow plots showing the frequency of IgA+ and IgG (IgA−) SM B cells gated on live SM B cells in biopsies of an HC and a patient with UC with inflammation. i, Frequency of IgG, IgA SM B cells and IgG:IgA ratio in SM B cells in HCs (n = 21) and in inflamed (n = 24) and non-inflamed (n = 18) biopsies from patients with UC. Data are shown as individual data and mean, Mann–Whitney test, two-tailed. P values are as indicated in the figure.
Overall, these data confirm major changes in the intestinal B cell compartment identified by scRNA-seq and demonstrate an acute ‘systemic-like’ B cell response skewed toward the production of pro-inflammatory IgG most pronounced in the inflamed colon.
Reduced somatic hypermutation and diversity of colonic PCs in UC.
To define the clonotypic architecture of PCs in patients with UC, we single-cell sorted short-lived CD38++CD27+IgD−CD19+ CD45+ PCs from the inflamed colon and gut-homing β7+CD19+/int CD38++CD27+IgD− PBs from the peripheral blood of four patients with active UC and four HCs (Supplementary Table 6). A total of 862 clones from the colon and 1,129 clones from the blood were analyzed.
In HCs, the dominant Ig isotype among colonic PCs and circulating gut-homing β7+ PBs was IgA, which accounted for 90.9% ± 6.6% of the clones and 62.2% ± 5.7% of the clones for colonic PCs and circulating β7+ PBs, respectively (Fig. 3a,b). Consistent with both scRNA-seq and flow cytometric data, the IgG clone frequency was substantially increased in the colon of patients with UC, with 5.8% ± 5.2% IgG+ PCs from total PC clones in HCs versus 39.6% ± 12.9% of IgG+ PCs from total PC clones in patients with UC (Fig. 3a). This increase reflected a substantial expansion of IgG1-expressing cells among IgG+ PCs, as IgG1-expressing PCs represented 72.1% of total IgG+ colonic PC clones in patients with UC compared to 40% in HCs (Fig. 3c). Similarly, 72% of circulating β7+ PBs were IgG1+ in patients with UC compared to 46.0% in HCs (Fig. 3c). Notably, the generation of IgG1 is largely associated with B cell responses to T-dependent protein antigens, and IgG1 antibodies exhibit potent pro-inflammatory effector functions11.
Fig. 3 |. Colonic PCs have reduced diversity, longer CDR3 regions and fewer somatic mutations in patients with UC.
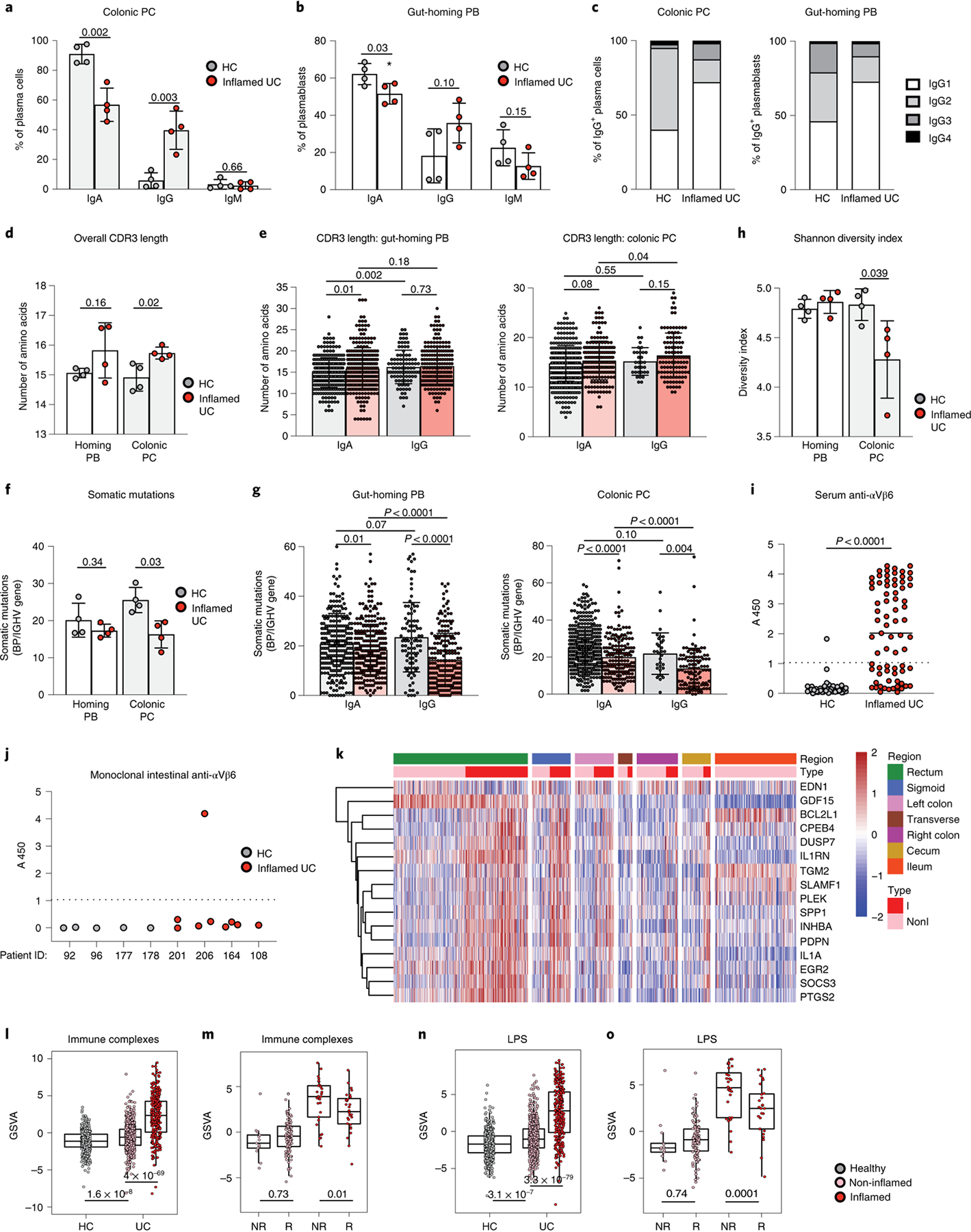
a, b, Isotype frequencies in short-lived colonic PCs (CD27+CD38++CD19+CD45+) (a) and circulating gut-homing PBs (β7-integrin+CD19+/−CD38++CD27+IgD-) (b), Mann–Whitney test, two-tailed. c, Proportion of each IgG subclass in HCs and patients with UC; chi-squared test. d, e, Mean IgH amino acid CDR3 length per patient (d) for each IgA and IgG clone (e) of gut-homing PBs and short-lived colonic PCs; Mann–Whitney test, two-tailed. f, g, Mean (f) V gene somatic mutations for each IgA and IgG clone (g) of gut-homing PBs and short-lived colonic PCs, Mann–Whitney test, two-tailed. h, Shannon diversity index of gut-homing PBs and short-lived colonic PCs. Mann–Whitney test, two-tailed. a–h are based on four HCs and four patients with active UC. i, Serum anti-αvβ6 integrin antibodies of 45 HCs and 73 patients with UC. The cutoff absorbance value for positivity (A450, mean + 3 s.d. of the control sera), indicated by a dashed line. Mann–Whitney U-test. j, Reactivity of clones from HCs (n = 4 clones) and patients with UC (n = 9 clones) against integrin αvβ6. Dashed line is the cutoff for positivity (defined in i). k, Heat map depicting the standardized expression profiles of genes in the macrophage Fc IC signature on inflamed and non-inflamed tissue for different intestinal regions (n = 421 patients with UC). l, n, GSVA-derived scores for macrophage Fc IC and LPS (n) gene signatures in inflamed and non-inflamed biopsies (n = 421 patients with UC and 243 non-IBD controls). Mann–Whitney test, two-tailed. Effect size is shown in Supplementary Table 12. m, o, Macrophage Fc IC (m) and LPS (o) gene scores of responders (R) and non-responders (NR) in TNF inhibitor (infliximab)-treated patients with IBD before treatment initiation, Mann–Whitney test, two-tailed. Effect sizes are shown in Supplementary Table 13. For l–o, box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range. In a, b, d, e, f, g, h, l, m, n and o, data are shown as individual data and mean, and P values are as indicated.
VH3 and JH4 genes were dominant and expressed in more than 50% of both HCs and patients with active UC. JH6 usage was increased in colonic PCs of patients with active UC when compared to HCs (Extended Data Fig. 4a–d). CDR3 length was significantly greater in colonic PCs of patients with UC when compared to HCs, with a trend for a longer CDR3 among IgA+ clones in both the peripheral blood and colon (Fig. 3d,e). We repeated this analysis after excluding JH6 clones and found no difference in CDR3 length (Extended Data Fig. 4e), suggesting that the increase of CDR3 length in circulating and colonic B cells from patients with UC is largely driven by an increased JH6 gene usage.
All IgH genes showed high numbers of somatic mutations as previously reported29. In HCs, VH genes from circulating gut-homing PBs carried on average 20.1 ± 4.6 mutations, whereas VH genes from colonic PCs had 25.4 ± 3.4 mutations. Notably, in patients with UC, the number of somatic mutations was reduced in both IgA+ and IgG+ clones, both in circulation and colon (Fig. 3f,g). Among short-lived PCs, clonally related sequences with identical VH and JH genes, identical CDR3 length and at least 80% homology in the amino acid sequence were found in three of four HCs and in all patients with UC. Of all colonic PC clones, 6.5% belonged to expanded clones (found at least two times) in HCs, whereas this frequency amounted to 11.2% in patients with UC (Extended Data Fig. 4f). Accordingly, the clonal diversity assessed by the Shannon diversity index was decreased in colonic short-lived PCs from patients with UC (Fig. 3h). Additionally, clones shared by blood and mucosal compartments were found in all patients with UC but in only one of four HCs (Extended Data Fig. 4f). Together, these data indicate a substantial increase in IgG PC clones from the inflamed colon, with reduced diversity and fewer somatic mutations.
We next sought to determine the antigenic specificity of this potentially pathogenic B cell response (Supplementary Table 7 and Methods). In patients with UC, serological IgA and IgG reactivity against microbial antigens was limited to three microbial antigens (14–2F, M18–1 and EcLW; Extended Data Fig. 5 and Supplementary Table 8). We then explored the reactivity to a recently described UC-associated endogenous autoantigen, the αvβ6 integrin30. We found that patients with UC had significantly higher levels of circulating IgG antibodies reactive to αvβ6 than HCs (Fig. 3i and Supplementary Table 9). Subsequently, we generated monoclonal antibodies by single-cell sorting and molecular cloning of intestinal short-lived PCs (Supplementary Table 10 and Methods). One of nine monoclonal antibodies that were produced from patients with UC demonstrated high reactivity against αvβ6. In contrast, none of the five antibodies produced from HCs reacted against this integrin (Fig. 3j). Although limited, these data suggest that the pathogenic mucosal B cell response associated with UC might be abnormally targeting self-antigens, including αvβ6.
IgG-mediated macrophage activation associates with colonic inflammation.
IgG elicits potent effector functions, partly by binding to Fcγ receptors (FcγRs) on myeloid effector cells, and potentially drives intestinal inflammation12. We generated a molecular signature of IgG-activated macrophages using transcriptomic analysis of murine colonic macrophages stimulated ex vivo with IgG immune complexes (ICs) (Extended Data Fig. 6a and Methods). Gene signatures of macrophages exposed to IgG-ICs or lipopolysaccharide (LPS) were enriched in inflamed UC biopsies compared to non-inflamed UC biopsies and HCs in our Validation Cohort 1 (VCo1) dataset across intestinal tissue sites (Fig. 3k l,n and Supplementary Tables 11 and 12). Notably, despite a minimal overlap in constituent genes (Extended Data Fig. 6b), both macrophage gene signatures induced by IgG-ICs and LPS were elevated in pre-treatment biopsies from patients with UC who were non-responsive (NR) to infliximab treatment (Fig. 3m,o and Supplementary Table 13). This suggests that IgG and Fc receptor signaling contribute to exacerbated macrophage activation that is an important component of the lack of response to anti-TNF therapy in patients with UC. As a caveat, we were unable to distinguish between ‘bona fide’ TNF non-responders and those for whom the non-response could have been related to low drug levels or the presence of antibodies. Such data were not captured in VCo1 in accordance with the clinical practices when it was assembled.
Using scRNA-seq, we identified ‘resident-like’ macrophages in both healthy and inflamed UC colons expressing tissue macrophage-associated genes, including C1qc, DNASE1L13, MRC1 (CD206), MAFB and CSF1R. We also identified a macrophage population (‘inflammatory macrophages’) enriched in pro-inflammatory genes such as NF-κB, IL-23 and TNF, which was increased in inflamed colons (Extended Data Fig. 7a). The macrophage gene signature induced by IgG-ICs was highly enriched in these inflammatory macrophages, which also expressed the LPS-induced signature. This suggests that inflammatory macrophages express different pro-inflammatory gene programs that possibly emerge in response to distinct microenvironmental cues simultaneously present in inflamed colons, including microbial Toll-like receptor ligands such as LPS and IgG-ICs (Extended Data Fig. 7b)
Therefore, in addition to an IgG-enriched B cell response, we have identified evidence of an IgG-mediated macrophage activation signature that is associated with colonic inflammation and, a priori, the lack of response to anti-TNF treatment.
CXCL13+ TPH cells are expanded in UC.
To assess whether T cell alterations might underpin acute B cell responses in UC, we next analyzed single colonic T cell transcriptomes. We identified 15 T cell clusters (Extended Data Fig. 8 and Supplementary Tables 2 and 3), and further curation revealed nine T cell subtypes and/or states (Methods), the global composition of which differed in UC versus HC (Fig. 4a and Extended Data Fig. 8b; Wilcoxon rank-sum test of Euclidean distances of log cell composition vector between UC samples versus distances between UC and HC samples, P = 8.69 × 10−6). Activated T cells expanded in UC and had high expression of Th17 and Th1 effector cytokines, including IFN-γ (Fig. 4a,b). This suggests that activated T cells contribute to establish the type 1 microenvironment associated with the IFN signature that we described in a subset of naive B cells-1 (cluster 12), IgG+ PBs and IgG+ PCs (Fig. 1). We also identified TPH expressing high levels of the follicular B-cell-attracting chemokine CXCL13 (ref.16) (Fig. 4b). Colonic CXCL13+ T cells expressed genes such as PDCD1 (PD-1), CD200, BCL6, TIGIT, ICOS and BATF but no CXCR5 transcripts (Fig. 4c). In addition, colonic TPH cells expressed several genes in common with regulatory T (Treg) cells, such as TIGIT, CTLA4, TNFRSF4 and MAF, whereas key Treg-defining genes, such as FOXP3 and IKZF2, were not detected (Fig. 4c). Although CXCL13 expression was restricted to TPH cells, Treg cells expressed the immunoregulatory cytokine IL-10 (Fig. 4b). Concordant with previous reports31, IL-17 was also detected in Tregs, although at lower levels than IL-10 (Fig. 4b). TPH cells, in turn, uniquely expressed the TFH program as well as previously unreported genes, such as NMB, FABP5 and PGM2L1 (Fig. 4c). The proportion of TPH cells was significantly increased in inflamed UC colon compared to HCs (Fig. 4d; Wilcoxon test, P = 0.008).
Fig. 4 |. CXCL13+ TPH cells are expanded in the inflamed mucosa of patients with UC.
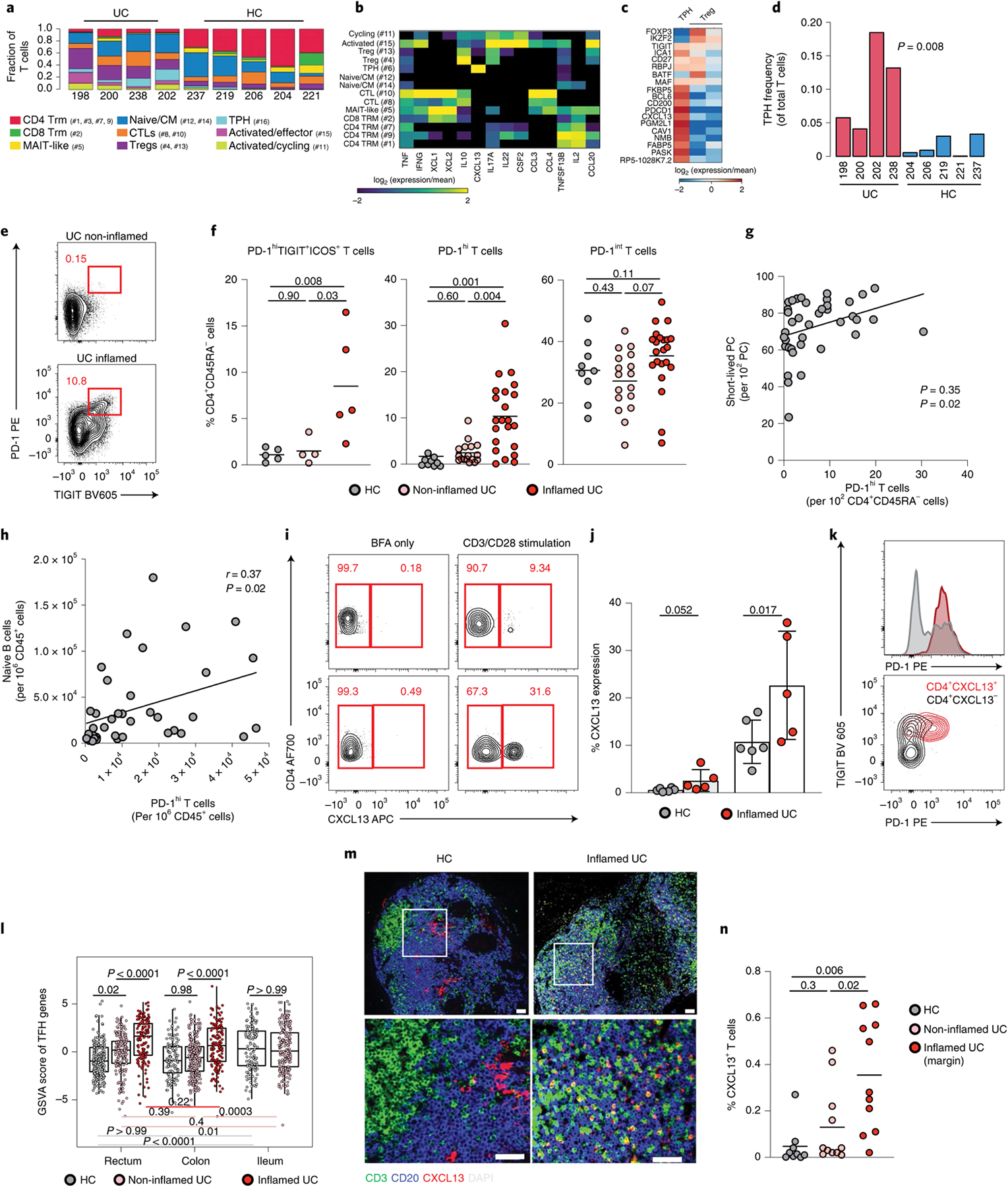
a, scRNA-seq-derived T cell frequencies within total T cells (n = 11,281 cells; chi-squared = 3,727.0; P < 0.01). Respective cluster numbers are indicated. b, c, Heat maps showing log (2-based) relative expression values of genes (rows) enriched in all T lymphocyte clusters (b) or in TPH cells and Tregs (c) (columns). d, TPH cell frequencies within total T cells in individual sample (Wilcoxon test, two-tailed). e, Representative flow plots showing PD-1hiTIGIT+ cells among live CD45+CD3+CD4+CD45RA− cells. f, Frequency of PD-1hiTIGIT+ICOS+ cells (n = 5 HC, n = 4 non-inflamed and n = 5 inflamed), PD-1hi cells and PD-1int cells among memory (CD45RA−) CD4 T cells (n = 9 HC, n = 17 non-inflamed and n = 22 inflamed), Mann–Whitney test, two-tailed. g, h, Linear regression (using Pearson test) showing the relationship between the frequencies of PD-1hi CD4+CD45RA− cells and short-lived PCs (g) and PD-1hi CD4+CD45RA− cells and naive B cells (h) in colonic biopsies of patients with UC (n = 39). Data are shown as individual data. i, Representative flow plots showing CD4+CXCL13+ T cells. j, Frequency of CXCL13 expression within lamina propria CD4 T cells with or without CD3/CD28 stimulation, Mann–Whitney test, two-tailed, n = 6 HC and n = 5 inflamed colon. k, Representative flow plots showing PD1 and TIGIT expressions of CXCL13+ (red) and CXCL13− (gray) CD4 T cells. l, Overall TPH activity scores in 421 patients with UC and in 243 non-IBD controls where canonical TFH genes were examined within whole-biopsy RNA sequence data from VCo1. Box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range. Effect sizes are shown in Supplementary Table 14. m, Representative images from an HC and the sample of a patient with inflamed UC with CD3 (green), CD20 (blue) CXCL13 (red) and DAPI (gray) staining. n, Quantification of CD3+CXCL13+ in nine HCs and 11 patients with UC with active inflammation (at the edge of inflammation and in an upstream uninflamed area), Mann–Whitney test, two-tailed. For f, j and n, data are shown as individual data and mean. P values are as indicated throughout the figure.
Using multi-parameter flow cytometry, we confirmed the expansion of PD-1hiTIGIT+ICOS+CD45RA− T cells in inflamed colon of patients with UC (Fig. 4e,f). Virtually all PD-1hi cells co-expressed TIGIT and ICOS (Supplementary Fig. 2). We, therefore, used PD-1hi expression to further examine TPH cells in the primary cohort. PD-1hi cells were increased in inflamed versus non-inflamed colons from patients with UC and HCs, respectively, wheres PD-1int cells did not significantly differ (Fig. 4f). Moreover, significant correlations existed between frequencies of PD-1hiCD4+ T cells and short-lived PCs (Fig. 4g) as well as naive B cells (Fig. 4h), suggesting that the three populations were interdependent. Ex vivo stimulation experiments confirmed CXCL13 production by PD-1hiTIGIT+CD4+ T cells as well as an increased proportion of CXCL13+PD-1hiTIGIT+CD4+ T cells in inflamed UC colons (Fig. 4i–k).
Next, we examined TPH cell-associated genes (PDCD1, TIGIT, BATF, ICA1, CTLA4 and TNFRSF4) within VCo1. As expected, higher expression of TPH cell-associated genes was detected in inflamed UC biopsies than in HCs, both in rectum and colon (Fig. 4l). Consistent with the abundance of TFH cell-enriched Peyer’s patches in the terminal ileum, overall activity scores of TPH cell-associated genes were higher in HC ileum than in rectum (P < 0.001) and colon (P = 0.001) (Fig. 4l and Supplementary Table 14).
To define the spatial distribution of CXCL13-expressing T cells, we performed immunofluorescence staining of surgical tissues (Extended Data Fig. 9 and Methods). In all, CXCL13+CD3+ T cells were rare (Fig. 4m). In contrast to non-UC controls, UC samples showed a marked expansion of CXCL13+CD3+ cells just distal to the macroscopically diseased margin (Fig. 4m,n and Extended Data Fig. 9b). In the same individuals, TPH cells were significantly reduced in the proximal colon with no macroscopic disease (Fig. 4n), indicating that TPH cells were closely associated with anatomical regions of disease activity.
Altogether, these findings provide evidence of a population of TPH cells with the potential to support the recruitment of naive B cells, which, together with activated T cells expressing Th1 cytokines (that is, IFN-γ), could drive the expansion of a subset of IFNγ-responsive naive B cells and promote IgG class switching in active UC.
Circulating gut-homing PBs/PCs are increased in UC.
We next investigated whether intestinal B cell alterations were reflected in the blood of patients with UC. Naive B cells, SM B cells and PBs/PCs in circulation were defined based on canonical markers (Supplementary Fig. 3). Total CD19+ B cells, naive CD19+CD38−CD10−IgM+IgD+ B cells and SM CD19+CD38−CD27+IgM−IgD− B cells, which included both SM IgA+ B cells and SM IgG+ B cells, were similar in HCs and patients with quiescent or active UC (Fig. 5a). In contrast, there was a significant increase of CD19+/intCD38++CD27+IgD− PBs in patients with active UC (Fig. 5b,c). IgG+ and IgA+ but not IgM+ PBs/PCs were significantly increased compared to HCs (Fig. 5c). Most CD19+/intCD38++CD27+IgD− cells were Ki-67+ with low-to-no expression of CD138 (Supplementary Fig. 4), which reflected a PB phenotype. Notably, expression of the gut-homing integrin β7 was significantly increased in IgA+ and IgG+ PBs (Fig. 5d,e), which pointed to the intestinal tract as both their origin and destination32. Profiling PBs and PCs from a subset of patients with active UC for additional tissue homing molecules, including CCR9, GPR15, CXCR3, CXCR4 and integrins β7 and β1, revealed decreased β1 and increased CXCR3 expression (Supplementary Fig. 5). CXCR3 is a receptor for IFN-γ-induced CXCL9 and CXCL10 (ref.33), two chemokines expressed by inflammatory macrophages (Extended Data Fig. 7b), again pointing to type 1 immunity as a driver of disease pathogenesis.
Fig. 5 |. Circulating β7-integrin-expressing PBs are expanded in patients with active UC and relate to disease activity.
a, Frequency of total B cells (CD19+), naive B cells (CD19+CD38−CD10−IgM+IgD+), CD27+ SM B cells (CD19+CD38−CD10−IgM−IgD−) and IgA+ and IgG (IgA−) SM B cells in HCs (n = 21), in patients with quiescent UC (n = 10) and in patients with active UC of the primary cohort (n = 58), Mann–Whitney test, two-tailed, b, Representative flow plots showing the frequency of CD38++CD10− cells and CD38−CD10− cells among live CD19+ PBMCs in HCs and in a patient with active UC. c, Frequency of PBs/PCs (CD19+CD38++CD10−CD27+IgD−) and IgA+ PBs/PCs, IgG+ PBs/PCs (IgA−IgM−) and IgM+ PBs/PCs out of live PBMCs in HCs (n = 21), in patients with quiescent UC (n = 10) and in patients with active UC of the primary cohort (n = 58), Mann–Whitney test, two-tailed. d, Representative flow plots of the expression of β7-integrin on SM B cells, naive B cells and PBs/PCs. e, Level of expression of β7-integrin on total, IgA+ and IgG+ PBs/PCs in HCs (n = 32) (gray) and in patients with active UC (n = 62) (red) from the primary cohort, Mann–Whitney test, two-tailed. f, Consensus clustering on primary cohort patients defined three clusters of patients and controls according to immune-phenotyping of PBMCs. g, Comparison of clinical Mayo Scores across the three consensus clusters. Box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range, Mann–Whitney test, two-tailed. h, Frequency of β7+ gut-homing PBs/PCs (CD19+CD38++CD10−CD27+IgD−) according to their disease extent defined by the Montreal classification, Mann–Whitney test, two-tailed, n = 21 HCs, n = 9 quiescent, n = 6 active E1, n = 17 E2 and n = 35 E3. i, Frequency of β7+ gut-homing PBs/PCs according to the rectal bleeding score, Mann–Whitney test, two-tailed, n = 11, 25, 24 and 13 for rectal bleeding scores of 0, 1, 2 and 3, respectively. j, Frequency of β7+ gut-homing PBs/PCs in patients with UC with a CRP level of less than 5 mg L−1 (n = 25) or more than 5 mg L−1 (n = 31), Mann–Whitney test, two-tailed. For a, c, e, h, i and j, data are shown as individual data and mean; for h and i, data are shown as mean ± s.d. P values are as indicated throughout the figure.
To investigate the relationship between circulating PBs and the intestinal inflammatory burden with subsequent disease complications, we performed consensus clustering after grouping patients based on the extent of similarity in their immune profile (Fig. 5f). Patients with the highest frequency of circulating gut-homing PBs and PCs from both IgG+ and IgA+ populations clustered together (consensus cluster 1) and had the highest disease severity score, as evidenced by the partial Mayo Index (Fig. 5g; Mann–Whitney test, two-tailed). The association between partial Mayo Index and cluster groups remained significant after adjusting for C-reactive protein (CRP) levels using multivariate linear regression (P = 0.01). Furthermore, the frequency of circulating gut-homing PBs was highest in patients with E3 disease (pancolitis) (Fig. 5h) and higher rectal bleeding scores (Fig. 5i) and CRP levels (Fig. 5j).
β7+ PBs/PCs in blood correlate with UC activity.
We tested the relevance of circulating gut-homing PBs as a potential cellular biomarker of disease activity in two validation cohorts (VCo1 and VCo2) using complementary methodologies. In VCo1, using mass cytometry, we confirmed that PB frequencies were significantly increased in patients with active versus quiescent UC or HCs (Fig. 6a,b). We validated increased expression of β7 integrin and CXCR3 and decreased β1 integrin expression in PBs from patients with active UC (Fig. 6c). Next, we assessed whether levels of circulating PBs could help predict later disease complications. Patients with levels above the median had a significantly higher probability of disease complications over a median follow-up of 1.6 years (inter-quartile range = 0.4–2.8; log-rank test, P = 0.02) (Fig. 6d). PBs were associated with disease complications after adjusting for age, sex, disease extent and disease duration using Cox regression analysis (P = 0.004) (Supplementary Table 14).
Fig. 6 |. An increase in gut-homing PBs/PCs is associated with disease complications and activity in two validation cohorts of patients with active UC.

a–d, Mass cytometry (CyTOF)-derived data from VCo1 comparing PC parameters among HCs (n = 65), patients with quiescent UC (n = 52) and patients with inflamed UC (n = 31). a, Representative tSNE plots (top) demonstrating cellular subsets in the peripheral blood of one HC, one patient with quiescent UC and one patient with inflamed UC. The bottom panels compare the expression of β7-integrin on CD27+CD38hi PBs/PCs. b, Cumulative data from VCo1 comparing the frequency of PBs/PCs among HCs and patients with quiescent UC and patients with inflamed UC; Mann–Whitney test, two-tailed. c, Mean level of expression of β7-integrin (left), β1-integrin (middle) and CXCR3 (right) on circulating PBs/PCs of HCs and patients with quiescent UC and patients with inflamed UC. Data are shown as individual data, mean and standard deviation; Mann–Whitney test, two-tailed. d, Kaplan–Meier estimates showing survival free of occurrence of complications (that is, intestinal surgery, UC-related hospitalization or introduction of a new biologic treatment) for patients of VCo1 with PB/PC frequency below the median (red curve) or above the median (blue curve). Survival curves were compared using a log-rank test, n = 73 patients with UC. e–g, IgJ gene signatures, representing circulating PBs/PCs in VCo2 comprised of 36 HCs and 107 patients with UC. e, Relative expression of IgJ gene from whole blood of HCs, patients with quiescent UC and patients with inflamed UC. f, g, Relative expression of IgJ compared with rectal bleeding scores (f) and fecal lactoferrin (g). In b, e and f, box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range from the top or bottom of the box. Data are shown as individual data. Comparisons between indicated groups were made with two-tailed t-test. Correlation coefficient (rho) in g was calculated using Spearman’s test. P values are as indicated throughout the figure. tSNE, t-distributed stochastic neighbor embedding.
We evaluated gene expression of IgJ as a proxy for PBs abundance34,35 in VCo2 (Methods). A significant increase was noted in the relative expression of IgJ in patients with active UC (defined as partial Mayo Score of 2 or higher) when compared to patients with quiescent UC and HCs (Fig. 6e). Increased IgJ expression was also associated with rectal bleeding scores (Fig. 6f) and fecal lactoferrin levels (Fig. 6g). Thus, an increased frequency of circulating gut-homing PBs identified patients with UC with higher disease activity and higher risk of long-term disease complications in three independent cohorts.
Discussion
Our data demonstrate profound alterations of gut B cells in UC. Akin to HCs36, IgA+ PCs were the largest population within the gut PC compartment in UC, a finding consistent with similar results from recent scRNA-seq-based studies37,38. In agreement with previous reports12,39–41, we also found a substantial increase of IgG+ PCs in the inflamed gut lamina propria from patients with UC. Furthermore, a significant increase was observed in actively cycling PBs and recently recruited PCs. This IgG-enriched B cell response could amplify the local inflammatory cascade via FcγR-dependent recruitment of inflammatory monocytes and TH17 cells12 and could further sensitize the gut mucosa to microbial40, dietary42 and autologous antigens. Accordingly, we identified an autoreactive PC clone that recognized the anti-αvβ6 (ref.30) integrin in the intestinal mucosa of a patient with UC.
Substantial qualitative alterations were also noted in IgH sequences from patients with UC. Consistent with earlier work43, we detected a consistent relative increase of IgG1+ PC clonotypes in both gut mucosa and peripheral blood from patients. with UC. Colonic IgA+ PCs from healthy volunteers were highly mutated as previously reported29,44, with somatic hypermutations linked to cross-microbial species reactivity10. In contrast, colonic IgG+ PCs from patients with UC exhibited a striking reduction of VDJ gene mutations, which was consistent with a similar, but less pronounced, reduction of VDJ gene mutations in colonic PCs as well as circulating PBs. This finding might reflect a perturbed GC maturation of both IgG+ and IgA+ PCs, which could stem from a chronic antigen-mediated overstimulation of gut follicular B cells as a result of the local inflammatory environment. This could lead to an increased extrafollicular development and expansion of auto-reactive B cell clones45,46. Alternatively, there could be a higher recruitment rate of incompletely differentiated and intrinsically autoreactive B cells due to inflammation. Although less striking, non-inflamed tissue from patients with UC also showed differences compared to colonic tissue from HCs. Specifically, the frequency of IgG PCs and SM B cells remained higher in non-inflamed UC tissues as compared to colonic tissue from HCs. This finding could reflect either a past inflamed state or the dissemination of IgG+ clones from inflamed to non-inflamed segments of the colon, as was recently reported37.
We sought to determine the origins of the dysregulated B cell response in UC. We found a population of IFN-primed naive B cells to be expanded in UC and to be associated with an increased frequency of IgG+ PBs and IgG+ PCs, with only IgG+ demonstrating an IFN signature. Clusters of activated/effector T cells expressing high levels of effector cytokines, including IFN-γ (cluster 15), potentially serve as a source of IFN-γ. Additionally, we detected a population of inflammatory macrophages expressing CXCL9, in the inflamed UC colon. CXCL9, an IFN-induced gene47, serves as a selective ligand for CXCR3 (ref.33). Accordingly, we found a significant expansion of intestinal CXCR3-expressing PCs in patients with UC, which is in agreement with a previous report48. Altogether, these data provide compelling evidence in support of type 1 immunity as a driver of naive B cell molecular imprinting as well as skewing of acute B cell responses toward IgG production in the intestines of patients with UC.
Several studies have reported an increase in circulating TFH cells in patients with IBD15,49–51. However, despite the possibility of a significant role in disease pathogenesis, tissue TPH cells have not been well characterized in patients with IBD. We found CXCL13+ TPH cells in the inflamed intestines and at the advancing edge of inflammation, which points to TPH-cell-guided assembly of inflammation-associated lymphoid aggregates in patients with UC. Of note, the neo-lymphoid aggregates seemingly driven by CXCL13+ TPH cells lack organized GCs and, therefore, could account for the induction of the less-mutated PBs that were detected by single-cell Ig gene sequencing. Targeting the CXCL13+ T cells or CXCL13 itself might help reduce the production of pathogenic PBs and, therefore, attenuate colonic inflammation. An increased understanding of intestinal TFH and TPH cells might also unveil the cellular mechanisms underlying the beneficial effects of existing therapeutics, such as those targeting the IL-23-driven pathway.
We found that, although circulating total, naive and SM B cells were unchanged in patients with active UC, circulating β7+ PBs were strongly increased in these patients, and this increase closely correlated with colonic inflammation and was predictive of complicated disease.
In summary, these data provide detailed insights into the magnitude and breadth of B cell perturbations during active colitis, suggesting potential new biomarker and therapeutic strategies. Longitudinal prospective data will be necessary to address the etiological and pathogenic importance of potential B cell activation mechanisms and to distinguish between homeostatic and pathogenic B cell expansion in the mucosa.
Methods
Patients.
Patients from three distinct cohorts were recruited, and different techniques were applied on subsets of patients as described in Extended Data Fig. 1.
Patients and HCs of the primary cohort were recruited, from April 2016 to May 2019, from the Inflammatory Bowel Disease Center, the gastroenterology department and the digestive endoscopy unit at Mount Sinai Hospital. The protocol was approved, and informed consent was obtained from all participants in accordance with the institutional review board. Patients who were enrolled in the study were asked to provide blood and/or biopsies. The latter were collected during colonoscopies planned for regular care. The primary cohort included a total of 145 individuals: 88 patients with UC with active disease, 14 patients with quiescent UC and 43 HCs. Patients with active UC had pancolitis (57%), left-sided colitis (35%) or proctitis (8%) with a partial Mayo Score of 5.3 ± 2.0, a rectal bleeding score of 1.8 ± 0.8 and an endoscopic Mayo Score of 2 (58.3%) or 3 (20.8%). Clinical characteristics of patients with blood phenotyping and biopsy flow cytometry analyses are shown in Supplementary Table 16.
Vco1 consisted of patients from the Mount Sinai Health System and affiliated clinicians, recruited to be part of the Mount Sinai Crohn’s and Colitis Registry (MSCCR) between December 2013 and September 2016, using a protocol approved by the institutional review board. All patients gave written informed consent to participate in the study, which was conducted in accordance with the ethics principles of the Declaration of Helsinki. Patients, 18 years of age or older, were enrolled during an office visit for their standard of care colonoscopy, whereas the non-IBD controls were mainly patients undergoing colonoscopy as part of colon cancer screening. Clinical and demographic information was obtained from an extensive MSCCR questionnaire administered at the time of the colonoscopy. Blood was collected 1 h before the colonoscopy and processed for flow cytometry. In VCo1, 664 individuals were enrolled: 421 patients with UC and 243 healthy volunteers (Supplementary Table 17). In VCo1, 73 patients with UC had follow-up clinical data available (Supplementary Table 18).
VCo2 was part of an observational study with no therapeutic intervention that has been previously described52. Healthy volunteers and patients with a previous diagnosis of UC who consented in accordance with institutional review board standards were enrolled. Stool samples were collected at a screening visit, and peripheral blood was collected into a PAXgene tube at the ileocolonoscopy visit. Quiescent disease was defined as having a partial Mayo Score of 2 or less, with no individual subscore greater than 1. In VCo2, 143 individuals were enrolled: 66 patients with active UC, 41 patients with quiescent UC and 36 healthy volunteers as previously detailed52. The expression of IgJ, a gene strongly linked with plasmablast abundance34, was determined and compared with rectal bleeding score and fecal lactoferrin. Correlation coefficient (rho) was calculated using Spearman’s test
Blood collection and peripheral blood mononuclear cell isolation.
Ten to 30 ml of blood was drawn in EDTA tubes. Blood was freshly processed, under sterile conditions, within 2 h after collection. Plasma was collected and stored immediately at −80 °C. The blood was then diluted with PBS 1× (Gibco) and gently overlaid on lymphocyte separation medium (MP Biomedicals). After centrifugation, the phase containing peripheral blood mononuclear cells (PBMCs) was harvested and washed two times with sterile PBS. All flow cytometry data presented in the study were generated from freshly stained PBMCs.
Intestinal lamina propria cells isolation.
During the colonoscopy procedure, 8–12 biopsies from inflamed and/or non-inflamed parts of the colon of each patient were collected with forceps directly in RPMI, on wet ice. Biopsies were processed fresh within 2 h of collection. They were first transferred in 10 ml of HBSS (free of calcium and magnesium) containing EDTA (0.5 M, pH 8, Invitrogen) and HEPES (1 M, Lonza) and incubated for 20 min at 37 °C. The biopsies were then vortexed, washed with HBSS and transferred in a digestion media (10 ml of RPMI containing 0.005 mg of collagenase IV (Sigma-Aldrich) and DNAse I (Sigma-Aldrich)). After an incubation of 40 min at 37 °C in a rotating incubator (215 r.p.m.), biopsies were physically disrupted by pipetting up and down ten times through a 25-gauge needle, filtered through a 100-μm and 40-μm cell strainer and washed with RPMI twice.
Flow cytometry.
Multi-parametric flow cytometry was performed on intestinal and PBMCs derived from HCs and patients with UC, sampling the non-inflamed and inflamed sites. The gating strategy for colonic B cells is shown in Supplementary Fig. 1, and the gating strategy for peripheral-blood-derived B cells is shown in Supplementary Fig. 3.
Isolated cells were incubated with PBS containing the flow antibody cocktail for 20 min at 4 °C in the dark. Cells were washed two times with PBS and then fixed using Cytofix/Cytoperm buffer (BD Biosciences) and washed two times with Perm/Wash buffer (BD Biosciences). Finally, cells were harvested in FACS buffer (PBS without Ca2+and Mg2+ with 5 mM EDTA) before acquisition. In case of intracellular staining, cells were permeabilized for 15 min in Cytofix/Cytoperm buffer, washed with Perm/Wash buffer and incubated with the intracellular antibody for 30 min. Samples were acquired using LSR Fortessa (BD Biosciences), and data were analyzed using FlowJo version 10 software. Dead cells and doublets were excluded from all analyses. The list of used antibodies is displayed in Supplementary Table 19.
Mass cytometry.
In VCo1, mass cytometry was performed on whole blood obtained from 177 individuals (53 patients with active UC, 59 patients with quiescent UC and 65 healthy volunteers).
All mass cytometry antibodies used in this study were conjugated in-house using Maxpar X8 conjugation kits (Fluidigm), and sample processing was performed as previously described53. In brief, a titrated antibody panel (Supplementary Table 20) was added directly to 400 μl of whole blood and incubated for 20 min at room temperature. The stained whole blood was then lysed and fixed using FACS lysing buffer (BD Biosciences) and then incubated in Fix/Perm buffer (Fluidigm) containing 0.125 nM Ir nucleic acid intercalator (Fluidigm). The samples were then washed and frozen in FBS containing 10% DMSO and stored at −80 °C until acquisition. Immediately before acquisition, the samples were thawed, washed with deionized water and resuspended at a concentration of 1 million cells per milliliter in deionized water containing a 1/20 dilution of EQ 4 element beads (Fluidigm). The samples were then acquired on a CyTOF2 (Fluidigm) equipped with a Super Sampler fluidics system (Victorian Airships) at a flow rate of 0.045 ml min−1 and an event rate of less than 400 events per second. After acquisition, the data were normalized using the bead-based Fluidigm normalization algorithm before data analysis, and normalization beads and debris were excluded on the basis of Ce140 and DNA.
Mass cytometry data analysis.
The CyTOF panel included 37 antibodies (detailed in Supplementary Table 20) with samples run in three batches of pre-mixed lyophilized antibody. Representative samples were analyzed and visualized using a combination of manual gating and viSNE dimensionality reduction as implemented in Cytobank. For semi-automated analysis of the whole subject cohort, CyTOF-defined populations were detected using a semi-supervised approach through the Astrolabe Cytometry Platform (Astrolabe Diagnostics). In summary, single-cell data were clustered using the R package FlowSOM and then labeled using the Ek’Balam algorithm based on canonical markers as previously reported54. Cell subset definitions follow the schema defined by Maecker et al.55 and Finak et al.56. Secondary statistics, such as population frequency and median marker expression, were then exported for secondary analyses. We interrogated subtype differences (control and quiescent and active UC) in the percentage of PBs for patients of VCo1, adjusting for age and gender and processing batch in a linear model.
Lamina propria cell stimulation.
Cells obtained from the lamina propria were resuspended at a concentration of 1 million cells per milliliter of R10 solution (RPMI with 10% heat-inactivated FBS and antifungal and antibiotic solutions containing streptomycin, amphotericin B and penicillin (1×, Gibco, 15240062)). Cells were then cultured in a U-bottomed 96-well plate with each well containing 200,000 cells. The cells were incubated overnight for 18 h with 2.5 μl of CD3CD28 beads (Gibco) or 2.5 μl of PBS in 200 μl as control. Brefeldin A (1,000×, BioLegend) was added in all wells at a final concentration of 1×, 4 h before harvesting the cells. Cells were then stained extra-cellularly and intra-cellularly and analyzed by flow cytometry as previously described.
Immunofluorescence staining from colonic biopsies (CD79a, IgA and Ki67).
Immunofluorescence staining was performed on five patients with untreated active colitis and five HCs. Formalin-fixed, paraffin-embedded human tissue sections 3 μm in thickness were treated in xylene, a decreasing alcohol gradient and distilled water to achieve de-waxing and rehydration of the tissue. Heat-induced epitope retrieval was performed for 15 min in Tris-EDTA buffer (pH 9). After epitope retrieval, tissue sections were permeabilized with 0.2% Triton X-100 in PBS, blocked with 5% BSA and 5% Fc receptor blocking reagant (Miltenyi Biotec) and stained with polyclonal rabbit anti-human IgA (A0262, DAKO, 1:1,000 dilution), monoclonal mouse anti-human CD79a (clone JCB117, DAKO, 1:25 dilution) and monoclonal rat anti-human Ki67 (SolA15, Invitrogen, 1:50 dilution). Staining with primary antibodies was followed by detection with polyclonal donkey Alexa Fluor 546-conjugated anti-mouse IgG antibody (Invitrogen, 1:200), Alexa Fluor 647-conjugated anti-rabbit IgG antibody (Invitrogen, 1:500) and Alexa Fluor 488-conjugated anti-rat IgG antibody (Invitrogen, 1:500). Nuclear DNA was visualized with DAPI, and coverslips were applied with FluorSave reagent (Merck Millipore). Images were acquired with either a Leica TCS SP5 upright confocal microscope or a Nikon Eclipse Ni-E microscope and were further analyzed with ImageJ software.
Surgical tissue analyses of TPH cells using immunofluorescence microscopy (CD3, CD20 and CXCL13).
Surgical tissues were obtained from patients with UC undergoing colectomy and compared to surgical tissues obtained from age-matched HCs who had partial colon resections owing to penetrating wounds (for example, gunshot wound). UC specimens with partial colonic involvement were selected such that a clear demarcation of the disease border was evident on macroscopic inspection (Extended Data Fig. 9). This allowed us to confirm whether there was an expansion of TPH cells in the entire colon or only at sites with macroscopic inflammation. Control colonic sections contained well-organized isolated lymphoid follicles that differed among individuals with respect to overall B and T cell content. Nine HCs and 11 patients with UC (clinical characteristics in Supplementary Table 21) were studied, and their intestinal tissues were stained for CD3, CD20 and CXCL13. For patients with UC, the colonic tissue was characterized at the margin of inflammation and upstream the inflamed mucosa (Extended Data Fig. 9).
Paraffin sections were prepared and immunostained after a citric acid antigen retrieval (Diva Decloaker, Biocare Medical). Overnight staining with primary antibodies to CD3 (Abcam, ab5690), CD20 (Abcam, ab9475) and CXCL13 (R&D Systems, AF801). Secondary antibodies used in detection were purchased from Jackson ImmunoResearch as conjugates to Cy2, Cy3, Cy5 or horseradish peroxidase (HRP). Detection with HRP was used in conjunction with the Opal 7-Color Kit from PerkinElmer, following the manufacturer’s directions. Images were acquired using a Leica SPE confocal microscope additionally outfitted with a Hamamatsu epifluorescence camera. Data analysis was carried out on the entire section after acquiring images using the tile scan feature of the microscope. Lamina propria area containing reactivity to these stains or co-localization of CD3 with CXCL13 to identify TPH cells was analyzed using Imaris and Fiji software (National Institutes of Health). Data are reported as fraction of lamina propria area stained for the indicated markers or co-localized marker.
Compared to normal colonic isolated lymphoid follicles (Extended Data Fig. 9c), many proximal colonic areas from patients with UC seemed normal, but, in some specimens, proximal tissue with a healthy macroscopic appearance contained either fibrotic scarring (Extended Data Fig. 9d), which suggested past disease involvement, or many inflammatory cells (Extended Data Fig. 9e), which reflected ongoing disease activity uncoupled from overt macroscopic changes.
IgH gene sequencing.
The IgH gene of cells of interest was sequenced from four patients with active UC and four HCs (clinical characteristics in Supplementary Table 6) using a method previously described57. In brief, we single-cell sorted short-lived PCs (live CD45+CD38++CD27+CD19+) and gut-homing PBs (CD19+/intCD38hiCD27+IgD−β7+) in 96-well polymerase chain reaction (PCR) plates in a lysis solution containing dithiothreitol (100 mM, Thermo Fisher Scientific) and RNAse inhibitor (RNAsin, Promega). PCR plates were immediately frozen on dry ice and stored at −80 °C until further processing. Single-cell cDNA synthesis was performed in the 96-well plates initially used for the sort, using random hexamer primer, dNTP mix (Denville), NP-40 (2%), RNAse inhibitors (RNAsin, Promega) and Maxima Reverse Transcriptase (RT Max, Thermo Fisher Scientific). IgH genes were then amplified with two rounds of nested PCRs (primer list in Supplementary Table 22). All PCR products were then sequenced (Genewiz) and analyzed for VH and JH usage, CDR3 mutations, V gene mutations and isotype. Sequences were analyzed by IMGT (www.imgt.org) to identify germline VDJ gene segments with the highest homology. Two clones were considered as the same clonotype when they had the same VH and JH families as well as the same CDR3 length and at least 80% homology in the amino acid sequence of the CDR3. Diversity was assessed using the Shannon diversity index calculated as:
Production of monoclonal antibodies.
We generated monoclonal antibodies by single-cell sorting and molecular cloning of intestinal short-lived PCs from four HCs and four patients with UC (same donors as those studied to analyze the B cell repertoire). Single-cell cDNA synthesis, nested PCR amplification and sequencing of IgK and IgL genes were then performed for clones of interests (12 clones from UC and eight clones from HC, with nine and five passing the quality control, respectively) as described previously57. Clones with highest frequencies and/or clones shared between circulating and colonic cells in each individual were selected. Gene blocks corresponding to variable regions of IgH, IgK and IgL genes of clones of interest were ordered from Twist Bioscience. Gene blocks corresponding to IgH V(D)J, Igk or Igl VJ variable regions were cloned into expression vectors encoding for the constant regions of the human IgG1, IgK and IgL, respectively. All antibodies are expressed as fully human Fc-IgG1 antibodies.
Expression and purification of recombinant antibodies.
239T HEK cells were transiently transfected with equal amounts of both corresponding IgH and IgL or IgK chain plasmids using polyethylenimie. Transfected HEK cells were cultured in OptiPRO SFM medium (Gibco). Supernatants were collected 6–8 d after transfection and filtered through a 0.45-μm pore size, radio-sterilized PVDF membrane and stored at 4 °C. Igs were purified from supernatants using protein G-based affinity chromatography (Protein G Sepharose TM 4 Fast Flow, GE Healthcare). Polyprophylene reservoir PKG50 columns (SPE Accessories) were loaded with supernatant. Antibodies were eluted using glycine 0.1 M (pH 2.7), and pH was neutralized with 1 M Tris-HCl (pH 9.0). Collected supernatants were concentrated and washed with PBS, and antibodies were collected in PBS.
Microbiota antigen microarray and detection.
We tested serologic IgA and IgG reactivity to a panel of 32 representative microbial antigens (Supplementary Table 8) in 22 HCs, 53 patients with active UC (from the primary cohort) and 28 patients with active Crohn’s disease with a microbiota antigen microarray58.
Bacterial proteins and extracts were arrayed in triplicate on 16 pad nitrocellulose slides (Maine Manufacturing) at a concentration of 0.2 mg ml−1 using a SpotBot Personal Microarrayer (Arrayit). Antigens were diluted from stocks to 10 mM Tris (pH 7.4), 20% glycerol and 0.1% SDS. To probe the microarray, pads were blocked for 1 h in SuperBlock (Thermo Fisher Scientific) and then probed with indicated sera diluted 1:100 in SuperBlock for 1 h. The pads were washed three times in PBS with 0.05% Tween 20; then, a mixture of anti-human IgA (ImmunoReagents) and anti-human IgG (Invitrogen) secondary antibodies labeled with Dylight 550 and Dylight 650, respectively, were applied at 1:1,000 (~0.5 μg ml−1) for 1 h in SuperBlock. The pads were then washed three times with PBS-Tween, air dried and scanned using a GenePix 4000B imager (Axion).
ELISA for detection of anti-integrin αvβ6 antibodies.
Total plasma IgG was measured using Mabtech human IgG ELISA kit following the manufacturer’s protocol. Once total plasma IgG was quantified, plasma samples were diluted to a concentration of 10 μg ml−1, and the clones produced from intestinal B cells were diluted 1:500 in PBS for subsequent testing for anti-integrin αvβ6 as described below.
For detection of IgG against integrin αvβ6, MaxiSorp immuno microtiter plates (Thermo Fisher Scientific) were coated with 100 μl per well of 1.5 μg ml−1 human recombinant integrin αvβ6 (R&D Systems) at room temperature for 60 min using coating buffer from the ELISA Starter Accessory Kit (Bethyl Laboratories). Next, 1 mM MgCl2 and CaCl2 was added to the incubation buffers to stabilize the αvβ6 integrin heterodimer as described by Kuwada et al.30. The plates were subsequently washed with ELISA washing buffer three times and blocked with 200 μl per well blocking buffer for 30 min at room temperature (all buffers purchased from Bethyl Laboratories). The plates were then washed three times and incubated with 100 μl per well of samples prepared as described above and with mouse anti-human αvβ6 (Millipore, MAB2077Z) from 312.5 ng ml−1 to 1.22 ng ml−1 to produce standard curve for 60 min at room temperature. Plates were then washed five times and incubated with secondary antibodies. For the human samples, anti-human IgG secondary conjugated to HRP (Invitrogen, 31410) diluted 1:4,000 was used; for the standards, anti-mouse IgG secondary conjugated to HRP (Invitrogen, 62–6520) diluted 1:2,000 was used. Plates were washed again five times and then developed by incubating with 100 μl per well of 3,3′,5,5′-tetramethylbenzidine for 4 min, at which time the reaction was stopped with 100 μl per well of 0.18 M H2SO4 and immediately read on the POLARstar Omega plate reader (BMG LABTECH).
Droplet-based scRNA-seq.
scRNA-seq was performed using the 10x Genomics platform (version 2, 3′ kit) as described previously17. In total, 10,000 cells at a concentration of 1.106 per milliliter in PBS were loaded in the Chromium Controller instrument within 15 min after completion of the cell suspension preparation using GemCode Gel Bead and Chip, all from 10x Genomics. Manufacturer recommendations were followed. Cells were partitioned into gel beads in emulsion in the Chromium Controller instrument where cell lysis and barcoded reverse transcription of RNA occurred. Library preparation was made with 10x Genomics Library Kits and sequenced using an Illumina NextSeq 500/550 with the 75-cycle kit and the parameters of 28 × 8 × 55. Read depth of more than 100 million reads per library, or an approximate average of 30,000 reads per cell, was obtained.
scRNA-seq clustering analysis.
Sequenced reads were processed by Cell Ranger version 2.0, and raw UMI matrices were generated for each sample. Single cells with more than 1,000 UMIs were classified as T or B PCs by a maximum likelihood classifier, which was trained on an independent IBD dataset17. Specifically, the cell type model parameters of the established model are assumed while a noise mixture parameter is fitted for each of the samples of the current study. Maximum likelihood mapping between the single cells to cell types in the model allows classification of the broad B cells, T cells and myeloid lineages. Separate subclustering of the B cell/PC and T cell compartments were performed using our previously published batch-aware iterative clustering algorithm17 with minor adjustments. The clustering did not use genes with strong lateral expression or substantial background noise levels. Training set size was 1,500 and 2,000 for the B/plasma and T cell clustering, respectively. For both the B/plasma and T cells, the best clustering initiation was selected from 1,000 instead of 10,000 k-means + runs, and the following parameters were used: Kreg_ds = 0.1; (P1,P2) = (10th,30th) percentiles; Kreg = 1 · 10−6; k = 20. Transcriptional profiles were analyzed by scDissector, an exploratory data analysis tool for scRNA-seq datasets59.
Correlation matrix of gene modules defined by gene-to-gene correlation analysis of single cells within the B lymphocyte clusters.
We downsampled the cells to 2,000 UMIs per cell and selected variable genes similarly to the seeding step of the clustering. Similarly to other publications60–62, we selected genes with variability that could not be explained by multinomial sampling variance. We calculated a loess curve for the log(variance/mean) versus log(mean) distribution and binned the log(variance/mean) values by intervals of 0.2 of log(mean). We selected genes with more than 50 UMIs in U’L from the 8th percentile of each bin and also required that their log(variance/mean) is 0.1 or higher above the loess curve. To focus on biologically relevant gene-to-gene correlation, we calculated a Pearson correlation matrix between genes for each sample. For that purpose, expression values were log-transformed log(1 + UMI(gene,cell), and genes with fewer than five UMIs were excluded. Correlation matrices were averaged following z-transformation. The averaged z-matrix was then transformed back to correlation coefficients. We grouped the genes into gene ‘modules’ by complete linkage hierarchical clustering.
For the scRNA-seq analyses, two R packages were used:
mixtools – normal mixture model
scDissector (https://github.com/effiken/scDissector/)—scRNA-seq visualization
Calculation of joint distributions of IgG and IgA scores per PB from scRNA-seq data.
PB cell where the scores are defined as:
Score(IgG):= log2(#UMIs IgG)
Score(IgA):= log2(#UMIs IgA)
We modeled the difference between the scores Score(IgG) − Score(IgA) as a mixture of two Gaussians and fitted the model using the normalmixEM function in the mixtools R package with the parameters k = 2 ; mu = c(−2,2). Cells were assigned to the IgG group or IgA group according to the posterior probability estimated by the mixture model with significance of 0.05. Cells with scores that could not be associated with IgG or IgA were assigned to the None/Both groups.
Identification of T cell subsets from scRNA-seq data.
To characterize the different T cell clusters, we first defined gene expression programs using literature-based knowledge and our previous work17. We defined five gene programs related to cell cycling (STMN1, CLSPN, CKS1B, NUF2, ASPM, UBE2T, CENPF, RRM2, DTYMK, SGOL1, CENPE, MAD2L1, CCNA2, CENPU, CCNB1, DHFR, HMMR, MXD3, GMNN, CENPW, MCM7, EZH2, SMC2, CCDC34, CDCA5, H2AFX, ZWINT, CDK1, HELLS, MKI67, TMEM106C, CDKN3, NUSAP1, CCNB2, KIAA0101, PRC1, CENPN, CDT1, AURKB, TOP2A, TK1, BIRC5, TYMS, CDC25B, PCNA, TPX2, UBE2C, ASF1B and GTSE1); immunoregulation (TNFRSF18, TNFRSF4, TNFRSF9, TNFRSF1B, AC133644.2, CTLA4, TIGIT, ICA1, RHBDD2, MAGEH1, IL2RA, TBC1D4, BATF, IKZF2 and FOXP3); naive/central memory (CM) (SELL, LEF1, SOX4, SC5D, CCR7, TOB1 and NOSIP); CD8/cytotoxic (CD8A, CD8B, DHRS3, GZMA, GFOD1, IFITM3, PRF1, KLRD1, GZMB, CCL5, NKG7, FGR, CD160, FCER1G, XCL2, XCL1, GNLY, EOMES, CMC1, DTHD1, AOAH, CLIC3, CTSW, KLRF1, KLRC2, KLRC1, PTGDR, MCTP2, CCL3, CCL4, CCL3L3, CCL4L2, MATK, MAPK1 and IL2RB); and resident memory T cell (JUN, KLF6, FOSB, PTGER2, FOS, SYTL3, SPRY1, ANKRD28, GPR171, PDE4D, JAML, IL7R, GLIPR1, CD69, NFKBIA, PPP1R15A, NFKBIZ, TNFAIP3, PTGER4, ANXA1, ID2, ATF3, MGAT4A, AC092580.4, KLRB1, RORA, IL18R1, STAT4, IFNGR1, PFKFB3 and GPR65) states. The total UMI counts of each of the five transcriptional programs were calculated for each cell and divided by the total number of UMIs per cell. The five scores were log-transformed after division by their 75% percentiles. These scores demonstrated the different distributions of the expression programs across T cell states (Extended Data Fig. 8).
T cell cluster 11 expressed a strong cell cycle score and genes encoding for membrane proteins associated with human T cell activation, such as HLA-DRA, CD38 and the high-affinity IL-2 receptor chain IL2RA (CD25) and, thus, was annotated as activated/cycling T cells. Cells in this cluster included both CD4 and CD8 T cells, indicating that T cells clustered according to their highly activated state over the CD4/CD8 lineage specification. Cells in cluster 15 had lower expression of the cycling program but expressed effector molecules associated with CD4+ T helper cell function—that is, CD40LG or CD8+ T cell cytotoxicity (GZMA, GZMB and PRF1) (Extended Data Fig. 8b)—as well as the highest levels of effector cytokines (TNF, IFN-γ, IL-17, IL-22 and GM-SCF), as compared to other T cell clusters (Fig. 4b), prompting us to annotate this cluster as activated/effector T cells. T cell clusters 8 and 10 expressed high levels of the cytotoxic program, including granzymes and perforin-encoding genes, as well as low levels of the resident memory, naive/CM and cell cycling programs and low expression of the IL-7 receptor gene IL7R (CD127) and, thus, were referred to as effector memory cytotoxic T lymphocytes (CTLs)63. T cell cluster 5 expressed CD8A and CD8B but lower level of the CD8/cytotoxic program. Cells in cluster 5 had strong expression of KLRB1 (CD161) and the resident memory T cell program, leading us to annotate them as MAIT-like T cells, as 3′ scRNA-seq does not inform on invariant TCR expression, such as Vα7.2-Jα33.
T cell clusters 4 and 13 expressed high levels of Treg-associated genes, including FOXP3, IL2RA (CD25) and CTLA4, together with genes whose high expression was consistently associated with Tregs by recent scRNA-seq studies, including TNFRSF18, TNFRSF4 and LAYN64–66 and BATF, which was recently identified as a critical transcription factor for tissue Tregs67, and were thus referred to as Tregs. Cells in cluster 6 were referred to as TPH cells, as described in the main text. Two cell clusters (12 and 14) expressed high levels of the naive/CM signature, including the chemokine receptor CCR7 and the selectin CD62L (SELL), which is known to promote the recruitment of naive and CM T cells to the T cell areas of secondary lymphoid organs through the high endothelial venule68. In agreement with the naive and/or CM state of T cells in these clusters, high expression of IL-7 receptor IL7R (CD127) was also detected63. The first of these two clusters (cluster 12) exhibited higher levels of the TF LEF1, preferentially expressed by naive T cells, before antigen exposure69, and low-to-no expression of genes associated with CD4 and CD8 effector T cell functions or tissue-directing chemokine receptors. As for activated T cells, CD4+ and CD8+ T cells were mixed in this cluster, supporting dominance of the naive state at driving the clustering, an observation already reported for blood naive T cells66.
In addition to cluster 5 described above, five clusters (1, 2, 3, 7 and 9) shared strong expression of the tissue resident memory (Trm) T cell program, which included the tissue retention molecule CD69; TFs such as ATF3, FOS, FOSB and KLF6; the integrin ITGA1 (CD49a); and the TF PRDM1, previously shown to be associated with gut Trm70–74. In support of the resident memory state of these cells, the Trm clusters accounted for the large majority of the T cells in non-inflamed tissues. Additional expressed genes included the prostaglandin E2 receptor (PTGER4), ANKRD28, SPRY1, G-protein-coupled receptor GPR171, the TF ID2, TNFAIP3 and an NF-κB inhibitor induced by TNF and protecting T cells against TNF-induced apoptosis. Cluster 2 corresponded to CD8 Trm. The remaining Trm clusters (1, 3, 7 and 9) expressed multiple genes associated with Th17 T cells (KLRB1, CCL20, IL17, IL22, MAF and CCR6), CD40LG (CD154) and the cytokine IL-2, thus suggesting antigen-dependent activation75.
Bulk RNA extraction from intestinal biopsies.
RNA was isolated from frozen tissue using Qiagen QIAsymphony RNA Kit on the QIAsymphony. Eluate volume was measured with VolumeCheck (BioMicroLab), with RNA concentration determined using the Trinean DropSense 96 (RNA quality number was generated using an Advanced Analytical Fragment Analyzer with Agilent’s standard sensitivity).
RNA sequencing of bulk biopsies.
One microgram of total RNA was used for the preparation of the sequencing libraries using the RNA TruSeq Kit (Illumina). Ribosomal RNA from biopsy tissue was depleted from total RNA using the Ribo-Zero Kit (Illumina) to enrich polyadenylated coding RNA as well as non-coding RNA. The rRNA-depleted RNA from biopsy total RNA was used for preparation of the sequencing library using RNA TruSeq Kit supplied by Illumina. In the first step, rRNA-depleted total RNA was fragmented in the presence of divalent cations at 94 °C. The fragmented RNA was converted into double-stranded cDNA. After polishing the ends of the cDNA, adenine base was added at the 3′ ends, after which Illumina-supplied specific adaptors are ligated. The adaptor-ligated DNA was size selected to get an average of 250 bp insert size using AMPure beads and amplified by 15-cycle PCR. The PCR-amplified DNA was then purified using AMPure beads to get the final sequencing library. The insert size and DNA concentration of the sequencing library were determined on an Agilent Bioanalyzer. The Ribo-Zero RNA-seq libraries were sequenced on the Illumina HiSeq 2500 platform using 100-bp paired-end protocol following the manufacturer’s procedure. Base calling from images and fluorescence intensities of the reads was done in situ on the HiSeq 2500 computer using Illumina software. Various quality control parameters, such as intensities of individual bases, visual and graphic focus quality of the images, sequence quality measured in terms of colored graphic representation of Q30 values (which is a measure of errors per thousand bases) and error rates at 35 and 75 cycles of sequencing, were monitored periodically to assess the quality of an ongoing run.
Macrophage activation signature analysis.
The macrophage IC stimulation signature was created using a previously generated RNA-seq dataset of murine colonic macrophages stimulated ex vivo with IgG ICs (GSE109040) (Extended Data Fig. 6). Gene set enrichment analysis of the top 200 upregulated and downregulated macrophage genes by IgG stimulation was performed on a pre-ranked RNA-seq dataset of human monocytes stimulated with complexed IgG (GSE102728) to further select highly conserved relevant genes that are enriched in the leading edge of the analysis. The signature was refined to obtained better predictors for disease-relevant networks using elastic net regularized logistic regression implemented in the glmnet R package with the α parameter set as 0.5 and leave-one-out cross-validation procedure applied76 by training it on a publicly available microarray dataset of colonic mucosal biopsies from human patients with UC and controls (GSE38713). The fitted model was tested internally with the training set and achieved 96.29% classification accuracy. Genes from the fitted model were then used to score additional datasets using gene set variation analysis (GSVA) with default parameters. We also generated a second microbial gene signature using transcriptomic analysis of colonic macrophages from mice treated with dextran sodium sulfate and refined on LPS-treated human monocytes (Extended Data Fig. 6)
RNA-seq alignment and feature quantitation of Validation Study 1 (MSSCR subcohort).
Genomic alignment to GRCh37 of single-end RNA-seq reads was performed using two-pass STAR77. Default parameters for STAR were used, as were those for the quantitation of aligned reads to GRCh37.75 gene features via featureCounts78. Multi-mapping reads were flagged and discarded.
Raw count data were pre-filtered to keep genes with counts per million greater than 0.5 for at least 3% of the samples. After filtering, count data were normalized via the weighted trimmed mean of M values79, where the weights originate from the delta method on the binomial data, and further variance is stabilized using a logarithmic transformation. Normalized counts were further transformed into normally distributed expression values via the voom-transformation using a model that included technical covariates (processing batch, RNA integrity number, exonic rate and rRNA rate) while accounting for the intra-patient correlation across regions. The voom-transformed expression data were then adjusted for technical covariates and the final input for statistical modeling.
Overall activity scores for the macrophage Fc IC signatures were obtained for this cohort using GSVA80. To evaluate differences in gene set activity scores across different conditions, linear mixed-effect models were used considering a random intercept for each patient and a compound symmetry correlation structure to model the correlation of multiple regional biopsies per patient. Fixed factors included disease (UC versus control) and anti-response (responder/non-responder), as appropriate to the comparison; its interaction with tissue (inflamed/non-inflamed); as well as the covariates gut region, age, gender and ethnicity (Supplementary Table 11)
Consensus clustering based on peripheral blood immunological data and statistical analyses.
We included in the analyses a total of 96 patients from the primary cohort, for whom complete phenotypic data from circulating T and B cells were available. Clinical variables including sex, extent of disease, disease duration, rectal bleeding score, clinical Mayo Index and CRP level were considered.
Marker abundance was log (x + 1) transformed. T and B cell markers were considered to cluster samples into different groups with similar immune infiltration profiles. For this purpose, the package ConsensusClusterPlus81 from Bioconductor82 was used. Specifically, at each iteration, 80% of the features were randomly subsampled without replacement, and samples were clustered via k-means clustering. This procedure was repeated for 1,000 iterations, and the number of clusters resulting in less variability across iterations was selected. Once the consensus clusters were derived, markers differentially expressed across clusters were identified. Upregulation/downregulation of each marker in different consensus groups was assessed using a linear regression where the marker abundance was modeled as a function of consensus clusters. First, every marker was normalized to z-score across samples. Then, the abundance of the j-th marker for sample i was modeled as:
with ϵi,j ~ N (0, σj), Ik being the set of samples belonging to the k-th consensus cluster, 1(A) being an indicator function equal to 1 if the event A occurs and 0 otherwise and βk,j being the coefficient capturing the association between marker j and the k-th group. P values were derived via t-test, and Benjamini-adjusted P values can be found in the Supplementary Data. Plots were drawn using GraphPad Prism software. Statistical significance of data between groups was assessed using the Mann–Whitney test and the two-sample paired Student t-test when appropriate (Supplementary Table 15). Correlations were assessed using the Spearman test.
Prediction of disease complications using Kaplan–Meier analyses.
Because the duration of follow-up for patients recruited in our primary cohort was short (less than 1 year for most patients), we focused on VCo1, where patients had been followed for up to 4 years after the index blood draw. Clinical follow-up information was available for 73 patients with UC from VCo1 (Supplementary Table 18). Complicated disease was defined as the occurrence of one of the following events: change in biologic treatment (identifying non-responsive disease), UC-related hospitalization or UC-related surgery. Thirty patients (41%) had an event over the time of follow-up. Patients were segregated into two groups based on the median frequency of PBs in the circulation, and Kaplan–Meier curves were generated to estimate the incidence of disease complications over time.
Statistics and reproducibility.
Patients were recruited prospectively into the principal cohort. As no a priori comparaison and hypothesis were made on group differences, no sample size calculation was made. For the validation cohorts, patient recruitment had been done before the analyses. Therefore, a priori sample size calculations were not performed. Both validation cohorts comprised more than 100 patients, allowing for accurate validation. Five samples from HCs and four samples from inflamed UC were chosen for single-cell sequencing, and four samples from HCs and four samples from inflamed UC were chosen for single-cell IgH gene sequencing, to allow for patient variability. Details of the statistical tests used are provided in the manuscript and figure legends. No data were excluded from the analyses. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Fig. 1 |. Outline of the patients and samples included in the study.
HC: Healthy controls; aUC: active ulcerative colitis; qUC: quiescent ulcerative colitis; PBMC = peripheral blood mononuclear cells.
Extended Data Fig. 2 |. Lineage grouping within 7 major cellular compartments from single-cell RNA sequencing of colonic biopsies.
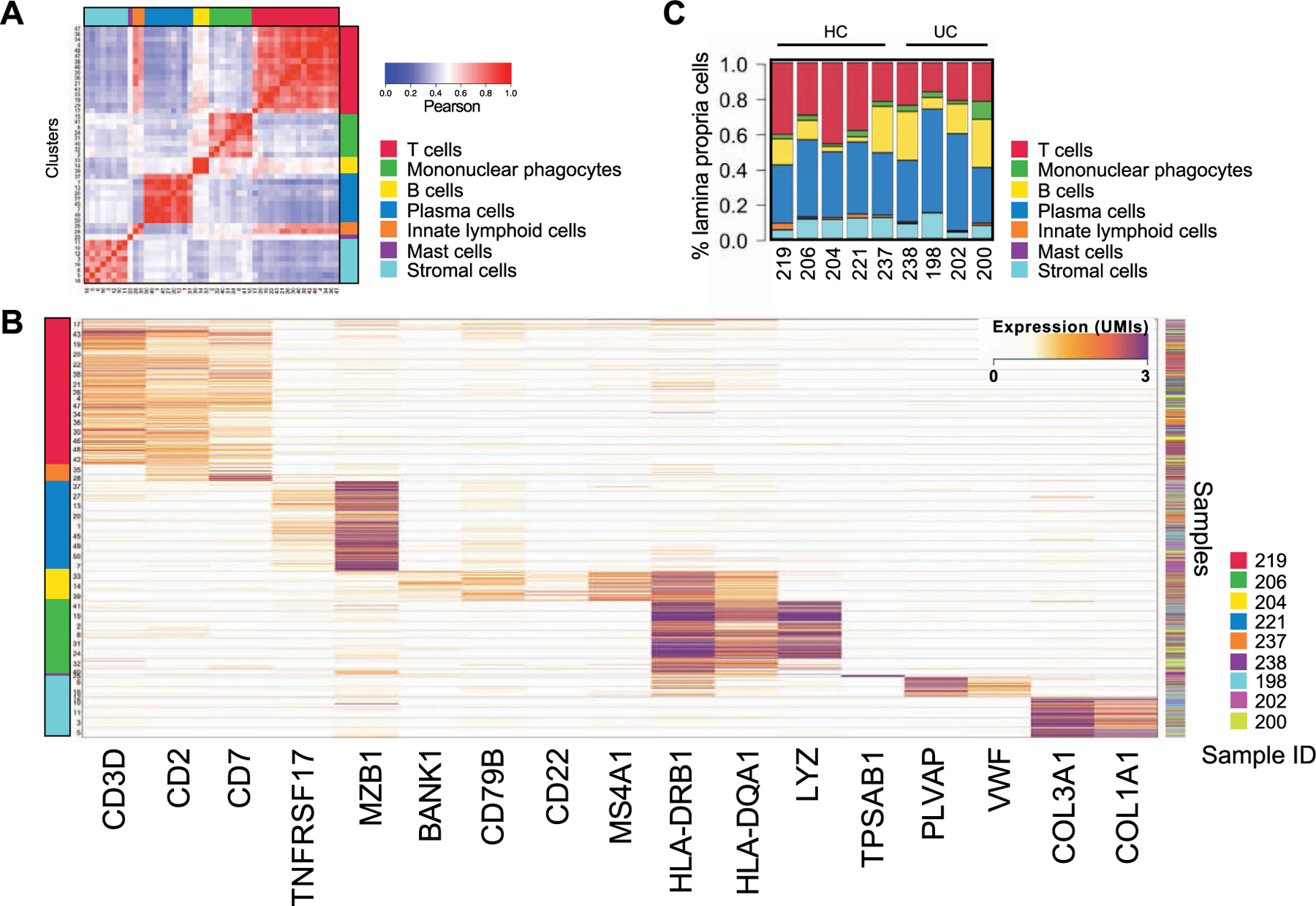
a, Grouping single-cell RNA sequencing cell clusters by similarity. Heatmap showing Pearson correlation coefficients between the log-averaged expression profiles of clusters. Ordering was determined by hierarchical clustering, which unbiasedly grouped the clusters by cellular lineage (color-coded bar). b, Heatmap showing color-coded down-sampled UMI counts of representative lineage marker genes in 100 randomly selected cells per cluster. The color code on the left indicates the lineage annotation of the different clusters as follows: red=T cells; orange=innate lymphoid cells; blue= plasma cells; yellow= B cells; green= mononuclear phagocytes; purple- mast cells; teal= stromal cells. c, Major cellular compartment subtypes frequencies within total lamina propria cells. The number of cells in each cellular subtype is divided by the total number of lamina propria cells and visualized in a stacked bar for each sample (HC and patients with UC).
Extended Data Fig. 3 |. Characterization of the 20 colonic clusters of B cells from single-cell RNA sequencing of colonic biopsies.
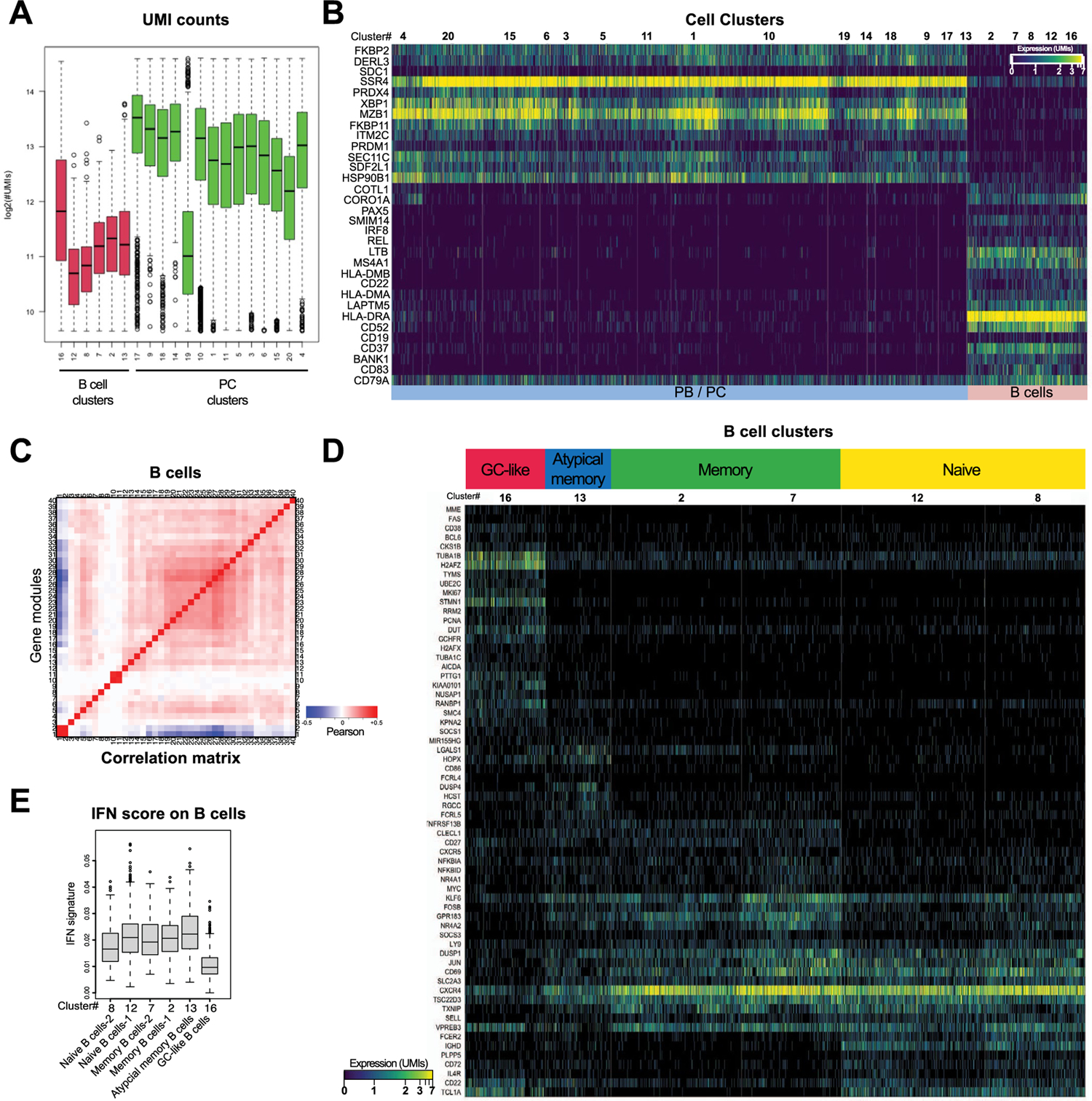
a, UMI counts in B lymphocyte and plasma cell clusters. box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range from the top or bottom of the box. b, Heat-map showing color-coded down-sampled UMI counts of highly-variable genes between clusters of B cells (n = 7621 cells) and PC (n = 21425 cells). Clusters are demarcated by gray bars. Cells were down-sampled to 2000 UMIs/cell. Detailed cell counts per B and PB/PC cluster per sample are accessible in Supplementary Table 2. c, Correlation matrix of gene modules defined by gene-to-gene correlation analysis of single cells within the B lymphocyte clusters. d, Heat-map showing color-coded down-sampled UMI counts of gene programs identifying naïve, memory and germinal center-like B lymphocytes. Clusters are demarcated by gray bars. Cells were down-sampled to 2000 UMIs/cell. e,I FN signature scores on the indicated cell types. Supplementary Table 2 shows cell-counts per cluster, box plots represent the third quartile (top of the box), median (center lines) and first quartile (bottom of the box) of measurements, and the whiskers represent 1.5 times the interquartile range from the top or bottom of the box.
Extended Data Fig. 4 |. JH, VH family usage and clonal relationships.
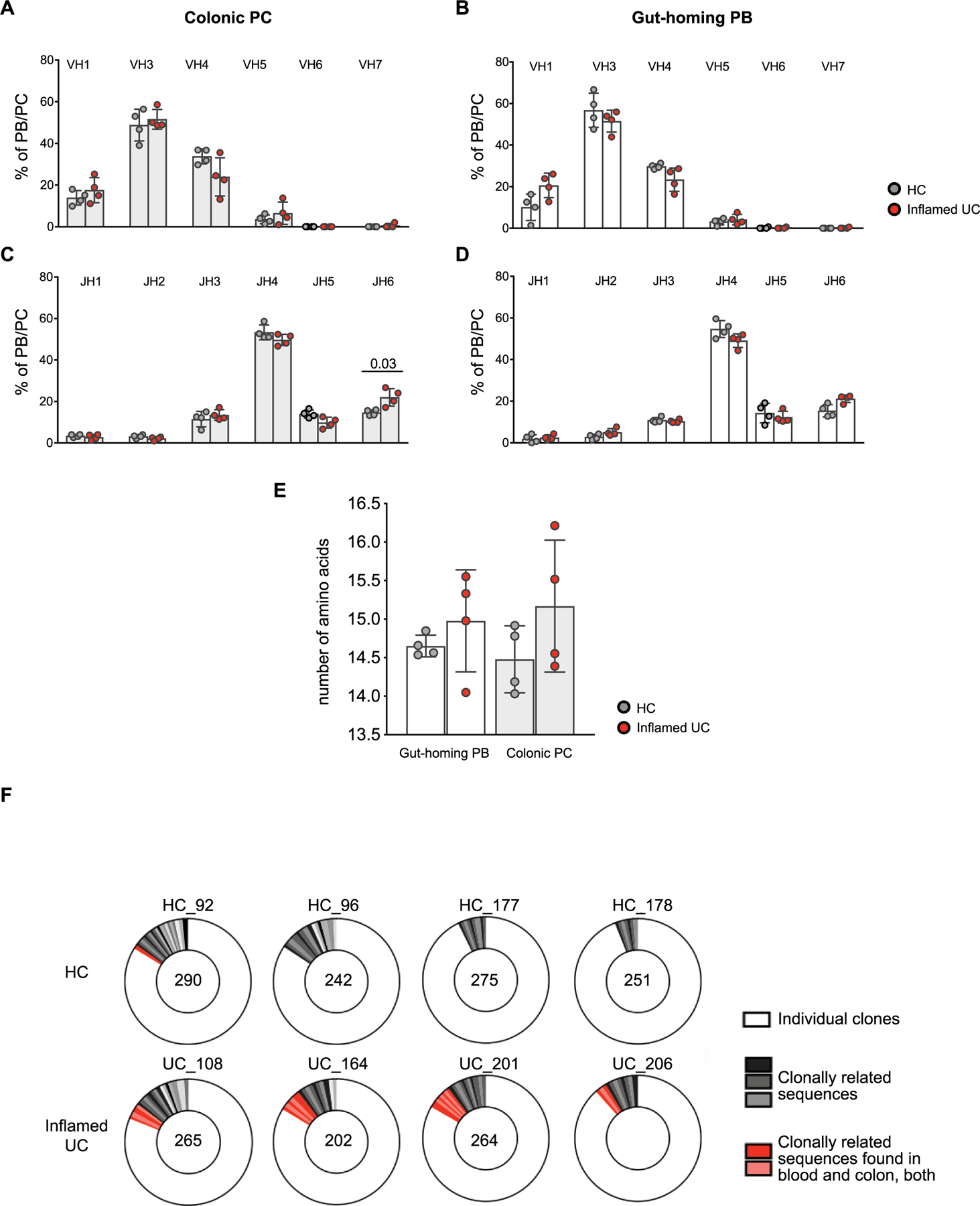
a-d, IgH V and J gene family usage in short-lived colonic PC (a,c) and in circulating gut-homing PBs (b,d). Data are shown as individual data, mean and standard deviation; Mann- Whitney test, two tailed. e, Mean IgH amino acid CDR3 length per patient excluding JH6 + clones. Mann-Whitney test, two tailed, no significant difference. Panels a-e are based on 4 HC and 4 active UC patients f, Clonal relationships of all clones analyzed for each HC and UC patient. Numbers within circles stand for total number of clones.
Extended Data Fig. 5 |. Serological reactivity against microbiota antigens.
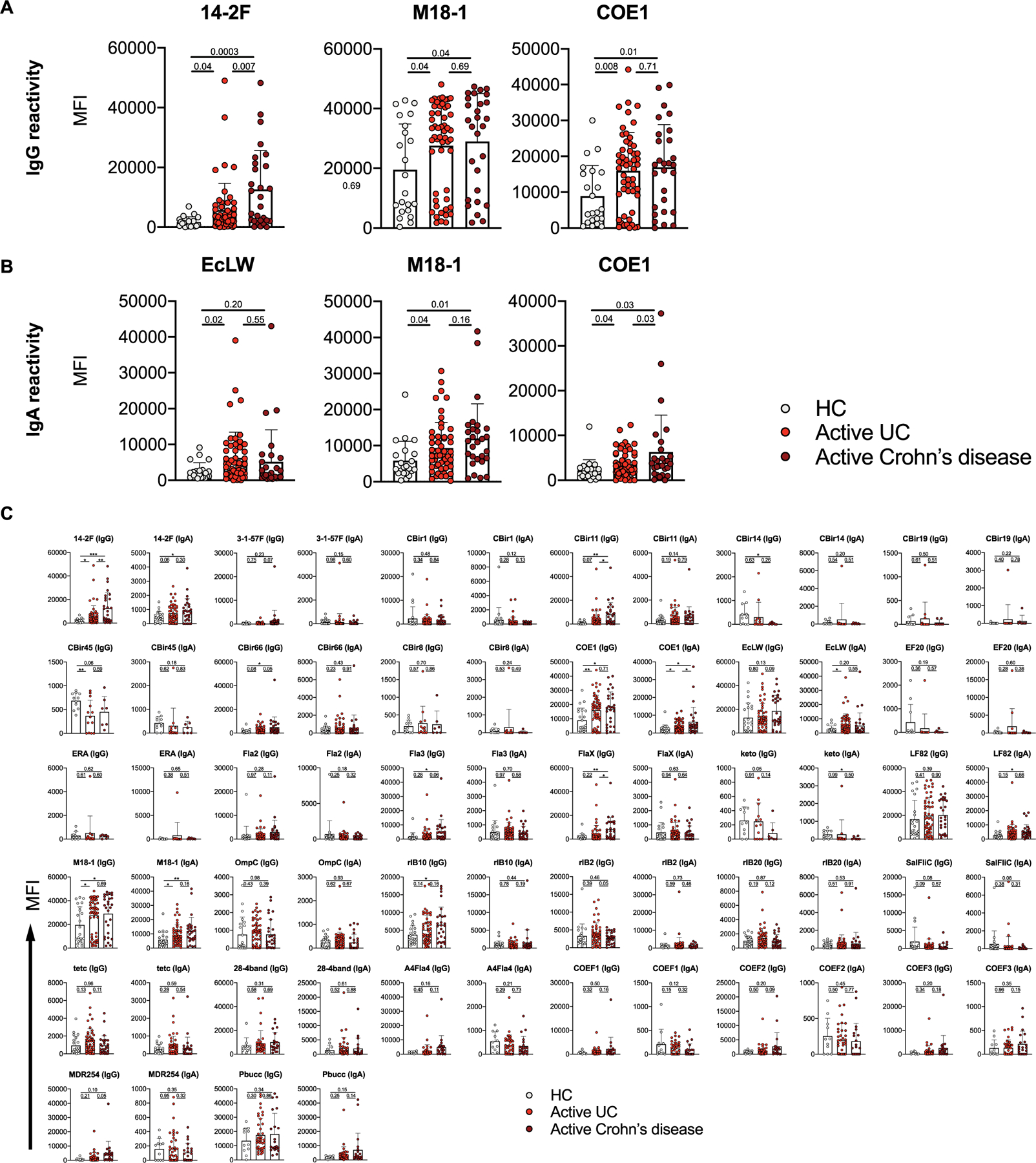
Thirty-two bacterial proteins and extracts (described in detail in Supplementary Table 8) were arrayed in triplicate on 16 pad nitrocellulose slides. After blocking, patient serum was added to the respective pads to determine reactivity against these antigens. a-b, Representation of increased IgG (a) and IgA (b) reactivity against microbial antigens 14–2 F, M18–1, COE1 and EcLW in patients with UC. c, IgG and IgA serological reactivity against 32 microbial antigens. Data are shown as individual data, mean and standard deviation, t-test, two-tailed. Details of the data from panel c are also provided in Supplementary Table 7.
Extended Data Fig. 6 |. Generation of a molecular signature of an IgG-activated or LPS colonic macrophage activation response.
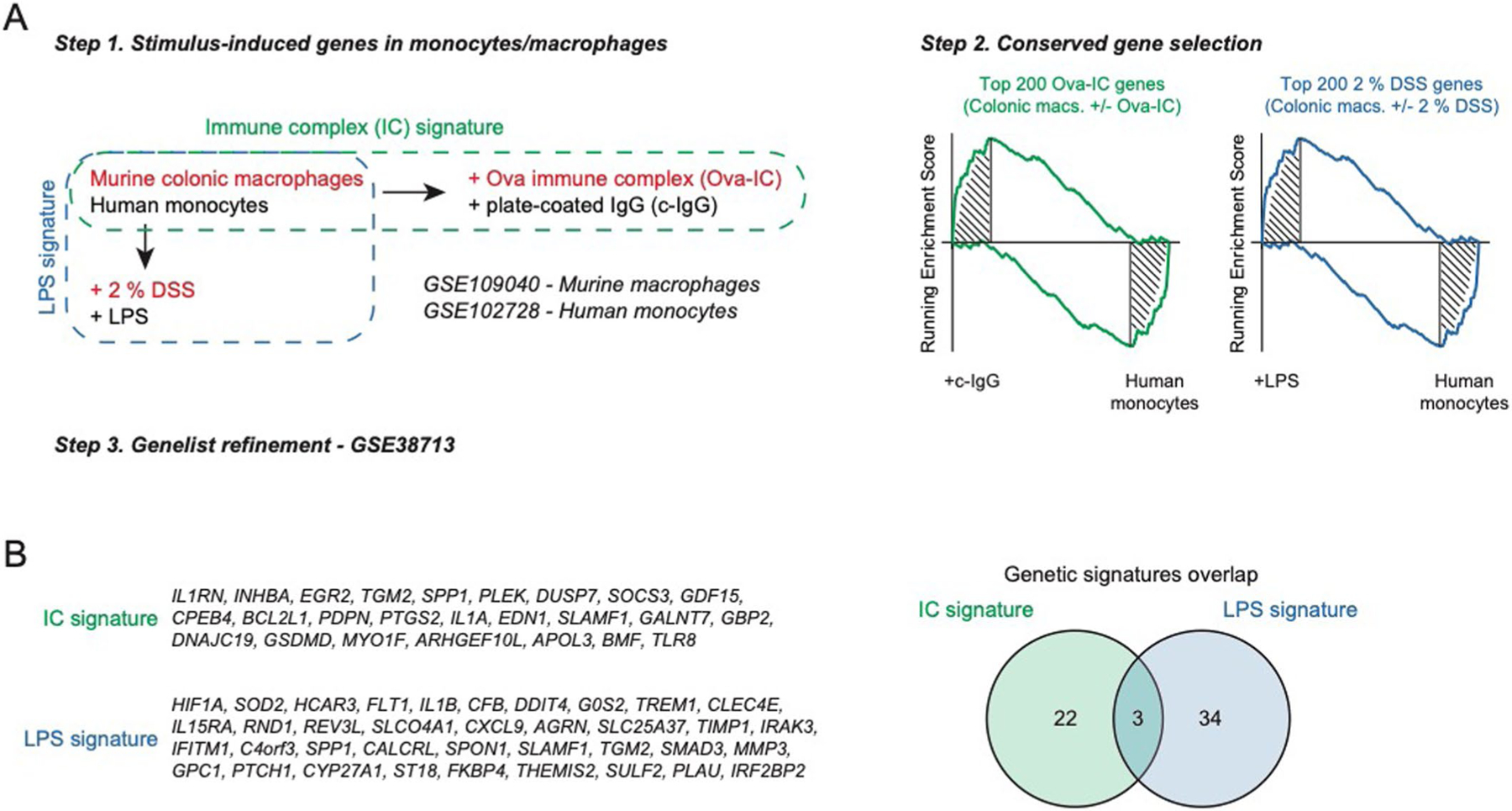
a, Generation of a molecular signature of IgG-activated macrophages using transcriptomic analysis of murine colonic macrophages stimulated ex vivo with IgG immune complexes. The signature was restricted to human- and disease-relevant genes using a second dataset of IgG IC-stimulated human monocytes (GSE102728) and elastic-net regularized logistic regression on a training mucosal UC dataset (GSE38713), respectively. A second microbial gene signature using transcriptomic analysis of colonic macrophages from DSS-treated mice and refined on LPS-treated human monocytes was also generated. b, List of constituent genes for the immune complexes signature and the LPS signature. The right panel shows the number of exclusive and shared genes between the 2 signatures.
Extended Data Fig. 7 |. Proportion of “resident-like” and inflammatory macrophages and their respective molecular signature.
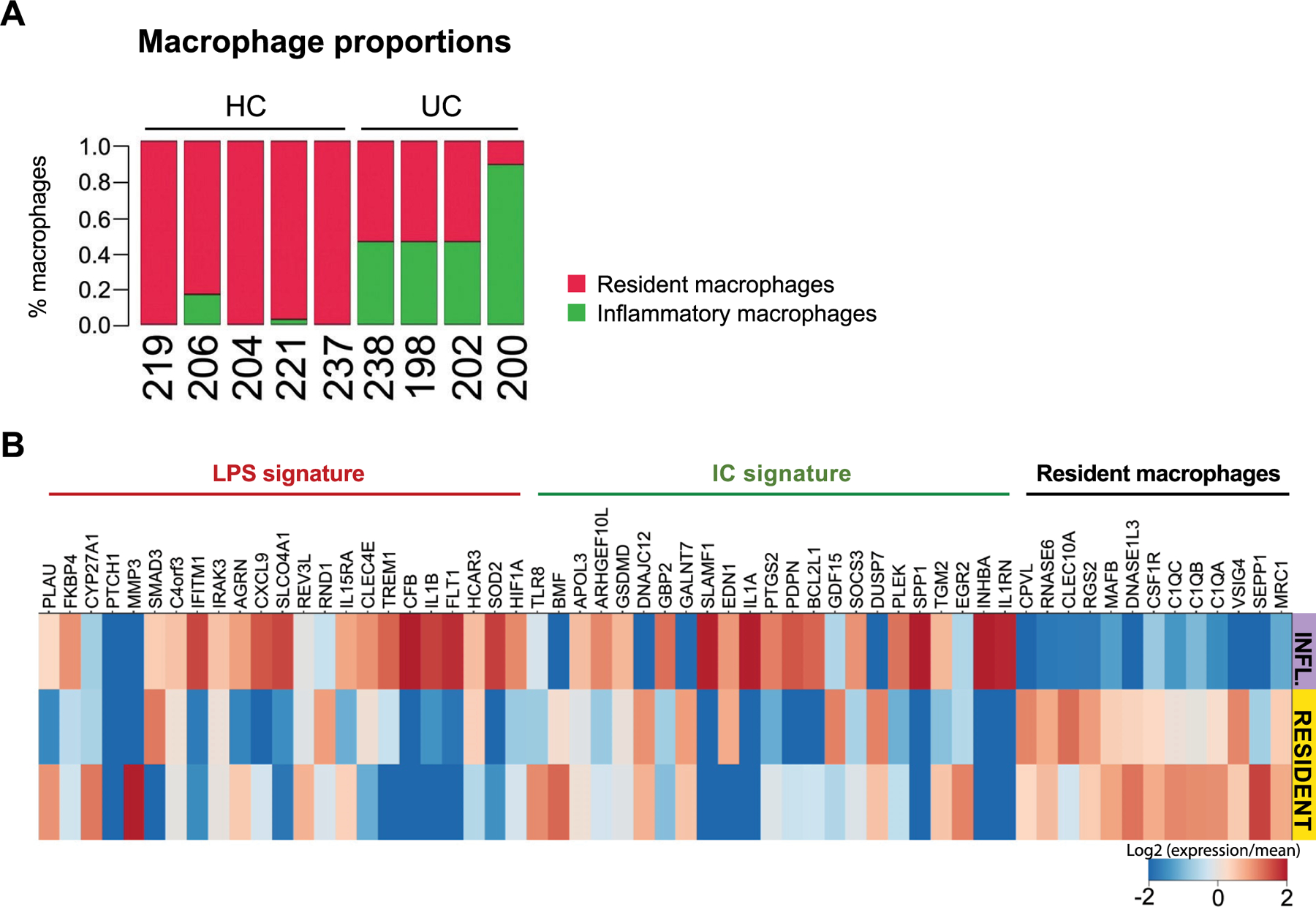
a, Proportion of “resident-like” macrophages and inflammatory macrophages among total colonic macrophages represented in individual samples of healthy controls UC and patients. b, Heat-map showing color-coded down-sampled UMI counts of gene programs identifying LPS signature (red), immune complexes (IC) signature (green) and resident macrophage genes signatures (black).
Extended Data Fig. 8 |. Characterization of T cell clusters from single-cell RNA sequencing of colonic biopsies.
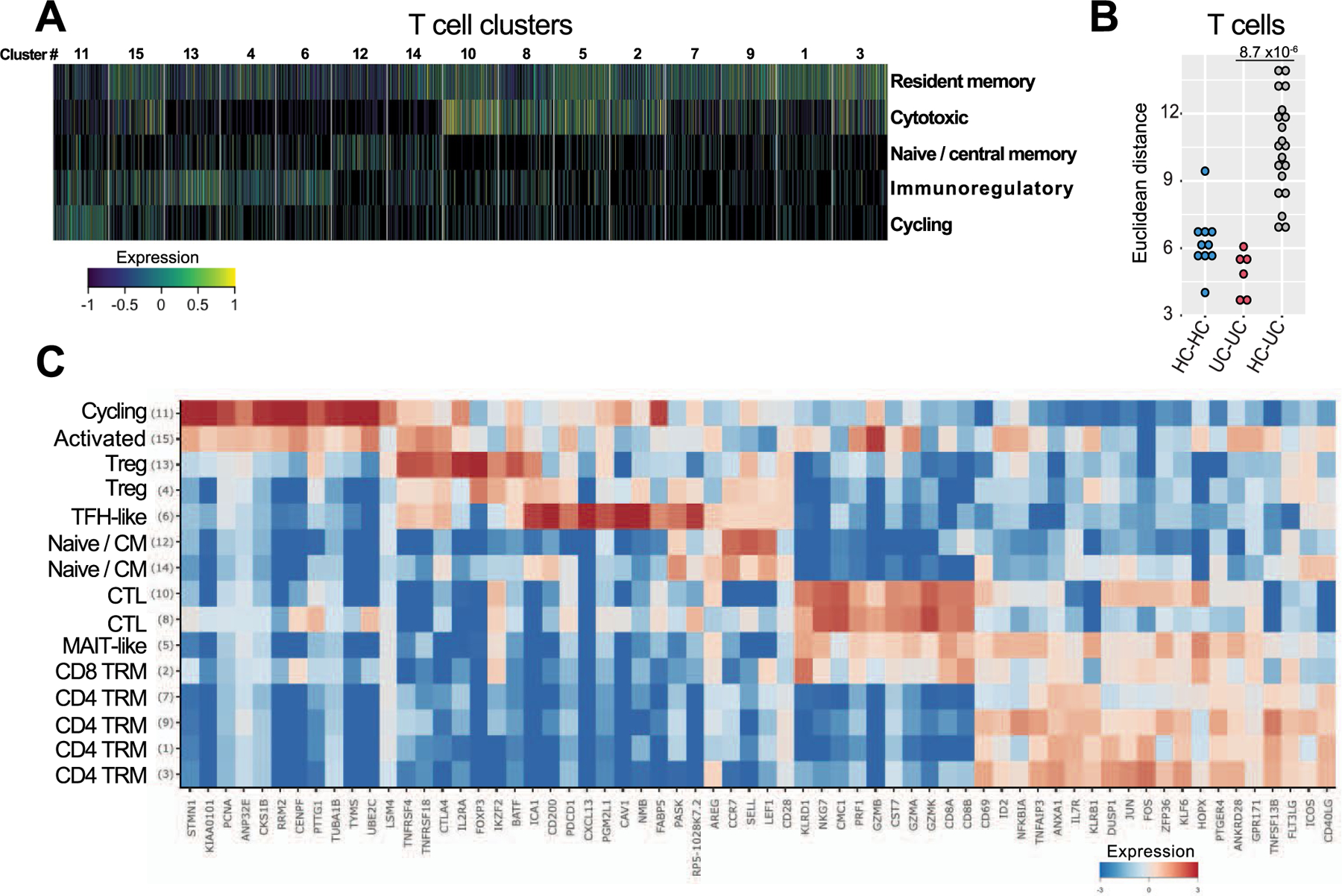
a, Heat-map showing the relative expression of 5 transcriptional scores (rows) (see Methods) in single T cells (n = 11281 cells) (columns). Cell clusters are demarcated by gray bars. Detailed cell counts per T cluster per sample are accessible in Supplementary Table 2. b, Euclidean distance for lineage normalized cell type frequencies was compared within healthy controls (HC), (HC-HC, blue), patients with UC (UC-UC, red) and between HC and patients with UC (HC-UC, grey) for T cells, Wilcoxon rank-sum test. c, Relative expression of genes identifying T cell subsets. Heatmap is showing log-2 transformed expression per subset divided by the mean expression across all subsets.
Extended Data Fig. 9 |. Representative histological examination of a surgical specimen from a UC patient before immunofluorescent staining.
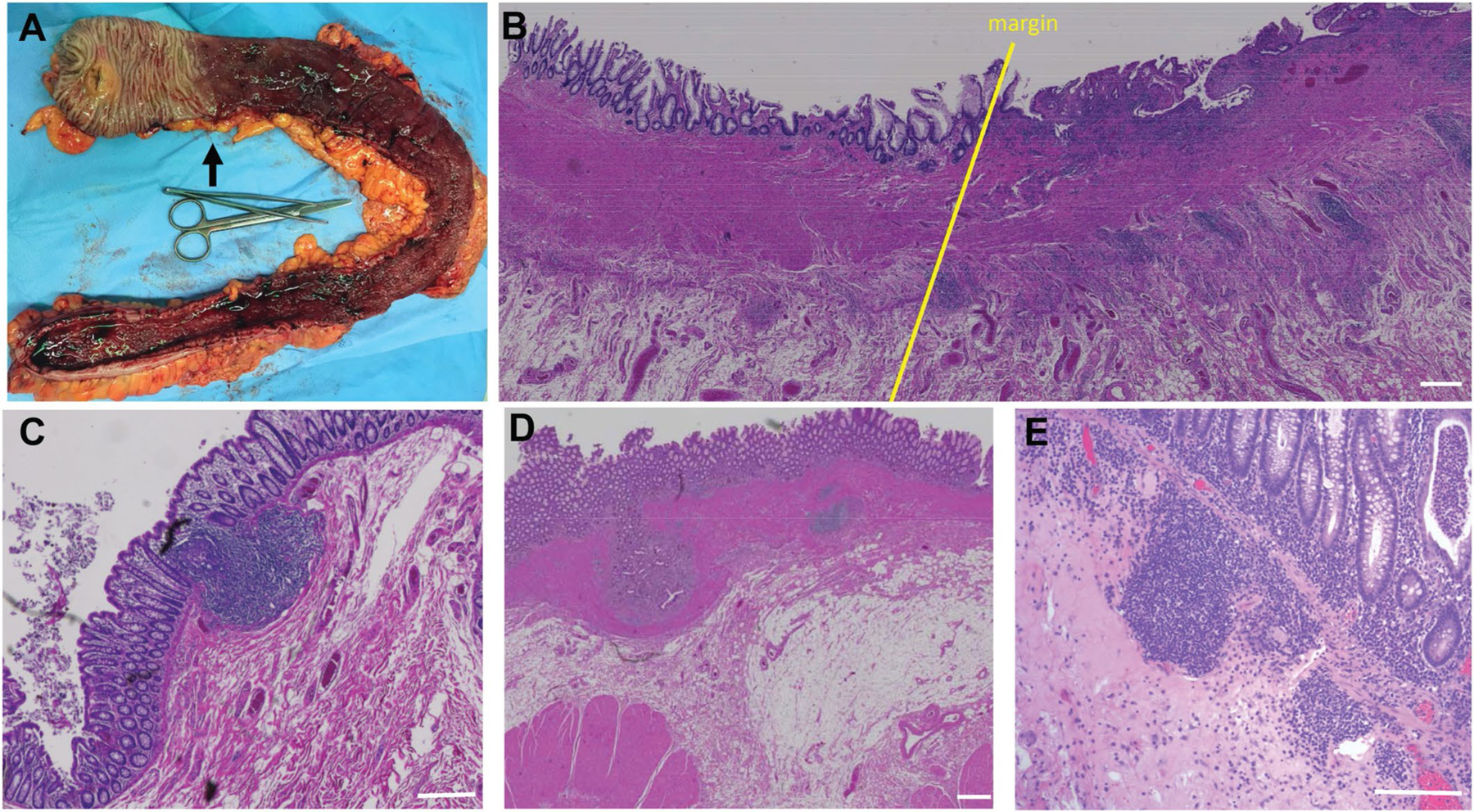
Macroscopic view of colon after resection. Black arrow delineates disease margin, which runs strikingly anti-parallel to the length of the colon. b, H&E section showing disease margin (yellow) line with proximal region on the left and margin progressing toward disease distally on the right. Note the increased number of small follicles on the diseased side. c, Normal isolated lymphoid follicle in the colon. d, Proximal colon from UC patient with delimited disease in the region that appears macroscopically healthy, with abnormal fibrotic accumulation beneath the epithelial lining. e, Macroscopically normal proximal specimen from a UC patient that contains diffuse inflammatory infiltrate despite a healthy macroscopic appearance. For panels b-e, white bars = 200 μm.
Supplementary Material
Acknowledgements
We thank the patients who participated in the study. The authors would like to thank the Biorepository and Pathology Core at the Icahn School of Medicine at Mount Sinai for carrying out some of the immunostaining experiments. This work was supported by NIH/NIDDK grant R01 112296 (S.M.). Additional support was provided by a Rainin Foundation Synergy Grant to S.M., G.J.R. and J.F.C. A.C. was supported by P01 AI061093 and Ministerio de Ciencia, Innovación y Universidades grants RTI2018-093894-B-I00 and RTI2018-093894-B-I00. Other sources of support included K23KD111995 (R.C.U.), a Career Development Award from the Crohn’s and Colitis Foundation (R.C.U.), R01 AI119006 (G.D.V.), R01 AI157137 (G.D.V.), DK121009 (S.A.L.) and DK110352 (S.A.L.). M.U. was supported by an ECCO-IOBD fellowship, by a grant from the French National Society of Gastroenterology (Bourse Robert Tournut) and by the Fondation pour la Recherche Médicale (FDM 41552). J.C.M. was supported by ‘Prix pour les jeunes chercheurs’ de la Fondation Bettencourt-Schueller, the Philippe Foundation, NExT ‘Junior Talent’ and ANR JCJC (ANR-20-CE17-0009). E.K.G. was supported by a postdoctoral fellowship from the Swedish Research Council (2015-06486). Q.Z. is the recipient of a research fellowship from Takeda Pharmaceuticals. M.R.C. and T.D.C. were supported by a Medical Research Council New Investigator Research Grant (MR/N024907/1) and by a Wellcome Trust Investigator Award (220268/Z/20/Z). The sampling of the IBD cohort (Crohn’s disease and UC) was jointly designed as part of the research alliance between Janssen Biotech and the Icahn School of Medicine at Mount Sinai. Beyond this exception, no other funders had a role in analyses, design or interpretation.
Competing interests
S.M. and J.F.C. have an unrestricted, investigator-initiated grant from Takeda Pharmaceuticals to examine novel homing mechanisms to the gastrointestinal tract. R.C.U. has served as an advisory board member or consultant for Eli Lilly, Janssen, Pfizer and Takeda. Mount Sinai coauthors (from Genetics and Genomics, Icahn Institute for Data Science and Genomic Technology, Human Immune Monitoring Center, Population Health Science and Policy, Division of Gastroenterology, Pediatric GI and Hepatology, Susan and Leonard Feinstein IBD Clinical Center at the Icahn School of Medicine at Mount Sinai) were partially funded as part of the research alliance between Janssen Biotech and the Icahn School of Medicine at Mount Sinai. J.P. and C.B. are employees of Janssen Research and Development, and J.R.F. is a former employee of Janssen Research and Development and is currently employed at Alnylam Pharmaceuticals. M.K., N.R., W.O. and J.H. are all employees of Genentech. M.D. is a consultant for Janssen, Abbvie, Arena, Bristol Meyers Squibb, Boehringer Ingelheim, Eli Lilly, Janssen, Pfizer, Prometheus Labs and Takeda. C.E. has received research support from Merck. R.U. has served as an advisory board member or consultant for AbbVie, Bristol Meyers Squibb, Janssen, Pfizer and Takeda and has received research support from AbbVie, Boehringer Ingelheim, Eli Lilly and Pfizer. J.F.C. reports receiving research grants from AbbVie, Janssen Pharmaceuticals and Takeda; receiving payment for lectures from AbbVie, Amgen, Allergan, Ferring Pharmaceuticals, Shire and Takeda; receiving consulting fees from AbbVie, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene Corporation, Eli Lilly, Ferring Pharmaceuticals, Galmed Research, GlaxoSmithKline, Geneva, Iterative Scopes, Janssen Pharmaceuticals, Kaleido Biosciences, Landos, Otsuka, Pfizer, Prometheus, Sanofi, Takeda and TiGenix; and holding stock options in Intestinal Biotech Development. B.L.C. receives the following financial support: advisory boards and consultant for Abbvie, Celgene/Bristol Myers Squibb, Eli Lilly, Pfizer, Sublimity Therapeutics, Takeda and TARGET RWE; CME companies: Cornerstones and Vindico; speaking fees: Abbvie; and educational grant: Pfizer. B.S. discloses research grants from Takeda, Pfizer, Theravance Biopharma R&D and Janssen; consulting fees from 4D Pharma, Abivax, Abbvie, Alimentiv, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Bacainn Therapeutics, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Myers Squibb, Calibr, Capella Bioscience, Celgene, Celltrion Healthcare, ClostraBio, Enthera, F. Hoffmann-La Roche, Ferring, Galapagos, Gilead, GlaxoSmithKline, GossamerBio, Immunic, Index Pharmaceuticals, Innovation Pharmaceuticals, Ironwood Pharmaceuticals, Janssen, Kaleido, Kallyope, Eli Lilly, MiroBio, Morphic Therapeutic, Oppilan Pharma, OSE Immunotherapeutics, Otsuka, Palatin Technologies, Pfizer, Progenity, Prometheus Biosciences, Prometheus Laboratories, Protagonist Therapeutics, Q32 Bio, Redhill Biopharma, Rheos Medicines, Salix Pharmaceuticals, Seres Therapeutics, Shire, Sienna Biopharmaceuticals, Sun Pharma, Surrozen, Takeda, Target PharmaSolutions, Teva Branded Pharmaceutical Products R&D, Thelium, Theravance Biopharma R&D, TLL Pharma, USWM Enterprises, Ventyx Biosciences, Viela Bio, Vivante Health and Vivelix Pharmaceuticals; and stock in Vivante Health and Ventyx Biosciences. All other authors declare no competing interests.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-022-01680-y.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The R code developed for clustering and analyses in this study is available at our GitHub page: https://github.com/effiken/Uzzan_et_al. Source data are provided with this paper.
Extended data is available for this paper at https://doi.org/10.1038/s41591-022-01680-y.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-022-01680-y.
Data availability
All requests for raw and analyzed data and materials will be promptly reviewed by the corresponding author and the study team. We have provided source data files for all the figures. Raw sequencing reads of scRNA-seq samples, as well as UMI tables, are available in the Gene Expression Omnibus under accession number GSE182270. Additional raw data are provided in Supplementary Tables 1–22. Source data are provided with this paper.
References
- 1.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L & Colombel J-F Ulcerative colitis. Lancet 389, 1756–1770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macpherson AJ & Uhr T Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Peterson DA, McNulty NP, Guruge JL & Gordon JI IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2, 328–339 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Bollinger RR et al. Secretory IgA and mucin-mediated biofilm formation by environmental strains of Escherichia coli: role of type 1 pili. Mol. Immunol 43, 378–387 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Geissmann F et al. A subset of human dendritic cells expresses IgA Fc receptor (CD89), which mediates internalization and activation upon cross-linking by IgA complexes. J. Immunol 166, 346–352 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Pilette C, Detry B, Guisset A, Gabriels J & Sibille Y Induction of interleukin-10 expression through Fcα receptor in human monocytes and monocyte-derived dendritic cells: role of p38 MAPKinase. Immunol. Cell Biol 88, 486–493 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Phalipon A & Corthésy B Novel functions of the polymeric Ig receptor: well beyond transport of immunoglobulins. Trends Immunol. 24, 55–58 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Donaldson GP et al. Gut microbiota utilize immunoglobulin a for mucosal colonization. Science 360, 795–800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLoughlin K, Schluter J, Rakoff-Nahoum S, Smith AL & Foster KR Host selection of microbiota via differential adhesion. Cell Host Microbe 19, 550–559 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Kabbert J et al. High microbiota reactivity of adult human intestinal IgA requires somatic mutations. J. Exp. Med 217, e20200275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castro-Dopico T & Clatworthy MR IgG and Fcγ receptors in intestinal immunity and inflammation. Front. Immunol 10, 805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castro-Dopico T et al. Anti-commensal IgG drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity 50, 1099–1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDermott RP et al. Alterations in serum immunoglobulin G subclasses in patients with ulcerative colitis and Crohn’s disease. Gastroenterology 96, 764–768 (1989). [PubMed] [Google Scholar]
- 14.Ueno H, Banchereau J & Vinuesa CG Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol 16, 142–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue G et al. Aberrant alteration of follicular T helper cells in ulcerative colitis patients and its correlations with interleukin-21 and B cell subsets. Medicine (Baltimore) 98, e14757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rao DA et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin JC et al. Single-cell analysis of crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castro-Dopico T, Colombel JF & Mehandru S Targeting B cells for inflammatory bowel disease treatment: back to the future. Curr. Opin. Pharmacol 55, 90–98 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 1–12 (2017). 2017 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Portugal S et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. eLife 4, e07218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenks SA et al. Distinct effector B cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic Lupus erythematosus. Immunity 49, 725–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dann E, Henderson NC, Teichmann SA, Morgan MD & Marioni JC Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol 10.1038/s41587-021-01033-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kett K & Brandtzaeg P Local IgA subclass alterations in ulcerative colitis and Crohn’s disease of the colon. Gut 28, 1013–21 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snapper CM & Paul WE Interferon-γ and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 236, 944–947 (1987). [DOI] [PubMed] [Google Scholar]
- 25.Toellner KM et al. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J. Exp. Med 187, 1193–1204 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gene Set Enrichment Analysis. Molecular Signatures Database (MSigDB). https://www.gsea-msigdb.org/gsea/msigdb
- 27.Obeng-Adjei N et al. Malaria-induced interferon-γ drives the expansion of Tbethi atypical memory B cells. PLoS Pathog. 13, e1006576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landsverk OJB et al. Antibody-secreting plasma cells persist for decades in human intestine. J. Exp. Med 214, 309–317 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benckert J et al. The majority of intestinal IgA+ and IgG+ plasmablasts in the human gut are antigen-specific. J. Clin. Invest 121, 1946–1955 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwada T et al. Identification of an anti-integrin αvβ6 autoantibody in patients with ulcerative colitis. Gastroenterology 160, 2383–2394 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Hovhannisyan Z, Treatman J, Littman DR & Mayer L Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140, 957–965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habtezion A, Nguyen LP, Hadeiba H & Butcher EC Leukocyte trafficking to the small intestine and colon. Gastroenterology 150, 340–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cole KE et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med 187, 2009–2021 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katewa A et al. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNα-driven lupus nephritis. JCI insight 2, e90111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owczarczyk K et al. A plasmablast biomarker for nonresponse to antibody therapy to CD20 in rheumatoid arthritis. Sci. Transl. Med 3, 101ra92 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Brandtzaeg P et al. The B-cell system of human mucosae and exocrine glands. Immunol. Rev 171, 45–87 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.James KR et al. Distinct microbial and immune niches of the human colon. Nat. Immunol 21, 343–353 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boland BS et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol 5, eabb4432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baklien K & Brandtzaeg P Comparative mapping of the local distribution of immunoglobulin-containing cells in ulcerative colitis and Crohn’s disease of the colon. Clin. Exp. Immunol 22, 197–209 (1975). [PMC free article] [PubMed] [Google Scholar]
- 40.Macpherson A, Khoo UY, Forgacs I, Philpott-Howard J & Bjarnason I Mucosal antibodies in inflammatory bowel disease are directed against intestinal bacteria. Gut 38, 365–375 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uo M et al. Mucosal CXCR4+ IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcγR-mediated CD14 macrophage activation. Gut 62, 1734–1744 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Van Den Bogaerde J et al. Gut mucosal response to food antigens in Crohn’s disease. Aliment. Pharmacol. Ther 16, 1903–1915 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Kett K, Rognum TO & Brandtzaeg P Mucosal subclass distribution of immunoglobulin G-producing cells is different in ulcerative colitis and Crohn’s disease of the colon. Gastroenterology 93, 919–924 (1987). [DOI] [PubMed] [Google Scholar]
- 44.Dunn-Walters DK, Isaacson PG & Spencer J Sequence analysis of human IgVH genes indicates that ileal lamina propria plasma cells are derived from Peyer’s patches. Eur. J. Immunol 27, 463–467 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Ekland EH, Forster R, Lipp M & Cyster JG Requirements for follicular exclusion and competitive elimination of autoantigen-binding B cells. J. Immunol 172, 4700–4708 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Yurasov S & Nussenzweig MC Regulation of autoreactive antibodies. Curr. Opin. Rheumatol 19, 421–426 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Schoenborn JR & Wilson CB Regulation of interferon-γ during innate and adaptive immune responses. Adv. Immunol 96, 41–101 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Hosomi S et al. Increased numbers of immature plasma cells in peripheral blood specifically overexpress chemokine receptor CXCR3 and CXCR4 in patients with ulcerative colitis. Clin. Exp. Immunol 163, 215–224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X et al. The shifted balance between circulating follicular regulatory T cells and follicular helper T cells in patients with ulcerative colitis. Clin. Sci 131, 2933–2945 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Long Y et al. The imbalance of circulating follicular helper T cells and follicular regulatory T cells is associated with disease activity in patients with ulcerative colitis. Front. Immunol 11, 104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z et al. Circulating follicular helper T cells in Crohn’s disease (CD) and CD-associated colorectal cancer. Tumor Biol. 35, 9355–9359 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Faubion WA et al. EMerging BiomARKers in Inflammatory Bowel Disease (EMBARK) study identifies fecal calprotectin, serum MMP9, and serum IL-22 as a novel combination of biomarkers for Crohn’s disease activity: role of cross-sectional imaging. Am. J. Gastroenterol 108, 1891–1900 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Amir EadD., Guo XV, Mayovska O & Rahman AH Average overlap frequency: a simple metric to evaluate staining quality and community identification in high dimensional mass cytometry experiments. J. Immunol. Methods 453, 20–29 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Van Gassen S et al. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Maecker HT, McCoy JP & Nussenblatt R Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol 12, 191–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finak G et al. Standardizing flow cytometry immunophenotyping analysis from the Human ImmunoPhenotyping Consortium. Sci. Rep 6, 20686 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wardemann H & Kofer J Expression cloning of human B cell immunoglobulins. Methods Mol. Biol 971, 93–111 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Alexander KL et al. Human microbiota flagellins drive adaptive immune responses in Crohn’s disease. Gastroenterology 161, 522–535 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.effiken/scDissector. https://github.com/effiken/scDissector
- 60.Baran Y et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaitin DA et al. Massively parallel single cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paul F et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163, 1663–1677 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Mahnke YD, Brodie TM, Sallusto F, Roederer M & Lugli E The who’s who of T-cell differentiation: human memory T-cell subsets. Eur. J. Immunol 43, 2797–2809 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Savas P et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med 24, 986–993 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Zemmour D et al. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR article. Nat. Immunol 19, 291–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng C et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Hayatsu N et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity 47, 268–283 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Förster R, Davalos-Misslitz AC & Rot A CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol 8, 362–371 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Willinger T et al. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J. Immunol 176, 1439–1446 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Hombrink P et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol 17, 1467–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Kumar BV et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Thome JJC et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med 22, 72–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong MT et al. A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity 45, 442–456 (2016). [DOI] [PubMed] [Google Scholar]
- 75.Chattopadhyay PK, Yu J & Roederer M A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat. Med 11, 1113–1117 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Friedman J, Hastie T & Tibshirani R Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 77.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liao Y, Smyth GK & Shi W FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hänzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilkerson MD & Hayes DN ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gentleman RC et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All requests for raw and analyzed data and materials will be promptly reviewed by the corresponding author and the study team. We have provided source data files for all the figures. Raw sequencing reads of scRNA-seq samples, as well as UMI tables, are available in the Gene Expression Omnibus under accession number GSE182270. Additional raw data are provided in Supplementary Tables 1–22. Source data are provided with this paper.