

Abstract
Objective.
Immune checkpoint blockade (ICB) therapy shows limited efficacy in ovarian cancers due to the “cold” immune phenotype surrounding these tumors. Previous studies have shown that in ovarian cancer Wnt/β-catenin pathway activation contributes to this immune phenotype. Here, we evaluated the anti-tumor and immune-enhancing properties of the Wnt inhibitor, CGX-1321, used alone or in combination with either DKN-01 or anti-PD-1 therapy, in pre-clinical ovarian cancer models.
Methods.
The parental ID8 murine ovarian cancer model harboring a knock-out of p53 (ID8p53−/−) and MISIIR-Tag spontaneous ovarian cancer models were used to test the effects of CGX-1321 alone or in combination therapies on tumor burden and immune cell landscape in the tumor microenvironment (TME). Flow cytometry and NanoString analyses were used to characterize the changes in tumor-intrinsic signaling and immune-related profiles in the TME of ovarian cancer in response to treatments.
Results.
CGX-1321 significantly reduced tumor burden and constrained tumor progression in the ID8p53−/− and MISIIR-Tag models. Furthermore, CGX-1321 increased infiltrating CD8+ T cells in the TME. Combining CGX-1321 with either DKN-01 or anti-PD-1 therapy also decreased tumor burden and increased CD8+ T cell infiltration in the omentum TME but did not do so to a greater extent that CGX-1321 monotherapy.
Conclusions.
CGX-1321 significantly reduced tumor burden and enhanced CD8+ T cell levels in ovarian cancer, nevertheless the addition of DKN-01 or anti-PD-1 therapies did not enhance these effects of CGX-1321. Further investigation is needed to determine if CGX-1321 + DKN-01 combination treatment sensitizes pre-clinical ovarian cancer to ICB therapy.
Keywords: Ovarian cancer, Wnt/β-catenin signaling, Immune checkpoint blockade (ICB) therapy, Tumor microenvironment (TME)
1. Introduction
An emerging paradigm has classified tumor microenvironments (TMEs) into different immunological phenotypes including hot, alteredexcluded, altered-immunosuppressed, and cold [1]. Cancers that are considered immunologically “cold” have a low immunoscore (low T and cytotoxic T (Tc) cell density in TME), low tumor mutational burden, poor antigen presentation, and/or a lack of T cell killing [1]. As the majority of ovarian cancers are considered “cold,” understanding how to reverse this phenotype is critical in order to develop new approaches to sensitize these tumors to ICB therapy in hopes to provide more durable therapeutic responses. The Wnt/β-catenin tumor-intrinsic signaling pathway has emerged as an immune evasion pathway, and data by our group and others show that Wnt/β-catenin signaling and increased β-catenin levels correlates with a lack of T cell-mediated killing in ovarian cancer [2,3].
The Wnt/β-catenin pathway is dysregulated in several cancers, including ovarian cancer [4,5]. Activated Wnt/β-catenin signaling corresponds to non-T cell inflamed tumors [6] and chemo-resistance [7]. Animal studies from our group using a murine ovarian cancer model, highlighted a negative correlation between the degree of Wnt/β-catenin signaling and T cell activity, and impairment of this signaling pathway increased T cell function, decreased tumor burden, and improved survival [3,8,9]. The Wnt/β-catenin pathway is activated by Wnt ligands, which is secreted from cells after modification by the porcupine (PORCN) enzyme [10], and PORCN inhibitors (PORCNi’s) can block the biogenesis of Wnt ligands thus suppressing Wnt/β-catenin-dependent signaling (Fig. 1A).
Fig. 1.
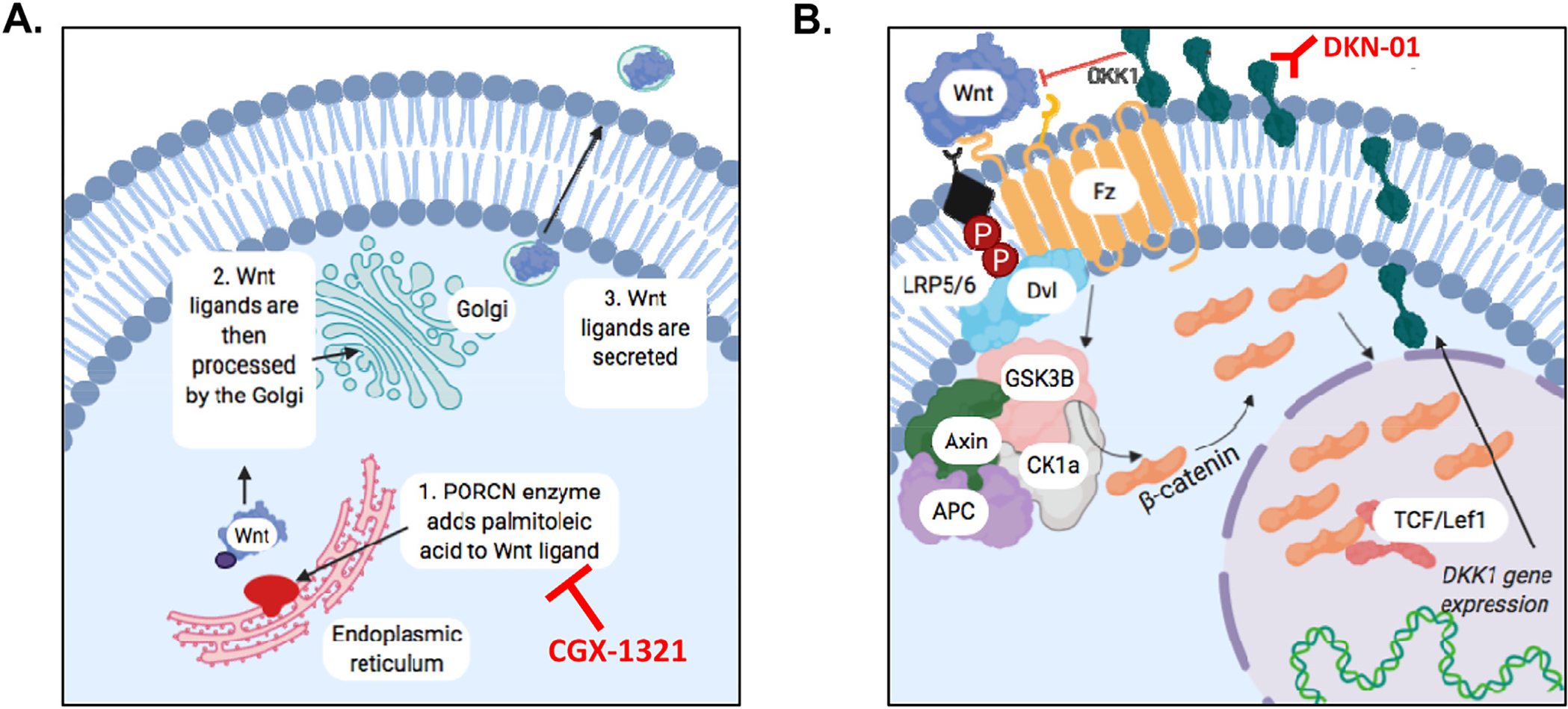
Role of PORCN and DKK1 in Wnt/β-catenin pathway signaling. (A) The porcupine (PORCN) enzyme is necessary for proper Wnt signaling to occur. PORCN is located within the endoplasmic reticulum and modifies Wnt ligands via the addition of adding palmitoleic acid. Ligands are then processed by the golgi body and ultimately secreted. Secretion of Wnt ligand can be blocked by PORCN inhibitors (e.g. CGX-1321). (B) Dickkopf-1 (DKK1) is a Wnt-target gene that encodes the DKK1 protein. As levels of DKK1 protein increase, Wnt signaling is eventually inhibited via a negative feedback mechanism. DKK1 can be inhibited by DKK1 antibody (e.g. DKN-01).
Dickkopf-1 (DKK1) is a Wnt-target gene that encodes the DKK1 protein that competes with Wnt ligand for LRP5/6 binding [11], thus creating a negative-feedback mechanism to suppress Wnt/β-catenin-dependent signaling (Fig. 1B). Even though DKK1 serves as a negative regulator of the Wnt/β-catenin pathway, increased levels of DKK1 corresponds to poor prognosis in various cancer types, including cervical [12] and ovarian cancer [13,14]. A prior study from our lab found that the over-expression of DKK1 in ovarian cancer promotes immune evasion by fostering myeloid-derived suppressor cells (MDSCs) within the TME of ovarian cancer [2]. These results suggest that blocking DKK1’s function would hinder immune evasion in ovarian cancer, but this would also leave Wnt/β-catenin signaling unregulated.
Both PORCN and DKK1 inhibitors have been investigated in clinical trials, (PORCN inhibitors: NCT02675946, NCT01351103; DKK1 inhibitor: NCT03395080, NCT02013154), but no studies have investigated dual inhibition of PORCN and DKK1. Therefore, we postulated that the combination of DKK1 inhibition and/or ICB therapy would increase the efficacy of PORCN inhibition to optimally decrease ovarian tumor progression.
2. Materials and methods
2.1. Cell lines and culture
The ID8 murine epithelial ovarian parental cancer cell line was provided by Dr. Yancey Gillespie (University of Alabama at Birmingham, Birmingham, Alabama). The ID8p53−/− cell line is a derivative of the ID8 parental cell line with CRISPR/Cas9 gene editing for a single gene mutation (Trp53−/−) and was provided by Dr. Iain McNeish (Wolfson Wohl Cancer Research Centre, Institute of Cancer Sciences, University of Glasgow, Glasgow, United Kingdom). Cells were maintained at 37 °C and 5% CO2 and cultured in DMEM medium (Corning) supplemented with 4% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, USA), 1× insulin/selenium/transferrin (Invitrogen), penicillin/streptomycin, and 2 mM L-glutamine. Cells were maintained in culture fewer than 10 passages from the parent stock. Experiments were performed when cells were at 70–80% confluency.
2.2. Mouse studies
All animal studies were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC). C57BL/6 mice were obtained from NCI Charles River (Wilmington, MA, USA), and 7–8 week old C57BL/6 mice were challenged with 7 million ID8p53−/− cells intraperitoneally (i.p.) on day 0. Treatment groups began with the same number of mce in each group. Treatments began either 10-, 14- or 28-days post-tumor challenge, and lasted until day 42 post-tumor challenge. Mice were treated with 1 mg/kg CGX-1321 (daily; oral gavage), 10 mg/kg DKN-01 (twice per week; i.p. injection), 250 μg/mouse anti-PD-1 antibody (every 3 days; i.p. injection), or vehicle/control. Doses were decided upon based on recommendations from the drug companies. The C57BL/6-MIISIR-Tag mice that spontaneously develop ovarian carcinoma due to expression of Simian Virus 40 T Antigen (SV40-TAg) under the control of the Mullerian Inhibitory Substance Type II Receptor promoter was provided by Dr. Troy Randall (University of Alabama at Birmingham). In this model, tumors spontaneously develop in the bilateral ovaries and metastasize to the omentum resulting in ascites [15]. Treatment with 1 mg/kg CGX-1321 (daily) was initiated when tumors had developed (approximately at 10 weeks of age) and continued for a total of eight weeks. Omentums and ascites were collected for endpoint analyses. Notably, mice intermittently died throughout the course of the experiments due to drug/experimental factors. This was variable between groups and thus impacted the group sizes reflected in the paper.
2.3. NanoString nCounter mRNA analysis
Omentum tumors were harvested and stored in RNALater (Invitrogen). RNA was isolated using Trizol Plus RNA Purification Kit (Invitrogen) according to the manufacturer’s instructions. Aqueous phase solutions were transferred to a spin cartridge found in the PureLink RNA Mini Kit and processed similarly. Samples of high purity (A260/A280 > 2, A260/A230 > 1.4) were processed on the NanoString nCounter Flex system (NanoString Technologies, Seattle, WA, USA) using the mouse PanCan Pathways and mouse PanCan Immune premade panels. Briefly, 100 ng of purified RNA was hybridized for at least 16 h with the respective Reporter Code set and Capture Probes for each panel separately. The samples were purified and immobilized on the NanoString Prep Station and counted on the NanoString Digital Analyzer. Analysis was performed using nSolver 3.0 software (NanoString Technologies). Cluster 3.0 and Java TreeView-1.1.6r4 were used to create heat maps. Expression levels were normalized to housekeeping genes that were not discarded by the gNorm program in the advanced analysis module.
2.4. Flow cytometry
Omentum tumors were harvested and placed in digestion buffer (DMEM 25 Mm HEPES, 3.5% fatty acid free BSA plus 0.5 mg/ml collagenase +70μg/ml DNAse I) (all from Corning) and fragmented with scissors. Tumor fragments were placed in an orbital shaker at 250–300 rpm for 30 min at 37 °C and subsequently filtered through 70 μm nylon mesh, washed with staining buffer (1× PBS, 2% BCS, and 2 mM EDTA) (all from Corning), and centrifuged at 800 xg for 10 min. The following antibodies were used for staining: CD3 (100214CD3), NK1.1 (108716) CD45.2 (109824), (all from Biolegend, San Diego, CA, USA), CD8a (551,162 A 1), CD5 (553023), CD19 (11–0191–85), zombie aqua (423101) (all from BD Biosciences, San Jose, CA, USA), CD4 (Q10092) (Invitrogen, Carlsbad, CA, USA) and live-dead discriminator 7-Amino-AMD (7AAD) (129935) (Calbiochem/Millipore Sigma, Burlington, MA, USA). Samples were run using a BD FACS Canto II (BD Biosciences), DIVA software 6.1.3. Data were analyzed with FlowJo version 10.9.6.
2.5. Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8.0 (San Diego, CA, USA). NanoString data was analyzed separately. Determination of statistical significance between group means was done using multiple unpaired independent t-tests with an alpha of 0.05. For determination of significance between more than two group means, one-way or two-way ANOVA’s were used correcting for multiple comparisons using Tukey’s multiple comparisons test.
3. Results
3.1. CGX-1321 monotherapy decreases tumor burden in the ID8p53−/− model, and ablated tumor progression in the MISIIR-Tag model
Previously, our group investigated the effects of PORCN inhibitor, CGX-1321, on tumor growth in ID8 parental and ID8p53−/− models [3]. To make our current study more translatable, we used the ID8p53−/− model since the majority of ovarian cancers harbor p53 mutations [16], and also lengthened CGX-1321 treatment. Here, treatment began 14-days post-tumor challenge and lasted for a total of 31 days. In doing so, we found that CGX-1321 significantly decreased omental weights (p = 0.008) compared to vehicle/control-treated mice when treated for 31 days (Fig. 2A). There were no ascites present in mice receiving treatment with CGX-1321 compared to vehicle/control-treated mice (Fig. 2B). Additionally, we studied the effects of CGX-1321 treatment on tumor burden in the MISIIR-Tag model. Treatment with CGX-1321 ablated the progression of tumors, as no increases were seen in omentum (Fig. 2C) or ovarian (Fig. 2D) weights compared to vehicle/control-treated mice.
Fig. 2.
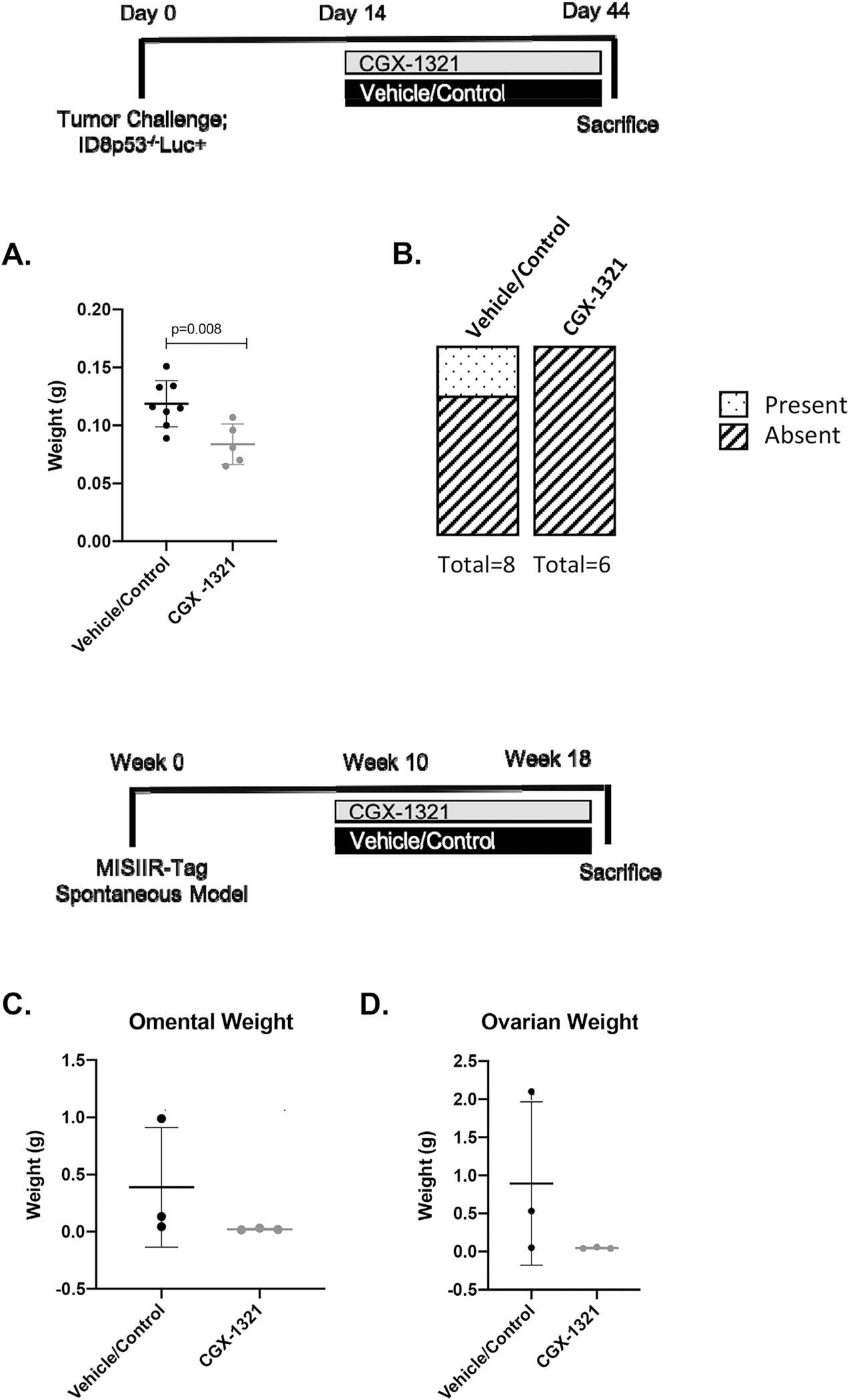
CGX-1321 monotherapy decreases tumor burden in the ID8p53−/− model, and ablated tumor progression in the MISIIR-Tag model. Female, 7–8 weeks old, C57BL/6 mice were challenged with 7 million ID8p53−/− cells on day 0. Treatment consisted of 1 mg/kg CGX-1321 (oral gavage) or vehicle/control beginning 14 days post-tumor challenge. Mice were treated daily for 30 days until day 44. Mice were monitored throughout treatment and sacrificed on day 45, and (A) omentum weights (g) and (B) presence of ascites were measured. MISIIR-Tag spontaneous ovarian cancer model was also used, and treatments (as previously described) began when mouse omentums had tumors (~10 weeks of age) and treatment lasted for 8 weeks, and (C) omentum and (D) ovary weights were measured at the end of treatment.
3.2. The addition of DKN-01 does not enhance CGX-1321-mediated anti-tumor effects in the ID8p53−/− model
Our previous study investigating the effects of DKK1 inhibitor, DKN-01, in the ID8 parental model suggested that DKN-01 does not directly impact tumor growth, but rather the immune components in the TME. Based on this, we believed that PORCN and DKK1 inhibition might have a synergistic anti-tumor effect through impacting both tumor intrinsic and immune components in the TME. In order to investigate the effects of CGX-1321 and DKN-01 treatments in the ID8p53−/− model, we first performed western blot analysis on ID8p53−/− whole cell lysates treated with control, CGX-1321, DKN-01, or CGX-1321 + DKN-01 to determine the effects of treatment on the phosphorylation of LRP6 (p-LRP6), a marker of Wnt signaling activation. We found that treatment with CGX-1321 (CGX) did not significantly alter p-LRP6 compared to control, but WNT3a (Wnt ligand) treatment increased p-LRP6 (activated Wnt signaling) compared to control (Fig. 3A). Similar to WNT3A, DKN-01 treatment increased p-LRP6 confirming that this antibody successfully blocked DKK1 negative regulation of Wnt signaling pathway (Fig. 3A). The combined treatment of CGX-1321 + DKN-01 decreased p-LRP6 as compared to DKN-01 single therapy (Fig. 3A). These results suggest that in combination, CGX-1321 can inhibit Wnt signaling by overriding effects of DKN-01, resulting in Wnt signaling inhibition.
Fig. 3.
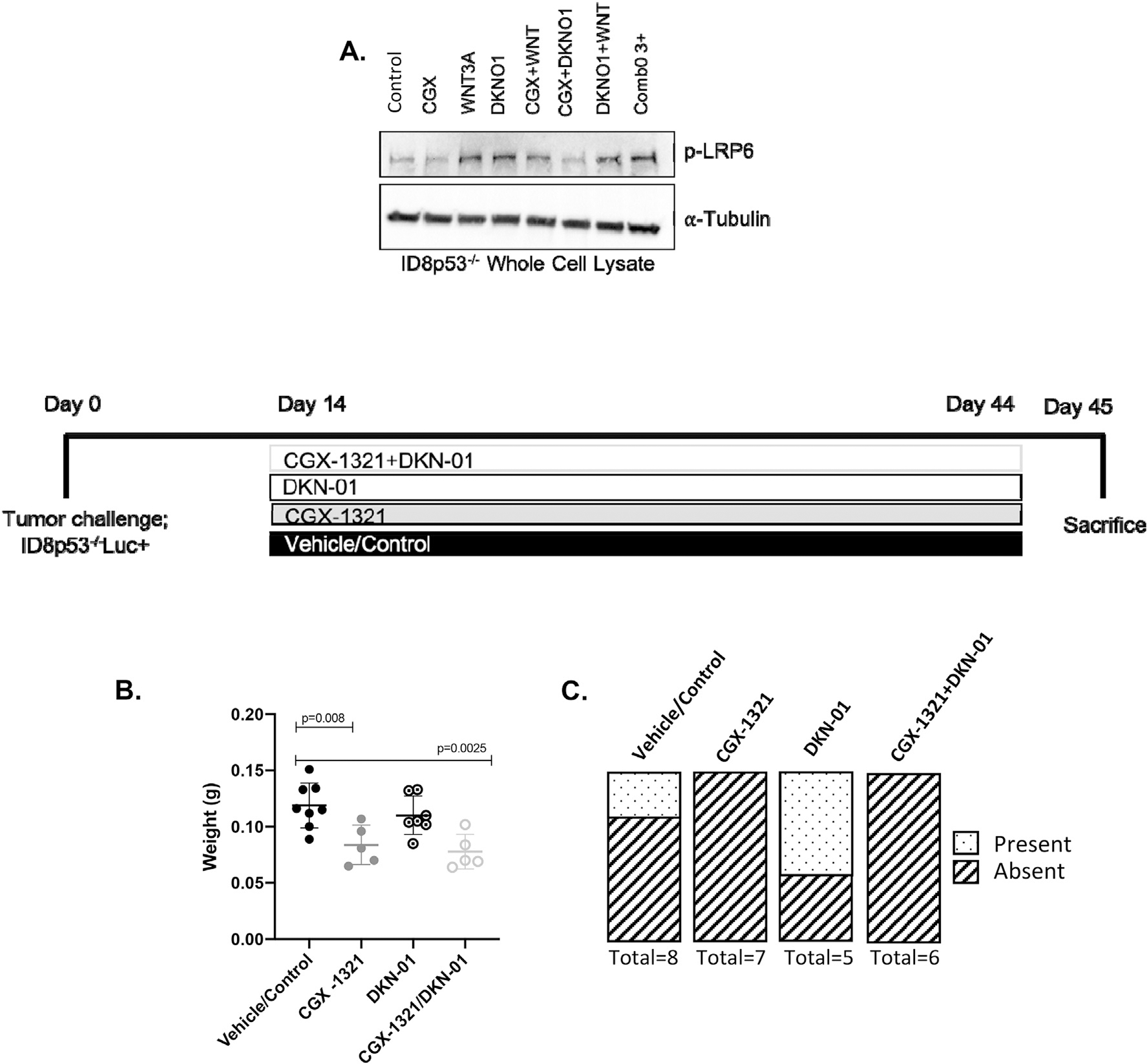
The addition of DKN-01 does not enhance CGX-1321-mediated anti-tumor effects in the ID8p53−/− model. (A) Western blot analysis on protein expression of phosphorylated LRP6 (p-LRP6) and α-Tubulin in ID8p53−/− whole cell lysates. ID8p53−/− model was initiated and treatment with 1 mg/kg CGX-1321 (oral gavage; daily), 10 mg/kg DKN-01 (intraperitoneal injection; 2×/week), and vehicle/control started day-14 post-tumor challenge and continued through day 44, and (B) omental weights (g) and (C) presence of ascites were measured on day 45.
Next, we sought to investigate the effects of DKN-01 alone and in combination with CGX-1321 on tumor growth in the ID8p53−/− model. Following 31 days of treatment, CGX-1321 monotherapy significantly decreased omental weights (p = 0.008) compared to vehicle/control-treated mice (Fig. 3B). However, no significant difference in omental weights (p = 0.40) was observed in mice treated with DKN-01 compared to vehicle/control-treated mice (Fig. 3B). Combination treatment with CGX-1321 + DKN-01 significantly decreased omental weights (p = 0.003) compared to vehicle/control-treated mice, however no difference was observed between CGX-1321- and CGX-1321 + DKN-01-treated mice (Fig. 3B). Mice treated with CGX-1321 or CGX-1321 + DKN-01 did not develop any ascites, whereas four DKN-01-treated mice developed ascites although the volume was unmeasurable in two of these mice, and two vehicle/control-treated mice developed ascites (Fig. 3C).
We performed full RNA sequencing on 3 representative omental tumors from each treatment group, and heatmaps were generated to highlight differences in gene expression for a panel of known Wnt-related genes [17] in response to CGX-1321, DKN-01, and CGX-1321 + DKN-01 treatment. The CGX-1321 + DKN-01-treated tumors had overall decreased expression of Wnt-related genes compared to vehicle/control-treated samples, while also having a distinct profile from tumors treated with DKN-01 or CGX-1321 monotherapies (Supplemental Fig. 1A). These results suggest that the anti-tumor effects seen were CGX-1321-driven but did not address the effects that these therapies might have on immune landscape in the omentum TME.
3.3. CGX-1321 promote CD8+ T cell levels, while DKN-01 decreases MDSC-related gene signatures in ID8p53−/− model
Our previous studies investigating PORCN’s effects on the immune landscape of ovarian tumors found that treatment with PORCNi increased T cell gene signatures, infiltration, and function [3,9]. Even though DKN-01 did not affect tumor growth, our previous data suggested it may improve anti-tumor immune response by decreasing MDSCs and increasing natural killer (NK) cells and CD8+ T cells in the TME of ovarian cancer [2]. Therefore, we next utilized NanoString analysis to screen for patterns of gene expression changes in immune-related pathways following treatment with vehicle/control, CGX-1321, DKN-01, and CGX-1321 + DKN-01 (Fig. 4A). When utilizing the immune pathway score as a means for identifying groups of genes affected by treatment, DKN-01 monotherapy did not demonstrate notable changes compared to vehicle/control-treated mice, although scores for dendritic cell (DC), NK, and major histocompatibility complex (MHC) functions were decreased in mice treated with DKN-01 compared to vehicle/control-treated mice (Fig. 4A). Both CGX-1321- and CGX-1321 + DKN-01-treated mice had increased scores for antigen processing and leukocyte function (Fig. 4A). These data suggest that the two therapies work differently in altering immune response, and together, elicit a diverse anti-tumor immune response.
Fig. 4.
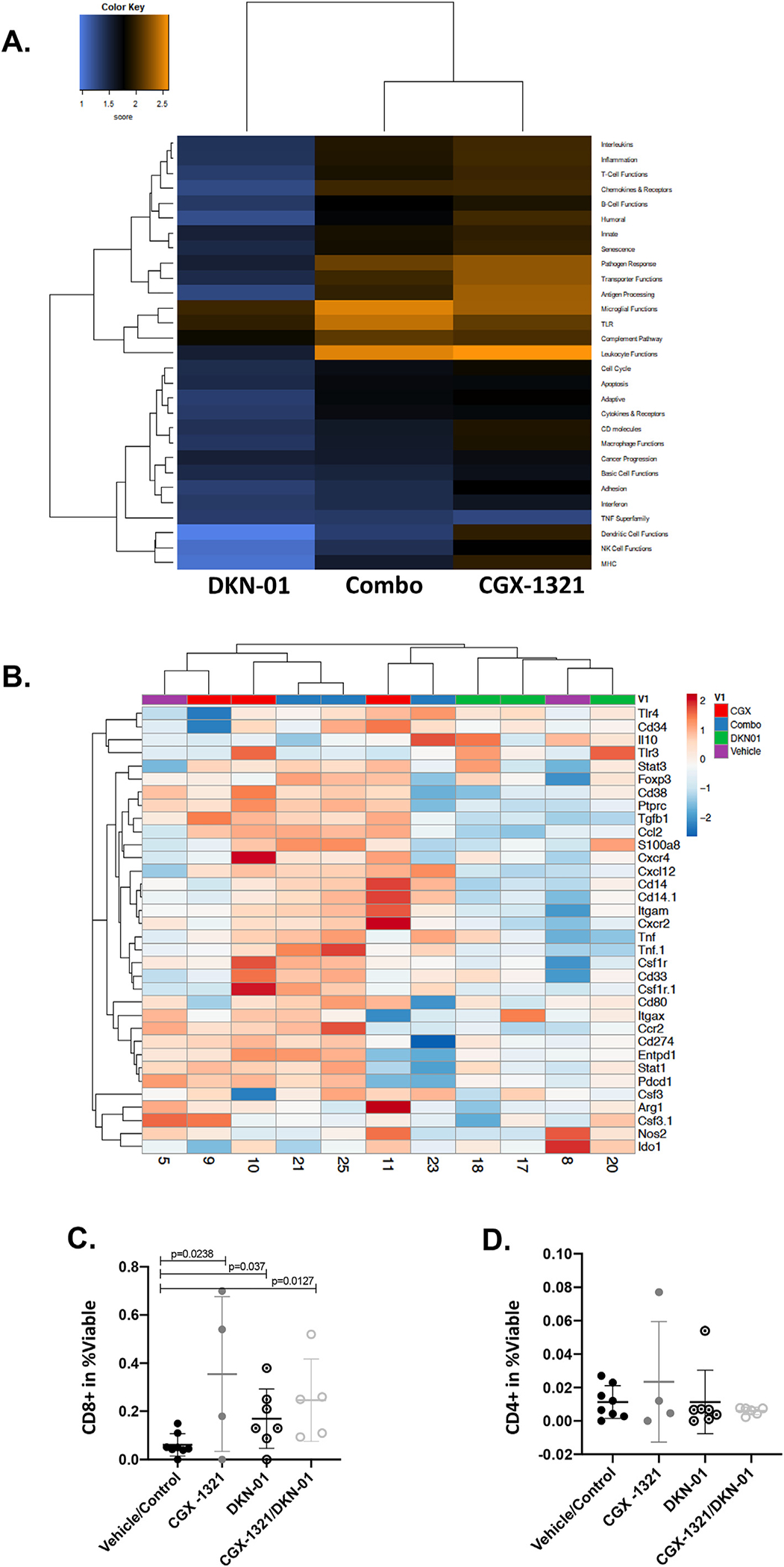
CGX-1321 promotes CD8+ T cell levels, while DKN-01 decreases MDSC-related gene signatures in ID8p53−/− model. (A) NanoString analysis of omentum samples from mice treated with CGX-1321, DKN-01, and DKN-01 + CGX-1321 (Combo) in ID8p53−/− model. Heat map depicts changes in immune pathway scores compared to vehicle/control-treated mice. (B) MDSC-related gene expression changes in ID8p53−/− model where mice were treated with vehicle/control, DKN-01, CGX-1321, or DKN-01 + CGX-1321 (Combo). Flow cytometry analysis was performed on omentums to determine the levels of infiltrating (C) cytotoxic T (Tc; CD8+) and (D) helper T (Th; CD4+) cells in omentum TME.
Following NanoString analysis, we analyzed specific changes in MDSC-related gene expression following treatment with vehicle/control, CGX-1321, DKN-01, or CGX-1321 + DKN-01. Heatmaps were generated to highlight differences in gene expression for a panel of known MDSC-related genes [18], and consistent with previous findings, DKN-01 treatment decreased the expression of various MDSC-related genes (Cd38, Ptprc, Tgfb1, Cxcr2, Tnf, Tnf.1, Ccr2, Entpd1, Nos2, and Arg1) compared to vehicle/control-, CGX-1321-, or CGX-1321 + DKN-01-treated mice (Fig. 4B). Mice treated with CGX-1321 and CGX-1321 + DKN-01 had increased expression of the majority of these genes compared to vehicle/control-treated mice (Fig. 4B). These data suggest that DKN-01 efficiently suppresses pro-tumor-related immune gene signatures in the omentum TME.
Flow cytometry was utilized to characterize immune cell populations within the omental TME following treatment with vehicle/control, CGX-1321, DKN-01, and CGX-1321 + DKN-01. Compared to vehicle/control treatment, each treatment increased the percentage of CD8+ T cells (Fig. 4C), but not CD4+ helper T cells (Fig. 4D), in the omental TME. However, the combination of CGX-1321 + DKN-01 had a similar effect on CD8+ T cell infiltration as CGX-1321 monotherapy (Fig. 4C). These results suggest that CD8+ T cell infiltration in the omentum TME was driven primarily by CGX-1321 monotherapy. Altogether, these data suggest that CGX-1321 and DKN-01 therapies elicit different immune responses in the omentum TME that need to be further characterized.
3.4. The addition of anti-PD-1 therapy does not enhance CGX-1321-mediated anti-tumor effects in the ID8p53−/− model
The programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) axis is a common mechanism employed by cancer cells to evade the immune system; PD-1 on T cells binds PD-L1 on the tumor cell, and this interaction prevents anti-tumor T cell activity [19]. This axis is a common target in ICB therapy but has seen limited success in gynecologic malignancies due to the lack of cytotoxic CD8+ T cell infiltration into the TME. Since our current and previous [3,9] data suggest that PORCNi increases CD8+ T cell infiltration, we next asked whether this therapy would increase sensitivity to anti-PD-1 therapy in the ID8p53−/− model.
Here, ID8p53−/− tumors were initiated as previously described and 10-days post-tumor challenge treatment began. Following 33 days of treatment, there was a significant decrease in omental weights (p = 0.0067; p = 0.008) (Fig. 5A) and ascites volumes (p < 0.0001; p = 0.0063) (Fig. 5B) in CGX-1321- and CGX-1321 + anti-PD-1-treated mice compared to vehicle/control-treated mice. However, there was no difference observed in omental weights (Fig. 5A) or ascites volumes (Fig. 5B) between anti-PD-1-treated and vehicle/control-treated mice. In addition, there were no differences observed in omental weights (Fig. 5A) or ascites volumes (Fig. 5B) between CGX-1321- and CGX-1321 + anti-PD-1-treated mice. These results suggest that the anti-tumor effects seen were CGX-1321-driven, and CGX-1321 administration did not sensitize tumors to anti-PD-1 therapy.
Fig. 5.
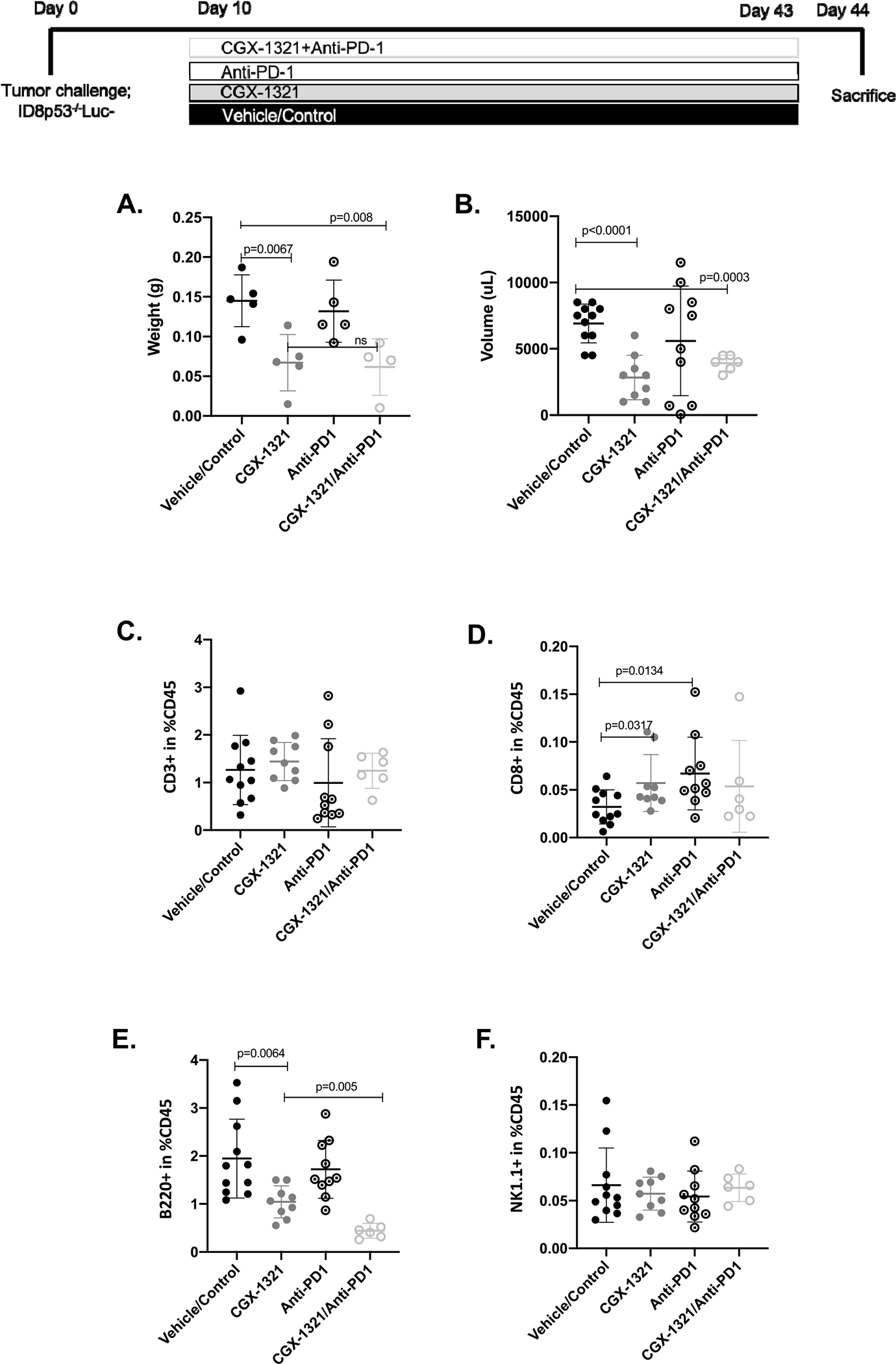
The addition of anti-PD-1 does not enhance CGX-1321-mediated anti-tumor effects in the ID8p53−/− model. ID8p53−/− model was initiated and treatment with 1 mg/kg CGX-1321 (oral gavage; daily), 250 μg/mouse anti-PD-1 (intraperitoneal injection; every 3 days), and vehicle/control started 10–days post-tumor challenge. (A) Omental weights (g) and (B) ascites volumes (uL) were measured at day 44. Flow cytometry analysis was performed on omentums to determine the levels of infiltrating of (C) T (CD3+) cells, (D) cytotoxic T (Tc; CD8+) cells, (E) B (B220+) cells, and (F) natural killer (NK1.1+) cells in TME.
To evaluate broad transcriptomic changes that may have arisen in response to CGX-1321 mono- or combinatorial therapies, we performed full RNA sequencing on 3 representative omentum tumors from each treatment group. Heatmaps were generated to highlight differences in gene expression for Wnt-signaling related genes [17] in response to vehicle/control, CGX-1321, anti-PD-1 therapy, and CGX-1321 + anti-PD-1 therapy. The general decrease in Wnt-related gene expressions by CGX-1321 was largely consistent with or without the addition of the anti-PD-1 therapy, whichdid not result in significant alternations of Wnt-related gene expressions (Supplemental Fig. 1B).
When investigating the effects of anti-PD-1 and CGX-1321 + anti-PD-1 on the immune landscape in the omental TME with flow cytometry, we found no change in total T cells (CD3+) in response to therapy (Fig. 5C). However, there was a significant increase in CD8+ T cells in omentum TME from mice treated with CGX-1321 (p = 0.03) or anti-PD-1 therapy (p = 0.01) (Fig. 5D). However, there was no significant difference in CD8+ T cell infiltration in the omentums of mice treated with CGX-1321 + anti-PD-1 therapy compared to either monotherapy (Fig. 5D). There was a significant decrease in B cells (B220+) in mice treated with CGX-1321 (p = 0.0064) and CGX-1321 + anti-PD-1 therapy (p = 0.005) compared to vehicle/control-treated mice (Fig. 5E). Furthermore, there was no difference NK cells (Fig. 5F) following treatments. These results confirm a CGX-1321-mediated CD8+ T cell response and highlighted a potential B cell response to CGX-1321 treatment.
4. Discussion
The interplay of Wnt/β-catenin signaling and immune exclusion is increasingly complicated in ovarian cancer and pre-clinical ovarian cancer models. PORCN and DKK1 play differing roles in the Wnt/β-catenin pathway; PORCNi, CGX-1321, inhibits Wnt/β-catenin signaling by impairing Wnt ligand biosynthesis, while DKK1i, DKN-01, promotes Wnt/β-catenin signaling by blocking DKK1 negative regulation. When analyzing Wnt signaling activity, we found that treatment with DKN-01 increased Wnt signaling activity (measured via p-LRP6) (Fig. 3A), confirming results that our group had previously obtained [2]. Since DKK1 is a negative regulator of Wnt signaling, these results confirm that the inhibition of DKK1 activity causes Wnt signaling to go unregulated in ovarian cancer. Treatment with CGX-1321 alone did not decrease Wnt signaling as measured by p-LRP6; we speculate that this is due to the inherent levels of baseline Wnt signaling in the ID8p53−/− model. However, the addition of CGX-1321 to DKN-01 restored Wnt signaling inhibition (Fig. 3A). These results were supported by data obtained analyzing the change in Wnt-related gene expression in omental tumors where CGX-1321 and CGX-1321 + DKN-01 treatments resulted in an overall reduction in Wnt-related gene expression compared to mice treated with DKN-01 (Supplemental Fig. 1A).
Previously, our lab found that CGX-1321 did not decrease tumor burdens in the ID8p53−/− model when given for 14 days [3], but herein we found that lengthening the course of CGX-1321 treatment to 31 days significantly decreased tumor burden, as assessed by both omental weights and ascites volumes (Figs. 2A–B). However, DKN-01 monotherapy was not sufficient to decrease tumor burden in the ID8p53−/− model. This data supports our previous observation where DKN-01 had no effect on tumor growth in the ID8 parental model [2]. Additionally, our data showed that CGX-1321 monotherapy ablated tumor progression in MISIIR-Tag model when given for 8 weeks (Figs. 2C–D). These results imply that CGX-1321 has a more direct anti-tumor effect than DKN-01, which could be explained by DKN-01’s role in immune landscape regulation in the TME. We did not observe the degree of synergism in reducing tumor burden (via omental weight) that was expected when treating with both agents. It is possible that enhanced tumor killing may occur if mice are first treated with DKN-01 to upregulate Wnt signaling, thereby making the tumor more susceptible to treatment with CGX-1321 (Fig. 6).
Fig. 6.
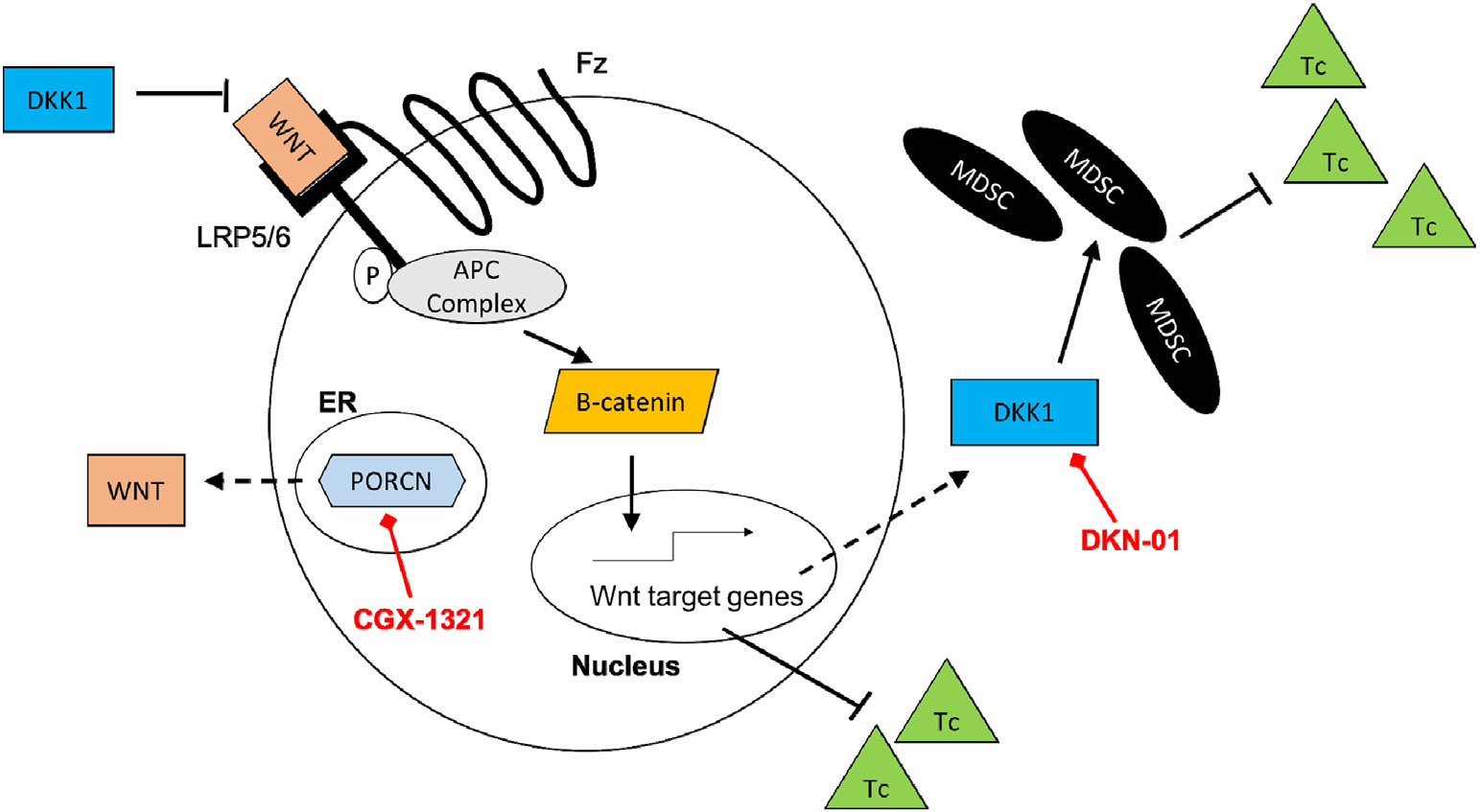
Dual inhibition of PORCN and DKK1 inhibition in ovarian cancer. CGX-1321 inhibits Wnt/β-catenin signaling by impairing Wnt ligand biosynthesis, thus decreasing tumor progression and increasing Tc cell infiltration into the TME. DKN-01 inhibits DKK1 causing a reduction in MDSC infiltration and increasing Tc cell infiltration into the TME. (Tc) cytotoxic T cell; (MDSC) myeloid-derived suppressor cell; (DKK1) dickkopf-1; (PORCN) porcupine; (Fz) frizzled receptor; (LRP5/6) low-density lipoprotein receptor-related protein 5/6; (ER) endoplasmic reticulum.
Since DKN-01 monotherapy had minimal effects on tumor growth, we wanted to investigate its effects on the immune landscape in the omentum TME of the ID8p53−/− model. Previously, our lab found that DKN-01 did not alter the immune landscape in the omentum TME of the ID8 parental model [2]. However, DKK1 over-expression in the ID8 parental model resulted in decreased anti-tumor immune cell infiltration (e.g. NK and CD8+ T cells) in omentum TME, suggesting DKK1 prevents immunosurveillance in ovarian cancer [2]. Additionally, previous reports have shown that down-regulation of β-catenin in MDSCs was necessary for their accumulation in the TME and resultant tumor growth due to their immunosuppressive effects [20,21]. In our current study, we found that DKN-01 did not have any significant effects on gene expression in immune-related pathways (Fig. 4A), but mice treated with DKN-01 had decreased expression of MDSC-related genes (Fig. 4B), with no significant changes in CD8+ T cell infiltration observed (Fig. 4B). Conversely, mice treated with CGX-1321 and CGX-1321 + DKN-01 had an increase in gene expression in immune pathways related to antigen presentation and leukocyte function (Fig. 4A), and significantly higher CD8+ T cell infiltration in the omental TMEs (Fig. 4C). Importantly, this supports the claim by our group, and others, that increased β-catenin levels are associated with T cell exclusion and tumor growth. In addition, these results suggest that each inhibitor is affecting the immune landscape in the TME differently, and further investigation is needed to characterize more immune cell populations and their function in response to treatments. The combination of DKN-01 with anti-PD1 may prove effective in improving the immune response and is an on-going area of research in our group.
Due to the fact that treatment with CGX-1321 promotes CD8+ T cell infiltration into the omentum TME, we wanted to determine if this effect would sensitize the tumors to ICB therapy. When investigating ICB therapy, anti-PD-1, in combination with CGX-1321, our results were consistent with single agent anti-PD-L1 therapy in the clinic, finding that anti-PD-1 therapy did not decrease ovarian tumor burden in the ID8p53−/− model (Figs. 5A–B). While CGX-1321 addition to anti-PD-1 therapy decreased tumor burden compared to anti-PD-1 monotherapy, the combination was no better than CGX-1321 monotherapy (Figs. 5A–B). Similar to our results seen with DKN-01, CGX-1321 and CGX-1321 + anti-PD-1 treatment regimens caused an overall reduction in Wnt-related gene expression as compared to vehicle/control- and anti-PD-1-treated mice (Supplemental Fig. 1B).
One thing to consider in this study is only a single treatment regimen investigating the effects of CGX-1321 + anti-PD-1 therapy was tested, with treatments starting simultaneously. It has been established that a positive response to anti-PD-1 therapy requires a robust adaptive immune response within the TME at baseline [22], which is generally not the case in ovarian cancers. Moving forward, prior treatment with CGX-1321 might be required to provide an adaptive immune response to then sensitize tumors to anti-PD-1 therapy by enhancing CD8+ T cell infiltration within the tumor.
While the addition of CGX-1321 to anti-PD-1 therapy did not affect CD8+ T cell infiltration (Fig. 5D), the percentage of B cells (Fig. 5E) in omentum TME was further decreased when CGX-1321 was combined with anti-PD-1 therapy. To date, the data regarding the role of B cells in gynecologic cancers is extremely limited. In ovarian cancer, both pro- and anti-tumor B cell responses have been identified [23]. Specifically, a subset of regulatory B cells (Bregs) has been found to be immunosuppressive by secreting cytokines that convert cytotoxic T (Tc) cells to Tregs [24], and increased levels of these Bregs is negatively associated with patient survival [25]. Our data regarding B cells is limited given we did not utilize more specific markers to further distinguish specific B cell populations within our tumors, which will be critical in further studies to elucidate the contributions of B cells to tumor immunity in gynecologic cancers.
Prior to initiating our study, we anticipated to see enhanced tumor cell death and increased anti-tumor immune infiltration and function when CGX-1321 was combined with either DKN-01 and/or anti-PD-1 therapy. We found that the combination therapies did not enhance the anti-tumor effects of CGX-1321 monotherapy. Importantly, this was the first study to investigate PORCN and DKK1 inhibition in the context of ovarian cancer, and more in-depth investigation is needed to further characterize immune landscape in response to these treatments. Moving forward, we consider that the simultaneous inhibition of PORCN and DKK1 prior to ICB therapy (sequential therapy), will better sensitize ovarian tumors to this therapeutic regimen resulting in the most robust anti-tumor effects.
Supplementary Material
HIGHLIGHTS.
DKN-01 suppresses MDSC gene signature.
CGX-1321 increases CD8+ T cell infiltration into the omentum TME.
Neither DKN-01 nor anti-PD-1 monotherapies enhanced the anti-tumor effects of CGX-1321.
Funding
This work was supported by grants from DOD (W81XWH-18–1-0231 - RCA) and NCI (T32CA183926 - WJT).
Footnotes
Declaration of Competing Interest
RCA has participated in Advisory Boards for Leap Therapeutics, AstraZeneca, GSK, Merck, VBL Therapeutics, and Caris Life Sciences.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygyno.2020.10.031.
References
- [1].Galon J, Bruni D, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies, Nat. Rev. Drug Discov. 18 (3) (2019) 197–218. [DOI] [PubMed] [Google Scholar]
- [2].Betella I, Turbitt WJ, Szul T, Wu B, Martinez A, Katre A, et al. , Wnt signaling modulator DKK1 as an immunotherapeutic target in ovarian cancer, Gynecol. Oncol. 157 (3) (2020) 765–774. [DOI] [PubMed] [Google Scholar]
- [3].Goldsberry WN, Meza-Perez S, Londono AI, Katre AA, Mott BT, Roane BM, et al. , Inhibiting WNT ligand production for improved immune recognition in the ovarian tumor microenvironment, Cancers 12 (3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nusse R, Clevers H, Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities, Cell 169 (6) (2017) 985–999. [DOI] [PubMed] [Google Scholar]
- [5].Wang B, Tian T, Kalland KH, Ke X, Qu Y, Targeting Wnt/beta-catenin signaling for Cancer immunotherapy, Trends Pharmacol. Sci. 39 (7) (2018) 648–658. [DOI] [PubMed] [Google Scholar]
- [6].Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF, WNT/beta-catenin pathway activation correlates with immune exclusion across human cancers, Clin. Cancer Res. 25 (10) (2019) 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arend RC, Londono-Joshi AI, Samant RS, Li Y, Conner M, Hidalgo B, et al. , Inhibition of Wnt/beta-catenin pathway by niclosamide: a therapeutic target for ovarian cancer, Gynecol. Oncol. 134 (1) (2014) 112–120. [DOI] [PubMed] [Google Scholar]
- [8].Boone JD, Arend RC, Johnston BE, Cooper SJ, Gilchrist SA, Oelschlager DK, et al. , Targeting the Wnt/beta-catenin pathway in primary ovarian cancer with the porcupine inhibitor WNT974, Lab. Investig. 96 (2) (2016) 249–259. [DOI] [PubMed] [Google Scholar]
- [9].Doo DW, Meza-Perez S, Londono AI, Goldsberry WN, Katre AA, Boone JD, et al. , Inhibition of the Wnt/beta-catenin pathway enhances antitumor immunity in ovarian cancer, Ther. Adv. Med. Oncol. 12 (2020) 1758835920913798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhong Z, Sepramaniam S, Chew XH, Wood K, Lee MA, Madan B, et al. , PORCN inhibition synergizes with PI3K/mTOR inhibition in Wnt-addicted cancers, Oncogene. 38 (40) (2019) 6662–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, et al. , LDL-receptor-related protein 6 is a receptor for Dickkopf proteins, Nature. 411 (6835) (2001) 321–325. [DOI] [PubMed] [Google Scholar]
- [12].Jiang T, Huang L, Zhang S, DKK-1 in serum as a clinical and prognostic factor in patients with cervical cancer, Int. J. Biol. Markers 28 (2) (2013) 221–225. [DOI] [PubMed] [Google Scholar]
- [13].Shizhuo W, Tao J, Shulan Z, Bing Z, The expression and significance of Dickkopf-1 in epithelial ovarian carcinoma, Int. J. Biol. Markers 24 (3) (2009) 165–170. [DOI] [PubMed] [Google Scholar]
- [14].Kagey MH, He X, Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology, Br. J. Pharmacol. 174 (24) (2017) 4637–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. , Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer, Cancer Res. 63 (6) (2003) 1389–1397. [PubMed] [Google Scholar]
- [16].Kurman RJ, Shih Ie M, The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory, Am. J. Surg. Pathol. 34 (3) (2010) 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B, et al. , Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling, BMC Genomics 15 (2014) 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. , Targeting YAP-dependent MDSC infiltration impairs tumor progression, Cancer Discov. 6 (1) (2016) 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. , Cold tumors: a therapeutic challenge for immunotherapy, Front. Immunol. 10 (2019) 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Capietto AH, Kim S, Sanford DE, Linehan DC, Hikida M, Kumosaki T, et al. , Downregulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer, J. Exp. Med. 210 (11) (2013) 2257–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].D’Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. , Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer, J. Exp. Med. 213 (5) (2016) 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ, Resistance to PD1/PDL1 checkpoint inhibition, Cancer Treat. Rev. 52 (2017) 71–81. [DOI] [PubMed] [Google Scholar]
- [23].Gupta P, Chen C, Chaluvally-Raghavan P, Pradeep S, B cells as an immune-regulatory signature in ovarian cancer, Cancers (Basel) 11 (7) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rodriguez GM, Galpin KJC, McCloskey CW, Vanderhyden BC, The tumor microenvironment of epithelial ovarian cancer and its influence on response to immunotherapy, Cancers (Basel) 10 (8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lundgren S, Berntsson J, Nodin B, Micke P, Jirstrom K, Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer, J. Ovarian Res. 9 (2016) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.