

Abstract
New, more effective therapeutics are required for the treatment of paediatric cancers. Current treatment protocols of cytotoxic treatments including chemotherapy trigger cancer-cell death by engaging the apoptosis pathway, and chemotherapy efficacy is frequently impeded by apoptosis dysregulation. Apoptosis dysregulation, through genetic or epigenetic mechanisms, is a feature of many cancer types, and contributes to reduced treatment response, disease progression and ultimately treatment resistance. Novel approaches are required to overcome dysregulated apoptosis signalling, increase the efficacy of cancer treatment and improve patient outcomes. Here, we provide an insight into current knowledge of how the apoptosis pathway is dysregulated in paediatric nervous system tumours, with a focus on TRAIL receptors, the BCL-2 proteins and the IAP family, and highlight preclinical evidence demonstrating that pharmacological manipulation of the apoptosis pathway can restore apoptosis signalling and sensitise cancer cells to treatment. Finally, we discuss the potential clinical implications of these findings.
Subject terms: Cancer therapeutic resistance, Paediatric cancer, CNS cancer, Chemotherapy
Facts
Apoptosis pathways are frequently dysregulated in cancer, including in paediatric nervous system malignancies and chemoresistance remains a significant clinical challenge
Reactivation of apoptosis pathways represents a rational strategy to sensitise cancer cells to death-inducing treatments, and the development of targeted agents enables the pharmacological modulation of anti-apoptotic proteins
A growing body of preclinical evidence highlights the utility of targeted agents such as BH3 mimetics and IAP inhibitors in paediatric oncology
Techniques such as BH3 profiling will facilitate the clinical translation of some of these agents
Open questions
Can novel apoptosis modulators increase the sensitivity of paediatric nervous system cancers to apoptosis-inducing treatment?
What biomarkers can identify patients likely to benefit from such treatments?
Which combinations of targeted agents are most beneficial in these cancers?
How can the apoptosis pathway be optimally targeted without resulting in on-target effects in non-disease tissue?
Introduction
After accidents, cancer is the leading cause of death in children, as well as the leading cause of illness-related death [1, 2]. Between 2001 and 2010, ~400,000 children and adolescents were diagnosed with cancer worldwide [3]. Tumours of the nervous system are among the most commonly occurring paediatric cancers, with brain tumours the second most common solid malignancy [4]. Within malignant brain tumours, medulloblastoma is the most frequent, accounting for 20% of all cases [5]. Less common paediatric brain tumours include glioblastoma, astrocytoma, choroid plexus carcinoma, primitive neuro-ectodermal tumours (PNET), meningioma and ependymoma [6]. Tumours of the sympathetic nervous system such as neuroblastoma also occur frequently in young children, with 90% of neuroblastoma cases diagnosed in patients aged <5 years [7]. In the majority of these malignancies, chemotherapy is a mainstay of treatment [8–10], and is of particular importance in patients <3 years for whom radiation therapy is avoided due to its associated side effects [11].
Apoptosis is a tightly regulated form of programmed cell death carried out by multicellular organisms as part of normal development. The two main pathways of apoptosis are the intrinsic and extrinsic pathways, and these differ in their triggers and the means by which the apoptotic signal is transduced (Fig. 1). Both pathways converge on caspase activation, which leads to cell death execution [12]. Intrinsic apoptosis is initiated by alterations in the intracellular environment and is defined by mitochondrial outer membrane permeabilization (MOMP) [12]. The extrinsic pathway is triggered by activation of cell surface receptors by external signalling molecules or perturbations of the extracellular environment, and is propagated by caspase 8 activation [12].
Fig. 1. Schematic representation of the extrinsic and intrinsic apoptosis pathways, indicating pharmacological agents and their therapeutic targets.

Proteins mediating similar functions are indicated by shading, while green shaded boxes highlight pharmacological agents. ↑ and ↓ indicates that the target is upregulated or downregulated by the drug, respectively. --| indicates inhibition of the target protein by the specified protein/drug. Created with BioRender.com.
Chemotherapy, and indeed most cancer therapies, target the apoptosis pathway to mediate their effects. Treatment success, however, is frequently hampered by apoptosis resistance. Indeed, evading apoptosis is one of the Hallmarks of Cancer, contributing to both tumour progression and treatment resistance [13]. Apoptosis resistance is either intrinsically present in cancer cells, for example through genetic mutations, or is acquired in response to cellular stress by upregulation or downregulation of anti- or pro-apoptotic proteins, respectively [13]. Characterising apoptosis defects can identify ways to restore apoptosis signalling, inform the development of novel anti-tumour strategies, and enable reduction of toxic chemotherapy regimens. This is of particular importance for paediatric tumours which are frequently associated with defective apoptotic machinery [14]. In this review, we focus on the ability of paediatric nervous system tumours to evade apoptosis, and highlight potential strategies to overcome the associated therapeutic resistance.
Targeting the extrinsic apoptosis pathway in paediatric nervous system tumours
The extrinsic apoptosis pathway
Extrinsic apoptosis is initiated by extracellular perturbations, generating a signal that is transduced by receptors located on the plasma membrane. Death receptors are activated by ligand binding. TRAIL, for instance, binds TRAIL-R1 and TRAIL-R2 (also known as DR4 and DR5) to initiate extrinsic apoptosis signalling [15]. In contrast, dependence receptors are activated when ligand levels drop below certain thresholds [16]. Death receptors are characterised by a cytoplasmic ‘death domain’ to relay the signal from the cell surface. TRAIL receptor ligation enables association of the FADD adapter molecule at the TRAIL-R1/R2 death domain [17], leading to formation of the death-inducing signalling complex (DISC) which recruits and activates caspase 8 [18]. Proteolytic activation of caspase 8 via interactions at the DISC initiates the caspase cascade and apoptosis execution. Cells are considered as type I or type II based on how they execute extrinsic apoptosis [19]. In type I cells, levels of activated caspase 8 are sufficient to drive apoptosis via activation of executioner caspases 3 and 7, and death is independent of the mitochondria [19]. In contrast, type II cells require involvement of the mitochondrial pathway to execute apoptosis, as DISC formation is lower in these cells [20]. Mitochondrial pathway involvement is mediated by caspase 8 truncation of BID, a pro-apoptotic protein [21], with truncated BID (tBID) subsequently engaging BAX and/or BAK to induce MOMP and the release of pro-apoptotic mediators, thereby amplifying the death signal (Fig. 1). Finally, cFLIP is a non-proteolytically active homologue of caspase 8 [22]. Its recruitment to the DISC forms an apoptosis inhibitory complex in conjunction with FADD [22], preventing caspase 8 activation. cFLIP therefore controls the susceptibility of tumour cells to TRAIL (Fig. 1).
Targeting TRAIL receptors and downstream signalling
Exogenous TRAIL holds promise as a potential cancer therapeutic due to its selectivity for malignant cells with minimal toxicity against normal tissue [23], and its translational relevance has been increased by the development of new variants such as IZI1551 [24] and ABV-2661, a TRAIL-R agonist fusion protein [25] which is currently in a Phase 1 clinical trial for adult patients with previously-treated malignancies (NCT03082209).
The efficacy of TRAIL against cancer is limited, by frequent dysregulation of the extrinsic apoptosis pathway, including downregulation of TRAIL receptors and downstream signalling mediators, for example by epigenetic silencing [26]. Therefore, agents that restore expression of these key molecules have potential as TRAIL sensitisers, and the reversibility of epigenetic silencing is of clinical importance as epigenetic remodelling can be targeted pharmacologically.
Upregulation of TRAIL-R2/DR5 in neuroblastoma cells by etoposide [27, 28] or doxorubicin treatment [27] has been documented to increase TRAIL sensitivity. In medulloblastoma, TRAIL-R2/DR5 is commonly expressed, while TRAIL-R1/DR4 is frequently absent [29, 30]. This silencing of DR4 is understood to be mediated by aberrant histone deacetylation [30], and treatment of medulloblastoma cell lines with the histone deacetylase (HDAC) inhibitor MS275 increased DR4 expression, via increased acetylation of H3 and H4 at its transcriptional start site, and enhanced TRAIL-induced apoptosis [30].
Other potential strategies to sensitise cells to TRAIL treatment have been identified, including modulation of the BCL-2 protein family which is of particular relevance in type II cells [20]. The combination of TRAIL with the proteasome inhibitor Bortezomib was highlighted as an effective strategy for inducing apoptosis in cell line models and primary cultures of neuroblastoma [31] and meningioma [32]. Bortezomib enhanced TRAIL-induced, caspase 8-mediated tBID levels, and induced mitochondrial apoptosis and caspase activation through a mechanism involving p53 and upregulation of NOXA, a pro-apoptotic protein [31] whose role is described further in section 3.1. Encouragingly, this effect was also observed in an in vivo model of neuroblastoma [31], underscoring the potential clinical relevance of this combination. However, no clinical trials have been undertaken to assess the efficacy of this, as Bortezomib lacks the ability to cross the blood-brain barrier, limiting its relevance in the context of CNS tumours [33]. In this regard, another proteasome inhibitor, Marizomib, has increased potential as it can penetrate the blood-brain barrier, and is currently in a phase III trial in adult GBM patients (NCT03345095).
Targeting caspase 8 to increase sensitivity to TRAIL
Weak caspase 8 expression is predictive of unfavourable progression-free survival (PFS) in medulloblastoma, where patients with low caspase 8 tumour levels had a 5-year PFS of 31%, compared with 73% in those with moderate or strong expression [34]. Additionally, loss or silencing of caspase 8 expression is associated with MYCN-amplified and aggressive neuroblastomas [35, 36].
Moreover, caspase 8 expression is key in determining TRAIL sensitivity, and loss of expression is frequent in the development of TRAIL resistance. Increased methylation of the caspase 8 promoter mediates its transcriptional silencing in medulloblastoma, PNET, and neuroblastoma [29, 34, 37–39]. Indeed, caspase 8 promoter methylation and subsequent silencing are associated with tumour aggressiveness in ganglioneuromas [39], and with invasiveness in TRAIL-resistant neuroblastoma cell lines [40, 41].
In this context, methylation inhibitors may be beneficial. In vitro treatment with 5-Aza-2’-deoxycytidine, a demethylating agent, successfully restored caspase 8 expression and sensitivity to TRAIL-induced apoptosis in cell line models of PNET [37], neuroblastoma [38, 40, 42] and medulloblastoma [42].
Furthermore, interferon-γ (IFNγ) treatment of caspase 8 deficient cells restored caspase 8 expression and TRAIL sensitivity in TRAIL-resistant medulloblastoma [34, 42] and neuroblastoma cell lines [27, 28, 43–46]. This was due to IFNγ-mediated regulation of an interferon-sensitive response element within the caspase 8 promoter [43, 47, 48]. IFNγ-mediated caspase 8 upregulation was also shown to enhance sensitivity of medulloblastoma cells to standard chemotherapeutic agents including cisplatin, doxorubicin, and etoposide, as well as to ionising radiation [49]. Together, these findings highlight that resistance to TRAIL-induced death may be overcome by targeting the epigenome to upregulate caspase 8 expression. The clinical importance of these findings is enhanced by the capability of 5-Aza-2’-deoxycytidine to readily penetrate the blood-brain barrier [50], and a phase I trial is underway investigating the safety of directly infusing this into the fourth ventricle in paediatric ependymoma patients (NCT02940483).
Targeting the BCL-2 protein family in paediatric nervous system tumours
The BCL-2 protein family
The intrinsic pathway of apoptosis is characterised by MOMP, which facilitates the cytosolic release of cytochrome c, SMAC, and other intermembrane space proteins, enabling apoptosome formation, caspase activation, and apoptosis execution. MOMP is tightly regulated by the BCL-2 protein family, whose members share homology in at least one BCL-2 homology (BH) domain (BH1, BH2, BH3 and BH4). On this basis they are classified into three groups, shown in Fig. 2: the anti-apoptotic proteins BCL-2, BCL-XL, MCL-1, BCL-W and BFL-1/A1 with sequence homology in all four BH regions, the pro-apoptotic proteins (termed multi-domain effectors) BAX, BAK and BOK with sequence homology at BH1, BH2 and BH3, and pro-apoptotic proteins such as BIM, BID, PUMA and NOXA, which only possess the short BH3 domain. This final group are designated as BH3-only proteins and can be further sub-classified as either ‘activators’ (BIM, BID and PUMA) or ‘sensitisers’, (BAD, BIK, BMF, HRK and NOXA) [51].
Fig. 2. The structure of BCL-2 protein family members.
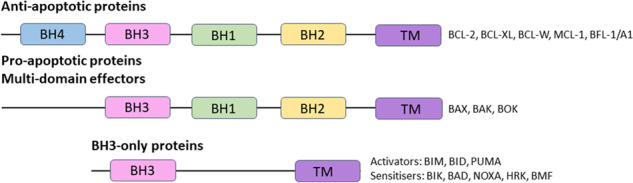
TM denotes the transmembrane domain.
BAX and BAK are MOMP effectors which dimerise via their exposed BH3 domains [52] and form higher-order structures through cross-linking of cysteine residues in their N-terminal epitopes [52]. These oligomers insert into the mitochondrial outer membrane lipid bilayer to form pores [53], leading to loss of mitochondrial membrane potential and disruption of mitochondrial structure, and culminating in release of cytochrome c and SMAC from the mitochondria [54]. While BOK is classified as a pro-apoptotic multi-domain effector protein, its role in apoptosis remains the subject of uncertainty and it is understood to be the least potent of the MOMP effector family [55]. As shown in Fig. 3, BAX/BAK activation is antagonised via direct interactions with the anti-apoptotic proteins BCL-XL, BCL-2, MCL-1, BCL-W and BFL-1/A1, which sequester pro-apoptotic proteins and prevent MOMP [51, 56–58]. Activator BH3-only proteins have the ability to bind non-activated BAX/BAK to directly trigger their activation [51]. Sensitiser BH3-only proteins mediate their pro-apoptotic functions indirectly, by binding anti-apoptotic proteins and competitively displacing BAX/BAK monomers or activator BH3-only proteins from the anti-apoptotic proteins [58, 59].
Fig. 3. BCL-2 protein family interactions.
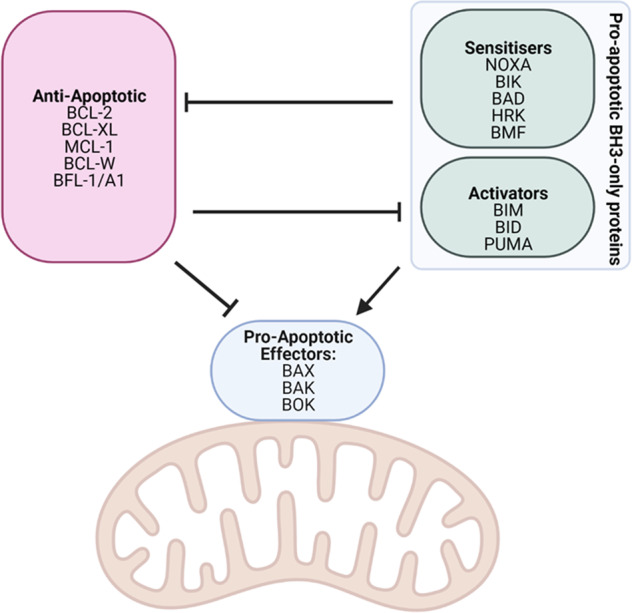
The BCL-2 protein family interactions mediate BAX/BAK-induced mitochondrial outer membrane permeabilisation (MOMP). Created with BioRender.com.
BCL-2 proteins as therapeutic targets in cancer
Dysregulation of BCL-2 protein family members is reported in many cancers, and hence, their targeting may sensitise cancer cells towards apoptotic stimuli. For example, tumours frequently depend on an overabundance of anti-apoptotic signals to sustain their survival in the presence of death-inducing agents. In this regard, BH3 mimetics, a class of highly specific small molecule inhibitors that mimic the function of the pro-apoptotic BH3-only proteins, hold great potential.
ABT-737 was one of the earliest BH3 mimetics designed to inhibit the anti-apoptotic BCL-2 protein family. With nanomolar affinity for BCL-2, BCL-XL and BCL-W [60], ABT-737 demonstrated single-agent efficacy against lymphoma and small-cell lung cancer (SCLC), and synergy in combination with chemotherapy and radiation in vitro and in vivo. However, in vivo ABT-737 treatment resulted in platelet apoptosis [61] due to the exquisite anti-apoptotic dependence of platelets on BCL-XL [62], leading to reduced platelet and lymphocyte counts [60].
The clinical utility of ABT-737 was also hindered by its lack of oral bioavailability, prompting the development of ABT-263/Navitoclax, an orally available BH3 mimetic with a similar binding profile to ABT-737 [63]. Mechanistically, ABT-263 induces apoptosis by disrupting BIM:BCL-XL and BIM:BCL-2 interactions, releasing BIM and resulting in BAX-dependent MOMP [63]. ABT-263 demonstrated activity as a single agent in SCLC and haematological malignancies, resulted in tumour regression in SCLC and acute lymphoblastic leukaemia (ALL) xenograft models [63], and enhanced the activity of chemotherapeutic agents in vivo [63]. However, ABT-263 also induced platelet apoptosis and thrombocytopenia in vivo [61, 63]. Nevertheless, a Phase 1 study showed Navitoclax to be well tolerated in patients with SCLC and other solid malignancies [64], although a Phase II study showed limited efficacy as a single agent [65]. Results from the Paediatric Preclinical Testing Programme, which tested the efficacy of ABT-263 in a panel of 23 cell lines and 44 xenograft models, showed that ABT-263 had activity in approximately 50% of cell lines, but only limited single-agent activity in solid tumour xenograft models [66]. Further studies therefore focused on the effect of ABT-263 administered concurrently with other agents such as carboplatin and paclitaxel, though only modest anti-tumour effects were observed [67]. Nevertheless, a current clinical trial is evaluating its efficacy in combination with the MEK inhibitor trametinib in adult patients with advanced solid tumours (NCT02079740). It is possible that the identification of biomarkers of ABT-263 response could be used to identify patients likely to respond to ABT-263 treatment.
Specific inhibitors of individual anti-apoptotic proteins have also been developed, such as the BCL-2 inhibitor ABT-199/Venetoclax [68], BCL-XL inhibitors A1331852 [69] and WEHI-539 [70], and MCL-1 inhibitors S63845 [71], AMG-176 [72] and AZD5991 [73] (Fig. 1). These inhibitors have potential utility in the treatment of both paediatric and adult cancers, in blood and solid malignancies.
BCL-2 proteins as therapeutic targets in paediatric tumours of the nervous system
With regards to paediatric tumours of the nervous system specifically, the anti-apoptotic BCL-2 protein family members are also potential therapeutic targets as their overexpression is commonly reported in these tumours, and correlates with clinical parameters such as therapeutic responsiveness and disease progression. BCL-2 expression in neuroblastoma [74–79] is associated with drug resistance, unfavourable histology and N-MYC amplification [74, 75], and a high-risk phenotype [76]. BCL-2 expression is also associated with poor outcome in paediatric glioblastoma [80] and ependymoma [81]. Interestingly, in medulloblastoma BCL-2 is the least frequently expressed of the anti-apoptotic BCL-2 proteins [82, 83], although expression is associated with poorly differentiated and highly proliferative tumour regions, and tends to correlate with poor outcome [84]. BCL-2 has also been identified as an important mediator of Hedgehog activity in the Sonic Hedgehog (SHH) subtype [85], and Gli1 and Gli2, essential transcriptional drivers of the SHH subtype of medulloblastoma, regulate BCL-2 transcription [86]. Indeed, mouse studies have shown that postnatal overexpression of BCL-2 promotes medulloblastoma tumour formation in cooperation with Sonic hedgehog pathway activation [87], highlighting a subtype-specific role for BCL-2 in medulloblastoma.
BCL-XL and MCL-1 are also of potential therapeutic interest. BCL-XL is the most frequently expressed anti-apoptotic BCL-2 protein in medulloblastoma tumours [83] and is expressed in the majority of medulloblastoma cell lines [88]. It is also expressed in most neuroblastoma cell lines [89], where its overexpression inhibits chemotherapy-induced apoptosis. Similarly, MCL-1 is also frequently expressed in medulloblastoma [~50% of tumours, [83]] and in neuroblastoma, where high expression is associated with a high-risk phenotype [76].
The identification of the anti-apoptotic BCL-2 proteins as therapeutic targets has led to the development of BH3 mimetics (Fig. 1), which have demonstrated efficacy in multiple cancer types in vitro as single agents or as sensitising agents. Early studies using the pan-BCL-2 inhibitor ABT-737 in neuroblastoma cell lines revealed a synergistic cytotoxicity with fenretinide, a synthetic retinoid derivative that inhibits cancer-cell line growth, while the combination also increased event-free survival in an orthotopic mouse model of neuroblastoma [90]. Subsequently, BCL-2 silencing in neuroblastoma cells was shown to induce apoptotic cell death [77] while ABT-199 also synergised with fenretinide in both neuroblastoma cell lines and PDX models expressing high BCL-2 levels, where improved event-free survival was demonstrated [91]. Mechanistically, neuroblastoma cell lines identified as BCL-2 dependent possess high levels of BCL-2 [92] and BIM:BCL-2 complexes, and ABT-199 mediates its effects by displacing BIM from BCL-2 [78, 79, 93], allowing mitochondrial apoptosis to proceed. MYCN-amplified neuroblastoma has been identified as particularly sensitive to ABT-199 treatment, and, in xenograft models, ABT-199 induced apoptosis and tumour regression in combination with the Aurora-A inhibitor MLN8237 [94]. In other xenograft models of BCL-2 dependent neuroblastoma [78], ABT-199 exhibited its effects through inhibition of tumour growth, rather than by inducing apoptosis. Results with ABT-263 [77] also suggested that ABT-263 mediates its effects in neuroblastoma through BCL-2 inhibition. Collectively, these studies identified a significant role for BCL-2 in mediating neuroblastoma resistance apoptosis, which could be overcome by utilising combinational treatment strategies.
Targeting MCL-1 or BCL-XL has also proven successful. Treatment with S63845 (MCL-1 inhibitor) or A1331852 (BCL-XL inhibitor) reduced viability and induced apoptosis in a subset of neuroblastoma cell lines, due to the release of pro-apoptotic proteins from BCL-XL- or MCL-1-mediated sequestration [92]. Furthermore, MCL-1 knockdown induced apoptosis and increased sensitivity to both etoposide and doxorubicin in cell line models of neuroblastoma [76], in a mechanism also understood to involve the release of BIM from MCL-1 sequestration [93]. The MCL-1 inhibitor A-1210477 is shown to synergise with the Hedgehog pathway inhibitor GANT61 in a subset of medulloblastoma cell lines [95]. BCL-XL targeting has additionally been demonstrated to sensitise medulloblastoma cells to MLN8237 treatment [96] as well as to ionising radiation [97], while synergy between ABT-263 and vincristine has also been attributed to its BCL-XL-targeting ability [98].
Combining BH3 mimetics has also emerged as a promising strategy to target BCL-2 protein-mediated resistance and enhance tumour cell killing, with early in vitro studies showing that MCL-1 knockdown overcomes ABT-737 resistance [76]. In some neuroblastoma mouse models, it was shown that ABT-199 treatment induced growth inhibition rather than apoptosis, due to MCL-1 upregulation and subsequent BIM sequestration by MCL-1 [78]. In vitro, MCL-1 inhibition sensitised neuroblastoma cells to ABT-199 treatment, highlighting the protective role MCL-1 mediates in cells with high BCL-2:BIM complex levels [78]. Furthermore, recent research demonstrated that dual inhibition of BCL-XL and MCL-1 synergistically reduces viability and induces death in neuroblastoma cell lines displaying treatment resistance to individual BCL-2, BCL-XL, or MCL-1 inhibitors [99]. Specifically, the efficacy of BCL-XL inhibition is limited by MCL-1, and the use of S63845 can therefore be used to abolish MCL-1-mediated resistance to A1331852 treatment by preventing MCL-1 binding pro-apoptotic the pro-apoptotic proteins displaced from BCL-XL by A1331852 [99]. The dual BCL-2/MCL-1 inhibitor TW-37 also successfully induces apoptosis in N-MYC-amplified neuroblastoma in vitro and in vivo [100], further emphasising that targeting multiple anti-apoptotic BCL-2 family members is necessary to induce apoptosis in some scenarios.
Clearly, the variable dependence of cancer cells on different anti-apoptotic proteins complicates the selection of the target protein. This is a particular concern in developing therapeutic combinations for paediatric patients, given that paediatric organs and tissues are more highly primed for apoptosis than the corresponding tissues of adults [101]. Fortunately, BH3 profiling represents a high-throughput experimental approach that can be used to elucidate dependencies of cancer-cell lines and patient tumour samples on individual anti-apoptotic proteins [102–104]. Studies in neuroblastoma, for instance, utilised BH3 profiling to predict responses to ABT-737 [105]. Alcon and colleagues have highlighted the potential of this approach in paediatric cancers [103], although further work is required to confirm the utility of BH3 profiling in the clinical paediatric cancer setting, and specifically in tumours of the paediatric nervous system.
Encouragingly, the lack of cancer-cell specificity of current BH3 mimetics is beginning to be addressed with the development of targeted inhibitors with a reduced toxicity profile. The dose-limiting platelet toxicity associated with BCL-XL inhibitors has led to the development of DT2216, a proteolysis-targeting chimera (PROTAC) that targets the BCL-XL protein for degradation via the Von Hippel-Lindau (VHL) (E3) ligase [106] in a cancer-cell specific manner. In vitro studies showed that DT2216 could sensitise prostate, TNBC, liver and colorectal cancer-cell lines to standard chemotherapeutic agents such as doxorubicin, docetaxel and vincristine [106], and this effect was also observed in a xenograft model of TNBC. Furthermore, DT2216 administered with ABT-199 completely suppressed tumour growth in a SCLC xenograft model [106], and also led to reduced disease burden and improved survival in a T-cell lymphoma PDX model [107]. DT2216 has entered a first-in-human clinical trial in patients with advanced/metastatic treatment-refractory solid or haematological malignancies (NCT04886622). A second compound selectively targeting BCL-XL in cancer is mirzotamab clezutoclax/ABBV-155 [108], an antibody-drug conjugate consisting of a BCL-XL inhibitor conjugated to a monoclonal anti-B7H3 antibody that targets the drug to cancer cells due to their selective B7H3 expression [109]. ABBV-155 is currently in dose-escalation studies in patients with relapsed or refractory solid tumours (NCT03595059). Trial results are eagerly awaited and potentially provide hope for the paediatric cancer community.
Targeting IAP signalling in paediatric cancers of the nervous system
The IAP family
The inhibitor of apoptosis (IAP) family of anti-apoptotic regulators prevent cell death by inhibiting caspases [110]. The IAP family has eight members, which are characterised by possession of at least one baculoviral IAP repeat (BIR) domain that mediates interactions with other proteins [111], as shown in Fig. 4. These are X-linked inhibitor of apoptosis protein (XIAP), cellular IAP 1 (cIAP1), cellular IAP 2 (cIAP2), IAP-like protein 2 (ILP-2), melanoma IAP (ML-IAP), neuronal apoptosis inhibitory protein (NAIP), Apollon and Survivin. XIAP is both the most potent and best studied member of the IAP family, and is the only member with the ability to directly inhibit activation of caspase 3, 7 and 9 [111]. The BIR2 domain has the ability to bind and inhibit caspase 3 and 7, while its BIR3 domain confers the ability to inhibit caspase 9 [112]. While cIAP1/2 and NAIP also possess all three BIR domains, they do not directly bind and inhibit caspases. cIAP1/2 indirectly inhibits caspase 3 and 7 by tagging them for proteasomal degradation, while NAIP constitutes part of the inflammasome, with roles in mediating innate immunity [113]. SMAC/DIABLO is an endogenous IAP antagonist which opposes the anti-apoptotic function of IAPs by interacting with and inhibiting their BIR2 and BIR3 domains via its N-terminal domain [114].
Fig. 4. The structure of IAP family members.
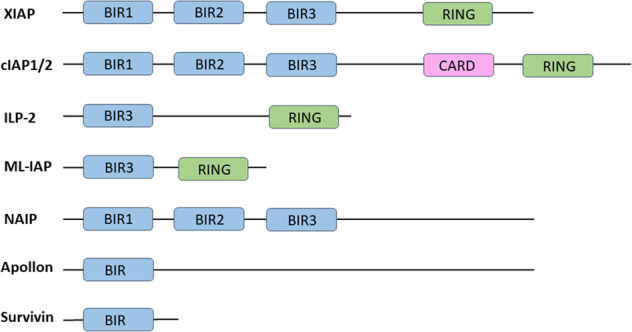
IAP members all have at least one baculoviral IAP repeat (BIR) domain that facilitates their interaction with other proteins. Some IAPs contain a Really Interesting New Gene (RING) domain that mediates E3 ligase activity. cIAP1/2 additionally contains a CAspase Recruitment Domain (CARD) which functions in protein-protein interactions.
The IAP family as therapeutic targets
IAP protein family members are dysregulated in both paediatric and adult malignancies. Medulloblastoma cell lines express higher IAP levels than normal astrocytes and brain tissue [115, 116], while XIAP is overexpressed in neuroblastoma cells compared with healthy adrenal gland tissue [117]. Additionally, cIAP1 and XIAP expression levels increase between primary and recurrent neuroblastoma in vitro, confirming their roles in aggressive recurrent disease [118]. Radiation and cisplatin treatment also upregulate protein and mRNA levels of cIAP1/2 and XIAP as a resistance mechanism [119].
IAPs, therefore, represent rational targets to directly sensitise cancer cells to apoptosis-inducing stimuli, and there are a diversity of pharmacological approaches capable of targeting IAPs. One potential mechanism is via SMAC mimetics which are based on the SMAC structure [120]. IAP suppression using SMAC mimetics effectively sensitises malignant cells to treatment with some classes of drugs in a cell line dependent manner [121]. For instance, treatment with the SMAC mimetic LCL161 activates both the extrinsic and intrinsic apoptosis pathways to overcome vincristine resistance in neuroblastoma cell lines [117, 118, 122]. Furthermore, LCL161 suppressed cell proliferation in medulloblastoma [116], and sensitised cells to both vincristine- and cisplatin-induced apoptosis with similar results observed with the IAP inhibitor, LBW242 [115, 116, 119]. Encouragingly, the combination of LBW242 and cisplatin was effective at inducing apoptosis in primary patient samples and a medulloblastoma xenograft model [119], as well as in CD133+ stem-like cancer cells [115] which are likely to be responsible for tumour initiation, maintenance and relapse [123]. Treatment with LCL161 was also shown to increase event-free survival in glioblastoma xenograft models [124]. While trials in xenograft models of paediatric cancers including neuroblastoma, medulloblastoma, ependymoma and glioblastoma have demonstrated limited activity of LCL161 as a single agent [124], combinatorial approaches may yet prove efficacious when tested in appropriate models.
Survivin is a member of the IAP family encoded by the BIRC5 gene, and is an apoptosis suppressor expressed primarily in embryonal development, and upregulated in cancer [125]. In cell line models of neuroblastoma, Survivin expression is linked to high proliferation rates and resistance to drug-induced death [126]. Similar associations have been made clinically, where its expression tends to be associated with poor prognosis and high-risk disease [126, 127]. For example, gain of 17q25 which contains the Survivin locus occurs frequently in neuroblastoma [127, 128]. Indeed, gain of 17q25 is especially common in advanced neuroblastoma and correlates with worse outcome [127]. Survivin overexpression is also common in medulloblastoma [129], correlating with poor prognosis, lower survival and recurrence [129–133], and is predictive of poor clinical outcome independent of clinical staging [129]. High expression has been linked to various negative clinical features in a variety of nervous system cancers. Specifically, in choroid plexus carcinoma and ependymoma, high expression levels have been associated with increased tumour grade [134] and cell proliferation and poor patient outcome [135], respectively, while high expression has also been identified in paediatric glioma [136]. Therefore, Survivin expression has potential as both a prognostic marker and therapeutic target.
A number of approaches have been utilised to elucidate the effects of targeting Survivin. siRNA-mediated Survivin depletion decreased proliferation and promoted cell cycle arrest via accumulation of cells in G2/M [137], while disruption of the chromosomal passenger complex containing Survivin resulted in mitotic catastrophe and apoptotic death [127]. The miRNA miR-542-3p post-transcriptionally inhibits Survivin expression, and its expression correlates with better prognosis in neuroblastoma [138]. Xenograft models of neuroblastoma treated with miR-542-3p-loaded nanoparticles also had reduced Survivin expression, decreased proliferation and increased apoptosis [138].
YM155, also known as sepantronium bromide, suppresses Survivin expression by downregulating transcriptional activity at the Survivin promoter [139] and inhibits proliferation and promotes apoptosis in medulloblastoma [140], glioma [141, 142] and neuroblastoma cell lines in vitro [143, 144]. In xenograft models of neuroblastoma YM155 increases apoptosis and tumour regression, both independently and in combination with cisplatin [143]. Interestingly, however, cell lines that are sensitive to Survivin knockdown do not necessarily respond to YM155 [145]. A large-scale study using >100 neuroblastoma cell lines identified that ABCB1 or P-glycoprotein expression determines resistance to YM155 by their efflux pump activity [144, 145]. Consistent with this, ABCB1 inhibition using shRNA, cyclosporine or lapatinib sensitised cells to YM155 [145, 146]. YM155 and lapatanib treatment also decreased tumour size in an in vivo neuroblastoma model [146]. Additional evidence highlights that the anti-tumour effects of YM155 are not solely mediated by its modulation of Survivin, but also via its transcriptional downregulation of MCL-1 [141, 147, 148] and induction of DNA damage [148, 149]. In this context, YM155 has been shown to synergise with both BCL-2 family inhibitors ABT-737 [142] and ABT-263 [147] to induce apoptosis via MCL-1 downregulation. Furthermore, the combination of YM155 and TRAIL synergistically induced caspase-dependent cell death via downregulation of both MCL-1 and Survivin [141], highlighting how YM155 may effectively target multiple resistance mechanisms within cancer cells. A number of trials are in progress to evaluate the clinical activity of YM155 in adult solid and blood cancers. Results of these trials in combination with the encouraging preclinical results above could potentially lead to clinical trials in paediatric patients. Finally, troglitazone, an anti-hyperglycemic and anti-inflammatory drug used in the management of type 2 diabetes, has been shown to reduce expression of both cFLIP and Survivin, resulting in TRAIL sensitisation in neuroblastoma cells [150]. As troglitazone is commonly used in the clinic, it would be of great interest to assess its effects as a sensitiser in combination with other approved chemotherapeutic agents in the paediatric setting.
Lastly, HDAC inhibitors including NaB, SAHA and TSA have been shown to increase the sensitivity of neuroblastoma cells to TRAIL-induced death, through downregulation of XIAP and Survivin [36]. Currently, four HDAC inhibitors, vorinostat, belinostat, panobinostat, and romidepsin, are approved by the FDA for cancer treatment, with several others under clinical investigation. Of note, two clinical trials are ongoing to examine the activity of vorinostat and valproic acid in paediatric high-grade glioma in combination with temozolomide (NCT03243461, NCT01236560). Results from these trials could pave the way for trials in other paediatric malignancies.
Conclusions
Tumours of the nervous system are among the most frequent malignancies in paediatric patients, and new treatment strategies are required to improve responsiveness and reduce the significant side effects associated with current treatment. Evasion of apoptosis is a Hallmark of cancer, with therapy resistance frequently associated with aberrant apoptosis signalling. A vast array of studies have characterised alterations in the apoptosis pathway that may contribute to defective apoptosis signalling, leading to the identification of druggable targets. The FDA approval of the BCL-2 inhibitor ABT-199/Venetoclax in 2016 was a critical development in the field of apoptosis-targeted therapy, highlighting the validity of this approach. The development of companion technologies such as BH3 profiling represents a means to personalise such therapeutic approaches.
Therapies targeting apoptosis have yet to translate to the paediatric setting, though many are under clinical trial investigation in adult patients. We here summarised the growing body of preclinical evidence that targeting the apoptosis pathway represents a promising strategy to mediate effective cancer-cell killing for the treatment of paediatric nervous system cancers. Current clinical trials in adults may open new avenues for more effective treatment of paediatric patients by identifying promising treatment combinations, while the development of specific cancer-targeting agents may further reduce the toxicity profile associated with current therapeutics and improve their potential clinical utility.
Acknowledgements
This research was funded by National Children’s Research Centre, Crumlin, grant number NCRC A/18/1. MCF kindly acknowledges support for her work from the Irish Association for Cancer Research (IACR Educational Grant).
Author contributions
Conceptualisation, MCF, PJOH, NMCC and BMM; literature search, MCF; writing—original draft preparation, MCF; writing—review and editing, MCF, PJOH, NMCC and BMM; supervision, NMCC and BMM; funding acquisition, PJO’H and BMM. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Edited by Dr. Cristina Munoz-Pinedo
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ward E, DeSantis C, Robbins A, Koheler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham RM, Walton MA, Carter PM. The major causes of death in children and adolescents in the United States. N. Engl J Med. 2018;379:2468–75. doi: 10.1056/NEJMsr1804754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol. 2017;18:719–31. doi: 10.1016/S1470-2045(17)30186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramanian S, Ahmad T. Cancer, childhood brain tumors. StatPearls. 2018. [PubMed]
- 5.Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(Supplement_1):iv1–iv96. doi: 10.1093/neuonc/noaa200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaderali Z, Lamberti-Pasculli M, Rutka JT. The changing epidemiology of paediatric brain tumours: a review from the hospital for sick children. Child’s Nerv Syst. 2009;25:787–93. doi: 10.1007/s00381-008-0771-9. [DOI] [PubMed] [Google Scholar]
- 7.Esiashvili N, Anderson C, Katzenstein HM. Neuroblastoma. Curr Probl Cancer. 2009;33:333–60. doi: 10.1016/j.currproblcancer.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Wolff JEA, Driever PH, Erdlenbruch B, Kortmann RD, Rutkowski S, Pietsch T, et al. Intensive chemotherapy improves survival in pediatric high-grade glioma after gross total resection: results of the HIT-gBM-c protocol. Cancer. 2010;116:705–12. doi: 10.1002/cncr.24730. [DOI] [PubMed] [Google Scholar]
- 9.Sun MZ, Oh MC, Ivan ME, Kaur G, Safaee M, Kim JM, et al. Current management of choroid plexus carcinomas. Vol. 37. Neurosurg Rev. 2014;37:179–92. doi: 10.1007/s10143-013-0499-1. [DOI] [PubMed] [Google Scholar]
- 10.Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Prim. 2019;5:11. doi: 10.1038/s41572-019-0063-6. [DOI] [PubMed] [Google Scholar]
- 11.White L, Johnston H, Jones R, Mameghan H, Nayanar V, McWhirter W, et al. Postoperative chemotherapy without radiation in young children with malignant non-astrocytic brain tumours - a report from the Australia and New Zealand Childhood Cancer Study Group (ANZCCSG) Cancer Chemother Pharmacol. 1993;32:403–6. doi: 10.1007/BF00735928. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 14.Johnsen JI, Kogner P, Albihn A, Henriksson MA. Embryonal neural tumours and cell death. Apoptosis. 2009;14:424–38. doi: 10.1007/s10495-009-0325-y. [DOI] [PubMed] [Google Scholar]
- 15.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 16.Gibert B, Mehlen P. Dependence receptors and cancer: addiction to trophic ligands. Cancer Res. 2015;75:5171–5. doi: 10.1158/0008-5472.CAN-14-3652. [DOI] [PubMed] [Google Scholar]
- 17.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 18.Oberst A, Pop C, Tremblay AG, Blais V, Denault JB, Salvesen GS, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285:16632–42. doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Semin Immunol. 2003;15:185–93. doi: 10.1016/S1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 20.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–87. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/S0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 22.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–40. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 23.Ralff MD, El-Deiry WS. TRAIL pathway targeting therapeutics. Expert Rev Precis Med Drug Dev. 2018;3:197–204. doi: 10.1080/23808993.2018.1476062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutt M, Marquardt L, Seifert O, Siegemund M, Müller I, Kulms D, et al. Superior properties of Fc-comprising scTRAIL fusion proteins. Mol Cancer Ther. 2017;16:2792–2802. doi: 10.1158/1535-7163.MCT-17-0551. [DOI] [PubMed] [Google Scholar]
- 25.Ratain MJ, Doi T, De Jonge MJ, LoRusso P, Dunbar M, Chiney M, et al. Phase 1, first-in-human study of TRAIL receptor agonist fusion protein ABBV-621. J Clin Oncol. 2019;37(15_suppl):3013. doi: 10.1200/JCO.2019.37.15_suppl.3013. [DOI] [Google Scholar]
- 26.Deng D, Shah K. TRAIL of hope meeting resistance in cancer. Trends Cancer. 2020;6:989–1001. doi: 10.1016/j.trecan.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong HX, Lu CW, Wang QS, Ma LY. Combination of IFNγ and chemotherapeutic agents increase trail sensitivity of neuroblastoma cell lines. Eur J Pediatr Surg. 2011;21:304–9. doi: 10.1055/s-0031-1279762. [DOI] [PubMed] [Google Scholar]
- 28.Kim HR, Lee MW, Kim DS, Jo HY, Lee SH, Chueh HW, et al. Etoposide sensitizes neuroblastoma cells expressing caspase 8 to TRAIL. Cell Biol Int Rep. 2012;19:e00017. doi: 10.1042/CBR20110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuzak TJ, Steinhoff DF, Sutton LN, Phillips PC, Eggert A, Grotzer MA. Loss of caspase-8 mRNA expression is common in childhood primitive neuroectodermal brain tumour/medulloblastoma. Eur J Cancer. 2002;38:83–91. doi: 10.1016/S0959-8049(01)00355-0. [DOI] [PubMed] [Google Scholar]
- 30.Aguilera DG, Das CM, Sinnappah-Kang ND, Joyce C, Taylor PH, Wen S, et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J Neurooncol. 2009;93:303–18. doi: 10.1007/s11060-008-9788-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naumann I, Kappler R, Von Schweinitz D, Debatin KM, Fulda S. Bortezomib primes neuroblastoma cells for TRAIL-induced apoptosis by linking the death receptor to the mitochondrial pathway. Clin Cancer Res. 2011;17:3204–18. doi: 10.1158/1078-0432.CCR-10-2451. [DOI] [PubMed] [Google Scholar]
- 32.Koschny R, Boehm C, Sprick MR, Haas TL, Holland H, Xu LX, et al. Bortezomib sensitizes primary meningioma cells to TRAIL-induced apoptosis by enhancing formation of the death-inducing signaling complex. J Neuropathol Exp Neurol. 2014;73:1034–46. doi: 10.1097/NEN.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 33.Huehnchen P, Springer A, Kern J, Kopp U, Kohler S, Alexander T, et al. Bortezomib at therapeutic doses poorly passes the blood–brain barrier and does not impair cognition. Brain Commun. 2020;2:fcaa021. doi: 10.1093/braincomms/fcaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, Shalaby T, et al. Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin Cancer Res. 2003;9:6401–9. [PubMed] [Google Scholar]
- 35.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. 2001;79:428–36. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 36.Mühlethaler-Mottet A, Flahaut M, Bourloud KB, Auderset K, Meier R, Joseph JM, et al. Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL-induced apoptosis by a caspases-dependent increase of the pro- to anti-apoptotic proteins ratio. BMC Cancer. 2006;6. [DOI] [PMC free article] [PubMed]
- 37.Grotzer MA, Eggert A, Zuzak TJ, Janss AJ, Marwaha S, Wiewrodt BR, et al. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene. 2000;19:4604–10. doi: 10.1038/sj.onc.1203816. [DOI] [PubMed] [Google Scholar]
- 38.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, et al. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced Apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–19. [PubMed] [Google Scholar]
- 39.Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002;62:5897–5901. [PubMed] [Google Scholar]
- 40.Hopkins-Donaldson, et al. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–9. [PubMed] [Google Scholar]
- 41.Mühlethaler-Mottet A, Balmas K, Auderset K, Joseph JM, Gross N. Restoration of TRAIL-induced apoptosis in a caspase-8-deficient neuroblastoma cell line by stable re-expression of caspase-8. Ann N. Y Acad Sci. 2003;1010:195–9. doi: 10.1196/annals.1299.033. [DOI] [PubMed] [Google Scholar]
- 42.Fulda S, Debatin KM. 5-Aza-2′-deoxycytidine and IFN-γ cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene. 2006;25:5125–33. doi: 10.1038/sj.onc.1209518. [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Merchant MS, Romero ME, Tsokos M, Wexler LH, Kontny U, et al. Induction of caspase 8 by interferon γ renders some neuroblastoma (NB) cells sensitive to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but reveals that a lack of membrane TR1/TR2 also contributes to TRAIL resistance in NB. Cancer Res. 2003;63:1122–9. [PubMed] [Google Scholar]
- 44.Kim S, Kang J, Evers BM, Chung DH. Interferon-γ induces caspase-8 in neuroblastomas without affecting methylation of caspase-8 promoter. J Pediatr Surg. 2004;39:509–15. doi: 10.1016/j.jpedsurg.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Johnsen JI, Pettersen I, Ponthan F, Sveinbjørnsson B, Flaegstad T, Kogner P. Synergistic induction of apoptosis in neuroblastoma cells using a combination of cytostatic drugs with interferon-gamma and TRAIL. Int J Oncol. 2004;25:1849–57. [PubMed] [Google Scholar]
- 46.Tong H, Lu C, Yang Y, Zhang J, Zhang J. Role of caspase 8 as a determinant in trail sensitivity of neuroblastoma cell lines. Pediatr Hematol Oncol. 2009;26:549–59. doi: 10.3109/08880010903271663. [DOI] [PubMed] [Google Scholar]
- 47.Casciano I, De Ambrosis A, Croce M, Pagnan G, Di Vinci A, Allemanni G, et al. Expression of the caspase-8 gene in neuroblastoma cells is regulated through an essential interferon-sensitive response element (ISRE) Cell Death Differ. 2004;11:131–4. doi: 10.1038/sj.cdd.4401327. [DOI] [PubMed] [Google Scholar]
- 48.De Ambrosis A, Casciano I, Croce M, Pagnan G, Radic L, Banelli B, et al. An interferon-sensitive response element is involved in constitutive caspase-8 gene expression in neuroblastoma cells. Int J Cancer. 2007;120:39–47. doi: 10.1002/ijc.22173. [DOI] [PubMed] [Google Scholar]
- 49.Meister N, Shalaby T, von Bueren AO, Rivera P, Patti R, Oehler C, et al. Interferon-γ mediated up-regulation of caspase-8 sensitises medulloblastoma cells to radio- and chemotherapy. Eur J Cancer. 2007;43:1833–41. doi: 10.1016/j.ejca.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 50.Chabot GG, Rivard GE, Momparler RL. Plasma and cerebrospinal fluid pharmacokinetics of 5-aza-2‘-deoxycytidine in rabbits and dogs. Cancer Res. 1983;43:592–7. [PubMed] [Google Scholar]
- 51.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/S1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 52.Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their α6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 53.Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–76. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/S0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 55.Ke F, Voss A, Kerr JB, O’Reilly LA, Tai L, Echeverry N, et al. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 2012;19:915–25. doi: 10.1038/cdd.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell. 1993;74:609–19. doi: 10.1016/0092-8674(93)90509-O. [DOI] [PubMed] [Google Scholar]
- 57.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl- 2 regulation of apoptosis. Science. 1997;275:1132–6. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 58.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 60.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 62.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–74. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 63.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 64.Gandhi L, Camidge DR, De Oliveira MR, Bonomi P, Gandara D, Khaira D, et al. Phase I study of navitoclax (ABT-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudin CM, Hann CL, Garon EB, Ribeiro De Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–9. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lock R, Carol H, Houghton PJ, Morton CL, Anders Kolb E, Gorlick R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1181–9. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- 67.Vlahovic G, Karantza V, Wang D, Cosgrove D, Rudersdorf N, Yang J, et al. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest N. Drugs. 2014;32:976–84. doi: 10.1007/s10637-014-0116-3. [DOI] [PubMed] [Google Scholar]
- 68.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 69.Shoemaker AR, Oleksijew A, Bauch J, Belli BA, Borre T, Bruncko M, et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006;66:8731–9. doi: 10.1158/0008-5472.CAN-06-0367. [DOI] [PubMed] [Google Scholar]
- 70.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al. Structure-guided design of a selective BCL-XL inhibitor. Nat Chem Biol. 2013;9:390–7. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 71.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 72.Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8:1582–97. doi: 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- 73.Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9:5341. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nunez G. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol. 1993;143:1543–50. [PMC free article] [PubMed] [Google Scholar]
- 75.Merchant AK, Maybaum J, Rode CK, Bloch CA, Castle VP. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994;54:3253–9. [PubMed] [Google Scholar]
- 76.Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8:1587–95. doi: 10.4161/cbt.8.16.8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lamers F, Schild L, Den Hartog IJM, Ebus ME, Westerhout EM, Ora I, et al. Targeted BCL2 inhibition effectively inhibits neuroblastoma tumour growth. Eur J Cancer. 2012;48:3093–103. doi: 10.1016/j.ejca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 78.Bate-Eya LT, den Hartog IJM, van der Ploeg I, Schild L, Koster J, Santo EE, et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget. 2016;7:27946–58. doi: 10.18632/oncotarget.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanos R, Karmali D, Nalluri S, Goldsmith KC. Select Bcl-2 antagonism restores chemotherapy sensitivity in high-risk neuroblastoma. BMC Cancer. 2016;16. [DOI] [PMC free article] [PubMed]
- 80.Ganigi PM, Santosh V, Anandh B, Chandramouli BA, Sastry Kolluri VR. Expression of p53, EGFR, pRb and bcl-2 proteins in pediatric glioblastoma multiforme: a study of 54 patients. Pediatr Neurosurg. 2005;41:292–9. doi: 10.1159/000088731. [DOI] [PubMed] [Google Scholar]
- 81.Zamecnik J, Snuderl M, Eckschlager T, Chanova M, Hladikova M, Tichy M, et al. Pediatric intracranial ependymomas: prognostic relevance of histological, immunohistochemical, and flow cytometric factors. Mod Pathol. 2003;16:980–91. doi: 10.1097/01.MP.0000087420.34166.B6. [DOI] [PubMed] [Google Scholar]
- 82.Reed JC, Meister L, Tanaka S, Cuddy M, Yum S, Geyer C, et al. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991;51:6529–38. [PubMed] [Google Scholar]
- 83.Krajewski S, Krajewska M, Ehrmann J, Sikorska M, Lach B, Chatten J, et al. Immunohistochemical analysis of Bcl-2, Bcl-X, Mcl-1, and Bax in tumors of central and peripheral nervous system origin. Am J Pathol. 1997;150:805–14. [PMC free article] [PubMed] [Google Scholar]
- 84.Schüller U, Schober F, Kretzschmar HA, Herms J. Bcl-2 expression inversely correlates with tumour cell differentiation in medulloblastoma. Neuropathol Appl Neurobiol. 2004;30:513–21. doi: 10.1111/j.1365-2990.2004.00553.x. [DOI] [PubMed] [Google Scholar]
- 85.Bar EE, Chaudhry A, Farah MH, Eberhart CG. Hedgehog signaling promotes medulloblastoma survival via BclII. Am J Pathol. 2007;170:347–55. doi: 10.2353/ajpath.2007.060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bigelow RLH, Chari NS, Undén AB, Spurgers KB, Lee S, Roop DR, et al. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J Biol Chem. 2004;279:1197–205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- 87.McCall TD, Pedone CA, Fults DW. Apoptosis suppression by somatic cell transfer of Bcl-2 promotes Sonic hedgehog-dependent medulloblastoma formation in mice. Cancer Res. 2007;67:5179–85. doi: 10.1158/0008-5472.CAN-06-4177. [DOI] [PubMed] [Google Scholar]
- 88.Othman RT, Kimishi I, Bradshaw TD, Storer LCD, Korshunov A, Pfister SM, et al. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol Commun. 2014;2. [DOI] [PMC free article] [PubMed]
- 89.Dole MG, Jasty R, Cooper MJ, Thompson CB, Nuñez G, Castle VP. Bcl-xL Is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 1995;55:2576–82. [PubMed] [Google Scholar]
- 90.Fang H, Harned TM, Kalous O, Maldonado V, DeClerck YA, Reynolds CP. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin Cancer Res. 2011;17:7093–104. doi: 10.1158/1078-0432.CCR-11-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nguyen TH, Koneru B, Wei SJ, Chen WH, Makena MR, Urias E, et al. Fenretinide via NOXA induction, enhanced activity of the BCL-2 inhibitor venetoclax in high BCL-2-expressing neuroblastoma preclinical models. Mol Cancer Ther. 2019;18:2270–82. doi: 10.1158/1535-7163.MCT-19-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bierbrauer A, Jacob M, Vogler M, Fulda S. A direct comparison of selective BH3-mimetics reveals BCL-XL, BCL-2 and MCL-1 as promising therapeutic targets in neuroblastoma. Br J Cancer. 2020;122:1544–51. doi: 10.1038/s41416-020-0795-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goldsmith KC, Gross M, Peirce S, Luyindula D, Liu X, Vu A, et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012;72:2565–77. doi: 10.1158/0008-5472.CAN-11-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, et al. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell. 2016;29:159–72. doi: 10.1016/j.ccell.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meister MT, Boedicker C, Linder B, Kögel D, Klingebiel T, Fulda S. Concomitant targeting of Hedgehog signaling and MCL-1 synergistically induces cell death in Hedgehog-driven cancer cells. Cancer Lett. 2019;465:1–11. doi: 10.1016/j.canlet.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 96.Levesley J, Steele L, Brüning-Richardson A, Davison A, Zhou J, Lawler S, et al. P11.15 Selective inhibition of BCL-XL increases sensitivity of pediatric tumour models to the anti-mitotic agent MLN8237. Neuro Oncol. 2017;19(suppl_3).
- 97.Kelley KD, Powell C, Chakraborty S, Wright AC, Al Dimassi SB, Symons M, et al. Bcl-XL as a Radiosensitizer in the Treatment of Group C Medulloblastoma. Int J Radiat Oncol. 2016;20:203–14. [Google Scholar]
- 98.Westhoff M-A, Schuler-Ortoli M, Zerrinius D, Hadzalic A, Schuster A, Strobel H, et al. Bcl-XL but not Bcl-2 is a potential target in medulloblastoma therapy. Pharmaceuticals. 2022;15:91. doi: 10.3390/ph15010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kehr S, Haydn T, Bierbrauer A, Irmer B, Vogler M, Fulda S. Targeting BCL-2 proteins in pediatric cancer: dual inhibition of BCL-XL and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 2020;482:19–32. doi: 10.1016/j.canlet.2020.02.041. [DOI] [PubMed] [Google Scholar]
- 100.Klenke S, Akdeli N, Stelmach P, Heukamp L, Schulte JH, Bachmann HS. The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines. BMC Cancer. 2019;19:243. doi: 10.1186/s12885-019-5439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2017;31:142–56. doi: 10.1016/j.ccell.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25:56–64. doi: 10.1038/cdd.2017.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alcon C, Manzano-Muñoz A, Prada E, Mora J, Soriano A, Guillén G, et al. Sequential combinations of chemotherapeutic agents with BH3 mimetics to treat rhabdomyosarcoma and avoid resistance. Cell Death Dis. 2020;11:634. doi: 10.1038/s41419-020-02887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glavey SV, Flanagan L, Bleach R, Kelly C, Quinn J, Ní Chonghaile T, et al. Secondary plasma cell leukaemia treated with single agent venetoclax. Br J Haematol. 2020;190:e242–e245. doi: 10.1111/bjh.16858. [DOI] [PubMed] [Google Scholar]
- 105.Goldsmith KC, Lestini BJ, Gross M, Ip L, Bhumbla A, Zhang X, et al. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ. 2010;17:872–82. doi: 10.1038/cdd.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938–47. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He Y, Koch R, Budamagunta V, Zhang P, Zhang X, Khan S, et al. DT2216 - a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J Hematol Oncol. 2020;13:95. doi: 10.1186/s13045-020-00928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tolcher AW, Carneiro BA, Dowlati A, Abdul Razak AR, Chae YK, Villella JA, et al. A first-in-human study of mirzotamab clezutoclax as monotherapy and in combination with taxane therapy in relapsed/refractory solid tumors: dose escalation results. J Clin Oncol. 2021;39(15_suppl):3015. doi: 10.1200/JCO.2021.39.15_suppl.3015. [DOI] [Google Scholar]
- 109.Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci. 2020;16:1767–73. doi: 10.7150/ijbs.41105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/S0092-8674(00)00009-X. [DOI] [PubMed] [Google Scholar]
- 111.Deveraux QL, Reed JC. IAP family proteins - suppressors of apoptosis. Genes Dev. 1999;13:239–52. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 112.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–51. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Abadía-Molina F, Morón-Calvente V, Baird SD, Shamim F, Martín F, MacKenzie A. Neuronal apoptosis inhibitory protein (NAIP) localizes to the cytokinetic machinery during cell division. Sci Rep. 2017;7:39981. doi: 10.1038/srep39981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao XY, Wang XY, Wei QY, Xu YM, Lau ATY. Potency and selectivity of SMAC/DIABLO mimetics in solid tumor therapy. Cells. 2020;9:1012. doi: 10.3390/cells9041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen SM, Li YY, Tu CH, Salazar N, Tseng YY, Huang SF, et al. Blockade of inhibitors of apoptosis proteins in combination with conventional chemotherapy leads to synergistic antitumor activity in medulloblastoma and cancer stem-like cells. PLoS One. 2016;11:e0161299. doi: 10.1371/journal.pone.0161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen SM, Lin TK, Tseng YY, Tu CH, Lui TN, Huang SF, et al. Targeting inhibitors of apoptosis proteins suppresses medulloblastoma cell proliferation via G2/M phase arrest and attenuated neddylation of p21. Cancer Med. 2018;7:3988–4003. doi: 10.1002/cam4.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Eschenburg G, Eggert A, Schramm A, Lode HN, Hundsdoerfer P. Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-α-independent apoptosis. Cancer Res. 2012;72:2645–56. doi: 10.1158/0008-5472.CAN-11-4072. [DOI] [PubMed] [Google Scholar]
- 118.Frommann K, Appl B, Hundsdoerfer P, Reinshagen K, Eschenburg G. Vincristine resistance in relapsed neuroblastoma can be efficiently overcome by Smac mimetic LCL161 treatment. J Pediatr Surg. 2018;53:2059–64. doi: 10.1016/j.jpedsurg.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 119.Keating J, Tsoli M, Hallahan AR, Ingram WJ, Haber M, Ziegler DS. Targeting the inhibitor of apoptosis proteins as a novel therapeutic strategy in medulloblastoma. Mol Cancer Ther. 2012;11:2654–63. doi: 10.1158/1535-7163.MCT-12-0352. [DOI] [PubMed] [Google Scholar]
- 120.Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharm Ther. 2014;144:82–95. doi: 10.1016/j.pharmthera.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Najem S, Langemann D, Appl B, Trochimiuk M, Hundsdoerfer P, Reinshagen K, et al. Smac mimetic LCL161 supports neuroblastoma chemotherapy in a drug class-dependent manner and synergistically interacts with ALK inhibitor TAE684 in cells with ALK mutation F1174L. Oncotarget. 2016;7:72634–53. doi: 10.18632/oncotarget.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Langemann D, Trochimiuk M, Appl B, Hundsdoerfer P, Reinshagen K, Eschenburg G. Sensitization of neuroblastoma for vincristine-induced apoptosis by Smac mimetic LCL161 is attended by G2 cell cycle arrest but is independent of NFκB, RIP1 and TNF-α. Oncotarget. 2017;8:87763–72. doi: 10.18632/oncotarget.21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang GH, Xu QF, Cui YH, Li N, Bian XW, Lv SQ. Medulloblastoma stem cells: promising targets in medulloblastoma therapy. Cancer Sci. 2016;107:583–9. doi: 10.1111/cas.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Houghton PJ, Kang MH, Reynolds CP, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58:636–9. doi: 10.1002/pbc.23167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: a unique target for tumor therapy. Cancer Cell Int. 2016;23:49. doi: 10.1186/s12935-016-0326-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Azuhata T, Scott D, Takamizawa S, Wen J, Davidoff A, Fukuzawa M, et al. The inhibitor of apoptosis protein survivin is associated with high-risk behavior of neuroblastoma. J Pediatr Surg. 2001;36:1785–91. doi: 10.1053/jpsu.2001.28839. [DOI] [PubMed] [Google Scholar]
- 127.Lamers F, Van Der Ploeg I, Schild L, Ebus ME, Koster J, Hansen BR, et al. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocr Relat Cancer. 2011;18:657–68. doi: 10.1530/ERC-11-0207. [DOI] [PubMed] [Google Scholar]
- 128.Islam A, Kageyama H, Takada N, Kawamato T, Takayasu H, Isogai E, et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–23. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 129.Li XN, Shu Q, Su JM, Adesina AM, Wong KK, Perlaky L, et al. Differential expression of survivin splice isoforms in medulloblastomas. Neuropathol Appl Neurobiol. 2007;33:67–76. doi: 10.1111/j.1365-2990.2006.00782.x. [DOI] [PubMed] [Google Scholar]
- 130.Pizem J, Cör A, Zadravec-Zaletel L, Popovic M. Survivin is a negative prognostic marker in medulloblastoma. Neuropathol Appl Neurobiol. 2005;31:422–8. doi: 10.1111/j.1365-2990.2005.00664.x. [DOI] [PubMed] [Google Scholar]
- 131.Fangusaro JR, Jiang Y, Holloway MP, Caldas H, Singh V, Boué DR, et al. Survivin, survivin-2B, and survivin-deltaEx3 expression in medulloblastoma: biologic markers of tumour morphology and clinical outcome. Br J Cancer. 2005;92:359–65. doi: 10.1038/sj.bjc.6602317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Haberler C, Slavc I, Czech T, Gelpi E, Heinzl H, Budka H, et al. Histopathological prognostic factors in medulloblastoma: high expression of survivin is related to unfavourable outcome. Eur J Cancer. 2006;42:2996–3003. doi: 10.1016/j.ejca.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 133.Abdel-Aziz A, Mohamed MAA, Akl FMF, Taha ANM. Survivin expression in medulloblastoma: a possible marker for survival. Pathol Oncol Res. 2013;19:413–9. doi: 10.1007/s12253-012-9594-9. [DOI] [PubMed] [Google Scholar]
- 134.Altura RA, Olshefski RS, Jiang Y, Boué DR. Nuclear expression of Survivin in paediatric ependymomas and choroid plexus tumours correlates with morphologic tumour grade. Br J Cancer. 2003;89:1743–9. doi: 10.1038/sj.bjc.6601334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Preusser M, Wolfsberger S, Czech T, Slavc I, Budka H, Hainfellner JA. Survivin expression in intracranial ependymomas and its correlation with tumor cell proliferation and patient outcome. Am J Clin Pathol. 2005;124:543–9. doi: 10.1309/PP2G5GAAFKV82DTG. [DOI] [PubMed] [Google Scholar]
- 136.Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88:245–50. doi: 10.1007/s11060-008-9566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chandele A, Prasad V, Jagtap JC, Shukla R, Shastry PR. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia. 2004;6:29–40. doi: 10.1016/S1476-5586(04)80051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Althoff K, Lindner S, Odersky A, Mestdagh P, Beckers A, Karczewski S, et al. miR-542-3p exerts tumor suppressive functions in neuroblastoma by downregulating survivin. Int J Cancer. 2015;136:1308–20. doi: 10.1002/ijc.29091. [DOI] [PubMed] [Google Scholar]
- 139.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, et al. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int J Biochem Mol Biol. 2012;3:179–97. [PMC free article] [PubMed] [Google Scholar]
- 140.Brun SN, Markant SL, Esparza LA, Garcia G, Terry D, Huang JM, et al. Survivin as a therapeutic target in Sonic hedgehog-driven medulloblastoma. Oncogene. 2015;34:3770–9. doi: 10.1038/onc.2014.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guo H, Wang Y, Song T, Xin T, Zheng Z, Zhong P, et al. Silencing of survivin using YM155 inhibits invasion and suppresses proliferation in glioma cells. Cell Biochem Biophys. 2015;71:587–93. doi: 10.1007/s12013-014-0238-4. [DOI] [PubMed] [Google Scholar]
- 142.Woo SM, Min KJ, Seo BR, Seo YH, Jeong YJ, Kwon TK. YM155 enhances ABT-737-mediated apoptosis through Mcl-1 downregulation in Mcl-1-overexpressed cancer cells. Mol Cell Biochem. 2017;429:91–102. doi: 10.1007/s11010-016-2938-0. [DOI] [PubMed] [Google Scholar]
- 143.Liang H, Zhang L, Xu R, Ju XL. Silencing of survivin using YM155 induces apoptosis and chemosensitization in neuroblastomas cells. Eur Rev Med Pharm Sci. 2013;17:2909–15. [PubMed] [Google Scholar]
- 144.Michaelis M, Voges Y, Rothweiler F, Weipert F, Zia‐ahmad A, Cinatl J, et al. Testing of the survivin suppressant YM155 in a large panel of drug‐resistant neuroblastoma cell lines. Cancers (Basel) 2020;12:577. doi: 10.3390/cancers12030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lamers F, Schild L, Koster J, Versteeg R, Caron HN, Molenaar JJ. Targeted BIRC5 silencing using YM155 causes cell death in neuroblastoma cells with low ABCB1 expression. Eur J Cancer. 2012;48:763–71. doi: 10.1016/j.ejca.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 146.Radic-Sarikas B, Halasz M, Huber KVM, Winter GE, Tsafou KP, Papamarkou T, et al. Lapatinib potentiates cytotoxicity of YM155 in neuroblastoma via inhibition of the ABCB1 efflux transporter. Sci Rep. 2017;7:3091. doi: 10.1038/s41598-017-03129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang H, Shao H, Yu C, Hou J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochem Pharmacol. 2011;82:1066–72. doi: 10.1016/j.bcp.2011.07.064. [DOI] [PubMed] [Google Scholar]
- 148.Voges Y, Michaelis M, Rothweiler F, Schaller T, Schneider C, Politt K, et al. Effects of YM155 on survivin levels and viability in neuroblastoma cells with acquired drug resistance. Cell Death Dis. 2016;7:e2410. doi: 10.1038/cddis.2016.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wani TH, Surendran S, Jana A, Chakrabarty A, Chowdhury G. Quinone-based antitumor agent sepantronium bromide (YM155) causes oxygen-independent redox-activated oxidative DNA damage. Chem Res Toxicol. 2018;31:12–618. doi: 10.1021/acs.chemrestox.8b00094. [DOI] [PubMed] [Google Scholar]
- 150.Schultze K, Böck B, Eckert A, Oevermann L, Ramacher D, Wiestler O, et al. Troglitazone sensitizes tumor cells to TRAIL-induced apoptosis via down-regulation of FLIP and survivin. Apoptosis. 2006;11:1503–12. doi: 10.1007/s10495-006-8896-3. [DOI] [PubMed] [Google Scholar]