

Abstract
Background:
Sturge-Weber Syndrome (SWS) is a sporadic, neurocutaneous syndrome involving the skin, brain, and eyes. Given the variability of clinical outcome and the lack of prospective studies, consensus recommendations for management and treatment have not reached a conclusion.
Objective:
This consensus statement aims to consolidate the current literature with expert opinion to make recommendations that will guide treatment and referral, particularly regarding the neurological and ophthalmological features of the disease as well as neuroimaging workup.
Methods:
Thirteen national peer-recognized experts in neurology, radiology, and ophthalmology with experience treating SWS patients were assembled. Key topics and questions were formulated for each group and included: (a) risk stratification; (b) indications for referral; and (c) optimum treatment strategies. An extensive PubMed search was performed of English language articles published in 2008–2018, as well as recent studies identified by the expert panel. Clinical practice guidelines were recommended.
Conclusions:
Any child with a high-risk facial port-wine birthmark (PWB) should be referred to a pediatric neurologist and pediatric ophthalmologist for a baseline neurological evaluation and ophthalmic evaluation, respectively, with periodic follow-up. In newborns and infants with a high-risk PWB and no history of seizures or neurological symptoms, routine screening for brain involvement is not recommended, but brain imaging can be performed in select cases. Routine follow-up neuroimaging is not recommended in children with SWS and stable neurocognitive symptoms. The treatment of ophthalmologic complications, such as glaucoma, differs based on the age and clinical presentation of the patient. These recommendations will help facilitate coordinated care for patients with SWS and may improve patient outcomes.
Keywords: Consensus statement, Sturge-Weber Syndrome, Port-wine birthmark, seizures, glaucoma
Introduction
There is a critical need for a consensus statement regarding the management of Sturge-Weber Syndrome (SWS). In a prior manuscript, we provided a discussion of dermatologic management and treatment1. In the current manuscript, we aim to provide clinical practice guidelines to guide care of patients with SWS, while focusing on the neurological and ophthalmological features of the disease. The goals of this consensus are to 1) review the literature to provide an approach to risk stratification and evaluation of SWS patients; 2) offer guidance on what neurologic, neuroimaging, and ophthalmologic workup should consist of for patients with suspected or newly diagnosed SWS and indications for referral to an ophthalmologist or neurologist; and 3) assess the current treatment options for brain and eye manifestations with considerations for age and severity of disease.
Methods
Eight national experts in neurology and radiology, and five national experts in ophthalmology, were consulted to form a consensus statement on the management and treatment of SWS, as part of a larger consensus statement1. Three key topics were established: (1) evaluation of port-wine birthmark (PWB) and risk stratification; (2) consultation and referral; and (3) optimum treatment strategies. Questions were formulated for each key topic and an extensive literature review was performed using PubMed for English-language papers published between 2008 and 2018. Articles before 2008 or after 2018 were added by the expert panel based on importance. Search terms included “Sturge-Weber syndrome” and terms associated with each key topic: “clinical presentation”, “pathogenesis”, “risk prediction”, “port-wine birthmark or port-wine stain”, “diagnostic workup”, “triage”, “management”, “treatment”, “laser therapy”, “light-based therapy or treatment”, “photodynamic therapy”, “infantile hemangioma”, and “nevus simplex.” 112 total manuscripts were identified for consideration, of which 84 of those were relevant to neurology, radiology, and ophthalmology, and 47 manuscripts were directly referenced by the expert panel. Publications associated with each key topic were identified and distributed to each group, who then developed clinical practice guidelines (Tables 1–3). These guidelines were consolidated into key points that were then presented to all groups for electronic discussion and modification before achieving final consensus.
Table 1.
Key Points for Neurological Management and Treatment in SWS
| 1. Any child with a high-risk facial port wine birthmark should be referred to a pediatric neurologist for a baseline neurological evaluation and have periodical follow-up. |
Table 3.
Key Points for Ophthalmological Management and Treatment in SWS
| 1. Any child with a high-risk facial port wine birthmark should be referred to a pediatric ophthalmologist for a baseline eye evaluation and have periodical follow-up. |
| 2. The treatment of glaucoma in patients with SWS differs based on the age and clinical presentation of the patient. |
Neuroradiology
Neurological Manifestations and Risk Stratification
Key Point 1: Any child with a high-risk facial port wine birthmark should be referred to a pediatric neurologist for a baseline neurological evaluation and have periodic follow-up.
SWS is associated with neurological abnormalities, including seizures, stroke-like episodes, headaches, and developmental delays 2,3. Seizures occur in approximately 75–80% of SWS patients.3. A recent study of 277 SWS patients with brain involvement demonstrated epilepsy in 81.6% of those patients 4. The age of onset is variable, but is usually within the first year of life; however, cases of seizure onset in adulthood have also been reported 5,6. Recognition and management of seizures is essential, because early onset, frequent seizures can adversely affect cognitive and neurodevelopmental outcomes 7,8.
Risk of SWS with brain involvement is greater in patients with hemifacial, forehead, and median locations of their PWB, since those locations involve skin derived from the frontonasal placode, which shares common progenitor cells with the brain 9. Any child with a high-risk facial PWB should be referred to a pediatric neurologist for a baseline evaluation and have periodic follow-up. An electroencephalography (EEG) to assess abnormal brain activity and identify patients at risk for future neurologic events, although not diagnostic, may be useful in patients with neurologically asymptomatic PWBs 10. The pediatric neurologist may then make recommendations regarding imaging and management of neurological complications. In 2018, De la Torre et al. provided a discussion of management and treatment options for neurological complications, along with specific clinical guidelines for neurology11. These recommendations remain valid. This review will focus on additional clinical guidelines, specifically related to neuroimaging.
Neuroimaging
Key Point 1A: In newborns and infants with a high-risk PWB and no history of seizures or neurological symptoms, routine screening for brain involvement is not recommended.
Brain involvement may be detected by imaging in infants with a high-risk PWB even before the onset of neurological symptoms. However, negative neuroimaging in a normally developing asymptomatic infant with a facial PWB does not exclude SWS brain involvement. Such false negative findings have been reported in 3–23% of the cases in retrospective studies 12–14. Negative MRI may provide false reassurance. False positive MRI in this early disease stage has been reported anecdotally, although its likelihood is low. Even if the MRI is true positive, neurologically asymptomatic children are unlikely to undergo immediate therapeutic intervention (exceptions in Key Point 1B), although it can increase vigilance for subclinical seizures. Nevertheless, future research may elucidate a better understanding regarding the utility of screening in this age group.
Key Point 1B: In newborns and infants with a high-risk PWB and no history of seizures or neurological symptoms, brain imaging can be considered for more subtle symptoms and extensive PWB. Indirect signs of SWS brain involvement and susceptibility-weighted imaging can optimize the yield of MRI in this group.
A screening brain MRI may be considered in select children with suspected SWS, for example, when presymptomatic treatment is contemplated. A retrospective study suggested the potential benefit of presymptomatic antiepileptic treatment combined with aspirin 15, but no prospective studies have been completed. Some children with a particularly high risk for seizures may benefit from presymptomatic treatment, for example when bilateral SWS is suspected (e.g., extensive bilateral PWBs). Treatment should be preceded by MRI to establish presence and extent of early brain involvement. In a retrospective review, MRI features suggestive of SWS were detected in 43%−73% of individuals with high-risk PWB 14. In a review of 32 children with high-risk PWB (hemifacial, median, and forehead), screening MRI had a sensitivity of 25%, specificity of 100%, positive predictive value of 100%, and negative predictive value of 77% for the detection of SWS brain involvement 14.
Concerns regarding MRI in infants include the potential long-term cognitive effects of anesthesia and repeated gadolinium contrast administration with systemic deposition in tissues, including brain and bone marrow. The optimal MRI approach for presymptomatic screening has not been established; however, to minimize the risks of clinical MRI in presymptomatic children, a fast, non-sedated, non-contrast screening MRI can be considered, preferably including axial T1, T2 [or fluid-attenuated inversion recovery (FLAIR)], susceptibility-weighted imaging (SWI), with or without diffusion-weighted imaging (DWI) on a 3T scanner 11. These images may detect early venous vascular abnormalities or accelerated myelination and atrophy. SWI can be particularly sensitive to detect early deep medullary veins during the first few months of life, even before contrast enhancement is detectable [Figure 1] 12,16. Current research aims at development and validation of ultra-fast, non-contrast MRI acquisitions to provide accurate and safe presymptomatic imaging without sedation risks.
Figure 1.
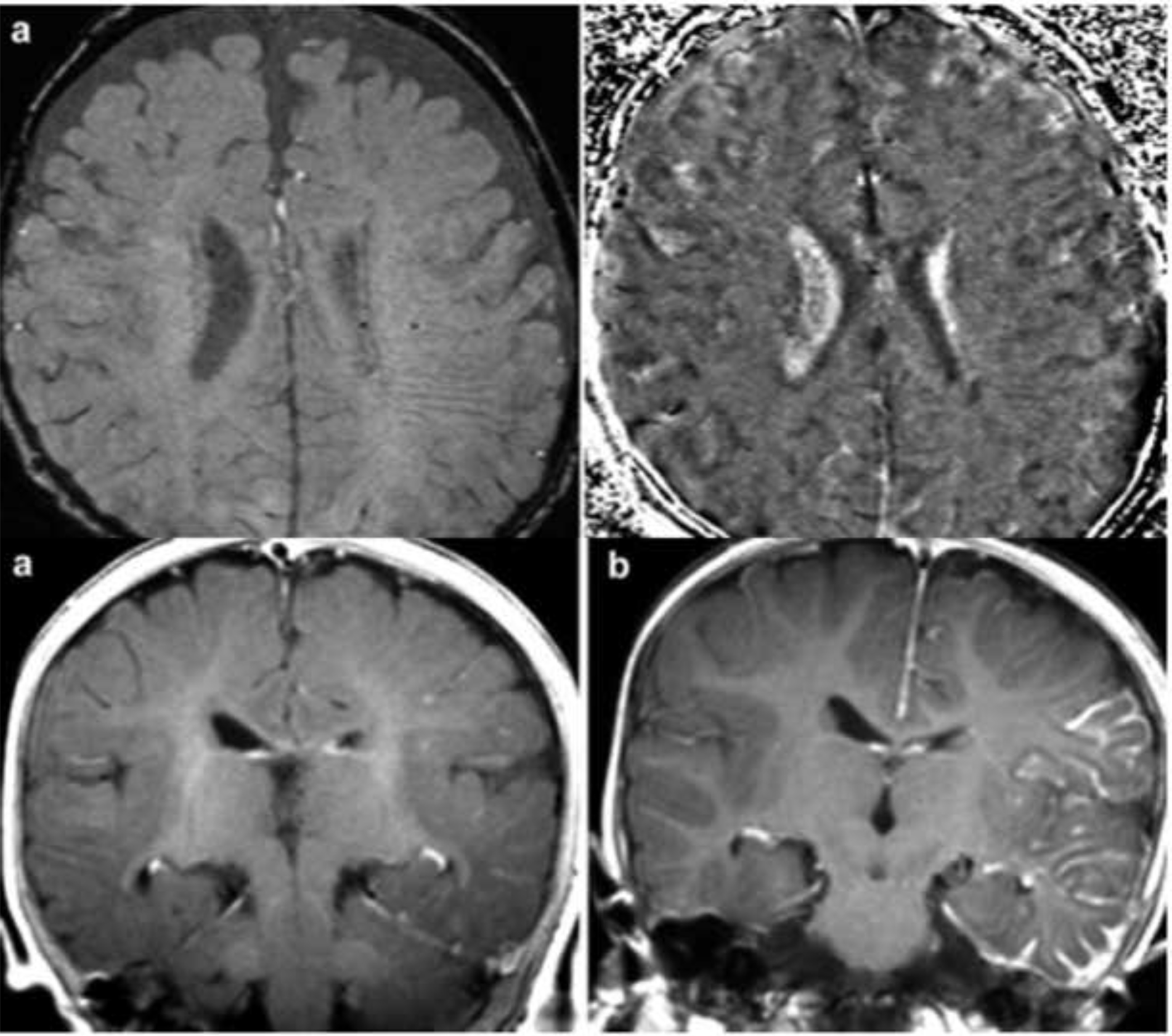
Detection of enlarged deep medullary veins by SWI in the left hemisphere of a 4-month old girl before the first seizure. Post-contrast T1-weighted MRI showed leptomeningeal enhancement only 8 months later, after the first clinical seizure (adopted from Mentzel et al., 2005).
Key Point 2A: In children with suspected SWS, the first post-symptomatic imaging should be an optimized pre- and post-contrast MRI sequence, focusing on the detection of both vascular and parenchymal abnormalities associated with SWS.
The routine use of computed tomography (CT) scans in children with new-onset SWS-related seizures or new neurological symptoms is not recommended due to low yield and the potential risks of radiation exposure. If a CT scan is done, an MRI with and without contrast administration should follow to establish the diagnosis of SWS and provide detailed insight on brain vascular and parenchymal abnormalities. A recent paper authored by a panel of experts recommends the use of pre- and post-contrast MRI, acquired under sedation (if needed) and including high-resolution volumetric sequences to optimize detection of subtle parenchymal changes and potential cortical malformations 11. The post-contrast MRI acquisition should include T1-weighted and FLAIR images 17, and the protocol should include SWI 18. SWI can depict small caliber venous abnormalities, such as deep medullary veins and is highly sensitive to visualize calcifications that are often not present or subtle in early disease but can be progressive 18–20. DWI can be useful to detect ischemic parenchymal changes, although these could be transient if MRI is done immediately after acutely developed clinical symptoms (see Key point 3A). The use of other, advanced MRI sequences is optional, their clinical yield remains to be established, and their potential benefit has to be weighed against the potential risks of prolonged sedation.
Key Point 2B: Routine follow-up neuroimaging is not recommended in children with established SWS and stable neurocognitive symptoms. In case of progressive neuro-cognitive symptoms during follow-up, multisequence brain MRI comparable to previous imaging should be performed for an accurate comparison.
If the child’s cognitive development is steady, seizures are controlled, and there are no progressive neurological symptoms, routine follow-up neuroimaging is not necessary. On the other hand, delayed or declining cognitive function, worsening seizures, or development of new neurological symptoms (hemiparesis, visual field defect, etc.) can prompt a follow-up MRI to evaluate if there is progression in structural brain abnormalities. Follow-up MRI should include optimized multisequence acquisitions so interval changes can be assessed accurately. While the extent of leptomeningeal enhancement is usually stable, underlying brain parenchymal abnormalities such as atrophy and calcifications often show interval progression, especially during the first few post-symptomatic years 20. Progressive atrophic changes have also been reported in adults with SWS, sometimes associated with late-onset new clinical symptoms, such as migraine attacks 21. Recent studies demonstrated that post-symptomatic expansion of deep medullary veins can occasionally be detected during early disease by SWI 22,23. Although data are preliminary, it is likely that expansion of these deep veins may be beneficial and represent an effective compensatory mechanism to offset the detrimental effects of impaired venous drainage.
Key Point 2C: Neuroimaging should be obtained in adults with PWB with or without glaucoma who have not had prior imaging; however, follow-up neuroimaging is not recommended in adults with established SWS and stable neurocognitive symptoms. Pre- and post-contrast brain MRI is recommended in case of new-onset or progressive symptoms.
Reports focusing on neuroimaging in adults with SWS are scarce. A recent review identified 31 reported cases where SWS was diagnosed in adulthood 24, but the incidence of such a late manifestation is not known. Imaging in these cases is typically prompted by new-onset symptoms and, in rare cases, a first-ever neurological manifestation of SWS, such as seizure(s). SWS may be missed in cases with no facial PWB, such as the case of a 55-year old patient with a history of episodes of transient weakness in the right extremities 25. Post-contrast FLAIR demonstrated fronto-parietal leptomeningeal capillary malformations, and SWI showed enlarged deep medullary veins and calcifications. Similar cases with adult-onset seizures along with the new diagnosis of SWS by neuroimaging have been reported 26,27. Despite the scarce data, most of the SWS pathology reported in pediatric populations is present in adult patients. Routine imaging, however, is not justified in adults with previously documented SWS brain involvement, unless new symptoms emerge. In adult patients with new-onset symptoms or progression of previous symptoms, pre- and post-contrast MRI including SWI and DWI should be the choice of imaging. At this point there are no data to support that the optimal MRI sequences for adults should be different from those recommended in pediatric SWS patients.
Key Point 3A: SWS patients rarely hemorrhage and there is little evidence for acute ischemia. The available data do not show bleeding or clear strokes in SWS patients who present with acute neurological symptoms; however, prolonged new-onset or acutely/subacutely deteriorating and non-resolving neurological ‘stroke-like’ episodes justify repeat neuroimaging.
In current clinical practice, children and adults with known SWS and new or progressive acute symptoms often undergo emergent imaging during an emergency room visit. However, the clinical value of this imaging in such a setting is questionable. One issue is that after prolonged or repeated seizures, status epilepticus or stroke-like episodes, MRI may show transient abnormalities 28–31 that could prompt follow-up MRI that shows normalization. In a recent retrospective study of 35 SWS patients, who presented to an emergency department with acute neurological symptoms, 89 urgent neuroimaging studies were reviewed, and none showed acute hemorrhagic or ischemic strokes 32. The authors concluded that urgent imaging after breakthrough seizures does not result in a significant change of clinical management. Similarly, cerebral angiograms are often performed on SWS patients, have little value, and significant risk. A sudden, severe deterioration of the neurologic status, including prolonged loss of consciousness, has been reported in a few SWS cases, where acute imaging revealed acute thalamic hemorrhage 33,34. It is therefore important to note that as SWS patients age, particularly with other health co-morbidities, they should be fully evaluated like any other patient for strokes and hemorrhages due to other causes and treated appropriately. The use of thrombolytics has not been investigated in SWS patients and needs to be carefully evaluated and could potentially be used if there is a strong clinical indication.
Key Point 4A: Presurgical evaluation in SWS can benefit from the use of advanced structural and functional imaging modalities. These studies and the subsequent surgery should be performed in specialized pediatric epilepsy centers that are experienced in processing and interpretation of advanced imaging and also in surgical techniques used in SWS (such as hemispherectomy).
Epilepsy surgery can be considered in patients (usually children) with SWS and drug-resistant seizures. The surgery is mostly reserved for those with unilateral involvement, with rare exceptions. The most common surgery type is hemispherectomy (structural or functional), while a posterior resection (preserving the frontal lobe including the motor cortex) is reserved for those with an intact frontal lobe. While multimodal MRI is essential to delineate the type, extent, and severity of vascular and brain parenchymal structural abnormalities in SWS, patients with drug-resistant seizures can benefit from additional, functional neuroimaging techniques during presurgical evaluation. Functional MRI of motor and language functions can be performed in non-sedated patients (such as older children) 35. Motor mapping by fMRI can be useful in children where a posterior resection, preserving the primary motor cortex, is being considered. Language mapping by fMRI can evaluate the risk for post-surgical language deficit in those with left hemispheric surgery. The structural integrity of critical pathways involved in motor, language, and visual functions can be evaluated by diffusion tensor imaging tractography that can be performed in sedated subjects 36,37. The data from such studies can predict the expected functional deficit from resecting brain tissue encompassing such pathways.
Ophthalmology
Ocular Manifestations and Risk Stratification
Key Point 1: Any child with a high-risk facial PWB should be referred to a pediatric ophthalmologist for a baseline eye evaluation and have periodic follow-up.
Approximately 50% of SWS patients show pathologic ocular changes, usually ipsilateral to the PWB, involving the eyelid, conjunctiva, episclera, anterior chamber, cornea, choroid, and retina 38. Patients may present with anterior segment alterations such as cataract or glaucoma, and often with posterior segment alterations such as choroidal hemangiomas 38. In darkly pigmented children in whom the PWB is difficult to discern, a conjunctival ‘blush’ may be the only sign of ocular involvement visible to the non-ophthalmologist. Patients with PWB overlying the eyelids or eye have a higher incidence of glaucoma during infancy and childhood 39. A PWB in a high-risk distribution, defined as on the forehead from the midline to an imaginary line between the outer canthus of the eye and the top of the ear including the upper eyelids, was highly associated with the development of glaucoma in a cohort of 192 children with PWB 40. Rarely, glaucoma may be bilateral, even in the presence of a unilateral PWB.
Because of the risk of preventable visual loss, every child diagnosed with SWS or with periocular vascular anomalies involving the eyelids should be referred to a pediatric ophthalmologist for examination. Should a pediatric neurologist be the first to diagnose a patient with SWS, this is an indication for referral to ophthalmology. The two most common causes of vision loss are glaucoma and amblyopia - both can be treated if detected early. Some children can have signs that prompt an ophthalmology referral such as enlarged corneal diameter in one eye, excessive tearing or rubbing, excessive light sensitivity, and/or cloudy appearance of the cornea, but a clinician should not wait for a child to have these signs to refer to a pediatric ophthalmologist. A baseline exam within the first few months of life will help to diagnose ocular involvement in SWS and will determine the timing of treatment or follow-up. The baseline eye exam should include visual acuity measurement, intraocular pressure (IOP) check in the clinic and a full dilated eye exam. If the IOP cannot be measured in the office, and there is a high suspicion for glaucoma, arrangements may need to be made for sedated examination.
The iCare™ rebound tonometer†, introduced a decade ago, can measure IOP in many children without the need of an exam under anesthesia or even topical (eyedrop) anesthesia. A dilated eye exam can assess whether the child has a choroidal hemangioma, with its “tomato ketchup” fundus appearance, and assess the optic nerve for glaucomatous optic neuropathy [Figure 2]. The pediatric ophthalmologist may then make further recommendations regarding the treatment of possible amblyopia, glaucoma or retinal disease. The risk of not treating amblyopia, glaucoma or retinal disease is permanent visual impairment in the affected eye. In the case of glaucoma in a young child, this may also cause severe ocular pain and discomfort. Discussion of specific risks or benefits should occur between the treating physician and the appropriate subspecialized ophthalmologist as the spectrum of ocular disease can vary greatly and is tailored to the individual patient.
Figure 2.
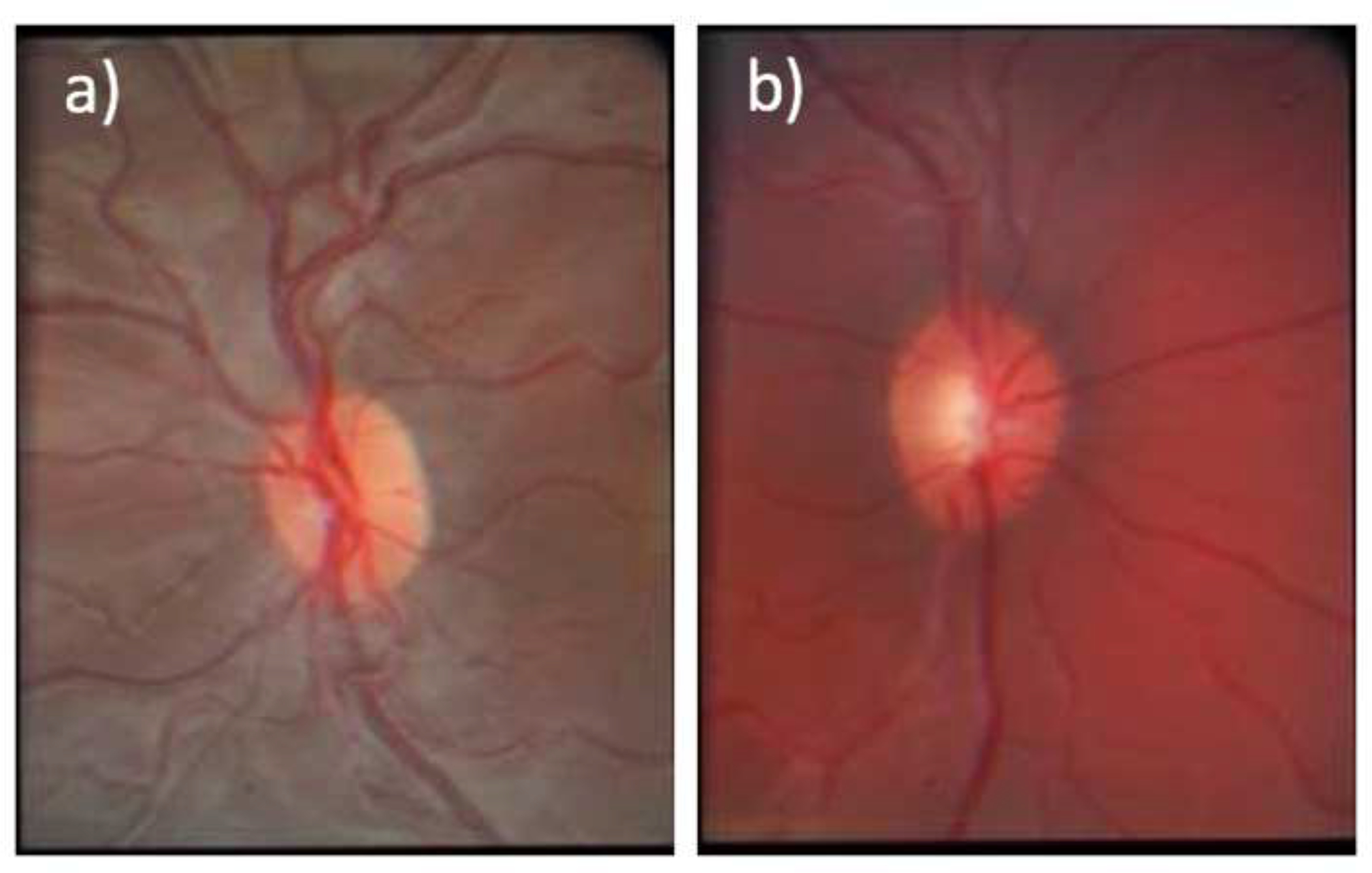
a) Normal fundus photo of the left eye; b) Fundus photo of the right eye demonstrating choroidal hemangioma and increased cup to disc ratio, consistent with glaucoma.
Determination of the Optimum Treatment
Key Point 2: The treatment of glaucoma in patients with SWS differs based on the age and clinical presentation of the patient.
In SWS, the ocular issues that arise are thought to be due to the increased venous pressure from episcleral and choroidal hemangiomas that “back up” the normal drainage pathways for the eye 41. The increased downstream venous pressure leads to increased intraocular pressure (IOP), and can also cause fluid buildup under the retina resulting in serous retinal detachments 41,42. Reyes-Capo et al report that the glaucoma associated with SWS or PWB’s occurs in 30–50% of affected cases in children younger than 36 months, however other references cite a lifetime risk of glaucoma upwards of 70% 41,42. Glaucoma, caused by high pressure or wide IOP fluctuations, damages the structures of the eye resulting in an irreversible optic neuropathy and subsequent progressive visual impairment. Thus, early detection and treatment is vital. The glaucoma associated with a PWB has a bimodal presentation, with some patients presenting in infancy (0–3 years) and others that present later in life 41. Glaucoma may present early during infancy and childhood in about 60% of patients or later during childhood and adolescence in about 40% of cases, with age of onset reported up to 41 years 5,38.
The glaucoma that presents in infancy is likely related to both the increased episcleral venous pressure as well as an anatomic developmental abnormality of the infant trabecular meshwork system 41,42. In a child younger than 3 years old, glaucoma may present acutely with enlargement of the cornea and globe, clouding of the cornea due to edema, pain and light sensitivity. In some patients the onset can be more insidious with ipsilateral ocular enlargement the only clue that glaucoma is present. In young children, the disease is almost always treated surgically 41,42, followed by medications and/or laser.
The glaucoma presenting in older individuals likely occurs as a result of the increased episcleral venous pressure with otherwise normal anatomic development 41. In these patients, the glaucoma may first be managed with medications, either topical or systemic. Surgery or laser treatment is often necessary if medications are not sufficient to control the disease. Surgical options need to be carefully considered as these eyes are more at risk for intraoperative and postoperative complications because of the increased pressure gradient across the choroidal vasculature, which can result in suprachoroidal hemorrhage or recalcitrant serous retinal detachments 41,42.
Psychosocial outcomes and quality of life in patients with SWS
Quality of life (QoL) is a construct that encompasses many components of well-being, including physical, functional, emotional, social, and family 43. Living with diseases like SWS is more complex than clinical findings alone. The QoL of individuals with facial PWB is affected by the skin condition and the procedures used to treat the PWB. In addition, caregivers’ QoL may be significantly impacted by the facial PWB. As research on QoL issues for SWS is limited, we provide an overview of the impact of any type of visible skin disease on QoL [Table 4] 44,45. These are likely representative of many aspects of the disease experience for individuals with PWB.
Table 4.
Psychosocial Outcomes and Quality of Life in Patients with SWS
| Impact of Visible Skin Disease on Patients (Ablett and Thompson, 2016) | ■ Feeling a sense of being different from others due to their appearance leading to isolation, especially if they are teased about their birthmark ■ Avoiding intimate behaviors due to embarrassment about their appearance ■ Need to educate others due to a lack of sympathy or underestimation of the impact of the skin condition on the individual’s life by medical professionals ■ Feeling a sense of powerlessness and separation as many individuals have been viewed as having “special needs” due to their skin condition |
| Impact of Visible Skin Disease on Parents and Caregivers (Ablett and Thompson, 2016) | ■ Treatments for skin disease are often time consuming, which takes away from spending time with spouses, other family members, and work ■ Feeling psychological strain when strangers make comments about their child’s skin condition and during painful treatments for the skin condition ■ Feeling blamed for not preventing sequela of the skin disease |
| Negative Impact of Treatments for Visible Skin Disease on Patients (Ablett and Thompson, 2016 and Bemmels et al., 2013) | ■ Addiction to attaining a perfect result ■ Missing school or work for treatments ■ Adjusting to an evolving appearance ■ Wondering when treatments will end ■ Experiencing stigma related to undergoing surgery due to missing school or work and their changing appearance ■ Strains on the parent (or caregiver) and child relationship as some resent their parent’s suggestion that there was something about their appearance that needs to be changed |
| Positive Impact of Treatments for Visible Skin Disease on Patients (Ablett and Thompson, 2016 and Bemmels et al., 2013) | ■ Improved self-esteem and reduced stigmatization when the appearance is more “normal” after treatments ■ Less staring, questioning, and teasing |
In addition to the presence of a facial PWB, epilepsy has a significant negative impact on QoL, and these patients may experience stigmatization and social isolation46. In the United States, 13.1 percent of children with epilepsy have depression, and 23.3 percent have anxiety47. Intellectual disability is also common in SWS, noted in 60% of patients11. In addition to decreased QoL of the patient with intellectual handicaps, family members also experience more stress and face many challenges to support the patient’s needs and transition to adulthood48. Therefore, physicians caring for these patients should inquire about QoL 49. It is important to note that addressing QoL and psychosocial issues depends on a strong patient- and family-physician relationship. Maintaining a positive attitude and providing reasonable hope through all medical care can often lessen the emotional toll of body image issues due to visible skin disease 50. For individuals suffering from psychosocial issues, social workers, psychologists, psychiatrists, and patient support/advocacy groups are often able to help patients and families who need community and support to reach their full potential 51.
Conclusion
This consensus statement reflects the current state of knowledge on the non-cutaneous manifestations of SWS and is meant to guide clinical decision-making. This document highlights the importance of consultation with other members of the SWS patients’ care team, including neurologists, ophthalmologists, and dermatologists, to manage SWS collaboratively and ensure proper assessment and treatment of patients to improve patient outcomes. A natural history study currently underway through the Brain Vascular Malformation Consortium, part of the Rare Diseases Clinical Research Network (RDCRN) of NIH, will obtain parallel longitudinal clinical and imaging data that will add to our knowledge base and improve clinical decision-making for SWS in the future.
Table 2.
Key Points for Neuroradiological Management and Treatment in SWS
| 1A. In newborns and infants with a high-risk PWB and no history of seizures or neurological symptoms, routine screening for brain involvement is not recommended. |
| 1B. In newborns and infants with a high-risk PWB and no history of seizures or neurological symptoms, brain imaging can be performed in select cases, e.g., in those where presymptomatic treatment is being considered. Indirect signs of SWS brain involvement and susceptibility-weighted imaging can optimize the yield of MRI in this group. |
| 2A. In children with suspected SWS, the first post-symptomatic imaging should be an optimized pre- and post-contrast MRI sequence, focusing on the detection of both vascular and parenchymal abnormalities associated with SWS. |
| 2B. Routine follow-up neuroimaging is not recommended in children with established SWS and stable neurocognitive symptoms. In case of progressive neuro-cognitive symptoms during follow-up, multisequence brain MRI comparable to previous imaging should be performed for an accurate comparison. |
| 2C. Neuroimaging should be obtained in adults with PWB with or without glaucoma who have not had prior imaging; however, follow-up neuroimaging is not recommended in adults with established SWS and stable neurocognitive symptoms. Pre- and post-contrast brain MRI is recommended in case of new-onset or progressive symptoms. |
| 3A. SWS patients rarely hemorrhage and there is little evidence for acute ischemia. The available data do not show bleeding or clear strokes in SWS patients who present with acute neurological symptoms; however, prolonged new-onset or acutely/subacutely deteriorating and non-resolving neurological ‘stroke-like’ episodes justify repeat neuroimaging. |
| 4A. Presurgical evaluation in SWS can benefit from the use of advanced structural and functional imaging modalities. These studies and the subsequent surgery should be performed in specialized pediatric epilepsy centers that are experienced in processing and interpretation of advanced imaging and also in surgical techniques used in SWS (such as hemispherectomy). |
Acknowledgments
Funding/Support:
This study was supported by The Sturge-Weber Foundation. The Brain Vascular Malformation Consortium (U54NS065705) is a part of the NIH Rare Diseases Clinical Research Network (RDCRN), supported through the collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS) and the NINDS.
Role of the Sponsors:
The Sturge-Weber Foundation was involved in review and approval of the manuscript.
Disclosures of the Authors:
Ms. Sara Sabeti: Research supported by The Sturge Weber Foundation.
Dr. Kristen Kelly: Drug donated by Allergan. Equipment provided by Solta; Syneron/Candela; Thermi RF; Vivosight; R2 Derm. Consultant for Shanghai Fudan-Zhangjiang Biopharmaceutical Co., Ltd, Syneron-Candela, Allergan, Sciton. Research Supported by Allergan, ASLMS; NIH; The Sturge Weber Foundation, UC Irvine ICTS.
Dr. Eric Segal: Speakers honarium - Neurelis, Zogenix, Eisai, Lundbeck, GW Pharmaceutical, Nutricia; Scientific Advisory - GW Pharmaceutical, Aquestive, Zogenix, Neurelis
Dr. Jonathan Salvin: Employment - Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA
Footnotes
References
- 1.Sabeti S; Ball KL; Burkhart C; Eichenfield L; Fernandez Faith E; Frieden IJ; Geronemus R; Gupta D; Krakowski AC; Levy ML; Metry ,D; Nelson ,JS; Tollefson ,MM; Kelly K Consensus Statement for the Management and Treatment of Port-Wine Birthmarks in Sturge-Weber Syndrome. JAMA Dermatology. 2020. doi:doi: 10.1001/jamadermatol.2020.4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosser T Neurocutaneous Disorders. Contin Lifelong Learn Neurol. 2018. doi: 10.1212/CON.0000000000000562 [DOI] [PubMed] [Google Scholar]
- 3.Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012. doi: 10.1111/j.1469-8749.2011.04169.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day AM, McCulloch CE, Hammill AM, et al. Physical and Family History Variables Associated With Neurological and Cognitive Development in Sturge-Weber Syndrome. Pediatr Neurol. 2019. doi: 10.1016/j.pediatrneurol.2018.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sujansky E, Conradi S. Sturge-Weber Syndrome: Age of Onset of Seizures and Glaucoma and the Prognosis for Affected Children. J Child Neurol. 1995. doi: 10.1177/088307389501000113 [DOI] [PubMed] [Google Scholar]
- 6.Traub R, Riley C, Horvath S. Teaching NeuroImages: Sturge-weber syndrome presenting in a 58-year-old woman with seizures. Neurology. 2010. doi: 10.1212/WNL.0b013e3181f39a52 [DOI] [PubMed] [Google Scholar]
- 7.Luat AF, Behen ME, Chugani HT, Juhász C. Cognitive and motor outcomes in children with unilateral Sturge–Weber syndrome: Effect of age at seizure onset and side of brain involvement. Epilepsy Behav. 2018. doi: 10.1016/j.yebeh.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosnyák E, Behen ME, Guy WC, Asano E, Chugani HT, Juhász C. Predictors of Cognitive Functions in Children With Sturge–Weber Syndrome: A Longitudinal Study. Pediatr Neurol. 2016. doi: 10.1016/j.pediatrneurol.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zallmann M, Leventer RJ, Mackay MT, Ditchfield M, Bekhor PS, Su JC. Screening for Sturge-Weber syndrome: A state-of-the-art review. Pediatr Dermatol. 2018. doi: 10.1111/pde.13304 [DOI] [PubMed] [Google Scholar]
- 10.Ewen JB, Kossoff EH, Crone NE, et al. Use of quantitative EEG in infants with port-wine birthmark to assess for Sturge-Weber brain involvement. Clin Neurophysiol. 2009. doi: 10.1016/j.clinph.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De la Torre AJ, Luat AF, Juhász C, et al. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr Neurol. 2018. doi: 10.1016/j.pediatrneurol.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams ME, Aylett SE, Squier W, Chong W. A Spectrum of unusual neuroimaging findings in patients with suspected Sturge-Weber syndrome. Am J Neuroradiol. 2009. doi: 10.3174/ajnr.A1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piram M, Lorette G, Sirinelli D, Herbreteau D, Giraudeau B, Maruani A. Sturge-Weber syndrome in patients with facial port-wine stain. Pediatr Dermatol. 2012. doi: 10.1111/j.1525-1470.2011.01485.x [DOI] [PubMed] [Google Scholar]
- 14.Zallmann M, Mackay MT, Leventer RJ, Ditchfield M, Bekhor PS, Su JC. Retrospective review of screening for Sturge-Weber syndrome with brain magnetic resonance imaging and electroencephalography in infants with high-risk port-wine stains. Pediatr Dermatol. 2018. doi: 10.1111/pde.13598 [DOI] [PubMed] [Google Scholar]
- 15.Day AM, Hammill AM, Juhász C, et al. Hypothesis: Presymptomatic treatment of Sturge-Weber Syndrome With Aspirin and Antiepileptic Drugs May Delay Seizure Onset. Pediatr Neurol. 2019. doi: 10.1016/j.pediatrneurol.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mentzel HJ, Dieckmann A, Fitzek C, Brandl U, Reichenbach JR, Kaiser WA. Early diagnosis of cerebral involvement in Sturge-Weber syndrome using high-resolution BOLD MR venography. Pediatr Radiol. 2005. doi: 10.1007/s00247-004-1333-2 [DOI] [PubMed] [Google Scholar]
- 17.Griffiths PD, Coley SC, Romanowski CAJ, Hodgson T, Wilkinson ID. Contrast-enhanced fluid-attenuated inversion recovery imaging for leptomeningeal disease in children. Am J Neuroradiol. 2003. [PMC free article] [PubMed] [Google Scholar]
- 18.Hu J, Yu Y, Juhasz C, et al. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber syndrome. J Magn Reson Imaging. 2008. doi: 10.1002/jmri.21435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Tarabishy B, Hu J, et al. Cortical calcification in sturge-weber syndrome on MRI-SWI: Relation to brain perfusion status and seizure severity. J Magn Reson Imaging. 2011. doi: 10.1002/jmri.22687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pilli VK, Behen ME, Hu J, et al. Clinical and metabolic correlates of cerebral calcifications in Sturge–Weber syndrome. Dev Med Child Neurol. 2017. doi: 10.1111/dmcn.13433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Planche V, Chassin O, Leduc L, Regnier W, Kelly A, Colamarino R. Sturge-Weber syndrome with late onset hemiplegic migraine-like attacks and progressive unilateral cerebral atrophy. Cephalalgia. 2014. doi: 10.1177/0333102413505237 [DOI] [PubMed] [Google Scholar]
- 22.Pilli VK, Chugani HT, Juhász C. Enlargement of deep medullary veins during the early clinical course of Sturge-Weber syndrome. Neurology. 2017. doi: 10.1212/WNL.0000000000003455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John F, Maqbool M, Jeong JW, Agarwal R, Behen ME, Juhász C. Deep cerebral vein expansion with metabolic and neurocognitive recovery in Sturge–Weber syndrome. Ann Clin Transl Neurol. 2018. doi: 10.1002/acn3.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao Z, Lai X. Sturge-weber syndrome coexisting with moyamoya disease in the Fifth Decade: A case report and literature review. Neurologist. 2019. doi: 10.1097/NRL.0000000000000207 [DOI] [PubMed] [Google Scholar]
- 25.Ishikawa H, Ii Y, Niwa A, Matsuura K, Maeda M, Tomimoto H. A case of 55-year-old man with first-ever generalized seizure diagnosed with Sturge-Weber syndrome type III by characteristic MRI findings. Clin Neurol. 2017. doi: 10.5692/clinicalneurol.cn-001006 [DOI] [PubMed] [Google Scholar]
- 26.Zhang R, Chen W, Hu Q, Shrestha S. Adult Sturge-Weber syndrome without facial hemangioma: report of one case. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2014. [DOI] [PubMed] [Google Scholar]
- 27.Serindağ HC, Eren F, Karahan MG, et al. Sturge Weber type 3 presenting with occipital epileptic seizure: case report. Ideggyogy Sz. 2019. doi: 10.18071/isz.72.0131 [DOI] [PubMed] [Google Scholar]
- 28.Shin RK, Moonis G, Imbesi SG. Transient focal leptomeningeal enhancement in Sturge-Weber syndrome. J Neuroimaging. 2002. doi: 10.1177/10528402012003010 [DOI] [PubMed] [Google Scholar]
- 29.Udani V, Pujar S, Munot P, Maheshwari S, Mehta N. Natural history and magnetic resonance imaging follow-up in 9 sturge-weber syndrome patients and clinical correlation. J Child Neurol. 2007. doi: 10.1177/0883073807300526 [DOI] [PubMed] [Google Scholar]
- 30.Sasaki M, Igarashi K, Suzuki S, Saito K. A case of atypical type of Sturge-Weber syndrome demonstrated reversible change by MRI FLAIR method in ictus and in postictal state. Brain and Nerve. 1999. [PubMed] [Google Scholar]
- 31.Kumar KR, Hon K, Schultz D, Agzarian MJ, Jones DN, Thyagarajan D. Transient changes on brain magnetic resonance imaging in a patient with sturge-weber syndrome presenting with hemiparesis. Neurologist. 2009. doi: 10.1097/NRL.0b013e3181940244 [DOI] [PubMed] [Google Scholar]
- 32.Jülich K, Neuberger I, Sahin M, Takeoka M, Pinto A, Prabhu SP. Yield of Emergent Neuroimaging in Patients With Sturge-Weber Syndrome Presenting With Acute Neurologic Symptoms. J Child Neurol. 2019. doi: 10.1177/0883073818801635 [DOI] [PubMed] [Google Scholar]
- 33.Nakajima M, Sugano H, Iimura Y, et al. Sturge-Weber syndrome with spontaneous intracerebral hemorrhage in childhood: Case report. J Neurosurg Pediatr. 2014. doi: 10.3171/2013.9.PEDS133 [DOI] [PubMed] [Google Scholar]
- 34.Chonan M, Suzuki Y, Haryu S, Mashiyama S, Tominaga T. Sturge–Weber syndrome with intracerebral hemorrhage: a case report. Springerplus. 2016. doi: 10.1186/s40064-016-3439-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juhász C, John F. Utility of MRI, PET, and ictal SPECT in presurgical evaluation of non-lesional pediatric epilepsy. Seizure. 2019. doi: 10.1016/j.seizure.2019.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeong JW, Chugani HT, Juhász C. Localization of function-specific segments of the primary motor pathway in children with Sturge-Weber syndrome: A multimodal imaging analysis. J Magn Reson Imaging. 2013. doi: 10.1002/jmri.24076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong JW, Tiwari VN, Shin J, Chugani HT, Juhász C. Assessment of brain damage and plasticity in the visual system due to early occipital lesion: Comparison of FDG-PET with diffusion MRI tractography. J Magn Reson Imaging. 2015. doi: 10.1002/jmri.24556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mantelli F, Bruscolini A, La Cava M, Abdolrahimzadeh S, Lambiase A. Ocular manifestations of Sturge–Weber syndrome: Pathogenesis, diagnosis, and management. Clin Ophthalmol. 2016. doi: 10.2147/OPTH.S101963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Yu RJ, Chen D, et al. Glaucoma in patients with eyes close to areas affected by port-wine stain has lateral and gender predilection. Chin Med J (Engl). 2017. doi: 10.4103/0366-6999.220319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port-wine stains: Improving prediction of Sturge-Weber risk. Br J Dermatol. 2014. doi: 10.1111/bjd.13203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allingham RR. The clinical forms of glaucoma. In: Shields TextBook of Glaucoma. 2011. doi: 10.1097/IJG.0b013e31822f4641 [DOI] [Google Scholar]
- 42.Reyes-Capó D, Cavuoto KM, Chang TC. Outcomes of infantile-onset glaucoma associated with port wine birthmarks and other periocular cutaneous vascular malformations. Asia-Pacific J Ophthalmol. 2018. doi: 10.22608/APO.2017447 [DOI] [PubMed] [Google Scholar]
- 43.Schipper H, Clinch J, Olweny CL Quality of life studies: Definitions and conceptual issues. In: Quality of Life and Pharmacoeconomics in Clinical Trial, 2nd Ed.; 1996. [Google Scholar]
- 44.Ablett K, Thompson AR. Parental, child, and adolescent experience of chronic skin conditions: A meta-ethnography and review of the qualitative literature. Body Image. 2016. doi: 10.1016/j.bodyim.2016.10.001 [DOI] [PubMed] [Google Scholar]
- 45.Bemmels H, Biesecker B, Schmidt JL, Krokosky A, Guidotti R, Sutton EJ. Psychological and social factors in undergoing reconstructive surgery among individuals with craniofacial conditions: An exploratory study. Cleft Palate-Craniofacial J. 2013. doi: 10.1597/11-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gholami A, Salarilak S, Lotfabadi P, et al. Quality of life in epileptic patients compared with healthy people. Med J Islam Repub Iran. 2016. [PMC free article] [PubMed] [Google Scholar]
- 47.LaGrant B, Marquis BO, Berg AT, Grinspan ZM. Depression and anxiety in children with epilepsy and other chronic health conditions: National estimates of prevalence and risk factors. Epilepsy Behav. 2020. doi: 10.1016/j.yebeh.2019.106828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenaro C, Flores N, Gutiérrez-Bermejo B, Vega V, Pérez C, Cruz M. Parental stress and family quality of life: Surveying family members of persons with intellectual disabilities. Int J Environ Res Public Health. 2020. doi: 10.3390/ijerph17239007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hagen SL, Grey KR, Korta DZ, Kelly KM. Quality of life in adults with facial port-wine stains. J Am Acad Dermatol. 2017. doi: 10.1016/j.jaad.2016.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perry M, Streusand WC. The Role of Psychiatry and Psychology Collaboration in Pediatric Dermatology. Dermatol Clin. 2013. doi: 10.1016/j.det.2012.12.012 [DOI] [PubMed] [Google Scholar]
- 51.Loewenstein J, Sutton E, Guidotti R, et al. The art of coping with a craniofacial difference: Helping others through “Positive Exposure.” Am J Med Genet Part A. 2008. doi: 10.1002/ajmg.a.32344 [DOI] [PMC free article] [PubMed] [Google Scholar]