

Abstract
Sarcomas are difficult to treat and the therapy, even when effective, is associated with long-term and life-threatening side effects. In addition, the treatment regimens for many sarcomas, including Ewing sarcoma, rhabdomyosarcoma, and osteosarcoma, are relatively unchanged over the past two decades, indicating a critical lack of progress. Although differentiation-based therapies are used for the treatment of some cancers, the application of this approach to sarcomas has proven challenging. Here, using a CRISPR-mediated gene knockout approach, we show that Inhibitor of DNA Binding 2 (ID2) is a critical regulator of developmental-related genes and tumor growth in vitro and in vivo in Ewing sarcoma tumors. We also identified that homoharringtonine, which is an inhibitor of protein translation and FDA-approved for the treatment of leukemia, decreases the level of the ID2 protein and significantly reduces tumor growth and prolongs mouse survival in an Ewing sarcoma xenograft model. Furthermore, in addition to targeting ID2, homoharringtonine also reduces the protein levels of ID1 and ID3, which are additional members of the ID family of proteins with well-described roles in tumorigenesis, in multiple types of cancer. Overall, these results provide insight into developmental regulation in Ewing sarcoma tumors and identify a novel, therapeutic approach to target the ID family of proteins using an FDA-approved drug.
INTRODUCTION
Ewing sarcoma is an aggressive bone and soft tissue sarcoma (1). Although the initiating oncogene in Ewing sarcoma tumors, EWS-FLI, was identified over two decade ago, the cell-of-origin and the cell lineage that gives rise to Ewing sarcoma tumors has been a source of debate (2–6). EWS-FLI1 functions, in part, as an aberrant transcription factor and this oncoprotein is reported to regulate, via direct and indirect mechanisms, multiple genes and pathways related to cell lineage, differentiation, and development (5,7–14). Consequently, the phenotype of Ewing sarcoma tumors likely depends on both intrinsic effects of the EWS-FLI1 oncogene and contributions from the cell- or lineage-of-origin. For example, expression of EWS-FLI1 in murine marrow stromal cells inhibits adipogenic and osteogenic differentiation (8,10). Similarly, EWS-FLI1 blocks the myogenic differentiation of myoblasts and upregulates the expression of neural crest genes (14). However, despite the well-described role of EWS-FLI1 in regulating differentiation and developmental pathways, the critical mediators downstream of EWS-FLI1 are unknown. Furthermore, an incomplete understanding of the proteins and pathways that regulate the differentiation and lineage commitment of Ewing sarcoma tumors has restricted efforts to identify differentiation-based therapies for the treatment of Ewing sarcoma, like the approaches used to treat acute promyelocytic leukemia and neuroblastoma (15).
We previously developed a genetically-defined and isogenic model of Ewing sarcoma using human embryonic stem cells, the inducible expression of EWS-FLI1, and an in vitro differentiation approach (16). In the current study, we used gene expression data from this model system to identify that Inhibitor of DNA Binding 2 (ID2) (previously known as Inhibitor of Differentiation 2), which is a well-described regulator of differentiation and developmental pathways, is significantly upregulated in cells expressing EWS-FLI relative to isogenic, differentiated fibroblasts (17,18). Ewing sarcoma cell lines and primary tumors also express high levels of ID2 and EWS-FLI1 is reported to bind to the ID2 gene promoter and up-regulate the transcription of ID2 (19–23).
The ID family of proteins (ID1–4), which bind to basic helix-loop-helix (bHLH) transcription factors and prevent bHLH-directed transcription, are critical regulators of the differentiation and chemoresistance of cancer cells derived from multiple cellular lineages (17,18,24–41). In particular, ID2 was previously shown to impair the in vitro differentiation of human mesenchymal stem cells (42). However, the functional role, if any, of ID2 in regulating differentiation, developmental pathways, and the oncogenic phenotype of Ewing sarcoma tumors is unknown. In addition, although a number of different strategies have been used to interfere with the function of ID proteins in the laboratory, including the use of targeted antisense delivery and deubiquitinase inhibitors, these approaches have not yet advanced to the clinic (17,43).
In the current study, we used CRISPR/Cas9 to knockout ID2 in Ewing sarcoma cell lines and identified that the loss of ID2 significantly decreases cell growth in vitro and in vivo in a xenograft experiment. Conversely, the overexpression of ID2 in NIH3T3 cells, which are mouse embryonic fibroblasts used in transformation assays to test sarcoma oncogenes, increases growth in vitro and vivo (44). Using RNA-seq and gene set enrichment analysis we found that ID2 regulates genes related to differentiation and development. Next, to address the current challenges of targeting ID proteins in the clinic, we identified that homoharringtonine (HHT), an inhibitor of protein translation that is FDA-approved for the treatment of chronic myeloid leukemia, decreases the level of the ID2 protein in vitro and in vivo in sarcoma cell lines (45). Furthermore, we show that HHT reduces tumor growth and significantly prolongs mouse survival in a xenograft experiment. HHT also decreases the levels of the ID1 and ID3 proteins, in addition to ID2, in cancer cell lines and inhibits the in vitro proliferation of multiple sarcoma subtypes. Overall, this work identifies a critical regulator of growth and differentiation in Ewing sarcoma tumors and develops a novel therapeutic approach for targeting ID proteins in multiple cancer types.
MATERIALS AND METHODS
Cell lines and culture:
The A673, TC32, TC71, SKNEP, and EW8 cell lines were provided by Dr. Kimberly Stegmaier (Dana-Farber Cancer Institute, Boston, MA). The HEK-293T, HT1080, and U2OS cell lines were obtained from ATCC. The RH30, RD, NIH3T3 cell lines were provided by Dr. Munir Tanas (University of Iowa, Iowa City, IA). The S462 cell line was provided by Dr. Dawn Quelle (University of Iowa, Iowa City, IA). The MIA PaCa and PANC-1 cell lines were provided by Dr. Garry Buettner (University of Iowa, Iowa City, IA). The ES1 and ES6 cell lines were kindly provided by the St. Jude Childhood Solid Tumor Network. The AGPN, E352, CHLA-9, and CHLA-10 cell lines were provided by the Childhood Cancer Repository (Children’s Oncology Group). The Jurkat, K562, and Nalm6 cell lines were obtained from ATCC and DSMZ. The cell lines were cultured as previously described (47, 48). Cell lines were maintained at 37° C in a 5% CO2 atmosphere. Cell lines were authenticated by DNA fingerprinting using the short tandem repeat (STR) method and used within 8–10 passages of thawing.
Chemical compounds:
Chemical compounds were purchased from MedChemExpress (homoharringtonine), Thermo Fisher Scientific (puromycin), and Sigma (MG132 and hygromycin).
ID2-knockout cell lines:
CRISPR/Cas9-mediated knockout of ID2 was performed using a pLentiCRISPR v2 plasmid (GenScript; Piscataway, NJ) that co-expresses Cas9 and a gRNA (CAATAGTGGGATGCGAGTCC) targeting ID2. Lentivirus was prepared as described in previous publications and cells were selected in 1 μg/mL puromycin starting 48 hours after transduction. The knockout cell lines were then single-cell cloned using flow cytometry (Becton Dickinson FACS Aria). Knockout of ID2 in multiple clones was then validated using immunoblotting.
shRNA knockdown of ID2:
The TET-pLKO.1-PURO-shID2-#2 vector was a gift from Kevin Janes (Addgene plasmid #83087) (46). Lentivirus was prepared as described above and cells were selected in 1 μg/mL puromycin starting 48 hours after viral transduction.
Cell viability assay:
Cell viability was measured using the AlamarBlue (resazurin) fluorescence assay, as previously described (47). Approximately 5,000 cells were plated in each well of a 96-well plate. The next day the cells were treated with a range of drug concentrations for 72 hours. Fluorescence readings were then obtained after adding AlamarBlue (Sigma) using a FLUOstar Omega microplate reader (BMG Labtech). IC50 values were calculated using log-transformed and normalized data (GraphPad Prism 9).
Colony formation assay:
Clonogenic assays were performed as described (48). Cells were plated in 6-well plates in triplicate and then continuously treated with drug or vehicle for 10–14 days. Colonies were then stained with crystal violet and counted using an inverted Olympus CKX41 microscope.
Anchorage-independent growth assay:
Soft agar growth assays were performed as previously described (48). Alternatively, cells (2,500) were suspended in 1 mL of ClonaCell medium (Stem Cell Technologies) in 12-well plates in triplicate. Plates were incubated at 37° C for 10 days and then colonies were counted using an inverted Olympus CKX41 microscope.
RNA sequencing and analysis:
RNA was isolated from cell lines using RNeasy Plus Mini Kit (Qiagen) and submitted to the Iowa Institute of Human Genetics Core Facility for analysis. Samples were barcoded, pooled, and sequenced on an Illumina NovaSeq 6000 (Illumina; San Diego, CA) to obtain a minimum of 30 million, paired-end, 100 bp reads per sample. FastQC was used to assess the quality of the sequencing reads. Reads were then mapped against the human reference genome (hg38) using the STAR aligner. The raw counts were normalized and transformed using the rlog function and principal components analysis (PCA) was performed to visualize sample clusters. No outlier samples were identified or removed from the analysis. The gene expression data were deposited in the Gene Expression Omnibus Repository under the accession number GSE183658. The DESeq2 package was used for the identification of differentially expressed (DE) genes. DE gene expression data were then analyzed using Cytoscape v3.8.2 (cytoscape.org) with the BINGO plugin to identify enriched gene ontology categories (49). The significance of gene set enrichment was assessed using a hypergeometric test and Benjamini-Hochberg false discovery rate for multiple testing correction. Transcription factor enrichment analysis was performed using ChIP-X Enrichment Analysis Version 3 (ChEA3; https://maayanlab.cloud/chea3/).
Protein isolation and immunoblotting:
Protein extracts for immunoblotting were prepared by incubating cells in RIPA buffer (Boston Bioproducts; Ashland, MA), supplemented with protease and phosphatase inhibitors (Halt Protease & Phosphatase Inhibitor Cocktail, EDTA-free; ThermoFisher Scientific), for 20 min. Supernatants were collected after centrifugation at 4° C. The BCA reagent (ThermoFisher Scientific) was used to determine the protein concentrations in the samples. SDS-PAGE was used to separate proteins, which were then transferred to polyvinylidene difluoride membranes (Sigma). Antibodies to the following proteins were used in the immunoblots: ID1 (Biocheck, #195–14, 1:1000), ID2 (Biocheck, #9–2-8, 1:1000), ID2 (Cell Signaling, #D39E8, 1:1000), ID3 (Biocheck, #17–3, 1:2000), puromycin (DSHB, PMY-2A4, 1:500), FLAG (Sigma, F1804, 1:1000), MYC (Proteintech, 60003–2, 1:2000), V5 (Proteintech, 14440–1, 1:2000), and Actin (Cell Signaling, #4970, 1:1000).
Doxycycline-inducible expression of ID1–3:
The FLAG-ID1-T2A-MYC-ID2-P2A-V5-ID3 construct was obtained as a gene block (IDT; Coralville, IA) and inserted into the Lenti-X-Tet-One vector (Takara Biology; Mountain View, CA) using NEBuilder HiFi DNA Assembly (NEB; Ipswich, MA). After verification by sequencing, the plasmid was used to make lentivirus, as described in previous publications (47,50).
Doxycycline-inducible expression of OLIG2:
An OLIG2 cDNA construct was obtained from IDT and inserted into the Lenti-X-Tet-One vector, as described above.
ID2-overexpression:
The pLV-ID2-hygromycin plasmid was obtained from VectorBuilder (Chicago, IL). Lentivirus was prepared as described in previous publications and infected cells were selected using hygromycin 0.4 mg/mL for seven days.
Puromycin labeling:
Protein synthesis was assessed using a puromycin labeling assay described in previous publications (50,51).
Xenograft:
The Institutional Animal Care and Usage Committee at the University of Iowa approved the animal studies and the studies were conducted in adherence with the NIH Guide for the Care and Use of Laboratory Animals. For all the xenograft experiments, 1.0 × 106 tumor cells were mixed with 30% matrigel and injected subcutaneously into the flanks of 6-week-old, female NCr mice. For the drug treatment study, mice were randomized to treatment with vehicle or HHT (2 mg/kg/day, IP, days 1–28) when tumors were palpable (~150–200 mm3). Tumor volumes were measured, without blinding of the investigators, every 2–3 days using calipers (volume = 0.5 x length x width2). Animals were sacrificed when a tumor reached 2 cm in any dimension.
Statistical Analysis:
Student’s t-test two-tailed was used to calculate P-values for the comparison of two groups. Analyses for more than two groups were conducted with a one-way ANOVA followed by Dunnett multiple comparisons test. The Log-rank (Mantel-Cox) test was used to calculate P-values comparing the mouse survival curves. Differences in tumor growth rates were assessed using a two-way ANOVA. Statistical analyses were conducted using GraphPad Prism 9.
RESULTS
Knockout of ID2 in Ewing sarcoma cell lines impairs growth in vitro and in vivo.
We previously developed a genetically-defined and isogenic model of Ewing sarcoma using human embryonic stem cells and an in vitro differentiation approach (16). Using gene expression data derived from this model system, we identified that Inhibitor of DNA Binding 2 (ID2), previously known as Inhibitor of Differentiation 2, is significantly upregulated in cells expressing EWS-FLI relative to isogenic, differentiated fibroblasts (Supplementary Figure 1). ID2 is also highly expressed in Ewing sarcoma cell lines, compared to other cancer types, and primary Ewing sarcoma tumors (Supplementary Figure 2) (19–22).
ID2 is a member of a family of proteins (ID1–4) that bind and sequester basic helix-loop-helix (bHLH) transcription factors and block bHLH-directed transcription (25). Consequently, to investigate the functional role of ID2 in Ewing sarcoma cells, we used CRISPR/Cas9 to knockout ID2 (ID2-KO) in EW8 and TC71 cell lines (Figure 1A). Knockout of ID2 significantly reduced colony formation in multiple clones in both cell lines (Figure 1B–C). The ID2-KO cell lines also proliferated more slowly than the parental cells in a daily growth assay (Supplementary Figure 3). We then used a lentiviral vector to re-express ID2 in the ID2-KO cells lines. As shown in Figures 1D–F, the re-expression of ID2 in the ID2-KO cell lines rescued the detrimental effect of gene knockout on colony formation in vitro. To test whether ID2-KO also impairs the growth of Ewing sarcoma cells in vivo we implanted TC71 cells and two different TC71-ID2-KO clonal cell lines in immunodeficient mice. The TC71-ID2-KO cell lines, relative to the parental cells, grew more slowly in vivo (Figure 1G), which resulted in a significant prolongation of mouse survival (Figure 1H). Similarly, we found that the knockout of ID2 in the EW8 cell line significantly impaired cell growth under anchorage-independent conditons in a suspension growth assay (Supplementary Figure 4). Finally, as a complementary approach to gene knockout, we used a validated, doxycycline-inducible shRNA to reduce levels of ID2 in the EW8 cell line (Figure 1I) (46). shRNA-mediated knockdown of ID2 reduced colony formation and impaired proliferation in a growth assay (Figure 1J and Supplementary Figure 5).
Figure 1. The CRISPR/Cas9-mediated knockout of ID2 in Ewing sarcoma cell lines impairs growth in vitro and in vivo.
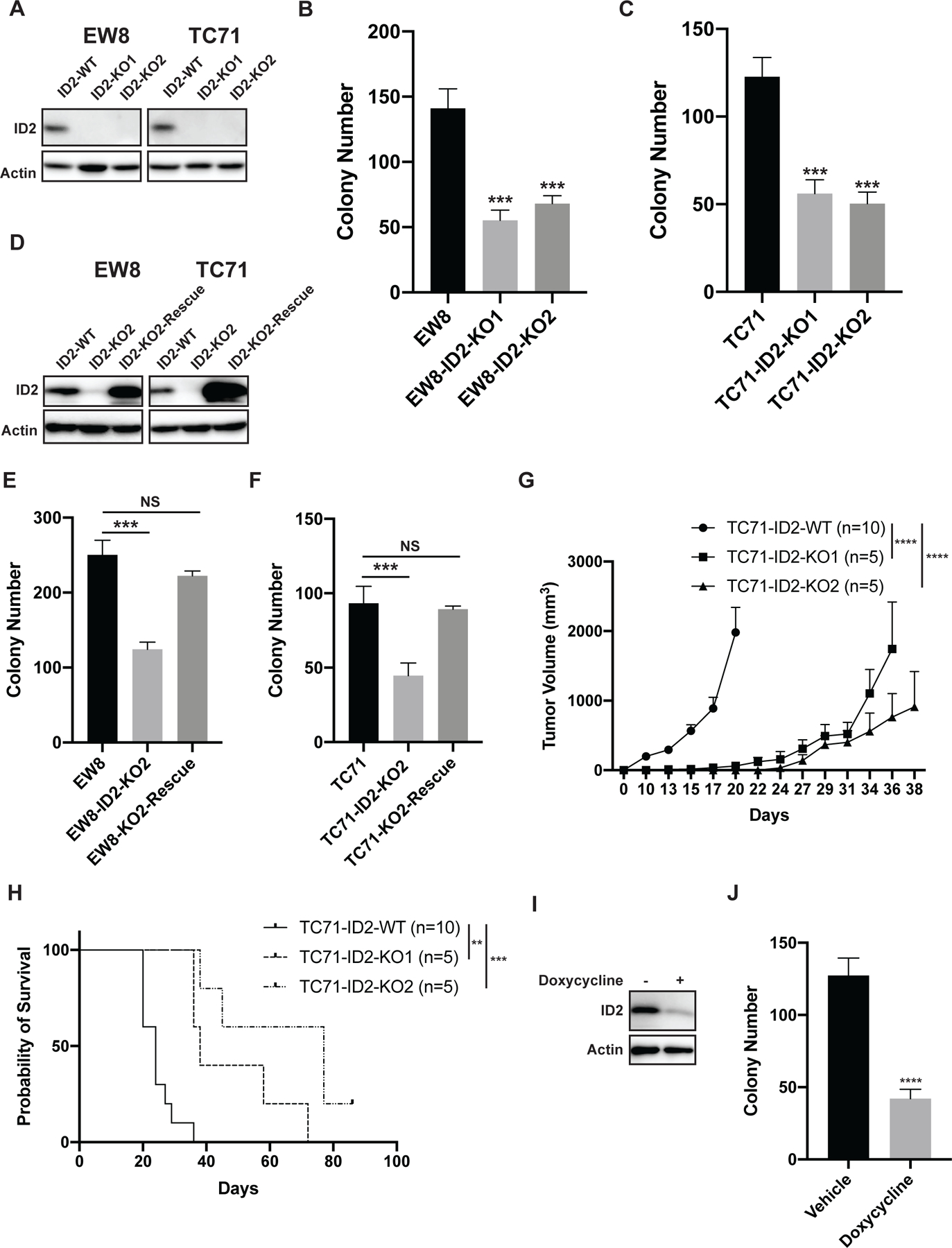
(A) Immunoblot showing ID2 expression level after CRISPR/Cas9-mediated gene knockout in EW8 and TC71 cell lines. Results for two independent clones are shown for each cell line. (B-C) Colony formation assay with the ID2-KO and parental cell lines. Error bars represent the mean ± SD of three technical replicates. (D) Immunoblot showing re-expression of ID2 in the ID2-KO cell lines. (E-F) Colony formation assay with the parental cells (ID2-WT), ID2-KO cells, and the ID2-KO-Rescue cells. Error bars represent the mean ± SD of three technical replicates. (G) TC71 cells and two TC71-ID2-KO clones were implanted subcutaneously in NCr mice. Tumor size was quantified every 2–3 days using caliper measurements. Growth curves for each mouse cohort are shown until mice were removed from that cohort due to tumor size. (H) Kaplan-Meier survival curves for the different mouse cohorts. Mice were sacrificed when tumor reached >2 cm in any dimension. Log-rank (Mantel-Cox) test was used to calculate P-values comparing the survival curves for the mice with knockout cell lines compared to mice with the parental cell line. (I) Immunoblot showing the doxycycline-inducible, shRNA-mediated knockdown of ID2 in TC71 cells. (J) Colony formation assay with the TC71-shID2 cells in presence and absence of doxycycline. ** indicates P < 0.01, *** indicates P < 0.001, **** indicates P < 0.0001.
Overexpression of ID2 in NIH3T3 cells increases tumorigenesis in vitro and in vivo.
ID2 is upregulated in Ewing sarcoma tumors and is a direct transcriptional target of the EWS-FLI1 oncoprotein (19–23). To determine whether upregulation of ID2 increases tumorigenesis we overexpressed ID2 in NIH3T3 cells (Figure 2A), which are mouse embryonic fibroblasts frequently used in transformation assays (44). Overexpression of ID2 in the NIH3T3 cells (NIH3T3-ID2) significantly increased colony formation (Figure 2B), as well as cell growth in suspension in soft agar (Figure 2C). Next, we assessed the growth of the NIH3T3 and NIH3T3-ID2 cells in vivo in a xenograft assay. Of note, as previously reported, we did observe tumor growth with the parental NIH3T3 cell line in immunodeficient mice (44,52). However, overexpression of ID2 in NIH3T3 cells significantly increased tumor growth in vivo compared to the parental NIH3T3 cells (Figure 2D) and shortened mouse survival with a reduction in median survival from 46 days to 32 days (Figure 2E).
Figure 2. Overexpression of ID2 in NIH3T3 cells increases tumorigenesis in vitro and in vivo.
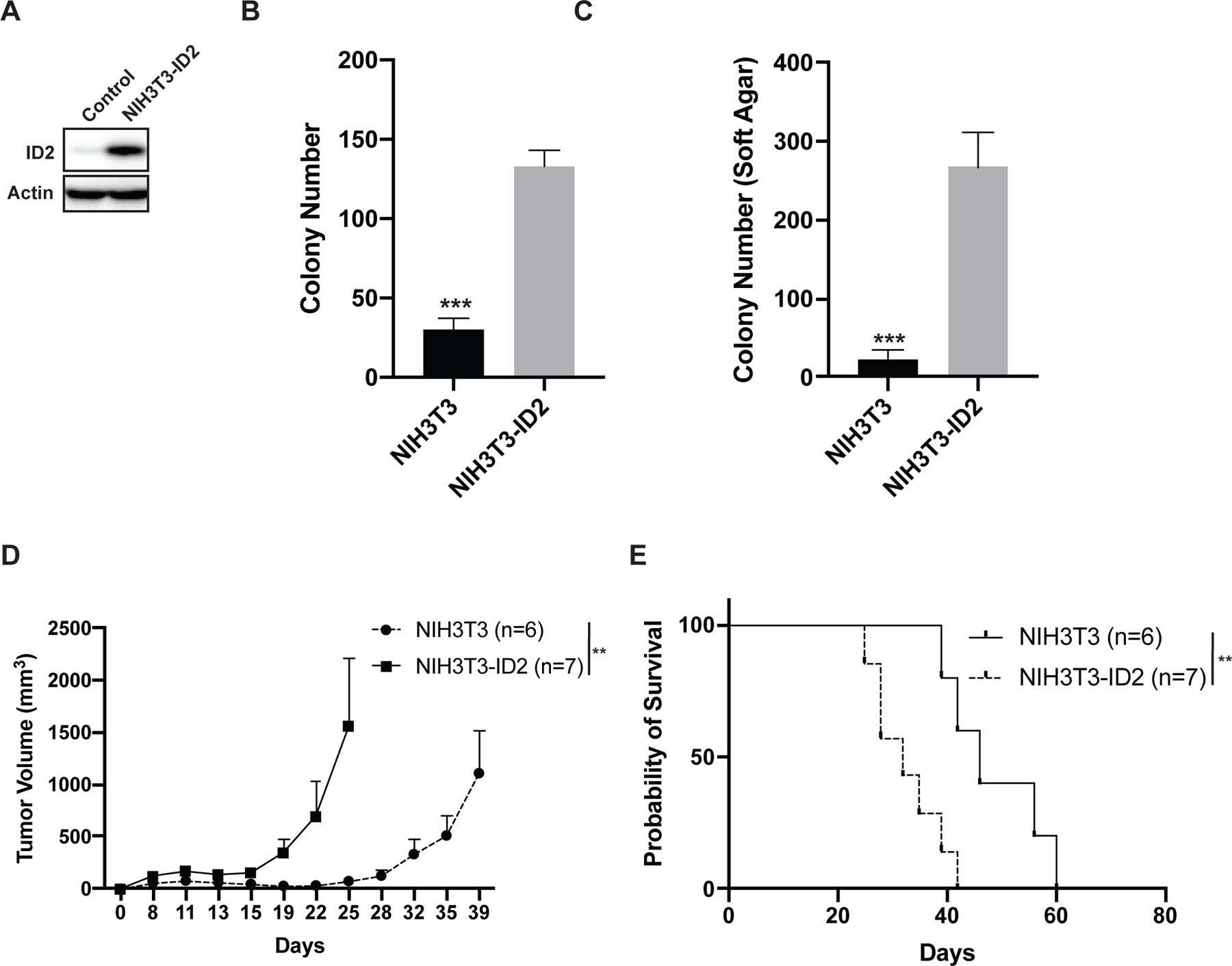
(A) Immunoblot showing the lentiviral-mediated overexpression of ID2 in NIH3T3 cells. (B) Colony formation assay for NIH3T3 and NIH3T3-ID2 cells. Error bars represent the mean ± SD of three technical replicates. (C) Growth in soft agar of NIH3T3 and NIH3T3-ID2 cells. Error bars represent the mean ± SD of three technical replicates. (D) In vivo growth of NIH3T3 and NIH3T3-ID2 cells in a xenograft experiment. Tumor size was quantified every 2–3 days using caliper measurements. Growth curves for each mouse cohort are shown until mice were removed from that cohort due to tumor size. (E) Kaplan-Meier survival curves for the NIH3T3 and NIH3T3-ID2 mouse cohorts. Mice were sacrificed when tumor reached >2 cm in any dimension. Log-rank (Mantel-Cox) test was used to calculate P-values comparing the survival curves. ** indicates P < 0.01, *** indicates P < 0.001.
ID2 regulates the expression of developmental-related genes in Ewing sarcoma.
The ID family of proteins inhibit the function of basic helix-loop-helix (bHLH) transcription factors (17,18,29). Consequently, in order to investigate the effects of ID2 on the transcriptome of Ewing sarcoma tumors, we performed RNA-seq analysis with the clonal cell lines, TC71-ID2-KO2 and TC71-ID2-KO2-Rescue. Notably, the re-expression of ID2 in the TC71-ID2-KO2 cell line resulted in an excess of downregulated genes compared to upregulated genes, as might be expected based on the well-established function of ID2 as a repressor of bHLH transcription factors (Figure 3A) (53). Next, we used gene set enrichment analysis to identify biological processes enriched in the genes downregulated by ID2 in the ID2-KO2-Rescue cell line (49). Figure 3B shows that multiple gene sets related to development are significantly (FDR p-value < 0.05) enriched in the set of genes downregulated by ID2, consistent with the role of ID proteins in regulating differentiation and developmental processes. We also compared the transcriptomes of the clonally-derived TC71-ID2-KO1 and TC71-ID2-KO2 cell lines to the parental TC71 cell line (Figures 3C and 3E). The transcriptomes of both the TC71-ID2-KO1 and TC71-ID2-KO2 cell lines, relative to the parental TC71 cells, demonstrated an excess of upregulated genes compared to downregulated genes. Gene expression changes in the TC71-ID2-KO cell lines were validated in the EW8-ID2-KO cell lines using RT-qPCR (Supplementary Figure 6).
Figure 3. ID2 regulates the expression of developmental genes in Ewing sarcoma cells.
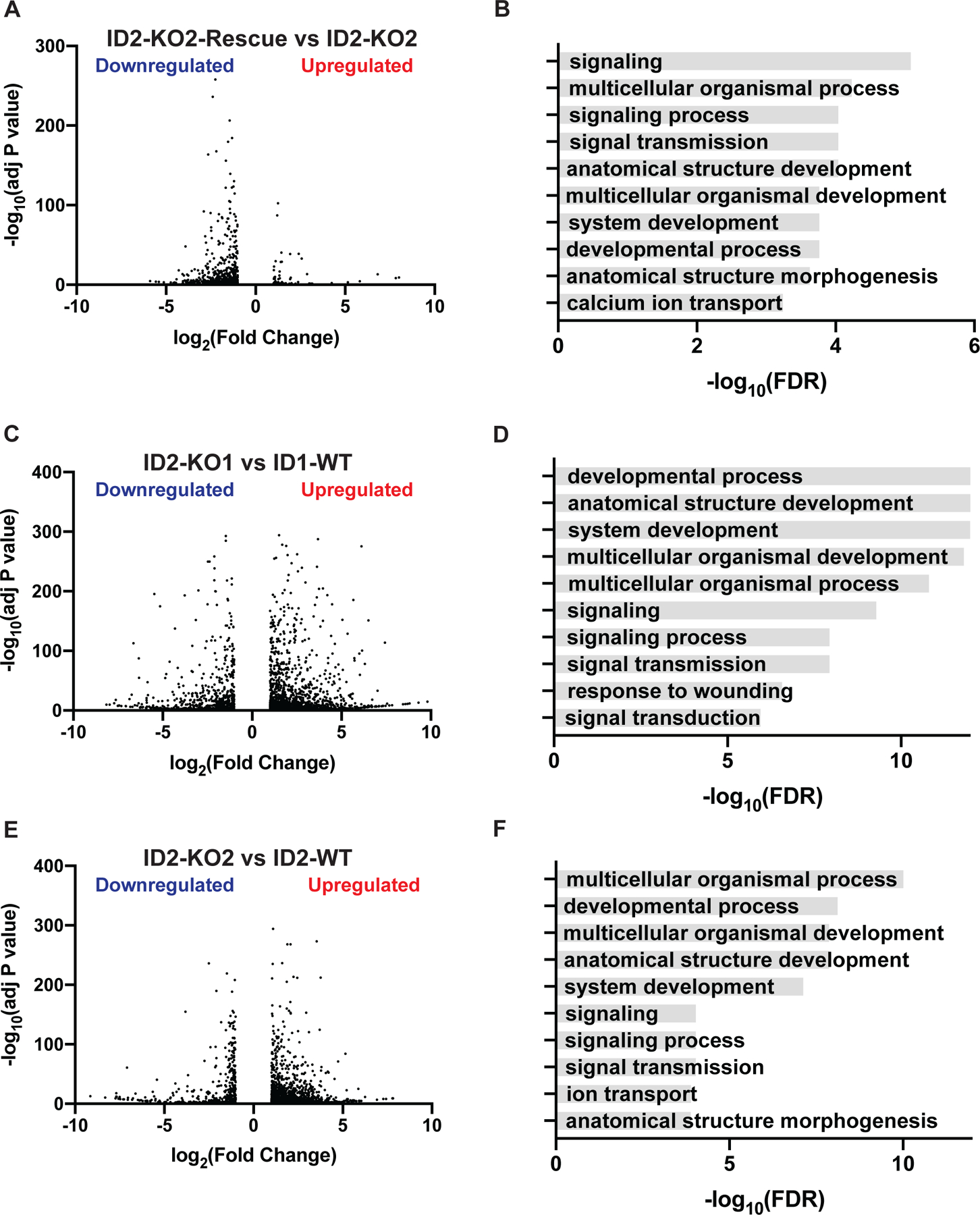
(A) Volcano plot of differentially expressed genes (Fold >2, adjusted P-value <0.05) in the TC71-ID2-KO2 and TC71-ID2-KO2-Rescue cell lines. (B) Top ten gene sets (biological processes) enriched in the upregulated genes in the TC71-ID2-KO2 compared to TC71-ID2-KO2-Rescue cells. (C) Volcano plot of differentially expressed genes (Fold >2, adjusted P-value <0.05) in the TC71-ID2-KO1 and parental TC71 cell lines. (D) Top ten gene sets (biological processes) enriched in the upregulated genes in the TC71-ID2-KO1 cells compared to the parental TC71 cells. (E) Volcano plot of differentially expressed genes (Fold >2, adjusted P-value <0.05) in the TC71-ID2-KO2 and parental TC71 cell lines. (F) Top ten gene sets (biological processes) enriched in the upregulated genes in the TC71-ID2-KO2 cells compared to the parental TC71 cells.
Gene set enrichment analysis with the upregulated genes in both ID2-knockout cell lines identified significant (FDR p-value < 0.05) enrichment for genes related to differentiation and development, in agreement with the results obtained with the knockout and rescue cell lines (Figures 3D and 3F). We also analyzed the differentially expressed genes between the ID2-KO and parental cells for enrichment for specific tissue types. Notably, the genes repressed by ID2 were significantly enriched for anatomical sites and structures in the neural lineage (Supplementary Figure 7A–B). Next, we overlapped the three gene expression datasets and identified a core set of 227 genes that are upregulated in TC71-ID2-KO1, upregulated in TC71-ID2-KO2, and downregulated in TC71-ID2-KO2-Rescue cells (Supplementary Figure 7C). We analyzed these 227 genes with transcription factor enrichment analysis (TFEA) (ChEA3, ChIP-X Enrichment Analysis Version 3) and identified that the top two transcription factors with significant enrichment for genes in this group were OLIG1 and OLIG2 (Supplementary Figures 7D–F), which are bHLH transcription factors and known targets of ID2 (54–56). In addition, we found that the overexpression of OLIG2 in Ewing sarcoma cell lines reproduced gene expression changes caused by ID2-KO, as assessed using RT-qPCR of target genes (Supplementary Figure 8). Finally, we compared a core EWS-FLI gene expression signature, developed by Hancock et al., to genes regulated by ID2, as identified in the RNA-seq experiment (57). We found that ID2 represses the expression of approximately 10% of the genes downregulated by EWS-FLI1, demonstrating that ID2 only accounts for a limited portion of the transcriptional effects of EWS-FLI1 (Supplementary Figure 9).
Homoharringtonine (HHT) inhibits protein synthesis and reduces levels of the ID proteins in Ewing sarcoma cell lines.
The ID1–3 proteins are ubiquitinated and rapidly degraded by the proteasome with half-lives of less than one hour (Supplementary Figure 10A–B) (42,58,59). Consequently, we hypothesized that the level of the ID2 protein in Ewing sarcoma cells may be highly dependent on active protein synthesis. In previous work, we identified that Ewing sarcoma cell lines are sensitive in vitro to homoharringtonine (HHT), an inhibitor of protein translation that is FDA-approved for the treatment of chronic myeloid leukemia (45,47). Figures 4A–B demonstrate that HHT inhibits general protein synthesis, assessed using a puromycin protein synthesis assay, and reduced the level of ID2 in a dose-dependent fashion (Figure 4B). Consistent with the rapid degradation of ID2 via the proteasome, treatment of Ewing sarcoma cells with HHT (50 nM) reduced levels of ID2 within two hours of adding the drug (Figure 4C). Similarly, addition of a proteasome inhibitor (MG132) partially rescued the level of ID2 in cell lines treated with HHT. Next, we treated four additional Ewing sarcoma cell lines with HHT (50 nM) and observed a reduction in ID2 levels, in agreement with the results with the EW8 and TC71 cell lines (Figure 4 E).
Figure 4. Homoharringtonine (HHT) inhibits protein synthesis and reduces levels of ID proteins in Ewing sarcoma cell lines.
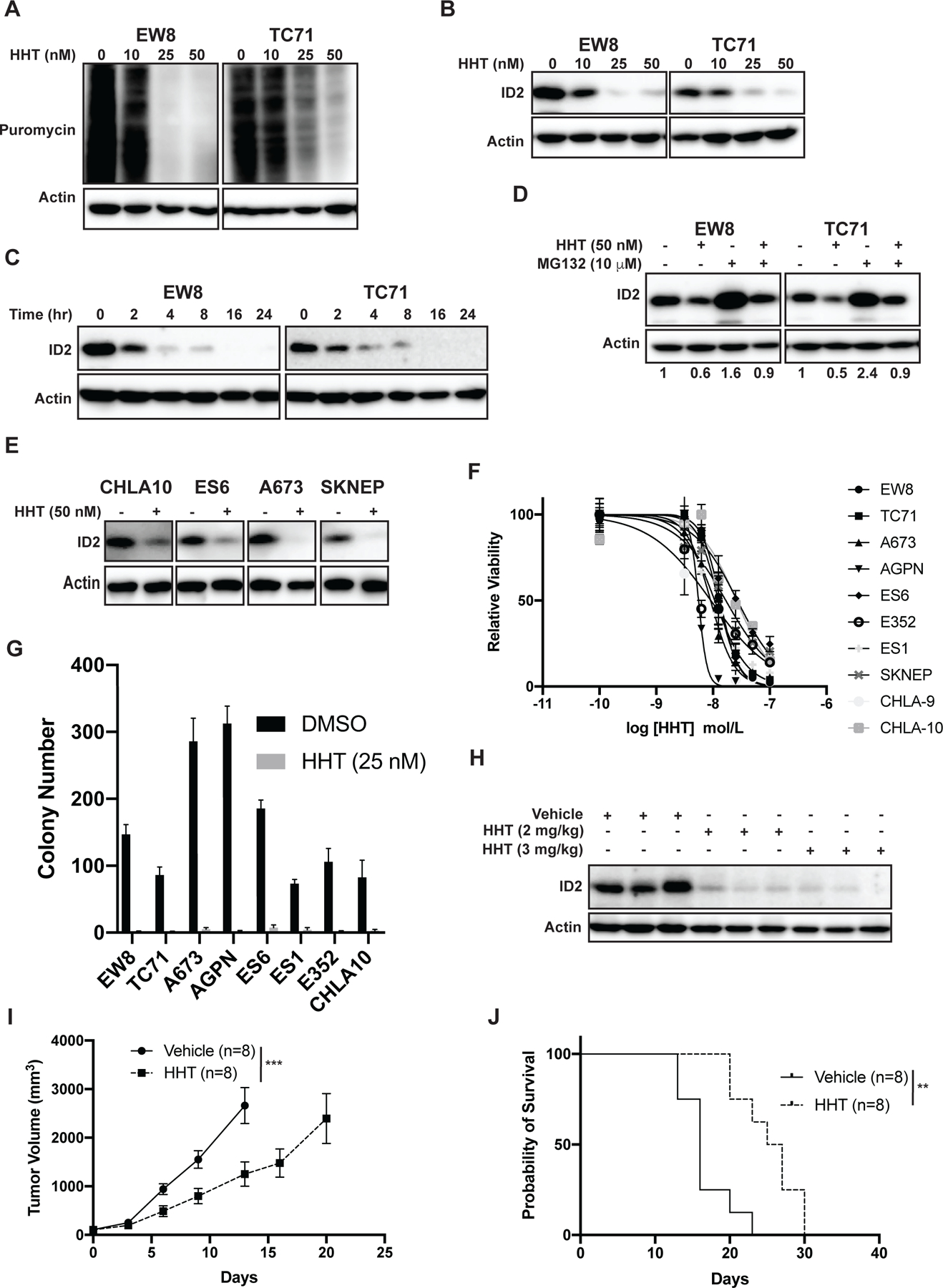
(A) Ewing sarcoma cells were treated with different doses of HHT for 24 hours. Puromycin was then added to the cells for 30 minutes to label newly synthesized proteins. (B) Ewing sarcoma cells were treated with different doses of HHT for 24 hours and lysates were then immunoblotted for ID2. (C) Ewing sarcoma cells were treated with HHT 50 nM for different amounts of time and then protein lysates were collected immunoblotted for ID2. (D) Ewing sarcoma cells were treated with HHT 50 nM in the presence or absence of the proteasome inhibitor MG132. Lysates were then collected and immunoblotted for ID2. (E) Additional Ewing sarcoma cell lines were treated with HHT 50 nM for 24 hours and lysates were then immunoblotted for ID2. (F) Dose-response curves for Ewing sarcoma cell lines treated with different concentrations of HHT for 72 hours. Cell viability was assessed using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (G) Colony formation assay for Ewing sarcoma cell lines treated with HHT 25 nM or vehicle. Error bars represent the mean ± SD of three technical replicates. (H) Mice with TC71 xenograft tumors were treated with HHT (2 mg/kg or 3 mg/kg) and tumors were then collected four hours after drug administration. Immunoblots for ID2 were performed. (I) TC71 Ewing sarcoma cells were engrafted in NCr mice. After developing tumors, the mice were divided into two cohorts and treated with either vehicle or HHT (2 mg/kg/day, IP, days 1–28). Tumor size was quantified using caliper measurements. Growth curves for each mouse cohort are shown until mice were removed from that cohort due to tumor size. (J) Kaplan-Meier survival curves for the TC71 xenograft mice treated with either vehicle or HHT. Mice were sacrificed when tumor reached >2 cm in any dimension. Log-rank (Mantel-Cox) test was used to calculate P-values comparing the survival curves. ** indicates P < 0.01.
To test the effect of HHT on the growth of Ewing sarcoma cell lines we performed dose response experiments with ten cell lines (Figure 4F). Notably, HHT reduced the growth of the cell lines with IC50 values ranging from ~10–40 nM (Table 1). HHT (25 nM) also reduced the clonogenic growth of the Ewing sarcoma cell lines in colony formation assays (Figure 4G). Next, to determine whether HHT reduces ID2 levels in vivo, we treated mice engrafted with TC71 xenograft tumors with HHT and then collected tumors four hours after drug administration. Figure 4H shows that HHT reduced ID2 levels in vivo in the xenograft tumors. We then evaluated whether HHT inhibits the growth of tumor cells in a mouse xenograft experiment. NCr mice were engrafted with TC71 cells and allowed to develop measurable tumors. The mice were then divided into two cohorts and treated with either vehicle or HHT (2 mg/kg/day, days 1–28). Treatment of the mice with HHT, compared to vehicle, reduced tumor growth (Figure 4I) and significantly increased the median mouse survival from 16 days to 26 days (Figure 4J).
Table 1.
Summary of IC50 values for HHT with Ewing sarcoma cell lines.
| Cell Line | IC50 (nM) | 95% CI (nM) |
|---|---|---|
| TC71 | 13 | 12–15 |
| A673 | 10 | 8–11 |
| AGPN | 6 | 5–7 |
| ES6 | 33 | 27–41 |
| E352 | 13 | 10–17 |
| ES1 | 12 | 10–14 |
| SKNEP | 19 | 16–22 |
| CHLA-9 | 29 | 22–40 |
| CHLA-10 | 34 | 27–44 |
| EW8 | 14 | 13–16 |
HHT is an inhibitor of general protein translation and is not specific for the ID2 protein. In previous work, we used reverse phase protein arrays to identify that HHT reduces the levels of many proteins in Ewing sarcoma cell lines, including ribonucleotide reductase M2 (RRM2) (47). RRM2 is a subunit of ribonucleotide reductase (RNR), which is the rate limiting enzyme in the synthesis of deoxyribonucleotides, and a therapeutic target in Ewing sarcoma tumors (47,48,60–62). Overexpression of ID2 in TC71 cells partially rescued the effects of HHT on the level of ID2, but did not impact the effect of HHT on the level of RRM2 or the sensitivity of the cells to HHT (Supplementary Figure 11A–B).
HHT reduces the levels of the ID1, ID2, and ID3 proteins in cell lines.
ID1 and ID3, similar to ID2, are rapidly degraded by the proteasome and function as oncogenes in some tumors (17,29). In addition, ID proteins are often co-expressed and demonstrate overlapping functions, in particular related to differentiation and development (17). Notably, in Ewing sarcoma tumors, SMAD1 signaling is reported to upregulate the mRNA levels of both ID1 and ID3 (63). Consistent with this data, immunoblotting shows variable expression of ID1 and ID3 in Ewing sarcoma cell lines (Supplementary Figure 12A). In addition, overexpression of either ID1 or ID3 in NIH3T3 cells increases clonogenic growth, similar to the effect of ID2 (Supplementary Figure 12B). Consequently, we wanted to determine whether HHT reduces levels of ID1 and ID3, in addition to ID2, in sarcoma cell lines. Due to variations in the expression levels and patterns of the different ID proteins in different cell lines, we used a lentiviral approach to express FLAG-ID1, MYC-ID2, and V5-ID3 from a polycistronic mRNA transcript in 293T cells (Figure 5A). Treatment of the cells with HHT reduced levels of the ID1, ID2, and ID3 proteins in dose-dependent fashion (Figures 5B–C). Addition of a proteasome inhibitor (MG132) partially rescued the level of ID1–3 in cell lines treated with HHT (Figure 5D). Similar results were obtained when we expressed FLAG-ID1, MYC-ID2, and V5-ID3 in Ewing sarcoma (TC71) and osteosarcoma (U2OS) cell lines (Figures 5E–F)
Figure 5. HHT reduces the levels of the ID1, ID2, and ID3 proteins in sarcoma cell lines.
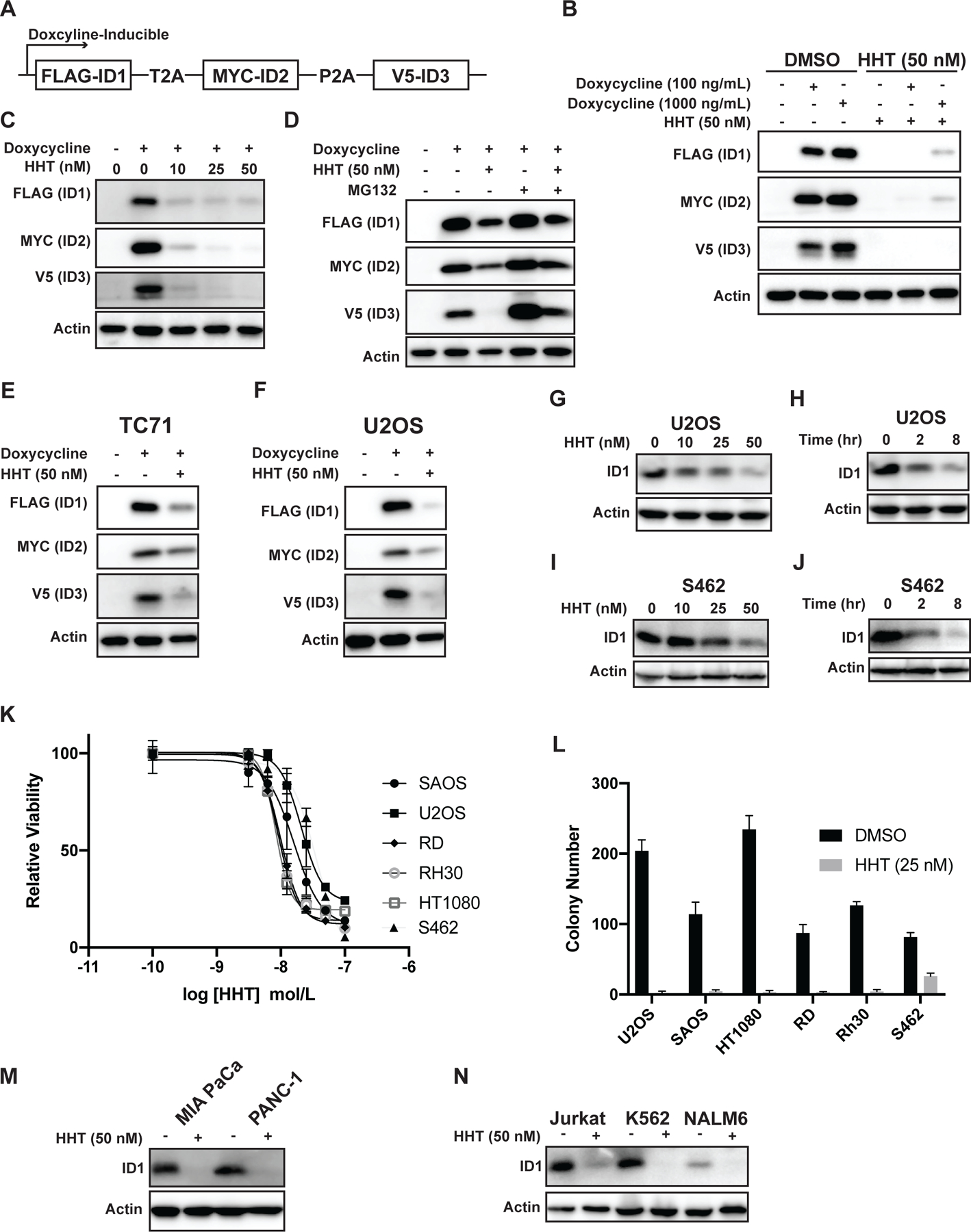
(A) Schematic of a doxycycline-inducible, lentiviral expression vector that expresses Flag-ID1, Myc-ID2, and V5-ID3 from a single mRNA transcript using P2A/T2A elements. (B) Immunoblot of 293T cells with doxycycline-inducible expression of Flag-ID1, Myc-ID2, and V5-ID3 (293T-ID1–3) treated with HHT 50 nM for 24 hours. (C) 293T-ID1–3 cells were treated with different doses of HHT for 24 hours and then lysates were immunoblotted for the epitope tags. (D) 293T-ID1–3 cells were treated with HHT 50 nM in the presence or absence of the proteasome inhibitor MG132. Lysates were then collected and immunoblotted for the epitope tags for ID1–3. (E-F) Immunoblot of Ewing sarcoma (E) and osteosarcoma (F) cell lines expressing Flag-ID1, Myc-ID2, and V5-ID3 treated with HHT (50 nM) for 24 hours. (G) The osteosarcoma cell line U2OS was treated with HHT for 24 hours and lysates were then immunoblotted for ID1. (H) U2OS osteosarcoma cells were treated with HHT 50 nM for different amounts of time and lysates were then immunoblotted for ID1. (I) The MPNST cell line S462 was treated with HHT for 24 hours and lysates were then immunoblotted for ID1. (J) S462 cells were treated with HHT 50 nM for different amounts of time and lysates were then immunoblotted for ID1. (K) Dose-response curves for osteosarcoma (U2OS, SAOS), rhabdomyosarcoma (RD, RH30), fibrosarcoma (HT1080), and MPNST (S462) cell lines treated with different concentrations of HHT for 72 hours. Cell viability was assessed using the AlamarBlue Fluorescence Assay. The results are representative of two independent experiments. Error bars represent mean ± SD of three technical replicates. (L) Colony formation assay for sarcoma cell lines treated with HHT 25 nM or vehicle. Error bars represent the mean ± SD of three technical replicates. (M-N) Pancreatic cancer (M) and leukemia (N) cell lines were treated with HHT (50 nM) for 24 hours and then immunoblotted for ID1.
ID1 is reported to contribute to the pathogenesis of osteosarcoma (OS) and malignant peripheral nerve sheath tumors (MPNST) (42,64–66). Consequently, we tested the effect of HHT on the expression level of endogenous ID1 in OS (Figures 5G–H) and MPNST (Figures 5I–J) cell lines. HHT reduced the level of the ID1 protein in both sarcoma cell lines. We then performed dose response experiments with HHT with additional sarcoma cell lines, SAOS (osteosarcoma), U2OS (osteosarcoma), RD (rhabdomyosarcoma), Rh30 (rhabdomyosarcoma), HT1080 (fibrosarcoma), and S462 (MPNST). Figure 5K demonstrates that HHT impaired the growth of the sarcoma cell lines with IC50 values ranging from ~10–35 nM. HHT (25 nM) also reduced the clonogenic growth of these sarcoma cell lines (Figure 5L). Finally, we tested the effect of HHT (50 nM) on the level of ID1 in pancreatic cancer (Figure 5M) and leukemia cell lines (Figure 5N), as ID1 is also reported to contribute to tumorigenesis in these cancers (67–70). HHT reduced the level of ID1 in these cell lines, in agreement with the results obtained with the other tumor types.
DISCUSSION
EWS-FLI1, the driver oncogene in Ewing sarcoma tumors, regulates multiple pathways related to cellular differentiation, development, and lineage specificity (5,7–14). The effects of EWS-FLI1 are also dependent on cellular context, which suggests a complex interplay between EWS-FLI1 and the cell-of-origin in Ewing sarcoma tumors. Of note, there is experimental support for both a mesenchymal and neural crest origin of Ewing sarcoma tumors (3,5,6,14). Similarly, from a clinical perspective, Ewing sarcoma and peripheral primitive neuroectodermal (PNET) tumors were originally described as distinct clinicopathologic entities. However, it is now known that Ewing sarcoma and PNET share the same driver oncogene (EWS-FLI1) and both tumors, despite histological differences, are classified as Ewing sarcoma tumors (71). In this work, we identified that ID2, which is overexpressed in Ewing sarcoma tumors, is a critical regulator of developmental genes and pathways in Ewing sarcoma tumors. Of note, the ID2 gene is a direct transcriptional target of EWS-FLI1, but expression of ID2 is also reported to be regulated by other mechanisms and transcription factors, including the c-Myc and SMAD1 signaling pathways (20,21,23,27,38,72,73).
The ID family of proteins are well-described regulators of development and differentiation in multiple cellular lineages, including mesenchymal stem cells (17,25,27,29,42). To test the functional role of ID2 in Ewing sarcoma tumors, we used CRISPR/Cas9 to knockout ID2 and found that loss of this protein significantly reduces cell growth in vitro and in vivo. Furthermore, using RNA-seq with ID2-knockout and rescue cell lines, we identified that ID2 regulates genes and pathways related to development and differentiation in Ewing sarcoma cells. In addition, using transcription factor enrichment analysis (TFEA) in combination with the RNA-seq data from the ID2-KO and ID2-KO-Rescue cell lines, we identified the bHLH transcription factors OLIG1 and OLIG2 as the top, putative targets of ID2 in Ewing sarcoma cells. OLIG1/2 are normally expressed in both the developing and mature central nervous system (CNS) and regulate cellular specification and differentiation. Numerous studies have demonstrated functional roles for OLIG1/2 in directing neuronal and glial formation during different stages of development (74–76). Notably, the overexpression of OLIG2 in neural precursor cells (NPC) enhances differentiation and the generation of oligodendrocytes (55,77–81). Consequently, we hypothesize that ID2-mediated inhibition of OLIG1/2 activity could modulate the histological phenotype of Ewing sarcoma/PNET tumors (75). However, only 13 genes regulated by either OLIG1 or OLIG2 overlap with the 227 ID2-regulated genes, suggesting the effects of ID2 on the transcriptome are unlikely to be explained by the inhibition of a single transcription factor. We also recognize that ID proteins have additional functions independent from the inhibition of bHLH transcription factors. For example, ID2 is a dominant-negative antagonist of proteins from the retinoblastoma (Rb) family (24,82). Consequently, identification of the relevant downstream targets of ID2 in Ewing sarcoma tumors, as well as the interplay between the different targets, will be a focus of further investigation.
In this work, we also identified that homoharringtonine (HHT), an FDA-approved drug that inhibits protein translation, decreases the level of the ID2 protein in vitro and in vivo in sarcoma cell lines. HHT reduced the in vivo growth of Ewing sarcoma cells and prolonged mouse survival in a xenograft experiment. In addition, although targeting a single member of the ID family of proteins can result in resistance via upregulation and compensation by other members of the ID family of proteins, we found that HHT reduced protein levels of ID1 and ID3 as well as ID2. However, although the inhibition of differentiation is a biological function that is shared by all members of the ID protein family, ID1–3 are also known to have additional, unique, and non-redundant functions (17). Consequently, defining the individual and overlapping functions of the ID1–3 proteins in Ewing sarcoma, as well as other sarcoma subtypes, will be a focus of future investigation. Of note, we did not evaluate the impact of HHT on ID4 in our work because this member of the ID family of proteins, unlike ID1–3, is not reported to be regulated by proteasomal degradation (58,83,84).
We also recognize that HHT is an inhibitor of general protein translation and reduces the levels of many proteins in addition to ID1–3. For example, HHT is reported to down-regulate the antiapoptotic protein MCL-1 in rhabdoid tumors and FLT3 in leukemia (85,86). In addition, in previous work, we used reverse phase protein arrays (RPPA) to identify that HHT downregulates the levels of numerous proteins, including ribonucleotide reductase M2 (RRM2), in Ewing sarcoma cell lines (47). Consequently, the mechanism of action of HHT in inhibiting the growth of sarcoma cells cannot be attributed solely to a reduction of the levels of ID proteins. However, we believe that the lack of specificity of HHT for ID2 is balanced by the extensive clinical experience using this drug as a single agent and in combination therapies in children and adults (87–89). Furthermore, from a clinical perspective, the lack of specificity of HHT and the ability of the drug to target additional oncogenic proteins and pathways, such as RRM2, may be an advantage in the clinic. The ID2-knockout and ID2-overexpressing cells lines provide a complementary approach to investigate the functions of the ID2 protein in a more specific and defined fashion.
The ID family of proteins function as oncogenes in a variety of cancer types and we hypothesize that HHT could have therapeutic efficacy in additional cancers that are reliant on ID proteins. For example, ID proteins are reported to contribute to leukemogenesis and we speculate that the clinical efficacy of HHT in treating leukemia may, in part, be due to effects on the levels of ID proteins (68,69,90). Similarly, rhabdoid tumors are reported to express the ID1–3 proteins and this aggressive pediatric tumor was recently described to be sensitive in vitro and in vivo to HHT (85,91). In addition, from the standpoint of drug resistance and combination therapies, HHT is frequently used with other chemotherapy drugs, including anthracyclines, nucleoside analogues, and tyrosine kinase inhibitors, in children and adults with leukemia (45,86,88,89). For example, the combination of HHT and sorafenib, a multitargeted tyrosine kinase used in patients with sarcomas, has shown clinical efficacy in patients with AML (86,92). Consequently, in future work, we plan to investigate HHT in combination with these other drugs for the treatment of Ewing sarcoma.
Additional approaches have been used to interfere with the function of ID proteins, including the use of targeted antisense delivery, cell-permeable peptides, antagonists of ID expression, and deubiquitinase inhibitors (17,43). These strategies have not yet advanced to the clinic, but provide further avenues for future investigation. For example, AGX51 is small-molecule compound that binds to ID proteins, interferes with the interaction between ID proteins and bHLH transcription factors, and increases the degradation of unbound ID proteins (93,94). Similarly, inhibitors of the deubiquitinase USP1, which deubiquitinates ID proteins and decreases the proteasomal degradation of this family of proteins, also reduce levels of ID proteins in vitro (42,68).
In conclusion, despite the use of aggressive therapies, neither the treatment regimens nor the outcomes of most pediatric sarcomas have changed in several decades. In this work, we identified that ID2 regulates the expression of developmental genes in Ewing sarcoma tumors and promotes tumorigenesis in vitro and in vivo in xenograft experiments. In addition, from a translational standpoint, we identified that the FDA-approved drug HHT downregulates levels of ID1–3 proteins in multiple types of cancer. Overall, our work provides novel mechanistic insight into the aberrant regulation of differentiation and development in Ewing sarcoma tumors and identifies a novel therapeutic approach, using a repurposed drug, to target ID proteins in cancer cells.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge Henry Keen for assistance with the analysis of the RNA-seq data. DJG is supported by a University of Iowa Dance Marathon Award, a Holden Comprehensive Cancer Center Sarcoma Multidisciplinary Oncology Group Seed Grant, Sammy’s Superheroes, The Matt Morrell and Natalie Sanchez Pediatric Cancer Research Foundation, Aiming for a Cure, and NIH Grant R37-CA217910. This work was also supported by a grant (P30CA086862) from the NCI, administered through the Holden Comprehensive Cancer Center at The University of Iowa. The authors would like to acknowledge use of the University of Iowa Flow Cytometry Facility and the DNA sequencing facility (Genomics Division of the Iowa Institute of Human Genetics) which are supported, in part, by the University of Iowa Carver College of Medicine and the Holden Comprehensive Cancer Center (NCI P30CA086862).
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing interests.
REFERENCES CITED
- 1.Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010. Feb;11(2):184–92. [DOI] [PubMed] [Google Scholar]
- 2.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992. Sep 10;359(6391):162–5. [DOI] [PubMed] [Google Scholar]
- 3.Riggi N, Suva M-L, Stamenkovic I. Ewing’s sarcoma origin: from duel to duality. Expert Rev Anticancer Ther 2009. Aug;9(8):1025–30. [DOI] [PubMed] [Google Scholar]
- 4.Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol 2012;7:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007. May;11(5):421–9. [DOI] [PubMed] [Google Scholar]
- 6.Sole A, Grossetête S, Heintzé M, Babin L, Zaïdi S, Revy P, et al. Unraveling Ewing sarcoma tumorigenesis originating from patient-derived Mesenchymal Stem Cells. Cancer Res 2021. Aug 2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leacock SW, Basse AN, Chandler GL, Kirk AM, Rakheja D, Amatruda JF. A zebrafish transgenic model of Ewing’s sarcoma reveals conserved mediators of EWS-FLI1 tumorigenesis. Dis Model Mech 2012. Jan;5(1):95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eliazer S, Spencer J, Ye D, Olson E, Ilaria RL. Alteration of mesodermal cell differentiation by EWS/FLI-1, the oncogene implicated in Ewing’s sarcoma. Mol Cell Biol 2003. Jan;23(2):482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potikyan G, France KA, Carlson MRJ, Dong J, Nelson SF, Denny CT. Genetically defined EWS/FLI1 model system suggests mesenchymal origin of Ewing’s family tumors. Lab Invest 2008. Dec;88(12):1291–302. [DOI] [PubMed] [Google Scholar]
- 10.Torchia EC, Jaishankar S, Baker SJ. Ewing tumor fusion proteins block the differentiation of pluripotent marrow stromal cells. Cancer Res 2003. Jul 1;63(13):3464–8. [PubMed] [Google Scholar]
- 11.Riggi N, Cironi L, Provero P, Suvà M-L, Kaloulis K, Garcia-Echeverria C, et al. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res 2005. Dec 15;65(24):11459–68. [DOI] [PubMed] [Google Scholar]
- 12.Riggi N, Suvà M-L, Suvà D, Cironi L, Provero P, Tercier S, et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 2008. Apr 1;68(7):2176–85. [DOI] [PubMed] [Google Scholar]
- 13.Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, von Levetzow C, et al. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014. Dec;9(12):1613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, et al. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One 2011. Apr 29;6(4):e19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behjati S, Gilbertson RJ, Pfister SM. Maturation block in childhood cancer. Cancer Discov 2021. Mar;11(3):542–4. [DOI] [PubMed] [Google Scholar]
- 16.Gordon DJ, Motwani M, Pellman D. Modeling the initiation of Ewing sarcoma tumorigenesis in differentiating human embryonic stem cells. Oncogene 2016. Jun 16;35(24):3092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer 2014. Feb;14(2):77–91. [DOI] [PubMed] [Google Scholar]
- 18.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer 2005. Aug;5(8):603–14. [DOI] [PubMed] [Google Scholar]
- 19.Park H-R, Jung WW, Kim HS, Santini-Araujo E, Kalil RK, Bacchini P, et al. Upregulation of the oncogenic helix-loop-helix protein Id2 in Ewing sarcoma. Tumori 2006. Jun;92(3):236–40. [PubMed] [Google Scholar]
- 20.Fukuma M, Okita H, Hata J-I, Umezawa A. Upregulation of Id2, an oncogenic helix-loop-helix protein, is mediated by the chimeric EWS/ets protein in Ewing sarcoma. Oncogene 2003. Jan 9;22(1):1–9. [DOI] [PubMed] [Google Scholar]
- 21.Nishimori H, Sasaki Y, Yoshida K, Irifune H, Zembutsu H, Tanaka T, et al. The Id2 gene is a novel target of transcriptional activation by EWS-ETS fusion proteins in Ewing family tumors. Oncogene 2002. Nov 28;21(54):8302–9. [DOI] [PubMed] [Google Scholar]
- 22.Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, et al. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res 2006. Jun 1;66(11):5574–81. [DOI] [PubMed] [Google Scholar]
- 23.Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suvà ML, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014. Nov 10;26(5):668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iavarone A, Garg P, Lasorella A, Hsu J, Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev 1994. Jun 1;8(11):1270–84. [DOI] [PubMed] [Google Scholar]
- 25.Roschger C, Cabrele C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun Signal 2017. Jan 25;15(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J, Kim K, Kim JH, Jin HM, Choi HK, Lee S-H, et al. Id helix-loop-helix proteins negatively regulate TRANCE-mediated osteoclast differentiation. Blood 2006. Apr 1;107(7):2686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng Y, Kang Q, Luo Q, Jiang W, Si W, Liu BA, et al. Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J Biol Chem 2004. Jul 30;279(31):32941–9. [DOI] [PubMed] [Google Scholar]
- 28.Kreider BL, Benezra R, Rovera G, Kadesch T. Inhibition of myeloid differentiation by the helix-loop-helix protein Id. Science 1992. Mar 27;255(5052):1700–2. [DOI] [PubMed] [Google Scholar]
- 29.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol 2003. Aug;13(8):410–8. [DOI] [PubMed] [Google Scholar]
- 30.Ogata T, Wozney JM, Benezra R, Noda M. Bone morphogenetic protein 2 transiently enhances expression of a gene, Id (inhibitor of differentiation), encoding a helix-loop-helix molecule in osteoblast-like cells. Proc Natl Acad Sci USA 1993. Oct 1;90(19):9219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 1990. Apr 6;61(1):49–59. [DOI] [PubMed] [Google Scholar]
- 32.Lasorella A, Rothschild G, Yokota Y, Russell RG, Iavarone A. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol 2005. May;25(9):3563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nair R, Teo WS, Mittal V, Swarbrick A. ID proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol Ther 2014. Aug;22(8):1407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Z, Bo Z, Gong W, Guo Y. Inhibitor of differentiation 1 (id1) in cancer and cancer therapy. Int J Med Sci 2020. Apr 6;17(8):995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jen Y, Weintraub H, Benezra R. Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev 1992. Aug;6(8):1466–79. [DOI] [PubMed] [Google Scholar]
- 36.Meng J, Liu K, Shao Y, Feng X, Ji Z, Chang B, et al. ID1 confers cancer cell chemoresistance through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis 2020. Feb 20;11(2):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bae WJ, Koo BS, Lee SH, Kim JM, Rho YS, Lim JY, et al. Inhibitor of DNA binding 2 is a novel therapeutic target for stemness of head and neck squamous cell carcinoma. Br J Cancer 2017. Dec 5;117(12):1810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 2000. Oct 5;407(6804):592–8. [DOI] [PubMed] [Google Scholar]
- 39.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 1999. Oct 14;401(6754):670–7. [DOI] [PubMed] [Google Scholar]
- 40.Sachdeva R, Wu M, Smiljanic S, Kaskun O, Ghannad-Zadeh K, Celebre A, et al. ID1 is critical for tumorigenesis and regulates chemoresistance in glioblastoma. Cancer Res 2019. Aug 15;79(16):4057–71. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Z, He H, Wang C, Tao B, Zhou H, Dong Y, et al. Downregulation of Id2 increases chemosensitivity of glioma. Tumour Biol 2015. Jun;36(6):4189–96. [DOI] [PubMed] [Google Scholar]
- 42.Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, et al. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 2011. Sep 16;146(6):918–30. [DOI] [PubMed] [Google Scholar]
- 43.Benezra R The Id proteins: targets for inhibiting tumor cells and their blood supply. Biochim Biophys Acta 2001. Oct 1;1551(2):F39–47. [DOI] [PubMed] [Google Scholar]
- 44.Greig RG, Koestler TP, Trainer DL, Corwin SP, Miles L, Kline T, et al. Tumorigenic and metastatic properties of “normal” and ras-transfected NIH/3T3 cells. Proc Natl Acad Sci USA 1985. Jun;82(11):3698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winer ES, DeAngelo DJ. A review of omacetaxine: A chronic myeloid leukemia treatment resurrected. Oncol Ther 2018. Jun;6(1):9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bajikar SS, Wang C-C, Borten MA, Pereira EJ, Atkins KA, Janes KA. Tumor-Suppressor Inactivation of GDF11 Occurs by Precursor Sequestration in Triple-Negative Breast Cancer. Dev Cell 2017. Nov 20;43(4):418–435.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goss KL, Koppenhafer SL, Waters T, Terry WW, Wen K-K, Wu M, et al. The translational repressor 4E-BP1 regulates RRM2 levels and functions as a tumor suppressor in Ewing sarcoma tumors. Oncogene 2021. Jan;40(3):564–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goss KL, Koppenhafer SL, Harmoney KM, Terry WW, Gordon DJ. Inhibition of CHK1 sensitizes Ewing sarcoma cells to the ribonucleotide reductase inhibitor gemcitabine. Oncotarget 2017. Oct 20;8(50):87016–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005. Aug 15;21(16):3448–9. [DOI] [PubMed] [Google Scholar]
- 50.Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. mTORC1/2 and Protein Translation Regulate Levels of CHK1 and the Sensitivity to CHK1 Inhibitors in Ewing Sarcoma Cells. Mol Cancer Ther 2018. Dec;17(12):2676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 2009. Apr;6(4):275–7. [DOI] [PubMed] [Google Scholar]
- 52.Merritt N, Garcia K, Rajendran D, Lin Z-Y, Zhang X, Mitchell KA, et al. TAZ-CAMTA1 and YAP-TFE3 alter the TAZ/YAP transcriptome by recruiting the ATAC histone acetyltransferase complex. Elife 2021. Apr 29;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benezra R, Davis RL, Lassar A, Tapscott S, Thayer M, Lockshon D, et al. Id: a negative regulator of helix-loop-helix DNA binding proteins. Control of terminal myogenic differentiation. Ann N Y Acad Sci 1990;599:1–11. [DOI] [PubMed] [Google Scholar]
- 54.Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res 2019. Jul 2;47(W1):W212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development 2004. Sep;131(17):4131–42. [DOI] [PubMed] [Google Scholar]
- 56.Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 2018. Apr 10;9(1):1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hancock JD, Lessnick SL. A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle 2008. Jan 15;7(2):250–6. [DOI] [PubMed] [Google Scholar]
- 58.Bounpheng MA, Dimas JJ, Dodds SG, Christy BA. Degradation of Id proteins by the ubiquitin-proteasome pathway. FASEB J 1999. Dec;13(15):2257–64. [PubMed] [Google Scholar]
- 59.Trausch-Azar JS, Lingbeck J, Ciechanover A, Schwartz AL. Ubiquitin-Proteasome-mediated degradation of Id1 is modulated by MyoD. J Biol Chem 2004. Jul 30;279(31):32614–9. [DOI] [PubMed] [Google Scholar]
- 60.Goss KL, Gordon DJ. Gene expression signature based screening identifies ribonucleotide reductase as a candidate therapeutic target in Ewing sarcoma. Oncotarget 2016. Sep 27;7(39):63003–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohmura S, Marchetto A, Orth MF, Li J, Jabar S, Ranft A, et al. Translational evidence for RRM2 as a prognostic biomarker and therapeutic target in Ewing sarcoma. BioRxiv 2021. Mar 4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waters T, Goss KL, Koppenhafer SL, Terry WW, Gordon DJ. Eltrombopag inhibits the proliferation of Ewing sarcoma cells via iron chelation and impaired DNA replication. BMC Cancer 2020. Nov 30;20(1):1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwentner R, Herrero-Martin D, Kauer MO, Mutz CN, Katschnig AM, Sienski G, et al. The role of miR-17–92 in the miRegulatory landscape of Ewing sarcoma. Oncotarget 2017. Feb 14;8(7):10980–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel AV, Chaney KE, Choi K, Largaespada DA, Kumar AR, Ratner N. An shrna screen identifies MEIS1 as a driver of malignant peripheral nerve sheath tumors. EBioMedicine 2016. Jul;9:110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao G-S, Zhang Q, Cao Y, Wang Y, Lv Y-F, Zhang Z-S, et al. High expression of ID1 facilitates metastasis in human osteosarcoma by regulating the sensitivity of anoikis via PI3K/AKT depended suppression of the intrinsic apoptotic signaling pathway. Am J Transl Res 2019. Apr 15;11(4):2117–39. [PMC free article] [PubMed] [Google Scholar]
- 66.Hao L, Liao Q, Tang Q, Deng H, Chen L. Id-1 promotes osteosarcoma cell growth and inhibits cell apoptosis via PI3K/AKT signaling pathway. Biochem Biophys Res Commun 2016. Feb 12;470(3):643–9. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Man N, Sun X-J, et al. Regulation of AKT signaling by Id1 controls t(8;21) leukemia initiation and progression. Blood 2015;126(5):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]; Blood 2016. Sep 29;128(13):1778. [Google Scholar]
- 68.Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther 2013. Dec;12(12):2651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tam WF, Gu T-L, Chen J, Lee BH, Bullinger L, Fröhling S, et al. Id1 is a common downstream target of oncogenic tyrosine kinases in leukemic cells. Blood 2008. Sep 1;112(5):1981–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang Y-H, Hu J, Chen F, Lecomte N, Basnet H, David CJ, et al. ID1 Mediates Escape from TGFβ Tumor Suppression in Pancreatic Cancer. Cancer Discov 2020. Jan;10(1):142–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khoury JD. Ewing sarcoma family of tumors. Adv Anat Pathol 2005. Jul;12(4):212–20. [DOI] [PubMed] [Google Scholar]
- 72.Lasorella A, Boldrini R, Dominici C, Donfrancesco A, Yokota Y, Inserra A, et al. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res 2002. Jan 1;62(1):301–6. [PubMed] [Google Scholar]
- 73.Veerasamy M, Phanish M, Dockrell MEC. Smad mediated regulation of inhibitor of DNA binding 2 and its role in phenotypic maintenance of human renal proximal tubule epithelial cells. PLoS One 2013. Jan 8;8(1):e51842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ligon KL, Fancy SPJ, Franklin RJM, Rowitch DH. Olig gene function in CNS development and disease. Glia 2006. Jul;54(1):1–10. [DOI] [PubMed] [Google Scholar]
- 75.Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 2002. Apr 5;109(1):61–73. [DOI] [PubMed] [Google Scholar]
- 76.Szu J, Wojcinski A, Jiang P, Kesari S. Impact of the olig family on neurodevelopmental disorders. Front Neurosci 2021. Mar 30;15:659601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maire CL, Buchet D, Kerninon C, Deboux C, Baron-Van Evercooren A, Nait-Oumesmar B. Directing human neural stem/precursor cells into oligodendrocytes by overexpression of Olig2 transcription factor. J Neurosci Res 2009. Nov 15;87(15):3438–46. [DOI] [PubMed] [Google Scholar]
- 78.Hwang DH, Kim BG, Kim EJ, Lee SI, Joo IS, Suh-Kim H, et al. Transplantation of human neural stem cells transduced with Olig2 transcription factor improves locomotor recovery and enhances myelination in the white matter of rat spinal cord following contusive injury. BMC Neurosci 2009. Sep 22;10:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maire CL, Wegener A, Kerninon C, Nait Oumesmar B. Gain-of-function of Olig transcription factors enhances oligodendrogenesis and myelination. Stem Cells 2010. Sep;28(9):1611–22. [DOI] [PubMed] [Google Scholar]
- 80.Islam MS, Tatsumi K, Okuda H, Shiosaka S, Wanaka A. Olig2-expressing progenitor cells preferentially differentiate into oligodendrocytes in cuprizone-induced demyelinated lesions. Neurochem Int 2009. Apr;54(3–4):192–8. [DOI] [PubMed] [Google Scholar]
- 81.Ehrlich M, Mozafari S, Glatza M, Starost L, Velychko S, Hallmann A-L, et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc Natl Acad Sci USA 2017. Mar 14;114(11):E2243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol 1996. Jun;16(6):2570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jen Y, Manova K, Benezra R. Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Dev Dyn 1996. Nov;207(3):235–52. [DOI] [PubMed] [Google Scholar]
- 84.Riechmann V, van Crüchten I, Sablitzky F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res 1994. Mar 11;22(5):749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howard TP, Oberlick EM, Rees MG, Arnoff TE, Pham M-T, Brenan L, et al. Rhabdoid Tumors Are Sensitive to the Protein-Translation Inhibitor Homoharringtonine. Clin Cancer Res 2020. Sep 15;26(18):4995–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lam SSY, Ho ESK, He B-L, Wong W-W, Cher C-Y, Ng NKL, et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci Transl Med 2016. Oct 5;8(359):359ra129. [DOI] [PubMed] [Google Scholar]
- 87.Bell BA, Chang MN, Weinstein HJ. A phase II study of Homoharringtonine for the treatment of children with refractory or recurrent acute myelogenous leukemia: a pediatric oncology group study. Med Pediatr Oncol 2001. Aug;37(2):103–7. [DOI] [PubMed] [Google Scholar]
- 88.Chen X, Tang Y, Chen J, Chen R, Gu L, Xue H, et al. Homoharringtonine is a safe and effective substitute for anthracyclines in children younger than 2 years old with acute myeloid leukemia. Front Med 2019. Jun 11;13(3):378–87. [DOI] [PubMed] [Google Scholar]
- 89.Jin J, Wang J-X, Chen F-F, Wu D-P, Hu J, Zhou J-F, et al. Homoharringtonine-based induction regimens for patients with de-novo acute myeloid leukaemia: a multicentre, open-label, randomised, controlled phase 3 trial. Lancet Oncol 2013. Jun;14(7):599–608. [DOI] [PubMed] [Google Scholar]
- 90.Wang L, Man N, Sun X-J, Tan Y, García-Cao M, Liu F, et al. Regulation of AKT signaling by Id1 controls t(8;21) leukemia initiation and progression. Blood 2015. Jul 30;126(5):640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Venneti S, Le P, Martinez D, Xie SX, Sullivan LM, Rorke-Adams LB, et al. Malignant rhabdoid tumors express stem cell factors, which relate to the expression of EZH2 and Id proteins. Am J Surg Pathol 2011. Oct;35(10):1463–72. [DOI] [PubMed] [Google Scholar]
- 92.Zhang C, Lam SSY, Leung GMK, Tsui S-P, Yang N, Ng NKL, et al. Sorafenib and omacetaxine mepesuccinate as a safe and effective treatment for acute myeloid leukemia carrying internal tandem duplication of Fms-like tyrosine kinase 3. Cancer 2020. Jan 15;126(2):344–53. [DOI] [PubMed] [Google Scholar]
- 93.Wojnarowicz PM, Lima E, Silva R, Ohnaka M, Lee SB, Chin Y, Kulukian A, et al. A Small-Molecule Pan-Id Antagonist Inhibits Pathologic Ocular Neovascularization. Cell Rep 2019. Oct 1;29(1):62–75.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wojnarowicz PM, Escolano MG, Huang Y-H, Desai B, Chin Y, Shah R, et al. Anti-tumor effects of an ID antagonist with no observed acquired resistance. NPJ Breast Cancer 2021. May 24;7(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.