

Abstract
Mitochondrial calcium (mCa2+) uptake couples changes in cardiomyocyte energetic demand to mitochondrial ATP production. However, excessive mCa2+ uptake triggers permeability transition and necrosis. Despite these established roles during acute stress, the involvement of mCa2+ signaling in cardiac adaptations to chronic stress remains poorly defined. Changes in NCLX expression are reported in human heart failure (HF) and models of cardiac hypertrophy. Therefore, we hypothesized that altered mCa2+ homeostasis contributes to the hypertrophic remodeling of the myocardium that occurs upon a sustained increase in cardiac workload. The impact of mCa2+ flux on cardiac function and remodeling was examined by subjecting mice with cardiomyocyte-specific overexpression (OE) of the mitochondrial Na+/Ca2+ exchanger (NCLX), the primary mediator of cardiac mCa2+ efflux, to several well-established models of hypertrophic and non-ischemic HF. Cardiomyocyte NCLX-OE preserved contractile function, prevented hypertrophy and fibrosis, and attenuated maladaptive gene programs in mice subjected to chronic pressure overload. Hypertrophy was attenuated in NCLX-OE mice, prior to any decline in cardiac contractility. NCLX-OE similarly attenuated deleterious cardiac remodeling in mice subjected to chronic neurohormonal stimulation. However, cardiomyocyte NCLX-OE unexpectedly reduced overall survival in mice subjected to severe neurohormonal stress with angiotensin II + phenylephrine. Adenoviral NCLX expression limited mCa2+ accumulation, oxidative metabolism, and de novo protein synthesis during hypertrophic stimulation of cardiomyocytes in vitro. Our findings provide genetic evidence for the contribution of mCa2+ to early pathological remodeling in non-ischemic heart disease, but also highlight a deleterious consequence of increasing mCa2+ efflux when the heart is subjected to extreme, sustained neurohormonal stress.
Keywords: mitochondria, calcium, HF, NCLX
Graphical abstract
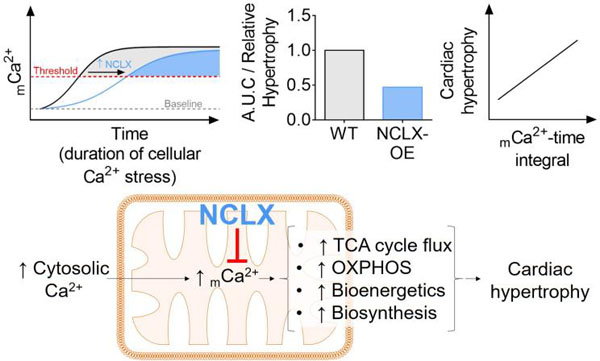
1. INTRODUCTION
Cardiomyocytes continuously fine-tune their metabolism to ensure an adequate ATP supply to fuel myofilament cross-bridge cycling and maintain ionic homeostasis in the face of constantly changing workloads. Calcium (Ca2+) released from the sarcoplasmic reticulum (SR) following each cardiac action potential activates myofilament cross-bridge cycling to enable contraction of the heart. Simultaneously, Ca2+ is transmitted to mitochondria to stimulate TCA cycle dehydrogenases and ATP synthesis. Thus, Ca2+ is proposed as the key second messenger that coordinates the parallel activation of myocardial energy consumption and production [1, 2].
Ca2+ released from the SR rapidly enters the mitochondrial matrix via the mitochondrial calcium uniporter channel complex (mtCU), a multi-protein channel that spans the inner mitochondrial membrane [3, 4]. The mitochondrial calcium uniporter protein (MCU) forms the Ca2+-selective pore of the mtCU and enables the efficient uptake of Ca2+ near SR release sites when the concentration rises above ~400nM [5, 6]. The mtCU is regulated by a number of subunits including EMRE, MICU1, MICU2, MICU3, MCUR1, and MCUB, which together control the permeation and Ca2+-sensitive gating of the mtCU [7–19]. Recent investigations employing genetic manipulation of MCU or the regulatory subunits of the mtCU have revealed that acute Ca2+ uptake through the uniporter is necessary to increase mitochondrial metabolism to support a sudden increase in cardiac work rate [20–25]. This phenomenon is critical for situations that require a rapid but transient increase in cardiac output, such as sympathetic fight-or-flight responses. In contrast, excessive mCa2+ uptake through the mtCU is detrimental in settings of acute pathological cellular stress, such as occurs during cardiac ischemia-reperfusion (IR). Here, excessive mCa2+ uptake and matrix Ca2+ accumulation triggers mitochondrial permeability transition and necrotic cell death [26] (reviewed in [27, 28]). Blockade of mtCU-dependent mCa2+ uptake in the adult heart via inducible genetic disruption of MCU [21, 22] or inducible overexpression of the inhibitory mtCU subunit, MCUB [25], is sufficient to protect the heart from IR injury and cardiomyocyte death.
Precisely how altered mCa2+ homeostasis affects the heart’s adaption to chronic rather than acute stress remains controversial. Altered cytosolic Ca2+ cycling including increased cellular Ca2+ influx; increased SR Ca2+ leak leading to diminished SR Ca2+ load and decreased cytosolic Ca2+ transient amplitude; slowed Ca2+ transient decay; and increased diastolic Ca2+ levels are reported in settings of cardiac pressure overload caused by aortic stenosis or chronic hypertension [29–39]. These same features of aberrant Ca2+ handling are observed in chronic heart diseases such as hypertrophic cardiomyopathy [40–48]. These reports show a clear association between altered cardiomyocyte calcium signaling, cardiac hypertrophy, and impaired contractile function. However, much less is known regarding whether such long-term alterations in cytosolic Ca2+ handling lead to coordinate changes in mitochondrial Ca2+ homeostasis, and what impact any chronic changes in mCa2+ may have on the development or progression of non-ischemic heart disease.
Several studies suggest that mtCU activity is impaired in the end-stage failing heart [49, 50], and that moderately increasing mCa2+ uptake in this context can improve contractile function [51]. In contrast, other reports indicate that cardiac MCU protein expression increases in experimental pressure overload-induced cardiac hypertrophy and in patients with aortic stenosis [52, 53]. These observations raise the possibility that mCa2+ uptake and mitochondrial matrix Ca2+ are elevated when cardiac workload and cytosolic Ca2+ levels are chronically elevated, just as they are when SR Ca2+ leak is increased in the post-infarction failing heart [54]. The findings that cardiac mCa2+ content is enhanced due to increased MCU expression and mCa2+ uptake along with reduced NCLX expression and mCa2+ efflux, and that heart size is increased in a mouse model of mitochondrial cardiomyopathy due to Tfam deletion [55], provide additional support for an association between mCa2+ signaling and cardiac hypertrophy. Indeed, increased mCa2+ content may help to fuel cardiac growth because it can stimulate TCA cycle flux [56–62] and respiratory complex activity [63, 64] to increase cellular ATP production needed to power anabolic reactions including protein synthesis. Chronic elevation of mCa2+ in the face of sustained cellular Ca2+ stress can also contribute to deleterious myocardial remodeling and cardiac dysfunction by predisposing to mCa2+ overload, cardiomyocyte death, and subsequent replacement fibrosis [28, 65–67]. Further complicating the question of how altered mCa2+ homeostasis impacts the heart’s response to chronic pathological stress is evidence that diminished mCa2+ content resulting from increased Na+-driven mitochondrial Na+/Ca2+ exchange in end-stage heart failure (HF) limits mitochondrial energetics, perturbs redox state, and further impairs cardiac contractility [68, 69].
Early experiments aiming to clarify the role of mCa2+ signaling in the heart’s adaptation to chronic stress attempted to limit mCa2+ accumulation by targeting rapid mCa2+ uptake through the mtCU. However, neither constitutive nor inducible genetic Mcu deletion showed any effect to prevent cardiac hypertrophy or preserve contractile function in mice subjected to chronic pressure overload with transverse aortic constriction [22, 70]. These studies also yielded inconsistent results on the efficacy of genetic Mcu disruption in altering steady-state mCa2+ content over the long-term, and so left the question of mCa2+’s role in the heart’s response to chronic stress unresolved. Chronic pharmacological inhibition of MCU with ruthenium red was recently reported to attenuate cardiac hypertrophy and improve contractile function in response to transverse aortic constriction [53]. Interpretation of these findings is confounded, though, by the fact that ruthenium red can affect the activity of other cellular Ca2+ channels such as the L-type calcium channel and ryanodine receptor [71–74]. Given the challenges in specifically manipulating mCa2+ homeostasis over chronic time scales via genetic or pharmacological targeting of MCU, we sought an alternative approach to perturb mCa2+ signaling within the heart.
Our laboratory recently demonstrated that the mitochondrial Na+/Ca2+ exchanger, NCLX, is the primary physiological route for mitochondrial Ca2+ efflux in cardiomyocytes [75]. We also reported that NCLX expression modestly increases in end-stage ischemic hearts at the time of transplantation [75]. NCLX extrudes 1 Ca2+ ion from the mitochondrial matrix in exchange for the import of 3 Na+ ions, and constitutes the rate-limiting step of mitochondrial Ca2+ exchange [76–79]. Modulation of net mitochondrial NCLX activity therefore can exert a substantial influence over mitochondrial Ca2+ homeostasis, and so offers an alternative strategy to MCU modulation for investigating the role of mCa2+ in cardiac responses to chronic stress. Here, we used mice with cardiomyocyte-specific overexpression of NCLX to test the hypothesis that mCa2+ signaling contributes to pathological cardiac remodeling and HF in response to sustained elevations in cardiac workload and cytosolic Ca2+ cycling. We employed three surgical models of chronically increased hemodynamic or neurohormonal load on the heart and assessed the impact of cardiomyocyte NCLX overexpression on contractile function, cardiac hypertrophy, and fibrotic remodeling of the myocardium. We then utilized an in vitro model of cardiomyocyte hypertrophy to examine the mechanism by which NCLX overexpression limits pathological cardiac growth. Our findings support a critical role for mCa2+ in driving early hypertrophic remodeling and subsequent contractile dysfunction in non-ischemic heart disease, but also indicate that limiting mCa2+ accumulation can be fatal under conditions of extreme neurohormonal stress.
2. RESULTS
2.1. Transgenic expression of NCLX in cardiomyocytes protects against pressure overload-induced cardiac remodeling and failure.
We recently developed and characterized mice with cardiomyocyte-specific, doxycycline-controlled overexpression of NCLX [75] (Fig. 1A). This model exhibits a ~2-fold increase in cardiac NCLX protein expression in adult mice (Fig. 1B–C; full-length blots shown in Extended Fig. 1) and results in a ~38% increase in the mCa2+ efflux rate during electrical pacing in intact, isolated adult cardiomyocytes, with no significant effect on the rate of mCa2+ influx [75]. To test how increased capacity for mCa2+ efflux affects the heart’s response to chronic hemodynamic stress, following the removal of doxycycline to allow for transgene activation we subjected mice with cardiomyocyte-specific NCLX overexpression (TRE-NCLX x αMHC-tTA, NCLX-Tg) and control mice (αMHC-tTA) to 12 weeks of pressure overload induced by transverse aortic constriction (TAC) (Fig. 1D). No significant differences in survival were observed among experimental groups (Supplemental Fig. S1A). αMHC-tTA mice exhibited a gradual increase in left ventricular end systolic dimension (LVESD) and a corresponding decrease in left ventricular fractional shortening (%FS) during the 12 weeks of TAC (Fig. 1 E–G). Both the increase in LVESD and decrease in %FS were attenuated in NCLX-Tg mice (Fig. 1 E–G). These findings suggest that enhancing cardiomyocyte mCa2+ efflux is sufficient to prevent the decline in contractile function that typically occurs in response to a sustained increase in cardiac afterload. We further examined the impact of NCLX overexpression on pathological cardiac remodeling during pressure overload. The TAC-mediated increase in the heart weight-to-tibia length (HW/TL) ratio (Fig. 1H) and increase in cardiomyocyte cross-sectional area (CSA) (Fig. 1 I–J) observed in αMHC-tTA control mice, were both attenuated with NCLX overexpression (36% increase in αMHC-tTA HW/TL and 16% increase in NCLX-Tg HW/TL vs. sham; 63% increase in αMHC-tTA CSA and 26% increase in NCLX-Tg CSA vs. sham). Likewise, the extent of myocardial fibrosis resulting from 12 weeks of TAC was diminished with NCLX overexpression (Fig. 1 K–L). Together, these data indicate that increasing cardiomyocyte NCLX activity protects against pressure overload-induced pathological cardiac remodeling.
Figure 1: Cardiomyocyte-specific NCLX overexpression protects against pressure overload-induced HF and pathological remodeling.
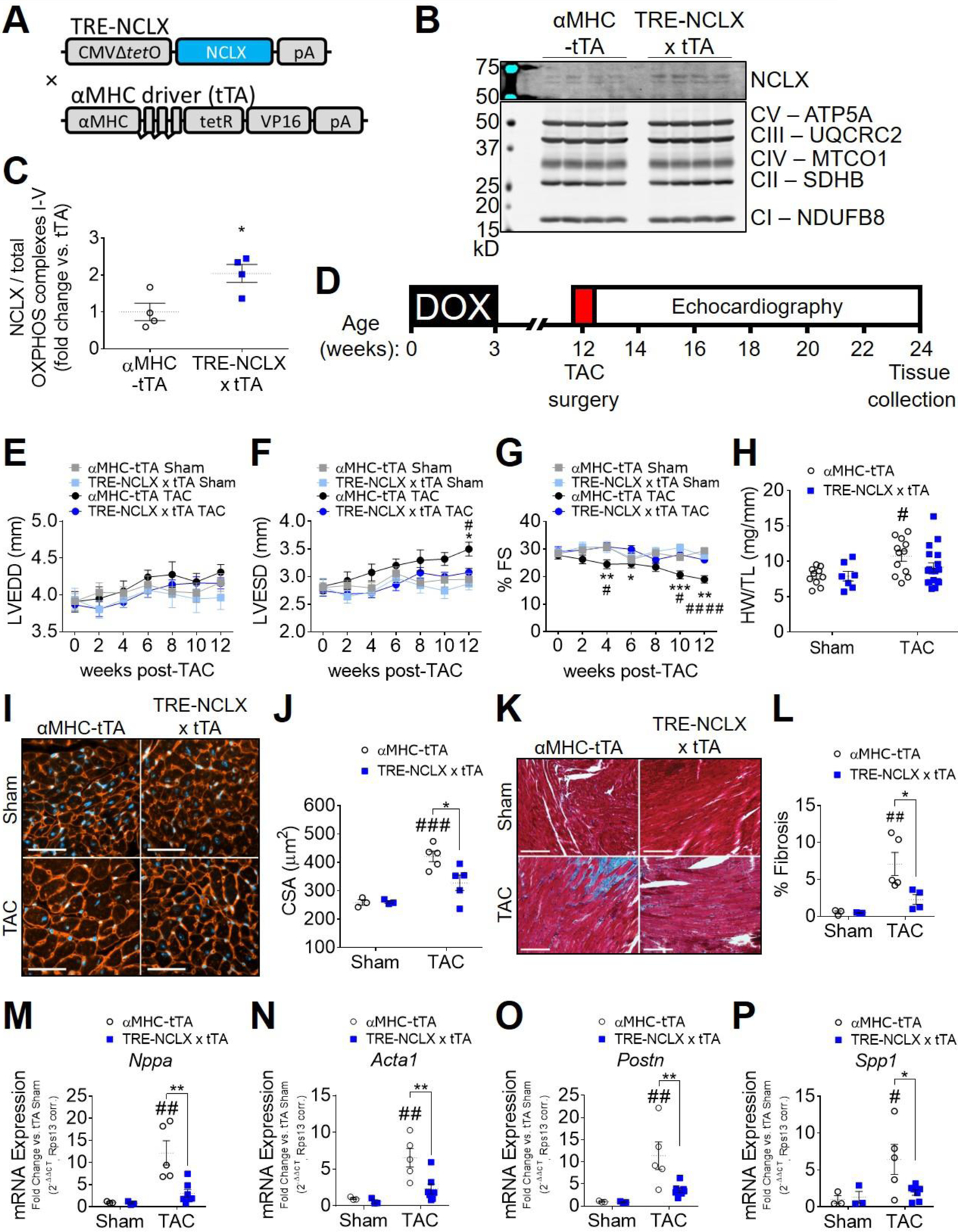
A) Genetic approach for doxycycline-controlled, cardiomyocyte-specific expression of a human NCLX transgene in mice. TRE-NCLX mice expressing human NCLX cDNA under the control of a tetracycline-responsive promoter were crossed to αMHC-tTA (cardiomyocyte-specific, doxycycline-off) transgenic mice [75]. All mice were fed doxycycline-containing chow during development and through three weeks of age to allow for normal cardiac development. NCLX transgene expression was induced by removal of doxycycline at the time of weaning at 3 weeks of age. B-C) Western blots for NCLX protein expression in heart lysates from adult αMHC-tTA and TRE-NCLX x αMHC-tTA mice, and quantification of NCLX expression normalized to total OXPHOS complexes I-V as a mitochondrial loading control. Data analyzed by t-test. *P<0.05. (n=4 mice/group). D) Timeline of transverse aortic constriction (TAC) experimental protocol. DOX, doxycycline administration. Left ventricular end diastolic dimension (LVEDD) (E), end systolic dimension (LVESD) (F), and percent fractional shortening (%FS) (G) over 12 weeks after TAC or sham surgery. Data analyzed by 2-way ANOVA with Tukey’s post-hoc test. *P <0.05, **P<0.01, ***P<0.001 αMHC-tTA TAC vs. TRE-NCLX x αMHC-tTA TAC. #P <0.05, ####P<0.0001 αMHC-tTA sham vs. TAC. (n=7–18 mice / group). H) Heart weight-to-tibia length ratio (HW/TL) 12 weeks post sham or TAC surgery. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05 vs. sham. (n=7–18 mice / group). I) Left ventricular tissue at 12 weeks post sham or TAC surgery, stained with wheat germ agglutinin (WGA, red) to delineate sarcolemma and with DAPI (blue). Scale bars = 50 µm. J) Quantification of cardiomyocyte cross-sectional area (CSA). Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ###P<0.001 vs. sham; *P < 0.05 αMHC-tTA vs. TRE-NCLX x αMHC-tTA. (n=3–5 mice / group). K) Masson’s trichrome stain for myocardial collagen deposition (blue) at 12 weeks post-surgery. Scale bars = 100 µm. L) Quantification of fibrotic area as percent of tissue area. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01 vs. sham; *P < 0.05 αMHC-tTA vs. TRE-NCLX x αMHC-tTA. (n=3–5 mice / group). qPCR quantification of mRNA expression of fetal (M-N) and pro-fibrotic (O-P) genes in hearts of mice 12 weeks after sham or TAC surgeries. Nppa, natriuretic peptide type A; Acta1, α-skeletal muscle actin; Postn, periostin; Spp1, osteopontin. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05, ##P<0.01 vs. sham; *P < 0.05, **P < 0.01 αMHC-tTA vs. TRE-NCLX x αMHC-tTA. (n=3–7 mice / group).
Examination of cardiac gene expression revealed that 12 weeks of TAC increased the expression of fetal genes such as atrial natriuretic peptide (Nppa or ANP), α-skeletal muscle actin (Acta1), and brain natriuretic peptide (Nppb or BNP) (Fig. 1 M–N; Supplemental Fig. S1B) in control αMHC-tTA mice. The ratio of α-myosin heavy chain (Myh6) to β-myosin heavy chain (Myh7) gene expression in αMHC-tTA hearts was also decreased by 12 weeks of TAC (Supplemental Fig. S1D). The expression of such developmental genes is consistent with the re-activation of the fetal gene program that is classically observed in pathological cardiac hypertrophy [80]. The induction of Nppa, Acta1, and Nppb by 12 weeks of TAC was attenuated in NCLX overexpressor mice (Fig. 1 M–N; Supplemental Fig. S1B). This result is in agreement with our observation that NCLX overexpression limited the extent of TAC-induced cardiac hypertrophy (Fig. 1 H–J). Consistent with our observation that NCLX overexpression limited TAC-induced cardiac fibrosis (Fig. 1 K–L), the expression of pro-fibrotic genes such as periostin (Postn) and osteopontin (Spp1) with 12 weeks of TAC was reduced in NCLX-Tg mice (Fig. 1 O–P).
2.2. Cardiomyocyte NCLX overexpression attenuates early pressure overload-induced cardiac hypertrophy
To better understand the temporal dynamics of how enhanced cardiomyocyte NCLX expression and mCa2+ efflux capacity alters the heart’s response to chronic pressure overload, we also evaluated cardiac pathology at an earlier time point, 2 weeks after TAC-induced pressure overload (Fig. 2A). No significant contractile dysfunction or changes in left ventricular dimensions were yet apparent at this early stage in the hearts of either control αMHC-tTA mice or TRE-NCLX x αMHC-tTA NCLX overexpressor mice subjected to TAC (Fig. 1 E–G). However, the hearts of αMHC-tTA mice did exhibit hypertrophy, indicated by a significant increase in heart weight-to-tibia length ratio (53% increase in αMHC-tTA HW/TL) and cardiomyocyte cross-sectional area (49% increase in αMHC-tTA CSA) as compared to sham animals (Fig. 2 B–D). In contrast, no significant increase in either the heart weight-to-tibia length ratio or cardiomyocyte cross-sectional area was detected in NCLX-Tg mice (Fig. 2B–D).
Figure 2: Cardiomyocyte NCLX overexpression attenuates early pressure overload-induced cardiac hypertrophy.
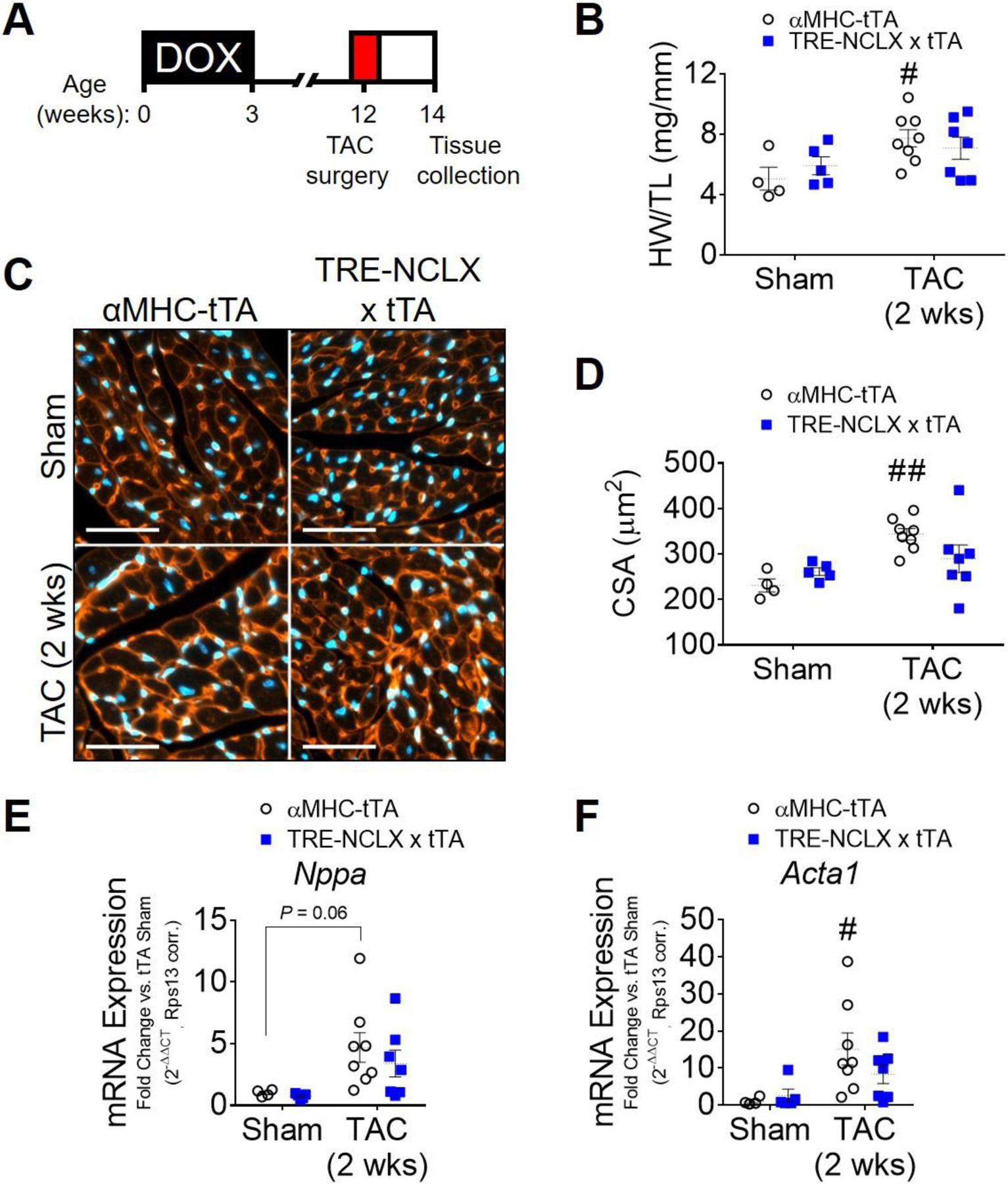
A) Timeline of 2-week transverse aortic constriction experimental protocol. DOX, doxycycline administration. B) Heart weight-to-tibia length ratio (HW/TL) of αMHC-tTA and TRE-NCLX x αMHC-tTA mice at 2 weeks post sham or TAC surgery. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05 vs. sham. (n=4–8 mice / group). C) Left ventricular tissue at 2 weeks post sham or TAC surgery, stained with wheat germ agglutinin (WGA, red) to delineate sarcolemma and with DAPI (blue). Scale bars = 50 µm. D) Quantification of cardiomyocyte cross-sectional area (CSA). Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01 vs. sham. (n=4–8 mice / group). E-F) qPCR quantification of mRNA expression of fetal genes in hearts of mice 2 weeks after sham or TAC surgeries. Nppa, natriuretic peptide type A; Acta1, α-skeletal muscle actin. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05 vs. sham. (n=4–8 mice / group).
We observed a significant induction of the expression of the fetal gene Acta1 and a trend towards increased expression of Nppa, in the hearts of αMHC-tTA mice following 2-week TAC (Fig. 2 E–F). In contrast, the expression of Nppa and Acta1 was not increased in NCLX overexpressor mice (Fig. 2 E–F). Consistent with this 2-week timepoint falling within the early stages of cardiac stress responses and remodeling, the expression of Npr1 was not yet changed after 2-weeks of TAC (Supplemental Fig. S2A); however, in agreement with our results for the 12-week TAC study (Supplemental Fig. S1D), the ratio of Myh6/Myh7 expression was significantly reduced by 2-week TAC in animals of both genotypes (Supplemental Fig. S2B). Nppb expression was likewise increased after 2-weeks of TAC in NCLX-Tg mice (Supplemental Fig. S2C). These results collectively suggest that NCLX overexpression and enhanced capacity for cardiomyocyte mCa2+ efflux attenuates the early hypertrophic response to chronic pressure overload, and abrogates or delays the activation of certain specific stress-responsive gene programs within the heart. Protein expression of the mtCU components MCU, EMRE, and MICU1 did not differ between sham αMHC-tTA and NCLX-Tg hearts under basal conditions, although MCU expression tended to increase in NCLX-Tg hearts with 2 weeks of TAC (Supplemental Fig. S3A–B). This may represent a compensatory response that develops over time during hypertrophic stimulation in the NCLX-Tg hearts in order to increase net mCa2+ accumulation in the face of increased NCLX-dependent mCa2+ extrusion. Interestingly, EMRE protein expression decreased significantly with 2 weeks of TAC only in the hearts of αMHC-tTA mice (Supplemental Fig. S3C), consistent with the notion that these hearts are experiencing some degree of mCa2+ overload that then elicits a compensatory downregulation of mCa2+ uptake. That we did not detect a significant decrease in EMRE expression in NCLX-Tg hearts with 2 weeks of TAC further supports the idea that NCLX overexpression is sufficient to limit net mCa2+ accumulation in response to pressure overload.
We previously demonstrated that NCLX overexpression is a powerful approach to minimize mCa2+ overload and cell death during acute cellular Ca2+ stress that occurs in cardiac IR injury and myocardial infarction [75]. Like IR injury, chronic pressure overload increases cytosolic Ca2+ levels [33, 38] and can culminate in cardiomyocyte dropout [65–67]. Therefore, we hypothesized that augmenting NCLX function in this context would be cardioprotective. In contrast to the appreciable fibrosis observed in αMHC-tTA hearts at 12 weeks post-TAC (Fig. 1 K–L), when examined at 2 weeks post-TAC neither genotype yet showed an increase in cardiac fibrosis (Supplemental Fig. S2D). The expression of Postn, a marker of activated fibroblasts [81], Spp1, Tgfb1, and α-smooth muscle actin (Acta2) were not yet significantly increased by this short-term stress (Supplemental Fig. S2 E–H). This result supports the notion that this 2-week TAC timepoint is prior to any substantial increase in myofibroblast activation or differentiation [82]. This suggests that NCLX overexpression limits pressure overload-induced cardiomyocyte hypertrophy, prior to any distinct role in preventing cardiomyocyte death and replacement fibrosis.
2.3. Cardiomyocyte NCLX overexpression attenuates neurohormonal-induced cardiac hypertrophy.
Next, to validate the effects of NCLX overexpression on cardiac hypertrophy in a second, independent in vivo model of sustained cellular Ca2+ stress, we implanted NCLX-Tg and control mice with osmotic minipumps to deliver a low dose of angiotensin II (AngII) for 2 weeks (1.44mg/kg/day, Fig. 3A). This treatment paradigm causes an increase in blood pressure and induces cardiac hypertrophy in mice [83, 84]. Angiotensin II also directly enhances cytosolic Ca2+ cycling in cardiomyocytes via activation of angiotensin II type 1 receptors [85, 86], which contributes to an increase in mCa2+ content [87].
Figure 3: Cardiomyocyte NCLX overexpression attenuates hypertrophy in mice infused with angiotensin II.
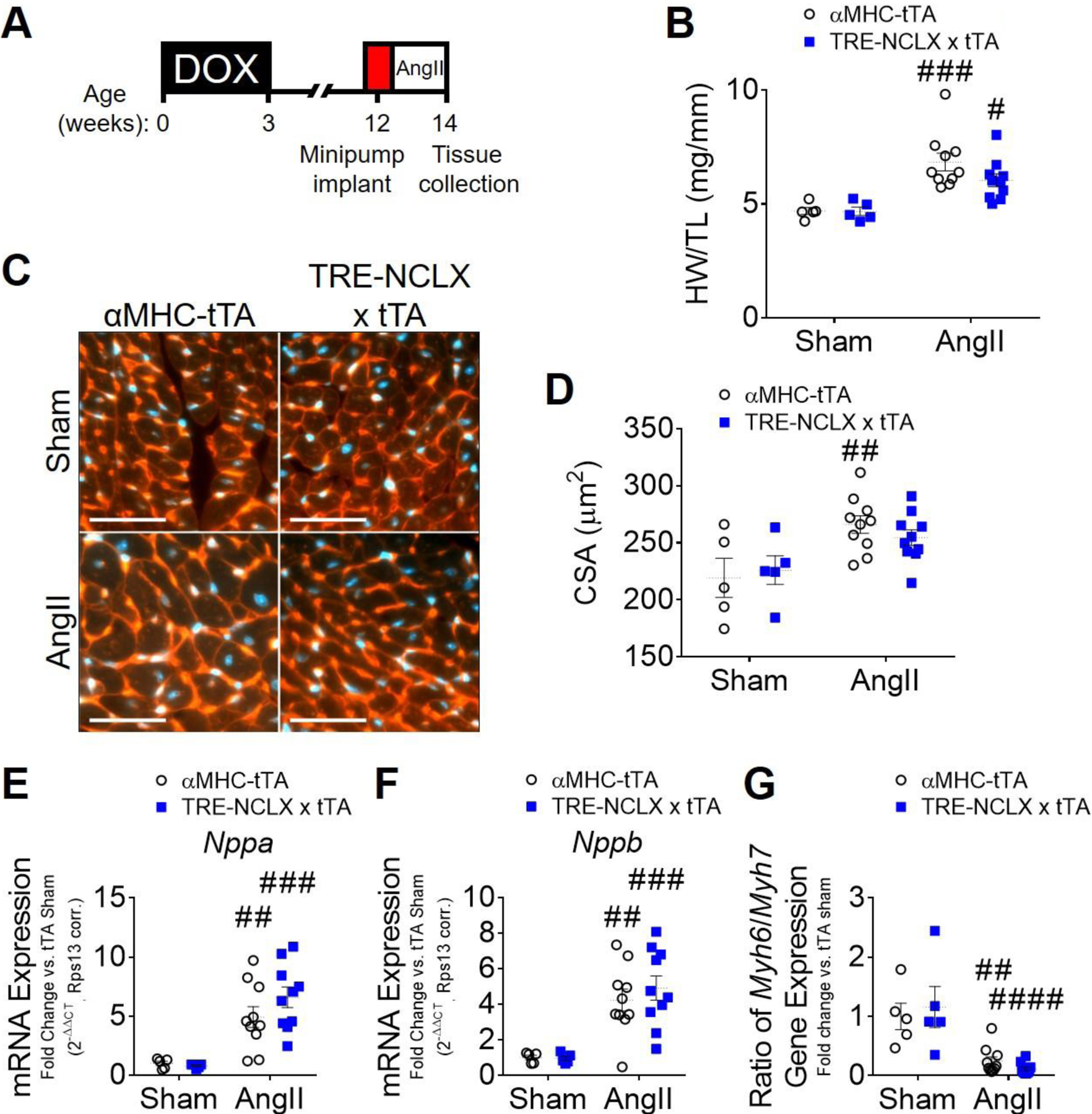
A) Timeline of angiotensin II (AngII) infusion experimental protocol. DOX, doxycycline administration. B) Heart weight-to-tibia length ratio (HW/TL) of αMHC-tTA and TRE-NCLX x αMHC-tTA mice 2 weeks after sham or angiotensin II minipump implantation surgery. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05, ###P<0.001 vs. sham. (n=5–10 mice / group). C) Left ventricular tissue 2 weeks after sham or angiotensin II minipump implantation surgery, stained with wheat germ agglutinin (WGA, red) to delineate sarcolemma and with DAPI (blue). Scale bars = 50 µm. D) Quantification of cardiomyocyte cross-sectional area (CSA). Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01 vs. sham. (n=5–10 mice / group). E-G) qPCR quantification of mRNA expression of fetal genes in hearts of αMHC-tTA and TRE-NCLX x αMHC-tTA mice 2 weeks after sham or angiotensin II minipump implantation surgery. Nppa, natriuretic peptide type A; Nppb, natriuretic peptide type b; Myh6, α-myosin heavy chain; Myh7, β-myosin heavy chain. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01, ###P<0.001, ####P<0.0001 vs. sham. (n=5–10 mice / group).
Two weeks of low-dose AngII infusion had no detrimental effect on survival or cardiac function (Supplemental Fig. S4 A–D). AngII treatment was sufficient to cause cardiac hypertrophy as indicated by an increase in the heart weight-to-tibia length ratio (46% increase in αMHC-tTA vs. sham; compared to 29% increase in NCLX-Tg vs. sham); and by an increase in cardiomyocyte cross-sectional area (21% increase in αMHC-tTA vs. sham) in control αMHC-tTA mice (Fig. 3 B–D). We again noted a trend towards increased MCU protein expression in the hearts of NCLX-Tg mice subjected to AngII infusion (Supplemental Figure S5). Consistent with our findings in pressure overload (Fig. 1–2), cardiomyocyte cross-sectional area was not significantly increased by 2-week AngII infusion in NCLX overexpressor mice (Fig. 3 C–D). Interestingly, despite this attenuation of AngII-induced cardiomyocyte hypertrophy in NCLX overexpressor mice, we still observed a significant induction of fetal genes including Nppa, Nppb, and Acta1, and a significant reduction in the Myh6/7 ratio, in the hearts of both αMCH-tTA and NCLX-Tg mice with 2 weeks of AngII infusion (Fig. 3 E–G; Supplemental Fig. S4 E–F). These findings indicate that NCLX overexpressor hearts still sense and respond to angiotensin II stimulation with expected changes in gene expression. This observation raises the intriguing possibility that enhanced mCa2+ efflux may slow stress-induced cardiomyocyte growth by limiting anabolic processes needed for an increase in cell mass, despite preserved activation of mitochondrial-independent signaling pathways classically associated with hypertrophy.
2.4. Cardiomyocyte NCLX overexpression attenuates pathological remodeling but reduces survival in mice infused with high-dose angiotensin II + phenylephrine.
Apart from attenuating cardiac hypertrophy, our findings indicated that the other major protective effect of cardiomyocyte NCLX overexpression on cardiac remodeling following TAC was to minimize the extent of fibrosis throughout the heart (Fig. 1 K–L), likely by minimizing mCa2+ overload-induced cardiomyocyte dropout and replacement fibrosis. To further explore this notion, as well as the proposed effect of Na+-driven mCa2+ extrusion through NCLX to starve the mitochondria of Ca2+ and compromise cellular energetics in settings of severe cardiac stress such as end-stage HF [50, 68, 69], we next evaluated cardiac function and remodeling using a more extreme chronic cytosolic Ca2+ stress model. Here, we subjected αMHC-tTA and TRE-NCLX x αMHC-tTA mice to 4-week osmotic minipump infusion of combined high doses of the Gq-coupled receptor agonists angiotensin II (10mg/kg/day) and phenylephrine (50mg/kg/day) (AngII/PE) (Fig 4A).
Figure 4: Cardiomyocyte NCLX overexpression attenuates remodeling but reduces survival in mice infused with chronic high-dose angiotensin II + phenylephrine.
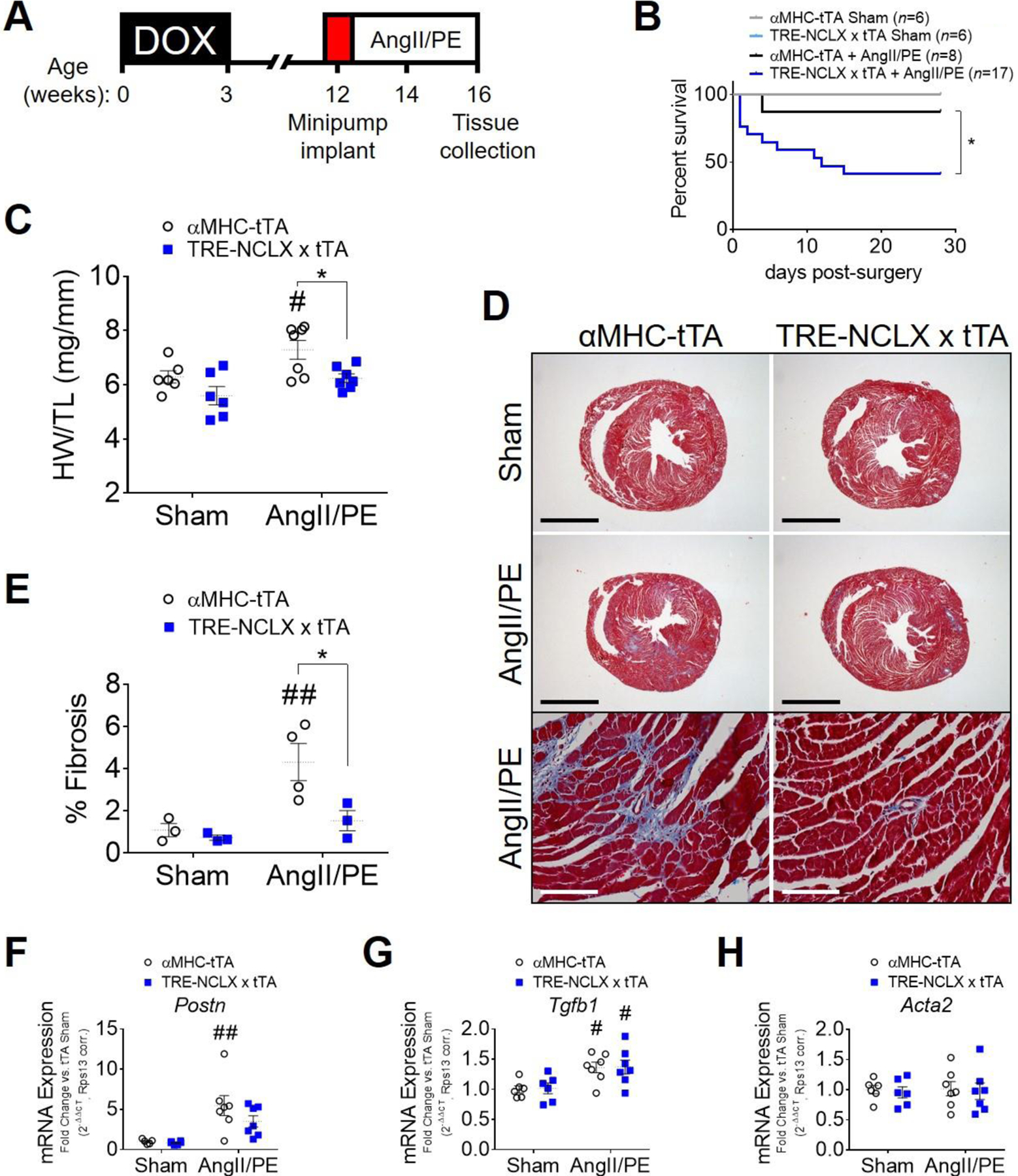
A) Timeline of angiotensin II + phenylephrine (AngII/PE) infusion experimental protocol. DOX, doxycycline administration. B) Kaplan-Meier survival curve of αMHC-tTA and TRE-NCLX x αMHC-tTA mice for 4 weeks after sham or AngII/PE osmotic minipump implantation surgeries. The number of animals in each group at the start of the study is indicated in parentheses. Data analyzed by log-rank (Mantel-Cox) test. *P<0.05 αMHC-tTA + AngII/PE vs. TRE-NCLX x αMHC-tTA + AngII/PE. C) Heart weight-to-tibia length ratio (HW/TL) of αMHC-tTA and TRE-NCLX x αMHC-tTA mice 4 weeks after sham or AngII/PE minipump implantation surgery. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05 vs. sham; *P < 0.05 αMHC-tTA vs. TRE-NCLX x αMHC-tTA. (n=5–7 mice / group). D) Masson’s trichrome stain for myocardial collagen deposition (blue) in αMHC-tTA and TRE-NCLX x αMHC-tTA hearts 4 weeks after sham or AngII/PE minipump implantation surgery. Black scale bars for whole-heart cross sections = 2mm. White scale bars for higher magnification micrographs (bottom row) = 100 µm. E) Quantification of fibrotic area as percent of tissue area. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01 vs. sham; *P < 0.05 αMHC-tTA vs. TRE-NCLX x αMHC-tTA. (n=3–4 mice / group). F-H) qPCR quantification of mRNA expression of pro-fibrotic genes in hearts of mice after 4 weeks of AngII/PE infusion. Postn, periostin; Tfgb1, transforming growth factor β−1; Acta2, α-smooth muscle actin. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05, ##P<0.01 vs. sham. (n=5–7 mice / group).
In stark contrast to the other models examined above, high-dose AngII/PE infusion caused a significant increase in mortality, specifically among NCLX overexpressor mice, with most death occurring during the first 2 weeks of infusion (Fig. 4B). Echocardiography over this time span revealed a transient decrease in left ventricular end diastolic and end systolic dimensions, with a corresponding increase in fractional shortening, in TRE-NCLX x αMHC-tTA mice after 1 week of AngII/PE infusion (Supplemental Fig. S6 A–C). The functional difference subsided by 2 weeks and little death was observed through the remainder of the study (Supplemental Fig. S6C; Fig. 4B). We were able to harvest cardiac tissue from a small number of the TRE-NCLX x αMHC-tTA mice that died spontaneously during the first two weeks of AngII/PE infusion. von Kossa staining of these hearts revealed substantial Ca2+ deposition throughout the myocardium (Supplemental Fig. S6D), consistent with cellular Ca2+ overload and necrosis as has been reported in reported previously in other mouse models of pathological cardiomyocyte Ca2+ loading [88]. We did not detect any positive von Kossa staining in control TRE-NCLX x tTA hearts collected up to 24 hours post-euthanasia, indicating that such Ca2+ deposition was not merely a post-mortem artifact. We also found no evidence for aortic aneurysm among animals that died spontaneously. These incidental findings suggest that when paired with extreme, sustained cytosolic Ca2+ stress, cardiac NCLX overexpression can be detrimental, perhaps because it compromises mitochondrial Ca2+ buffering capacity and/or mitochondrial bioenergetics within the heart.
In control αMHC-tTA mice that survived for the duration of the 4-week study, the combined high dose of AngII/PE elicited significant cardiac hypertrophy (16% increase in αMHC-tTA HW/TL vs. sham) (Fig. 4C). Consistent with our previous models, cardiac hypertrophy was attenuated in NCLX-Tg mice that survived to study termination (11% increase in NCLX HW/TL vs. sham) (Fig. 4C). MCU protein expression was increased in the hearts of NCLX-Tg mice subjected to AngII/PE infusion (Supplemental Figure S7 A–B), once again suggestive of an eventual compensatory upregulation of mCa2+ uptake in response to hypertrophic stimulation. Such upregulation of MCU may counteract mCa2+ depletion or exaggerated cytosolic Ca2+ overload resulting from diminished mCa2+ buffering in the NCLX-Tg hearts. Given enough time, it may also allow sufficient mCa2+ accumulation to allow for some pathological cardiac growth in NCLX-Tg hearts, albeit at slower rate as compared to the control genotype.
Chronic AngII/PE infusion caused cardiac injury as indicated by patchy fibrotic scarring throughout the myocardium of αMHC-tTA mice that survived to study termination, and this fibrotic effect was attenuated in in surviving NCLX-Tg mice (Fig. 4 D–E). Chronic AngII/PE infusion significantly increased the expression of Postn and Tgfb1 in αMHC-tTA mice (Fig. 4 F–H). The finding that Acta2 expression did not increase with AngII/PE infusion (Fig. 4H), despite increased cardiac fibrosis, is in agreement with our findings in the 12-week TAC model (Supplemental Fig. S1F). This observation may reflect that hearts subjected to 12-week TAC or 4-week AngII/PE treatment are in a more chronic phase of the fibrotic response, where the transient expression of some fibrotic genes such as α-smooth muscle actin subsides as myocardial scar tissue matures [81, 89]. Consistent with our finding of reduced fibrotic deposition in NCLX-Tg hearts after 4 weeks of AngII/PE infusion (Fig. 4 D–E), Postn expression in the hearts of TRE-NCLX x αMHC-tTA mice was not significantly increased by AngII/PE treatment (Fig. 4F). Together, these findings support the notion that increasing mCa2+ efflux is sufficient to limit myocardial fibrosis in settings of chronic Ca2+ overload, likely by minimizing cardiomyocyte death and subsequent replacement scarring of the myocardium. The results of this high-dose AngII/PE model further reveal that although increased cardiomyocyte NCLX activity consistently mitigates pathological cardiac remodeling over chronic time scales, it also has deleterious effects that negatively impact overall survival in the context of extreme neurohormonal stress that may cause severe cytosolic Ca2+ overload.
2.5. NCLX expression attenuates mCa2+ accumulation, oxidative metabolism, and cardiomyocyte hypertrophy in vitro.
Finally, we employed a reductionist in vitro model of cardiac hypertrophy using neonatal rat ventricular myocytes (NRVMs) transduced with adenovirus encoding human NCLX (Ad-NCLX) or control adenovirus encoding β-galactosidase (Ad-LacZ) to investigate the cellular mechanism by which NCLX overexpression limits pathological cardiomyocyte growth. 48hrs of phenylephrine stimulation increased steady-state mitochondrial Ca2+ content as indicated by an increase in resting mito-R-GECO1 fluorescence in NRVMs transduced with Ad-LacZ (Fig. 5A and Supplemental Fig. S8A). Adenoviral NCLX expression attenuated the phenylephrine-induced increase in mCa2+ (Fig. 5A and Supplemental Fig. S8A), consistent with an increased capacity for mCa2+ efflux. Phenylephrine treatment increased cytosolic Ca2+ concentration, as measured by the ratiometric Ca2+-sensitive dye Fura-2, AM, in NRVMs transduced with either Ad-LacZ or Ad-NCLX (Fig. 5B and Supplemental Fig. S8B). Notably, cytosolic Ca2+ was significantly higher in PE-treated NRVMs transduced with Ad-NCLX than those transduced with Ad-LacZ, indicating that the reduction in mCa2+ accumulation with exogenous NCLX expression is not attributable to diminished cytosolic Ca2+ loading. This finding further supports the notion that during hypertrophic stimulation, NCLX overexpression limits or delays mitochondrial Ca2+ accumulation. Phenylephrine stimulation likewise increased mitochondrial superoxide production in NRVMs transduced with Ad-LacZ, and this effect was significantly reduced in NRVMs transduced with Ad-NCLX (Fig. 5C and Supplemental Fig. S8C).
Figure 5: NCLX expression limits oxidative capacity and biosynthetic potential of cardiomyocytes during hypertrophic stimulation in vitro.
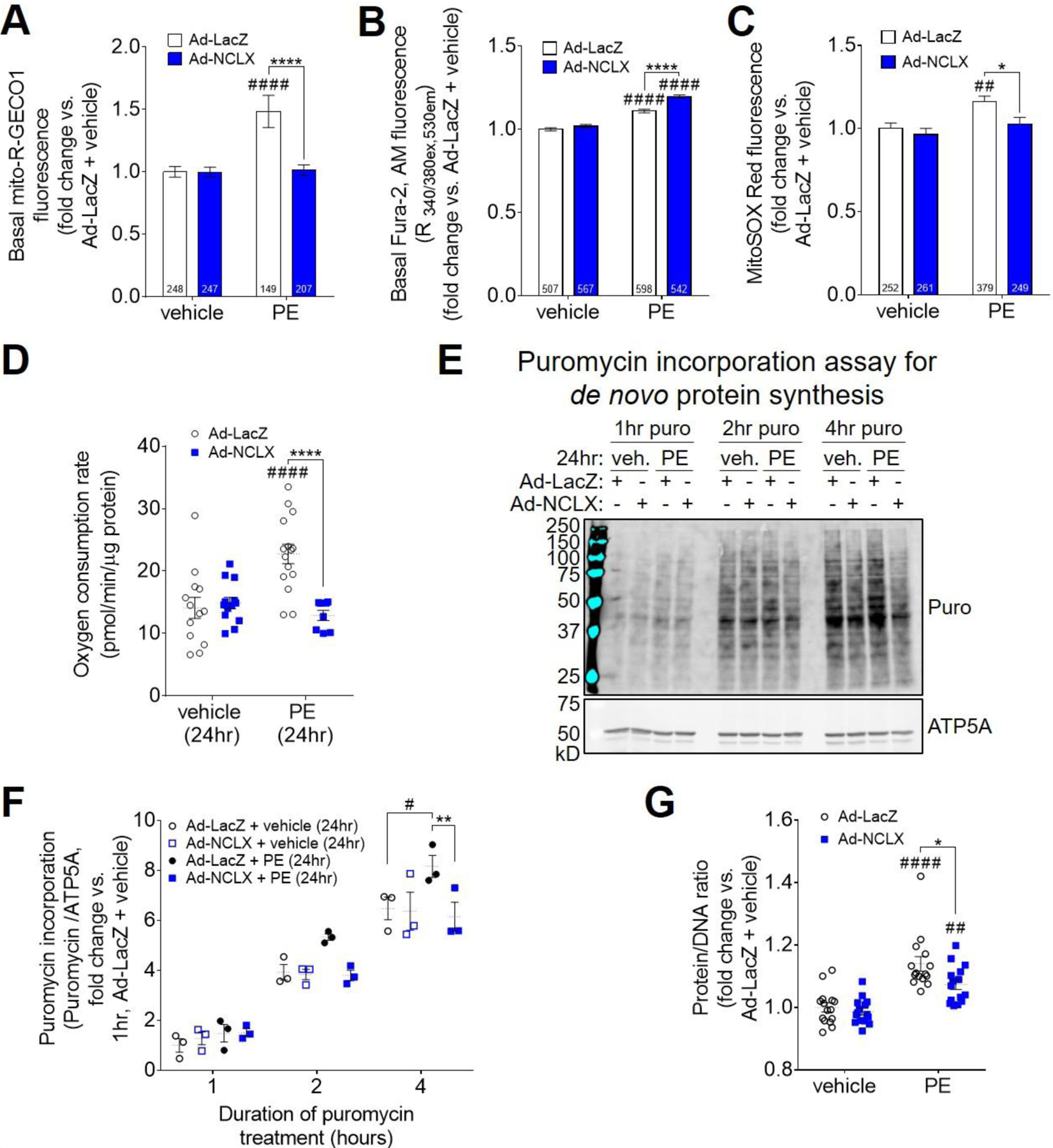
A) Basal, steady-state mCa2+ content in neonatal rat ventricular myocytes (NRVMs) transduced with adenovirus encoding β-galactosidase (Ad-LacZ) or human NCLX (Ad-NCLX) and treated with vehicle or phenylephrine (PE), as indicated by fluorescence of the genetically-encoded mitochondrial Ca2+ reporter, mito-R-GECO1. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ####P<0.0001 vs. vehicle; ****P<0.0001 Ad-LacZ vs. Ad-NCLX. Cell number per experimental group (n) is indicated within the corresponding bar of the graph. Individual data points are depicted in Supplemental Fig. S8A. B) Basal, steady-state cytosolic Ca2+ level in NRVMs, as indicated by fluorescence of the ratiometric Ca2+ reporter dye, Fura-2, AM. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ####P<0.0001 vs. vehicle; ****P<0.0001 Ad-LacZ vs. Ad-NCLX. Cell number per experimental group (n) is indicated within the corresponding bar of the graph. Individual data points are depicted in Supplemental Fig. S8B. C) Mitochondrial superoxide production in NRVMs, as indicated by fluorescence of the dye MitoSOX Red. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01 vs. vehicle; *P<0.05 Ad-LacZ vs. Ad-NCLX. Cell number per experimental group (n) is indicated within the corresponding bar of the graph. Individual data points are depicted in Supplemental Fig. S8C. D) Basal oxygen consumption rate per unit cellular protein in NRVMs after 24 hours of treatment with vehicle or phenylephrine. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ####P<0.0001 vs. vehicle; ****P<0.0001 Ad-LacZ vs. Ad-NCLX. (n=8–15 replicates / group). E) Western blots for puromycin incorporation, and mitochondrial loading control ATP5A, in NRVMs treated with puromycin (puro) for 1–4 hours starting at 24 hours of phenylephrine stimulation. Corresponding full-length western blots are shown in Extended Fig. 2. F) Quantification of cellular puromycin incorporation normalized to loading control, ATP5A. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. #P<0.05 vs. vehicle; **P<0.01 Ad-LacZ vs. Ad-NCLX. (n=3 replicates / group). G) Protein/DNA ratio in NRVMs after 48 hours of vehicle or PE treatment. Data analyzed by 2-way ANOVA with Sidak’s post-hoc test. ##P<0.01, ####P<0.001 vs. vehicle; *P<0.05 Ad-LacZ vs. Ad-NCLX. (n=15 replicates / group).
We next examined NRVMs at 24 hours of phenylephrine stimulation, a timepoint at which we could not yet detect cardiomyocyte hypertrophy, to investigate how limiting mCa2+ accumulation affects cellular metabolism that may be required for cardiomyocyte growth. Whereas phenylephrine treatment increased oxygen consumption per unit cellular protein in NRVMs transduced with Ad-LacZ, this effect was abrogated in NRVMs transduced with Ad-NCLX (Fig. 5D). This finding is consistent with the notion that increased mCa2+ stimulates TCA cycle flux and oxidative phosphorylation, as well as with our finding that adenoviral NCLX expression attenuates the phenylephrine-induced increase in mCa2+ (Fig. 5A and Supplemental Fig. S8A). Together, these results suggest that increased NCLX expression limits the oxidative capacity of cardiomyocytes during hypertrophic stimulation by suppressing mCa2+-dependent stimulation of metabolism. Measurement of cellular puromycin incorporation showed that 24 hours of PE stimulation increased the rate of de novo protein synthesis in NRVMs transduced with Ad-LacZ, and that this PE-dependent effect was abolished in NRVMs transduced with Ad-NCLX (Fig. 5E–F; full-length blots shown in Extended Fig. 2). This observation indicates that by limiting the ability for cardiomyocytes to increase oxidative metabolism in response to hypertrophic stimulation, NCLX overexpression consequently limits the cells’ capacity for anabolic growth. Indeed, fitting with this model and in agreement with our chronic in vivo studies, the increase in the protein/DNA ratio of NRVMs after 48 hours of phenylephrine stimulation was significantly reduced in cells transduced with Ad-NCLX (Fig. 5G).
3. DISCUSSION AND CONCLUSIONS
The net increase in mCa2+ concentration is a key mechanism that increases cardiac ATP production to augment an increase in cardiac workload and ATP consumption. At the same time, excessive mCa2+ accumulation has long been recognized as a driver of cardiomyocyte death and the resulting contractile dysfunction following myocardial infarction. A key unanswered question in the HF field is what causative role, if any, altered mCa2+ homeostasis plays in the development of non-ischemic heart disease and failure. Here, we report that cardiomyocyte-specific, transgenic overexpression of the mitochondrial sodium-calcium exchanger, NCLX, attenuates cardiac hypertrophy and fibrosis and improves contractile function in a mouse model of chronic pressure overload via 12-week transverse aortic constriction (Fig. 1). We demonstrate that cardiomyocyte-specific NCLX overexpression mitigates early hypertrophic remodeling that occurs in response to 2-week TAC or 2-week angiotensin II stimulation, prior to the manifestation of contractile dysfunction (Fig. 2–3). NCLX overexpression also reduces cardiac hypertrophy myocardial fibrosis in mice subjected to chronic neurohormonal overload with infusion of a combined high dose of angiotensin II and phenylephrine (Fig. 4). This provides genetic evidence that enhancing mCa2+ efflux mitigates maladaptive cardiac remodeling and the development of contractile failure when hearts are subjected to sustained hemodynamic and neurohormonal load, and supports the hypothesis that mCa2+ accumulation contributes to early pathological remodeling and subsequent contractile dysfunction in non-ischemic heart disease. We further demonstrate that NCLX limits mCa2+ accumulation, oxidative metabolism, and protein biosynthesis during hypertrophic stimulation in vitro (Fig. 5), suggesting that an increase in mCa2+ content drives increased mitochondrial metabolism that is required to fuel cardiomyocyte growth. However, our in vivo findings also suggest the existence of a lower threshold or “floor” for matrix Ca2+ content, below which interventions that increase cardiomyocyte mCa2+ efflux switch from being adaptive to being maladaptive and increase the likelihood of death. This finding emphasizes that therapies targeting mCa2+ exchange to either increase or decrease net mCa2+ accumulation in chronic heart disease may need to be matched to the specific nature of Ca2+ dysregulation and the patient’s stage of disease.
Several prior studies have attempted to explore the relationship between altered cardiomyocyte mCa2+ homeostasis and the development of maladaptive remodeling and contractile dysfunction in response to chronic hemodynamic stress. Mcu deletion has proven ineffective at limiting cardiac hypertrophy or contractile dysfunction with chronic pressure overload [22, 70], possibly because alternative, slower routes of mCa2+ uptake are sufficient to increase mCa2+ content upon chronic elevations in cytosolic Ca2+ [21, 22, 90]. The NCLX overexpression models used in the current study overcome this technical limitation and provide novel insight into the contribution of mCa2+ signaling to pathological cardiac remodeling and the development of non-ischemic HF.
Our findings indicate that cardiomyocyte NCLX overexpression mitigates early pathological cardiac remodeling and the development of contractile dysfunction via two distinct mechanisms. First, the observation that cardiomyocyte NCLX overexpression minimizes myocardial fibrosis in response to 12-week TAC and 4-week infusion with angiotensin II + phenylephrine is consistent with our earlier report that cardiomyocyte NCLX overexpression protects against cell death in response to acute cellular Ca2+ overload [75]. This result suggests that increased capacity for mCa2+ efflux similarly lowers the overall risk of mCa2+ overload and necrotic cardiomyocyte death during chronic stress. Our finding of reduced fibrotic deposition in the hearts of NCLX-Tg mice subjected to 4-week AngII/PE infusion, despite similar activation of the expression of some pro-fibrotic genes such as transforming growth factor β−1, also fits with a model of a net delay in fibrotic remodeling with cardiomyocyte NCLX overexpression. That is, with a delay in mCa2+ overload and cardiomyocyte death in NCLX-Tg mice, less total time would have passed between the initial stimulus for the induction of pro-fibrotic gene expression, subsequent synthesis of extracellular matrix proteins, and study termination.
Second, cardiomyocyte NCLX overexpression attenuates cardiomyocyte hypertrophy. This result was consistent across all in vivo models examined (Fig. 1–4) and was recapitulated in NRVMs (Fig. 5). The observation that cardiomyocyte hypertrophy was still attenuated in NCLX-Tg mice even in instances where there was similar activation of the fetal gene program suggests that the rate of cardiomyocyte growth in response to the same initial stressor was slowed by increased mCa2+ efflux activity. That NCLX overexpression alone does not affect heart weight, cardiomyocyte cross sectional area, protein synthesis rates, protein/DNA ratio, nor mitochondrial or cytosolic Ca2+ content under homeostatic conditions is consistent with the view that NCLX overexpression specifically mitigates stress-induced cardiac hypertrophy by limiting net mCa2+ accumulation in response to elevations in cytosolic Ca2+ concentration. This suggests that an increase in net mCa2+ accumulation during chronic stress directly contributes to the process of pathological cardiac hypertrophy. We acknowledge that we consistently observed trends towards upregulation of MCU in NCLX-Tg hearts when subjected to chronic stress (Supplemental Figs. 3, 5, and 7). This indicates that given enough time, hearts have some capacity to compensate for NCLX overexpression, particularly when subjected to perturbations that increase cardiac workload. Such compensatory mechanisms to augment mCa2+ accumulation may help to explain the somewhat modest effects of NCLX overexpression on cardiac remodeling in our chronic in vivo studies.
Our in vivo results raised the question of how precisely mCa2+ contributes to cardiomyocyte hypertrophy. Adenoviral-mediated NCLX expression in neonatal rat ventricular cardiomyocytes in vitro revealed that limiting mCa2+ accumulation during pro-hypertrophic stimulation is associated with an impaired ability to increase mitochondrial oxidative metabolism. These observations fit with the notion that increased mCa2+ levels enhance metabolic flux by stimulating the activity of pyruvate dehydrogenase and TCA cycle dehydrogenases [57, 62, 91, 92]. Such increased mitochondrial metabolism is likely required to provide the energy and to generate the macromolecules that are required for cell growth. Fitting with this view, elevated mCa2+ concentration in mPTP-deficient, cyclophilinD-null hearts is associated with an increase in net glucose oxidation [93]. The Tian laboratory recently described a mechanism by which a shift from fatty acid oxidation to glucose oxidation increases anapleurotic input of pyruvate into the TCA cycle and thereby drives an increase in de novo aspartate biosynthesis [94]. Such increased aspartate production supports nucleotide and protein synthesis to enable anabolic growth of the heart in response to hypertrophic stimulation. These reports establish a model in which increased mCa2+ signaling drives increased glucose oxidation, to increase biosynthetic pathways that fuel biomass deposition within the heart. Our current results indicate that cardiomyocyte NCLX overexpression limits mCa2+ accumulation when the heart is subjected to sustained stress, and so limits the increase in oxidative metabolism that is needed to provide the energy and molecular building blocks required for cardiomyocyte growth. Indeed, we find that exogenous NCLX expression in vitro mitigates the phenylephrine-induced increase in the rate of de novo protein synthesis and cardiomyocyte growth, processes that ultimately depend upon the integration of cellular bioenergetic status (availability of ATP to invest in energy-consuming anabolic reactions such as protein translation) and the availability of macromolecules such as amino acids, nucleotides, and lipids, whose synthesis is coupled to TCA cycle flux. We therefore propose that NCLX overexpression limits mCa2+ accumulation when the heart is subjected to chronic stress, thereby limiting the oxidative capacity of the cardiomyocytes and so limiting the cells’ potential for anabolic growth.
The fact that NCLX mediates mitochondrial Na+ influx in exchange for mCa2+ efflux can help to reconcile the deleterious effect of cardiomyocyte NCLX overexpression on overall animal survival we observed in the model of extreme neurohormonal overload with combined high-doses of angiotensin II + phenylephrine (Fig. 4B) with the cardioprotective phenotype we observed in all other stress conditions. Cytosolic Na+ concentration is increased in the failing heart [95, 96]. Elevated cytosolic Na+ levels drive increased mitochondrial Na+/Ca2+ exchange through NCLX, such that the mitochondrial matrix may become deficient in Ca2+ [50, 68]. Under these conditions, ATP production can be impaired, and mitochondrial redox balance can be disrupted, resulting in increased ROS stress [97]. These combined consequences of mCa2+ depletion in the failing heart can limit cardiac contractility and increase the susceptibility to fatal cardiac arrhythmia [51, 69]. We found that a significant number of NCLX overexpressor animals receiving AngII/PE died within the first two weeks of treatment. The abrupt onset of this mortality, along with the absence of overt systolic dysfunction, are in line with possibility that NCLX-Tg mice exhibit a greater propensity for arrhythmia under extreme adrenergic stress, leading to sudden cardiac death. These findings could be explained by increased intracellular Na+ accumulation due to increased plasma membrane Na+/Ca2+ and Na+/H+ exchange in the mice treated with AngII/PE [98, 99], as compared to the less severe models we tested. Such increased cytosolic Na+ would favor mitochondrial Na+/Ca2+ exchange, limit mCa2+ accumulation, and augment increases in cytosolic Ca2+ levels, effects that would have been exacerbated in NCLX-Tg hearts. Our anecdotal observations of pronounced Ca2+ deposition in the hearts of NCLX-Tg mice that died spontaneously following AngII/PE infusion (Supplemental Fig. 6D) agree with this interpretation and suggest that impaired mCa2+ accumulation compromises mitochondrial Ca2+ buffering capacity, possibly leading to energetic impairment and cytosolic Ca2+ overload that culminate in necrosis. Such tissue-level damage likely disrupts the electrical syncytium of the myocardium, resulting in aberrant propagation of electrical activity and increasing the chance for fatal cardiac arrhythmia. Liu et al’s finding that NCLX inhibition protects against sudden cardiac death in a guinea pig HF model [69] lends further support to the view that excessive NCLX activity in the failing or extremely-stressed heart increases the overall risk for arrhythmia, separable from the protective effects of increased NCLX activity against deleterious cardiac remodeling that develops over chronic time scales.
In conclusion, our data provide evidence for enhancing NCLX efflux as an effective strategy to mitigate pathological mCa2+ overload and minimize detrimental hypertrophic remodeling as chronically-stressed hearts are beginning to progress towards failure. Thus, increasing NCLX function within the heart may be a suitable therapeutic strategy to slow or delay the onset of cardiac pathology and contractile dysfunction that develops in non-ischemic heart disease and inherited cardiomyopathies. Our findings further demonstrate that although increasing mCa2+ efflux is protective early in the disease process, if applied in settings of extreme stress—perhaps less clinically appropriate—it can become deleterious. Our results combined with earlier reports [51, 69] suggest a threshold or switch for net mCa2+ exchange in chronic heart disease, where increasing mCa2+ content is maladaptive and contributes to pathological remodeling and dysfunction early in the heart’s response to increased workloads, but later on in the disease process or with more severe perturbation of cellular ion homeostasis—where the mitochondria could become starved of Ca2+—increasing mCa2+ content instead becomes adaptive in order to augment mitochondrial energetics and correct mitochondrial redox balance. Our study thus adds nuance and complexity to our understanding of the multifarious roles of mCa2+ signaling as the heart remodels in response to chronic stress and progresses towards failure.
Future research to fully elucidate the specific molecular mechanisms linking increased mCa2+ to cardiomyocyte growth will offer insight into novel therapeutic strategies to mitigate pathological hypertrophy in chronic heart disease. Further work will also be necessary to distinguish any specific effects of altered mNa+ content, as opposed to altered mCa2+ homeostasis, in mediating pathological remodeling and contractile dysfunction in HF, as well as the anti-hypertrophic effects of NCLX overexpression. Finally, research into the molecular mechanisms regulating NCLX activity is needed to identify novel, tractable ways to enhance or inhibit NCLX-dependent mCa2+ exchange as appropriate for the treatment of HF and other human disease.
4. MATERIALS AND METHODS
Mice
The generation of mice with doxycycline-controlled, cardiomyocyte-restricted overexpression of NCLX was described previously [75]. In brief, TRE-NCLX mice expressing human SLC8B1 cDNA under the control of a Ptight Tet-responsive promoter were crossed to α-myosin heavy chain tetracycline transactivator (αMHC-tTA) transgenic mice. This strategy allows inducible expression of human NCLX within the cardiomyocytes upon removal of doxycycline (DOX) from the animals’ diet. Breeder cages were provided with chow containing DOX (TestDiet, #MRMH1500/625PPM DOXY 1/2 IRR) and litters were maintained on DOX until weaning at 3 weeks of age in order to prevent transgenic NCLX overexpression during development. Pups were genotyped for the TRE-NCLX transgene using the forward primer: 5’-TGTCGGCCATTTCTACCACACTGA-3’ and the reverse primer: 5’-ACACATACAAGCCCAGGTAACCCA-3’. PCR reaction mixture contained 1uL tail DNA in DirectPCR Lysis reagent (Viagen Biotech #102-T), 1x Taq buffer (Syd Labs #MB042-EUT), 80µM each dNTPs (New England Biolabs #N0447L), 800nM each forward and reverse primers, 200µM betaine (Affymetrix #77507), and 1.25 U Taq polymerase (Syd Labs #MB042-EUT). The PCR conditions were: denaturation at 95°C for 3 minutes, followed by 35 cycles (95°C for 30 seconds, 62°C for 30 seconds, 72°C for 40 seconds), followed by 10 minutes at 72°C. Pups were genotyped for the αMHC-tTA transgene using the forward primer: 5’-AGCGCATTAGAGCTGCTTAATGAGGTC-3’ and the reverse primer: 5’-GTCGTAATAATGGCGGCATACTATC-3’. PCR reaction mixture contained 1uL tail DNA in DirectPCR Lysis reagent, 1x Taq buffer, 80µM each dNTPs, 800nM each forward and reverse primers, and 1.25U Taq polymerase. The PCR conditions were: denaturation at 95°C for 3 minutes, followed by 35 cycles (95°C for 30 seconds, 66°C for 30 seconds, 72°C for 1min), followed by 10 minutes at 72°C.
Adult TRE-NCLX x αMHC-tTA mice and age-matched αMHC-tTA controls were used in surgical studies between 8–25 weeks of age. Both male and female mice were included. Experiments were performed using a numbered ear-tagging system in order to blind the experimenter to mouse genotype and experimental group. All animal experiments followed AAALAC guidelines and were approved by Temple University’s IACUC.
Western blot analysis
Hearts were excised from mice, rinsed in ice-cold phosphate buffered saline, immediately snap frozen using liquid nitrogen-cooled metal tongs, and stored at −80°C until use. Neonatal rat ventricular cardiomyocytes (NRVMs) were trypsinized off of tissue culture plates, washed once in phosphate buffered saline, pelleted, and snap frozen in liquid nitrogen. Heart tissue was homogenized in ice-cold 1X RIPA buffer (Millipore #20–188) supplemented with 1X SigmaFast protease inhibitor cocktail (Sigma-Aldrich #S8830–20TAB) and 1X PhosStop phosphatase inhibitor (Roche #04 906 845 001) using a bead mill homogenizer (VWR, #75840–022). NRVM pellets were lysed in this same RIPA buffer. Crude homogenates were sonicated for 10 seconds and then centrifuged for 5 minutes at 5,000 x g to pellet insoluble material. Lysate supernatants were collected and protein concentration was determined using a bicinchoninic acid protein assay (bioWORLD #20831001–1). Samples were mixed 4:1 with 5x SDS sample buffer (250µM Tris-HCl, pH 7.0; 40% (volume/volume) glycerol; 8% (weight/volume) sodium dodecyl sulfate; 20% (volume/volume) β-mercaptoethanol; 0.1% (weight/volume) bromophenol blue) and boiled for 5 minutes. For western blots for NCLX, 50µg of protein lysate per well was loaded on 10% (weight/volume) polyacrylamide Tris-glycine SDS gels. 20µg NRVM protein lysate per well was used for western blotting for puromycin. For large-format western blots of mouse heart tissue from surgical cohorts, 25µg protein per well was loaded on NuPAGE 4–12% Bis-Tris gels (Thermo Fisher, # WG1403BOX). Proteins were separated by electrophoresis and then transferred to polyvinyldienefluoride membranes (Millipore #IPFL00010), which were then blocked for 1 hour with blocking buffer (Rockland #MB-070). Membranes were incubated overnight at 4°C with primary antibodies diluted in 50% blocking buffer / 50% Tris-buffered saline (bioWORLD #42020056–3) + 0.1% TWEEN (Sigma-Aldrich #P9416) (TBS-T). Membranes were washed 3 times in TBS-T, incubated for 1.5 hours at room temperature with secondary antibodies diluted in 50% blocking buffer / 50% TBS-T, and washed 3 times in TBS-T before imaging on a LI-COR Odyssey infrared imager system. Primary antibodies and dilutions used in this study included: rabbit polyclonal against NCLX (GeneTex #GTX87452), 1:1000; mouse monoclonal total OXPHOS rodent antibody cocktail (Abcam #ab110413), 1:1000; rabbit monoclonal against MCU (Cell Signaling Technology #14997), 1:1000; rabbit antibody against EMRE (Bethyl Laboratories #A300-BL19208), 1:1000; custom-made rabbit polyclonal against MICU1[18] 1:500; mouse monoclonal against ATP5A (Abcam #ab14748), 1:2000; and mouse monoclonal against puromycin (EMD Millipore #MABE343), 1:5000. Secondary antibodies and dilutions included: IRDye 680RD goat anti-rabbit (LI-COR #926–68071), 1:10,000; IRDye 800CW goat anti-mouse (LI-COR #926–32210), 1:10,000; IRDye 800CW goat anti-rabbit (LI-COR #925–32210), 1,10,000; IRDye 680RD goat anti-mouse (LI-COR #925–68070), 1:10,000. All full-length western blots are shown in Extended Figs. 1–2, Supplemental Fig. S3, Supplemental Fig. S5, and Supplemental Fig. S7. Western blot densitometry was measured using LI-COR Image Studio software (LI-COR, version 2.0.38).
Transverse aortic constriction
Transverse aortic constriction was performed as described previously [100]. Briefly, mice were anesthetized with 3% isoflurane. Anesthetized mice were intubated and the chest cavity entered via partial thoracotomy to the second rib. The aortic arch was exposed and the transverse aorta was constricted by tying a 6–0 suture against a 27½ -gauge needle, which was then promptly removed. For sham mice, the chest was entered and the aorta exposed, but no suture was secured around the transverse aorta.
Left ventricular echocardiography
Transthoracic echocardiography of the left ventricle was performed on a Vevo 2100 imaging platform (VisualSonics) as described elsewhere [21]. In brief, mice were anesthetized with 1.5% isoflurane in 100% oxygen during imaging and M-mode images were collected in the short-axis view. Recordings were analyzed using VisualSonics Vevo LAB software (VisualSonics version 3.1.1). For TAC studies, the efficacy of aortic constriction was confirmed using pulsed wave Doppler echocardiography to measure blood flow velocity through the site of constriction. The aortic pressure gradient was calculated according to the formula: pressure gradient = 4 X V max2 (Li 2016) [101]. For mice subjected to TAC surgery, animals with an aortic pressure gradient <30mmHg were excluded from subsequent analysis [102].
Tissue gravimetrics and histology
Hearts were collected at the indicated time points and massed. Mouse tibia length was measured for normalization of heart mass. The atria were removed from the ventricles, and the ventricles were divided into thirds for further analysis. Ventricle base samples were rinsed in ice-cold phosphate buffered saline, then immediately snap frozen using liquid nitrogen-cooled metal tongs and stored at −80°C until use. Ventricle apex samples were rinsed in ice-cold phosphate buffered saline, incubated in RNAlater (Thermo Fisher, #AM7020) for 24 hours at 4°C, and then stored at −80°C until use. Mid-ventricle samples were rinsed in ice-cold phosphate buffered saline and then fixed in 10% buffered formalin (EKI, #4498–1GL) before dehydration and embedding in paraffin, as described previously [75]. Paraffin-embedded ventricle samples were cut to 5-μm sections using a microtome and placed on glass slides. For measurement of cardiomyocyte cross-sectional area, heart sections were stained with TRITC-conjugated wheat germ-agglutinin (Sigma Aldrich #L-5266) at 100μg/mL to label the sarcolemma. Coverslips were mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen #P36935). The left ventricle was imaged on an Axio Observer Z1 fluorescence microscope (Zeiss) and cardiomyocyte cross sectional area quantified using ImageJ (National Institutes of Health). A minimum of 80 cardiomyocytes were measured per mouse. For measurement of myocardial fibrosis, heart sections were stained with Masson’s trichrome (Sigma-Aldrich #HT15). Images of the myocardium were captured at 20x magnification using an Eclipse Ni-E light microscope (Nikon). The percent fibrosis was quantified in ImageJ by dividing the area of tissue staining blue (collagen) by the total tissue area. For assessment of cellular calcium overload, heart sections were stained with von Kossa stain (Abcam #ab150687) according to the manufacturer’s instructions. All images of whole heart cross sections were captured on a SMZ1000 stereomicroscope (Nikon).
qPCR gene expression analysis
mRNA was isolated from ventricle apex samples preserved in RNAlater using a RNeasy Fibrous Tissue Kit (Qiagen, #74704) according to the manufacturer’s instructions. cDNA was prepared from mRNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher #4368813) following the manufacturer’s instructions. qPCR was performed on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) using PowerUp SYBR Green Master Mix (Applied Biosciences #100029283). The real-time PCR conditions were: UDG activation at 50°C for 2 minutes, initial denaturation at 95°C for 10 minutes, followed by 40 cycles (95°C for 15 seconds, 60°C for 30 seconds, 72°C for 30 seconds). qPCR primers against mouse transcripts are listed in Table 1.
Table 1:
Sequences of qPCR primers for mouse genes.
| Gene | Forward Primer (5’→3’) | Reverse Primer (5’→3’) |
|---|---|---|
| Nppa | GGGTAGGATTGACAGGATTGG | CTCCTTGGCTGTTATCTTCGG |
| Nppb | GCACAAGATAGACCGGATCG | CCCAGGCAGAGTCAGAAAC |
| Npr1 | CACAGTCAACACAGCTTCAAG | TTCATTTCCACGTCACCTCG |
| Acta1 | TGAACCCCAAAGCTAACCG | CCCCAGAATCCAACACGATG |
| Myh6 | GCAGAACAGTAAAATTGAGGACG | CGCAGCTTCTCCACCTTAG |
| Myh7 | GACGACGTCACCTCCAACA | TGCTCCGGTGCTCATTCATC |
| Postn | AAGAGATGGTCACTTCACGC | GCACTGGAGGGTATTTAGGATG |
| Spp1 | GCTTGGCTTATGGACTGAGGTC | CCTTAGACTCACCGCTCTTCATG |
| Acta2 | GTGAAGAGGAAGACAGCACAG | GCCCATTCCAACCATTACTCC |
| Tgfb1 | CCTGAGTGGCTGTCTTTTGA | CGTGGAGTTTGTTATCTTTGCTG |
| Rps13 | GCACCTTGAGAGGAACAGAA | GAGCACCCGCTTAGTCTTATAG |
Osmotic minipump infusion studies
Osmotic minipump implantation surgeries were performed as described previously [103]. Mice were anesthetized with 3% isoflurane and a small midline incision was made in the back. After subcutaneous insertion of the minipump, the incision was closed with 5–0 absorbable suture. For sham mice, an incision was made in the back and then closed with 5–0 absorbable suture. All mice were treated preemptively for infection via subcutaneous injection of the antibiotic cefazolin (Sandoz, #007813450) at a dose of 40mg/kg. Osmotic minipumps (Alzet Model 1004, #000992) were set to deliver the Gq-coupled receptor agonist angiotensin II (Sigma-Aldrich #A9525) dissolved in sterile saline at a dose of 1.44mg/kg/day for 2 weeks [83, 84]. In a separate study, osmotic minipumps (Alzet Model 2004, #0000298) were set to deliver a combination of high doses of the Gq-coupled receptor agonists angiotensin II and phenylephrine hydrochloride (Sigma-Aldrich #P6126) dissolved in sterile saline at a dose of 10mg/kg/day angiotensin II + 50mg/kg/day phenylephrine hydrochloride [104] for 4 weeks.
Neonatal rat ventricular myocyte hypertrophy in vitro
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1 day-old Sprague-Dawley rats as described previously [105]. NRVMs were plated at 100,000 cells per 35mm collagen-coated glass-bottomed dish (MatTek #P35GCOL-1.5–10-C) for imaging assays; at 200,000 cells/well of 6-well tissue culture plates for puromycin incorporation assay and measurement of protein/DNA ratio; or 20,000 cells/well of XF96 cell culture microplates (Agilent #101085–004) for measurement of oxygen consumption. NRVMs were plated and maintained in DMEM (Corning, #10–013-CV) + 5% fetal bovine serum (FBS) (Peak Serum, #PS-FB3) + 1% penicillin/streptomycin (Sigma-Aldrich #P0781–100ML) + 0.1mM 5’-bromo-2’-deoxyuridine (BrdU) (Sigma-Aldrich #B5002–1G) + 2µg/mL vitamin B12 (Sigma-Aldrich #V2876–1G) for 4 days. 4 days after isolation, the media was changed to DMEM + 5% FBS 1% penicillin/streptomycin + 2µg/mL vitamin B-12. The following day, the media was changed to serum-free DMEM + 1% penicillin/streptomycin + 2µg/mL vitamin B12 containing control adenovirus encoding β-galactosidase (Ad-LacZ) or adenovirus encoding human NCLX (Ad-NCLX), either alone or in combination with adeno-associated virus 6 encoding the genetic mitochondrial Ca2+ reporter, mito-R-GECO1 (AAV6-mito-R-GECO1). After 24 hours of viral transduction, the media was changed to serum-free DMEM + 1% penicillin/streptomycin + 2µg/mL vitamin B12 supplemented with 100µM phenylephrine hydrochloride (PE) (Sigma-Aldrich #P6126–5G) or vehicle control. Subsequent experiments were conducted at 24 or 48 hours after the beginning of PE treatment as indicated.
Ca2+ and mitochondrial superoxide imaging
Cytosolic and mitochondrial Ca2+ content and mitochondrial superoxide production in NRVMs were assessed after 48 hours of treatment with phenylephrine or vehicle control. For cytosolic Ca2+ assessment, NRVMs were loaded with 1µM Fura-2, AM (Thermo Fisher Scientific #F1221) for 15 minutes at room temperature, washed twice in Tyrode’s buffer (150 mM NaCl, 5.4 mM KCl, 1.2mM MgCl2, 10 mM glucose, 2 mM sodium pyruvate, 5 mM HEPES, 2 mM CaCl2, pH 7.4), and then imaged at 340nm and 380nm excitation/510±40nm emission. For mitochondrial Ca2+ assessment, NRVMs transduced with AAV6-mito-R-GECO1 were washed twice in Tyrode’s buffer and imaged at 572±17.5nm excitation/632±30nm emission. For detection of mitochondrial superoxide production, NRVMs were loaded with 5µM MitoSOX Red (Thermo Fisher Scientific #M36008) for 15 minutes at 37°C, washed twice in Tyrode’s buffer, and then imaged at 490±10nm excitation and 585±20nm emission. All imaging was performed in a 37°C heated chamber in fresh Tyrode’s buffer on an Axio Observer Z1 fluorescence microscope (Zeiss).
Oxygen consumption measurements
After 24 hours of stimulation with phenylephrine or vehicle control, NRVMs were changed to basal DMEM (Corning # 90–113-PB), pH 7.4, supplemented with 25mM glucose, 4mM L-glutamine, 1mM sodium pyruvate, 0.2mM bovine serum albumin-conjugated sodium palmitate, 0.2mM L-carnitine hydrochloride, and 100µM phenylephrine or vehicle control. Basal oxygen consumption rate (OCR) was measured in NRVMs using a Seahorse Bioscience XF96 extracellular flux analyzer. The cells were then washed with phosphate buffered saline, and lysed by adding 1x RIPA buffer directly to the XF96 cell culture microplates. Protein concentration per well was determined using a bicinchoninic acid protein assay. The OCR measured for each well was normalized to the µg protein per well.
Puromycin incorporation assay for de novo protein synthesis
After 24 hours of stimulation with phenylephrine or vehicle control, NRVMs were changed to fresh media containing phenylephrine or vehicle and supplemented with 1µg/mL puromycin dihydrochloride (Sigma-Aldrich # P8833–25MG). Cells were collected 1, 2, or 4 hours later for western blotting for puromycin incorporation, as described above.
Assessment of NRVM hypertrophy
After 48 hours of stimulation with phenylephrine or vehicle control, NRVMs were trypsinized off of tissue culture plates, washed once in phosphate buffered saline, pelleted, snap frozen in liquid nitrogen, and stored at −80°C until analysis. Cell pellets were lysed using the CyQUANT Cell Proliferation Assay Kit (Thermo Fisher Scientific #C7026), and then sonicated. Protein concentration in the lysates was determined using a bicinchoninic acid protein assay. An aliquot of each lysate was treated with DNAse-free RNAse A (Thermo Fisher Scientific #EN0531) and the DNA concentration determined with the CyQUANT Cell Proliferation Assay Kit following the manufacturer’s instructions. NRVM hypertrophy was assessed by normalizing the protein concentration of each sample to its DNA concentration.
Statistical Analysis
All results are presented as mean ± S.E.M. Statistical analysis was performed using Prism 6.0 (GraphPad Software). Direct comparisons between two groups used a two-tailed t-test. The log-rank (Mantel-Cox) test was used for comparison of Kaplan-Meier survival curves. For analysis of longitudinal echocardiographic studies, 2-way ANOVA was used with Tukey post-hoc analysis. Grouped data were analyzed by 2-way ANOVA with Sidak’s post-hoc analysis. For all comparisons, P values less than 0.05 were considered significant.
Extended Data
Extended Figure 1: Full-length blots for Figure 1B.
Arrow indicates full-length NCLX.
Extended Figure 2: Full-length blots for Figure 5E.
Arrow indicates ATP5A band.
Supplementary Material
Highlights.
The mitochondrial Na+/Ca2+ exchanger, NCLX, mediates mitochondrial calcium efflux.
NCLX limits mitochondrial Ca2+ accumulation upon hypertrophic stimulation.
NCLX limits oxidative metabolism and protein synthesis during hypertrophy.
NCLX overexpression attenuates pathological cardiac hypertrophy in vivo.
FUNDING
The research was supported by the NIH (T32HL091804 to J.F.G; F32HL151146 to J.F. G.; K99AG065445 to P.J.; P01HL147841, R01HL142271, R01HL136954, P01HL134608, and R01HL123966 to J.W.E.) and the American Heart Association (20EIA35320226 to J.W.E.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE AND CONFLICT OF INTEREST
J.W.E. is a paid consultant for Mitobridge and Janssen.
REFERENCES
- [1].McCormack JG, Halestrap AP, Denton RM, Role of calcium ions in regulation of mammalian intramitochondrial metabolism, Physiological reviews 70(2) (1990) 391–425. [DOI] [PubMed] [Google Scholar]
- [2].Griffiths EJ, Rutter GA, Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells, Biochim Biophys Acta 1787(11) (2009) 1324–33. [DOI] [PubMed] [Google Scholar]
- [3].Garbincius JF, Luongo TS, Elrod JW, The debate continues - What is the role of MCU and mitochondrial calcium uptake in the heart?, Journal of molecular and cellular cardiology 143 (2020) 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Garbincius JF, Elrod JW, Mitochondrial calcium exchange in physiology and disease, Physiological reviews 102(2) (2022) 893–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature 476(7360) (2011) 341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R, A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter, Nature 476(7360) (2011) 336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK, EMRE is an essential component of the mitochondrial calcium uniporter complex, Science 342(6164) (2013) 1379–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK, MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake, Nature 467(7313) (2010) 291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M, MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival, Cell 151(3) (2012) 630–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G, MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter, Cell Metab 17(6) (2013) 976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK, MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling, PLoS One 8(2) (2013) e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kamer KJ, Mootha VK, MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter, EMBO Rep 15(3) (2014) 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R, MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity, Molecular cell 53(5) (2014) 726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D, MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake, Cell death and differentiation 26(1) (2019) 179–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M, MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism, Nature cell biology 14(12) (2012) 1336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Vais H, Tanis JE, Muller M, Payne R, Mallilankaraman K, Foskett JK, MCUR1, CCDC90A, Is a Regulator of the Mitochondrial Calcium Uniporter, Cell Metab 22(4) (2015) 533–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE, Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition, Proceedings of the National Academy of Sciences of the United States of America 113(13) (2016) E1872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M, MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics, Cell Rep 15(8) (2016) 1673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R, The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit, The EMBO journal 32(17) (2013) 2362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nature cell biology 15(12) (2013) 1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW, The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition, Cell Rep 12(1) (2015) 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD, The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart, Cell Rep 12(1) (2015) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME, The mitochondrial uniporter controls fight or flight heart rate increases, Nat Commun 6 (2015) 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME, Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart, Proceedings of the National Academy of Sciences of the United States of America 112(29) (2015) 9129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW, MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress, Circulation 140(21) (2019) 1720–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Baumgartner HK, Gerasimenko JV, Thorne C, Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJ, Gerasimenko OV, Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening, J Biol Chem 284(31) (2009) 20796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL, Mitochondrial calcium and the permeability transition in cell death, Biochim Biophys Acta 1787(11) (2009) 1395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rasola A, Bernardi P, Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis, Cell Calcium 50(3) (2011) 222–33. [DOI] [PubMed] [Google Scholar]
- [29].Shannon TR, Pogwizd SM, Bers DM, Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure, Circ Res 93(7) (2003) 592–4. [DOI] [PubMed] [Google Scholar]
- [30].Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Forster A, Schmitto JD, Gummert J, Schondube FA, Hasenfuss G, Maier LS, Sossalla S, Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology, Circulation 128(9) (2013) 970–81. [DOI] [PubMed] [Google Scholar]
- [31].Volk T, Noble PJ, Wagner M, Noble D, Ehmke H, Ascending aortic stenosis selectively increases action potential-induced Ca2+ influx in epicardial myocytes of the rat left ventricle, Exp Physiol 90(1) (2005) 111–21. [DOI] [PubMed] [Google Scholar]
- [32].Ito K, Yan X, Tajima M, Su Z, Barry WH, Lorell BH, Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts, Circ Res 87(7) (2000) 588–95. [DOI] [PubMed] [Google Scholar]
- [33].Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, Samarel AM, Bers DM, Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure-overload hypertrophy, Am J Physiol 272(5 Pt 2) (1997) H2425–35. [DOI] [PubMed] [Google Scholar]
- [34].Keung EC, Calcium current is increased in isolated adult myocytes from hypertrophied rat myocardium, Circ Res 64(4) (1989) 753–63. [DOI] [PubMed] [Google Scholar]
- [35].Moore RL, Yelamarty RV, Misawa H, Scaduto RC Jr., Pawlush DG, Elensky M, Cheung JY, Altered Ca2+ dynamics in single cardiac myocytes from renovascular hypertensive rats, Am J Physiol 260(2 Pt 1) (1991) C327–37. [DOI] [PubMed] [Google Scholar]
- [36].Wang J, Flemal K, Qiu Z, Ablin L, Grossman W, Morgan JP, Ca2+ handling and myofibrillar Ca2+ sensitivity in ferret cardiac myocytes with pressure-overload hypertrophy, Am J Physiol 267(3 Pt 2) (1994) H918–24. [DOI] [PubMed] [Google Scholar]
- [37].Kuramochi T, Honda M, Tanaka K, Enomoto K, Hashimoto M, Morioka S, Calcium transients in single myocytes and membranous ultrastructures during the development of cardiac hypertrophy and heart failure in rats, Clin Exp Pharmacol Physiol 21(12) (1994) 1009–18. [DOI] [PubMed] [Google Scholar]
- [38].Zhang XQ, Moore RL, Tenhave T, Cheung JY, [Ca2+]i transients in hypertensive and postinfarction myocytes, Am J Physiol 269(3 Pt 1) (1995) C632–40. [DOI] [PubMed] [Google Scholar]
- [39].Bentivegna LA, Ablin LW, Kihara Y, Morgan JP, Altered calcium handling in left ventricular pressure-overload hypertrophy as detected with aequorin in the isolated, perfused ferret heart, Circ Res 69(6) (1991) 1538–45. [DOI] [PubMed] [Google Scholar]
- [40].Okuda S, Sufu-Shimizu Y, Kato T, Fukuda M, Nishimura S, Oda T, Kobayashi S, Yamamoto T, Morimoto S, Yano M, CaMKII-mediated phosphorylation of RyR2 plays a crucial role in aberrant Ca(2+) release as an arrhythmogenic substrate in cardiac troponin T-related familial hypertrophic cardiomyopathy, Biochem Biophys Res Commun 496(4) (2018) 1250–1256. [DOI] [PubMed] [Google Scholar]
- [41].Sakai T, Naito AT, Kuramoto Y, Ito M, Okada K, Higo T, Nakagawa A, Shibamoto M, Yamaguchi T, Sumida T, Nomura S, Umezawa A, Miyagawa S, Sawa Y, Morita H, Lee JK, Shiojima I, Sakata Y, Komuro I, Phenotypic Screening Using Patient-Derived Induced Pluripotent Stem Cells Identified Pyr3 as a Candidate Compound for the Treatment of Infantile Hypertrophic Cardiomyopathy, Int Heart J 59(5) (2018) 1096–1105. [DOI] [PubMed] [Google Scholar]
- [42].Zhou W, Bos JM, Ye D, Tester DJ, Hrstka S, Maleszewski JJ, Ommen SR, Nishimura RA, Schaff HV, Kim CS, Ackerman MJ, Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with MYL2-R58Q-Mediated Apical Hypertrophic Cardiomyopathy Show Hypertrophy, Myofibrillar Disarray, and Calcium Perturbations, J Cardiovasc Transl Res 12(5) (2019) 394–403. [DOI] [PubMed] [Google Scholar]
- [43].Harding SE, MacLeod KT, Jones SM, Vescovo G, Poole-Wilson PA, Contractile responses of myocytes isolated from patients with cardiomyopathy, Eur Heart J 12 Suppl D (1991) 44–8. [DOI] [PubMed] [Google Scholar]
- [44].Kim SJ, Iizuka K, Kelly RA, Geng YJ, Bishop SP, Yang G, Kudej A, McConnell BK, Seidman CE, Seidman JG, Vatner SF, An alpha-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes, Am J Physiol 276(5) (1999) H1780–7. [DOI] [PubMed] [Google Scholar]
- [45].Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M, Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling, Circ Res 92(4) (2003) 428–36. [DOI] [PubMed] [Google Scholar]
- [46].Olsson MC, Palmer BM, Stauffer BL, Leinwand LA, Moore RL, Morphological and functional alterations in ventricular myocytes from male transgenic mice with hypertrophic cardiomyopathy, Circ Res 94(2) (2004) 201–7. [DOI] [PubMed] [Google Scholar]
- [47].Robinson P, Liu X, Sparrow A, Patel S, Zhang YH, Casadei B, Watkins H, Redwood C, Hypertrophic cardiomyopathy mutations increase myofilament Ca(2+) buffering, alter intracellular Ca(2+) handling, and stimulate Ca(2+)-dependent signaling, J Biol Chem 293(27) (2018) 10487–10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wu H, Yang H, Rhee JW, Zhang JZ, Lam CK, Sallam K, Chang ACY, Ma N, Lee J, Zhang H, Blau HM, Bers DM, Wu JC, Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients, Eur Heart J 40(45) (2019) 3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC, Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels, Circulation 119(18) (2009) 2435–43. [DOI] [PubMed] [Google Scholar]
- [50].Liu T, O’Rourke B, Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching, Circ Res 103(3) (2008) 279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu T, Yang N, Sidor A, O’Rourke B, MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca(2+) Leak, Circ Res (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, Prando V, Coppini R, Vida V, Stolen TO, Ulrik W, Cerbai E, Stellin G, Faggian G, De Stefani D, Sandri M, Rizzuto R, Di Lisa F, Pozzan T, Catalucci D, Mongillo M, Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy, Proceedings of the National Academy of Sciences of the United States of America 114(43) (2017) E9006–E9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yu Z, Chen R, Li M, Yu Y, Liang Y, Han F, Qin S, Chen X, Su Y, Ge J, Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement, Int J Cardiol 271 (2018) 161–168. [DOI] [PubMed] [Google Scholar]
- [54].Santulli G, Xie W, Reiken SR, Marks AR, Mitochondrial calcium overload is a key determinant in heart failure, Proceedings of the National Academy of Sciences of the United States of America 112(36) (2015) 11389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, Winge DR, Villanueva CJ, Chaudhuri D, Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium, Journal of molecular and cellular cardiology 113 (2017) 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rossi CS, Lehninger AL, Stoichiometry of Respiratory Stimulation, Accumulation of Ca++ and Phosphate, and Oxidative Phosphorylation in Rat Liver Mitochondria, J Biol Chem 239 (1964) 3971–80. [PubMed] [Google Scholar]
- [57].Denton RM, Randle PJ, Martin BR, Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase, Biochem J 128(1) (1972) 161–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Denton RM, Richards DA, Chin JG, Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues, Biochem J 176(3) (1978) 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].McCormack JG, Denton RM, The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex, Biochem J 180(3) (1979) 533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lawlis VB, Roche TE, Effect of micromolar Ca2+ on NADH inhibition of bovine kidney alpha-ketoglutarate dehydrogenase complex and possible role of Ca2+ in signal amplification, Molecular and cellular biochemistry 32(3) (1980) 147–52. [DOI] [PubMed] [Google Scholar]
- [61].Rutter GA, Denton RM, Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios, Biochem J 252(1) (1988) 181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Denton RM, Regulation of mitochondrial dehydrogenases by calcium ions, Biochim Biophys Acta 1787(11) (2009) 1309–16. [DOI] [PubMed] [Google Scholar]
- [63].Territo PR, Mootha VK, French SA, Balaban RS, Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase, Am J Physiol Cell Physiol 278(2) (2000) C423–35. [DOI] [PubMed] [Google Scholar]
- [64].Glancy B, Willis WT, Chess DJ, Balaban RS, Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria, Biochemistry 52(16) (2013) 2793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Capasso JM, Palackal T, Olivetti G, Anversa P, Left ventricular failure induced by long-term hypertension in rats, Circ Res 66(5) (1990) 1400–12. [DOI] [PubMed] [Google Scholar]
- [66].Li Z, Bing OH, Long X, Robinson KG, Lakatta EG, Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat, Am J Physiol 272(5 Pt 2) (1997) H2313–9. [DOI] [PubMed] [Google Scholar]
- [67].Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P, Apoptosis in pressure overload-induced heart hypertrophy in the rat, J Clin Invest 97(12) (1996) 2891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B, Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes, Circ Res 99(2) (2006) 172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP, O’Rourke B, Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure, Circ Res 115(1) (2014) 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E, Finkel T, Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter, Journal of molecular and cellular cardiology 85 (2015) 178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M, Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis, Cell Calcium 40(5–6) (2006) 553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Malecot CO, Bito V, Argibay JA, Ruthenium red as an effective blocker of calcium and sodium currents in guinea-pig isolated ventricular heart cells, Br J Pharmacol 124(3) (1998) 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Arduino DM, Perocchi F, Pharmacological modulation of mitochondrial calcium homeostasis, J Physiol 596(14) (2018) 2717–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hymel L, Schindler H, Inui M, Fleischer S, Reconstitution of purified cardiac muscle calcium release channel (ryanodine receptor) in planar bilayers, Biochem Biophys Res Commun 152(1) (1988) 308–14. [DOI] [PubMed] [Google Scholar]
- [75].Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW, The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability, Nature 545(7652) (2017) 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I, NCLX is an essential component of mitochondrial Na+/Ca2+ exchange, Proceedings of the National Academy of Sciences of the United States of America 107(1) (2010) 436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kim B, Matsuoka S, Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange, J Physiol 586(6) (2008) 1683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Dash RK, Beard DA, Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry, J Physiol 586(13) (2008) 3267–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Palty R, Hershfinkel M, Yagev O, Saar D, Barkalifa R, Khananshvili D, Peretz A, Grossman Y, Sekler I, Single alpha-domain constructs of the Na+/Ca2+ exchanger, NCLX, oligomerize to form a functional exchanger, Biochemistry 45(39) (2006) 11856–66. [DOI] [PubMed] [Google Scholar]
- [80].Dirkx E, da Costa Martins PA, De Windt LJ, Regulation of fetal gene expression in heart failure, Biochim Biophys Acta 1832(12) (2013) 2414–24. [DOI] [PubMed] [Google Scholar]
- [81].Tallquist MD, Molkentin JD, Redefining the identity of cardiac fibroblasts, Nat Rev Cardiol 14(8) (2017) 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R, Osteopontin expression is required for myofibroblast differentiation, Circ Res 102(3) (2008) 319–27. [DOI] [PubMed] [Google Scholar]
- [83].Frantz S, Klaiber M, Baba HA, Oberwinkler H, Volker K, Gabetaner B, Bayer B, Abebetaer M, Schuh K, Feil R, Hofmann F, Kuhn M, Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I, Eur Heart J 34(16) (2013) 1233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Matsumura N, Zordoky BN, Robertson IM, Hamza SM, Parajuli N, Soltys CM, Beker DL, Grant MK, Razzoli M, Bartolomucci A, Dyck JRB, Co-administration of resveratrol with doxorubicin in young mice attenuates detrimental late-occurring cardiovascular changes, Cardiovasc Res 114(10) (2018) 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Touyz RM, Sventek P, Lariviere R, Thibault G, Fareh J, Reudelhuber T, Schiffrin EL, Cytosolic calcium changes induced by angiotensin II in neonatal rat atrial and ventricular cardiomyocytes are mediated via angiotensin II subtype 1 receptors, Hypertension 27(5) (1996) 1090–6. [DOI] [PubMed] [Google Scholar]
- [86].Sempe S, Stuyvers B, Tariosse L, Gouverneur G, Besse P, Bonoron-Adele S, Effect of angiotensin II on calcium release phenomena in normal and hypertrophied single cardiac myocytes, Journal of molecular and cellular cardiology 26(12) (1994) 1649–58. [DOI] [PubMed] [Google Scholar]
- [87].Brandenburger Y, Kennedy ED, Python CP, Rossier MF, Vallotton MB, Wollheim CB, Capponi AM, Possible role for mitochondrial calcium in angiotensin II- and potassium-stimulated steroidogenesis in bovine adrenal glomerulosa cells, Endocrinology 137(12) (1996) 5544–51. [DOI] [PubMed] [Google Scholar]
- [88].Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD, Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure, J Clin Invest 117(9) (2007) 2431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J Clin Invest 128(5) (2018) 2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS, Identification of a ryanodine receptor in rat heart mitochondria, J Biol Chem 276(24) (2001) 21482–8. [DOI] [PubMed] [Google Scholar]
- [91].Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE, Monitoring phosphorylation of the pyruvate dehydrogenase complex, Analytical biochemistry 389(2) (2009) 157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gray LR, Tompkins SC, Taylor EB, Regulation of pyruvate metabolism and human disease, Cellular and molecular life sciences : CMLS 71(14) (2014) 2577–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD, Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice, J Clin Invest 120(10) (2010) 3680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, Hsu YA, Kolwicz SC Jr., Raftery D, Tian R, Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis, Circ Res 126(2) (2020) 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM, Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged, Circulation 105(21) (2002) 2543–8. [DOI] [PubMed] [Google Scholar]
- [96].Pogwizd SM, Sipido KR, Verdonck F, Bers DM, Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis, Cardiovasc Res 57(4) (2003) 887–96. [DOI] [PubMed] [Google Scholar]
- [97].Garbincius JF, Elrod JW, Is the Failing Heart Starved of Mitochondrial Calcium?, Circ Res 128(8) (2021) 1205–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Stengl M, Mubagwa K, Carmeliet E, Flameng W, Phenylephrine-induced stimulation of Na+/Ca2+ exchange in rat ventricular myocytes, Cardiovasc Res 38(3) (1998) 703–10. [DOI] [PubMed] [Google Scholar]
- [99].Ye M, Flores G, Batlle D, Angiotensin II and angiotensin-(1–7) effects on free cytosolic sodium, intracellular pH, and the Na(+)-H+ antiporter in vascular smooth muscle, Hypertension 27(1) (1996) 72–8. [DOI] [PubMed] [Google Scholar]
- [100].deAlmeida AC, van Oort RJ, Wehrens XH, Transverse aortic constriction in mice, J Vis Exp (38) (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Li L, Guo X, Chen Y, Yin H, Li J, Doan J, Liu Q, Assessment of Cardiac Morphological and Functional Changes in Mouse Model of Transverse Aortic Constriction by Echocardiographic Imaging, J Vis Exp (112) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Martini JS, Raake P, Vinge LE, DeGeorge BR Jr., Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ, Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes, Proceedings of the National Academy of Sciences of the United States of America 105(34) (2008) 12457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ, Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade, Circulation 98(17) (1998) 1783–9. [DOI] [PubMed] [Google Scholar]
- [104].Liu R, van Berlo JH, York AJ, Vagnozzi RJ, Maillet M, Molkentin JD, DUSP8 Regulates Cardiac Ventricular Remodeling by Altering ERK1/2 Signaling, Circ Res 119(2) (2016) 249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Simpson P, McGrath A, Savion S, Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines, Circ Res 51(6) (1982) 787–801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.