

Abstract
Introduction
We explored what combination of blood‐based biomarkers (amyloid beta [Aβ]1‐42/1‐40, phosphorylated tau [p‐tau]181, neurofilament light [NfL], glial fibrillary acidic protein [GFAP]) differentiates Alzheimer's disease (AD) dementia, frontotemporal dementia (FTD), and dementia with Lewy bodies (DLB).
Methods
We measured the biomarkers with Simoa in two separate cohorts (n = 160 and n = 152). In one cohort, Aβ1‐42/1‐40 was also measured with mass spectrometry (MS). We assessed the differential diagnostic value of the markers, by logistic regression with Wald's backward selection.
Results
MS and Simoa Aβ1‐42/1‐40 similarly differentiated AD from controls. The Simoa panel that optimally differentiated AD from FTD consisted of NfL and p‐tau181 (area under the curve [AUC] = 0.94; cohort 1) or NfL, GFAP, and p‐tau181 (AUC = 0.90; cohort 2). For AD from DLB, the panel consisted of NfL, p‐tau181, and GFAP (AUC = 0.88; cohort 1), and only p‐tau181 (AUC = 0.81; cohort 2).
Discussion
A combination of plasma p‐tau181, NfL, and GFAP, but not Aβ1‐42/1‐40, might be useful to discriminate AD, FTD, and DLB.
Keywords: amyloid beta, blood biomarker, glial fibrillary acidic protein, neurofilament light, phosphorylated tau
1. BACKGROUND
Dementia is one of the major causes of disability and dependency among older people and affects ≈50 million people worldwide. 1 Alzheimer's disease (AD) is the most common form of dementia and represents ≈70% of cases, 2 followed by frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB). To support the clinical AD diagnosis, amyloid beta (Aβ) and phosphorylated tau (p‐tau) are currently measured in the cerebrospinal fluid (CSF) or visualized on positron emission tomography (PET) scans. 2 Efforts are ongoing to translate these markers into blood‐based biomarkers, with promising results. 3 There are no blood‐based biomarkers available for specifically the FTD and DLB pathologies, but AD‐specific biomarkers or more general neurodegenerative and astrocyte activation biomarkers might be useful to differentiate patients with AD dementia from patients with other dementias. 4 Being able to measure different aspects of neurodegenerative dementias comprehensively in a minimally invasive and low‐cost approach, could contribute to accurate differential diagnosis in an early stage of the diagnostic process. This could accelerate drug development and help with timely treatment and patient management.
Plasma Aβ1‐42/1‐40 and p‐tau181 can be used to discriminate amyloid PET positive and negative individuals across the clinical AD continuum. 5 , 6 , 7 , 8 , 9 , 10 , 11 Furthermore, elevated plasma p‐tau181 levels could accurately differentiate patients with AD from FTD. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 Glial fibrillary acidic protein (GFAP) is a major constituent of reactive astrocytes, and reflects neuronal injury and immune‐related processes. 20 Combined analysis of plasma GFAP with Aβ1‐42/1‐40 improved the prediction of amyloid PET status. 6 , 7 Furthermore, blood‐based GFAP was increased in AD, DLB, and some variants of FTD compared to non‐demented controls, suggesting use as a cross‐dementia biomarker. 21 , 22 Neurofilament light (NfL) is a neurological damage biomarker, with especially high levels in FTD and to a lesser extent in AD, 23 , 24 suggesting NfL could contribute to the differential diagnosis between AD and FTD. 23
A new Simoa multiplex assay was developed that measures Aβ1‐42, Aβ1‐40, NfL, and GFAP simultaneously with high sensitivity and selectivity. 25 It may be interesting and informative to add p‐tau181 to this multiplex. How such a panel of combined markers could improve differential diagnoses, however, remains to be established. Furthermore, up to now, it seems that the Aβ1‐42/1‐40 ratio, alone or as a composite with amyloid beta precursor protein (APP669‐711), reaches a higher accuracy for AD when measured with immunoprecipitation mass spectrometry (IPMS) than immune assay based methods such as Simoa. 8 , 9 , 10 , 11 , 26 It remains to be established if a multiplex panel either solely based on Simoa, or in combination with an IPMS Aβ1‐42/1‐40 method, would optimally provide a differential diagnosis of AD, DLB, and FTD.
The main goal of this study was to explore the differential diagnostic value of combinations of Aβ1‐42/1‐40 ratio, measured by Simoa or IPMS, GFAP, NfL, and p‐tau181.
2. METHODS
2.1. Clinical samples
Cohort 1 consisted of a selection of 160 participants from the Amsterdam Dementia Cohort 27 based on equal diagnostic group size, availability of CSF biomarker data, and availability of ethylenediaminetetraacetic acid (EDTA) plasma samples in the Amsterdam Dementia Biobank. Forty participants were controls (21 with psychiatric symptoms and 19 with subjective cognitive decline), 40 had AD dementia, 40 had FTD (11 with FTD with motor neuron disease [FTD‐motor], 29 with FTD with primary progressive aphasia [FTD‐PPA]), and 40 had DLB. Cohort 2 consisted of 152 participants, also from the Amsterdam Dementia Cohort, based on equal diagnostic group sizes and availability of EDTA plasma in the Amsterdam Dementia Biobank, but were selected to have a more comparable age; 27 38 were controls (all with subjective cognitive decline), 38 had AD dementia, 38 had FTD (six with FTD‐motor, 32 with FTD‐PPA), and 38 had DLB. All visited the Alzheimer Center Amsterdam for extensive dementia screening, consisting of neurological, physical, and neuropsychological evaluation; electroencephalography; brain magnetic resonance imaging; and CSF AD biomarker analysis. 27 , 28 Diagnoses were made upon multidisciplinary consensus based on applicable clinical criteria. 2 , 29 , 30 , 31 , 32 All patients with AD dementia were CSF amyloid positive, and all controls were CSF amyloid negative.
RESEARCH IN CONTEXT
Systematic review: We reviewed the current literature on blood‐based biomarkers for dementia using PubMed, with a focus on the biomarkers amyloid beta (Aβ), phosphorylated tau (p‐tau), neurofilament light (NfL), and glial fibrillary acidic protein (GFAP). The value of combination of these blood‐based biomarkers in discriminating between Alzheimer's disease (AD) dementia and other types of dementia remained to be explored.
Interpretation: We observed that a combination of plasma p‐tau181, NfL, and GFAP, but not the Aβ1‐42/1‐40 ratio, was able to discriminate patients with AD dementia from patients with frontotemporal dementia or from patients with dementia with Lewy bodies, with high accuracy.
Future directions: Our proposed blood‐based biomarker panel should next be validated in a larger cohort, which should include the development and validation of cutoffs to make such a panel useful in daily clinical practice.
Written informed consent to use medical data and biomaterials for research purposes was in place. The study was approved by the VU University medical center ethical committee, and in accordance with the Declaration of Helsinki.
2.2. CSF Aβ1‐42 measurements
CSF Aβ1‐42 concentrations were available for all samples of cohort 1 and for n = 148 (97%) of cohort 2. CSF AD biomarkers of 120 and 140 participants were measured with Innotest enzyme‐linked immunosorbent assays and of 40 and 8 participants with Elecsys, for cohort 1 and cohort 2, respectively. Innotest Aβ1‐42 concentrations were corrected for the drift in biomarker concentrations that occurred over the years. 33 The drift‐corrected cut‐off for Innotest for CSF amyloid positivity was 813 pg/mL. 33 The cut‐off for Elecsys was 1000 pg/mL. 34 To determine the correlation with the plasma markers, Elecsys results were transformed to the predicted Innotest concentration using the formula Innotest Aβ 1‐42 = (Elecsys Aβ 1‐42 +365)/1.87. 34
2.3. Plasma biomarker measurements
K2EDTA‐plasma samples were obtained through venipuncture. After 10‐minute centrifugation at 1,800 x g within 2 hours, plasma was aliquoted in 0.5 mL aliquots in polypropylene tubes and stored at –80°C.
2.3.1. Simoa measurements
Samples were thawed at room temperature and centrifuged at 10,000 x g for 10 minutes. For cohort 1, samples were measured using a pre‐commercial Neurology 4‐plex E kit (Quanterix) that measures Aβ1‐42, Aβ1‐40, GFAP, and NfL simultaneously. This Neurology 4‐plex E was developed in a collaboration between Amsterdam University Medical Centers, and biotechnology companies ADx NeuroSciences and Quanterix. 35 For cohort 2, the commercial Neurology 4‐plex E kit (Quanterix) was used, which gave different absolute values than the pre‐commercial assay. In the next freeze–thaw cycle, p‐tau181 was measured in 149 of the 160 participant samples of cohort 1 and in all samples of cohort 2, using the pTau‐181 V2 Advantage kit (Quanterix; same kit batch for both cohorts). All measurements were performed on the Simoa HDx analyzer, according to manufacturer's instructions.
2.3.2. IPMS
Aβ1‐42, Aβ1‐40, and APP669‐711 were measured in a second zero‐thaw aliquot of the samples of cohort 1 with matrix‐assisted laser desorption/ionization–linear time‐of‐flight mass spectrometer (AXIMA Assurance, Shimadzu) after two consecutive IP steps with Dynabeads M‐270 Epoxy used as beads and mouse monoclonal anti‐Aβ antibodies 6E10 and 4G8 to coat the beads. 8 The composite marker was based on the average of the Z score of Aβ1‐40/1‐42 ratio and APP669‐711/Aβ1‐42 ratio (details in the supporting information). Centre Hospitalier Universitaire (CHU) of Montpellier performed the measurements and modified the normalization from the original method, and followed their own quality control. One APP669‐711 measurement failed; therefore, the composite is available for 159 of the 160 participants.
2.4. Statistics
The IPMS Aβ1‐42/1‐40 ratio, NfL, GFAP, and p‐tau181 were right skewed, thus natural log transformed. In cohort 1, one outlier IPMS Aβ1‐ 42/1‐40 ratio in the DLB group of 34x the mean was excluded, so was one CSF Aβ1‐ 42 concentration in the FTD group of 4x the mean (note: only excluded in analyses with continuous data). In cohort 1, one outlier NfL concentration in the FTD group of 15x the mean (351 pg/mL) and a GFAP outlier in the AD group of 6x the mean (613 pg/mL) were excluded. We used analysis of variance with Bonferroni correction to assess biomarker differences between the diagnostic groups. Pearson correlations were assessed for the Aβ1‐42/1‐40 ratios measured with IPMS and 4‐plex, and the difference in strength between two correlations was tested using methods described in Diedenhofen and Musch. 36 Receiver operating characteristic (ROC) analyses determined the differentiation accuracy of the plasma markers. Areas under the curve (AUCs) were compared with the DeLong test. 37 Sensitivity and specificity were determined at Youden indices. 38 We selected biomarker combinations using the automated backward elimination logistic regression procedure based on the Wald statistic. As a sensitivity analysis, we ran the same models including the other Aβ1‐42/1‐40 ratio or IPMS composite score, which did not change the results (data not shown). Statistical analyses were performed using SPSS (version 26) and R (version 3.6.1, packages Cocor, pROC).
3. RESULTS
3.1. Patient characteristics
Characteristics and fluid biomarker concentrations of the cohorts are shown in Table 1. In cohort 1, patients with FTD and DLB were older than the controls and patients with AD. In cohort 2, all four groups were of comparable age, but there were more males in the DLB group than in the other diagnostic groups. For both cohorts, Mini‐Mental State Examination scores of patients with FTD and DLB were between those of controls and patients with AD. In both cohorts, NfL (r = 0.35–0.37, both: P < .001) and GFAP (r = 0.27–0.39, both: P < .001) correlated with age, whereas p‐tau181 correlated with age only in cohort 2 (r = 0.22, P = .007; cohort 1: r = 0.08, P = .34) and 4‐plex Aβ1‐42/1‐40 correlated with age only in cohort 1 (r = –0.20, P = .01; cohort 2 –0.11, P = .18). The IPMS Aβ1‐42/1‐40 ratio (r = –0.08, P = .31) and IPMS composite (r = –0.04, P = .63) did not correlate with age. There were no differences in biomarker concentrations between males and females within the diagnostic groups in either of the cohorts.
TABLE 1.
Cohort characteristics
| Cohort 1 | Cohort 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Control n = 40 |
AD n = 40 | FTD n = 40 | DLB n = 40 | Total n = 160 |
Control n = 38 |
AD n = 38 | FTD n = 38 | DLB n = 38 | Total n = 152 | |
| Age | 56 (53–59) | 58 (55–59) | 64 (61–70) a , b | 70 (62–74) a , b | 59 (56–66) | 63 (59–66) | 63 (59–67) | 63 (59–67) | 67 (64–69) | 64 (60–68) |
| Female sex (%) | 20 (50%) | 20 (50%) | 22 (55%) | 12 (30%) | 74 (46%) | 20 (53%) | 20 (53%) | 20 (53%) | 3 (8%) a , b | 63 (41%) |
| Education | 5 (4–6) | 5 (4–6) | 5 (4–6) | 5 (4–6) | 5 (4–6) | 6 (5–6) | 5 (5–6) | 5 (4–6) | 5 (4–6) | 5 (4–6) |
| MMSE | 28 (25–29) b | 18 (13–22) a | 25 (20–27) a , b | 23 (17–26) a , b | 23 (18–27) | 29 (28–29) b | 20 (18–23) a | 24 (18–27) a | 23 (21–26) a , b | 24 (20–28) |
| CSF Aβ1‐42 (pg/mL)* | 1075 (962–1182) b | 576 (495–652) a | 950 (739–1159) b | 724 (615–991) a , b | 818 (616–1094) | 1092 (922–1271) b | 602 (512–692) a | 942 (761–1270) b | 839 (648–1094) a , b | 856 (648–1120) |
| CSF amyloid positive (%) | 0 (0%) | 40 (100%) | 10 (25%) | 24 (60%) | 74 (46%) | 0 (0%) | 38 (100%) | 10 (29%) | 18 (47%) | 66 (45%) |
| Mass spectrometry | ||||||||||
| Aβ1‐42/1‐40 ratio* | 0.044 (0.042–0.048) b | 0.037 (0.035–0.041) a | 0.041 (0.038–0.045) | 0.043 (0.038–0.044) b | 0.042 (0.037–0.045) | N.A. | N.A. | N.A. | N.A. | N.A. |
| Aβ1‐42 (pg/mL)* | 0.49 (0.42–0.53) b | 0.41 (0.36–0.45) a | 0.46 (0.42–0.52) b | 0.48 (0.44–0.56) b | 0.46 (0.42–0.52) b | N.A. | N.A. | N.A. | N.A. | N.A. |
| Aβ1‐40 (pg/mL)* | 10.8 (9.8–11.8) | 10.5 (9.5–12.1) | 11.5 (10.3–12.5) | 11.7 (10.4–13.1) | 11.1 (9.9–12.5) | N.A. | N.A. | N.A. | N.A. | N.A. |
| APP699‐711 (pg/mL)* | 0.46 (0.40–0.50) | 0.40 (0.37–0.50) | 0.42 (0.36–0.48) | 0.45 (0.38–0.53) | 0.43 (0.38–0.48) | N.A. | N.A. | N.A. | N.A. | N.A. |
| Simoa | ||||||||||
| Aβ1‐42/1‐40 ratio* | 0.14 (0.13–0.16) b | 0.12 (0.11–0.13) a | 0.13 (0.11–0.14) a | 0.13 (0.11–0.14) a | 0.13 (0.12–0.14) | 0.06 (0.05–0.07) b | 0.05 (0.05–0.06) a | 0.05 (0.05–0.06) | 0.05 (0.05–0.06) | 0.05 (0.05–0.06) |
| Aβ1‐42 (pg/mL)* | 12.7 (11.1–14.5) b | 10.9 (9.0–11.7) a | 11.6 (9.7–13.1) | 11.3 (10.6–13.1) | 11.5 (9.9–13.1) | 7.22 (6.48–7.73) b | 6.16 (5.47–6.73) a | 7.33 (6.47–8.34) b | 6.83 (6.25–8.20) b | 6.81 (6.10–7.74) |
| Aβ1‐40 (pg/mL)* | 88.4 (82.9–97.0) | 90.7 (78.0–97.3) | 93.3 (83.4–103.6) | 90.5 (85.5–102.9) | 90.4 (82.6–100.5) | 115 (105–137) | 119 (102–140) | 136 (117–154) a , b | 125 (114–147) | 124 (109–145) |
| NfL (pg/mL)* | 17.0 (13.4–23.6) a , b | 31.3 (25.2–40.1) a | 45.5 (35.3–86.5) a , b | 30.9 (22.1–53.3) a | 30.6 (20.8–44.6) | 11.0 (8.5–15.3) b | 18.8 (14.0–25.7) a | 28.7 (21.4–52.0) a , b | 15.5 (11.7–20.2) | 17.6 (12.0–26.8) |
| GFAP (pg/mL)* | 534 (342–693) b | 1580 (1091–1970) a | 943 (610–1231) a , b | 1047 (611–1580) a , b | 962 (563–1487) | 66.6 (47.1–85.9) b | 119 (99.4–178) a | 91.0 (54.2–128) b | 106 (65.6–142) a , b | 96.4 (59.0–129) |
| P‐tau181 (pg/mL), plasma | 1.18 (0.92–1.39) b | 2.94 (2.04–3.99) a | 1.35 (0.94–2.05) b | 1.81 (1.09–2.65) a , b | 1.61 (1.03–2.59) | 1.24 (1.09–1.64) b | 2.60 (2.01–3.03) a | 1.65 (1.09–2.71) b | 1.55 (1.07–2.04) b | 1.68 (1.20–2.55) |
Notes: Values shown as median (interquartile range). Aβ1‐42, Aβ1‐40, NfL, and GFAP were measured with the 4‐plex pre‐commercial assay in cohort 1, and with the 4‐plex commercial assay in cohort 2. These two assays give different absolute values. CSF Aβ1‐42 was missing for 4 FTD patients of cohort 2.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; p‐tau181, phosphorylated tau 181.
Indicates a statistically significant difference between groups (P < .05) with Control in post hoc pairwise comparisons.
P < .05 versus AD.
3.2. Comparison of Aβ1‐42/1‐40 ratio between the platforms: 4‐plex and IPMS
First, we assessed in cohort 1 how the Simoa and IPMS platforms for Aβ1‐42/1‐40 measurement compare. The Aβ1‐42/1‐40 ratio measured with 4‐plex and with IPMS were only moderately correlated (r = 0.34, P < .001, n = 159; Figure 1A). The 4‐plex Aβ1‐42/1‐40 ratio correlated with similar strength with CSF Aβ1‐ 42 (r = 0.34, P < .001, n = 159; Figure 1B) as the IPMS plasma Aβ1‐42/1‐40 ratio (r = 0.46, P < .001, n = 158, ∆r = 0.12, P = .14; Figure 1C). Assessing differences between CSF amyloid negative controls and CSF amyloid positive patients with AD, to decide which assay should be selected for our further analyses, we observed that with the 4‐plex, median Aβ1‐42/1‐40 ratio was decreased 16% in patients with AD (median 0.12 [interquartile range (IQR): 0.11–0.13] pg/mL) compared to controls (0.14 [0.13–0.16], P < .001; Table 1; Figure 2A), and with the IPMS Aβ1‐42/1‐40 ratio this decrease was 17% in patients with AD (0.037 [0.035–0.041] pg/mL) compared to controls (0.044 [0.042—0.048] pg/mL, P < .001; Table 1; Figure 2B). The 4‐plex Aβ1‐42/1‐40 ratio differentiated controls and patients with AD with AUC = 0.80 (95% confidence interval [CI]: 0.70–0.90, P < .001; Figure 2C) and a sensitivity of 0.90 and a specificity of 0.65. The IPMS Aβ1‐42/1‐40 ratio had a similar AUC of 0.82 (95% CI: 0.72–0.92, P < .001; Figure 2C), but a sensitivity of 0.78 and a specificity of 0.83. The performance of the 4‐plex and IPMS Aβ1‐42/1‐40 ratio did not differ in terms of AUC (∆AUC = 0.02, DeLong P = .83) and was similar to the IPMS composite (∆AUC = 0.01, DeLong P = .83, Figure S1 in supporting information). Given the similarity in AUCs and the value of the Simoa 4‐plex as a multiplex assay that measures several independent markers simultaneously and requires less volume than IPMS, we continued the analyses for differential diagnostic value with the 4‐plex Aβ1‐42/1‐40 ratio as amyloid marker.
FIGURE 1.
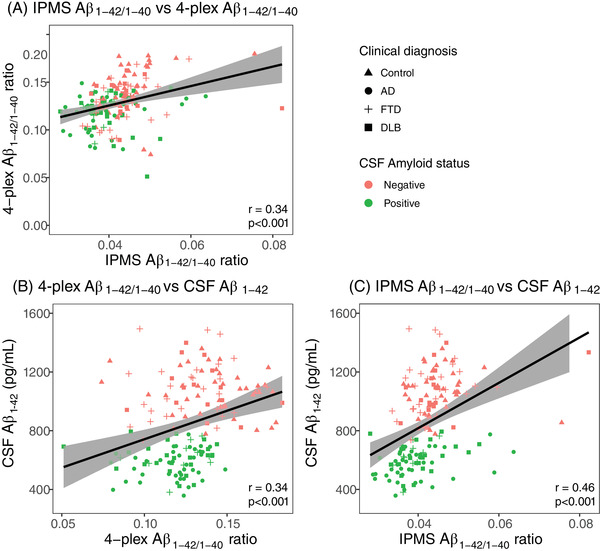
Correlations between amyloid plasma markers and CSF markers. A, Pearson correlation between the 4‐plex and IPMS Aβ1‐42/1‐40 ratio. B, Pearson correlation between the 4‐plex Aβ1‐42/1‐40 ratio and CSF Aβ1‐ 42. C, Pearson correlation between the IPMS Aβ1‐42/1‐40 ratio and CSF Aβ1‐ 42. Shape indicates clinical diagnosis, color indicates CSF amyloid status. Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; IPMS, immunoprecipitation mass spectrometry
FIGURE 2.
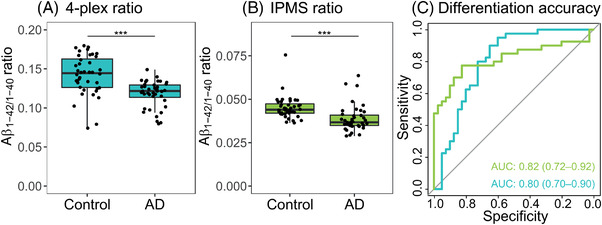
Differentiation accuracy of amyloid markers in CSF amyloid negative controls and CSF amyloid positive AD patients. A, The Aβ1‐42/1‐40 ratio measured with the Simoa 4‐plex. B, The Aβ1‐42/1‐40 ratio measured with IPMS. C, ROC curves of the two plasma amyloid marker for differentiation between control and AD (in blue: Simoa; in green: IPMS). There was no difference in accuracy between the two ratios. Aβ, amyloid beta; AD, Alzheimer's disease; AUC, area under the curve; CSF, cerebrospinal fluid; IPMS, immunoprecipitation mass spectrometry; ROC, receiver operating characteristic
3.3. Differential diagnosis
In addition to the Aβ1‐42/1‐40 ratio, we measured NfL, GFAP, and p‐tau181. In cohort 1, the Aβ1‐42/1‐40 ratio was decreased not only in patients with AD (P < .001), but also in patients with FTD (P = .010) and DLB (P = .002) compared to controls. The Aβ1‐42/1‐40 ratio did not differ among the AD, FTD, or DLB group in this cohort (P > .48). NfL concentrations were increased in all diagnostic groups compared to controls (all P < .001), and were highest in patients with FTD. GFAP concentrations were increased in all diagnostic groups compared to controls (all P < .001), and were highest in patients with AD. P‐tau181 concentrations were increased in patients with AD compared to the other dementia groups and controls (P≤.001; Figure 3, Table 1).
FIGURE 3.
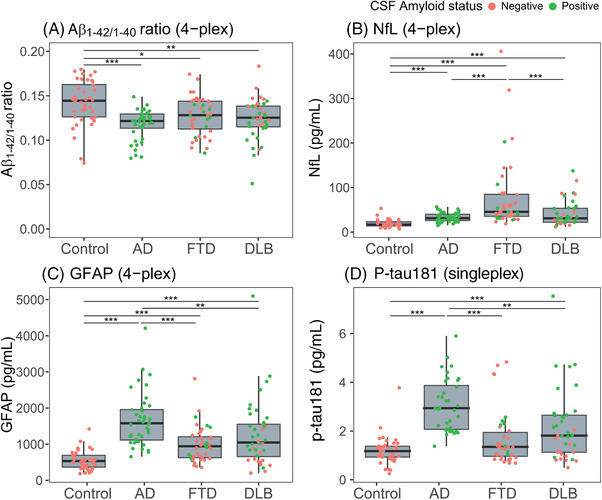
4‐Plex plasma (A) Aβ1‐42/1‐40, (B) NfL, (C) GFAP, and (D) Single‐plex p‐tau181 in cohort 1. Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; p‐tau, phosphorylated tau. ***P < .001, **P < .01, *P < .05
When measured in cohort 2, the Aβ1‐42/1‐40 ratio was decreased only in patients with AD (P = .002) compared to controls. Similar to cohort 1, the Aβ1‐42/1‐40 ratio did not differ among the AD, FTD, or DLB groups (all: P = 1.000). NfL concentrations were increased in all diagnostic groups compared to controls (all: P < .015). NfL concentrations were again highest in patients with FTD. Contrary to cohort 1, GFAP was increased in patients with AD (P < .001) and in patients with DLB (P = .042) compared to controls, but not in patients with FTD compared to controls (P = .060). GFAP was also increased in patients with AD compared to patients with FTD (P = .011) and patients with DLB (P = .017). In agreement with findings in cohort 1, p‐tau181 concentrations were increased in AD compared to the other dementia groups and compared to controls (all: P < .007; Figure S2 in supporting information; Table 1).
3.4. Classification of patients by the biomarkers
The AUCs of the ROC curve for differentiation between patients with AD and FTD and between patients with AD and DLB are detailed in Table 2. To define the panels, Wald's backward elimination logistic regression analysis was performed among all biomarkers (4‐plex Aβ1‐42/1‐40, NfL, GFAP, and single‐plex p‐tau181).
TABLE 2.
Diagnostic accuracy of the 4‐plex Aβ1‐42/1‐40 ratio, NfL, GFAP, p‐tau181, and optimal panel
| Cohort 1 | ||||||
|---|---|---|---|---|---|---|
| AD vs. FTD | n | AUC | 95% CI | P‐value | Sensitivity | Specificity |
| Aβ1‐42/1‐40 ratio | 40 vs 40 | 0.62 | 0.49–0.75 | .065 | 0.38 | 0.95 |
| NfL | 40 vs 40 | 0.79 | 0.69–0.89 | <.001 | 0.48 | 0.98 |
| GFAP | 40 vs 40 | 0.81 | 0.71–0.90 | <.001 | 0.75 | 0.73 |
| P‐tau181 | 36 vs 38 | 0.85 | 0.75–0.94 | <.001 | 0.74 | 0.97 |
| Panel (NfL, P‐tau181) | 36 vs 38 | 0.94 | 0.87–1.00 | <.001 | 0.87 | 1.00 |
| AD vs. DLB | ||||||
| Aβ1‐42/1‐40 ratio | 40 vs 40 | 0.60 | 0.47–0.73 | .124 | 0.38 | 0.95 |
| NfL | 40 vs 40 | 0.50 | 0.37–0.64 | .946 | 0.28 | 0.98 |
| GFAP | 40 vs 40 | 0.69 | 0.57–0.81 | .004 | 0.50 | 0.85 |
| P‐tau181 | 36 vs 37 | 0.75 | 0.63–0.87 | <.001 | 0.54 | 0.97 |
| Panel (NfL, GFAP, p‐tau181) | 36 vs 37 | 0.88 | 0.80–0.96 | <.001 | 0.84 | 0.81 |
| Cohort 2 | ||||||
|---|---|---|---|---|---|---|
| AD vs. FTD | n | AUC | 95% CI | P‐value | Sensitivity | Specificity |
| Aβ1‐42/1‐40 ratio | 38 vs 38 | 0.58 | 0.46–0.71 | .205 | 0.63 | 0.55 |
| NfL | 38 vs 37 | 0.78 | 0.68–0.88 | <.001 | 0.89 | 0.55 |
| GFAP | 37 vs 38 | 0.71 | 0.60–0.83 | .001 | 0.45 | 0.95 |
| P‐tau181 | 38 vs 38 | 0.71 | 0.59–0.83 | .002 | 0.66 | 0.76 |
| Panel (NfL, GFAP, p‐tau181) | 37 vs 37 | 0.90 | 0.82–0.98 | <.001 | 0.81 | 0.92 |
| AD vs. DLB | ||||||
| Aβ1‐42/1‐40 ratio | 38 vs 38 | 0.56 | 0.43–0.69 | .372 | 0.29 | 0.90 |
| NfL | 38 vs 38 | 0.63 | 0.51–0.76 | .045 | 0.55 | 0.71 |
| GFAP | 37 vs 38 | 0.65 | 0.52–0.77 | .027 | 0.40 | 0.87 |
| P‐tau181 | 38 vs 38 | 0.81 | 0.71–0.91 | <.001 | 0.76 | 0.76 |
| Panel (p‐tau181) | 38 vs 37 | 0.81 | 0.71–0.91 | <.001 | 0.76 | 0.76 |
Notes: The panels were selected with backward logistic regression based on Wald's statistics, among the plasma markers Aβ1‐42/1‐40, NfL, GFAP, and p‐tau181. Sensitivity and specificity are at Youden's indices.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; AUC, area under the curve; CI, confidence interval; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; p‐tau181, phosphorylated tau 181.
3.5. AD versus FTD
In cohort 1, Aβ1‐42/1‐40 had the lowest AUC (AUC = 0.62, 95% CI: 0.49–0.75, P = .065) and p‐tau181 had the highest AUC (AUC = 0.85, 95% CI: 0.75–0.94, P < .001, Figure 4A) for differentiating patients with AD from patients FTD. The combination of NfL and p‐tau181 was selected as the optimal differential diagnostic panel for AD versus FTD with an AUC of 0.94 (95% CI: 0.87–1.00, P < .001, sens: 0.87, spec: 1.00).
FIGURE 4.
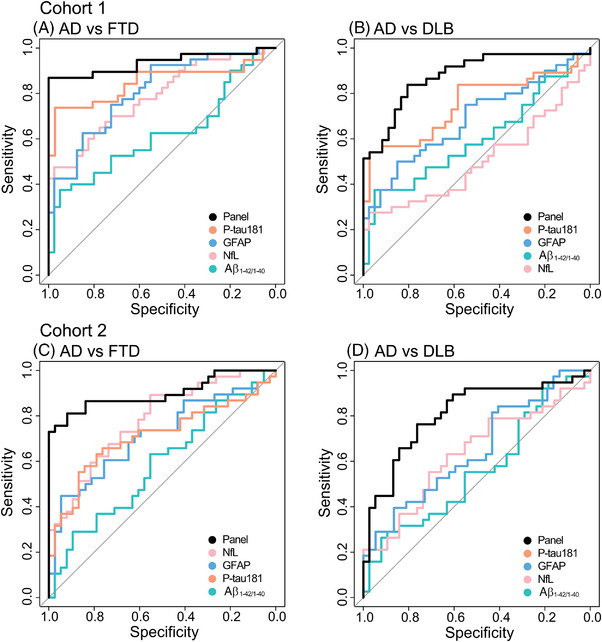
ROC AUCs for the different biomarkers. Cohort 1: (A) Differentiation between AD and FTD. The panel consists of NfL and p‐tau181. (B) Differentiation between AD and DLB. The panel consists of NfL, GFAP and P‐tau181. Cohort 2: (C) Differentiation between AD and FTD. The panel consists of NfL, GFAP, and P‐tau181. (D) Differentiation between AD and DLB. The panel consists of p‐tau. The panels were corrected for age and sex. Aβ, amyloid beta; AD, Alzheimer's disease; AUC, area under the curve; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies, FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; p‐tau181, phosphorylated tau 181; ROC, receiver operating characteristic
In cohort 2, Aβ1‐42/1‐40 had the lowest AUC (AUC = 0.58, 95% CI: 0.46–0.71, P = .205), similar to cohort 1, but NfL had the highest AUC (AUC = 0.78, 95% CI: 0.68–0.88, P < .001, Figure 4C) for differentiating patients with AD from patients with FTD. The combination of NfL, GFAP, and p‐tau181 was selected in the cohort 2 as the optimal differential diagnostic panel for AD versus FTD with an AUC of 0.88 (95% CI: 0.80–0.96, P < .001, sens: 0.84, spec: 0.81).
3.6. AD versus DLB
In cohort 1, NfL had the lowest AUC (AUC = 0.50, 95% CI: 0.37–0.64, P = .946) and p‐tau181 had the highest AUC (AUC = 0.75, 95% CI: 0.63–0.87, P < .001, Figure 4B) for differentiating patients with AD from patients with DLB. The combination of NfL, GFAP, and p‐tau181 was selected as the optimal differential diagnostic panel for AD versus DLB with an AUC of 0.88 (95% CI: 0.80–0.96, P < .001, sens: 0.84, spec: 0.81).
In cohort 2, Aβ1‐42/1‐40 had the lowest AUC (AUC = 0.56, 95% CI: 0.43–0.69, P = .372) and similar to cohort 1, p‐tau181 had the highest AUC (AUC = 0.81, 95% CI: 0.71–0.91, P < .001, Figure 4D) for differentiating patients with AD from patients with DLB. P‐tau181 alone was selected as the optimal differential diagnostic panel for AD versus DLB with an AUC of 0.81 (95% CI: 0.71–0.91, P < .001, sens: 0.76, spec: 0.76).
4. DISCUSSION
We found that the 4‐plex and IPMS Aβ1‐42/1‐40 ratio and IPMS composite performed similarly in differentiating CSF amyloid positive patients with AD and CSF amyloid negative controls. Furthermore, we showed that the plasma Aβ1‐42/1‐40 ratio, NfL, GFAP, and p‐tau181 differ between diagnostic groups. All markers showed a largely similar pattern of differences between the diagnostic groups in both cohort 1 and cohort 2, though the optimal panel for differential diagnosis differed between the cohorts. Our data suggest that, depending on patient characteristics (e.g., age, sex), a plasma biomarker panel for optimal differentiation between AD and FTD, and between AD and DLB, will include plasma p‐tau181 but not plasma Aβ1‐42/1‐40 ratio, and will likely include plasma GFAP and plasma NfL.
We hypothesized that the IPMS would perform better than the 4‐plex, because the differentiation accuracy for amyloid positivity in cohorts was higher in IPMS studies (AUC range 0.84–0.89) 8 , 9 , 10 , 39 than in Simoa studies (AUC range 0.73–0.77). 7 , 40 Also, two recent head‐to‐head studies showed that IPMS Aβ1‐42/1‐40 outperformed Simoa Aβ1‐42/1‐40. 11 , 26 The similarity in performance in our study could be due to our cohort selection, which includes two extremes; CSF amyloid positive symptomatic AD patients compared to CSF amyloid negative controls. In a preclinical cohort, the differences in plasma amyloid might be more subtle and better reflected with IPMS. In addition, the IPMS analyses were performed with the same method but by a different research group than described in this recent comparison study. 26 Inter‐laboratory variability and modifications in assay set‐up could be an explanation for the difference in performance. IPMS has the advantage of high assay specificity and no interference of the sample matrix. 41 The 4‐plex assay uses antibodies that are highly specific for full‐length Aβ1‐42 and Aβ1‐40, just like IPMS. 35
We found that p‐tau181 had often the highest accuracy in differentiating AD from FTD, and AD from DLB, and was included as a discriminatory biomarker in both panels for both cohorts. Earlier studies by us and others also convincingly showed that p‐tau181 is a highly specific marker for AD pathology. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 That p‐tau181 is useful to differentiate patients with AD from patients with DLB has not been reported extensively yet. In line with earlier publications, 23 , 42 , 43 , 44 we found that NfL is strongly increased in patients with FTD compared to AD, which explains that it was included in the AD versus FTD panel for both cohort 1 and cohort 2. GFAP showed a similar pattern to p‐tau181 for differential diagnoses, but with lower or comparable accuracy than p‐tau181. Specifically, GFAP was selected in only cohort 1 in the AD versus DLB panel and only in cohort 2 in the AD versus FTD panel. This suggests that GFAP might be of added value to p‐tau181 in a differential diagnostic panel, but it depends on the patient characteristics. FTD and DLB patients were older than AD dementia patients in cohort 1 but not in cohort 2, and over the cohorts, we observed relationships of both NfL and GFAP with age.
The observed high accuracy of GFAP as a standalone marker for AD suggests that this marker reflects the process of Aβ aggregates triggering a neuroinflammatory state and promoting glial cell activation. 45 The primary morphological change in astrogliosis is hypertrophy, which is linked to an increase in expression of intermediate filaments such as GFAP. 46 Astrocytes are the most prevalent cell types in the brain, thus even if only a small amount of GFAP is released per cell, this may still be a major response that may result in high GFAP concentrations in blood. In addition, in AD, GFAP was increased in DLB compared to controls in both cohorts, with concentrations between control and AD, 21 which was driven by the CSF amyloid positive patients with DLB. GFAP levels were also increased in FTD patients compared to controls in cohort 1, to a lesser extent than in AD. Of note, we could not reproduce this in our more age‐matched cohort 2. A recent study has shown that GFAP concentrations can be increased in symptomatic progranulin (GRN) mutation carriers. 47 We lack the genetic data to verify this in our cohort. The 4‐plex Aβ1‐42/1‐40 ratio showed a similar pattern of changes as p‐tau181 and GFAP, being deviated in patients with AD, though the differentiation accuracy was lower than for p‐tau181 and GFAP (AUC > 0.56, P ≤ .372). Few studies have focused on the Aβ1‐42/1‐40 ratio in neurodegenerative diseases outside of the AD spectrum thus far. One study measured Aβ1‐42/1‐40 with Simoa in controls, patients with mild cognitive impairment (MCI), AD, and across different FTD syndromes but did not find a difference among all groups. 12 Similarly, our results show no differential diagnostic value of Aβ1‐42/1‐40 across patients with AD, FTD, or DLB.
In both cohorts, the differentiation accuracy between AD and FTD strongly improved by using biomarker panels compared to single biomarkers, except that based on our cohort 2, p‐tau181 was sufficient as a single marker to differentiate between AD and DLB. This is likely the result of the larger number of CSF amyloid negative patients with DLB in cohort 2, which results in smaller overlap in the p‐tau181 concentrations of the AD and DLB groups. Every biomarker tested reflects a different aspect of AD pathology or brain damage in general. In the future, these markers should be tested in a larger and more heterogeneous population, for example, to define cut‐off values for clinical use. Our exploratory results, however, suggest that an algorithm based on NfL, GFAP, and p‐tau as plasma biomarkers could provide an indication of probability per clinical diagnosis. Combined with clinical assessment this could help provide differential diagnoses of AD and other neurodegenerative diseases and support clinical trial inclusion in a minimally invasive and low‐cost manner. 48
This study had several limitations. First, our cohorts were relatively small and were included from our memory clinic only and did not reflect a broader population of people with dementia, that is, with older patients with AD. As a result, our AD patients typically have less copathology, which might have influenced our panel selections. It would also be interesting to assess whether our panels are also succesful in differentiating among the MCI stages of DLB, FTD, and AD. Second, we used two different batches of the Neurology 4‐plex E kit, due to which absolute values were not comparabale. Third, due to the cohort characteristics (large difference in age for cohort 1 and large differences in sex in cohort 2) age and sex were not appropriate covariates. Last, because we did not know the apolipoprotein E (APOE) status of all participants, we did not include this as a variable in the regression analyses, while APOE is an important risk factor for AD and confounded blood biomarker results in some studies. 10 , 49
One of the strengths of our study was the diversity of our cohort, including different non‐AD dementias. Also, the Aβ1‐42/1‐40 measured with IPMS and immunoassay in the same cohort allowed us to robustly compare their performance. Last, the Simoa Neurology 4‐plex E kit is commercially available for immediate use by other groups, with the advantage that Simoa technology offers scalability for large‐scale validation and routine implementation.
In summary, the IPMS and 4‐plex immunoassay Aβ1‐42/1‐40 ratio showed similar performance in indicating amyloid pathology. Different combinations of NfL, GFAP, and p‐tau181 were valuable for differential diagnosis of AD, FTD, and DLB. Further research on cut‐off values would help implement these markers in clinical practice.
AUTHOR CONTRIBUTIONS
Design: ET, IV, CT; experiments: JK, AA; data analysis: ET, IV; manuscript writing: ET, IV with assistance of all authors; study supervision: JV, AB, YP, EL, WF, ES, CH, CT.
CONFLICTS OF INTEREST
ET, IV, JK, AA, and YP report no conflicts of interest. JV is an employee of ADx NeuroSciences. AB was an employee of Quanterix Corporation when the study was performed and holds shares in Quanterix. AL has received funding from stichting Dioraphte, Alzheimer Nederland, and ZonMW Memorabel (project #733050509). AL has performed contract research with Axovant, EIP Pharma, and Combinostics. All funding is paid to her institution. WF has performed contract research for Biogen MA Inc, and Boehringer Ingelheim. All funding is paid to her institution. WF has been an invited speaker at Boehringer Ingelheim, Biogen MA Inc, Danone, Eisai, WebMD Neurology (Medscape), Springer Healthcare. All funding is paid to her institution. WF is consultant to Oxford Health Policy Forum CIC, Roche, and Biogen MA Inc. All funding is paid to her institution. WF participated in advisory boards of Biogen MA Inc and Roche. All funding is paid to her institution. WF was associate editor of Alzheimer, Research & Therapy in 2020/2021. WF is associate editor at Brain. All funding is paid to her institution. ES is an employee and shareholder of ADx NeuroSciences, Gent, Belgium. CH has a collaboration contract with Shimadzu European Innovation Center including a PhD thesis. CT has a collaboration contract with ADx NeuroSciences and Quanterix, performed contract research for or received grants from AC‐Immune, Axon Neurosciences, Biogen, Brainstorm Therapeutics, Celgene, EIP Pharma, Eisai, PeopleBio, Roche, Toyama, Vivoryon. CT serves on editorial boards of Medidact Neurologi/Springer, Alzheimer Research and Therapy, Neurology: Neuroimmunology & Neuroinflammation, and is editor of a Neuromethods book for Springer. All funding is paid to her institution.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
IV is appointed on a research grant by Alzheimer Nederland (NL‐17004). JK and CH received funding from France Alzheimer (PI : Pr A. Gabelle). Research of CT is supported by the European Commission (Marie Curie International Training Network, grant agreement No 860197 [MIRIADE], and JPND), Health Holland, the Dutch Research Council (ZonMW), Alzheimer Drug Discovery Foundation, The Selfridges Group Foundation, Alzheimer Netherlands, Alzheimer Association. CT is a recipient of ABOARD, which is a public–private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). Research programs of Wiesje van der Flier have been funded by ZonMW, NWO, EU‐FP7, EU‐JPND, Alzheimer Nederland, CardioVascular Onderzoek Nederland, Health∼Holland, Topsector Life Sciences & Health, stichting Dioraphte, Gieskes‐Strijbis fonds, stichting Equilibrio, Pasman stichting, stichting Alzheimer & Neuropsychiatrie Foundation, Biogen MA Inc, Boehringer Ingelheim, Life‐MI, AVID, Roche BV, Fujifilm, Combinostics. WF holds the Pasman chair. WF is a recipient of ABOARD, which is a public–private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). All funding is paid to her institution.
Thijssen EH, Verberk IMW, Kindermans J, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alzheimer's Dement. 2022;14:e12285. 10.1002/dad2.12285
REFERENCES
- 1. WHO . Global action plan on the public health response to dementia 2017‐2025. World Health Organization; 2017. Licence: CC BY‐NC‐SA 3.0 IGO. [Google Scholar]
- 2. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11:e11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lleó A, Irwin DJ, Illán‐Gala I, et al. A 2‐Step Cerebrospinal Algorithm for the Selection of Frontotemporal Lobar Degeneration Subtypes. JAMA Neurol. 2018;75:738‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Meyer S, Schaeverbeke JM, Verberk IMW, et al. Comparison of ELISA‐ and SIMOA‐based quantification of plasma Abeta ratios for early detection of cerebral amyloidosis. Alzheimers Res Ther. 2020;12:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Translational Psychiatry. 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verberk IMW, Thijssen E, Koelewijn J, et al. Combination of plasma amyloid beta(1‐42/1‐40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res Ther. 2020;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐beta biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 9. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13:841‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma beta‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647‐e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Keshavan A, et al. Population‐based blood screening for preclinical Alzheimer's disease in a British birth cohort at age 70. Brain : a journal of neurology. 2021;144:434‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020:387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020:379‐386. [DOI] [PubMed] [Google Scholar]
- 14. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422‐433. [DOI] [PubMed] [Google Scholar]
- 15. Benussi A, Karikari TK, Ashton N, et al. Diagnostic and prognostic value of serum NfL and p‐Tau181 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2020;91:960‐967. [DOI] [PubMed] [Google Scholar]
- 16. Lantero Rodriguez J, Karikari TK, Suárez‐Calvet M, et al. Plasma p‐tau181 accurately predicts Alzheimer's disease pathology at least 8 years prior to post‐mortem and improves the clinical characterisation of cognitive decline. J. 2020;140:267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14:989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated‐tau isoforms track CNS change in Alzheimer's disease. J Exp Med. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative Accuracy of Plasma Phospho‐tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nature Neuroscience. 2021;24:312‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oeckl P, Halbgebauer S, Anderl‐Straub S, et al. Glial Fibrillary Acidic Protein in Serum is Increased in Alzheimer's Disease and Correlates with Cognitive Impairment. J Alzheimers Dis. 2019;67:481‐488. [DOI] [PubMed] [Google Scholar]
- 22. Benussi A, Ashton NJ, Karikari TK, et al. Serum Glial Fibrillary Acidic Protein (GFAP) Is a Marker of Disease Severity in Frontotemporal Lobar Degeneration. J Alzheimers Dis. 2020;77:1129‐1141. [DOI] [PubMed] [Google Scholar]
- 23. Illán‐Gala I, Lleo A, Karydas A, et al. Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease. Neurology. 2021;96:e671‐e683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bridel C, Van Wieringen WN, Zetterberg H, et al. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: a Systematic Review and Meta‐analysis. JAMA Neurol. 2019:1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thijssen EH, Verberk IMW, Vanbrabant J, et al. Highly specific and ultrasensitive plasma test detects Abeta(1‐42) and Abeta(1‐40) in Alzheimer's disease. Sci Rep. 2021;11:9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐Head Comparison of 8 Plasma Amyloid‐β 42/40 Assays in Alzheimer Disease. JAMA Neurol. 2021;78:1375‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Der Flier WM, Scheltens P. Amsterdam Dementia Cohort: performing Research to Optimize Care. J Alzheimers Dis. 2018;62:1091‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Der Flier WM, Pijnenburg YAL, Prins N, et al. Optimizing patient care and research: the Amsterdam Dementia Cohort. J Alzheimers Dis. 2014;41:313‐327. [DOI] [PubMed] [Google Scholar]
- 29. McKeith IG, et al. Diagnosis and management of dementia with Lewy bodies. Fourth consensus report of the DLB Consortium. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krudop WA, Kerssens CJ, Dols A, et al. Building a new paradigm for the early recognition of behavioral variant frontotemporal dementia: late Onset Frontal Lobe Syndrome study. Am J Geriatr Psychiatry. 2014;22:735‐740. [DOI] [PubMed] [Google Scholar]
- 32. Jessen F, Amariglio RE, Buckley RF, et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020;19:271‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tijms BM, Willemse EAJ, Zwan MD, et al. Unbiased Approach to Counteract Upward Drift in Cerebrospinal Fluid Amyloid‐beta 1‐42 Analysis Results. Clin Chem. 2018;64:576‐585. [DOI] [PubMed] [Google Scholar]
- 34. Willemse EAJ, Maurik IS, Tijms BM, et al. Diagnostic performance of Elecsys immunoassays for cerebrospinal fluid Alzheimer's disease biomarkers in a nonacademic, multicenter memory clinic cohort: the ABIDE project. Alzheimers Dement (Amst). 2018;10:563‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thijssen EH, Verberk IMW, Vanbrabant J, et al. Highly specific and ultrasensitive plasma test detects Abeta(1‐42) and Abeta(1‐40) in Alzheimer's disease. Scientific Reports. 2021;11:9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Diedenhofen B, Musch J. cocor: a comprehensive solution for the statistical comparison of correlations. PloS one. 2015;10:e0121945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delong ER, Delong DM, Clarke‐Pearson DL. Comparing the Areas under Two or More Correlated Receiver Operating Characteristic Curves: a Nonparametric Approach. Biometrics. 1988;44:837‐845. [PubMed] [Google Scholar]
- 38. Youden WJ. Index for rating diagnostic tests. Cancer;3:32‐35. [DOI] [PubMed] [Google Scholar]
- 39. Tosun D, Veitch D, Aisen P, et al. Detection of β‐amyloid positivity in Alzheimer's Disease Neuroimaging Initiative participants with demographics, cognition, MRI and plasma biomarkers. Brain Communications. 2021;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janelidze S, Stomrud E, Palmqvist S, et al. Plasma beta‐amyloid in Alzheimer's disease and vascular disease. Sci Rep. 2016;6:26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oeckl P, Otto M. A Review on MS‐Based Blood Biomarkers for Alzheimer's Disease. Neurol Ther. 2019;8:113‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zucchi E, Bonetto V, Sorarù G, et al. Neurofilaments in motor neuron disorders: towards promising diagnostic and prognostic biomarkers. Mol Neurodegener. 2020;15:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Wolf F, Ghanbari M, Licher S, et al. Plasma tau, neurofilament light chain and amyloid‐beta levels and risk of dementia; a population‐based cohort study. Brain. 2020;143:1220‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14:577‐589. [DOI] [PubMed] [Google Scholar]
- 45. Heneka MT, Carson MJ, Khoury JEl, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frost GR, Li Y‐M. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol. 2017;7:170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91:263‐270. [DOI] [PubMed] [Google Scholar]
- 48. Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer's disease dementia using plasma phospho‐tau combined with other accessible measures. Nat Med. 2021:1034‐1042. [DOI] [PubMed] [Google Scholar]
- 49. Palmqvist S, Janelidze S, Stomrud E, et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease‐Related beta‐Amyloid Status. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information