

Abstract
We report the convergent total synthesis of (±)-hamigeran M, enabled by five C–H functionalization reactions and proceeding in 11 steps in 3.9% overall yield. The C–H functionalizations include a hydroxy-directed C–H borylation, one C–H metalation-1,2-addition, one C–H metalation-Negishi coupling, a late-stage oxazole-directed C–H borylation-oxidation, and one electrophilic bromination. Two of these five C–H functionalizations forged strategic C–C bonds in the seven-membered ring of hamigeran M. The oxazole-directed C–H borylation-oxidation was unprecedented and ensured a late-stage hydroxylation. Other key steps include a tandem Suzuki reaction-lactonization to join the cyclopentane building block with the aromatic moiety and a hydrogen-atom transfer (HAT) reaction to reduce a challenging tetrasubstituted double bond.
Graphical Abstract
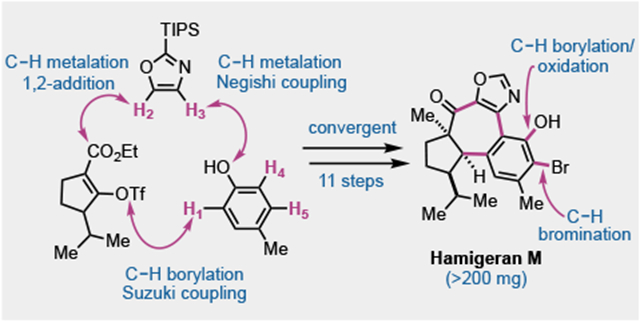
Paradigm-shifting synthetic methodologies such as the Diels-Alder reaction,1 the transition metal-catalyzed cross couplings,2 the olefin metathesis,3 and others, always bring revolutionary changes in the total synthesis of complex natural products. In the last two decades, many advances have been made in the field of C–H functionalization.4 Various C–H functionalization methodologies have been developed to improve synthetic efficiency and economy by avoiding functional group preinstallation and manipulation.5 Herein, we report how multiple well-orchestrated C–H functionalizations enabled the first total synthesis of the complex anticancer natural product hamigenran M in racemic form (>200 mg) in only 11 LLS steps.
The hamigerans are a family of diterpenoids with remarkable structural diversity and biological activities. Since the discovery of their first family members by Cambie and coworkers in 2000,6 a large number of hamigerans have been isolated.7 The hamigerans are characterized by a brominated and polysubstituted aromatic ring. Most of them feature either a 6-6-5 (cf. 1 and 2, Figure 1A) or 6-7-5 (cf. 3-6) tricyclic carbon skeleton. Hamigeran M (6) stands out with its unique oxazole moiety. Natural products with an oxazole moiety are very rare among terpenes of marine origin. Despite their rareness, the oxazole-containing marine terpenes have shown remarkable antimicrobial, anticancer, and other activities.8 Biologically, hamigeran B (2) showed 100% in vitro inhibition activity against the replication of herpes and poliovirus without any significant cytotoxicity against host cells.6 Hamigerans G (5)7b and M (6)7a demonstrated growth inhibition activity against HL-60 leukemia cell line with 2.5 μM and 6.9 μM IC50 values, respectively. Additionally, the hamigerans are structurally related to the gukulenin natural products (cf. 7),9 which contain unusual tropolone moieties and exhibited strong growth inhibition activity against various human cancer cell lines with nanomolar IC50 values.
Figure 1.

The hamigerans, plausible biosynthesis, and retrosynthetic analysis.
The hamigerans have garnered a significant amount of attention from the synthetic community. Most of the early efforts focus on hamigeran B (2) and its congeners with the 6-6-5 tricyclic core. The groups of Nicolaou,10 Clive,11 Trost,12 Taber,13 Stoltz,14 and other15 have reported elegant syntheses of hamigeran B (2). However, synthetic efforts toward the more challenging 6-7-5 tricyclic hamigerans are very limited. In 2016, Gao and coworkers reported the first total syntheses of hamigerans D (4), G (5), and N-Q in 24-25 steps from (R)-piperitone.16 In 2018 and 2020, the Stoltz group17 and our group18 reported synthetic studies to construct their 6-7-5 tricyclic carbon skeleton.
Biosynthetically, hamigeran M (6) was proposed to be synthesized from compound such as 8 and glycine by going through intermediates including 9, 10, 11, and 12 (Figure 1B).7a While the oxazole moiety was formed at the last stage of hamigeran M’s biosynthesis, we planned to introduce it at the very beginning by using an oxazole as one of the three building blocks (13, 14, 15, Figure 1C). Retrosynthetically, hamigeran M could be prepared from 16 via an electrophilic bromination. If the phenol hydroxyl group could not be preinstalled (X = H for 16), a late-stage oxazole-directed C–H oxidation or borylation-oxidation would be needed. While oxazole has been used as a directing group in C–H funtionalizations,19 there were no examples of C–H oxidation or borylation of 4-aryl oxazoles. For the formation of the central seven-membered ring, we proposed a direct transition metal-catalyzed intramolecular oxazole C–H arylation to forge the key C–C bond and close the seven-membered ring. Alternatively, a selective C–H metalation followed by intramolecular cross coupling would complete the same task. While such oxazole-based seven-membered ring closure via a direct C–H functionalization was unprecedented, it would be highly efficient, thus worth developing. Compound 17 could be derived from 18 via a 1,2-addition of a nucleophile (cf. oxazol-5-yllithium) generated from selective C5 deprotonation of 13a with a strong base. The acidity difference of the three positions of oxazole 13 [C2 (H) > C5 (H2) > C4 (H3)] would allow us to selectively differentiate these three protons (H, H2, H3) and execute the proposed sequential C–H functionalization. Since the C2 (H) position is the most active one, a C–H protection would be needed (cf. 13a). The all-carbon quaternary center of 18 could be installed via a stereoselective α-methylation of the corresponding lactone, which could be potentially prepared from stereoselective reduction of the tetrasubstituted double bond of 19. Reduction of a tetrasubstituted double bond has always been a synthetic challenge. We were hoping the newly developed HAT reductions20 may be our recourse. 19 itself could be quickly assembled via a tandem Suzuki reaction-lactonization between vinyltriflate 14 and aryl boronate 20. A phenol-directed C–H borylation21 could give access to 20 from 21. Triflate 14 could be prepared from the corresponding β-ketoester.
Our synthesis started with preparing triflate 14 (Scheme 1). Its precursor, β-ketoester 24, was synthesized from commercially available 22 (~$1/g). Alkylation of 22 with isopropyl iodide gave 23, which after a simple filtration to remove the inorganic solid, was treated with sodium ethoxide in ethanol to promote a retro-Dieckmann reaction followed by Dieckmann condensation to afford 24 in 64% yield. Deprotonation of 24 with LiHMDS followed by trapping the resulting enolate with triflic anhydride gave 14 in 85% yield. We then synthesized aryl boronate 26 from commercially available 2-methoxy-4-methylphenol 25 (~$1/g). The methoxy group of 25 would be demethylated to give the free phenol of hamigeran M. We were hoping that introduction of this group at the beginning would save us trouble in installing it toward the end, but as discussed later that the methoxy group brought an insurmountable problem to the synthesis. The iridium-catalyzed C–H borylation developed by Hartwig and coworkers converted 25 to 26 in high yield (95%).22 This reaction can be scaled up to multi-gram scale as well, thus providing a safe access to 26 without using strong and pyrophoric bases such as nBuLi or tBuLi. Suzuki cross coupling between 26 and 14 took place smoothly. Under the same reaction conditions, simultaneous lactonization occurred to afford 27 in 98% yield. With 27 in hand, we focused on stereoselective reduction of the challenging tetra-substituted double bond, but various catalytic hydrogenations, single-electrontransfer reductions, and hydride-mediated conjugate reductions failed. The breakthrough came when the newly developed HAT reductions with Shenvi’s silane (PhSi(OiPr)H2) were investigated.23 Product 28 was obtained as a single diastereomer in 34% or 66% yield with the use of Mn(dpm)3 or Co(dpm)2,24 respectively. α-Methylation of 28 gave 29 with a cis ring junction and an all-carbon quaternary center. At this stage, we prepared C2 TIPS-protected oxazole 30 via a C–H silylation. Selective C–H lithiation of 30 with nBuLi gave an oxazol-5-yllithium intermediate, which reacted with lactone 29 via a 1,2-addition process. After aqueous workup and concentration, the crude phenol was treated with triflic anhydride and pyridine to provide 31 in 62% yield over two steps.
Scheme 1.

Synthetic attempts toward 32.
We next focused on the formation of the seven-membered ring. To save precious substrate 31, model compound 34 was prepared and used for the explorations. We first tried to form the seven-membered ring with palladium-catalyzed direct C–H arylations,25 but were not successful. Only products with TIPS removal and/or triflate hydrolysis were detected. We then focused on the C–H metalation26 followed by intramolecular cross coupling. We identified that TMPZnCl·LiCl developed by Knochel and coworkers27 was effective to convert 34 to the corresponding oxazol-4-ylzinc reagent for a one-pot intramolecular Negishi cross coupling. A combination of the Buchwald’s 2nd generation preformed catalyst XPhos Pd G2 (10 mol%)28 and XPhos (10 mol%) was identified to be effective. Model product 35 was obtained in 67% at almost gram scale. Unfortunately, the success in the model study could not be transferred to 31 with an electron-donating methoxy group ortho to the triflate. While we were able to generate the oxazol-4-ylzinc as evidenced by the deuterium trapping reaction (31→33), no ring closing product 32 was obtained and most of the starting material was recovered. Apparently, the electronic and steric influences generated by the methoxy group significantly inhibited the oxidative addition. Thus, in order to close the seven-membered ring, we had to remove the methoxy group from 31 and introduce the oxygen functionality at a late stage via a C–H oxidation directed by the oxazole.
We then prepared a new ring closing precursor 41 from p-cresol (36) (Scheme 2). Without the methoxy group to block one of the two ortho positions, an alternative C–H borylation was needed to avoid bisborylation. After modifying the procedure developed by Smith et al.,29 we were able to prepare monoboronate 37 from 36 in 65% yield at multi-gram scale. The previously established tandem Suzuki reaction-lactonization worked smoothly to deliver 38 in 92% yield. The next Co(dpm)2-mediated HAT reduction gave 39 in 77% yield at gram scale. The following α-methylation (39→40), C–H metalation-1,2-addition and triflation (40→41) worked uneventfully to afford 41 in multi-gram scale. As we anticipated, removal of the methoxy group did help the ring closing reaction, after C–H metalation with TMPZnCl·LiCl, the subsequent intramolecular Negishi cross coupling afforded product 42 in 40% yield with the Buchwald XPhos Pd G2. To further improve the reaction conversion and yield, several third generation (G3) Buchwald precatalysts30 were explored because of their thermal stability and long life in solutions. RuPhos Pd G3 and Me4tBuXPhos Pd G3 both gave 42 in 52% yield at small scale, albeit a higher BRSM yield (67%) was provided by Me4tBuXPhos Pd G3. When the reaction was conducted at 0.227 g-scale (five reactions in parallel), 42 could be obtained in 48% yield (82% brsm). After the TIPS group was removed with HCl to produce 43, we focused on the oxazole-directed C–H oxidation. We first explored transition metal such as palladium, copper, or rhodium-catalyzed C–H oxygenations to install the hydroxy group or a protected hydroxy group directly with various oxidants, but these reactions either gave the starting material back or decomposed it mainly at the oxazole part. We then investigated the C–H borylation-oxidation process. The use of [Rh(cod)OH]2 in combination with silica-SMAP31 afforded the desired boronate 44 with B2pin2 as the borylating reagent. Under the [Rh(cod)OH]2/silica-SMAP conditions, we noted that one main byproduct was a secondary alcohol derived from reduction of the seven-membered ketone by the boron hydride species generated in situ. To suppress this reduction, sacrificial cyclohexanone or 1-octene was used to consume the boron hydride species. Both of them were effective and the use of 1-octene gave higher yield and cleaner reaction. The C–H borylation product 44 was then oxidized to phenol 45 via a one-pot procedure with 50% overall yield at 25 mg scale. When then reaction was conducted at 400 mg scale, purification of 44 was beneficial and 45 was obtained in 42% steps after oxidation. The last electrophilic bromination with pyridine·HBr3 completed the total synthesis of (±)-hamigeran M (6). Its structure was unambiguously confirmed by X-ray crystallography (CCDC 2114503). Our synthesis has accumulated over 200 mg of (±)-hamigeran M so far.
Scheme 2.

Total synthesis of (±)-hamigeran M.
In summary, we completed the first total synthesis of (±)-hamigeran M in 11 LLS steps and 3.9% overall yield. The synthesis was enabled by five well-orchestrated C–H funtionalizations. Other notable steps include a tandem Suzuki reaction-lactonization, a HAT reaction to reduce a highly challenging tetrasubstituted double bond, and a stereoselective α-methylation to build the all-carbon quaternary center. This synthesis is concise, convergent, and scalable, which is expected to facilitate the corresponding biological studies of hamigeran M.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by NIH GM128570. The NIH CA023168 is acknowledged for supporting shared NMR resources to Purdue Center for Cancer Research. The XRD data is collected on a new single crystal X-ray diffractometer supported by the NSF CHE 1625543 through the MRI Program.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectra data (PDF file)
Cystallographic data for 6 (cif file)
The authors declare no competing financial interest.
REFERENCES
- (1).Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed 2002, 41, 1668–1698. [DOI] [PubMed] [Google Scholar]
- (2).Nicolaou KC; Bulger PG; Sarlah D Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. Int. Ed 2005, 44, 4442–4489. [DOI] [PubMed] [Google Scholar]
- (3).Nicolaou KC; Bulger PG; Sarlah D Metathesis Reactions in Total Synthesis. Angew. Chem. Int. Ed 2002, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]
- (4).(a) Rogge T; Kaplaneris N; Chatani N; Kim J; Chang S; Punji B; Schafer LL; Musaev DG; Wencel-Delord J; Roberts CA; Sarpong R; Wilson ZE; Brimble AA; Johansson MJ; Ackermann L C–H activation. Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar]; (b) Davies HML; Bois JD; Yu J-Q C–H Functionalization in organic synthesis. Chem. Soc. Rev 2011, 40, 1855–1856. [DOI] [PubMed] [Google Scholar]
- (5).(a) For reviews:Gutekunst WR; Baran PS C–H functionalization logic in total synthesis. Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]; (b) Abrams DJ; Provencher PA; Sorensen EJ Recent applications of C–H functionalization in complex natural product synthesis. Chem. Soc. Rev 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]; (c) McMurray L; O’Hara F; Gaunt MJ Recent developments in natural product synthesis using metal-catalysed C–H bond functionalization. Chem. Soc. Rev 2011, 40, 1885–1898. [DOI] [PubMed] [Google Scholar]; (d) Baudoin O Multiple Catalytic C–H Bond Functionalization for Natural Product Synthesis. Angew. Chem. Int. Ed 2020, 59, 17798–17809. [DOI] [PubMed] [Google Scholar]; (e) Yamaguchi J; Yamaguchi A; Itami K C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]; (f) Hong B; Luo T; Lei X Late-Stage Diversification of Natural Products. ACS Cent. Sci 2020, 6, 622–635. For examples: [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wu F; Zhang J; Song F; Wang S; Guo H; Wei Q; Dai H; Chen X; Xia X; Liu X; Zhang L; Yu J-Q; Lei X Chrysomycin A Derivatives for the Treatment of Multi-Drug-Resistant Tuberculosis. ACS Cent. Sci 2020, 6, 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Leal RA; Bischof C; Lee YV; Sawano S; McAtee CC; Latimer LN; Russ ZN; Dueber JE; Yu J-Q; Sarpong R Application of a Palladium-Catalyzed C–H Functionalization/Indolization Method to Syntheses of cis-Trikentrin A and Herbindole B. Angew. Chem. Int. Ed 2016, 55, 11824–11828. [DOI] [PubMed] [Google Scholar]; (i) Hong B; Li C; Wang Z; Chen J; Li H; Lei X Enantioselective Total Synthesis of (−)-Incarviatone A. J. Am. Chem. Soc 2015, 137, 11946–11949. [DOI] [PubMed] [Google Scholar]
- (6).Wellington KD; Cambie RC; Rutledge PS; Bergquist PR Chemistry of Sponges. 19. Novel Bioactive Metabolites from Hamigera tarangaensis. J. Nat. Prod 2000, 63, 79–85. [DOI] [PubMed] [Google Scholar]
- (7).(a) Dattelbaum JD; Singh AJ; Field JJ; Miller JH; Northcote PT The Nitrogenous Hamigerans: Unusual Amino Acid-Derivatized Aromatic Diterpenoid Metabolites from the New Zealand Marine Sponge Hamigera tarangaensis. J. Org. Chem 2015, 80, 304–312. [DOI] [PubMed] [Google Scholar]; (b) Singh AJ; Dattelbaum JD; Field JJ; Smart Z; Woolly EF; Barber JM; Heathcott R; Miller JH; Northcote PT Structurally diverse hamigerans from the New Zealand marine sponge Hamigera tarangaensis: NMR-directed isolation, structure elucidation and antifungal activity. Org. Biomol. Chem 2013, 11, 8041–8051. [DOI] [PubMed] [Google Scholar]; (c) Woolly EF; Singh AJ; Russell ER; Miller JH; Northcote PT Hamigerans R and S: Nitrogenous Diterpenoids from the New Zealand Marine Sponge Hamigera tarangaensis. J. Nat. Prod 2018, 81, 387–393. [DOI] [PubMed] [Google Scholar]
- (8).(a) Kobayashi J; Madono T; Shigemori H Nakijinol, a Novel Sesquiterpenoid Containing a Benzoxazole Ring from an Okinawan Sponge. Tetrahedron Lett. 1995, 36, 5589–5590. [Google Scholar]; (b) Rodríguez AD; Rodríguez C; Rodríguez II; Rodríguez EG Novel Antimycobacterial Benzoxazole Alkaloids, from the West Indian Sea Whip Pseudopterogorgia elisabethae. Org. Lett 1999, 1, 527–530. [DOI] [PubMed] [Google Scholar]
- (9).Kats-Kagan R; Herzon SB The Discovery of a Novel Route to Highly Substituted α-Tropolones Enables Expedient Entry to the Core of the Gukulenins. Org. Lett 2015, 17, 2030–2033. [DOI] [PubMed] [Google Scholar]
- (10).Nicolaou KC; Gray DLF; Tae J Total Synthesis of Hamigerans and Analogues Thereof. Photochemical Generation and Diels–Alder Trapping of Hydroxy-o-quinodimethanes. J. Am. Chem. Soc 2004, 126, 613–627. [DOI] [PubMed] [Google Scholar]
- (11).Clive DLJ; Wang J Stereospecific Total Synthesis of the Antiviral Agent Hamigeran B—Use of Large Silyl Groups to Enforce Facial Selectivity and to Suppress Hydrogenolysis. Angew. Chem. Int. Ed 2003, 42, 3406–3409. [DOI] [PubMed] [Google Scholar]
- (12).Trost BM; Pissot-Soldermann C; Chen I; Schroeder GM An Asymmetric Synthesis of Hamigeran B via a Pd Asymmetric Allylic Alkylation for Enantiodiscrimination. J. Am. Chem. Soc 2004, 126, 4480–4481. [DOI] [PubMed] [Google Scholar]
- (13).Taber DF; Tian W Synthesis of (−)-Hamigeran B. J. Org. Chem 2008, 73, 7560–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stoltz BM; Mukherjee H; McDougal NT; Virgil SC A Catalytic, Asymmetric Formal Synthesis of (+)-Hamigeran B. Org. Lett 2011, 13, 825–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Cao B-C; Wu G-J; Yu F; He Y-P; Han F-S A Total Synthesis of (−)-Hamigeran B and (−)-4-Bromohamigeran B. Org. Lett 2018, 20, 3687–3690. [DOI] [PubMed] [Google Scholar]; (b) Lau SYW Concise and Protective Group-Free Syntheses of (±)-Hamigeran B and (±)-4-Bromohamigeran B. Org. Lett 2011, 13, 347–349. [DOI] [PubMed] [Google Scholar]; (c) Jiang B; Li M; Xing P; Huang Z A Concise Formal Synthesis of (−)-Hamigeran B. Org. Lett 2013, 15, 871–873. [DOI] [PubMed] [Google Scholar]; (d) Lin H; Xiao L-J; Zhou M-J; Yu H-M; Xie J-H; Zhou Q-L Enantioselective Approach to (−)-Hamigeran B and (−)-4-Bromohamigeran B via Catalytic Asymmetric Hydrogenation of Racemic Ketone To Assemble the Chiral Core Framework. Org. Lett 2016, 18, 1434–1437. [DOI] [PubMed] [Google Scholar]; (e) Miesch L; Welsch T; Rietsch V; Miesch M Intramolecular Alkynylogous Mukaiyama Aldol Reaction Starting from Bicyclic Alkanones Tethered to Alkynyl Esters: Formal Total Synthesis of (±)-Hamigeran B. Chem. Eur. J 2009, 15, 4394–4401. [DOI] [PubMed] [Google Scholar]; (f) Kuwata K; Fujita R; Hanaya K; Higashibayashi S; Sugai T Formal total synthesis of (−)-hamigeran B from a chemo-enzymatically prepared building block with quaternary chiral center. Tetrahedron 2018, 74, 740–745. [Google Scholar]
- (16).Li X; Xue D; Wang C; Gao S Total Synthesis of the Hamigerans. Angew. Chem. Int. Ed 2016, 55, 9942–9946. [DOI] [PubMed] [Google Scholar]
- (17).Duquette DC; Jensen T; Stoltz BM Progress towards the total synthesis of hamigerans C and D: a direct approach to an elaborated 6–7–5 carbocyclic core. J. Antibiotics 2018, 71, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jiang B; Dai M Synthetic Studies toward the Hamigerans with a 6-7-5 Tricyclic Core. Org. Lett 2020, 22, 4176–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Simonetti M; Cannas DM; Just-Baringo X; Vitorica-Yrezabal IJ; Larrosa I Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals. Nat. Chem 2018, 10, 724–731. [DOI] [PubMed] [Google Scholar]; (b) Ni S; Hribersek M; Baddigam SK; Ingner FJL; Orthaber A; Gates PJ; Pilarski LT Mechanochemical Solvent-Free Catalytic C–H Methylation. Angew. Chem. Int. Ed 2021, 60, 6660–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Crossley SWM; Obradors C; Martinez RM; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mkhalid IA; Barnard JH; Marder TB; Murphy JM; Hartwig JF C–H Activation for the Construction of C–B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (22).Timothy AB; Hartwig JF Silyl-Directed, Iridium-Catalyzed ortho-Borylation of Arenes. A One-Pot ortho-Borylation of Phenols, Arylamines, and Alkylarenes. J. Am. Chem. Soc 2008, 130, 7534–7535. [DOI] [PubMed] [Google Scholar]
- (23).Obradors C; Martinez RM; Shenvi RA Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J. Am. Chem. Soc 2017, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Strotman NA; Chobanian HR; Guo Y; He J; Wilson JE Highly Regioselective Palladium-Catalyzed Direct Arylation of Oxazole at C-2 or C-5 with Aryl Bromides, Chlorides, and Triflates. Org. Lett 2010, 12, 3578–3581. [DOI] [PubMed] [Google Scholar]; (b) Liégault B; Lapointe D; Caron L; Vlassova A; Fagnou K Establishment of Broadly Applicable Reaction Conditions for the Palladium-Catalyzed Direct Arylation of Heteroatom-Containing Aromatic Compounds. J. Org. Chem 2009, 74, 1826–1834. [DOI] [PubMed] [Google Scholar]
- (26).Haag B; Mosrin M; Ila H; Malakhov V; Knochel P Regio- and Chemoselective Metalation of Arenes and Heteroarenes Using Hindered Metal Amide Bases. Angew. Chem. Int. Ed 2011, 50, 9794–9824. [DOI] [PubMed] [Google Scholar]
- (27).Haas D; Mosrin M; Knochel P Regioselective Functionalization of the Oxazole Scaffold Using TMP-Bases of Mg and Zn. Org. Lett 2013, 15, 6162–6165. [DOI] [PubMed] [Google Scholar]
- (28).Yang Y; Oldenhuis NJ; Buchwald SL Mild and General Conditions for Negishi Cross-Coupling Enabled by the Use of Palladacycle Precatalysts. Angew. Chem. Int. Ed 2012, 52, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chattopadhyay B; Dannatt JE; Andujar-De Sanctis IL; Gore KA; Maleczka RE; Singleton DA; Smith MR Ir-Catalyzed ortho-Borylation of Phenols Directed by Substrate–Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc 2017, 139, 7864–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bruno NC; Tudge MT; Buchwald SL Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kawamorita S; Miyazaki T; Ohmiya H; Iwai T; Sawamura M Rh-Catalyzed Ortho-Selective C–H Borylation of N-Functionalized Arenes with Silica-Supported Bridgehead Monophosphine Ligands. J. Am. Chem. Soc 2011, 133, 19310–19313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.